

Abstract
Background
Chronic kidney disease (CKD) is characterized by increased myocardial mass despite near-normal blood pressure suggesting the presence of a separate trigger. A potential driver is signal regulatory protein alpha (SIRPα), a mediator impairing insulin signaling. The objective of this study is to assess the role of circulating SIRPα in CKD-induced adverse cardiac remodeling.
Methods
SIRPα expression was evaluated in mouse models and patients with CKD. Specifically, global (Mt), muscle (m) or cardiac muscle-specific (cs) SIRPα knockout (KO) mice were examined after subtotal nephrectomy. Cardiac function was assessed by echocardiography. Metabolic responses were confirmed in cultured muscle cells or cardiomyocytes.
Results
We demonstrate that SIRPα regulates myocardial insulin/IGF1 receptor signaling in CKD. First, in the serum of both mice and patients, SIRPα was robustly secreted in response to CKD. Secondly, cardiac muscle upregulation of SIRPα was associated with impaired insulin/IGF1 receptor signaling, myocardial dysfunction and fibrosis. However, both global and csSIRPα KO mice displayed improved cardiac function when compared to control mice with CKD. Thirdly, both m- or csSIRPα KO mice did not significantly activate fetal genes and maintained insulin/IGF1 receptor signaling with suppressed fibrosis despite the presence of CKD. Importantly, SIRPα directly interacted with the IGF1R. Next, recombinant SIRPα protein was introduced into mSIRPα KO mice re-establishing the insulin/IGF1 receptor signaling activity. Additionally, overexpression of SIRPα in myoblasts and cardiomyocytes impaired pAKT and insulin/IGF1 receptor signaling. Furthermore, myotubes and cardiomyocytes, but not adipocytes treated with high glucose, or cardiomyocytes treated with uremic toxins stimulated secretion of SIRPα in culture media, suggesting these cells are the origin of circulating SIRPα in CKD. Both intracellular and extracellular SIRPα exert biologically synergistic effects impairing intracellular myocardial insulin/IGF1 receptor signaling.
Conclusions
Myokine SIRPα expression impairs insulin/IGF1 receptor functions in cardiac muscle, affecting cardiometabolic signaling pathways. Circulating SIRPα constitutes an important readout of insulin resistance in CKD-induced cardiomyopathy.
Subject Terms: Nephrology and Kidney, Remodeling, Cell Signaling/Signal Transduction, Mechanisms, Metabolism, Cardiomyopathy, Cardiorenal Syndrome, Heart Failure, Hypertrophy
Keywords: Insulin resistance, chronic kidney disease, uremic cardiomyopathy, SIRPα
INTRODUCTION
Mortality associated with chronic kidney disease (CKD) is higher than that of the general population and worsens with advancing stages of the disease 1. Based on the Second National Health and Nutrition Examination Survey (NHANES II) cardiovascular causes attributed to a 51 percent increase risk of death when glomerular filtration rates fell below 70 mL/min/1.73m2 1. In fact, cardiovascular clinical trials often exclude patients with CKD 2. The pathologic mechanisms responsible for CKD-induced cardiomyopathy include hypertension, volume overload, anemia, abnormalities in mineral metabolism, and fibroblast growth factor 23 (FGF23) 3. In early kidney disease with near-normal blood pressure, there is evidence of increased myocardial mass even without significant changes in blood pressure 4, 5. We postulate that cardiac remodeling occurring in CKD may be the result of impairments in insulin/insulin growth factor-1 (IGF1R) receptor signaling pathways.
Insulin resistance is a critical feature of pathologic cardiac remodeling in patients with hypertrophic and dilated cardiomyopathy 6–9. The triggers of CKD-induced insulin resistance in the myocardium are not known. A potentially relevant protein implicated in this process is signal regulatory protein-alpha (SIRPα). We previously uncovered that upregulation of SIRPα is a mechanism responsible for CKD-induced insulin resistance in skeletal muscle 10. CKD stimulates inflammatory cytokines including NF-κB activation to induce SIRPα expression in skeletal muscles of mice with CKD 10. SIRPα is a transmembrane glycoprotein containing three extracellular immunoglobulin-like domains and a cytoplasmic region with src homology-2 (SH-2) binding motifs 11. SIRPα has been identified as a docking protein for tyrosine phosphatases (i.e. SHP1–2) 11, promoting insulin resistance in skeletal muscles and adipose tissues, while inducing cachexia in CKD 12. SIRPα regulation as a myokine with systemic responses to distant organs remains unclear. Myokines are either autocrine or paracrine in function regulating whole-body metabolic processes 13, 14. This suggests a central role of the muscle in the interorgan regulation of insulin responsiveness and energy homeostasis. In fact, uremic toxins directly influence insulin resistance in subtotal nephrectomy models of CKD or patients with advanced CKD 15, 16. In advanced CKD with evidence of insulin resistance, myokine SIRPα release occurs in response to CKD-induced cachexia 12.
Here we have examined in vivo and in vitro evidence to determine that impaired insulin/IGF1 receptor signaling contributes to CKD-induced cardiomyopathy. We hypothesized that SIRPα behaves as a myokine, specifically an anti-insulin/IGF1 receptor factor, contributing to adverse cardiac remodeling in CKD. The hypothesis is derived from the fact that the growth hormone/IGF1 axis is thought to be one of the causes of sarcopenia in CKD 17. Therefore, we evaluated circumstances responsible for release of SIRPα in CKD and its impact on myocardial intracellular insulin/IGF1 receptor signaling. To test this hypothesis, we utilized an insulin sensitive model (acute exercise) and a model of insulin resistance and cachexia (CKD). Additionally, muscle-specific SIRPα knockout (KO) mice were exposed to recombinant (r) SIRPα to assess for recapitulation of CKD-specific intracellular signaling changes in cardiac muscle. These findings suggest that SIRPα behaves as a myokine to antagonize myocardial insulin/IGF1 receptor signaling while stimulating CKD-induced cardiomyopathy.
METHODS
Data Availability.
The authors declare that all supporting data are available within the Online Data Supplement. The supporting data are also available from the corresponding author upon reasonable request.
Detailed Materials and Methods are available in the Online Data Supplement.
Transgenic mice
SIRPα Mt mice (exons 7 and 8 were replaced with a neomycin selection cassette) were obtained from Riken (Saitama, Japan) and backcrossed over five generations on C57BL/6 background. SIRPαfl/fl mice were obtained in conjunction with the BaSH/EUCOMM. Skeletal muscle-specific SIRPα KO (mSIRPα−/−), cardiac-myocyte specific SIRPα KO mice (csSIRPα−/−), and adipose-specific SIRPα KO mice (AD-SIRPα−/−) with deletion of exons 3 and 4 were generated using a Cre (muscle creatine kinase-Cre or Myh6 also known as αMyHC-Cre or adipoq-Cre respectively) mice from Jackson Laboratory (Bar Harbor, ME, USA) recombinase:loxP system as previously described 18; all of these mice were on C57BL/6 background and appeared to have a grossly normal phenotype. Mice were maintained in 12 h light/dark cycles (6 a.m. to 6 p.m.) at 24°C and fed diets of standard rodent chow.
CKD model
To generate CKD model 19, male mice at eight to ten weeks of age were subjected to a subtotal nephrectomy for 12–16 weeks.
Doppler ultrasound
Transthoracic echocardiography with pulse wave and tissue Doppler imaging was performed on male mice 10–12 weeks after subtotal nephrectomy under isoflurane anesthesia using a VisualSonics Vevo 3100 platform with a MX550D probe 20. Subsequent analyses were performed by an experienced sonographer who was blinded to the type of mouse model.
Voluntary wheel exercise model
To generate exercise model, mice were randomly assigned to individual cages containing either 4-inch exercise wheels (exercise group) or infrared (sedentary group) sensors, to measure voluntary exercise and basal activity respectively.
Hyperglycemia model
For hyperglycemia, mice received high-glucose treatment (2 g/kg, 3 repeats at 40-min intervals) 21.
Octet RED384 assay development
The binding affinity between IGF1R immunoprecipitated lysates and purified recombinant SIRPα protein (rSIRPα) was determined based on the association rate (ka) and the dissociation rate (kdis) constants using an Octet RED384 System (Fremont, CA, USA).
Study Approval
All procedures of experimental animals in protocols were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine and conformed to the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Human studies
The procedures for human samples were approved by the Ethics Committee of the Department of Internal Medicine of the University of Genoa, in accordance with the Declaration of Helsinki regarding ethics of human research. Serum samples from healthy controls were obtained from anonymous blood donors or from CKD patients prior to peritoneal dialysis catheter placement. Before the patients' participation, the nature, purpose, and risks of the study were reviewed with all of the participants and their voluntary consent was obtained. The detail characteristics of patients with CKD are available (Table S1).
Statistical analysis
Values are expressed as means ± SEM, unless otherwise specified. Statistical analyses were performed using two-tailed unpaired Student’s t-test for data from 2 groups. Differences in >2 groups were analyzed using one-way ANOVA followed by Bonferroni multiple comparisons test, unless otherwise specified in figure legends. A p < 0.05 was considered statistically significant. GraphPad Prism software was used for statistical analysis.
RESULTS
CKD-Induces SIRPα Expression in Both Cardiac Muscle and Serum
Suppressing SIRPα not only improves insulin signaling but also prevents skeletal muscle loss10,12. Therefore, to determine the physiologic impact of SIRPα in the myocardium, mice were subjected to acute exercise, an insulin sensitive model. After 6 days of voluntary exercise, the hearts of these mice were harvested. Transcription of SIRPα was suppressed in these exercised mice (Figure S1A). While immunoblots of exercised mice revealed improved insulin sensitivity (i.e. upregulation of pAKT, PI3K, and GLUT4) with myocardial suppression of SIRPα (Figure S1B). These results promoted further evaluations for the role of SIRPα in pathologic conditions.
SIRPα impacts survival in CKD, specifically global SIRPα knockout (KO) or mutant (Mt) mice exhibited a significant improvement in survival despite the presence of CKD 12. Therefore, we characterized cardiovascular responses of wild type (WT) mice with CKD and compared them to SIRPα Mt mice with CKD. After subtotal nephrectomy, WT and SIRPα Mt mice had similar serum creatinine and blood urea nitrogen (BUN) levels (2–3-fold higher; Figure 1A). Additionally, mice subjected to subtotal nephrectomy exhibit evidence of metabolic acidosis, increased parathyroid hormone levels, and muscle wasting 10, 12 which classically occurs in patients with advanced CKD 22 . Notably, in WT mice with CKD, SIRPα expression was markedly increased in ventricular cardiac muscle rising to 3.6-fold higher than values measured in hearts of WT, sham-operated, control mice (Figure 1B). Additionally, increased myocardial SIRPα expression was noted in WT mice with CKD via immunohistochemistry (Figure 1C). Likewise, serum SIRPα was identified in WT mice with CKD (Figure 1D). Importantly, a robust SIRPα release was detected in the serum of patients with advanced CKD (Figure 1E, Table S1). Thus, both circulating and endogenous myocardial SIRPα expression were noted in response to CKD.
Figure 1. CKD induces myocardial SIRPα expression and release into serum.
