

Abstract
Epidemiological studies identified alcohol use disorder (AUD) as a risk factor for Alzheimer’s disease (AD), yet there is conflicting evidence on how alcohol use promotes AD pathology. In this study, a 10-week moderate two-bottle choice drinking paradigm was used to identify how chronic ethanol exposure alters amyloid-β (Aβ)-related pathology, metabolism, and behavior. Ethanol-exposed APPswe/PSEN1dE9 (APP/PS1) mice showed increased brain atrophy and an increased number of amyloid plaques. Further analysis revealed that ethanol exposure led to a shift in the distribution of plaque size in the cortex and hippocampus. Ethanol-exposed mice developed a greater number of smaller plaques, potentially setting the stage for increased plaque proliferation in later life. Ethanol drinking APP/PS1 mice also exhibited deficits in nest building, a metric of self-care, as well as increased locomotor activity and central zone exploration in an open field test. Ethanol exposure also led to a diurnal shift in feeding behavior which was associated with changes in glucose homeostasis and glucose intolerance. Complementary in vivo microdialysis experiments were used to measure how acute ethanol directly modulates Aβ in the hippocampal interstitial fluid (ISF). Acute ethanol transiently increased hippocampal ISF glucose levels, suggesting that ethanol directly affects cerebral metabolism. Acute ethanol also selectively increased ISF Aβ40 but not ISF Aβ42, levels during withdrawal. Lastly, chronic ethanol drinking increased N-methyl-d-aspartate receptor (NMDAR) and decreased γ-aminobutyric acid type-A receptor (GABAAR) mRNA levels, indicating a potential hyperexcitable shift in the brain’s excitatory/inhibitory (E/I) balance. Collectively, these experiments suggest that ethanol may increase Aβ deposition by disrupting metabolism and the brain’s E/I balance. Furthermore, this study provides evidence that a moderate drinking paradigm culminates in an interaction between alcohol use and AD-related phenotypes with a potentiation of AD-related pathology, behavioral dysfunction, and metabolic impairment.
Keywords: Amyloid-β, Alcohol, Metabolism, Brain, Alzheimer’s disease
1. Introduction
Alzheimer’s disease (AD) is the most common form of dementia, accounting for 60–80% of dementia cases. In the US, ~6 million people have been diagnosed with AD, and those numbers are expected to increase to ~14 million by 2050 (Long and Holtzman, 2019). AD pathology is characterized by the aggregation of extracellular amyloid-β (Aβ) into amyloid plaques, the intracellular accumulation of tau into neurofibrillary tangles, and neurodegeneration (Jack et al., 2016). Aβ aggregation and other pathological events precede the onset of cognitive decline and clinical diagnosis by ~10–20 years (Jack Jr. et al., 2010). Thus, it is important to identify risk factors that accelerate the onset of AD. Epidemiological studies identified alcohol use disorder (AUD) as a risk factor for AD (Harwood et al., 2010; Xu et al., 2017; Schwarzinger et al., 2018; Zhornitsky et al., 2021), yet there is conflicting evidence on how alcohol use promotes AD pathology. Preclinical studies show that chronic ethanol administration increases amyloid plaque pathology and amyloidogenic processing of amyloid precursor protein (APP) (Huang et al., 2018; Hoffman et al., 2019). Conflicting studies suggest that low-to-moderate alcohol consumption may reduce the risk of AD in humans (Rehm et al., 2019). Thus, questions remain as to whether ethanol directly modulates Aβ levels, or how moderate ethanol consumption affects factors that contribute to amyloid pathology such as metabolic deficits.
Recognizing this gap in knowledge and the critical need to better understand how AUD increases the risk for AD, this study investigated how chronic ethanol consumption alters the behavioral and metabolic disturbances associated with AD pathogenesis. Here, a well-validated mouse model of AD-related pathology and Aβ overexpression (APPswe/PSEN1dE9; APP/PS1) (Jankowsky et al., 2004) was exposed to a moderate ethanol-drinking paradigm. The effects of ethanol on AD-related pathology, metabolism, anxiety- and depression-related behaviors, cognitive measures, and excitatory/inhibitory (E/I) receptors were then analyzed. In vivo microdialysis in APP/PS1 mice was also used to measure how an acute ethanol exposure directly impacts cerebral glucose metabolism, Aβ40, and Aβ42 levels in the hippocampal interstitial fluid (ISF).
Moderate ethanol consumption, via a two-bottle choice drinking paradigm, induced changes in brain atrophy, amyloid plaque number, and plaque size, without affecting APP levels or APP processing. Chronic and acute ethanol disrupted peripheral and cerebral glucose homeostasis, both of which are known to drive of Aβ-related pathology (Macauley et al., 2015; Stanley et al., 2016; Kavanagh et al., 2019). Moderate ethanol drinking also exacerbated behavioral deficits typically observed in APP/PS1 mice. An acute ethanol exposure selectively increased interstitial fluid (ISF) Aβ40 levels by ~20% during withdrawal, but not ISF Aβ42. Because Aβ40 is released in an activity-dependent manner (Bero, 2011a; Cirrito et al., 2008; Cirrito et al., 2005), this suggests that acute ethanol is altering neuronal activity. In support of this, chronic ethanol administration led to changes in NMDA and GABAA receptor subunit expression. This gives further credence to the notion that ethanol is driving amyloid pathology through changes in neuronal activity. Collectively, this study provides evidence that ethanol increases amyloid pathology through disruptions in glucose homeostasis and brain excitability. This study suggests that chronic ethanol consumption, even at moderate amounts, may exacerbate the development of AD-related pathology and AD-associated behavioral deficits.
2. Materials & methods
2.1. Animals
5.5-month-old male APPswe/PSEN1dE9 mice (Jankowsky et al., 2004) (APP/PS1; The Jackson Laboratory; n = 20) and age-matched wildtype B6C3 control mice (n = 20) were used for the chronic drinking studies. Six animals following baseline behavioral testing died resulting in a total of 17 APPswe/PSEN1dE9 mice and 17 age-matched wildtype mice. Group size was determined based on similar studies conducted by our labs and others. Since epidemiological studies have shown that men with AUDs develop dementia at higher rates than women with AUDs (Schwarzinger et al., 2018) and male and female APP/SP1 mice develop pathology at different rates, only male mice were used for these initial studies. All animals were housed individually in standard mouse cages under a 12-h artificial light–dark cycle. Room temperature and humidity were kept constant (temperature: 22 ± 1 °C; relative humidity: 55 ± 5%). Standard laboratory rodent chow (LabDiet 5P00 Prolab RMH 3000 rodent chow) and tap water were provided ad libitum throughout the experimental period. Mice underwent a battery of behavioral tests at baseline, and at various stages during ethanol exposure. A separate cohort of 3-month-old male APP/PS1 mice (n = 4–9; see Supplementary Tables 1–3) was used for acute ethanol exposure experiments. All experimental procedures were approved by the Committee on Animal Care and Use at Wake Forest School of Medicine.
2.2. Experimental design
At 3 months of age, APP/PS1 and control mice were run through a battery of behavioral tests to identify baseline differences in behavior. Following completion of this behavioral battery, 5.5-month-old wildtype and APP/PS1 mice were randomly assigned to drinking groups. Mice were exposed to ethanol for 10 weeks via a modified two-bottle choice paradigm (Huynh et al., 2019). Mice were weighed before and after each drinking session. Throughout the 10-week drinking period, mice were assessed for changes in anxiety and AD-related behaviors during the three-day abstinence period. At the end of the study, mice were euthanized within 24–48 h after the final ethanol exposure (see Fig. 1a for experimental timeline). Briefly, mice were anesthetized with an overdose of sodium pentobarbital and transcardially perfused with ice-cold 0.3% heparin in DPBS. Prior to perfusion ~200 μL of blood was collected from the left ventricle and transferred into EDTA-coated tubes and kept on ice. Tubes were centrifuged at 3000g for 5 min at 4 °C and plasma was removed then flash-frozen in dry ice and stored at −80 °C. After perfusion, brains were removed, weighed, and bisected. The left hemisphere was fixed in 4% paraformaldehyde at 4 °C, while the right hemisphere was dissected and flash-frozen in dry ice.
Fig. 1.
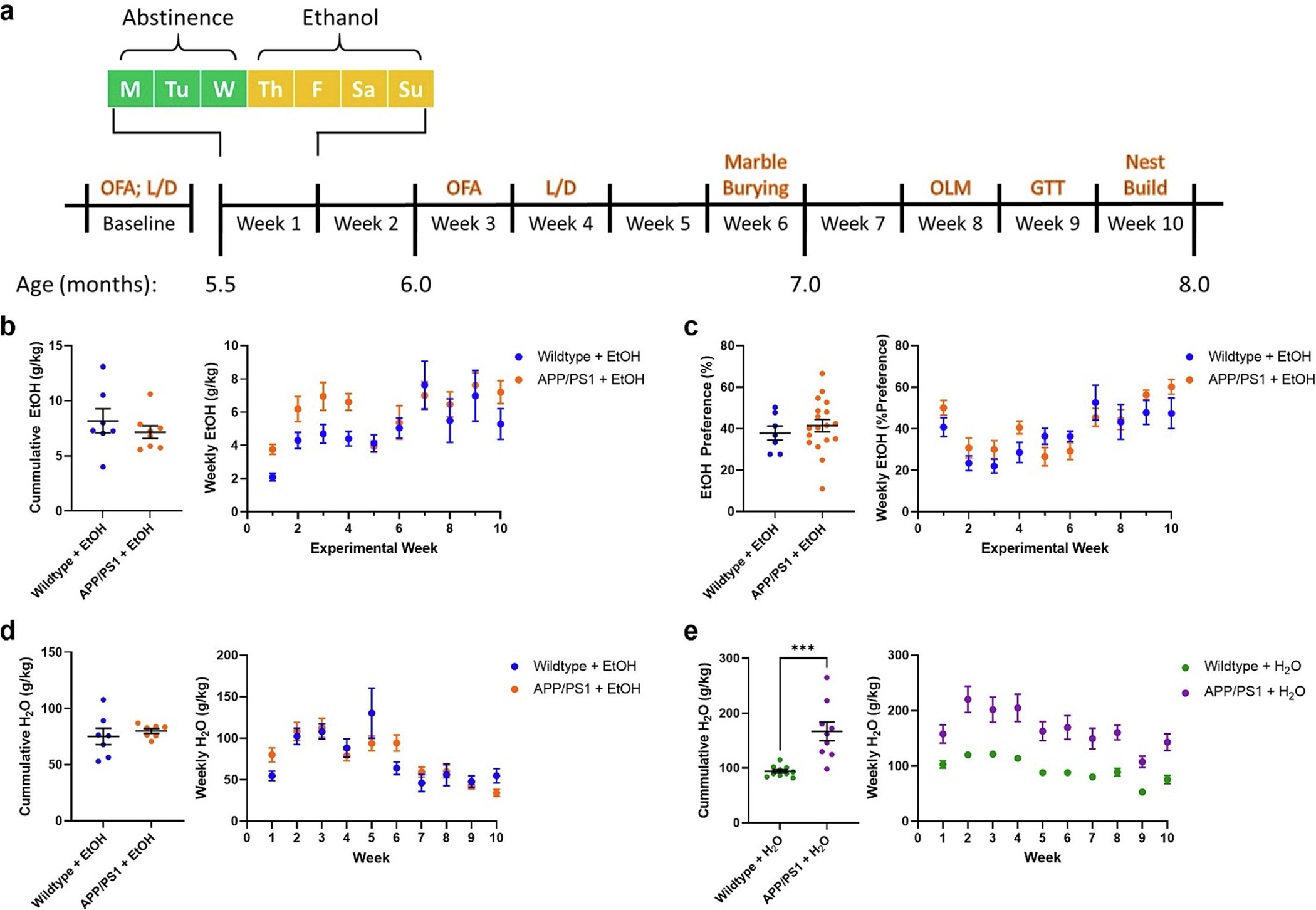
APP/PS1 mice do not consume more ethanol than control mice. a) Timeline for the experimental protocol. b) Cumulative and average weekly EtOH intake (g/kg) from the 10-week plotted as a function of genotype. APP/PS1 mice did not consume more EtOH than wildtype mice. c) Cumulative and average weekly EtOH preference (% total fluid) from the 10-week exposure period plotted as a function of genotype. No difference between wildtype and APP/PS1 mice was observed (unpaired t-test). d) Cumulative and average weekly water consumption across the 10-week EtOH exposure in EtOH-treated mice. No difference was seen in EtOH-treated wildtype or APP/PS1 mice (unpaired t-test). e) Cumulative and average weekly water consumption across the 10-week EtOH exposure period in water-treated mice. APP/PS1 mice consumed more water than wildtype mice (p < 0.0001, unpaired t-test). Wildtype + H2O, n = 10; APP/PS1 + H2O, n = 9; Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8. ***p < 0.001.
