

Abstract
Background
Mpox was declared a Public Health Emergency of International Concern (PHEIC) by the World Health Organization (WHO) on 23 July 2022, following the identification of thousands of cases in several non‐endemic countries in previous months. There are currently no licenced therapeutics for treating mpox; however, some medications may be authorized for use in an outbreak. The efficacy and safety of possible therapeutic options has not been studied in humans with mpox. There is a need to investigate the evidence on safety and effectiveness of treatments for mpox in humans; should any therapeutic option be efficacious and safe, it may be approved for use around the world.
Objectives
There are two parts to this Cochrane Review: a review of evidence from randomized controlled trials (RCTs), and a narrative review of safety data from non‐randomized studies.
Randomized controlled trials review
To systematically review the existing evidence on the effectiveness of therapeutics for mpox infection in humans compared to:
a) another different therapeutic for mpox, or
b) placebo, or
c) supportive care, defined as the treatment of physical and psychological symptoms arising from the disease.
Non‐randomized studies review
To assess the safety of therapeutics for mpox infection from non‐randomized studies (NRS).
Search methods
Randomized controlled trials review
We searched the following databases up to 25 January 2023: MEDLINE (OVID), Embase (OVID), Biosis previews (Web of Science), CAB Abstracts (Web of science), and Cochrane CENTRAL (Issue 1 2023). We conducted a search of trial registries (Clinicaltrials.gov and International Clinical Trials Registry Platform (ICTRP)) on 25 January 2023. There were no date or language limits placed on the search. We undertook a call to experts in the field for relevant studies or ongoing trials to be considered for inclusion in the review.
Non‐randomized studies review
We searched the following databases on 22 September 2022: Cochrane Central Register of Controlled Trials (CENTRAL; Issue 9 of 12, 2022), published in the Cochrane Library; MEDLINE (Ovid); Embase (Ovid); and Scopus (Elsevier). We also searched the WHO International Clinical Trials Registry Platform and ClinicalTrials.gov for trials in progress.
Selection criteria
For the RCT review and the narrative review, any therapeutic for the treatment of mpox in humans was eligible for inclusion, including tecovirimat, brincidofovir, cidofovir, NIOCH‐14, immunomodulators, and vaccine immune globulin.
Randomized controlled trials review
Studies were eligible for the main review if they were of randomized controlled design and investigated the effectiveness or safety of therapeutics in human mpox infection.
Non‐randomized studies review
Studies were eligible for inclusion in the review of non‐randomized studies if they were of non‐randomized design and contained data concerning the safety of any therapeutic in human mpox infection.
Data collection and analysis
Randomized controlled trials review
Two review authors independently applied study inclusion criteria to identify eligible studies. If we had identified any eligible studies, we planned to assess the risk of bias, and report results with 95% confidence intervals (CI). The critical outcomes were serious adverse events, development of disease‐related complications, admission to hospital for non‐hospitalized participants, pain as judged by any visual or numerical pain scale, level of virus detected in clinical samples, time to healing of all skin lesions, and mortality.
We planned to perform subgroup analysis to explore whether the effect of the therapeutic on the planned outcomes was modified by disease severity and days from symptom onset to therapeutic administration. We also intended to explore the following subgroups of absolute effects: immunosuppression, age, and pre‐existing skin disease.
Non‐randomized studies review
One review author applied study inclusion criteria to identify eligible studies and extracted data. Studies of a non‐randomized design containing data on the safety of therapeutics could not be meta‐analyzed due to the absence of a comparator; we summarized these data narratively in an appendix.
Main results
Randomized controlled trials review
We did not identify any completed RCTs investigating the effectiveness of therapeutics for treating mpox for the main review. We identified five ongoing trials that plan to assess the effectiveness of one therapeutic option, tecovirimat, for treating mpox in adults and children. One of these ongoing trials intends to include populations with, or at greater risk of, severe disease, which will allow an assessment of safety in more vulnerable populations.