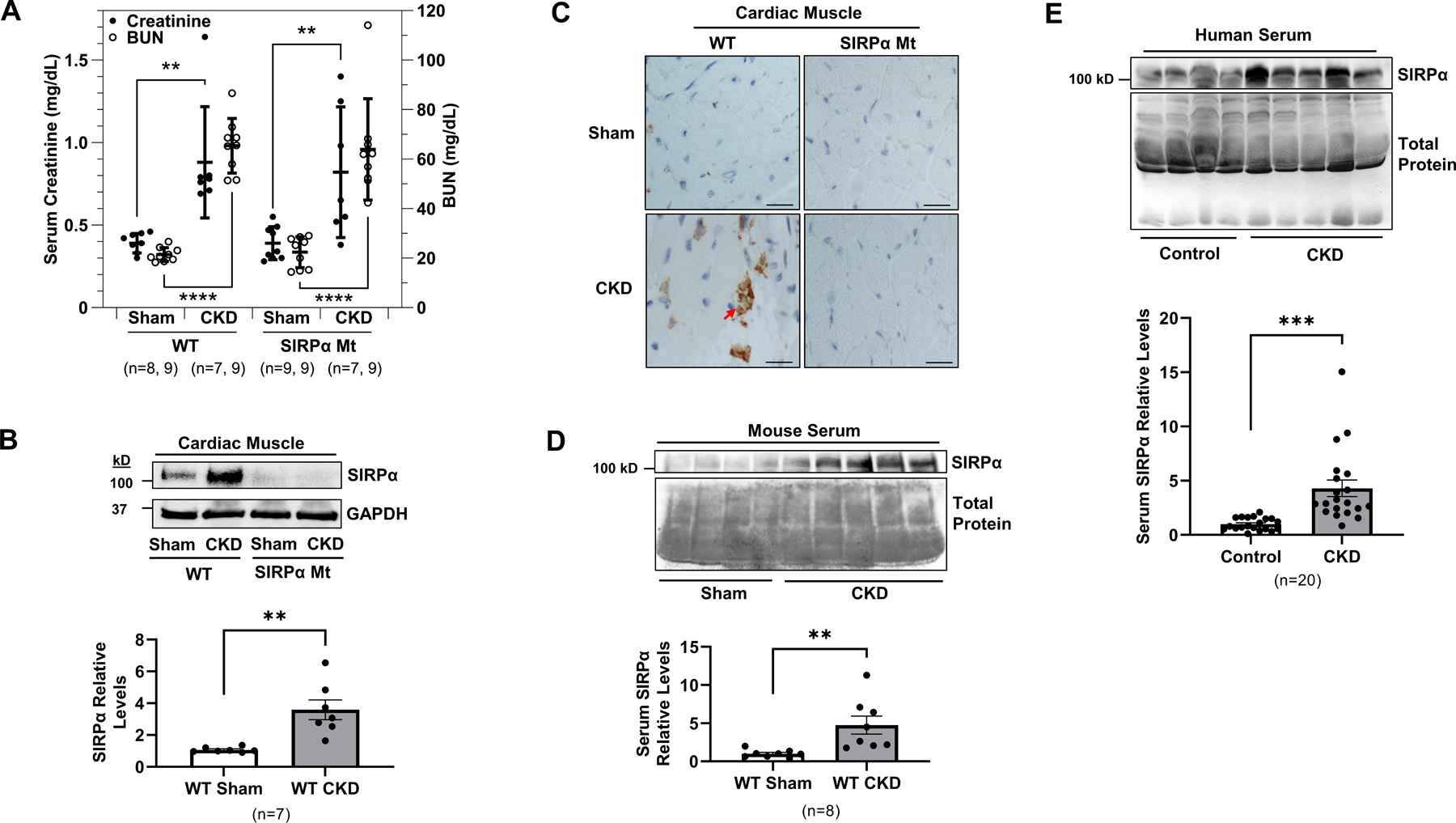
(A) Blood urea nitrogen (BUN) and creatinine were measured at 10–12 weeks after subtotal nephrectomy or sham operation in WT and SIRPα Mt mice with or without CKD. (B) At week 16 after subtotal nephrectomy, protein lysates of hearts were immunoblotted to detect SIRPα and GAPDH and representative immunoblots of averaged data (top panel) with relative densities of GAPDH (bottom panel) are shown. (C) Immunohistochemistry staining of antibody against SIRPα (arrows) from heart sections of WT and SIRPα Mt with or without CKD (scale bar =25 µm) and representative images of averaged data are shown. (D) Serum from Sham and CKD mice were immunoblotted for SIRPα and representative immunoblots of averaged data (top panel), with relative densities to total protein (bottom panel) are shown. (E) Serum was obtained from age-matched, healthy control subjects and patients with advanced CKD to detect circulating serum levels of SIRPα, representative immunoblots of averaged data (top panel), with relative densities to total protein (bottom panel) are shown. Statistical significance was calculated using one-way ANOVA with Bonferroni's multiple comparisons test (A) and unpaired two-tailed Studen's t-test (B, D-E). Values are means ± SEM. ** p<0.01, *** p<0.001, **** p<0.0001.
Absence of SIRPα Prevents Cardiac Dysfunction in CKD
Next, we examined cardiac function of SIRPα Mt mice which were compared to WT mice with CKD using evaluations of in vivo M-mode and Doppler echocardiography (Figure 2A, Table S2). WT mice with CKD displayed impaired left ventricular systolic function, with reduced ejection fraction (EF%; Figure 2B), reduced fractional shortening (FS%; Figure 2C), and impaired calculated cardiac output (CO mL/min/mm; Figure 2D). Conversely, hearts from SIRPα Mt mice with CKD displayed preserved left ventricular systolic function, FS%, and CO (Figure 2B–D). In addition, Doppler analysis revealed significantly suppressed E/A ratios in WT mice with CKD when compared to sham control mice (Table S2). However, no significant differences were noted in E/A ratios in SIRPα Mt mice despite the presence of CKD when compared to their respective sham control mice (Table S2). Additionally, no significant differences were noted in myocardial perfusion indices (MPI) or E/e’. Therefore, global SIRPα Mt mice with CKD were protected from myocardial systolic dysfunction despite the presence of CKD.
Figure 2. Absence of SIRPα prevents cardiac dysfunction in CKD.
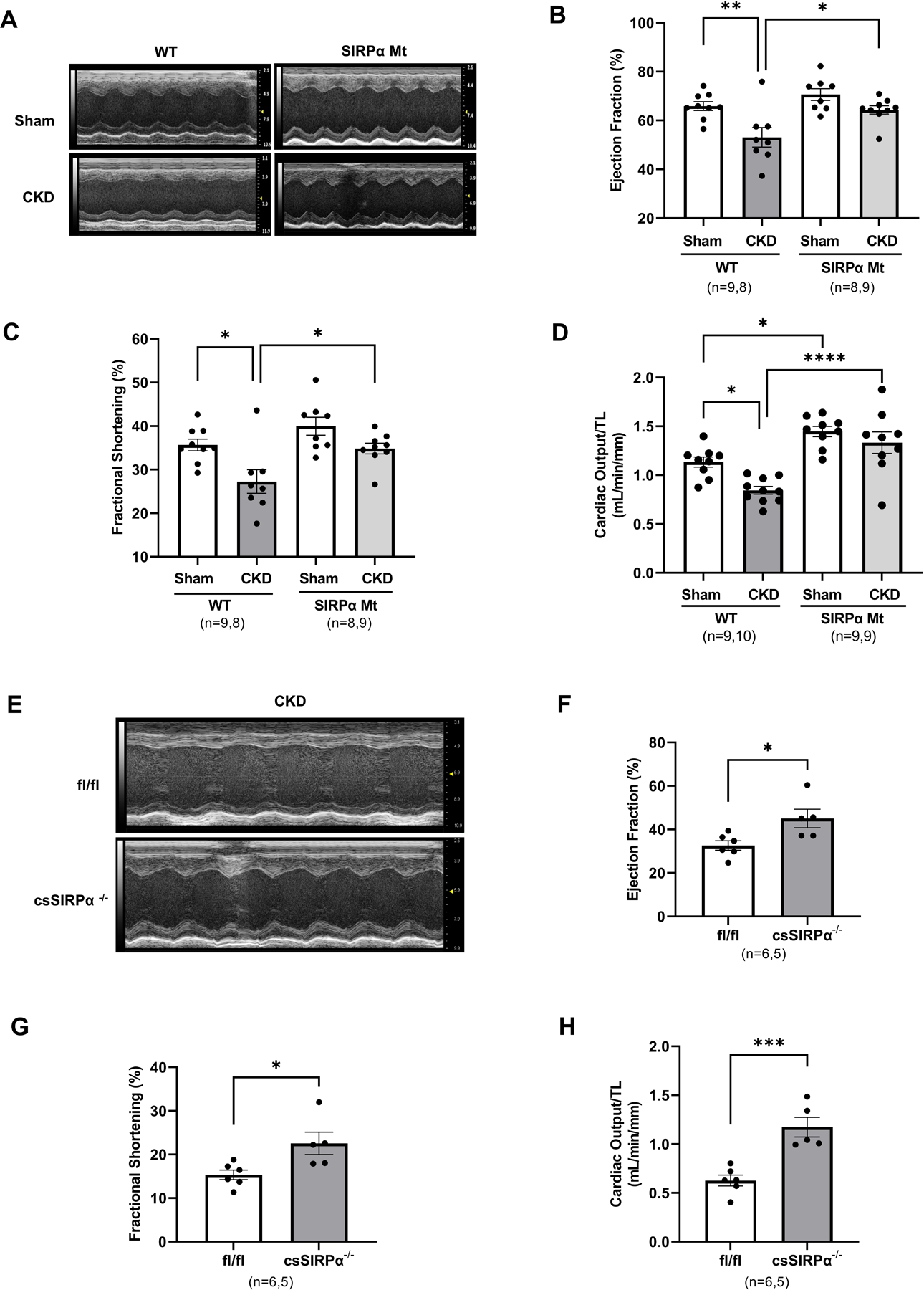
(A) At weeks 10–12 after subtotal nephrectomy on normal chow, hearts functions were evaluated with Doppler echocardiography and M-mode and representative images of the left ventricle are shown, and (B) ejection fraction %, (C) fractional shortening %, (D) cardiac output normalized to tibia length (mL/min/mm) were compared in WT and SIRPα Mt mice with or without CKD. After 10 weeks of subtotal nephrectomy on normal chow, cardiac-specific SIRPα KO mice (csSIRPα−/−) were fed a 40% high protein diet for 10 d. (E) Cardiac functions were evaluated by M-mode and representative images of the left ventricles are shown, and (F) ejection fraction %, (G) fractional shortening %, and (H) cardiac output normalized to tibia length (mL/min/mm) in SIRPα fl/fl and csSIRPα−/− mice with CKD were compared. Statistical significance was calculated using one-way ANOVA with Bonferroni's multiple comparisons test (B-D) and unpaired two-tailed Studen's t-test (F-H). Values are expressed as means ± SEM; *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
To determine myocardial-specific effects of suppressing SIRPα in CKD, cardiac-myocyte specific KO (csSIRPα−/−) mice were created and subjected to subtotal nephrectomy and compared to flox mice with CKD. In vivo M-mode echocardiography imaging was utilized to determine myocardial function (Figure 2E). csSIRPα−/− mice with CKD displayed improved EF% (Figure 2F), FS % (Figure 2G), and CO when compared to flox littermate control mice with CKD (Figure 2H, Table S3). These results suggest that the cardioprotection from CKD observed in the whole body SIRPα KO is, at least in part, due to myocardial suppression of SIRPα.
