

Abstract
Our understanding of cancer and the key pathways that drive cancer survival has expanded rapidly over the past several decades. However, there are still important challenges that continue to impair patient survival, including our inability to target cancer stem cells, metastasis and drug resistance. The transcription factor p63 is a p53 family member with multiple isoforms that carry out a wide array of functions. Here, we discuss the critical importance of the ΔNp63α isoform in cancer and potential therapeutic strategies to target ΔNp63α expression to impair the cancer stem cell population, as well as to prevent metastasis and drug resistance to improve patient survival.
Keywords: p63, cancer, signaling, therapeutic strategy
Cancer stem cells
In adult tissue, stem cells are essential for tissue homeostasis and regeneration. Stem cells are long-lived cells that generate progeny throughout life to regenerate multiple specialized, shorter-lived cells that are essential for various tissue-specific functions [1]. As stem cells are critical to the maintenance of normal tissue, so too are cancer stem cells (CSCs) critical to the maintenance of many tumors. CSCs are broadly defined as cells that possess the ability to initiate tumor growth, self-renew, and differentiate to give rise to the heterogeneous bulk tumor cell population [1]. The existence of CSCs explains many clinical observations and their challenges, such as recurrence following initially successful therapy, as well as metastasis, drug resistance and dormancy. While cancer treatment has made tremendous strides over the years, drug resistance, recurrence and metastasis remain key problems contributing to therapy failure. In many tumor types, these failures can be attributed to the inability to target the CSC population [1]. Therefore, understanding signaling essential to CSC survival and maintenance is of critical importance to improving therapeutic strategies and patient survival. One protein we believe is at the heart of CSC related signaling is the transcription factor ΔNp63α. It has long been known that ΔNp63α is critical for epithelial development and maintenance [2]. Recent advances in the field of p63 biology have demonstrated key roles for ΔNp63α in cancer progression, metastasis and drug resistance. Despite the importance of p63 in this context, therapeutic strategies to target ΔNp63α are limited because it is an essential transcription factor with a similar structure to family members with opposing functions to its own [3]. In this review, we look at ΔNp63α and its role in CSCs, metastasis and drug resistance, and highlight recent advances in our understanding of ΔNp63α-related signaling that provide exciting therapeutic opportunities in cancer.
ΔNp63α and stemness
In normal tissue, ΔNp63α is highly expressed in several stem cell compartments, particularly in stratified and glandular epithelial [5]. The critical role of ΔNp63α can be seen in p63 deficient mice, which display a lack of all squamous epithelia and their derivatives [2], as well as the severe human developmental defects that occur from germline mutations in p63 (reviewed in [6]). ΔNp63α is required to maintain the self-renewing capacity of epithelial stem cells and is critical for epithelial stem cell differentiation and proliferation through the regulation of a wide array of downstream targets. Based on its role in regulating normal stem cells homeostasis in epithelial tissues, it is not surprising that ΔNp63α is also a key driver of CSCs in multiple tumor types [5].
ΔNp63α in cancer stem cells
ΔNp63α expression has been linked to a CSC phenotype in a number of epithelial cancers, with increased ΔNp63α being associated with elevated numbers of tumor initiating cells, tumorsphere formation, invasive potential, and enhanced tumorigenicity [7, 8]. In squamous cell carcinoma (SCC) the gene encoding stem cell factor SOX2 is co-amplified along with the p63 locus, and preferentially interacts with the ΔNp63α protein [9]. The gene encoding the chromatin modifying protein ACTL6A is also co-amplified with the TP63 locus in head and neck squamous cell carcinoma (HNSCC), leading to a CSC phenotype and impaired terminal differentiation [10]. ΔNp63α and ACTL6A cooperate to decrease chromatin accessibility, which results in the repression of the metastasis suppressor gene WWC1 and the activation of YAP, an oncogene that regulates stemness [10]. Furthermore, YAP can bind to ΔNp63α directly to stabilize it, leading to enhanced cancer stem cell survival in SCC [11]. The Lymphoid-specific helicase (HELLS) is an additional chromatin-modifying protein that is regulated by ΔNp63α. HELLS expression is important for embryonic development and cellular senescence [12]. ΔNp63α is capable of binding to consensus p63 binding sites in the HELLS promoter, increasing expression and leading to senescence bypass during tumor initiation in squamous cell carcinoma[12]. ΔNp63α also induces the expression of genes encoding cell surface proteins involved in establishment of the CSC phenotype. CD44 is a cell surface antigen with roles in migration and adhesion, and is considered a marker of cancer stem cells in various epithelial tumors [13]. Overexpression of ΔNp63α enhances the CD44+/CD24− subpopulation and leads to increased proliferation, colony formation, spheroid formation, and tumor growth in xenografts derived from SCC and MCF-7 cells [14, 15]. ΔNp63α regulates the expression of not only CD44, but the hyaluronan synthase gene HAS3, allowing ΔNp63α to regulate CD44 expression and activation in both HNSCC and breast cancer cell lines [16, 17]. In addition to CD44, ΔNp63α regulates genes encoding integrins α6, β4 and α3 in breast epithelial cells [18]. α6β4 integrin is an essential component of hemidesmosomes, which provide stable adhesion to basal epithelial cells and the underlying basement membrane, and α6β4 integrin has been implicated as a key regulator of cancer stemness in several epithelial cancers [19]. Thus, ΔNp63α-induced expression of these cell surface markers increases cellular adhesion to the extracellular matrix (ECM) and confers resistance to anoikis [18]. In breast cancer, ΔNp63α drives WNT signaling, a critical regulator of epithelial stem cell homeostasis, by directly driving the expression of FZD7, a receptor for WNT ligands [20]. ΔNp63α can also transcriptionally activate NOTCH1, leading to enhanced CSC properties [15]. Finally, ΔNp63 enhances stemness through regulation of Hedgehog signaling by directly controlling the expression of SHH, GLI2 and PTCH1 in mammary CSCs [21].
Resistance to apoptosis is a critical feature of CSCs, and ΔNp63α plays a key role in that feature as well. ΔNp63α overexpression protects cells from oxidative stress induced by oxidants, DNA damage, anoikis, and ferroptosis-inducing agents [3, 22]. ΔNp63α regulates redox homeostasis through transcriptional control of glutathione biogenesis, utilization, and regeneration [22]. Overexpression of ΔNp63α promotes clonogenic survival of p53−/−; Bax−/−; Bak−/− cells against DNA damage, and coexpression of BCL-2 and ΔNp63α confers clonogenic survival against matrix detachment and promotes cancer metastasis in lung SCC [22]. Collectively, these unique capabilities clearly indicate that ΔNp63α is linked to multiple pathways that are central to regulating the CSC phenotype and CSC survival.
ΔNp63α in metastasis
Metastasis is the result of a multistep process by which cancer cells travel from the primary tumor through lymphatic or blood vessels to invade distant organs. This complex cascade of events involves a number of signaling pathways that allow for local invasion, survival in circulation, extravasation and ultimately proliferation at a distant site. CSCs are widely regarded as key drivers of metastasis, as many pathways involved in the CSC phenotype also contribute to the cells’ ability to metastasize, and several reports have indicated the CSC pool is critical for metastatic colonization [1, 23]. In line with this, numerous reports have implicated ΔNp63α as critical to driving the metastatic cascade at multiple levels.
Early in the metastatic cascade, ΔNp63α can contribute to local invasion in basal-like breast cancer through regulation of matrix metalloproteinases MT1-MMP and MMP13, important proteases involved in tumor invasion [24, 25]. Additionally, ΔNp63α directly regulates the transcription of genes encoding two chemokines, CXCL2 and CCL22, which drive the recruitment of myeloid-derived immunosuppressor cells (MDSCs) in triple-negative breast cancer [26]. MDSCs secrete prometastatic factors, including MMP9 to further facilitate invasion [26]. Another important aspect of cancer cell invasion is epithelial-mesenchymal transition (EMT), which confers greater metastatic potential on cells. Endogenous ΔNp63α induces several markers of EMT, including SNAIL, TWIST and VIMENTIN in esophageal squamous carcinoma cell lines, thereby promoting migration and invasion in a β-catenin-dependent manner [27]. And in breast cancer, ΔNp63α enhances cell invasion by transcriptionally regulating genes encoding the EMT-related markers SLUG, FAT2, and AXL [28, 29]. ΔNp63α also upregulates the TGF-β pathway by activating SMAD4 and TGF-βR2, thus facilitating EMT, invasion and migration in osteosarcoma cells [30]. The ability of ΔNp63α to regulate matrix metalloproteinases and EMT is likely why ΔNp63α is so robustly expressed at the edge of invasive tumors, as ΔNp63α activity might be locally upregulated in the migrating front of cells, enabling ECM degradation and invasion.