Non‐randomized studies review
Three non‐randomized studies met the inclusion criteria for the narrative review, concerning data on the safety of therapeutics in mpox. Very low‐certainty evidence from non‐randomized studies of small numbers of people indicates no serious safety signals emerging for the use of tecovirimat in people with mpox infection, but a possible safety signal for brincidofovir. All three participants who received brincidofovir had raised alanine aminotransferase (ALT), but not bilirubin, suggesting mild liver injury. No study reported severe drug‐induced liver injury with brincidofovir.
Authors' conclusions
Randomized controlled trials review
This review found no evidence from randomized controlled trials concerning the efficacy and safety of therapeutics in humans with mpox.
Non‐randomized studies review
Very low‐certainty evidence from non‐randomized studies indicates no serious safety signals emerging for the use of tecovirimat in people with mpox infection. In contrast, very low‐certainty evidence raises a safety signal that brincidofovir may cause liver injury. This is also suggested by indirect evidence from brincidofovir use in smallpox. This warrants further investigation and monitoring.
This Cochrane Review will be updated as new evidence becomes available to assist policymakers, health professionals, and consumers in making appropriate decisions for the treatment of mpox.
Keywords: Adult; Child; Humans; Immunoglobulins; Mpox, Monkeypox; Organophosphonates
Plain language summary
Therapeutics for treating mpox
What is the aim of this review?
There are no drugs licenced specifically for treating mpox, but some drugs that are licenced for treating similar viral infections, such as smallpox, are authorized for treating mpox during an outbreak. The effects of these drugs have not yet been studied in randomized trials in people with mpox. Randomized trials include at least two treatment groups, such as one receiving the drug and one receiving a placebo, where the people recruited for the trial are randomly allocated to either group. This review summarizes evidence on the safety and effectiveness of treatments for mpox in humans, which was completed in two parts: a review of evidence from randomized controlled trials (RCTs) and a review of safety data from non‐randomized studies.
Randomized controlled trials review
We searched for RCTs on both the safety and effectiveness of treatments for mpox.
Non‐randomized studies review
We searched for non‐randomized studies on the safety of treatments for mpox only.
Key messages
Randomized controlled trials review
When their results become available, data from five ongoing RCTs will allow us to compare safety and effectiveness outcomes between people who receive treatment for mpox and those who do not. We will be able to compare patient‐important outcomes for different treatment options.
Non‐randomized studies review
Tecovirimat, an oral anti‐viral drug, is safe and well tolerated in individuals with mpox, based on evidence from non‐randomized studies. Three people treated with brincidofovir, another oral anti‐viral drug, experienced a sudden increase in one liver enzyme called alanine transaminase (ALT), and their treatment was discontinued as a result. This result may indicate mild liver injury caused by the drug. More severe forms of drug‐induced liver injury can cause serious liver damage and even liver failure, so this should be closely monitored in people with mpox who are treated with brincidofovir.
What was studied in the review?
Randomized controlled trials review
This review identified five ongoing clinical trials assessing tecovirimat for treating people with mpox. They aim to recruit a total of 1750 people in the USA, Canada, Brazil, Switzerland, UK, and Democratic Republic of Congo.
Non‐randomized studies review
This review identified three non‐randomized studies conducted in the Central African Republic, UK and USA. A total of 355 people received tecovirimat, and three received brincidofovir as their treatment drug.
What are the main results of the review?
Randomized controlled trials review
There is currently no evidence from RCTs available, but the five ongoing trials identified will be assessed in future updates of this review.
Non‐randomized studies review
Three non‐randomized studies assessed safety in 358 people treated for mpox (355 received tecovirimat, 3 received brincidofovir). Very few of the people who received tecovirimat reported unwanted or harmful effects (16 out of 355 people). These included 11 mild unwanted or harmful effects, two mental health outcomes, and one case of increased liver enzymes. There was also one death and one case of anaemia, but neither of these was thought to be related to the study drug.