Suppressing SIRPα Prevents Pathologic Cardiac Hypertrophy
Next, we harvested hearts from WT and SIRPα Mt mice to evaluate cardiac remodeling in response to CKD. Hearts from WT mice with CKD displayed evidence of hypertrophy with increased myocardial weights when normalized to tibia length (Figure 3A) when compared to sham control mice. In fact, cardiomyocyte size was increased in WT mice with CKD (Figure 3B). However, in SIRPα Mt mice with CKD, neither heart weights nor cardiomyocyte size were significantly increased when compared to their respective sham control mice (Figure 3A–B). Additionally, SIRPα Mt sham mice displayed evidence of hypertrophy with a significant increase in cardiomyocyte size and cardiac output, when compared to WT sham mice (Figure 3A–B and Figure 2D). We also evaluated changes in systolic blood pressure (SBP) and heart rate (HR). WT mice with CKD displayed a significant difference in SBP (Figure 3C) and HR (Figure S2A), when compared to WT sham control mice and increased SBP when compared to SIRPα Mt mice with CKD (Figure 3C). Furthermore, in order to determine muscle-specific effects, both Mck-Cre or αMyHC (Myh6)-Cre SIRPα−/−, (mSIRPα−/− or csSIRPα−/− respectively) mice were subjected to CKD with similar BUN and creatinine levels in csSIRPα−/− mice when compared to SIRPαfl/fl with CKD (Figure S2B). Flox mice with CKD presented with higher SBP (Figure 3C) and increased heart weight normalized to tibia length (Figure 3D) but no differences in heart rate when compared to sham control mice or csSIRPα−/− mice with CKD (Figure S2A). Littermate flox sham mice and csSIRPα−/− sham mice displayed similar harvest heart weights (Figure 3D). Also, csSIRPα−/− mice with CKD did not display significant evidence of hypertrophy or changes in myocardial weight despite elevations in SBP and heart rate (which was similar in mSIRPα −/− mice with CKD) when compared to sham control mice (Figure 3D and Figure S2A). However, flox mice with CKD displayed significant evidence of cardiac hypertrophy with increased myocardial weight when compared to flox sham mice, csSIRPα−/− mice with CKD, or mSIRPα−/− mice with CKD (Figure 3D). These data indicate an important impact of SIRPα on myocardial mass.
Figure 3. Suppressing SIRPα prevents pathologic hypertrophy and inhibits fetal gene expression.
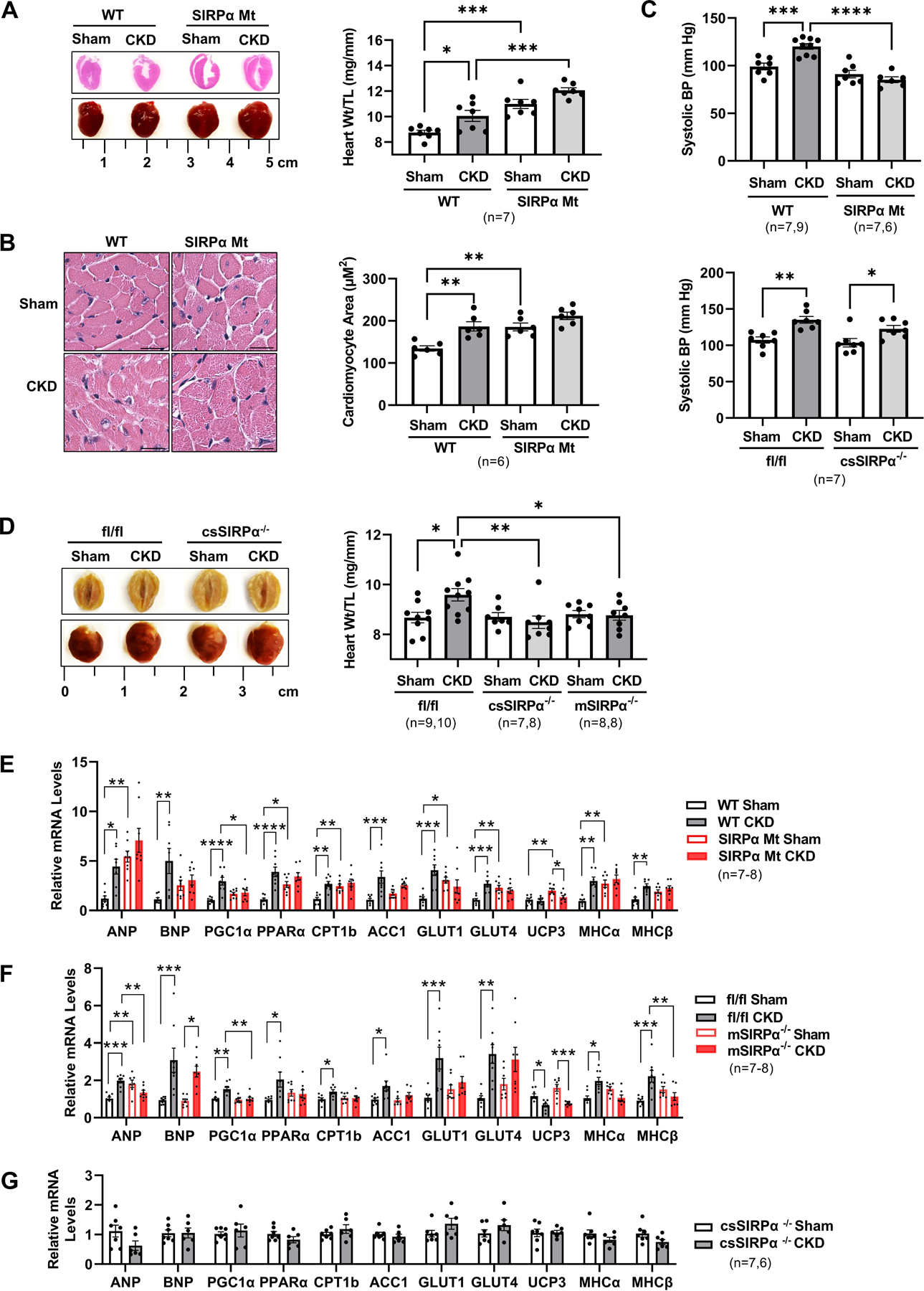
(A) Representative images of hearts and photomicrographs by H&E staining from WT and SIRPα Mt with or without CKD (left panel). The ratio of heart weight to tibia length (TL) were measured (right panel). (B) H&E staining of cardiomyocytes and representative images of averaged data (left panel, scale bar=25 µm) with measured cardiomyocyte area of WT and SIRPα Mt with or without CKD (right panel) are shown. (C) After subtotal nephrectomy (post-surgery weeks 6–9), systolic blood pressure (BP) from WT and SIRPα Mt (top panel) or SIRPαfl/fl and cardiac-specific SIRPα KO mice (csSIRPα−/−, post-surgery weeks 12–14) (bottom panel) were measured. (D) Representative images of averaged data are shown of SIRPαfl/fl and csSIRPα−/− with or without CKD (left panel) and muscle-specific KO (mSIRPα−/−) with or without CKD are compared and the ratio of heart weights to tibia length (TL) (right panel) are shown. (E-G) Relative mRNA levels of ANP, BNP and fetal genes were determined by quantitative real time-PCR analysis and normalized relative to cyclophilin. Statistical significance was calculated using one-way ANOVA with Bonferroni's multiple comparisons test (A-F) and unpaired two-tailed Studen's t-test (G). Values are expressed as means ± SEM; *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Return to the fetal gene program is a hallmark heralding a decline in cardiac function 23. Little is known about the fetal gene program responses in CKD-induced cardiomyopathy. We found evidence for activation of fetal genes in WT mice with CKD which was suppressed in SIRPα Mt mice with CKD when compared to their respective sham control mice. Specifically, natriuretic peptides, ANP and BNP, were stimulated in response to CKD in WT mice. However, in SIRPα Mt mice with CKD displayed no significant upregulation of ANP and BNP when compared to their respective controls (Figure 3E). Next, we analyzed if there was a muscle-specific effect on the fetal gene program in response to CKD. Muscle-specific mSIRPα−/− mice with CKD presented with similar fetal gene expression when compared to its respective sham control with the exception of BNP and UCP3 (Figure 3F). Similarly, csSIRPα−/− mice with CKD revealed no significant increases in ANP, BNP or most other fetal genes when compared to their respective controls (Figure 3G). These results suggest that SIRPα knockdown plays a critical role in suppressing fetal gene activation in response to CKD.
SIRPα Suppression Attenuates CKD-induced Myocardial Fibrosis
Myocardial biopsies and necropsies of patients with CKD reveal perivascular and interstitial fibrosis 24. To evaluate the effects of SIRPα on myocardial collagen deposition, picrosirius red staining of ventricular cardiac muscle was performed. In WT mice with CKD, the collagen deposition area was nearly 3-fold higher when compared to their sham control. However, in SIRPα Mt mice there was no significant increase in myocardial fibrosis despite the presence of CKD (Figure 4A). Furthermore, immunoblotting analysis of extracellular matrix proteins (ECM) in cardiac muscle revealed an increased expression of α-SMA and fibronectin in WT mice with CKD. Additionally, PI3 kinase (PI3K) was downregulated while pAKT was activated in WT CKD mice (Figure 4B). SIRPα Mt mice with CKD displayed similar protein levels of fibronectin, αSMA, PI3K and pAKT when compared to their respective sham control mice (Figure 4B). However, since SIRPα Mt sham mice fibronectin levels were elevated when compared to WT sham mice, therefore, we evaluated littermate flox mice and compared them to mSIRPα−/− mice. Additionally, csSIRPα−/− mice with or without CKD were evaluated.
Figure 4. Suppressing SIRPα prevents cardiac fibrosis.