2.3. Two bottle choice procedure
Following baseline behavioral testing, mice were exposed to a modified two bottle choice paradigm (Simms et al., 2008) for 10 weeks. Briefly, 5.5-month-old wildtype (n = 7), and APP/PS1 mice (n = 8) were provided free access ethanol (20% w/v in water) and water for 12 h/day for 4 consecutive days during their dark cycle. Ethanol and water positions were alternated daily to control for side preference. Control groups consisted of age-matched wildtype (n = 10) and APP/PS1 mice (n = 9), provided with two bottles of water during the same time periods. All mice were weighed prior to each drinking exposure. Bottles were weighed before and after each drinking period. Ethanol consumption data is presented as grams of ethanol per kilogram of body weight. Ethanol preference was calculated as percent of ethanol intake over total liquid consumption.
2.4. Open field assay (OFA)
The open field assay was performed as described previously (Ewin et al., 2019). Briefly, mice were placed in the center of a plexiglass chamber (40 cm × 40 cm × 30 cm) equipped with Omnitech Superflex Sensors (Omnitech Electronics, Inc). This box uses arrays of infrared photodectectors located at regular intervals along each wall of the chamber. The chamber walls were solid and were contained within sound-attenuating boxes with a 15-watt light bulb to illuminate the arena. Exploratory activity was measured for 15 min and quantified as locomotor activity and % time spent in the central zone. OFA activity was assessed at baseline when mice were ~ 3 months-old, and again after 3 weeks of ethanol exposure when mice were ~ 6 months-old.
2.5. Light/Dark assay (LD)
The light/dark box test was conducted as previously described (Miller et al., 2011). Control and APP/PS1 mice were placed into a polycarbonate box (40 cm × 40 cm) with two equally sized regions. One region was dark and concealed, while the other was open and light. A 10 cm opening allowed free movement between both regions. Mice were monitored for five minutes. Latency to enter the light side, number of light-side entries, and total time spent in the light-side of the box were recorded with EthoVision XT tracking software. Increased reluctance to venture into the light, uncovered, side was interpreted as anxiety-related behavior. LD activity was assessed at baseline when mice were ~ 3 months-old, and again after 3 weeks of ethanol exposure when mice were ~ 6 months-old.
2.6. Marble burying
The marble burying test was performed as previously described (Amodeo et al., 2012). Control and APP/PS1 mice were brought into a novel environment and habituated for one hour before behavioral testing. Mice were placed in a cage (19.56 cm × 30.91 cm × 13.34 cm) containing 12 marbles (13 mm diameter) on corncob bedding (5 cm depth). Mice were allowed to freely move within the cage for 30 min. Following the 30-min period, mice were removed from the cage and returned to their respective home cages. Images of each cage were recorded, and the number of marbles were counted. A marble was considered buried when >75% of the object was covered by bedding.
2.7. Object location memory task (OLM)
Object location memory task was conducted as previously described (Day et al., 2019). Mice were habituated to an opaque plastic chamber (40 cm × 40 cm) with visible spatial cues for 10 min. After 24 h, mice were returned to the chamber with two identical objects and were allowed to freely explore and interact with the objects for 10 min. Twenty-four hours later, mice were returned to the chamber again, where one of the two objects had been relocated to an adjacent position. Changes in objects and locations were randomized and counterbalanced. Time spent with each object was measured and calculated as a percentage of the total object interaction time. Relocated object preference of ~50% indicates memory impairments. Time with objects was measured both manually and with EthoVision XT tracking software. Mice with a total object interaction time of <5 s were excluded from analysis. Data collection and analysis were performed blinded to condition.
2.8. Glucose tolerance test
After 9 weeks of ethanol exposure, a glucose tolerance test was performed as previously described (Day et al., 2019). Briefly, mice were fasted for 4 h and 2.0 g/kg glucose was administered via i.p injection. Blood samples were taken from tail veins and blood glucose was measured at baseline, 15-, 30-, 45-, 60-, 90-, and 120 min from glucose injection using a glucometer (Bound Tree Medical Precision XTRA Glucometer; Fisher). Glucose tolerance tests were performed on non-drinking days.
2.9. Nest building
Nest building behavior was assessed as previously described (Deacon, 2006). 24 h following the last day of EtOH treatment during the dark cycle, control and APP/PS1 mice were provided fresh nesting material (a paper Bed-r’Nest (TheAndersons)) and cotton nestlet (Ancare) in their home cages. At the beginning of the light cycle, photos of the nests were recorded and rated on a 1–5 scale by two blinded analysts. A score of 1 was considered a completely unconstructed nest, while a 5 was considered a completed nest that integrated all available materials.
2.10. Brain mass, Aβ immunohistochemistry, and X34 staining
Prior to sectioning, brains were cryoprotected in 30% sucrose then sectioned on a freezing microtome at 50 μm. Three serial sections (300 μm apart) through the anterior-posterior aspect of the hippocampus were immunostained for Aβ deposition using a biotinylated, HJ3.4 antibody (anti-Aβ1–13, a generous gift from Dr. David Holtzman, Washington University). Sections were developed using a Vectastain ABC kit and DAB reaction. For fibrillary plaques, free floating sections were permeabilized with 0.25% Triton X-100 and stained with 10 μM X-34 in 40% ethanol +0.02 M NaOH in PBS (Ulrich et al., 2018). Brain sections were imaged using a NanoZoomer slide scanner and the percent area occupied by HJ3.4 or X34 was quantified using ImageJ software (National Institutes of Health) as previously described (Bero et al., 2011b; Roh et al., 2012). A histogram analysis was performed to quantify the frequency of each plaque by pixel size, excluding any plaques smaller than 10 pixels. Statistical significance was determined using a two-tailed, unpaired t-test (percent area) and a 2-way ANOVA with Šídás multiple comparisons post-hoc tests (size × frequency). Aβ deposition, amyloid plaque size, amyloid plaque, and neurofibrillary plaque area fraction were quantified by a blinded researcher. Because wildtype mice do not develop amyloid plaques, Aβ IHC and X34 staining was only performed in wildtype mice. Data is represented by means ±SEM.
2.11. Western blot
Western blot analysis was used to measure protein levels of APP processing enzymes and excitatory and inhibitory receptors. Because wildtype mice do not develop amyloid plaques, Western bot analysis for APP and APP processing enzymes were only performed in APP/PS1 mice. For APP processing enzymes, posterior cortical tissue was homogenized in 1× cell lysis buffer (Cell Signaling) supplemented with protease inhibitor cocktail (Roche), 1 mM PMSF (Cell Signaling), 1 mM DTT (Sigma-Aldritch), and a phosphatase inhibitor cocktail (Millipore) using a probe sonicator at 30% amplitude, 1 s pulse with a 5 s delay, 5 times while on ice. Tissue homogenates were then spun down at 10,000 g for 10 min at 4 °C and the supernatant was used for immunoblotting. Protein concentrations were analyzed using BCA protein assay kit (Pierce). For APP and APP c-terminal fragments (CTFs), 15 μg of protein were run in 15% tris-tricene gels to increase separation between CTF-β and CTF-α. All other proteins were run in 10% tris-trice gels. All gels were run using BioRad Protean mini then rapid-transferred to PVDF membranes using BioRad Semi-dry membranes (BioRad). Membranes were subsequently blocked using 5% BSA in 1× TBST for 1 h and then incubated with primary antibodies overnight at 4 °C. Secondary antibody conjugated with HRP-specific to primary antibody were incubated at room temperature for 1 h in 1× TBST. The following primary and secondary antibodies were used for this study: APP (including CTFβ and CTFα; Invitrogen; CT695; 1:1000), BACE1 (Cell Signaling; 5606S; 1:1000), ADAM10 (Millipore; AB19026; 1:1000), IDE (Abcam; ab232216; 1:1000), GluN2A (Cell Signaling; 4025; 1:1000), GluN2B (Cell Signaling; 4212; 1:1000), GABAAR α5 (Santa Cruz; Sc393921; 1:1000), and β-actin (Millipore; MAB1501; 1:50,000), anti-mouse (Cell Signaling; 7076S; 1:5000), anti-rabbit (Cell Signaling; 7074S; 1:5000). Protein bands were visualized using chemiluminescence using ECL (EMD Millipore). Densitometric analysis was performed using ImageJ, and data were normalized to β-actin (Millipore; MAB1501; 1:50,000). β-actin expression was not affected by genotype or ethanol exposure (supplementary fig. 1).
2.12. Plasma glucose and lactate measurements
Plasma was collected during euthanasia, as described above. Glucose and lactate concentrations were measured using the YSI 2900 analyzer (YSI incorporated) per the manufacturer’s instructions as previously described (Macauley et al., 2015). Data is represented by means ± SEM.
2.13. Insulin ELISA
Plasma was collected during euthanasia, as described above, and insulin was measured by ELISA (Alpco; 80-INSMSU-E10) according to manufacturer’s instructions (Stanley et al., 2016). Homeostasis model assessment of insulin resistance scores were then calculated (HOMA-IR = plasma glucose [mmol/L] × plasma insulin [U/mL]/22.5).
2.14. In vivo microdialysis
To determine how ethanol directly affects ISF glucose, ISF lactate, ISF Aβ40, and ISF Aβ42, a separate cohort of APP/PS1 mice was exposed to a single intoxicating dose of ethanol (2.0 g/kg, 15% w/v, i.p). ISF Aβ40 and Aβ42 concentrations in young mice corresponds to plaque deposition in aged animals (Bero et al., 2011b). Additionally, Aβ aggregates in a concentration-dependent manner. Therefore, 3-month-old APP/PS1 mice were used to measure how Aβ levels change prior to the onset of brain Aβ deposition (Jankowsky et al., 2004). Hippocampal ISF was continuously collected before and after ethanol exposure using in vivo microdialysis as previously described (Macauley et al., 2015). Five days prior to acute ethanol exposure, guide cannulas (BASi) were stereotaxically implanted into the hippocampus (from bregma, A/P: −3.1 mm; M/L: −2.5 mm; D/V: −1.2 mm; at 12° angle) and secured into place with dental cement. One day prior to ethanol, 3-month-old APP/PS1 mice were transferred to sampling cages (Bioanalytical Systems). Microdialysis probes (2 mm; 38 kDa molecular weight cut off; BR-style; BASi) were inserted into the guide cannula, connected to a syringe pump and infused with 0.15% bovine serum albumin (BSA, Sigma) in artificial cerebrospinal fluid (aCSF; 1.3 mM CaCl2, 1.2 mM MgSO4, 3 mM KCl, 0.4 mM KH2PO4, 25 mM NaHCO3 and 122 mM NaCl; pH = 7.35) at a flow rate of 1 μL/min. Hippocampal ISF was collected hourly, beginning in the early afternoon. Approximately 24 h later, mice were administered 3.0 g/kg ethanol via i.p. injection from a 15% ethanol (w/v; in 0.9% saline) and ISF was collected for another 24 h.
2.15. ISF glucose and ethanol measurements
ISF ethanol, glucose and lactate concentrations were measured in each ISF sample from 3-month-old APP/PS1 mice (n = 6–8, supplementary Tables 1–2) using the YSI 2900 analyzer (YSI incorporated) per the manufacturer’s instructions.
2.16. Aβ40 and Aβ42 ELISAs
ISF samples from 3-month-old APP/PS1 mice (n = 5) collected from in vivo microdialysis experiments were analyzed for Aβ40 using sandwich ELISAs as previously described (Bero et al., 2011b; Roh et al., 2012; Macauley et al., 2015). Briefly, Aβ40 and Aβ42 were quantified using monoclonal capture antibodies (generous gifts from Dr. David Holtzman, Washington University) targeted against amino acids 33–40 (HJ2, Aβ40) or amino acids 35–42 (HJ7.4, Aβ42). For detection, a biotinylated monoclonal antibody against the central domain amino acids 13–28 (HJ5.1B) was used, followed by streptavidin-poly-HRP-40. Assays were developed using Super Slow TMB (Sigma). Plates were read on a Bio-Tek Synergy 2 plate reader at 650 nm.
2.17. qPCR
RNA isolation from mouse brain was performed as previously described (Musiek et al., 2013). Briefly, anterior cortex was homogenized by trituration through a 23-gauge needle in TRIzol (Invitrogen). Chloroform (1:5) was added then samples were mixed, and centrifuged (13,000 g; 15 min; 4 °C). Chloroform was removed, and samples were diluted 1:1 in 70% ethanol and purified using RNeasy columns and reagents (QIAGEN). RNA concentration was measured using a NanoDrop spectrophotometer. Reverse transcription was performed using a high-capacity RNA-cDNA kit (Applied Biosystems [ABI]) with 1 μg RNA per 20 μL reaction. Real-time qPCR was performed with ABI TaqMan primers and reagents on an ABI Prizm 7500 thermocycler according to the manufacturer’s instructions. Primers used: Grin2a (TaqMan; Mm00433802_m1), Grin2b (TaqMan; Mm00433820_m1), Gabra5 (IDT; Mm.PT.58.5845925), Actb (TaqMan; Mm01205647_g1). All mRNA measurements were normalized to Actb (β-actin) then to wildtype + H2O group mRNA levels.