In support of this, ΔNp63α has been shown in breast cancer organoids to control the ‘collective invasion’ process, a type of cellular invasion in which tumor cells remain connected and invade as multicellular units [31]. These cells display a basal epithelial gene expression pattern that facilitates collective invasion. Particularly, the invading tumor cells activate expression of ΔNp63α and CK14, which are required for local invasion of breast cancer cells. By maintaining the basal epithelial state, the cells retain enhanced invasive properties characteristic of less differentiated epithelial cells, thus allowing for collective invasion [31].
Another key aspect of the metastatic cascade is survival in circulation. ΔNp63α contributes to this critical step by suppressing anoikis through regulation of integrins, BCL-2 and EGFR [18, 22, 32]. When cells reach the metastatic site, they must be able to engage the ECM and proliferate. To facilitate this process, primary tumors actively modify potential metastatic sites prior to dissemination through secretion of various factors [33]. ΔNp63α contributes to the formation of the metastatic niche by transcriptionally regulating ANGPTL2 [34]. ANGPTL2 is a secreted glycoprotein and pro-inflammatory and angiogenic factor that is capable of signaling through α5β1 integrins to contribute to metastatic niche formation [34]. When cancer cells arrive at the metastatic site, ΔNp63α likely further contributes to metastasis through transcriptional regulation of CYR61, a matricellular protein linked to extravasation during metastasis through engagement with integrins and heparin sulfate proteoglycans [35, 36].
All together, these data suggest that ΔNp63α exploits multiple pathways, including the induction of EMT-related factors, metalloproteinases, enhancement of collective invasion, anoikis resistance and metastatic colonization, all of which work together to enhance the metastatic potential of cancer cells. However, there is also evidence suggesting caution should be taken, as ΔNp63α depletion can have differing impacts under certain conditions. For instance, in certain SCC lines that predominantly express ΔNp63α, p63 depletion results in increased mesenchymal marker expression associated with invasion [37] and overexpression of ΔNp63α results in reduced Vimentin and ZEB1 expression [38]. In line with this, in two non-transformed mammary epithelial cell lines (MCF10A and MCF12A) expression of H-RasV12 reduces ΔNp63α expression and increases EMT and cell migration [39]. Work in the MCF10A cell line also showed that depletion of ΔNp63α and ΔNp63β leaving only ΔNp63γ resulted in TGFβ driven EMT [40]. In breast and prostate lines, ΔNp63α has been shown to impair invasion through the suppression of miR-205, a key regulator of EMT [41, 42] and in prostate cancer cell lines, miR-301 was shown to induce EMT through inhibition of p63 [43]. Beyond the differing impacts on cancer cells, the key role of ΔNp63α in senescence and aging in normal tissue should also be considered (reviewed in [44]). Thus, more work is needed to fully understand the cellular context in which ΔNp63α can suppress EMT and invasive behavior and therefore know when it is appropriate to target ΔNp63α therapeutically.
Drug resistance
Chemotherapy is one of the principal modes of treatment for cancer, but the effectiveness of chemotherapy is kept in check by drug resistance. Although combination therapies have become the standard for cancer therapy to help circumvent resistance against single-agent treatment, drug resistance continues to be a major obstacle [45], and recent work has linked ΔNp63α to drug resistance in several cell lines. ΔNp63α has been implicated in Cisplatin resistance through several mechanisms. In HNSCC, ΔNp63α has been shown to regulate the transcription of AKT1, leading to Cisplatin resistance [46]. In pancreatic cancer, ΔNp63α results in Cisplatin resistance through the transactivation of EGFR and 14-3-3σ [47]. In breast cancer, upregulation of ΔNp63α leads to an increase in the expression of EGFR and WIP1 to drive cisplatin resistance [48]. Finally, in oral cancer, ΔNp63α promotes the expression and nuclear translocation of PTEN, leading to Cisplatin resistance [49]. In addition to Cisplatin, ΔNp63α has been shown to induce resistance to Doxorubicin in hepatocellular carcinoma by downregulating CD95 and BAX gene activation, and Bortezomib resistance in HNSCC through regulation of CYGB-ROS signaling [50, 51]. Therefore, ΔNp63α is capable of regulating a multitude of targets involved in numerous aspects of cancer progression, including stem cell self-renewal, invasion, anoikis resistance, colonization and drug resistance.
Druggable targets upstream of ΔNp63α
Because of the difficulties in targeting ΔNp63α directly, we believe targeting upstream regulators of ΔNp63α is a potential therapeutic strategy. Below we discuss what we believe are exciting therapeutic targets upstream of ΔNp63α that could provide a means to reduce ΔNp63α expression and the CSC phenotype, metastasis and drug resistance associated with it. It is important to note that upstream regulators of ΔNp63α can vary in differing cell types, and the pathways discussed below may only be present in certain tissues or cell contexts.
Chromatin modifying proteins
BRD4/EZH2
A number of chromatin modifying proteins have been linked to ΔNp63α (summarized in Figure 2). In pancreatic cancer, loss of KDM6A results in squamous-like, metastatic cancers, which are selectively sensitive to Bromodomain and Extraterminal domain (BET) inhibitors including JQ1 [52]. Treatment with JQ1, which predominantly inhibits BRD4, reverses squamous differentiation. It was shown that BRD4 binds to ΔNp63α-regulating super enhancers, and treatment with JQ1 not only evicts BRD4 from these super enhancers, but also disrupts their long-range interaction with the ΔNp63α promoter [52]. BRD4 has also been linked to ΔNp63α in SCC, with genetic depletion or pharmacological inhibition of BRD4 using BET inhibitors JQ1 or MS436 reducing ΔNp63α protein levels and impairing cancer stem cell phenotypes [53]. In this context, BRD4 transcriptionally regulates C-MYC, leading to increased activity of EZH2. EZH2 then binds to STAT3, methylating and activating it, allowing STAT3 to bind to the ΔNp63α promoter. Furthermore, treatment with EZH2 or STAT3 inhibitors successfully reduce ΔNp63α expression and the CSC phenotype associated with it [53].
Figure 2. Mechanisms of chromatin modifying protein-mediated regulation of ΔNp63α.