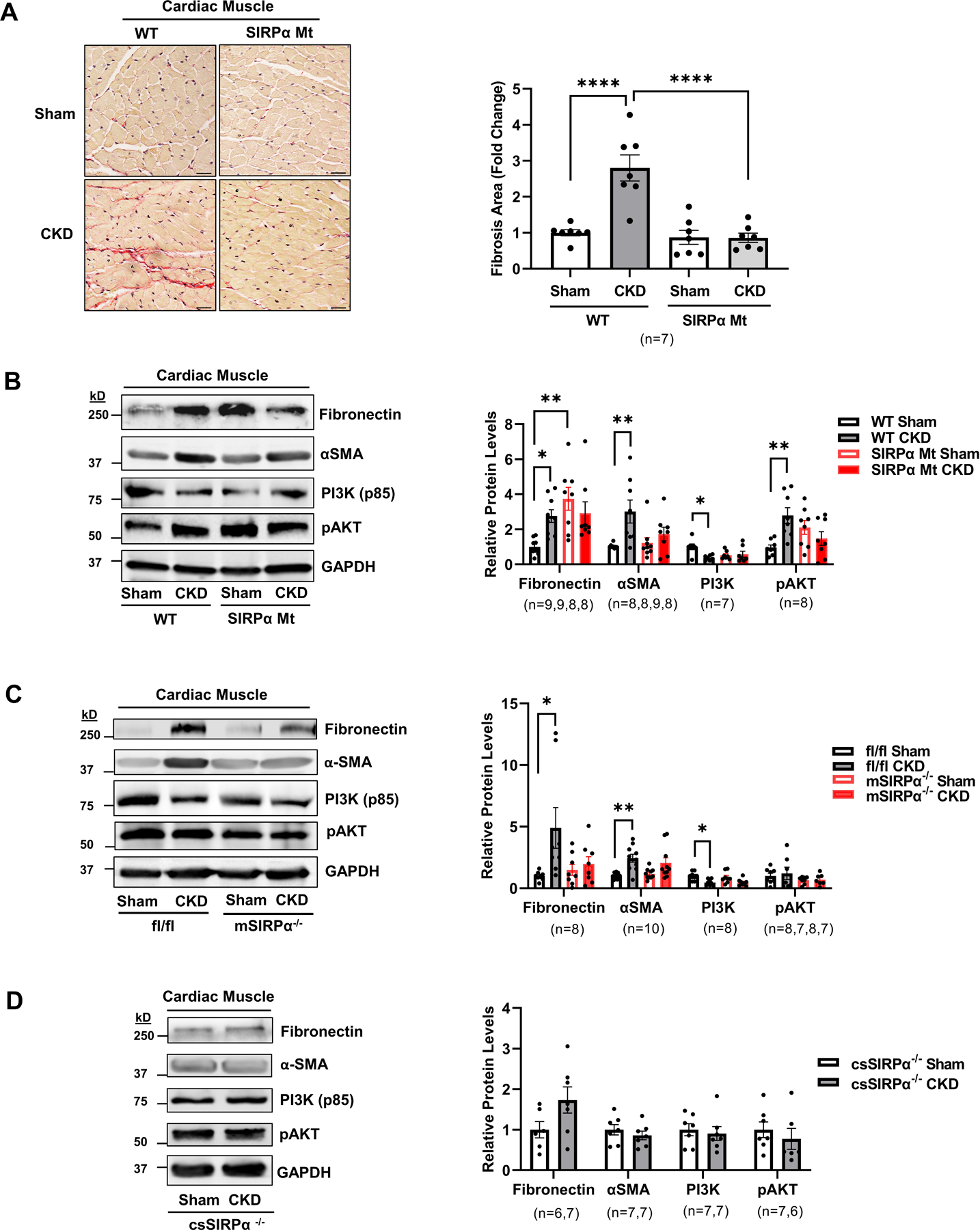
(A) Picrosirius red staining of myocardial sections for fibrosis which include representative images of averaged data are shown (left panel, scale bar=25 µm) with the fold change for fibrosis area which were analyzed in the WT and SIRPα Mt with or without CKD (right panel). After subtotal nephrectomy, heart lysates of (B) WT vs. SIRPα Mt with or without CKD, (C) muscle-specific KO (mSIRPα−/−) vs. flox (SIRPαfl/fl) with or without CKD or (D) cardiac-specific KO (csSIRPα−/−) mice with or without CKD were immunoblotted to detect fibronectin, α-SMA, PI3K (p85), and pAKT and representative immunoblots of averaged data (left panel) with relative densities to GAPDH (right panel) are shown. Statistical significance was calculated using one-way ANOVA with Bonferroni's multiple comparisons test (A-C) and unpaired two-tailed Studen's t-test (D). Values are expressed as means ± SEM; *p<0.05, ** p<0.01, **** p<0.0001.
Skeletal and Cardiac Muscle-Specific Suppression of SIRPα Improves Insulin Signaling and Myocardial Fibrosis Despite the Presence of CKD
In littermate flox vs. mSIRPα−/− sham mice there were no differences detected in insulin signaling, fibronectin and αSMA (Figure 4C). In SIRPαfl/fl mice with CKD, PI3K was significantly downregulated. Meanwhile extracellular matrix proteins, αSMA and fibronectin, were upregulated in these mice (Figure 4C). In both mSIRPα−/− and csSIRPα−/− mice with CKD, PI3K and pAKT were similar to their sham control. Additionally, both αSMA and fibronectin, were not stimulated in mSIRPα−/− or csSIRPα−/− mice in response to CKD (Figure 4C–D). These results indicate suppression of SIRPα improves myocardial intracellular insulin signaling while preventing myocardial fibrosis in CKD.
SIRPα Interacts with the IGF1R Impairing its Signaling in Cardiac Muscle
Since SIRPα interacts with mediators of insulin receptor signaling in skeletal muscles 10, we evaluated SIRPα interactions with the myocardial IGF1R. Specifically, we determined that flox mice with CKD displayed reduced myocardial levels of tyrosine phosphorylation of IGF1R. However, mSIRPα−/− mice with CKD displayed no significant change in IGF1R tyrosine phosphorylation despite the presence of CKD (Figure 5A). Next, we examined the interactions of these proteins by immunoprecipitation analysis. Coimmunoprecipitation assays revealed an interaction between SIRPα with total IGF1R (Figure 5B). To further validate this interaction between IGF1R and SIRPα, Octet RED384 systems (bio-layer interferometry) was utilized to assess protein quantities and characterization of kinetics. Specifically, IGF1R was immunoprecipitated (Figure S3A) from cardiac muscle of flox CKD mice and the binding kinetics of Fc-tagged-rSIRPα to the IGF1R was identified via bio-layer interferometry. We concluded that rSIRPα was bound to immunoprecipitated IGF1R with a kD of 147 μM (Figure 5C, Table S4). Thus, these results indicate that extracellular SIRPα binding to myocardial IGF1R occurs with a relevant affinity.
Figure 5. SIRPα interacts with the IGF1 Receptor.
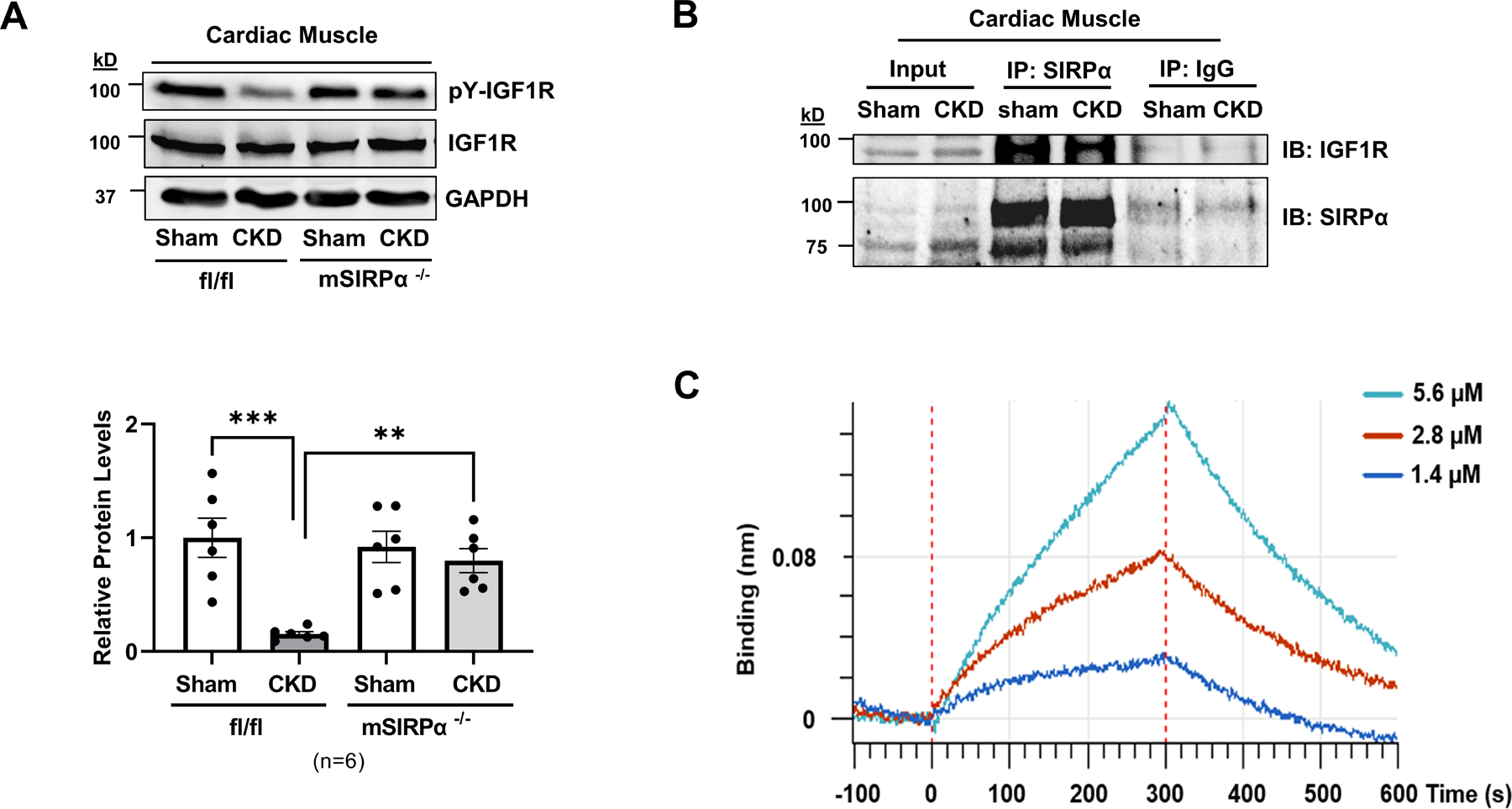
(A) After subtotal nephrectomy, heart lysates of flox (SIRPαfl/fl) and skeletal muscle-specific SIRPα KO (mSIRPα−/−) mice with or without CKD were immunoblotted to detect pY-IGF1R and total IGF1R and representative immunoblots of averaged data (top panel) with relative densities to total IGF1R (bottom panel) are shown. Statistical significance was calculated using one-way ANOVA with Bonferroni's multiple comparisons test (A). Values are expressed as a means ± SEM; ** p<0.01, *** p<0.001. (B) SIRPα was immunoprecipitated in heart lysates of SIRPαfl/fl mice with CKD vs. Sham and immunoblotted for total IGF1R. (C) In heart lysates of control CKD mice, IGF1R was immunoprecipitated. The binding response signals and kinetics of recombinant SIRPα (rSIRPα 2.5 µg/mL) with immunoprecipitated IGF1R are shown (n=3 with serial concentrations: 1.4, 2.8, 5.6 µM) which were evaluated by bio-layer interferometry, Octet RED384 platform.
Exogenous and Intracellular SIRPα Exacerbates Insulin/IGF1 Receptor Responses
Since SIRPα communicates distally with peripheral organs (i.e. adipose tissue12) and circulating SIRPα was found in serum of mice and patients with CKD (Figure 1D–E) we evaluated the effects of extracellular SIRPα on muscle cells. Thus, recombinant SIRPα (rSIRPα) was introduced to mice and myotubes in order to determine the exogenous effects of SIRPα on cardiac muscle or myotubes. Specifically, we examined if rSIRPα influenced insulin resistance and extracellular matrix protein deposition. Flox mice treated with rSIRPα displayed reduced levels of pAKT and tyrosine phosphorylation of IGF1R in the cardiac muscle (Figure 6A), as well as impaired intracellular insulin signaling (reduced pAKT) in gastrocnemius skeletal muscle (GAS), epididymal, and inguinal white adipose tissues (eWAT and iWAT; Figure S4A). Similarly, in mSIRPα−/− mice treated with rSIRPα, there was recapitulation of phenotype with reduced levels of pAKT relative to AKT and pY-IGF1R relative to total IGF1R when compared to control mice treated with diluent (Figure 6A). Next, myotubes were treated with exogenous rSIRPα for various time points up to 24 h which led to a reduction in pAKT and pY-IGF1R that persisted at 24 h, along with an upregulation in fibronectin (Figure 6B). Lastly, SIRPα plasmid overexpression in C2C12 myoblasts or HL-1 cardiomyocytes led to a reduction in pY-IGF1R and pAKT compared to GFP control (Figure 6C–D). These results suggest that both exogenous and endogenous SIRPα stimulates impaired insulin/IGF-1 receptor signaling activities in both skeletal muscle cells and cardiomyocytes.