All three people who received brincidofovir reported an increase in alanine transaminase (ALT; a liver enzyme released by liver cells when they are damaged) which led to discontinuation of their treatment. In at least two of the cases treated with brincidofovir, their increased alanine transaminase level may indicate mild drug‐induced liver injury. We do not know whether this would have progressed to become more serious if treatment had not been discontinued. Liver enzymes should be carefully monitored in individuals receiving this drug.
What are the limitations of the evidence?
Randomized controlled trials review
There is currently no evidence from RCTs available.
Non‐randomized studies review
People included in the studies all received the treatment, so we are unable to compare these results against people with a placebo or no treatment. The study investigating brincidofovir was also very small.
How up to date is this review?
Randomized controlled trials review
The review authors searched for studies available up to 25 January 2023.
Non‐randomized studies review
The review authors searched for studies available up to 22 September 2022.
Background
Description of the condition
Mpox (previously termed monkeypox) is a zoonosis, first identified in macaques transported to Denmark and discovered in the human population in 1970 in the Democratic Republic of the Congo. Until 2022, primary cases of human mpox were largely associated with animal contact, and secondary transmission to household contacts was estimated at around 8% (Beer 2019). The incidence of cases in endemic areas of central and West Africa has been increasing rapidly over the past decades, coinciding with the cessation of the smallpox vaccination and eradication programme (Rimoin 2010). Evidence of mpox infection has been identified in many species, including non‐human primates and monkeys, but the main reservoir species is suspected to be a rodent such as the rope squirrel (Funisciuris) (Falendysz 2017).
Mpox virus is an enveloped double‐stranded DNA virus that belongs to the Orthopoxvirus genus of the Poxviridae family. Two distinct clades of the virus exist: Clade I (former Congo Basin), and Clade IIa (former West African) (WHO 2022a). The clade circulating in the 2022 outbreak of mpox is known as Clade IIb.
Mpox was declared a Public Health Emergency of International Concern (PHEIC) by the World Health Organization (WHO) on 23 July 2022, following the identification of thousands of cases in several non‐endemic countries in previous months (WHO 2022b). A more recent meeting of the International Health Regulations (2005) (IHR) Emergency Committee on 9 February 2023 advised maintaining the PHEIC status of the outbreak (WHO 2023). The 2022 outbreak was associated with sustained human‐to‐human transmission, which had not previously been described (CDC 2023a).
Mpox infection has an incubation period ranging between five and 21 days, and typically presents symptoms in two stages (NHS inform 2023). The invasion period lasting for zero to five days is characterized by fever, headache, lymphadenopathy, back pain, myalgia, and asthenia; following this, skin presentations can appear between one and three days from onset of fever, with a rash evolving from macules (lesions with a flat base) to papules (slightly raised firm lesions), vesicles (lesions filled with clear fluid), pustules (lesions filled with yellowish fluid), and crusts, often affecting the face, extremities, oral mucous membranes, genitalia, and conjunctivae (WHO 2022c).
Compared to the closely related smallpox infection, mpox infection is less clinically severe with mortality rates for Clade I between one and ten per cent and mortality for Clade II at around three to six per cent, although severity and mortality are affected by clade and patient characteristics (Americo 2022; WHO 2022c). Disease severity has been demonstrated to vary between populations, but those at the highest risk of severe disease include immunocompromised people, paediatric populations, people who are pregnant or breastfeeding, and people with a condition affecting skin integrity (CDC 2023b). In severe cases of infection, people may have a very high number of lesions, necrotic lesions, severe lymphadenopathy, or develop complications such as pneumonitis, encephalitis, keratitis or abscesses. Over the past year, complications of mpox which had not previously been reported have included proctitis, tonsillitis, and myocarditis (Patel 2022).