2.18. Synaptoneurosome preparation
Synaptoneurosomes (SYNs) were prepared from whole hippocampal tissue as previously described (Sosanya et al., 2013; Ewin et al., 2019). Briefly, whole hippocampal tissue was homogenized in buffer (50 mM Tris, pH 7.35; protease and phosphatase inhibitors (Halt, Thermofisher). Homogenates were sequentially filtered through 100 μm and 5 μm filters to produce SYNs (Niere et al., 2016; Quinlan et al., 1999). SYNs were centrifuged (14,000 g; 20 min; 4 °C) to obtain a pellet that was solubilized in RIPA buffer (150 mM NaCl; 10 mM Tris, pH 7.4; 0.1% SDS; 1% Triton X-100; 1% deoxycholate 5 mM EDTA; Halt). Samples were then centrifuged (14,000 g; 20 min; 4 °C) and the soluble fraction was removed and used for Western blot analysis as described above.
2.19. Statistical analysis
Statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software, Inc., San Diego, CA). Two-way repeated-measures ANOVAs were used to analyze differences between-subject factors (genotype and time or genotype and alcohol exposure) and post hoc analyses (Šídák’s multiple comparisons) were performed for assessing specific group comparisons. Kruskal-Wallis ANOVA was used for nest building score analysis followed by post hoc analyses stated above. Two-way ANOVAs were employed for all other statistical analyses using Tukey’s HSD test for all post hoc analyses. Grubbs outlier tests were performed on all data (α = 0.05), and outliers were excluded from analyses. The level of statistical significance was set at p ≤ 0.05. Data were expressed as means ±SEM. Expanded statistical results can be found in supplementary Tables 4–10.
3. Results
3.1. APP/PS1 mice do not consume more ethanol than wildtype mice
Ethanol-consumption was characterized in APP/PS1 mice after an earlier study reported an initial increase in ethanol consumption in 3xTg-AD mice (Hoffman et al., 2019). First, weekly ethanol consumption and preference was assessed in wildtype and APP/PS1 mice. Over the course of the 10-week study, wildtype and APP/PS1 mice consumed similar amounts of ethanol and displayed a similar preference to ethanol (Fig. 1b–c). Likewise, ethanol-exposed wildtype and APP/PS1 mice consumed similar amounts of water throughout the course of the study (Fig. 1d). Of note, neither wildtype nor APP/PS1 mice did not reach clinically relevant blood ethanol concentrations (BECs, data not shown); however, this could be due to the moderate nature of the ethanol exposure paradigm as well as the rate at which mice metabolize ethanol. Compared to H2O-control wildtype mice, H2O-controlAPP/PS1 mice consistently consumed greater amounts of H2O each week (Fig. 1e, p < 0.0001). This resulted in an overall increase in water consumption over the 10-week experiment. (Fig. 1e, p < 0.0001). Thus, wildtype and APP/PS1 mice consume similar amounts of ethanol.
3.2. Ethanol treatment promotes neurodegeneration in APP/PS1 mice
Neurodegeneration is a major component of AD pathology and AUD (Jack et al., 2016; Rehm et al., 2019). Therefore, brain atrophy was measured in APP/PS1 and wildtype mice after 10 weeks of ethanol self-administration. APP/PS1 mice had lower brain masses than wildtype mice, and this effect was exacerbated by ethanol consumption (Fig. 2a). Ethanol consumption had no effect on brain mass in wildtype mice. There were no differences in cortical thickness (Fig. 2b) or in hippocampal area (Fig. 2c) in APP/PS1 mice, suggesting that other brain regions may be the cause of ethanol-induced brain atrophy.
Fig. 2.
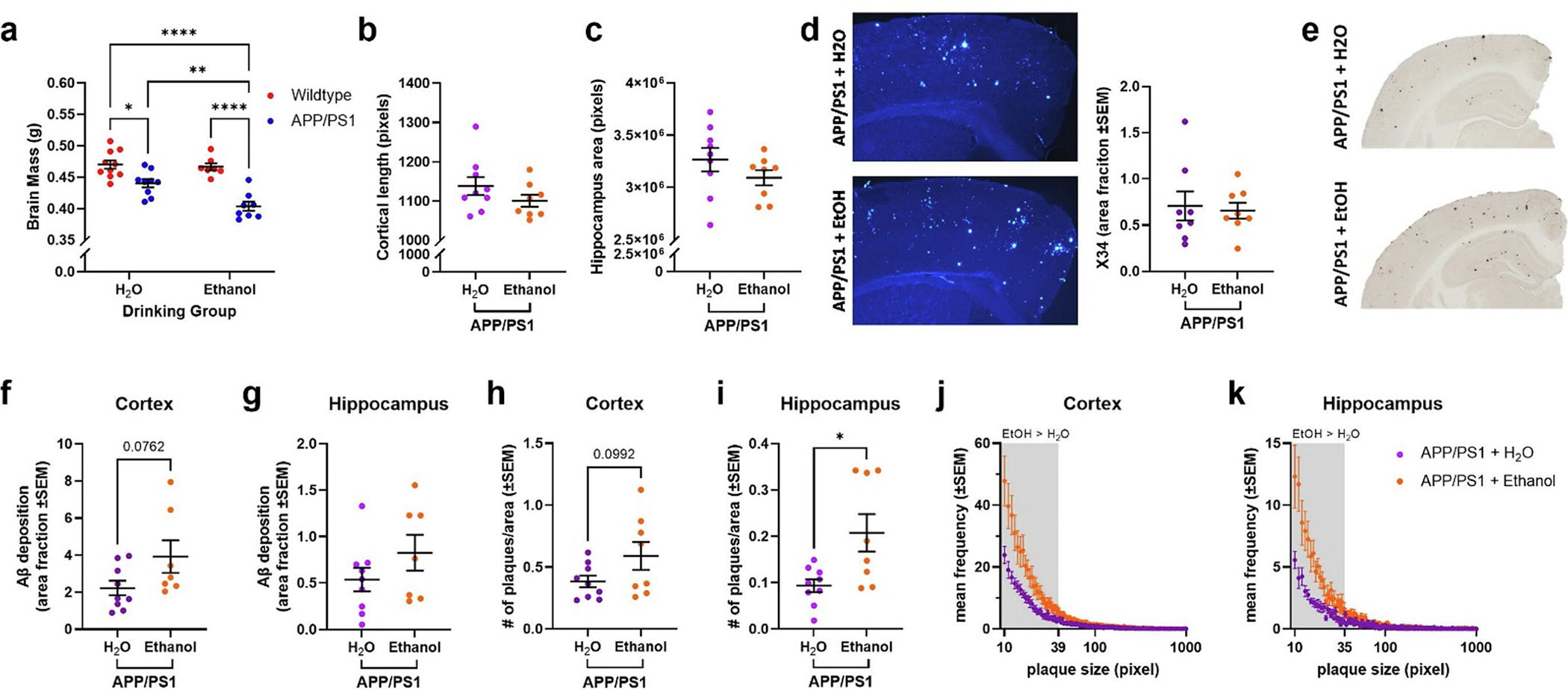
Ethanol exposure increases brain atrophy and amyloid pathology in APP/PS1 mice. a) Brain atrophy was increased in H2O-exposed APP/PS1 mice (p < 0.05), an effect that was exacerbated in ethanol-exposed APP/PS1 mice (p < 0.01). b) Cortical thickness was comparable between H2O- and ethanol-exposed APP/PS1 mice. c) Hippocampal volume was comparable between H2O- and ethanol-exposed APP/PS1 mice. d) Representative images of X34 staining in cortex of H2O- and ethanol-exposed APP/PS1 mice. There were no differences in X34+ amyloid plaques was found. e) Representative images of Aβ deposition in the cortex and hippocampus of H2O- and ethanol-exposed APP/PS1 mice. f) Ethanol-exposed APP/PS1 mice showed a trend towards increased Aβ deposition in the cortex compared to H2O-exposed APP/PS1 mice (p = 0.0762). g) No change in Aβ deposition in the hippocampus of H2O- and ethanol-exposed APP/PS1 mice. h) Ethanol-treated APP/PS1 mice had a trend towards increased cortical plaque number compared to H2O-exposed APP/PS1 mice (p = 0.0992). i) Ethanol-exposed APP/PS1 mice had increased hippocampal plaque number compared to H2O-exposed APP/PS1 mice (p < 0.05). j) Frequency distribution of cortical amyloid plaque size (in pixels). Ethanol-exposed APP/PS1 mice had more smaller plaques in the cortex compared to H2O-exposed APP/PS1 mice. k) Frequency distribution of hippocampal amyloid plaque size (in pixels). Ethanol-exposed APP/PS1 mice had more smaller plaques in the hippocampus compared to H2O-exposed APP/PS1 mice. Wildtype + H2O, n = 10; APP/PS1 + H2O, n = 9; Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8. *p < 0.05, **p < 0.01, ****p < 0.0001.
3.3. Ethanol treatment increases the frequency of smaller amyloid plaques
At high levels, ethanol consumption exacerbates AD-like pathology (Hoffman et al., 2019; Huang et al., 2018). Therefore, Aβ pathology was quantified in APP/PS1 mice after 10 weeks of ethanol self-administration. Quantification of Aβ deposition and amyloid plaques was performed in H2O- and ethanol-exposed APP/PS1 mice using HJ3.4B and X34 staining, respectively (Fig. 2d–e). While ethanol exposure had no effect on the percent area covered by Aβ deposition or amyloid plaques (Fig. 2d), there was a trend towards increased Aβ deposition in the cortex (Fig. 2f, p = 0.0762). There were a greater number of plaques in the hippocampus (Fig. 2i, p < 0.05) and a trend towards increased plaque number in the cortex (Fig. 2h, p = 0.0992). These changes in plaque number corresponded with differences in the size distribution of plaques. Ethanol-exposed mice had a greater number of smaller plaques in the cortex and hippocampus (Fig. 2j–k). Thus, moderate ethanol-exposure may promote Aβ pathology by generating a greater number of smaller plaques throughout the brain. These findings may also represent an intermediate stage of plaque proliferation. An ethanol exposure paradigm that promotes greater amounts of daily ethanol consumption (Huang et al., 2018; Hoffman et al., 2019) or exposes the animal to ethanol for a longer duration may induce greater plaque proliferation. Additional studies are needed to assess whether these changes in plaque size distribution can lead to increased Aβ pathology.
3.4. Ethanol exposure does not alter APP protein levels or metabolism
It is unclear whether these differences in the plaque size and number were due to changes in APP expression, APP processing, or Aβ proteostasis. Therefore, APP expression, APP processing, and Aβ degrading enzymes, such as insulin degrading enzyme (IDE), were quantified via Western blot analysis. APP, CTF-β, and CTF-α protein levels were comparable between H2O- and ethanol-exposed APP/PS1 mice (Fig. 3a–c). BACE-1 and ADAM-10 (β- and α-secretase, respectively) protein levels were also similar between H2O- and ethanol-exposed APP/PS1 mice (Figs. 3d–e). There was also a trend towards decreased insulin degrading enzyme (IDE) protein levels in ethanol-exposed APP/PS1 mice (Figs. 3f). Collectively, this indicates that ethanol exposure does not affect amyloidogenic or non-amyloidogenic APP processing but may lead to decreased Aβ degradation via IDE.
Fig. 3.