Figure 6. Both exogenous and intracellular SIRPα exacerbates insulin/IGF1 receptor responses.
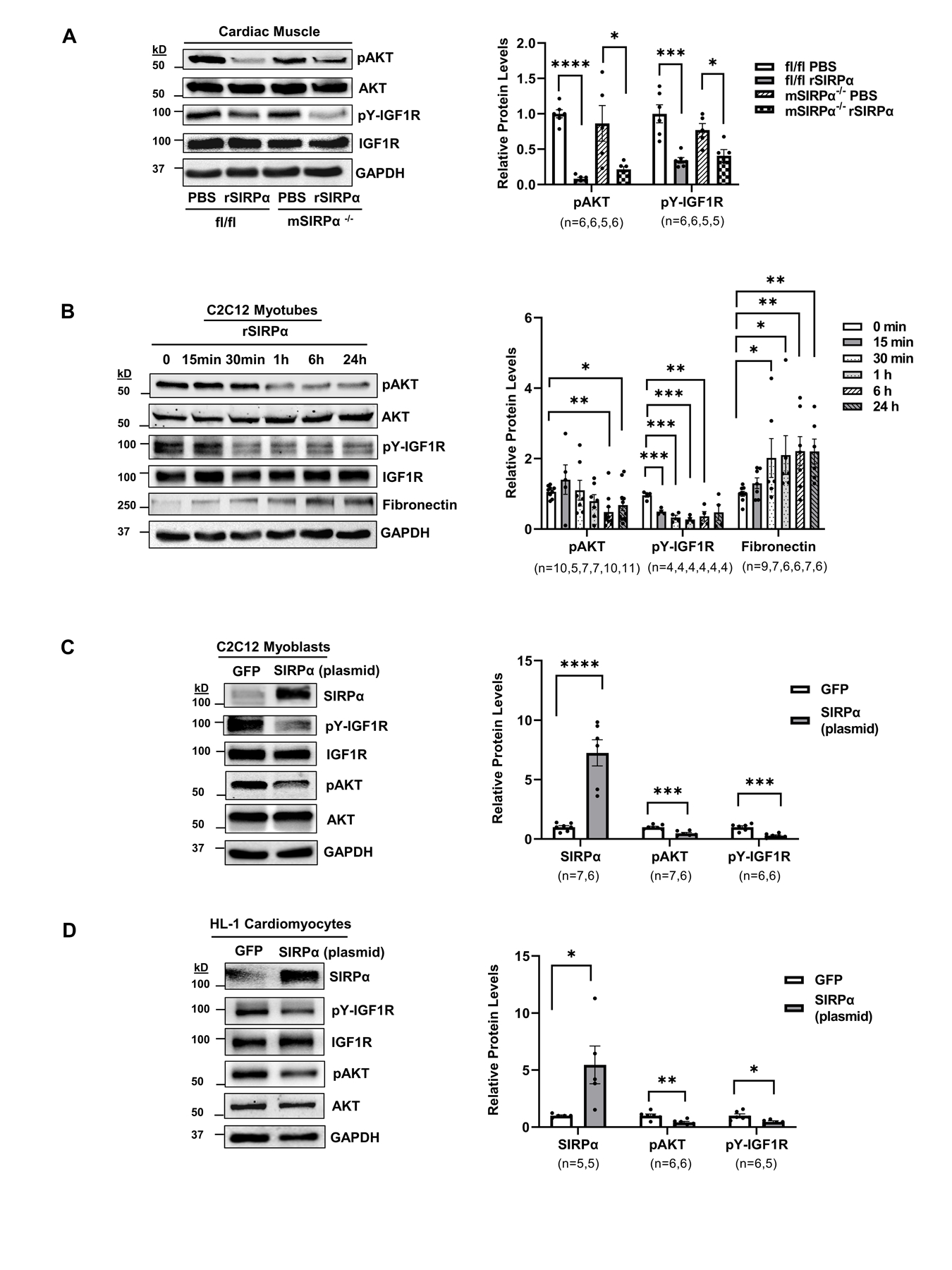
(A) Flox (SIRPαfl/fl) and skeletal muscle-specific SIRPα KO (mSIRPα−/−) mice were treated with recombinant SIRPα protein (rSIRPα, 1 µg/g) or with diluent PBS control via left ventricular injection which was allowed to circulate for 5 min. Protein lysates of cardiac muscle were immunoblotted and representative immunoblots of averaged data (left panel) to detect pAKT relative to AKT and pY-IGF1R relative to IGF1R with densities (right panel) are shown. (B) C2C12 myotubes were treated with rSIRPα (1 µg/mL) and cell lysates were immunoblotted to detect pAKT relative to AKT, fibronectin, relative to GAPDH and pY-IGF1R relative to IGF1R and representative immunoblots of averaged data (left panel) are shown. The relative protein densities are shown (right panel). Myocytes (C) C2C12 myoblasts and (D) HL-1 cardiomyocytes were electroporated with 1.5 μg of GFP or 1.5 μg of SIRPα plasmid, protein lysates from cells were immunoblotted to detect SIRPα relative to GAPDH, pAKT relative to AKT, pY-IGF1R relative to IGF1R and representative immunoblots of averaged data (left panel) with relative densities (right panel) are shown. Statistical significance was calculated using unpaired two-tailed Studen's t-test (A-D). Values are means ± SEM. *p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
Hyperglycemia or Uremic Toxin Stimulates Myocyte Release of SIRPα
Since mice with CKD display insulin resistance 12 and trigger release of SIRPα into the circulation (Figure 1D) to counteract activities of the insulin/IGF1R (Figure 5A–B), we examined whether acute exposure to high levels of glucose or uremic toxin stimulates SIRPα expression in myocytes, unrelated to changes in impaired renal clearance of SIRPα. Mice with cardiac myocyte-specific SIRPα KO mice (Myh6-Cre, cs-SIRPα−/−), muscle-specific SIRPα KO mice (Mck-Cre, mSIRPα−/− mice), adipose-specific SIRPα KO (adiponectin-Cre, AD-SIRPα−/− mice) vs. flox mice were each subjected to high glucose (2 g/kg intraperitoneal every 40 minutes × 3 doses 21). All groups of mice experienced an average serum glucose level of ~500–600 mg/dL. The responses of flox mice exposed to high concentrations of glucose stimulated a significant increase in serum SIRPα (~ 4-fold) when compared to the mice treated with PBS control. AD-SIRPα−/− mice displayed ~ 5-fold increase (Figure S5A). In contrast, mSIRPα−/− or cs-SIRPα−/− mice injected with high glucose did not display a significant increase in serum SIRPα. Additionally, hyperglycemia induced phosphorylation of AKT in the cardiac muscle and gastrocnemius skeletal muscle of flox mice (Figure S5B).
Next, C2C12 myotubes were exposed to high glucose media (HG; 25 mM) and compared to low glucose (LG; 5 mM) treated myotubes. In myotubes exposed to high glucose, SIRPα increased at 48 h after exposure (Figure 7A). Additionally, these myotubes displayed impaired IGF1R signaling with reduction in pY-IGF1R (Figure S5C). Subsequently, high glucose treated myotubes revealed SIRPα secretion into the media after 48 h hyperglycemia exposure (Figure 7B). Subsequently, HL-1 cardiomyocytes, were also treated with high glucose media which additionally triggered higher SIRPα expression and a reduction in pY-IGF1R in cardiomyocytes (Figure 7C, S5D) with SIRPα accumulation in cultured media at 24 h after hyperglycemia exposure (Figure 7D). The glucose effect was also studied in adipocytes. Despite a high level of adipocyte SIRPα expression (Figure 7E), high glucose treated adipocyte media did not display any evidence of SIRPα accumulation in its culture media (Figure 7F) nor changes in IGF1R activities in adipocytes (Figure S5E).
Figure 7. Hyperglycemia induces myocyte release of SIRPα.
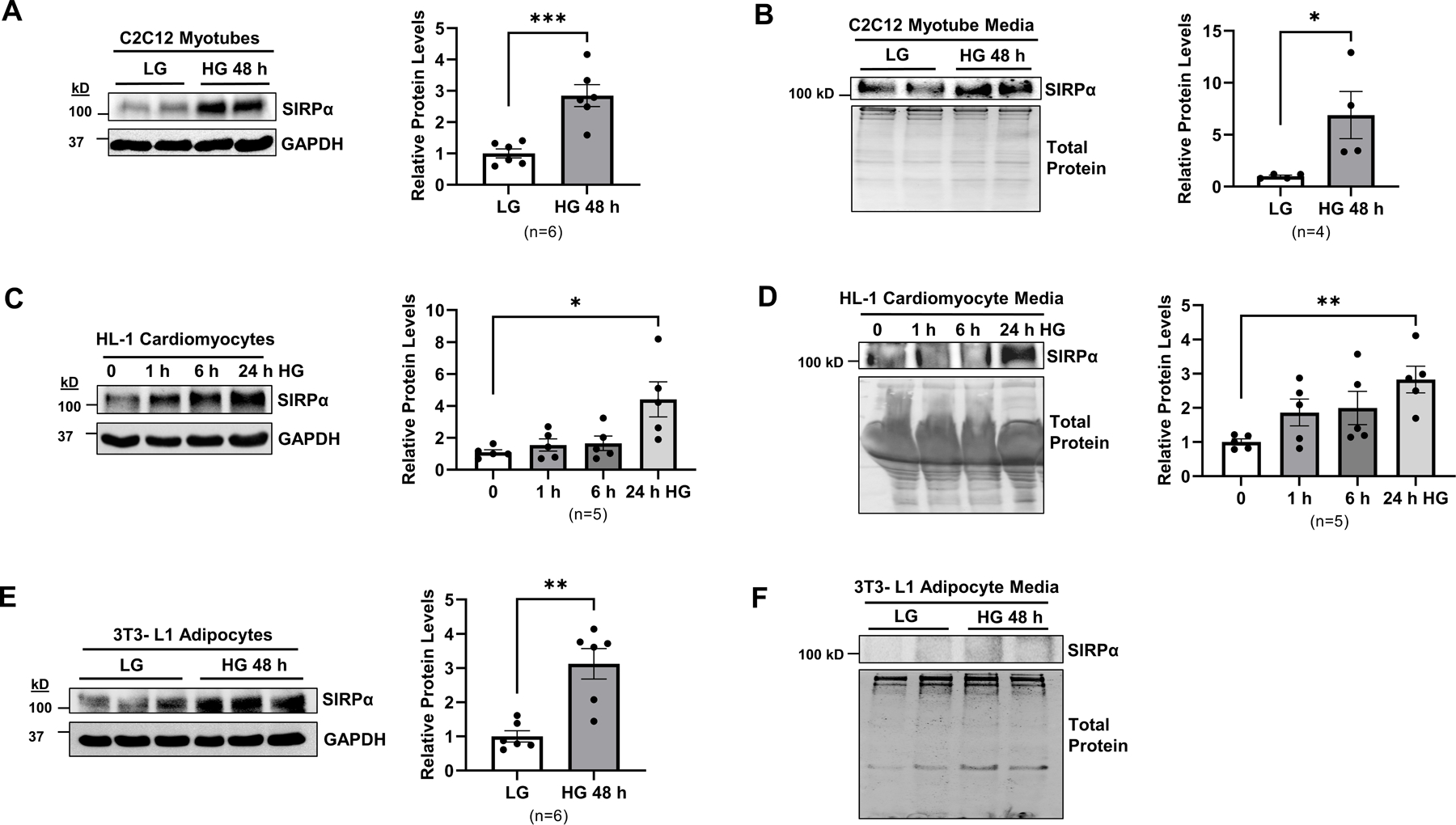
(A) C2C12 myotubes cultured in low glucose (LG) media (5 mM) were treated with high glucose (HG) 25 mM for 48 h. Protein lysates were immunoblotted to detect SIRPα and GAPDH and representative immunoblots of averaged data (left panel) with relative levels (right panel) are shown; (B) SIRPα was identified in C2C12 cultured media by immunoblot and representative immunoblots of averaged data (left panel) with relative levels to total protein (right panel) are shown. (C) HL-1 cardiomyocytes were cultured in Claycomb media and treated with high glucose (HG, 90 mM) for 1, 6, 24 h. Protein lysates were immunoblotted to detect SIRPα and GAPDH and representative immunoblots of averaged data (left panel) with relative levels (right panel) are shown; (D) SIRPα was identified in HL-1 cardiomyocyte cultured media and detected by immunoblot and representative immunoblots of averaged data (left panel) with relative levels to total protein (right panel) are shown; (E) 3T3-L1 adipocytes were treated with LG media (5 mM) or HG (25 mM) for 48 h. Protein lysates were immunoblotted to detect SIRPα and GAPDH and representative immunoblots of averaged data (left panel) with relative levels (right panel) are shown; (F) Adipocyte media was immunoblotted to detect SIRPα and total protein and representative immunoblots of averaged data are shown. Statistical significance was calculated using unpaired two-tailed Studen's t-test (A-E). Values are means ± SEM. *p<0.05, ** p<0.01, *** p<0.001.