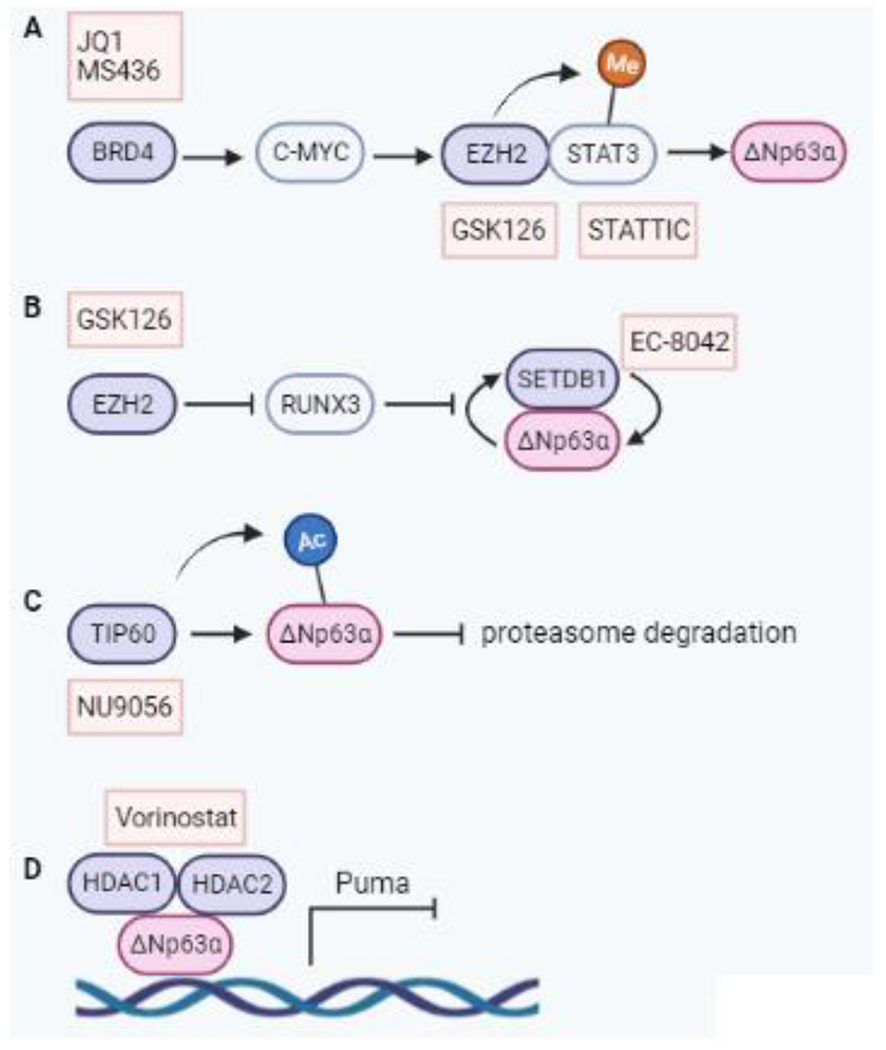
A BRD4-driven C-MYC leads to EZH2 binding to and methylating STAT3, activating it. This results in STAT3 transcriptionally activating ΔNp63α. Targeting BRD4 with JQ1 or MS436, EZH2 with GSK126 or STAT3 with STATTIC impair this pathway and subsequent ΔNp63α expression. B EZH2 suppression of RUNX3 leads to enhanced ΔNp63α and SETDB1 expression, which interact to stabilize expression of the other. The EZH2 inhibitor GSK126 can suppress EZH2 activity leading to increased RUNX3 and reduced SETDB1 and ΔNp63α. The SETDB1 inhibitor EC-8024 is an additional potential means of targeting this pathway. C TIP60 acetylates ΔNp63α to prevent ubiquitin-mediated degradation. Inhibiting TIP60 with NU9056 reduces ΔNp63α protein and transcript. D HDAC1 and HDAC2 bind to ΔNp63α to form an active transcriptional repressor complex. The HDAC inhibitor Vorinostat impairs activity of this complex, resulting in increased downstream activation of targets including Puma. Abbreviations: BRD4, bromodomain containing protein; C-MYC, cellular-myelocytomatosis; EZH2, enhancer of zeste homolog 2; HDAC, histone deacytylase; HDAC2, histone deacytylase 2; RUNX3, runt-related transcription factor 3; SETDB1, SET domain bifurcated histone lysine methyltrasferase 1; STAT3, signal transducer and activator of transcription 3; STATTIC, STAT3 inhibitory compound; TIP60 tat interactive protein 60.
In addition to regulating ΔNp63α through STAT3, EZH2 can also regulate ΔNp63α through RUNX3 in SCC. In multiple cancers, RUNX3 has been shown to be a direct target of EZH2-mediated repression via promoter hypermethylation. Pharmacological inhibition of EZH2 or CRISPR-mediated depletion significantly augments RUNX3 expression at both the mRNA and protein level [54]. This coincides with the loss of ΔNp63α. Direct activation of RUNX3 through either CRISPRa or cDNA overexpression leads to a significant compromise in ΔNp63α expression at both the protein and mRNA level.
SETDB1
The histone methyltransferase SETDB1 was shown to physically interact with the C-terminal TID domain of ΔNp63α in breast cancer [55]. Depletion of SETDB1 or ΔNp63α reduces expression of the other, indicating their reciprocal modes of regulation. SETDB1 depletion leads to upregulation of 30 targets of ΔNp63α repression, indicating a possible novel mechanism of ΔNp63α-mediated gene repression via SETDB1. Consequently, SETDB1 regulates ΔNp63α expression in breast cancer, as well as being a binding partner that may cooperate to repress ΔNp63α target genes [55].
The interaction between these proteins is also demonstrated in SCC. The loss of either protein results in a significant disruption of a CSC phenotype [54]. Additionally, the proteins regulate each other’s expression and reintroduction of ΔNp63α into SETDB1-deficient cells rescues the cancer stem cell phenotype. Likewise, SETDB1 reintroduction rescues CSC phenotypes in ΔNp63α-deficient cells, highlighting the intimate connection between these two proteins.
Therapeutic targeting of SETDB1 is a developing area of study that may hold great promise in disrupting ΔNp63α-driven cancers with high-level SETDB1 expression. To date, several compounds have been shown to have efficacy in targeting SETDB1, including mithramycin A, the mithramycin analog EC-8042, and a selective inhibitor of SETDB1’s tandem Tudor domains [56, 57].
TIP60
The histone acetyltransferase TIP60 activates ΔNp63α expression in SCC [58]. Upon TIP60 depletion, ΔNp63α is decreased at both the RNA and protein level. This is due to TIP60 directly acetylating ΔNp63α, thereby preventing its ubiquitin-mediated degradation. Importantly, the TIP60-selective inhibitor NU9056 produces a similar effect as TIP60 depletion, providing a potential means of targeting ΔNp63α in SCCs co-expressing ΔNp63α and TIP60 [58].
HDACs
Histone deacetylases (HDACs) play an important role in regulating transcription. HDACs represent potential anticancer targets, as their inhibition can induce apoptosis, differentiation, and growth arrest in cancer cells. In HNSCC, trichostatin A (TSA), an inhibitor of HDAC1 and 6, downregulates the expression of p63 and reduces invasion and migration [49] whereas treatment with suberoylanilide hydroxamic acid (SAHA) reduces EMT and ΔNp63α [59]. In SCC, ΔNp63α associates with HDAC1 and HDAC2 to form an active transcriptional repressor complex that can be targeted therapeutically with Vorinostat, which effectively reduces ΔNp63α expression [60].
Signals from the microenvironment
The tumor microenvironment (TME) consists of diverse cell types and extra-cellular matrix components that surround and support the tumor. There is growing interest in targeting the TME due to its critical role in regulating several aspects of cancer progression. Interleukins (IL) are a key component of the microenvironment, and several have been implicated in regulating ΔNp63α, including IL-1β in MCF7 cells, IL-6 in lung cancer, and IL-13, IL-17 and IL-22 in keratinocytes [48, 61–64], ΔNp63α also induces IL-6 and IL-1 in pancreatic cancer cells, providing potential for a positive feedback loop [65]. IL-17A produced by Th17 cells induces ΔNp63α in keratinocytes through aTRAF4/ERK-mediated pathway [66] and the type 2 interleukins IL-4/13, require ΔNp63α to block early keratinocyte differentiation [64]. In addition to interleukins, enhanced ECM content augments ΔNp63α expression, and inhibition of collagen synthesis reduces ΔNp63α levels. Altered ΔNp63α levels are also found in keratinocytes grown on different ECM components, with ΔNp63α levels in epithelial stem cells varying according to the particular matrix composition and stiffness. Activation of the laminin receptor, a key molecule involved in adhesion to the basement membrane, increases ΔNp63α levels in keratinocytes, as does the ECM component TGFBIp, and integrin-linked kinase (ILK), which is involved in integrin mediated signal transduction [67–69].
These data suggest that ΔNp63α is capable of regulating and being regulated by various aspects of the TME. With growing interest in targeting the TME and crosstalk between the TME and cancer cells, targeting interleukins upstream of ΔNp63α potentially represents an opportunity to target not only critical factors of the TME, but also a key regulator of cancer progression that the TME supports.
Cell Surface markers
In addition to the signals released from the TME, cancer cell surface markers are critical in crosstalk with the TME, as they relay those signals to the cancer cells. In line with the importance of signals emanating from the TME in regulating ΔNp63α, many cell surface markers involved in ‘outside in’ signaling have been linked to ΔNp63α as well.
EGFR
The tyrosine kinase receptor epidermal growth factor receptor (EGFR) is frequently overexpressed in squamous cell carcinomas, where it has been shown to induce ΔNp63α expression through activation of phosphatidylinositol 3-kinase (PI3K), in turn, activating mTOR-dependent activation of STAT3 [70]. ΔNp63α is also capable of regulating EGFR expression in cooperation with SOX2 and CCAT1 [71], suggesting a possible feedback loop between EGFR and ΔNp63α in squamous cell carcinoma. In basal-like triple negative breast cancer, ΔNp63α expression increases both EGFR mRNA and protein levels, as well as increasing its activity [32]. Silencing of ΔNp63α in epithelial cells reduces both the total- and phospho-EGFR levels, impairing the activation of EGFR signaling [32, 71].