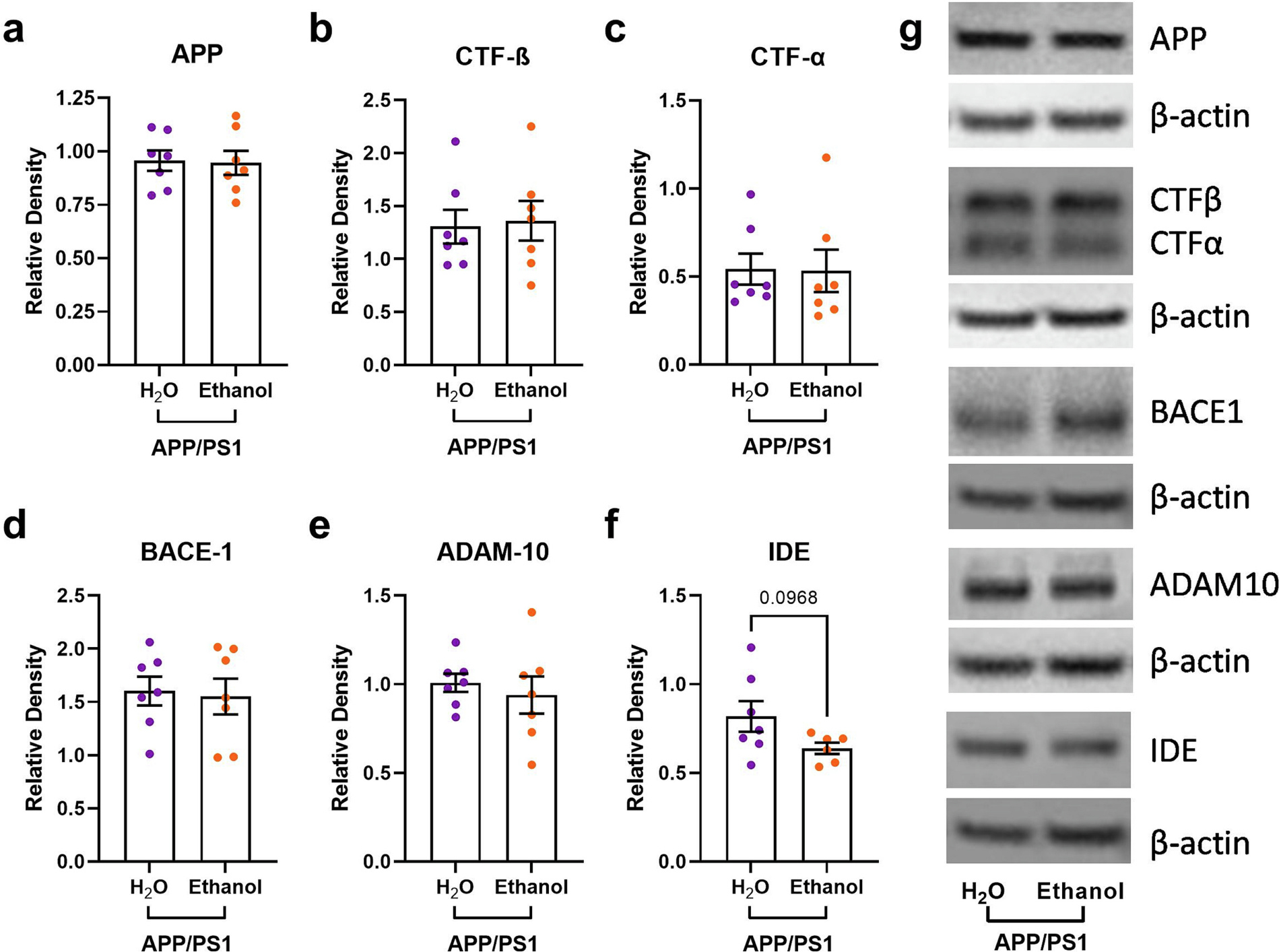
Moderate ethanol drinking does not alter cortical APP metabolism in APP/PS1 mice. There were no differences in a) cortical APP levels, b) cortical CTF -β levels, c) cortical CTF-α levels, d) cortical BACE-1 levels, or e) cortical ADAM-10 levels between H2O- and ethanol-treated APP/PS1 mice; f) There was a trend towards decreased IDE expression in ethanol-treated APP/PS1 mice compared to controls (unpaired t-test, p = 0.0968); g) Representative gels from Western blot experiments. Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8.
3.5. Ethanol exposure dysregulates diurnal food consumption in APP/PS1 mice
AD and AUD are characterized by disruptions in circadian rhythms (Carroll and Macauley, 2019; Koob and Colrain, 2020). Thus, starting at week 6 of ethanol exposure food consumption was measured every 12 h on drinking days (Fig. 4a–c). While food consumption was highest at night for all groups, there was a trend towards decreased food consumption during this period in ethanol-exposed APP/PS1 mice (Fig. 4b). Additionally, ethanol-drinking APP/PS1 mice consumed more food during their light cycle, when mice normally spend more time sleeping (Fig. 4c). Interestingly, this effect was not observed in ethanol-exposed wildtype mice (Fig. 4b–c). Total food consumption across the 24-h day was comparable between H2O-control APP/PS1 and wildtype mice (Fig. 4b–c). Because mice are nocturnal, the majority of food intake occurs during the dark cycle. Thus, the diurnal misalignment of food intake observed here suggests that chronic ethanol consumption may disrupt sleep, specifically in mice with Aβ overexpression.
Fig. 4.
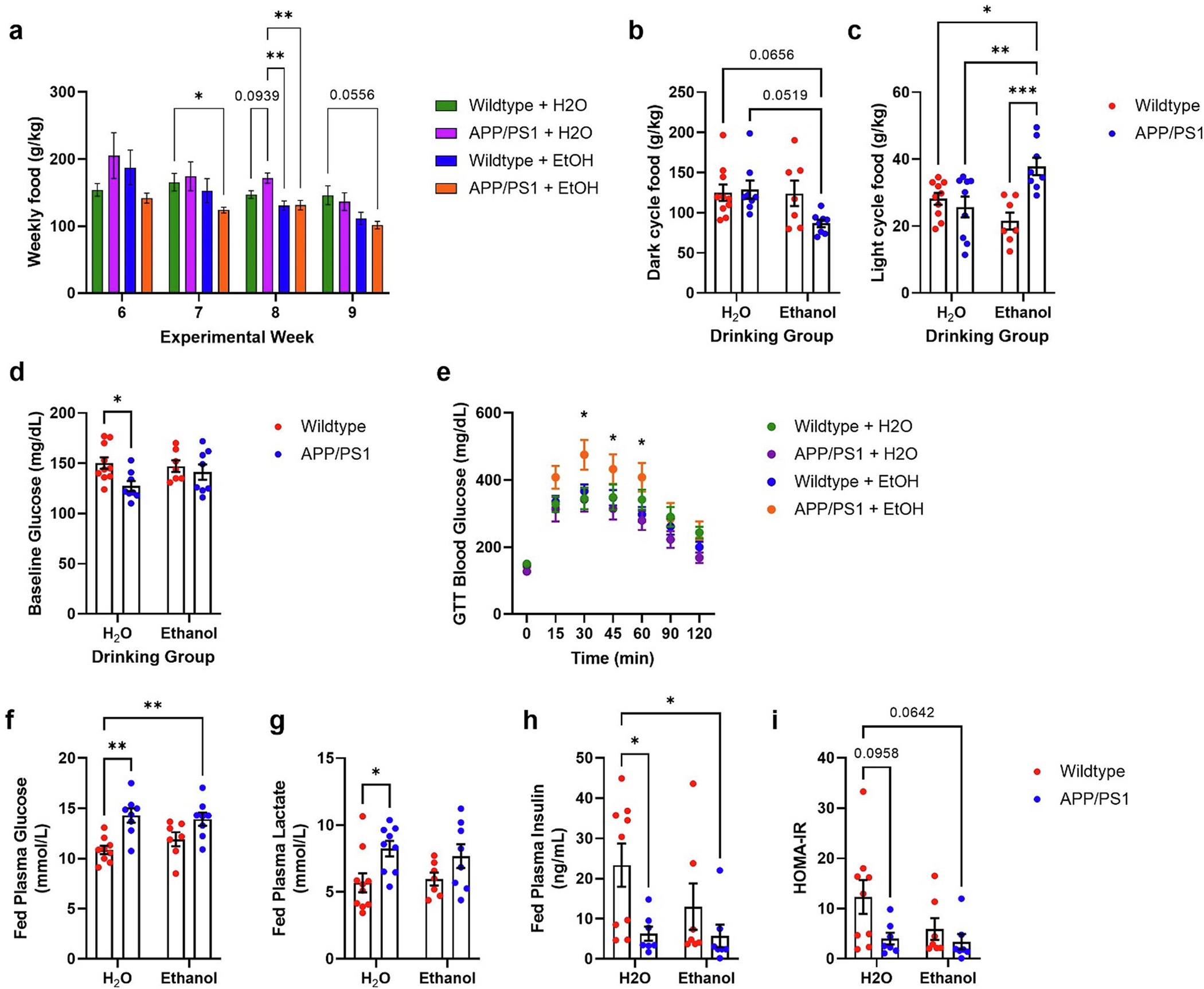
Ethanol exposure dysregulates diurnal feeding behavior and peripheral metabolism in APP/PS1 mice. a) Decreased weekly food consumption in APP/PS1 with or without ethanol treatment during experimental weeks 6–9. b) Ethanol-treated APP/PS1 mice showed decreased food consumption during the dark period. Two-way ANOVA revealed trend of differences between drinking group (p = 0.0580) and genotype × drinking group (p = 0.0697). c) Ethanol-treated APP/PS1 mice showed increased food consumption during light cycle. 2-way ANOVA revealed significant genotype (p = 0.0107) and genotype × drinking group (p = 0.0009) effects. d) H2O-treated APP/PS1 mice showed lower fasted blood glucose concentrations prior to glucose tolerance test. 2-way ANOVA revealed significant genotype effect (p = 0.0273). e) Ethanol-treated APP/PS1 mice displayed glucose intolerance during glucose tolerance test. Two-way ANOVA revealed significance over time (p < 0.0109), and time × group (p < 0.0001). Tukey’s multiple comparisons revealed that EtOH-treated APP/PS1 mice had significantly higher blood glucose concentrations at 30-, 45-, and 60-min post-glucose injection. f) H2O- and EtOH-treated APP/PS1 mice had higher plasma glucose at the terminal timepoint. 2-way ANOVA revealed genotype effect (p = 0.0001). g) H2O-treated APP/PS1 mice had higher plasma lactate levels at the terminal time point compared to H2O-treated wildtype mice. 2-way ANOVA revealed a genotype effect (p = 0.0049). h) H2O- and EtOH-exposed APP/PS1 mice had decreased fed insulin levels at the terminal timepoint, compared to H2O-treated wildtype mice. 2-way ANOVA revealed a genotype effect (p = 0.0123). i) Calculated HOMA-IR values showed a trend trended towards decreased in H2O- and ethanol-exposed APP/PS1 mice. 2-way ANOVA revealed a genotype effect (p = 0.0381). Wildtype + H2O, n = 10; APP/PS1 + H2O, n = 9; Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8. *p < 0.05, **p < 0.01, ***p < 0.001.
3.6. Chronic ethanol exposure alters glucose homeostasis in APP/PS1 mice
AUD and AD are associated with metabolic impairment and impaired glucose homeostasis (Macauley et al., 2015). Therefore, alterations in glucose metabolism were assessed in 7.5-month-old mice after 9 weeks of ethanol exposure. Fasted blood glucose levels were lower in H2O-control APP/PS1 mice compared to wildtype mice (Fig. 4d); however, this effect was reversed with ethanol exposure. In fact, fed plasma glucose levels were elevated in H2O- and ethanol-exposed APP/PS1 mice compared to wildtype mice, suggesting Aβ pathology differentially affects peripheral metabolism (Fig. 4f). Interestingly glucose intolerance was only observed in ethanol-exposed APP/PS1 mice during a glucose tolerance test, suggesting ethanol exposure exacerbates metabolic dysfunction and insulin resistance in APP/PS1 mice (Fig. 4e). In addition to changes in fed glucose levels, H2O-control APP/PS1 mice had elevated fed plasma lactate levels (Fig. 4g). Both H2O- and ethanol-exposed APP/PS1 mice had decreased fed insulin levels at a terminal timepoint (Fig. 4h). They also showed a trend towards increased insulin resistance as measured by HOMA-IR (Fig. 4i). Given these differences in peripheral metabolism, body weights were measured for all groups at the beginning and end of the study. At the terminal timepoint, there were no differences in body weights between groups (data not shown). Taken together these data indicate that moderate levels of chronic ethanol drinking induces metabolic dysfunction in APP/PS1 mice.
3.7. Chronic ethanol consumption alters activity- and dementia-related behaviors in APP/PS1 mice
Under basal conditions, APP/PS1 mice do not demonstrate differences in anxiety- and depression-related behaviors (Webster et al., 2013) but do show increased compulsive behaviors (Shepherd et al., 2021). Compared to wildtype mice, APP/PS1 mice also exhibit heightened depression- and anxiety-related behaviors in response to chronic mild stress (Gao et al., 2018). Ethanol consumption is known to cause a stress response (Lu and Richardson, 2014) and evoke depression- and anxiety-related behaviors (Gong et al., 2017). Certain forms of alcohol use increase the risk to develop AD and other forms of dementias. Thus, behavioral assays were used to determine whether Aβ deposition and ethanol consumption interacted to exacerbate AD-related behavioral phenotypes.