Finally, since uremia contributes to insulin resistance-induced cachexia and cardiomyopathy 12, 15, 25 we utilized p-cresol sulfate (PCS), a well-established uremic toxin, at a dose previously published to be biologically active in ESRD patients (PCS of 40 µg/mL (212 µM)), a concentration found in humans with ESRD 15. HL-1 cardiomyocytes were treated with uremic toxin, PCS, which stimulated myocardial SIRPα expression with a reduction in pAKT (Figure S5F). Importantly, SIRPα was noted to be released into the cardiomyocyte media after PCS treatment (Figure S5G). Taken together these results indicate that both hyperglycemia or uremia - induced SIRPα release originates from myocytes and are unrelated to impaired renal clearance.
DISCUSSION
Multiple lines of evidence suggest that SIRPα is a negative regulator of insulin/IGF1 receptor signaling, a potential myokine, responsible for pathologic myocardial remodeling and dysfunction. The evidence presented here supports the following: 1) SIRPα is increased in cardiac muscle in response to CKD. 2) SIRPα knockdown prevents CKD-induced cardiomyopathy. Specifically, systolic function was preserved in SIRPα Mt mice with CKD when compared to sham-control mice. Furthermore, global SIRPα KO (SIRPα Mt) mice displayed evidence of physiologic hypertrophy with increased cardiac output, trend towards increased EF% and FS% and no evidence of elevated SBP, fibrosis, collagen deposition, and αSMA when compared to WT control mice despite elevations in fibronectin. Importantly, mSIRPα−/− mice and csSIRPα−/− sham mice did not display any evidence of increased heart size or ECM matrix proteins when compared to littermate flox controls. Additionally, csSIRPα−/− mice were cardio-protected with improved cardiac function vs. littermate control flox mice despite the presence of CKD. Moreover, previous reports suggest that αMyHC-Cre mice display evidence of cardiac dysfunction and toxicity by greater than 3 months of age when compared to WT littermate controls 26, however these csSIRPα−/− mice subjected to CKD were protected in spite of the multiple stressors (i.e., Cre driver plus CKD). 3) Furthermore, unlike control mice with CKD, there was no significant evidence of myocardial fibrosis in SIRPα Mt, mSIRPα−/− or csSIRPα−/− mice with CKD. Notably, these cardiac fibrosis responses in WT mice with CKD were likely unrelated to hypertension since these mice did not display elevated SBP classically displayed in pressure overload, with only mild elevations in SBP in control mice with CKD. This is consistent with prior descriptions that noted no significant increases in blood pressure in the background strain of C57BL/6 mice 27, 28. 4) Fetal gene activation is a feature of heart failure 23. Here, we show that heart muscles of control mice with CKD exhibit return to the fetal gene program. Furthermore, we identified that mSIRPα−/− or csSIRPα−/− mice display no significant elevations in fetal genes when compared to sham control mice despite the presence of CKD. Therefore, since SIRPα KO mice generally did not display activation of fetal programming, this suggests that the adult gene program is preserved in these mice. 5) CKD stimulates the breakdown of muscle proteins via initiation of inflammatory mediators, NF-κB activation increases SIRPα expression in skeletal muscle 10, 12. In fact, CKD stimulates circulating SIRPα in serum suggesting a potential role for it affecting other organ systems. Our current proposal extends these findings since stimulation of circulating SIRPα triggers impaired myocardial intracellular insulin/IGF1 receptor activities. We explored the potential relevance in humans, by showing that SIRPα levels are increased in serum of patients with CKD. Since CKD stimulates SIRPα into circulation, its triggered release from muscle is related to a potential endocrine or autocrine communication to impair insulin/IGF1R activities. 6) As well, we determined that SIRPα interacts with the IGF1R in ventricular cardiac muscle. This suggests that if SIRPα is upregulated it may do so to counteract insulin/IGF1 receptor pathways leading to dysregulation of myocardial muscle metabolism. 7) We also examined whether hyperglycemia alone, or mediators of CKD (e.g. uremia), increase circulating SIRPα levels. Interestingly, hyperglycemia raised entry of SIRPα into serum in flox mice with normal renal function. These results suggest that changes in circulating SIRPα level were not the result of impaired clearance of SIRPα in flox mice exposed to hyperglycemia. 8) Additionally, we determined that SIRPα accumulation in culture media did not occur when adipocytes were exposed to hyperglycemia despite an increased endogenous level of SIRPα in these cells. We previously identified that in response to CKD adipose-specific SIRPα KO displayed higher levels of serum SIRPα 12. This response did not occur in muscle-specific SIRPα KO mice with CKD 12. Taken together, our previous and current results suggest that SIRPα behaves as a myokine after exposure to either CKD, uremia or hyperglycemia. 9) Furthermore, in vivo or in vitro exposure of exogenous SIRPα recombinant protein impaired insulin/IGF1R functions or increased expression of extracellular matrix proteins (i.e. fibronectin). Finally, these experiments illustrate that the role of intracellular and extracellular SIRPα activities which converge to behave synergistically as an anti-insulin/IGF1 receptor factor. This is relevant because targeting SIRPα could potentially improve cardiac function in CKD-induced cardiomyopathy or other metabolic heart diseases associated with insulin resistance-induced cardiomyopathy.
Heart failure is associated with insulin resistance which is accompanied by changes in myocardial structure and function 29–31. We demonstrate that CKD stimulates interactions between SIRPα and the IGF1R in cardiac muscle impairing IGF1R functions while interfering with cardiac function and exacerbating myocardial fibrosis. Specifically, the IGF1R activation is suppressed (reduction in phosphotyrosines) in CKD. Ling et al. identified that tyrosine phosphatases (SHP2)-SIRPα were relevant for downstream IGF1/MAPK signaling in smooth muscle cells 32 but did not demonstrate the interactions of SIRPα with the IGF1R and its intracellular relevance in insulin/IGF1 receptor signaling in cardiac muscle. Additionally, Umemori et al. determined that the extracellular domain of SIRPα was cleaved and released into skeletal muscle cell media in response to cellular differentiation 33. While Londino et al. determined that inflammation stimulates myeloid cell cleavage of the SIRPα extracellular domain 34. Further investigations are required to determine the relationship between transmembrane SIRPα and secretion of extracellular SIRPα in response to CKD.
We have previously shown a role of SIRPα in promoting insulin resistance in skeletal muscle and adipose tissues while inducing cachexia in CKD 12. This is now the first report detailing the role of SIRPα in CKD-induced cardiomyopathy. SIRPα upregulation in both skeletal and cardiac muscle in response to CKD is associated with skeletal muscle loss and cardiomyopathy. SIRPα may play a significant role in the phenomena of cardiac cachexia syndrome in CKD-induced heart failure. Typically, the syndrome of cardiac cachexia is associated with elevated sympathetic hormonal activation specifically with high renin–angiotensin–aldosterone system (RAAS) 35. It remains to be examined if improvements in insulin signaling along with suppressed RAAS activation are relevant for the protection against CKD-induced cardiomyopathy in SIRPα KO mice with CKD. In fact, SIRPα Mt mice displayed a lower aldosterone level when compared to WT mice with or without CKD (data not shown). Future studies are required to examine the role of RAAS activation in these mice. Additionally, triggering of intracellular mediators of beta adrenergic receptor signaling (i.e. PKA activation) occurred in adipose tissue of control mice with CKD but suppressed in SIRPα Mt mice with CKD 12. Further studies are required to fully delineate the link between SIRPα-induced cachexia and heart failure in CKD.
Jiang et al. documented that SIRPα may be protective rather than deleterious in a model of pressure-overload after aortic banding 36. However, the CKD model (subtotal nephrectomy) we utilized in WT or SIRPα Mt mice did not induce pressure overload or significant elevations in systolic blood pressure as seen in aortic constriction 37 (Figure 3C–D). Recent studies have suggested that secretion of glucose-sensitive myokines can play a critical role in insulin resistance to promote metabolic derangements. Overexpression of MG53 in mouse hearts increased circulating levels of MG53 and suppressed insulin responses directly 21. These results induce worsening cardiovascular complications. Regarding mechanisms for SIRPα findings, we propose that SIRPα disrupts insulin/IGF1 receptor signaling, potentially upstream of MG53, while disrupting protective pathways of the insulin/IGF1R signaling by tyrosine dephosphorylating these mediators. In this case, tyrosine dephosphorylation of the insulin receptor and IRS1 may cause IRS1 degradation by the UPS due to its increased susceptibility to E3 ubiquitin ligases (i.e. MG53). Similarly, myostatin, a muscle-specific myokine and member of the transforming growth factor-β has been implicated in impairing insulin signaling leading to muscle wasting 38. Myostatin suppression prevents insulin resistance while upregulation of myostatin leads to muscle atrophy 38, 39. Suppression of SIRPα prevents muscle atrophy similar to myostatin inhibition in stress states of CKD however myostatin does not directly impact tyrosine dephosphorylation of insulin mediators 39, 40. Additionally, myostatin regulation is differentially regulated in dilated cardiomyopathy versus ischemic cardiomyopathy in end stage heart failure 41. Moreover, cardiac activation of the IGF1R was found to be protective against the detrimental effects of myocardial infarction 42 or diabetes 42, 43, therefore SIRPα may play a pivotal role in diabetic cardiomyopathy. Finally, with mounting evidence for cardiovascular and reno-protective benefits of the sodium-glucose cotransporter-2 inhibitors (SGLT2i) or the glucagon-like peptide-1 (GLP1) receptor agonists 44–47, further examination of their role in suppressing circulating SIRPα in CKD-induced cardiomyopathy may provide insights into their beneficial effects.