Integrins/TG2/NRP1
Signaling through α6β4 integrin has also been shown to regulate ΔNp63α expression. In squamous cell carcinoma, the enzyme transglutaminase 2 (TG2) interacts with a6b4 integrin. This interaction leads to activation of FAK-SRC and PI3K-PDK1 kinases. Signaling through this cascade results in the inhibition of large tumor suppressor kinase 1 (LATS1), an integral component of the Hippo signaling pathway that suppresses YAP [72]. Signaling through this cascade results in the inhibition of large tumor suppressor kinase 1 (LATS1), an integral component of the Hippo signaling pathway that suppresses YAP. This frees YAP to enter the nucleus, where it binds to ΔNp63α and stabilizes its expression by impairing degradation of ΔNp63α by the proteasome [11].
Neuropilin-1 (NRP1) is another protein that can activate signaling through α6β4 integrin to regulate ΔNp63α. NRP1 is a transmembrane protein and co-receptor for a number of extracellular ligands. NRP1 interacts with GAIP C-terminus interacting protein 1 (GIPC1), a scaffolding protein, and α6β4 integrin. This complex activates a downstream kinase cascade that also leads to suppression of Hippo signaling and increased ΔNp63α [73]. YAP also mediates stabilization of ΔNp63α in response to DNA damage-induced p63 phosphorylation by c-Abl, leading to YAP/ΔNp63α binding [74]. As mentioned above, ΔNp63α transcriptionally regulates several integrin isoforms including α6, β4 and α3 [18]. This represents another feedback loop that can be targeted therapeutically, as small molecule inhibitors for TG2, NRP1 and YAP are available that have been shown to impair the CSC phenotype in various cancer types [72, 73, 75]. YAP in particular has generated significant clinical interest, with new small molecule inhibitors in development, as well as efforts to repurpose existing drugs like Verteporfin and Digitoxin [76].
Wnt/β-catenin pathway
Wnt/β-catenin signaling is a key regulator of stemness through the regulation of self-renewal, pluripotency, differentiation and migration. In cancer, abnormal activation of Wnt/β-catenin promotes a CSC phenotype and metastasis. [77]. ΔNp63α is under direct control of the WNT/β-catenin pathway through binding of lymphoid enhancer binding factor 1 (Lef1) and β-catenin between the promoters of TAp63 and ΔNp63 [78]. Another layer of regulation comes from a β-catenin responsive element within the proximal ΔNp63α promoter. In addition to direct regulation of ΔNp63α, WNT/β-catenin can also regulate the transcriptional co-factor limb-bud and heart (LBH). In mammary epithelial cells, LBH increases ΔNp63α transcription while downregulating transcription of TAp63α, resulting in enhanced replicative potential and stemness [79]. Together, these data suggest that in cancers with elevated ΔNp63α levels and active β-catenin signaling, targeting the β-catenin pathway may represent a means for impairing ΔNp63α expression.
STAT3
Of the seven members of the STAT protein family, STAT3 is arguably the most important for cancer progression [80]. STAT3 is not only critical for transducing signals from multiple receptor and non-receptor tyrosine kinases that are frequently activated in cancer cells, but STAT3 is also a transcription factor regulating the expression of a wide range of targets that contribute to tumor progression, most notably ΔNp63α [80]. STAT3 binds to the promoter of ΔNp63α in several cell types, and the dual-regulatory effect of ΔNp63α on its own promoter is dependent on STAT3 activation [81, 82]. STAT3 serves as a key mediator of ΔNp63α for several pathways mentioned above, including IL-6, EGFR, BRD4 and EZH2 [53, 70, 80]. In addition to these, there are likely numerous other activators of STAT3 that can be linked to ΔNp63α in cancer. Things like VEGFR, PDGFR, CXCR4, and S1PR1 that lead to STAT3 activation and CSC phenotypes, but have yet to be definitively shown to activate ΔNp63α, all represent interesting areas of investigation [80]. This also leaves STAT3 uniquely positioned directly upstream of ΔNp63α and important for its activation, while also being downstream of numerous signaling cascades critical for cancer biology. Combine that with a number of STAT3 inhibitors currently at various stages of clinical trials, and STAT3 appears to be a most exciting therapeutic opportunity for targeting ΔNp63α. The upstream regulators of ΔNp63α discussed in this section have been summarized in Figure 3.
Figure 3. Schematic representation of signaling cascades that regulate ΔNp63α and the drugs that have been shown to target them.
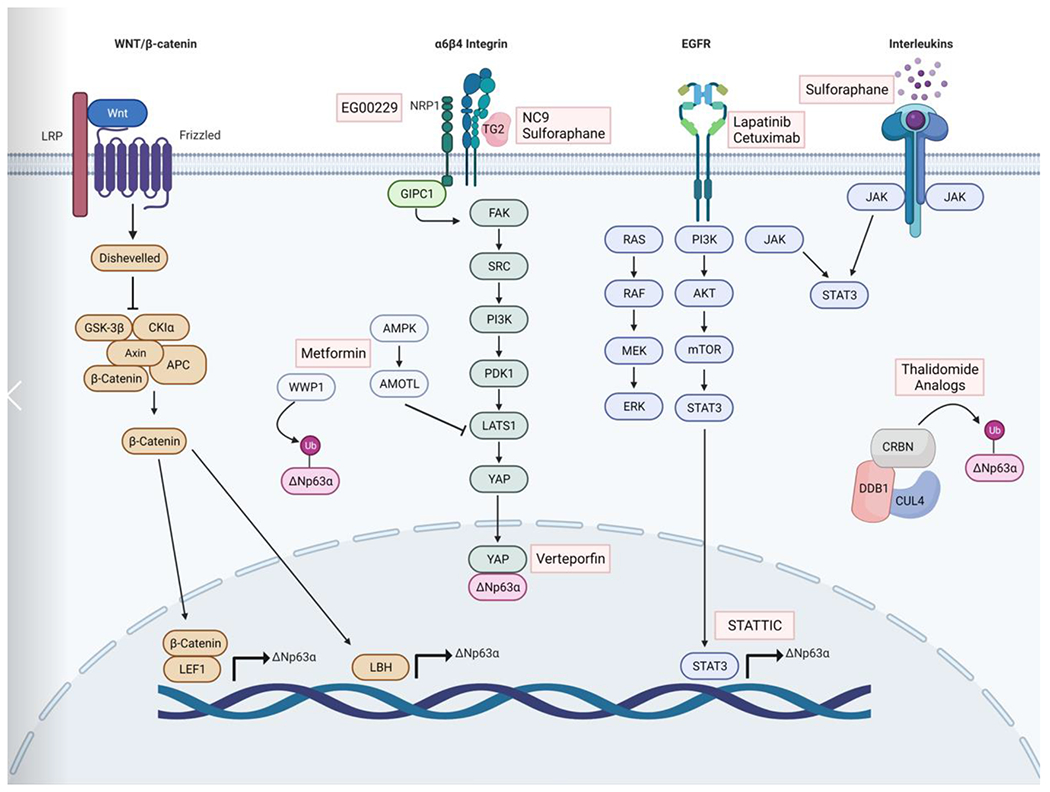
Several signaling cascades have been implicated in the regulation of ΔNp63α. In Wnt/β-catenin signaling, Wnt binds to Frizzled receptors, leading to the formation of a larger cell surface complex with LRP. Activation of the Wnt receptor complex triggers displacement of GSK-3β from the APC/Axin/GSK-3β-complex. β-catenin is translocated to the nucleus where it binds to LEF1 and transcriptionally activates ΔNp63α. α6β4 integrin interaction with TG2 or NRP1 leads to the activation of a kinase cascade that suppresses the Hippo signaling component LATS1, allowing YAP to enter the nucleus where it binds to ΔNp63α preventing proteasome degradation. Several compounds including the NRP1 inhibitor EG00229, the TG2 inhibitor NC9 or the diet derived compound Sulforaphane which inhibits TG2, and the YAP inhibitor Verteporfin can impair ΔNp63α protein expression. In addition to TG2, Sulforaphane can inhibit Interleukin driven JAK/STAT3 activation to suppress ΔNp63α expression. EGFR signaling also regulates ΔNp63α through STAT3. EGFR activation leads to phosphorylation and activation of the PI3K/AKT/mTOR pathway which phosphorylates STAT3, which binds to the promoter of ΔNp63α. CRBN, DDB1 and Cul4, form the E3 ubiquitin ligase complex CRL4CRBN. Thalidomide analogues alter the CRL4CRBN ubiquitin ligase to target ΔNp63α, resulting in its degradation in the presence of thalidomide. Abbreviations: AKT, alpha serine/threonine-protein kinase; AMOTL, angiomotin-like protein; AMPK, AMP-activated protein kinase; APC, adenomatous polyposis coli; CK1α, casein kinase 1α CRBN, cereblon; CUL4, cullin 4; DDB1, DNA damage binding protein 1; EGFR, epidermal growth factor receptor; ERK, extracellular signal related kinases; FAK, focal adhesion kinase; GIPC1m GIPC PDZ domain containing family member 1; JAK, janus kinase; LATS1, large tumor suppressor kinase 1; LRP, low-density lipoprotein receptor related protein; MEK, mitogen activated protein kinase kinase; mTOR, mammalian target of rapamycin; NRP1, Neuropilin 1; PDK1, pyruvate dehydrogenase kinase 1; PI3K, phosphatidylinositol 3-kinase; RAF, rapidly accelerated fibrosarcoma; RAS, rat sarcoma; STAT3, signal transducer and activator of transcription 3; STATTIC, STAT3 inhibitory compound; WNT, wingless and INT1; WWP1, WW domain containing E3 ubiquitin protein ligase 1.