Here, activity-related behaviors were measured using open field (OFA) and light/dark (LD) assays at baseline and after 3 weeks of ethanol exposure. Marble-burying, object location memory (OLM), and nest building tasks were only performed after ethanol exposure. At baseline, APP/PS1 mice showed a trend towards increased locomotor activity during the OFA (Fig. 5a, p = 0.0668), but not in the percent time spent in the center zone (Fig. 5b). After 3 weeks of ethanol exposure, there was increased locomotor activity in ethanol-exposed APP/PS1 mice when compared to H2O-control wildtype mice (Fig. 5a). Further, post-hoc tests showed that ethanol-drinking APP/PS1 mice spent more time in the central zone than H2O-control and ethanol-exposed wildtype mice (Fig. 5b). No differences in behavior were observed in LD at baseline or after 4 weeks of ethanol self-administration (Fig. 5c). After 5 weeks of ethanol drinking, mice were tested using the marble burying task, where increased marble burying is used as a measure of anxiety or compulsive-like behavior. Control APP/PS1 mice buried fewer marbles than wildtype mice, while ethanol-drinking APP/PS1 mice did not (Fig. 5d). This might suggest that H2O-control APP/PS1 exhibit decreased anxiety-like behavior or disengagement in the task. After 7 weeks of ethanol treatment, the OLM task evaluated the effects of ethanol drinking on memory. As expected, H2O-control wildtype mice spent more time interacting with the relocated object than with the object in the familiar location (Fig. 5e, p = 0.0078). Conversely, APP/PS1 mice and ethanol-exposed wildtype mice spent similar amounts of time investigating both objects (Fig. 5e). This indicates that impaired memory due to Aβ pathology was not exacerbated by ethanol exposure. This effect could be due to a ceiling effect as APP/PS1 mice exhibit maximal memory impairment on this assay. Lastly, no significant differences in nest building were observed between H2O-control wildtype and H2O-control APP/PS1 mice (Fig. 5f). In contrast, ethanol-drinking APP/PS1 mice had lower nest scores than wildtype mice. This suggests that chronic moderate ethanol consumption may exacerbate alterations in self-care in APP/PS1 mice.
Fig. 5.
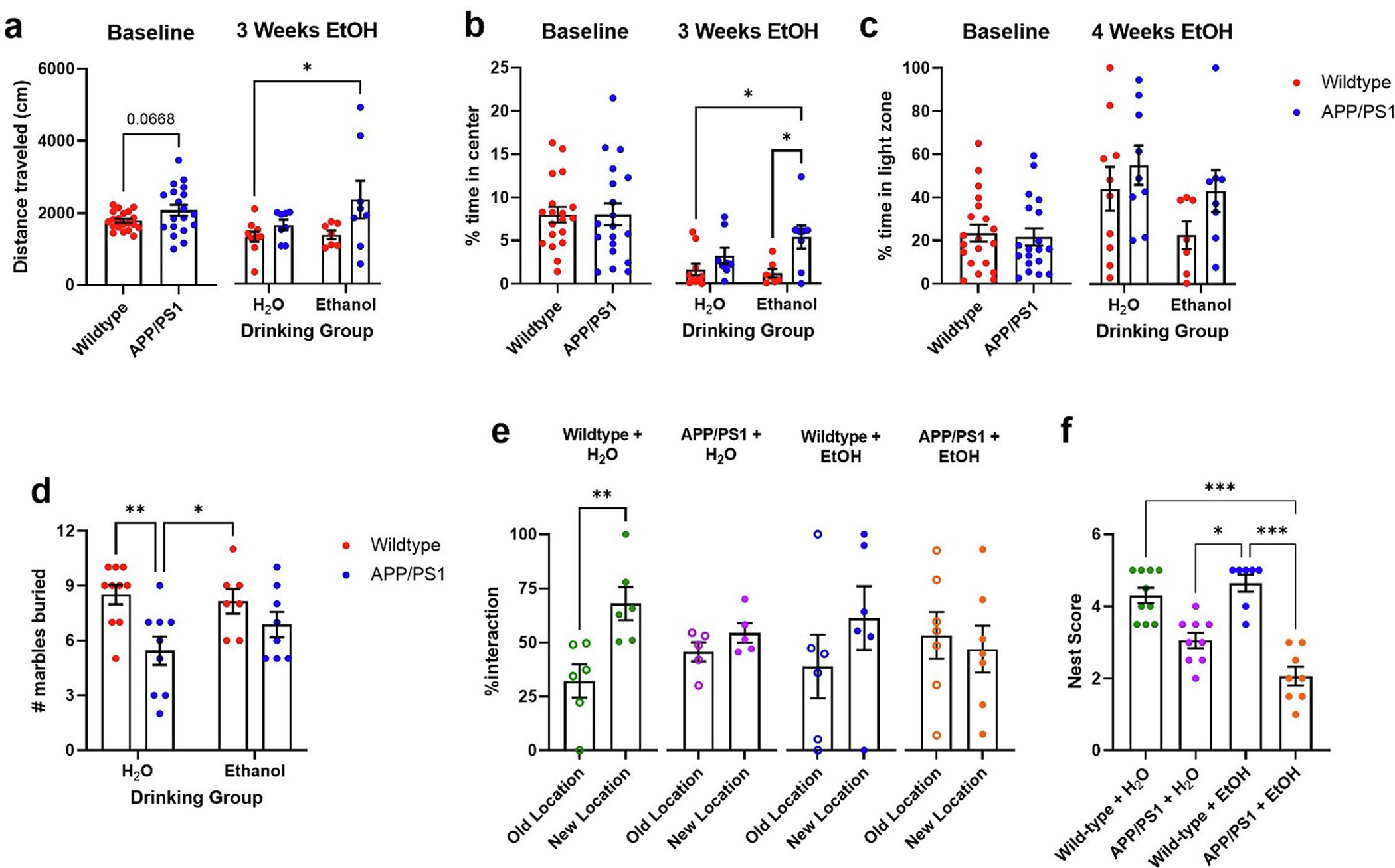
Chronic ethanol consumption alters anxiety-related and dementia-related behaviors in APP/PS1 mice. a) At baseline, APP/PS1 mice showed a trend towards increased locomotor activity during the OFA (unpaired t-test, p = 0.0668). After 3 weeks of ethanol exposure APP/PS1 mice showed more locomotor activity than other groups. b) There were no differences in the % time spent in the center zone at baseline. After 3 weeks of ethanol treatment, APP/PS1 mice spent more time in central zone than wildtype controls. c) Mice exhibited no differences spent in the light zone in the LD box at baseline or following treatment. d) H2O-treated APP/PS1 mice buried more marbles than wildtype controls. e) H2O-treated wildtype spent significantly more time interacting with the relocated object than with the object in the familiar location (unpaired t-test, p = 0.0078), while other groups spent similar amounts of time interacting with both objects. f) APP/PS1 mice + ETOH made poorer nests compared to wildtype mice. 2-way ANOVA revealed differences in nest building scores between groups after 9 weeks of EtOH treatment. (Kruskal-Wallis test: p < 0.0001). Wildtype + H2O, n = 10; APP/PS1 + H2O, n = 9; Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8. *p < 0.05, **p < 0.01, ***p < 0.001.
3.8. Ethanol acutely modulates ISF Aβ40, but not ISF Aβ42, and ISF glucose in APP/PS1 mice
Chronic ethanol exposure altered AD-related pathology and metabolism. Therefore, in vivo microdialysis was used to determine whether ethanol directly modulates hippocampal ISF ethanol, glucose, lactate, Aβ40, and Aβ42 in unrestrained, unanesthetized APP/PS1 mice (Fig. 6a). A moderate, acute dose of ethanol (2.0 g/kg, i.p.) rapidly increased ISF ethanol levels in 3-month-old wildtype (13.63 ± 1.45 mmol/L) and APP/PS1 mice (8.36 ± 1.25 mmol/L). ISF ethanol levels then returned to baseline over the next 4 h (Fig. 6b, Supplementary Table 1). There were no differences in the half life of ISF ethanol between wildtype and APP/PS1 mice (Fig. 6c). These data demonstrate that ethanol freely crosses the blood brain barrier into hippocampal ISF and is cleared from the ISF at similar rates in both mouse strains. Similarly, in both wildtype and APP/PS1 mice, ISF glucose levels increased while ethanol was in the brain and returned to baseline levels after ethanol was cleared from the ISF (Fig. 6d–e, supplementary Table 2). Meanwhile, ethanol had no effect on ISF lactate levels (Fig. 6f–g, supplementary Table 2).
Fig. 6.
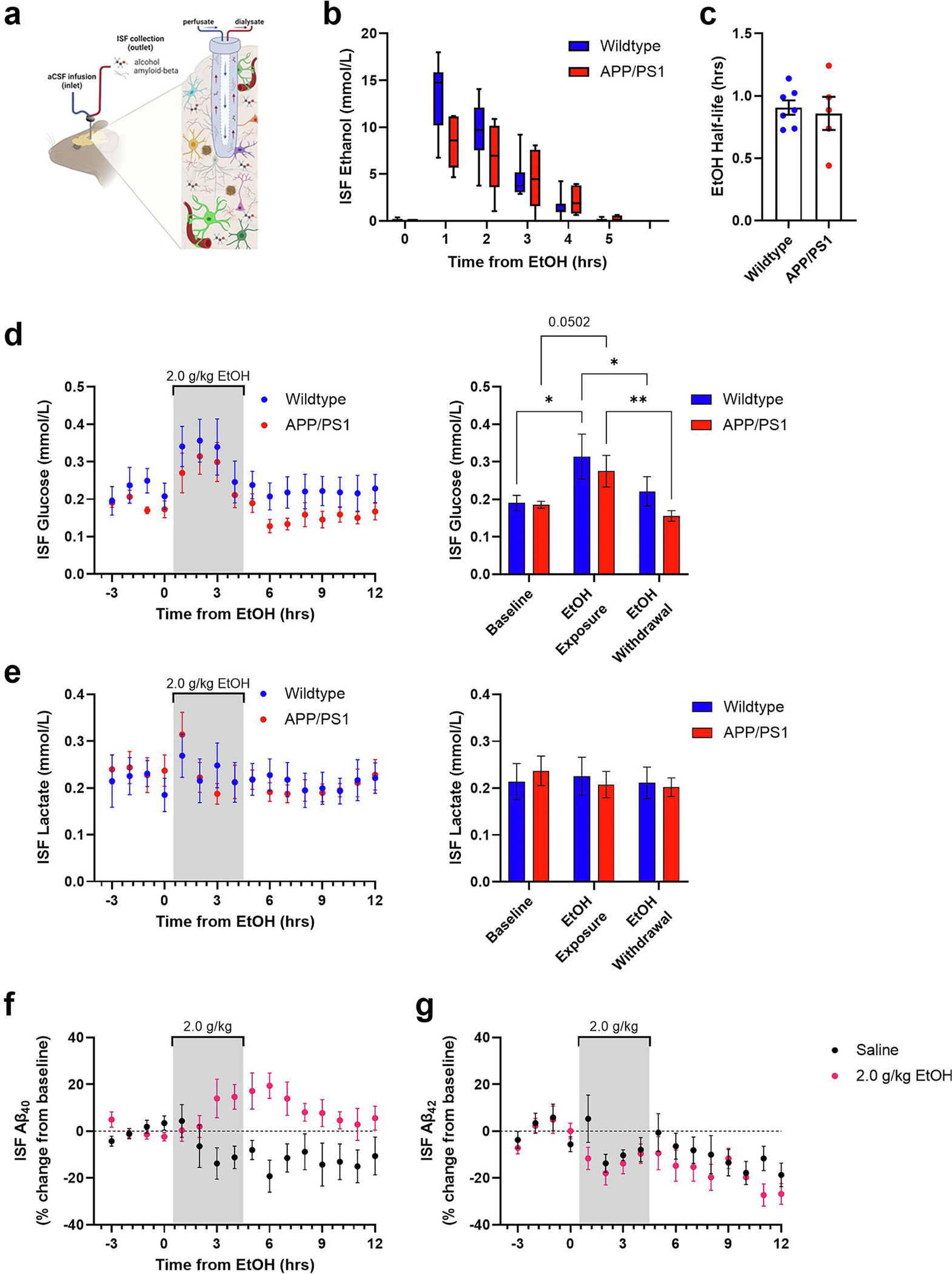
Ethanol acutely modulates ISF Aβ40, but not ISF Aβ42, and ISF glucose in 3-month-old APP/PS1 mice. a) Schematic of 38 kDa in vivo microdialysis to sample brain hippocampal interstitial fluid (ISF). b) Ethanol is detectable in the ISF for 4 h after acute ethanol exposure (2.0 g/kg, ip; wildtype, n = 9; APP/PS1, n = 6). c) ISF half-life is similar between 3-month-old wildtype and APP/PS1 mice (wildtype, n = 7; APP/PS1, n = 5). d-e) ISF glucose levels increase during an ethanol exposure (gray bar) and return to near-baseline levels during withdrawal (wildtype, n = 8; APP/PS1, n = 7). f-g) Acute ethanol does not affect ISF lactate levels (wildtype, n = 7; APP/PS1, n = 7). h) ISF Aβ40 levels increase during withdrawal from 2.0 g/kg ethanol (saline, n = 6; ethanol, n = 9). i) ISF Aβ42 levels are unaffected by 2.0 g/kg ethanol (saline, n = 4; ethanol, n = 7). *p < 0.05; **p < 0.01.