The present study suggests that SIRPα behaves as a possible myokine, released from muscles into serum in response to uremia or acute hyperglycemia. These stimuli lead to interactions with SIRPα and the myocardial IGF1R, impairing IGF1R activity while adversely influencing myocardial function and fibrosis (Figure 8). Both intracellular and extracellular SIRPα exert biologically synergistic effects on insulin/IGF1 receptor signaling to impair metabolic homeostasis. Our discovery suggests novel targets for therapies to potentially prevent CKD-associated heart failure.
Figure 8. Graphical Abstract:
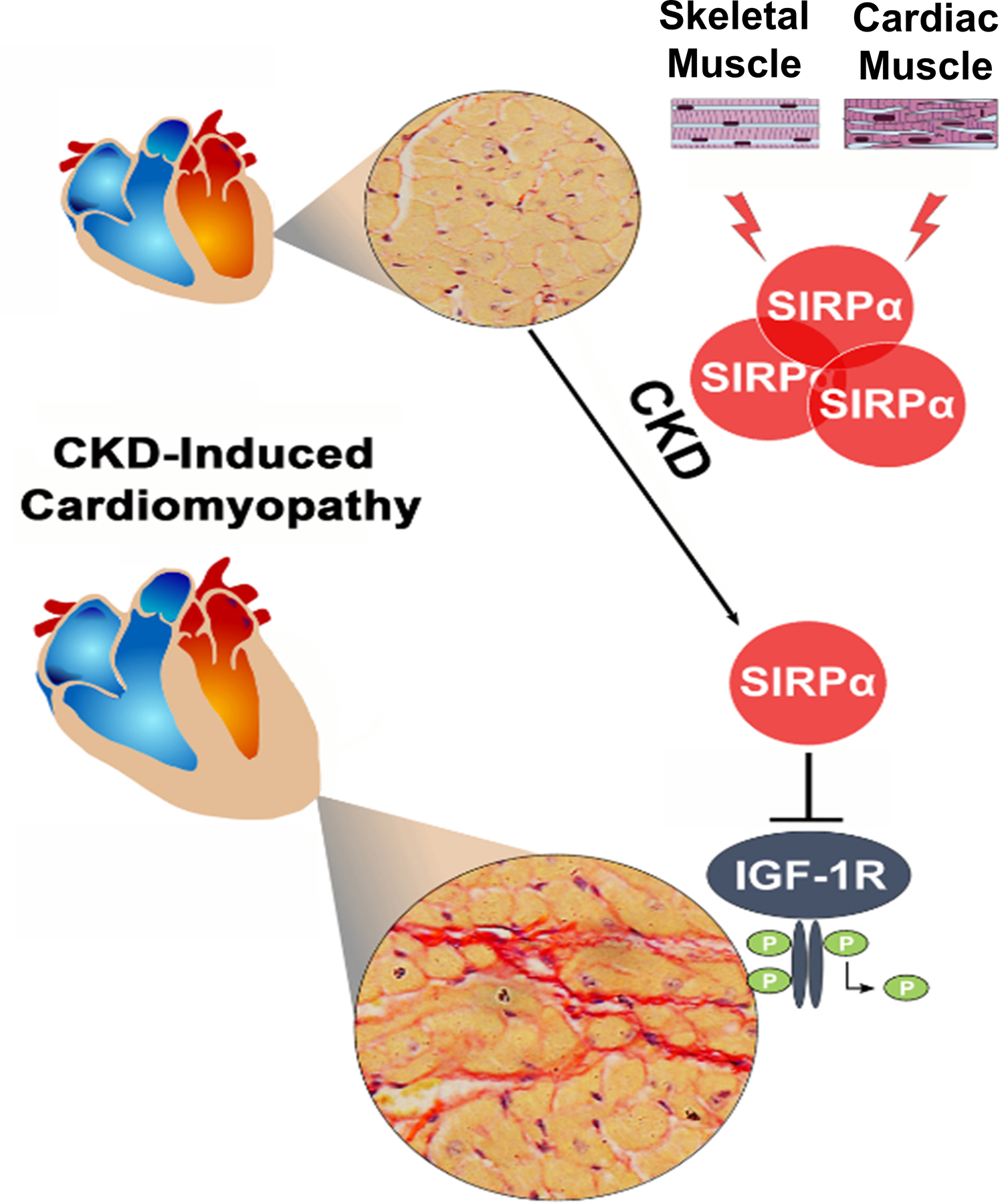
CKD-induces circulating SIRPα, a myokine, to impair myocardial IGF1R functions by promoting tyrosine dephosphorylation of the receptor, while inducing myocardial fibrosis and cardiac dysfunction.
Supplementary Material
Major Resources Table
| Animals (in vivo studies) | ||||
|---|---|---|---|---|
| Species | Vendor or Source | Background Strain | Sex | Persistent ID / URL |
| SIRPα Mt | Riken | C157BL/6 | M/F | https://knowledge.brc.riken.jp/resource/animal/card?_Lang_=en&brc_no=RBRC01544 |
| SIRPα tm1c (fl/fl) | Created from tm1a(EUCOMM) | C57BL/6 | M/F | https://www.mousephenotype.org/data/genes/MGI:108563#order |
| Myh6 Cre | Jackson Labs | https://www.jax.org/strain/011038 | ||
| Mck Cre | Jackson Labs | https://www.jax.org/strain/006475 | ||
| Adipoq Cre | Jackson Labs | https://www.jax.org/strain/028020 | ||
| DNA/cDNA Clones | |||
|---|---|---|---|
| Clone Name | Catalog# | Source / Repository | Persistent ID / URL |
| Green fluorescent protein (GFP) plasmid | VDC-1040 | Lonza | https://bioscience.lonza.com/lonza_bs/US/en/Transfection/p/000000000000191671/pmaxCloning%E2%84%A2-Vector |
| SIRPα plasmid cDNA | MMM1013–9201146 | Open Biosystems/Thermo Fisher | https://www.thermofisher.com/order/genome-database/details/gene-expression/Mm01268655_g1 |
| Recombinant Protein | ||
|---|---|---|
| Name | Source / Repository | Persistent ID / URL |
| SIRPα | R&D Systems | https://www.rndsystems.com/products/recombinant-mouse-sirp-alpha-cd172a-fc-chimera-protein-cf_7154-sa |
| Cultured Cells | |||
|---|---|---|---|
| Name | Vendor or Source | Sex (F, M, or unknown) | Persistent ID / URL |
| C2C12 | ATCC | unknown | https://www.atcc.org/products/crl-1772 |
| HL-1 | Dr. Jizhong Cheng-Baylor College of Medicine | unknown | |
| 3T3-L1 | ATCC | unknown | https://www.atcc.org/products/cl-173 |
| Reagents | ||
|---|---|---|
| Description | Source / Repository | Catalog# |
| Picrosirius Red Stain Kit | Polysciences | 24901 |
| Rneasy Fibrous Tissue Mini Kit | Qiagen | 74704 |
| Rnase-Free Dnase Set | Qiagen | 79254 |
| iScript cDNA Synthesis Kit | Bio-Rad | 1708891 |
| iQ SYBR Green Supermix | Bio-Rad | 1708880 |
| Phosphatase Inhibitor | Thermo Fisher Scientific | A32957 |
| Protease Inhibitor | Roche | A32955 |
| TRIzol | Thermo Fisher Scientific | 15596026 |
| RIPA Lysis & Extraction Buffer | G-Biosciences | 786–489 |
| Insulin | Eli Lilly | 0002–8215 |
| Randomization and Blinding |
|---|
| Experimental groups were allocated randomly, and investigators were blinded to group allocation when performing all data collection. |
NOVELTY AND SIGNIFICANCE.
What Is Known?
Insulin resistance occurs early in the course of kidney disease.
Impaired insulin signaling contributes to CKD-induced cardiomyopathy.
The metabolic milieu in CKD and events occurring in cardiomyocytes are distinct from events in other diseases. Little is known of the specific mediators associated with kidney failure that contribute to myocardial remodeling.
What New Information Does This Article Contribute?
SIRPα is a myokine secreted from muscles in response to CKD.
Acute exercise, a model of improved insulin sensitivity, suppresses SIRPα.
SIRPα inhibits insulin responses in cardiac muscle and other organs.
CKD stimulates circulating SIRPα to interact with insulin-like growth factor receptor 1 (IGF1R) to impair myocardial receptor signaling while promoting maladaptive cardiac remodeling.
SIRPα stimulates insulin resistance in skeletal muscles and adipose tissues which contribute to CKD-induced cachexia. SIRPα regulation in cardiac muscle and systemic responses to distant organs are scarcely known. To determine the role of SIRPα on cardiac remodeling in CKD, we created global, muscle-specific (Mck-Cre) and cardiac-specific (Myh6-Cre) SIRPα knockout (KO) mice. In SIRPα KO mice maladaptive myocardial remodeling was prevented despite the presence of CKD. In addition, SIRPα was found in circulation in response to CKD, uremia or hyperglycemia which promoted interactions with myocardial IGF1R to impair insulin/IGF1 receptor signaling in cardiac muscle. Impaired myocardial insulin signaling was “rescued” in SIRPα KO mice with exposure to exogenous SIRPα. These results suggest a novel response to CKD: myokine SIRPα releases into circulation in response to CKD impairing myocardial insulin/IGF1 receptor signaling while promoting CKD-induced cardiomyopathy.
Acknowledgments
We thank Rebecca Salazar from McGovern Medical School at The University of Texas Health Science Center for her excellent technical support and Dr. Jingbo Niu from Baylor College of Medicine for her statistical advice.
Nonstandard Abbreviations & Acronyms
- AKT
Protein kinase B
- BUN
Blood urea nitrogen
- CKD
Chronic kidney disease
- FGF23
Fibroblast growth factor 23
- GFR
Glomerular filtration rate
- IGF1R
Insulin growth factor-1 receptor
- IRS1
Insulin Receptor Substrate 1
- KO
Knockout
- PI3K
Phosphoinositide 3-kinases
- SH-2
Src homology-2
- SHP1/2
Src homology domain-containing protein tyrosine phosphatase 1/2
- SIRPα
Signal regulatory protein alpha
Footnotes
Sources of Funding
This study was supported by the VA Career Development Award IK2 BX002492 to SST from the United States Department of Veterans Affairs, Biomedical Laboratory Research and Development Program; and in part from the NIH R01DK037175 to WEM/ZH, R01HL061483 to HT, and from the American Heart Association 18SFRN34110369 to GD. We acknowledge the generous support of Dr. and Mrs. Harold Selzman and the Mike Hogg Fund.