Figure 1. p63 is the primordial member of the p53/p63/p73 family of transcription factors.
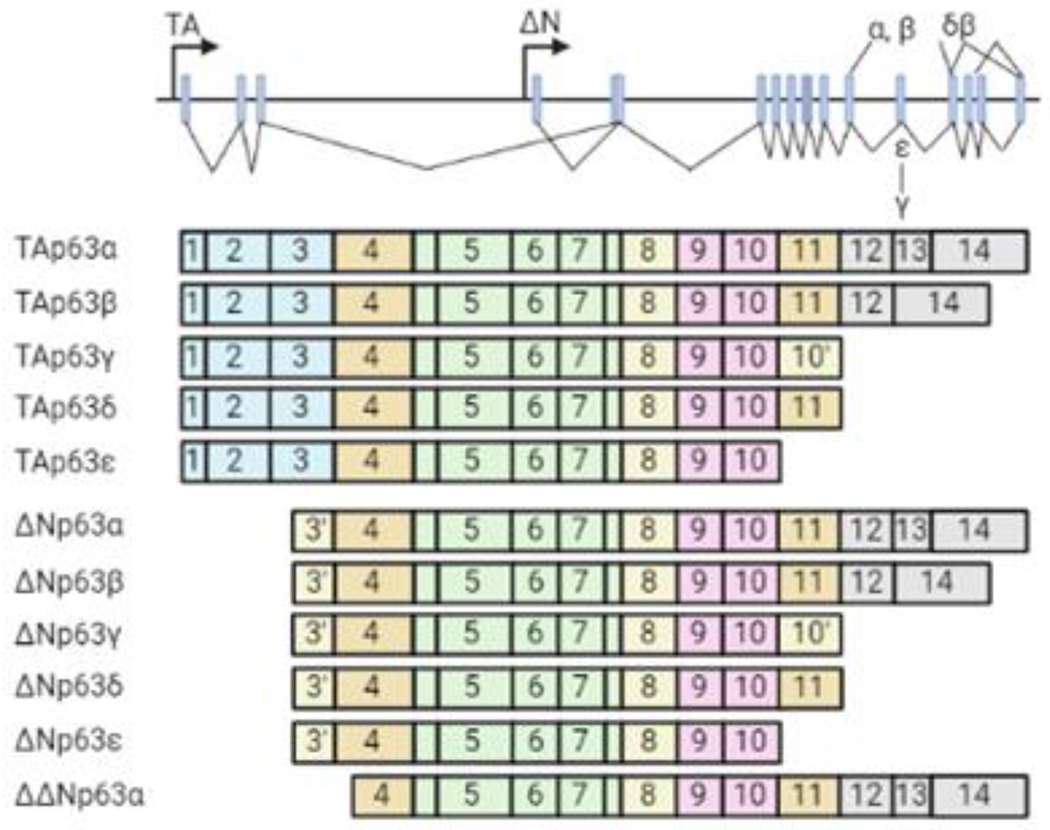
The human TP63 gene consists of 15 exons spanning ~270 kb and maps to chromosome 3q27 [3]. It encodes two classes of isoforms generated by alternative promoters: TAp63 transcripts, which possess an N-terminal transactivation domain, and ΔNp63 isoforms that lack the N-terminal transactivation domain but retain the ability to induce genes via a second transcription activation domain. Alternative splicing occurring at the 3’ end of p63 mRNAs generates multiple C-terminal variants (α, β, γ, δ and ε) for both TAp63 and ΔNp63 classes [3]. TAp63- and ΔNp63- isoforms have distinct tissue distributions. ΔNp63 but not TAp63 is present in basal and parabasal cells in squamous epithelium and urinary bladder, and in basal cells of breast and prostate. TAp63 is detected in lymphocytes and germ cell precursors and some mesenchymal cells and endothelial cells. The existence of multiple isoforms of TP63 with differing functions allows TP63 to regulate a wide array of biological processes such as development and differentiation, senescence, proliferation, stem cell maintenance, and apoptosis [4]. In the context of cancer, TAp63 isoforms are generally regarded as tumor suppressors, [4]. However, ΔNp63 isoforms—ΔNp63α in particular—frequently act as oncogenes.
Box 1. Additional therapeutic opportunities.
Metformin
Metformin is commonly used to increase insulin sensitivity in patients with type II diabetes, and has numerous known functions such as activating AMP-activated protein kinase (AMPK) and inhibiting glucagon-induced cAMP increases [83]. A recent study in SCC reveals an AMPK-independent mechanism for metformin by which treatment causes an increase in the E3 ubiquitin ligase WWP1, a known ΔNp63α E3 ligase [83]. Upon depletion of WWP1 in metformin-treated cells, expression of ΔNp63α protein is rescued. Further, it was shown that in combination with the glycolysis inhibitor, 2-Deoxy-d-glucose, metformin treatment significantly reduces tumor growth [83]. Multiple studies have also shown an effect of metformin on both YAP localization and expression levels [84–86] . This is linked to increased cytoplasmatic sequestration and inactivation of YAP by Angiomotin (AMOT) and Angiomotin-like proteins 1-2 (AMOTL1-2), representing another possible mechanism by which Metformin impairs ΔNp63α expression [84].
Sulforaphane
Sulforaphane (SFN) is a natural isothiocyanate derived from broccoli and other cruciferous vegetables that can act as a cancer preventative [87]. In cutaneous SCC, SFN treatment was shown to increase YAP1 phosphorylation and proteolytic degradation, thereby reducing ΔNp63α levels [87]. It was later found that SFN covalently and irreversibly binds to TG2 to inhibit transamidase activity and shift TG2 to an open/extended conformation, leading to a partial inhibition of GTP binding [88]. As inhibition of TG2 activity is linked to impaired YAP/ΔNp63α levels, this represents a likely mechanism for the SFN-induced reduction in ΔNp63α expression. Finally, in lung cancer, tobacco smoke is shown to induce a CSC phenotype driven by IL-6-mediated regulation of ΔNp63α. Treatment with SFN suppresses IL-6/ΔNp63α signaling and reduces the CSC phenotype [63].
Thalidomide analogues
Thalidomide, most known for its teratogenic effects, is approved for use in multiple myeloma patients [89]. Cereblon (CRBN), together with DDB1 and Cul4, forms an E3 ubiquitin ligase complex called cullin-ring ligase 4 (CRL4CRBN) [90]. Thalidomide analogues were recently found to alter the CRL4CRBN ubiquitin ligase to target a number of cellular proteins for ubiquitination and proteasome degradation. ΔNp63α is a neo-substrate of CRL4CRBN in response to Thalidomide treatment, and is targeted for degradation in the presence of thalidomide [90].
While the compounds discussed above have all shown to inhibit ΔNp63α in various cell lines, whether they will affect ΔNp63α in patients has yet to be established.