An acute ethanol dose increased ISF Aβ40 levels by 19.40% ± 5.46% at 6 h when compared to a saline control (Fig. 6f). ISF Aβ40 levels returned to baseline over the next 3–6 h. Conversely, ISF Aβ42 levels did not rise and were similar to changes post-saline injection, indicating that acute ethanol injection had no effect on ISF Aβ42 (Fig. 6g). These data show that ethanol selectively increases ISF Aβ40, but not Aβ42, during withdrawal. Thus, acute ethanol dysregulates brain metabolism while selectively increasing ISF Aβ40.
3.9. Ethanol exposure alters NMDA and GABAA receptor gene expression
Aβ40 is released into the ISF in response to glutamatergic neurotransmission (Bero, 2011a; Cirrito et al., 2008; Cirrito et al., 2005). Furthermore, ethanol exposure and withdrawal directly modulate neuronal excitability and inhibition in rodents. N-methyl-d-aspartate and γ-aminobutyric acid type-A receptors (NMDARs and GABAARs) play important roles in mediating excitability and inhibition during ethanol exposure and withdrawal. NMDAR and GABAAR subunit expression is altered in AUD patients and in rodents after chronic ethanol exposure (Farris and Mayfield, 2014; Roberto et al., 2006; Gruol et al., 2018). Therefore, we examined mRNA and protein levels for excitatory and inhibitory receptors in WT and APP/PS1 mice from the chronic ethanol drinking study. Specifically, we measured NMDAR subunits GluN2A (Grin2a) and GluN2B (Grin2b) as well as the α-5 subunit of GABAAR (GABAAα5; Gabra5) via qPCR and Western blot. There were no differences between groups in cortical Grin2a mRNA levels (Fig. 7a). Cortical Grin2b mRNA levels were elevated in ethanol-exposed APP/PS1 mice compared to ethanol-exposed wildtype mice (Fig. 7b, p = 0.0319), suggesting that moderate ethanol increases cortical GluN2B expression in APP/PS1 mice. Gabra5 mRNA levels were elevated in H2O-control APP/PS1 mice relative to H2O-control wildtype (Fig. 7c, p = 0.0388). Interestingly, ethanol-exposed APP/PS1 mice showed a trend towards lower Gabra5 mRNA levels compared to H2O-control APP/PS1 mice (Fig. 7c, p = 0.0687). This suggests that chronic ethanol decreases α-5 subunit-containing GABAARs in APP/PS1 mice. Next, synaptoneurosomes were isolated from the hippocampus and analyzed via Western blot to explore changes in NMDAR (e.g. GluN2A, GluN2B) and GABAAR (e.g. GABAAα5) subunit levels occurring at the synapse. In synaptoneurosomes, GluN2A and GluN2B levels were similar between groups (Fig. 7d–e). Ethanol exposure increased synaptic GABAAα5 protein levels in wildtype mice, but not in APP/SP1 mice (Fig. 7f). Together, these results suggest that ethanol-exposure affects NMDA and GABAA receptor subunits differentially across brain regions as well as trafficking to the synapse in APP/PS1 mice compared to wildtype.
Fig. 7.
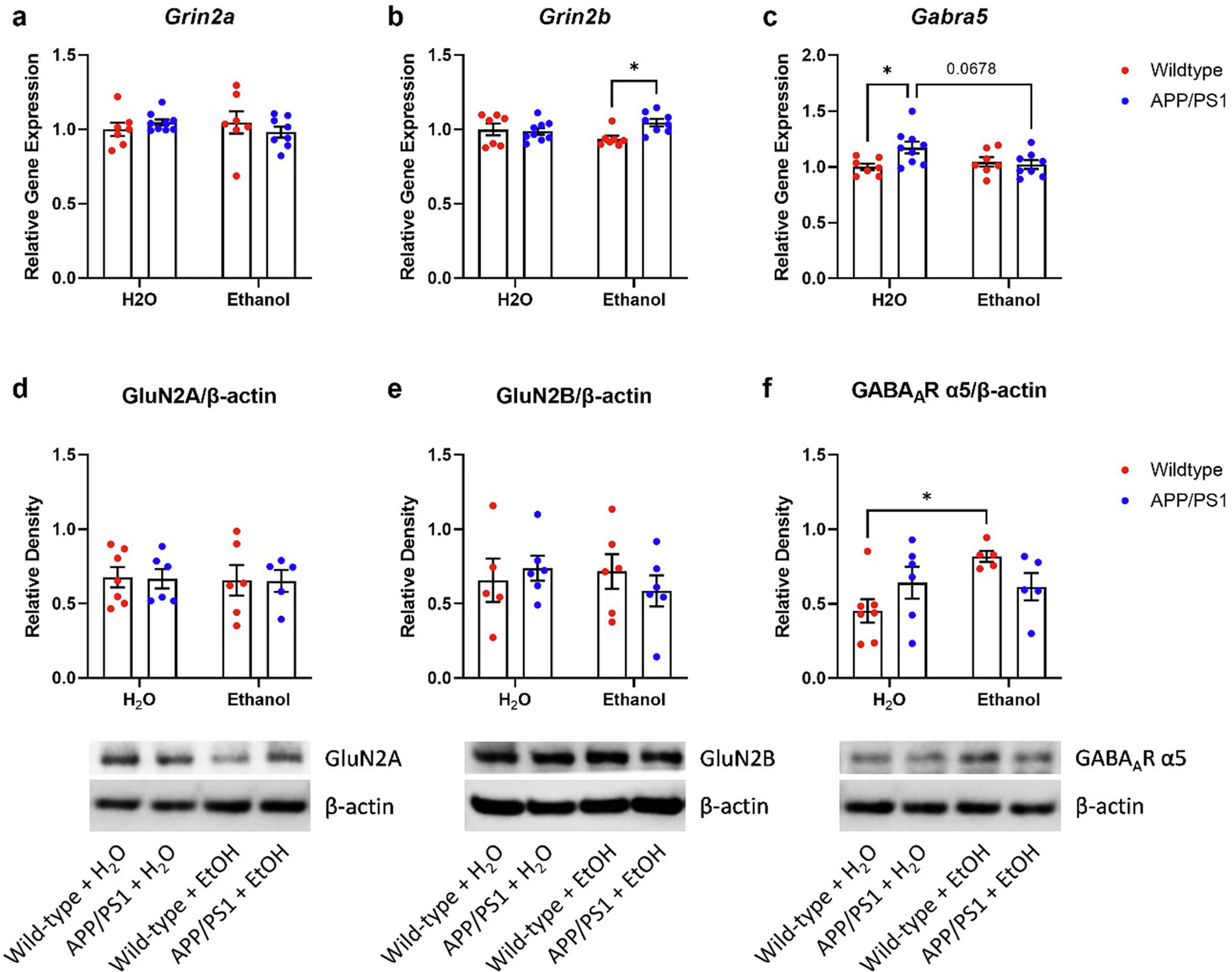
Chronic moderate drinking differentially alters NMDA and GABAA receptors in the cortex and hippocampus of APP/PS1 mice. a) Ethanol treatment did not alter cortical Grin2a expression in wildtype or APP/PS1 mice. b) Ethanol-treated APP/PS1 mice had higher cortical Grin2b expression compared to EtOH-treated wildtype mice. 2-way ANOVA revealed a significant treatment × genotype interaction (p = 0.0319). c) H2O-treated APP/PS1 mice showed increased cortical Gabra5 expression compared to H2O-exposed wildtype mice (p < 0.05). This effect was lost in EtOH-exposed APP/PS1 Gabra5 mRNA levels. 2-way ANOVA revealed a significant treatment × genotype interaction (p = 0.0249) and a trend in genotype effects (p = 0.0723). d) Synaptic GluN2A levels was unaltered in the hippocampus of H2O- or EtOH-treated wildtype or APP/PS1 mice. e) Synaptic GluN2B levels was unaltered in the hippocampus of H2O- or EtOH-treated wildtype or APP/PS1 mice. f) Ethanol-treated wildtype mice showed increased synaptic GABAAR α5 subunit levels compared to H2O-treated wildtype mice. Ethanol treatment had no effect on GABAAR α5 subunit levels in APP/PS1 mice. 2-way ANOVA revealed a significant treatment × genotype effect (p = 0.0347) and a trend in treatment effects (p = 0.0644). Wildtype + H2O, n = 10; APP/PS1 + H2O, n = 9; Wildtype + EtOH, n = 7; APP/PS1 + EtOH, n = 8. *p < 0.05.
4. Discussion
This study found that long-term, moderate ethanol self-administration increases AD-related pathology, alters peripheral metabolism, and exacerbates some behavioral deficits in APP/PS1 mice. Ethanol exposure consistently exacerbated phenotypes related to Aβ pathology, neuronal E/I balance, metabolism, and behavior. This suggests that early changes in Aβ pathology synergize with ethanol exposure to potentiate damage to the brain. During withdrawal after an acute ethanol exposure, hippocampal ISF Aβ40 levels were selectively increased. To our knowledge, this is the first study to show that a single exposure of ethanol directly modulates ISF Aβ40 but not Aβ42 levels. These findings also build upon existing studies to demonstrate that even at moderate intake levels, ethanol exposure can worsen AD-related pathology and behavioral impairment.
Chronic alcohol consumption can lead to increased anxiety and depression (Schuckit and Marc, 1996), which can be further exacerbated by changes in glucose metabolism (Bouwman et al., 2010). Further, mild cognitive impairment (MCI), which is an early indicator of dementia, is associated with increased rates of anxiety and depression. To determine whether chronic ethanol consumption would exacerbate these symptoms associated with MCI and dementia, a battery of behavioral assays was performed. During weekly withdrawal periods, ethanol-drinking APP/PS1 mice exhibited increased locomotor activity and central zone exploration in the OFA but not in LD exploration (Fig. 5c). Ethanol did not induce these behavioral changes in wildtype mice, suggesting that ethanol-drinking APP/PS1 mice may exhibit hyperactive, impulsive-, or compulsive-like behaviors. Conversely, ethanol treatment did not aggravate the deficits in the marble-burying task observed in APP/PS1 controls (Fig. 5d). Thus, ethanol drinking may not exacerbate anxiety or depression-related behaviors. Future studies will expound on and gain greater insight into the behavioral phenotypes reported here.
The effect of ethanol on cognition are well-documented (Sabia et al., 2014) and the present study indicates that chronic ethanol drinking may be detrimental to long-term memory in wildtype mice. In the object location memory task (OLM), ethanol exposure disrupted memory in wildtype mice but had no effect on APP/PS1 mice, possibly due to a ceiling effect (Fig. 5e). Future studies should employ more sensitive cognitive tasks to identify the degree to which ethanol affects cognition in APP/PS1 mice. One major deficit commonly observed in patients with mild cognitive impairment is a disruption in self-care behaviors (e.g. cleaning one’s room, showering, etc.) (Hirschfeld et al., 2000). In this study, ethanol drinking exacerbated deficits in self-care-related behaviors, as measured by nest building (Jirkof, 2014). While no differences in nest building were observed between wildtype and APP/PS1 H2O-control mice, ethanol-drinking APP/PS1 mice displayed reduced nest-building behaviors compared to H2O-control and ethanol drinking wildtype mice suggesting ethanol exposure affects self care (Fig. 5f).
APP/PS1 mice exhibited metabolic dysfunction after both acute and chronic ethanol exposure. An acute ethanol treatment increased ISF glucose levels in wildtype and APP/PS1 mice (Fig. 6d–e). Previous work shows that changes in ISF glucose are sufficient to drive changes in ISF Aβ levels (Macauley et al., 2015), offering a metabolic explanation for the relationship between AUD and AD. The data from the chronic ethanol drinking studies reinforce the idea that moderate ethanol exposure alters peripheral metabolism. APP/PS1 mice demonstrated alterations in fed glucose, lactate, and insulin levels when compared to wildtype mice (Fig. 4f–i). This suggests Aβ pathology disrupts glucose homeostasis independent of ethanol. Ethanol exposure also caused glucose intolerance but only in APP/PS1 mice, which may have been due to hypoinsulinemia. Furthermore, these changes were not observed in wildtype mice, suggesting that Aβ and ethanol interact to exacerbate changes in metabolism in an AD-specific manner. While metabolic diseases like type-2 diabetes are known to put the brain at risk for AD (Ott et al., 1999; Arnold et al., 2018), a growing body of evidence suggests that AD can also exacerbate metabolic dysfunction, glucose intolerance, or insulin resistance.
Ethanol intake causes a metabolic shift in brain metabolism, utilizing acetate instead of glucose for energy production (Volkow et al., 2013; Volkow et al., 2015). An acute ethanol exposure increased ISF glucose levels, which may be indicative of a glucose-to-acetate shift in energy utilization. Ethanol consumption also increases insulin sensitivity and glucose uptake in the periphery (Facchini et al., 1994), which may be a means by which ethanol consumption can reduce the risk to develop type 2 diabetes and alleviate glucose intolerance (Knott et al., 2015). Because APP/PS1 mice show glucose intolerance (Macklin et al., 2017), this study sought to identify whether ethanol could alter glucose tolerance. Instead, glucse intolerance was exacerbated in ethanol-exposed APP/PS1 mice when compared to control APP/PS1 mice. This suggests that amyloid pathology may interact with ethanol consumption in a maladaptive manner, worsening metabolic dysfunction.