This project utilized equipment/services from Baylor College of Medicine’s Mouse Metabolism and Phenotyping Core, which receives support from NIH UM1HG006348, R01DK114356, R01HL130249, and the Roderick D. MacDonald Research Fund at Baylor St. Luke's Medical Center.
Disclosures
Contents of this paper do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Supplemental Materials
References:
- 1.Go AS, Chertow GM, Fan D, McCulloch CE and Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004;351:1296–305. [DOI] [PubMed] [Google Scholar]
- 2.Charytan D and Kuntz RE. The exclusion of patients with chronic kidney disease from clinical trials in coronary artery disease. Kidney Int 2006;70:2021–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang X and Shapiro JI. Evolving concepts in the pathogenesis of uraemic cardiomyopathy. Nat Rev Nephrol 2019;15:159–175. [DOI] [PubMed] [Google Scholar]
- 4.Stewart GA, Gansevoort RT, Mark PB, Rooney E, McDonagh TA, Dargie HJ, Stuart R, Rodger C and Jardine AG. Electrocardiographic abnormalities and uremic cardiomyopathy. Kidney Int 2005;67:217–26. [DOI] [PubMed] [Google Scholar]
- 5.Stefanski A, Schmidt KG, Waldherr R and Ritz E. Early increase in blood pressure and diastolic left ventricular malfunction in patients with glomerulonephritis. Kidney Int 1996;50:1321–6. [DOI] [PubMed] [Google Scholar]
- 6.Razeghi P, Young ME, Cockrill TC, Frazier OH and Taegtmeyer H. Downregulation of myocardial myocyte enhancer factor 2C and myocyte enhancer factor 2C-regulated gene expression in diabetic patients with nonischemic heart failure. Circulation 2002;106:407–11. [DOI] [PubMed] [Google Scholar]
- 7.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC and Rosenzweig A. PI3K rescues the detrimental effects of chronic Akt activation in the heart during ischemia/reperfusion injury. J Clin Invest 2005;115:2128–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jia G, DeMarco VG and Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol 2016;12:144–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Banerjee D, Biggs ML, Mercer L, Mukamal K, Kaplan R, Barzilay J, Kuller L, Kizer JR, Djousse L, Tracy R, Zieman S, Lloyd-Jones D, Siscovick D and Carnethon M. Insulin resistance and risk of incident heart failure: Cardiovascular Health Study. Circ Heart Fail 2013;6:364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomas SS, Dong Y, Zhang L and Mitch WE. Signal regulatory protein-alpha interacts with the insulin receptor contributing to muscle wasting in chronic kidney disease. Kidney Int 2013;84:308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kharitonenkov A, Chen Z, Sures I, Wang H, Schilling J and Ullrich A. A family of proteins that inhibit signalling through tyrosine kinase receptors. Nature 1997;386:181–6. [DOI] [PubMed] [Google Scholar]
- 12.Wu J, Dong J, Verzola D, Hruska K, Garibotto G, Hu Z, Mitch WE and Thomas SS. Signal regulatory protein alpha initiates cachexia through muscle to adipose tissue crosstalk. J Cachexia Sarcopenia Muscle 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pedersen BK and Febbraio MA. Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 2012;8:457–65. [DOI] [PubMed] [Google Scholar]
- 14.Shimano M, Ouchi N and Walsh K. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation 2012;126:e327–32. [DOI] [PubMed] [Google Scholar]
- 15.Koppe L, Pillon NJ, Vella RE, Croze ML, Pelletier CC, Chambert S, Massy Z, Glorieux G, Vanholder R, Dugenet Y, Soula HA, Fouque D and Soulage CO. p-Cresyl sulfate promotes insulin resistance associated with CKD. J Am Soc Nephrol 2013;24:88–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mak RH. Insulin resistance in uremia: effect of dialysis modality. Pediatr Res. 1996;40:304–8. [DOI] [PubMed] [Google Scholar]
- 17.Mak RH, Cheung WW and Roberts CT, Jr. The growth hormone-insulin-like growth factor-I axis in chronic kidney disease. Growth Horm IGF Res 2008;18:17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF and Bradley A. A conditional knockout resource for the genome-wide study of mouse gene function. Nature 2011;474:337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kir S, Komaba H, Garcia AP, Economopoulos KP, Liu W, Lanske B, Hodin RA and Spiegelman BM. PTH/PTHrP Receptor Mediates Cachexia in Models of Kidney Failure and Cancer. Cell Metab 2016;23:315–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Respress JL and Wehrens XH. Transthoracic echocardiography in mice. J Vis Exp 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu HK, Zhang Y, Cao CM, Hu X, Fang M, Yao Y, Jin L, Chen G, Jiang P, Zhang S, Song R, Peng W, Liu F, Guo J, Tang L, He Y, Shan D, Huang J, Zhou Z, Wang DW, Lv F and Xiao RP. Glucose-Sensitive Myokine/Cardiokine MG53 Regulates Systemic Insulin Response and Metabolic Homeostasis. Circulation 2019;139:901–914. [DOI] [PubMed] [Google Scholar]
- 22.Sharma D, Hawkins M and Abramowitz MK. Association of sarcopenia with eGFR and misclassification of obesity in adults with CKD in the United States. Clin J Am Soc Nephrol 2014;9:2079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taegtmeyer H, Sen S and Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 2010;1188:191–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopez B, Gonzalez A, Hermida N, Laviades C and Diez J. Myocardial fibrosis in chronic kidney disease: potential benefits of torasemide. Kidney Int Suppl 2008:S19–23. [DOI] [PubMed] [Google Scholar]
- 25.Tonelli M, Karumanchi SA and Thadhani R. Epidemiology and Mechanisms of Uremia-Related Cardiovascular Disease. Circulation 2016;133:518–36. [DOI] [PubMed] [Google Scholar]
- 26.Pugach EK, Richmond PA, Azofeifa JG, Dowell RD and Leinwand LA. Prolonged Cre expression driven by the alpha-myosin heavy chain promoter can be cardiotoxic. J Mol Cell Cardiol 2015;86:54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma LJ and Fogo AB. Model of robust induction of glomerulosclerosis in mice: importance of genetic background. Kidney Int 2003;64:350–5. [DOI] [PubMed] [Google Scholar]
- 28.Han H, Zhu J, Zhu Z, Ni J, Du R, Dai Y, Chen Y, Wu Z, Lu L and Zhang R. p-Cresyl sulfate aggravates cardiac dysfunction associated with chronic kidney disease by enhancing apoptosis of cardiomyocytes. J Am Heart Assoc 2015;4:e001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castellano G, Affuso F, Conza PD and Fazio S. The GH/IGF-1 Axis and Heart Failure. Curr Cardiol Rev 2009;5:203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Riehle C and Abel ED. Insulin Signaling and Heart Failure. Circ Res 2016;118:1151–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Young ME, McNulty P and Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation 2002;105:1861–70. [DOI] [PubMed] [Google Scholar]
- 32.Ling Y, Maile LA, Lieskovska J, Badley-Clarke J and Clemmons DR. Role of SHPS-1 in the regulation of insulin-like growth factor I-stimulated Shc and mitogen-activated protein kinase activation in vascular smooth muscle cells. Mol Biol Cell 2005;16:3353–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Umemori H and Sanes JR. Signal regulatory proteins (SIRPS) are secreted presynaptic organizing molecules. J Biol Chem 2008;283:34053–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Londino JD, Gulick D, Isenberg JS and Mallampalli RK. Cleavage of Signal Regulatory Protein alpha (SIRPalpha) Enhances Inflammatory Signaling. J Biol Chem 2015;290:31113–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anker SD and Sharma R. The syndrome of cardiac cachexia. Int J Cardiol 2002;85:51–66. [DOI] [PubMed] [Google Scholar]
- 36.Jiang DS, Zhang XF, Gao L, Zong J, Zhou H, Liu Y, Zhang Y, Bian ZY, Zhu LH, Fan GC, Zhang XD and Li H. Signal regulatory protein-alpha protects against cardiac hypertrophy via the disruption of toll-like receptor 4 signaling. Hypertension 2014;63:96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nicks AM, Kesteven SH, Li M, Wu J, Chan AY, Naqvi N, Husain A, Feneley MP, Smith NJ, Iismaa SE and Graham RM. Pressure overload by suprarenal aortic constriction in mice leads to left ventricular hypertrophy without c-Kit expression in cardiomyocytes. Sci Rep 2020;10:15318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zimmers TA, Davies MV, Koniaris LG, Haynes P, Esquela AF, Tomkinson KN, McPherron AC, Wolfman NM and Lee SJ. Induction of cachexia in mice by systemically administered myostatin. Science 2002;296:1486–8. [DOI] [PubMed] [Google Scholar]
- 39.Verzola D, Barisione C, Picciotto D, Garibotto G and Koppe L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease Kidney Int 2019;95:506–517. [DOI] [PubMed] [Google Scholar]
- 40.Eilers W, Chambers D, Cleasby M and Foster K. Local myostatin inhibition improves skeletal muscle glucose uptake in insulin-resistant high-fat diet-fed mice. Am J Physiol Endocrinol Metab 2020;319:E163–E174. [DOI] [PubMed] [Google Scholar]
- 41.Baan JA, Varga ZV, Leszek P, Kusmierczyk M, Baranyai T, Dux L, Ferdinandy P, Braun T and Mendler L. Myostatin and IGF-I signaling in end-stage human heart failure: a qRT-PCR study. J Transl Med 2015;13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Troncoso R, Ibarra C, Vicencio JM, Jaimovich E and Lavandero S. New insights into IGF-1 signaling in the heart. Trends Endocrinol Metab 2014;25:128–37. [DOI] [PubMed] [Google Scholar]
- 43.Huynh K, McMullen JR, Julius TL, Tan JW, Love JE, Cemerlang N, Kiriazis H, Du XJ and Ritchie RH. Cardiac-specific IGF-1 receptor transgenic expression protects against cardiac fibrosis and diastolic dysfunction in a mouse model of diabetic cardiomyopathy. Diabetes 2010;59:1512–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wanner C, Inzucchi SE, Lachin JM, Fitchett D, von Eynatten M, Mattheus M, Johansen OE, Woerle HJ, Broedl UC, Zinman B and Investigators E-RO. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N Engl J Med 2016;375:323–34. [DOI] [PubMed] [Google Scholar]
- 45.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE and Investigators E-RO. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 46.Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, Steinberg WM, Stockner M, Zinman B, Bergenstal RM, Buse JB, Committee LS and Investigators LT. Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. N Engl J Med 2016;375:311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuttle KR, Lakshmanan MC, Rayner B, Busch RS, Zimmermann AG, Woodward DB and Botros FT. Dulaglutide versus insulin glargine in patients with type 2 diabetes and moderate-to-severe chronic kidney disease (AWARD-7): a multicentre, open-label, randomised trial. Lancet Diabetes Endocrinol 2018;6:605–617. [DOI] [PubMed] [Google Scholar]
- 48.Claycomb WC, Lanson NA Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ Jr. HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A 1998;95:2979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peng H, Wang Q, Lou T, Qin J, Jung S, Shetty V, Li F, Wang Y, Feng XH, Mitch WE, Graham BH and Hu Z. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun 2017;8:1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all supporting data are available within the Online Data Supplement. The supporting data are also available from the corresponding author upon reasonable request.
Detailed Materials and Methods are available in the Online Data Supplement.