Concluding Remarks
The transcription factor ΔNp63α is a key regulator of epidermal morphogenesis and epithelial tissue homeostasis. Here, we have discussed evidence supporting the notion that ΔNp63α regulates various aspects of cancer stemness, metastasis and drug resistance across a number of cancer types. ΔNp63α regulation of these critical features of cancer biology has been linked to the regulation of several pathways including HELLS, CD44, integrins, WNTs, interleukins and EMT markers. Therefore, impairing ΔNp63α in certain cancer contexts has the potential to have a profound effect on patient survival. There are a variety of therapeutic targets upstream of ΔNp63α ranging from chromatin modifying proteins to cell surface receptors, kinases and transcription factors. We believe there are still many regulators of ΔNp63α with therapeutic potential yet to be characterized. Further characterization of ΔNp63α interacting partners can allow for the disruption of signaling complexes that either indirectly interfere with ΔNp63α activity or result in proteasome degradation of ΔNp63α. In addition, we believe the role of ΔNp63α in crosstalk with the microenvironment is a particularly exciting area for future research. Several components of the microenvironment have been identified that regulate or are regulated by ΔNp63α, indicating ΔNp63α could be a potential hub for crosstalk with the microenvironment. This raises multiple potential interesting areas of investigation (see Outstanding Questions). Although there is evidence indicating ΔNp63α can transcriptionally regulate some cytokines and interleukins, and interleukins can in turn regulate ΔNp63α, the impact of ΔNp63α on modelling the immune-landscape has yet to be characterized. An immunosuppressive microenvironment facilitates cancer progression, and a substantial portion of SCC patients that frequently overexpress ΔNp63α do not respond to immunotherapies [91]. Understanding if and how ΔNp63α can contribute to resistance to immunotherapies could lead to better therapeutic options in these patients. It will also be interesting to see how therapeutic targeting of cancer associated fibroblasts (CAFs) alters ΔNp63α expression. CAFs are capable of stimulating multiple upstream regulators of ΔNp63α and the growing efforts to target CAF populations may represent an indirect method of reducing ΔNp63α expression. Therefore, we believe further investigations into how ΔNp63α crosstalks with the TME will help to continue to identify regulatory pathways with therapeutic potential.
Outstanding Questions.
What additional interacting partners of ΔNp63α have yet to be identified and what are their roles in regulating ΔNp63α?
Do upstream regulators of STAT3 like VEGFR, PDGFR, CXCR4 and S1PR1 regulate ΔNp63α expression through activation of STAT3?
What is the role of ΔNp63α in the immune-landscape? Does ΔNp63α regulate or get regulated by the immune-landscape, and can targeting ΔNp63α improve responses to immunotherapies in patients with high ΔNp63α?
Does therapeutic targeting of CAFs impact ΔNp63α expression in cancer cells? What components of CAF signaling regulate ΔNp63α?
Can suppressing ΔNp63α prevent/overcome drug resistance?
Highlights.
ΔNp63α is a p63 isoform in the p53 family that is a master regulator of epithelial stemness in normal tissue.
In cancer, ΔNp63α regulates a number of key aspects of cancer progression including CSC maintenance, metastasis and drug resistance through regulation of several downstream pathways.
ΔNp63α is difficult to target directly, but multiple pathways upstream of ΔNp63α with druggable targets have been identified that represent potential therapeutic opportunities in cancer.
Many pathways upstream of ΔNp63α are involved in crosstalk with the tumor microenvironment. With growing interest in targeting the tumor niche, further investigation into how ΔNp63α is involved in crosstalk with the microenvironment represents an exciting area of future investigation.
Acknowledgements
This work was supported by the Office of the Director, National Institutes of Health through award numbers 5P30CA045508 (Cancer Center Support Grant), CA225134 (to M.L. Fisher), CA247400 (to S.Balinth), as well as R01CA190997 and R21OD018332 (to A.A. Mills). This project was also supported through the Cold Spring Harbor Laboratory and Northwell Health Affiliation.
Glossary
- Tumorsphere
a spherical formation developed from the proliferation of a single cancer stem or progenitor cell in 3D culture.
- hyaluronan synthase
enzyme involved in the synthesis of unbranched glycosaminoglycan hyaluronan, or hyaluronic acid, a CD44 ligand.
- Hemidesmosomes
protein complexes that facilitate the stable adhesion of basal epithelial cells to the underlying basement membrane.
- Extracellular matrix
three-dimensional network of extracellular components including collagens, glycoproteins and proteoglycans, that provide structural and biochemical support to surrounding cells.
- Anoikis
apoptosis that results from loss of attachment to the extracellular matrix or neighboring cells
- Hedgehog signaling
signaling pathway critical during development for intercellular communication, and is frequently dysregulated in cancer. There are three mammalian Hedgehog proteins including Sonic Hedgehog, Indian Hedgehog and Desert Hedgehog
- Ferroptosis
a form of cell death driven by iron-dependent phospholipid peroxidation regulated by multiple cellular metabolic pathways.
- Clonogenic survival
an in vitro cell survival assay based on the ability of a single cell to grow into a colony, testing the ability of cells to undergo unlimited division. This method is frequently used to determine the effectiveness of cytotoxic agents.
- Matrix metalloproteinases
members of the metzincin group of proteases which share the conserved zinc-binding motif in their catalytic active site, and are involved in regulating various components of the extracellular matrix.
- Metastatic niche
an environment in a secondary organ that provides favorable growth conditions for cancer cells, allowing for the establishment of metastasis from a primary tumor.
- Cisplatin
an anti-cancer, antineoplastic or cytotoxic chemotherapy drug classified as an alkylating agent and works by interfering with DNA replication.
- Bortezomib
a dipeptide boronic acid derivative and proteasome inhibitor used to treat multiple myeloma and mantle cell lymphoma.
- JQ1
a potent inhibitor of the BET family of bromodomain proteins which include BRD2, BRD3, BRD4, and the testis-specific protein BRDT in mammals
- CRISPRa
a variant of CRISPR in which a catalytically dead (d) Cas9 is fused with a transcriptional effector to alter target gene expression. Once the guide RNA navigates to the genome locus along with the effector arm, the dCas9 is unable to make a cut, and instead, the effector activates the downstream gene expression
- Mithramycin A
an antibiotic with anti-tumor properties that binds to G-C rich DNA and displaces SP1 transcription factor from its sites in the promoters of selected oncogenes, such as c-Myc and c-Src
- Tudor domains
A Tudor domain is a protein region roughly 60 amino acids in length, which folds into an SH3-like structure with a five-stranded antiparallel beta-barrel form. Tudor domains recognize and bind methylated lysine and arginine residues, allowing them to function as histone readers in an epigenetic context
- EC-8042
a mithramycin analog (mithralog) with enhanced anti-tumor activity that inhibits SP1 activity.
- Vorinostat
an oral histone deacetylase inhibitor and antineoplastic agent that binds to the catalytic domain of the histone deacetylases (HDACs)
- Interleukins
a group of cytokines that play essential roles in the activation and differentiation of immune cells, as well as cell proliferation, maturation, migration, and adhesion.
- Hippo signaling
an evolutionarily conserved pathway that controls organ size by regulating cell proliferation, apoptosis, and stem cell self-renewal. In addition, dysregulation of the Hippo pathway contributes to cancer development.
- Transamidase
an enzyme that catalyzes the transfer of an amide group from one molecule to another
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflicts of interest.