Despite glucose intolerance, ethanol-exposed APP/PS1 mice did not eat more overall. Nevertheless, they did show shifts in diurnal eating patterns, with greater food consumption during their light cycle when they should be asleep (Fig. 4c). This diurnal mismatch in feeding behavior suggests that ethanol-exposed APP/PS1 mice have altered sleep/wake cycles, which may potentiate their metabolic dysfunction and plaque formation. A bidirectional relationship exists between AD and sleep impairment, where disrupted sleep increases AD risk, and increased Aβ and tau aggregation further disrupts sleep (Carroll and Macauley, 2019). Individuals with AUD also show disrupted sleep (Koob and Colrain, 2020). Thus, alterations in sleep and diurnal rhythms, like those observed with feeding behavior, may provide one explanation for why AUD increases AD risk. Interestingly, a recent study also demonstrated that nest-building increases during proximity to sleep in mice (Sotelo et al., 2022). Although further studies are needed, these findings suggest that chronic ethanol drinking exacerbates disruptions in metabolism and sleep that are frequently observed in AD.
In humans, amyloid pathology begins to accumulate ~10–20 years before the onset of clinical symptoms (Jack Jr. et al., 2010). In APP/PS1 mice, Aβ begins to aggregate into amyloid plaques from 6 to 9 months of age (Jankowsky et al., 2004). Previous studies consistently show that chronic ethanol exposure is sufficient to increase amyloid burden in mouse models of cerebral amyloidosis (Hoffman et al., 2019; Huang et al., 2018). Here, APP/PS1 mice showed signs of brain atrophy, as measured by decreased brain mass. Chronic ethanol consumption exacerbated this phenotype in APP/PS1 mice while ethanol had no effect on brain mass in wildtype mice (Fig. 2a). This could be due to an interaction between ethanol and the formation of amyloid plaques or through a mechanism independent from AD-like pathology. Interestingly, AUD can lead to different forms of dementia, including vascular dementia, Parkinson’s disease, Korsakoff syndrome, or alcohol-related dementia – all of which display alcohol-related brain damage, neurodegeneration, and brain atrophy (Hakon et al., 2018). Future studies are needed to tease apart whether the brain atrophy observed here was dependent on changes in the Alzheimer’s cascade.
While ethanol had modest effects on amyloid plaque burden, it increased the number of plaques in the cortex and hippocampus (Fig. 2h–i) and resulted in a greater number of small plaques in both the cortex and hippocampus (Fig. 2j–k). Ethanol exposure could be driving this effect either by initiating the formation of more plaques or by restricting plaque growth. First, chronic ethanol may exacerbate amyloid pathology by increasing the number of smaller plaques at an age that corresponds to presymptomatic AD. A greater number of smaller plaques could create multiple pro-aggregation sites or plaque seeds, which would ultimately lead to greater plaque proliferation later in life. Alternatively, the emergence of smaller amyloid plaques could suggest that low-to-moderate ethanol consumption somehow restricts plaque growth. Future studies should explore whether these changes in plaque size and plaque number are a harmful or protective response to moderate alcohol consumption. Nevertheless, because ethanol clearly drives brain atrophy in APP/PS1 mice, it is more likely that the increased plaque number and reduced plaque size are early aggregatory events that would be potentiated if ethanol dose and duration were increased. Because female mice show greater AD-like pathology after chronic ethanol exposure (Tucker et al., 2022), future studies will explore sex differences by utilizing both female and male mice.
In vivo microdialysis showed that a moderate dose of ethanol directly modulates ISF Aβ40 levels in unrestrained and unanesthetized APP/PS1 mice. Interestingly, ISF Aβ40 levels increased during ethanol withdrawal while ISF Aβ42 levels were unaffected by ethanol. (Fig. 6 f–g). Because Aβ42 is the more aggregate-prone species, ethanol’s selective effect on Aβ40 during withdrawal might provide an additional mechanism by which the moderate ethanol exposure had a mild effect on Aβ deposition. It might also explain why the changes in plaque number and sizes did not correspond with changes in APP, CTFα, CTFβ, APP secretase enzymes, or Aβ degrading enzymes (Fig. 3). By selectively increasing Aβ40 during withdrawal, moderate ethanol consumption could limit plaque growth, but not proliferation. While this has important implications for understanding the etiology of AUD-associated dementia, further studies are needed to understand and explore the mechanisms by which ethanol exposure preferentially increases Aβ40 over Aβ42.
One potential mechanism driving increased ISF Aβ40 during withdrawal relates to the relationship between ethanol and neuronal activity. Because Aβ40 is released from neurons in an activity-dependent manner (Cirrito et al., 2005; Bero et al., 2011b; Verges et al., 2011), the increase in ISF Aβ40 levels during withdrawal may be due to ethanol withdrawal-induced neuronal hyperactivity. Ethanol directly modulates neuronal activity by increasing GABA inhibition during exposure and increasing NMDA hyperexcitability during withdrawal (Weiner and Valenzuela, 2006; Ramachandran et al., 2015; Roberto and Varodayan, 2017; Ariwodola and Weiner, 2004; Slawecki et al., 2006; Cheaha et al., 2014; Wang et al., 2016). Preclinical and postmortem studies demonstrate that the NMDAR subunits, GluN2A and GluN2B, are upregulated in rodents after a chronic ethanol exposure as well as in humans with AUD (Farris and Mayfield, 2014; Roberto et al., 2004). The effects of chronic ethanol on GABAARs are also well-documented (Roberto and Varodayan, 2017), and α5 subunit-containing GABAARs (GABAAR-α5) are modulated by chronic ethanol in preclinical studies at the gene and protein level (Zeng et al., 2019; Gruol et al., 2018; Centanni et al., 2014). Moderate ethanol exposure led to changes in GluN2B and GABAAα5 mRNA and protein levels (Fig. 7). Cortical GluN2B mRNA levels were higher in ethanol-exposed APP/PS1 mice than in ethanol-exposed wildtype mice. Conversely, cortical GABAAα5 mRNA levels were lower in ethanol-exposed APP/PS1 mice than in APP/PS1 controls (Fig. 7). Though moderate, these changes correspond with a trend towards increased Aβ deposition and plaque number in the cortex of ethanol-exposed APP/PS1 mice (Fig. 2g, i). Furthermore, these changes could represent a disruption in the brain’s E/I balance that put the brain in a more hyperexcitable state. Furthermore, the ethanol-induced changes in E/I balance may drive the activity-dependent production of Aβ40. This may represent an early pathological mechanism by which ethanol increases plaque deposition. While this study is limited in that it does not include functional assays of brain excitability, it identifies key mechanisms by which ethanol drives AD-like pathology. Furthermore, this study lays the groundwork for future studies seeking to understand the relationship between AUD and AD-like pathology. Future studies are still needed to explore how higher doses of ethanol alters metabolism and neuronal E/I balance in APP/PS1 mice, and how modulating those factors ameliorates or exacerbates amyloid pathology.
5. Conclusions
Contrary to prior clinical and preclinical studies, this study demonstrates that chronic intake of moderate amounts of ethanol can exacerbate behavioral and pathological AD-like phenotypes in APP/PS1 mice. A chronic, moderate drinking paradigm leads to a shift in amyloid plaque development while acute ethanol increases ISF Aβ40, but not ISF Aβ42 levels. Moderate ethanol consumption altered NMDAR and GABAAR subunit mRNA levels, potentially disrupting the brain’s E/I balance. Taken together, these data suggest that ethanol may affect AD-like pathology through increased brain excitability. In addition to changes in the E/I balance, acute and chronic ethanol exposure profoundly impacted peripheral and CNS metabolism, both of which exacerbate AD-related pathology. These findings contribute to the growing body of evidence that suggests chronic alcohol consumption may represent an important, modifiable risk factor for AD. Future studies will further characterize the biological mechanisms by which chronic ethanol intake promotes and exacerbates AD-related pathology.
Supplementary Material
{kind=link}
Funding
We would like to acknowledge the following funding sources: T32AA007565 (SMD, SCG), K01AG050719 (SLM), R01AG068330 (SLM), BrightFocus Foundation A20201775S (SLM), Averill Foundation (SLM), P50AA26117 (JLW).
Abbreviations:
- AD
Alzheimer’s disease
- Aβ
Amyloid-β
- APP
Amyloid precursor protein
- CTF
C-terminal fragment
- E/I
Excitatory/inhibitory
- GABAAR
γ-aminobutyric acid type A receptor
- ISF
Interstitial fluid
- IDE
Insulin degrading enzyme
- LD
Light/dark assay
- NMDAR
N-methyl-d-aspartate receptor
- OFA
Open field assay
- OLM
Object location memory
- SYN
Synaptoneurosome
Footnotes
CRediT authorship contribution statement
Stephen C. Gironda: Conceptualization, Investigation, Formal analysis, Writing – original draft, Writing – review & editing. Stephen M. Day: Conceptualization, Investigation, Formal analysis, Writing – original draft, Writing – review & editing. Caitlin W. Clarke: Investigation. J. Andy Snipes: Investigation. Noelle I. Nicol: Investigation. Hana Kamran: Investigation. Warner Vaughan: Investigation. Shannon L. Macauley: Supervision, Conceptualization, Investigation, Formal analysis, Writing – original draft, Writing – review & editing, Funding acquisition. Jeff L. Weiner: Supervision, Conceptualization, Investigation, Formal analysis, Writing – original draft, Writing – review & editing, Funding acquisition.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105967.
Data availability
Data will be made available on request.
References
- Amodeo Dionisio A., Jones Joshua H., Sweeney John A., Ragozzino Michael E., 2012. Differences in BTBR T+ tf/J and C57BL/6J mice on probabilistic reversal learning and stereotyped behaviors. Behav. Brain Res. 227, 64–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariwodola OJ, Weiner JL, 2004. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABA(B) receptors. J. Neurosci. 24, 10679–10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold SE, Arvanitakis Z, Macauley-Rambach SL, Koenig AM, Wang HY, Ahima RS, Craft S, Gandy S, Buettner C, Stoeckel LE, Holtzman DM, Nathan DM, 2018. Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat. Rev. Neurol. 14, 168–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM, 2011a. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM, 2011b. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat. Neurosci. 14, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwman Vanessa, Adriaanse Marcel C., Riet Esther van’t, Snoek Frank J., Dekker Jacqueline M., Nijpels Giel, 2010. Depression, anxiety and glucose metabolism in the general dutch population: the new Hoorn study. PLoS One 5, e9971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll Caitlin M., Macauley Shannon L., 2019. The interaction between sleep and metabolism in Alzheimer’s disease: cause or consequence of disease?’, Frontiers in aging. Neuroscience 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centanni SW, Teppen T, Risher ML, Fleming RL, Moss JL, Acheson SK, Mulholland PJ, Pandey SC, Chandler LJ, Swartzwelder HS, 2014. Adolescent alcohol exposure alters GABAA receptor subunit expression in adult hippocampus. Alcohol. Clin. Exp. Res. 38, 2800–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheaha D, Sawangjaroen K, Kumarnsit E, 2014. Characterization of fluoxetine effects on ethanol withdrawal-induced cortical hyperexcitability by EEG spectral power in rats. Neuropharmacology 77, 49–56. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Yamada KA, Finn MB, Sloviter RS, Bales KR, May PC, Schoepp DD, Paul SM, Mennerick S, Holtzman DM, 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM, 2008. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron 58, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day Stephen M., Yang Wenzhong, Wang Xin, Stern Jennifer E., Zhou Xueyan, Macauley Shannon L., Ma Tao, 2019. Glucagon-like Peptide-1 cleavage product improves cognitive function in a mouse model of down syndrome. eneuro 6. ENEURO.0031–19.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deacon RM, 2006. Assessing nest building in mice. Nat. Protoc. 1, 1117–1119. [DOI] [PubMed] [Google Scholar]
- Ewin Sarah E., Morgan James W., Niere Farr, McMullen Nate P., Barth Samuel H., Almonte Antoine G., Raab-Graham Kimberly F., Weiner Jeffrey L., 2019. Chronic intermittent ethanol exposure selectively increases synaptic excitability in the ventral domain of the rat hippocampus. Neuroscience 398, 144–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchini F, Chen YD, Reaven GM, 1994. Light-to-moderate alcohol intake is associated with enhanced insulin sensitivity. Diabetes Care 17 (2), 115–119. 10.2337/diacare.17.2.115. [DOI] [PubMed] [Google Scholar]
- Farris SP, Mayfield RD, 2014. RNA-Seq reveals novel transcriptional reorganization in human alcoholic brain. Int. Rev. Neurobiol. 116, 275–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Jun-Ying., Chen Ying., Su Dong-Yuan., Marshall Charles., Xiao Ming., 2018. Depressive- and anxiety-like phenotypes in young adult APPSwe/PS1dE9 transgenic mice with insensitivity to chronic mild stress. Behav Brain Res 353, 114–123. 10.1016/j.bbr.2018.07.007. [DOI] [PubMed] [Google Scholar]
- Gong Mei-Fang., Wen Rui-Ting., Xu Ying., Pan Jian-Chun., Fei Ning., Zhou Yan-Meng., Xu Jiang-Ping., Liang Jian-Hui., Zhang Han-Ting., 2017. Attenuation of ethanol abstinence-induced anxiety- and depressive-like behavior by the phosphodiesterase-4 inhibitor rolipram in rodents. Psychopharmacology (Berl) 234 (20), 3143–3151. 10.1007/s00213-017-4697-3. [DOI] [PubMed] [Google Scholar]
- Gruol DL, Huitron-Resendiz S, Roberts AJ, 2018. Altered brain activity during withdrawal from chronic alcohol is associated with changes in IL-6 signal transduction and GABAergic mechanisms in transgenic mice with increased astrocyte expression of IL-6. Neuropharmacology 138, 32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakon J, Quattromani MJ, Sjolund C, Tomasevic G, Carey L, Lee JM, Ruscher K, Wieloch T, Bauer AQ, 2018. Multisensory stimulation improves functional recovery and resting-state functional connectivity in the mouse brain after stroke. Neuroimage Clin. 17, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood DG, Kalechstein A, Barker WW, Strauman S, St George-Hyslop P, Iglesias C, Loewenstein D, Duara R, 2010. The effect of alcohol and tobacco consumption, and apolipoprotein E genotype, on the age of onset in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 25, 511–518. [DOI] [PubMed] [Google Scholar]
- Hirschfeld Robert M.A., Montgomery Stuart A., Keller Martin B., Kasper Siegfried, Schatzberg Alan F., Moller Hans-Jurgen, Healy David, Baldwin David, Humble Mats, Versiani Marcio, 2000. Social functioning in depression: a review. J. Clin. Psychiatry 61, 0–0. [DOI] [PubMed] [Google Scholar]
- Hoffman JL, Faccidomo S, Kim M, Taylor SM, Agoglia AE, May AM, Smith EN, Wong LC, Hodge CW, 2019. Alcohol drinking exacerbates neural and behavioral pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Int. Rev. Neurobiol. 148, 169–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Yu M, Yang S, Lou D, Zhou W, Zheng L, Wang Z, Cai F, Zhou W, Li T, Song W, 2018. Ethanol alters APP processing and aggravates Alzheimer-associated phenotypes. Mol. Neurobiol. 55, 5006–5018. [DOI] [PubMed] [Google Scholar]
- Huynh N, Arabian NM, Asatryan L, Davies DL, 2019. Murine drinking models in the development of pharmacotherapies for alcoholism: drinking in the dark and two-bottle choice. J. Vis. Exp. 57027 (143) 10.3791/57027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ, 2010. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 9, 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack Clifford R., Bennett David A., Blennow Kaj, Carrillo Maria C., Feldman Howard H., Frisoni Giovanni B., Hampel Harald, Jagust William J., Johnson Keith A., Knopman David S., Petersen Ronald C., Scheltens Philip, Sperling Reisa A., Dubois Bruno, 2016. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR, 2004. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170. [DOI] [PubMed] [Google Scholar]
- Jeffrey A. Simms, Pia Steensland, Brian Medina, Kenneth E. Abernathy L. Judson Chandler, Roy Wise, Selena, E Bartlett, 2008. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcohol Clin. Exp. Res. 32 (10), 1816–1823. 10.1111/j.1530-0277.2008.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirkof P, 2014. Burrowing and nest building behavior as indicators of well-being in mice. J. Neurosci. Methods 234, 139–146. [DOI] [PubMed] [Google Scholar]
- Kavanagh K, Day SM, Pait MC, Mortiz WR, Newgard CB, Ilkayeva O, McClain DA, Macauley SL, 2019. Type-2-diabetes alters CSF but not plasma Metabolomic and AD risk profiles in Vervet monkeys. Front. Neurosci. 13, 843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott Craig., Bell Steven., Britton Annie., 2015. Alcohol consumption and the risk of type 2 diabetes: a systematic review and dose-response meta-analysis of more than 1.9 million individuals from 38 observational studies. Diabetes Care 38 (9), 1804–1812. 10.2337/dc15-0710. [DOI] [PubMed] [Google Scholar]
- Koob George F., Colrain Ian M., 2020. Alcohol use disorder and sleep disturbances: a feed-forward allostatic framework. Neuropsychopharmacology 45, 141–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JM, Holtzman DM, 2019. Alzheimer disease: an update on pathobiology and treatment strategies. Cell 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-L., Richardson HN., 2014. Alcohol, stress hormones, and the prefrontal cortex: a proposed pathway to the dark side of addiction. Neuroscience 277, 139–151. 10.1016/j.neuroscience.2014.06.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, Stanley M, Caesar EE, Yamada SA, Raichle ME, Perez R, Mahan TE, Sutphen CL, Holtzman DM, 2015. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J. Clin. Invest. 125, 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin Lauren., Griffith, Chelsea M., Cai Yan., Rose Gregory, M, Yan Xiao-Xin., Patrylo Peter R., 2017. Glucose tolerance and insulin sensitivity are impaired in APP/PS1 transgenic mice prior to amyloid plaque pathogenesis and cognitive decline. Exp. Gerontol. 88, 9–18. 10.1016/j.exger.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Miller Stephanie M., Piasecki Christopher C., Lonstein Joseph S., 2011. Use of the light-dark box to compare the anxiety-related behavior of virgin and postpartum female rats. Pharmacol. Biochem. Behav. 100, 130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Lim MM, Yang G, Bauer AQ, Qi L, Lee Y, Roh JH, Ortiz-Gonzalez X, Dearborn JT, Culver JP, Herzog ED, Hogenesch JB, Wozniak DF, Dikranian K, Giasson BI, Weaver DR, Holtzman DM, Fitzgerald GA, 2013. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Invest. 123, 5389–5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niere F, Namjoshi S, Song E, Dilly GA, Schoenhard G, Zemelman BV, Mechref Y, Raab-Graham KF, 2016. Analysis of proteins that rapidly change upon mechanistic/mammalian target of rapamycin complex 1 (mTORC1) repression identifies Parkinson protein 7 (PARK7) as a novel protein aberrantly expressed in tuberous sclerosis complex (TSC). Mol. Cell. Proteomics 15, 426–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM, 1999. Diabetes mellitus and the risk of dementia: the Rotterdam study. Neurology 53, 1937–1942. [DOI] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF, 1999. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo. Nat. Neurosci. 2, 352–357. [DOI] [PubMed] [Google Scholar]
- Ramachandran B, Ahmed S, Zafar N, Dean C, 2015. Ethanol inhibits long-term potentiation in hippocampal CA1 neurons, irrespective of lamina and stimulus strength, through neurosteroidogenesis. Hippocampus 25, 106–118. [DOI] [PubMed] [Google Scholar]
- Rehm J, Hasan OSM, Black SE, Shield KD, Schwarzinger M, 2019. Alcohol use and dementia: a systematic scoping review. Alzheimers Res. Ther. 11, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Varodayan FP, 2017. Synaptic targets: chronic alcohol actions. Neuropharmacology 122, 85–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Schweitzer P, Madamba SG, Stouffer DG, Parsons LH, Siggins GR, 2004. Acute and chronic ethanol alter glutamatergic transmission in rat central amygdala: an in vitro and in vivo analysis. J. Neurosci. 24, 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR, 2006. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology 31, 988–996. [DOI] [PubMed] [Google Scholar]
- Roh JH, Huang Y, Bero AW, Kasten T, Stewart FR, Bateman RJ, Holtzman DM, 2012. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer’s disease pathology. Sci. Transl. Med. 4, 150ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabia Séverine, Elbaz Alexis, Britton Annie, Bell Steven, Dugravot Aline, Shipley Martin, Kivimaki Mika, Singh-Manoux Archana, 2014. Alcohol consumption and cognitive decline in early old age. Neurology 82, 332–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuckit, Marc A, 1996. Alcohol, Anxiety, and Depressive Disorders. Alcohol Health Res World 20 (2), 81–85. [PMC free article] [PubMed] [Google Scholar]
- Schwarzinger M, Pollock BG, Hasan OSM, Dufouil C, Rehm J, Group QalyDays Study, 2018. Contribution of alcohol use disorders to the burden of dementia in France 2008–13: a nationwide retrospective cohort study. Lancet Public Health 3 e124–e32. [DOI] [PubMed] [Google Scholar]
- Shepherd Amy, Carlos May, Leonid Churilov, Adlard Paul A., Hannan Anthony J., Burrows Emma L., 2021. Evaluation of attention in APP/PS1 mice shows impulsive and compulsive behaviours. Genes Brain Behav 20 (1), e12594. 10.1111/gbb.12594. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Roth J, Gilder A, 2006. Neurobehavioral profiles during the acute phase of ethanol withdrawal in adolescent and adult Sprague-Dawley rats. Behav. Brain Res. 170, 41–51. [DOI] [PubMed] [Google Scholar]
- Sosanya NM, Huang PP, Cacheaux LP, Chen CJ, Nguyen K, Perrone-Bizzozero NI, Raab-Graham KF, 2013. Degradation of high affinity HuD targets releases Kv1.1 mRNA from miR-129 repression by mTORC1. J. Cell Biol. 202, 53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotelo MI, Tyan J, Markunas C, Sulaman BA, Horwitz L, Lee H, Morrow JG, Rothschild G, Duan B, Eban-Rothschild A, 2022. Lateral hypothalamic neuronal ensembles regulate pre-sleep nest-building behavior. Curr 32, 806–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley M, Macauley SL, Caesar EE, Koscal LJ, Moritz W, Robinson GO, Roh J, Keyser J, Jiang H, Holtzman DM, 2016. The effects of peripheral and central high insulin on brain insulin signaling and amyloid-β in young and old APP/PS1 mice. J. Neurosci. 36, 11704–11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker AE, Alicea Pauneto CDM, Barnett AM, Coleman LG Jr., 2022. Chronic ethanol causes persistent increases in Alzheimer’s tau pathology in female 3xTg-AD mice: a potential role for lysosomal impairment. Front. Behav. Neurosci. 16, 886634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Ulland TK, Mahan TE, Nystrom S, Nilsson KP, Song WM, Zhou Y, Reinartz M, Choi S, Jiang H, Stewart FR, Anderson E, Wang Y, Colonna M, Holtzman DM, 2018. ApoE facilitates the microglial response to amyloid plaque pathology. J. Exp. Med. 215, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verges DK, Restivo JL, Goebel WD, Holtzman DM, Cirrito JR, 2011. Opposing synaptic regulation of amyloid-beta metabolism by NMDA receptors in vivo. J. Neurosci. 31, 11328–11337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Kim SW, Wang GJ, Alexoff D, Logan J, Muench L, Shea C, Telang F, Fowler JS, Wong C, Benveniste H, Tomasi D, 2013. Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage 64, 277–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Shokri Kojori E, Fowler JS, Benveniste H, Tomasi D, 2015. Alcohol decreases baseline brain glucose metabolism more in heavy drinkers than controls but has no effect on stimulation-induced metabolic increases. J. Neurosci. 35, 3248–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Zhao J, Liu Z, Guo F, Wang Y, Wang X, Zhang R, Vreugdenhil M, Lu C, 2016. Acute ethanol inhibition of gamma oscillations is mediated by Akt and GSK3beta. Front. Cell. Neurosci. 10, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster Scott J., Bachstetter Adam D., Eldik Van, Linda J., 2013. Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer’s disease. Alzheimers Res. Ther 5 (3), 28. https://doi.org/10.1186/alzrt182. eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF, 2006. Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol. Ther. 111, 533–554. [DOI] [PubMed] [Google Scholar]
- Xu Wei, Wang Huifu, Yu Wan, Tan Chenchen, Li Jieqiong, Tan Lan, Jin-Tai Yu., 2017. Alcohol consumption and dementia risk: a dose–response meta-analysis of prospective studies. Eur. J. Epidemiol. 32, 31–42. [DOI] [PubMed] [Google Scholar]
- Zeng K, Xie A, Dong X, Jiang J, Hao W, Jiang M, Liu X, 2019. GABA-Aalpha5 might be involved in learning-memory dysfunction in the Offsprings of chronic ethanol-treated rats via GABA-Aalpha5 histone H3K9 acetylation. Front. Neurosci. 13, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhornitsky S, Chaudhary S, Le TM, Chen Y, Zhang S, Potvin S, Chao HH, van Dyck CH, Li CR, 2021. Cognitive dysfunction and cerebral volumetric deficits in individuals with Alzheimer’s disease, alcohol use disorder, and dual diagnosis. Psychiatry Res. Neuroimaging 317, 111380. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.