References
- 1.Batlle E and Clevers H, Cancer stem cells revisited. Nat Med, 2017. 23(10): p. 1124–1134. [DOI] [PubMed] [Google Scholar]
- 2.Mills AA, et al. p63 is a p53 homologue required for limb and epidermal morphogenesis. Nature, 1999. 398(6729): p. 708–13. [DOI] [PubMed] [Google Scholar]
- 3.Fisher ML, Balinth S, and Mills AA, p63-related signaling at a glance. J Cell Sci, 2020. 133(17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray-Zmijewski F, Lane DP, and Bourdon JC, p53/p63/p73 isoforms: an orchestra of isoforms to harmonise cell differentiation and response to stress. Cell Death Differ, 2006. 13(6): p. 962–72. [DOI] [PubMed] [Google Scholar]
- 5.Melino G, et al. Maintaining epithelial stemness with p63. Sci Signal, 2015. 8(387): p. re9. [DOI] [PubMed] [Google Scholar]
- 6.Osterburg C, et al. Isoform-Specific Roles of Mutant p63 in Human Diseases. Cancers (Basel), 2021. 13(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gatti V, et al. p63 at the Crossroads between Stemness and Metastasis in Breast Cancer. Int J Mol Sci, 2019. 20(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moses MA, et al. Molecular Mechanisms of p63-Mediated Squamous Cancer Pathogenesis. Int J Mol Sci, 2019. 20(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watanabe H, et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest, 2014. 124(4): p. 1636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saladi SV, et al. ACTL6A Is Co-Amplified with p63 in Squamous Cell Carcinoma to Drive YAP Activation, Regenerative Proliferation, and Poor Prognosis. Cancer Cell, 2017. 31(1): p. 35–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher ML, et al. Transglutaminase Interaction with alpha6/beta4-Integrin Stimulates YAP1-Dependent DeltaNp63alpha Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res, 2016. 76(24): p. 7265–7276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 12.Keyes WM, et al. DeltaNp63alpha is an oncogene that targets chromatin remodeler Lsh to drive skin stem cell proliferation and tumorigenesis. Cell Stem Cell, 2011. 8(2): p. 164–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Skandalis SS, et al. Hyaluronan-CD44 axis orchestrates cancer stem cell functions. Cell Signal, 2019. 63: p. 109377. [DOI] [PubMed] [Google Scholar]
- 14.Boldrup L, et al. DeltaNp63 isoforms regulate CD44 and keratins 4, 6, 14 and 19 in squamous cell carcinoma of head and neck. J Pathol, 2007. 213(4): p. 384–91. [DOI] [PubMed] [Google Scholar]
- 15.Du Z, et al. Overexpression of ΔNp63α induces a stem cell phenotype in MCF7 breast carcinoma cell line through the Notch pathway. Cancer Sci, 2010. 101(11): p. 2417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Compagnone M, et al. ΔNp63-mediated regulation of hyaluronic acid metabolism and signaling supports HNSCC tumorigenesis. Proc Natl Acad Sci U S A, 2017. 114(50): p. 13254–13259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatti V, et al. ΔNp63 regulates the expression of hyaluronic acid-related genes in breast cancer cells. Oncogenesis, 2018. 7(8): p. 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carroll DK, et al. p63 regulates an adhesion programme and cell survival in epithelial cells. Nat Cell Biol, 2006. 8(6): p. 551–61. [DOI] [PubMed] [Google Scholar]
- 19.Cooper J and Giancotti FG, Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell, 2019. 35(3): p. 347–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarti R, et al. ΔNp63 promotes stem cell activity in mammary gland development and basal-like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol, 2014. 16(10): p. 1004–15, 1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Memmi EM, et al. p63 Sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc Natl Acad Sci U S A, 2015. 112(11): p. 3499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang GX, et al. ΔNp63 Inhibits Oxidative Stress-Induced Cell Death, Including Ferroptosis, and Cooperates with the BCL-2 Family to Promote Clonogenic Survival. Cell Rep, 2017. 21(10): p. 2926–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peinado H, et al. Pre-metastatic niches: organ-specific homes for metastases. Nat Rev Cancer, 2017. 17(5): p. 302–317. [DOI] [PubMed] [Google Scholar]
- 24.Lodillinsky C, et al. p63/MT1-MMP axis is required for in situ to invasive transition in basal-like breast cancer. Oncogene, 2016. 35(3): p. 344–57. [DOI] [PubMed] [Google Scholar]
- 25.Celardo I, et al. p63 transcriptionally regulates the expression of matrix metallopeptidase 13. Oncotarget, 2014. 5(5): p. 1279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, et al. ΔNp63-driven recruitment of myeloid-derived suppressor cells promotes metastasis in triple-negative breast cancer. J Clin Invest, 2018. 128(11): p. 5095–5109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee KB, et al. p63-Mediated activation of the β-catenin/c-Myc signaling pathway stimulates esophageal squamous carcinoma cell invasion and metastasis. Cancer Lett, 2014. 353(1): p. 124–32. [DOI] [PubMed] [Google Scholar]
- 28.Dang TT, et al. ΔNp63α induces the expression of FAT2 and Slug to promote tumor invasion. Oncotarget, 2016. 7(19): p. 28592–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dang TT, et al. ΔNp63α Promotes Breast Cancer Cell Motility through the Selective Activation of Components of the Epithelial-to-Mesenchymal Transition Program. Cancer Res, 2015. 75(18): p. 3925–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rodriguez Calleja L, et al. ΔNp63α Silences a miRNA Program to Aberrantly Initiate a Wound-Healing Program That Promotes TGFβ-Induced Metastasis. Cancer Res, 2016. 76(11): p. 3236–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung KJ, et al. Collective invasion in breast cancer requires a conserved basal epithelial program. Cell, 2013. 155(7): p. 1639–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Holcakova J, et al. ΔNp63 activates EGFR signaling to induce loss of adhesion in triple-negative basal-like breast cancer cells. Breast Cancer Res Treat, 2017. 163(3): p. 475–484. [DOI] [PubMed] [Google Scholar]
- 33.Ganesh K and Massagué J, Targeting metastatic cancer. Nat Med, 2021. 27(1): p. 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Charan M, et al. Tumor secreted ANGPTL2 facilitates recruitment of neutrophils to the lung to promote lung pre-metastatic niche formation and targeting ANGPTL2 signaling affects metastatic disease. Oncotarget, 2020. 11(5): p. 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang YT, et al. The matricellular protein CYR61 promotes breast cancer lung metastasis by facilitating tumor cell extravasation and suppressing anoikis. Oncotarget, 2017. 8(6): p. 9200–9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu N, et al. p63 regulates human keratinocyte proliferation via MYC-regulated gene network and differentiation commitment through cell adhesion-related gene network. J Biol Chem, 2012. 287(8): p. 5627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbieri CE, et al. Loss of p63 leads to increased cell migration and up-regulation of genes involved in invasion and metastasis. Cancer Res, 2006. 66(15): p. 7589–97. [DOI] [PubMed] [Google Scholar]
- 38.Zhao W, et al. ΔNp63α attenuates tumor aggressiveness by suppressing miR-205/ZEB1-mediated epithelial-mesenchymal transition in cervical squamous cell carcinoma. Tumour Biol, 2016. 37(8): p. 10621–32. [DOI] [PubMed] [Google Scholar]
- 39.Yoh KE, et al. Repression of p63 and induction of EMT by mutant Ras in mammary epithelial cells. Proc Natl Acad Sci U S A, 2016. 113(41): p. E6107–e6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lindsay J, et al. Role of DeltaNp63gamma in epithelial to mesenchymal transition. J Biol Chem, 2011. 286(5): p. 3915–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tucci P, et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc Natl Acad Sci U S A, 2012. 109(38): p. 15312–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran MN, et al. The p63 protein isoform ΔNp63α inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J Biol Chem, 2013. 288(5): p. 3275–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nam RK, et al. MiR-301a regulates E-cadherin expression and is predictive of prostate cancer recurrence. Prostate, 2016. 76(10): p. 869–84. [DOI] [PubMed] [Google Scholar]
- 44.Keyes WM and Mills AA, p63: a new link between senescence and aging. Cell Cycle, 2006. 5(3): p. 260–5. [DOI] [PubMed] [Google Scholar]
- 45.Konieczkowski DJ, Johannessen CM, and Garraway LA, A Convergence-Based Framework for Cancer Drug Resistance. Cancer Cell, 2018. 33(5): p. 801–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sen T, et al. DeltaNp63alpha confers tumor cell resistance to cisplatin through the AKT1 transcriptional regulation. Cancer Res, 2011. 71(3): p. 1167–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danilov AV, et al. DeltaNp63alpha-mediated induction of epidermal growth factor receptor promotes pancreatic cancer cell growth and chemoresistance. PLoS One, 2011. 6(10): p. e26815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mendoza-Rodríguez MG, et al. IL-1β Inflammatory Cytokine-Induced TP63 Isoform ΔNP63α Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. Int J Mol Sci, 2019. 20(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hao T and Gan YH, ΔNp63α promotes the expression and nuclear translocation of PTEN, leading to cisplatin resistance in oral cancer cells. Am J Transl Res, 2020. 12(10): p. 6187–6203. [PMC free article] [PubMed] [Google Scholar]
- 50.Mundt HM, et al. Dominant negative (DeltaN) p63alpha induces drug resistance in hepatocellular carcinoma by interference with apoptosis signaling pathways. Biochem Biophys Res Commun, 2010. 396(2): p. 335–41. [DOI] [PubMed] [Google Scholar]
- 51.Zhou P, et al. ΔNp63α promotes Bortezomib resistance via the CYGB-ROS axis in head and neck squamous cell carcinoma. Cell Death Dis, 2022. 13(4): p. 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andricovich J, et al. Loss of KDM6A Activates Super-Enhancers to Induce Gender-Specific Squamous-like Pancreatic Cancer and Confers Sensitivity to BET Inhibitors. Cancer Cell, 2018. 33(3): p. 512–526.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fisher ML, et al. BRD4 Regulates Transcription Factor ΔNp63α to Drive a Cancer Stem Cell Phenotype in Squamous Cell Carcinomas. Cancer Res, 2021. 81(24): p. 6246–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Balinth S, et al. EZH2 regulates a SETDB1/ΔNp63α axis via RUNX3 to drive a cancer stem cell phenotype in squamous cell carcinoma. Oncogene, 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Regina C, et al. Setdb1, a novel interactor of ΔNp63, is involved in breast tumorigenesis. Oncotarget, 2016. 7(20): p. 28836–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Federico A, et al. Mithramycin A and Mithralog EC-8042 Inhibit SETDB1 Expression and Its Oncogenic Activity in Malignant Melanoma. Mol Ther Oncolytics, 2020. 18: p. 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guo Y, et al. Structure-Guided Discovery of a Potent and Selective Cell-Active Inhibitor of SETDB1 Tudor Domain. Angew Chem Int Ed Engl, 2021. 60(16): p. 8760–8765. [DOI] [PubMed] [Google Scholar]
- 58.Stacy AJ, et al. TIP60 up-regulates ΔNp63α to promote cellular proliferation. J Biol Chem, 2019. 294(45): p. 17007–17016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Citro S, et al. Synergistic antitumour activity of HDAC inhibitor SAHA and EGFR inhibitor gefitinib in head and neck cancer: a key role for ΔNp63α. Br J Cancer, 2019. 120(6): p. 658–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramsey MR, et al. Physical association of HDAC1 and HDAC2 with p63 mediates transcriptional repression and tumor maintenance in squamous cell carcinoma. Cancer Res, 2011. 71(13): p. 4373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kubo T, et al. IL-13 modulates ΔNp63 levels causing altered expression of barrier- and inflammation-related molecules in human keratinocytes: A possible explanation for chronicity of atopic dermatitis. Immun Inflamm Dis, 2021. 9(3): p. 734–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ekman AK, et al. IL-17 and IL-22 Promote Keratinocyte Stemness in the Germinative Compartment in Psoriasis. J Invest Dermatol, 2019. 139(7): p. 1564–1573.e8. [DOI] [PubMed] [Google Scholar]
- 63.Xie C, et al. Sulforaphane Inhibits the Acquisition of Tobacco Smoke-Induced Lung Cancer Stem Cell-Like Properties via the IL-6/ΔNp63α/Notch Axis. Theranostics, 2019. 9(16): p. 4827–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brauweiler AM, Leung DYM, and Goleva E, The Transcription Factor p63 Is a Direct Effector of IL-4- and IL-13-Mediated Repression of Keratinocyte Differentiation. J Invest Dermatol, 2021. 141(4): p. 770–778. [DOI] [PubMed] [Google Scholar]
- 65.Somerville TD, et al. Squamous trans-differentiation of pancreatic cancer cells promotes stromal inflammation. Elife, 2020. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu L, et al. A novel IL-17 signaling pathway controlling keratinocyte proliferation and tumorigenesis via the TRAF4-ERK5 axis. J Exp Med, 2015. 212(10): p. 1571–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nam SM, et al. Ex Vivo Expansion of Human Limbal Epithelial Cells Using Human Placenta-Derived and Umbilical Cord-Derived Mesenchymal Stem Cells. Stem Cells Int, 2017. 2017: p. 4206187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hsueh YJ, et al. Extracellular Matrix Protein Coating of Processed Fish Scales Improves Human Corneal Endothelial Cell Adhesion and Proliferation. Transl Vis Sci Technol, 2019. 8(3): p. 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ma DH, et al. Preservation of human limbal epithelial progenitor cells on carbodiimide cross-linked amniotic membrane via integrin-linked kinase-mediated Wnt activation. Acta Biomater, 2016. 31: p. 144–155. [DOI] [PubMed] [Google Scholar]
- 70.Ripamonti F, et al. EGFR through STAT3 modulates ΔN63α expression to sustain tumor-initiating cell proliferation in squamous cell carcinomas. J Cell Physiol, 2013. 228(4): p. 871–8. [DOI] [PubMed] [Google Scholar]
- 71.Jiang Y, et al. Co-activation of super-enhancer-driven CCAT1 by TP63 and SOX2 promotes squamous cancer progression. Nat Commun, 2018. 9(1): p. 3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fisher ML, et al. Transglutaminase Interaction with α6/β4-Integrin Stimulates YAP1-Dependent ΔNp63α Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res, 2016. 76(24): p. 7265–7276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 73.Grun D, Adhikary G, and Eckert RL, NRP-1 interacts with GIPC1 and α6/β4-integrins to increase YAP1/ΔNp63α-dependent epidermal cancer stem cell survival. Oncogene, 2018. 37(34): p. 4711–4722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yuan M, et al. c-Abl phosphorylation of ΔNp63α is critical for cell viability. Cell Death Dis, 2010. 1(1): p. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Eckert RL, et al. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog, 2015. 54(10): p. 947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elisi GM, et al. Repurposing of Drugs Targeting YAP-TEAD Functions. Cancers (Basel), 2018. 10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nusse R and Clevers H, Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell, 2017. 169(6): p. 985–999. [DOI] [PubMed] [Google Scholar]
- 78.Chu WK, et al. Glycogen synthase kinase-3beta regulates DeltaNp63 gene transcription through the beta-catenin signaling pathway. J Cell Biochem, 2008. 105(2): p. 447–53. [DOI] [PubMed] [Google Scholar]
- 79.Lindley LE, et al. The WNT-controlled transcriptional regulator LBH is required for mammary stem cell expansion and maintenance of the basal lineage. Development, 2015. 142(5): p. 893–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galoczova M, Coates P, and Vojtesek B, STAT3, stem cells, cancer stem cells and p63. Cell Mol Biol Lett, 2018. 23: p. 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hsueh YJ, et al. STAT3 regulates the proliferation and differentiation of rabbit limbal epithelial cells via a ΔNp63-dependent mechanism. Invest Ophthalmol Vis Sci, 2011. 52(7): p. 4685–93. [DOI] [PubMed] [Google Scholar]
- 82.Chu WK, et al. Transcriptional activity of the DeltaNp63 promoter is regulated by STAT3. J Biol Chem, 2008. 283(12): p. 7328–37. [DOI] [PubMed] [Google Scholar]
- 83.Yi Y, et al. Metformin Promotes AMP-activated Protein Kinase-independent Suppression of ΔNp63α Protein Expression and Inhibits Cancer Cell Viability. J Biol Chem, 2017. 292(13): p. 5253–5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.DeRan M, et al. Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep, 2014. 9(2): p. 495–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan X, et al. Metformin inhibits glioma cells stemness and epithelial-mesenchymal transition via regulating YAP activity. Biomed Pharmacother, 2018. 102: p. 263–270. [DOI] [PubMed] [Google Scholar]
- 86.Wu Y, et al. Metformin targets a YAP1-TEAD4 complex via AMPKα to regulate CCNE1/2 in bladder cancer cells. J Exp Clin Cancer Res, 2019. 38(1): p. 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fisher ML, et al. Sulforaphane reduces YAP/ΔNp63α signaling to reduce cancer stem cell survival and tumor formation. Oncotarget, 2017. 8(43): p. 73407–73418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Rorke EA, et al. Sulforaphane covalently interacts with the transglutaminase 2 cancer maintenance protein to alter its structure and suppress its activity. Mol Carcinog, 2022. 61(1): p. 19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Asatsuma-Okumura T, Ito T, and Handa H, Molecular mechanisms of cereblon-based drugs. Pharmacol Ther, 2019. 202: p. 132–139. [DOI] [PubMed] [Google Scholar]
- 90.Asatsuma-Okumura T, et al. p63 is a cereblon substrate involved in thalidomide teratogenicity. Nat Chem Biol, 2019. 15(11): p. 1077–1084. [DOI] [PubMed] [Google Scholar]
- 91.Davis RJ, Van Waes C, and Allen CT, Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and Tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol, 2016. 58: p. 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]