Description of the intervention
At present, there are no licenced treatments for mpox infection. Antiviral medications used for the treatment of smallpox, as well as medications for the treatment of similar infections and skin conditions, may have a role in the treatment of mpox infection (NIAID 2022). These antivirals include: tecovirimat or ST‐246 (TPOXX); brincidofovir (Tembexa); and cidofovir (Vistide). Additionally, intravenous vaccinia immune globulin (VIGIV), which is licensed for the treatment of complications from smallpox (vaccinia) vaccination, may be authorized for use to treat mpox and other pox viruses during an outbreak.
Both tecovirimat and brincidofovir have been approved for the treatment of smallpox in the USA, in 2018 and 2021 respectively (Chimerix 2022), but neither has yet been studied in human efficacy trials. Efficacy data from animal models suggest that these antivirals are effective in the treatment of orthopoxvirsuses (Grosenbach 2018; Grossi 2017; Hutson 2021; Smith 2011; Trost 2015).
How the intervention might work
Tecovirimat
Initially developed as a drug for the treatment of smallpox under the Food and Drug Administration’s (FDA) ‘animal rule’, tecovirimat (ST‐246, TPOXX) inhibits the activity of key proteins, preventing the formation of extracellular enveloped virion (EEVs) required for dissemination of the virus (Hoy 2018). Pre‐clinical studies have shown tecovirimat to be highly effective against mpox both in vitro and in vivo in a number of animal models of orthopoxviruses, including mice, rabbits, ground‐squirrels and non‐human primates (Jordan 2009; Sbrana 2007). The barrier to resistance for tecovirimat is considered to be low, and resistance mutations to tecovirimat under artificial culture pressure have been described (Frenois‐Veyrat 2022).
Brincidofovir
Brincidofovir (TEMBEXA) is a prodrug that is taken up via endogenous lipid uptake pathways, then converted intracellularly to cidofovir, which is subsequently phosphorylated to the active antiviral (LiverTox 2022). The lipid conjugation of brincidofovir improves oral bioavailability and reduces the nephrotoxicity seen with cidofovir. Pre‐clinical studies have shown brincidofovir to be highly effective against mpox virus in vitro and in vivo with significant improvement in mortality in animal models of orthopoxviruses, including rabbitpox and mousepox models (Siegrist 2022). Cell culture studies have shown that amino acid substitutions in the target viral DNA polymerase protein can confer resistance, although there are no known instances of naturally occurring brincidofovir resistant orthopoxviruses (Foster 2017).
Cidofovir
Cidofovir is currently licensed for use in the treatment of cytomegalovirus (CMV) retinitis in adults with AIDS. It shows broad spectrum antiviral activity against most DNA viruses, including orthopoxviruses (Seley‐Radtke 2021). It acts via selective inhibition of viral DNA synthesis. Preclinical studies demonstrate in vitro effectiveness against mpox virus and in vivo effectiveness in animal models of pox viruses (Kern 2003; Andrei 2010). It has been found to be effective clinically for the treatment of the poxviruses molluscum contagiosum virus and orf virus in immunocompromised patients (De Clerq 2002). Nephrotoxicity is the major dose limiting toxicity associated with cidofovir, and use of cidofovir must be accompanied by measures to reduce nephrotoxicity including pre‐hydration with intravenous saline and co‐administration of probenecid.
NIOCH‐14
NIOCH‐14 is a synthetic analogue of tecovirimat, which has undergone phase 1 trials. Pre‐clinical studies have shown effectiveness comparable to tecovirimat both in vitro and in vivo in mouse and marmot mpox virus and variola virus (VARV) models (Mazurkov 2016).
Vaccinia Immune Globulin Intravenous (VIGIV)
VIGIV is a purified IgG fraction of human plasma containing antibodies to vaccinia virus, collected from healthy donors following exposure to vaccinia virus (VV) vaccine. The exact mechanism of action is not known. It provides passive immunity and is indicated for the treatment of complications associated with vaccinia vaccination, including eczema vaccinatum, progressive vaccinia, and severe generalized vaccinia (Cangene 2010). Pre‐clinical studies using the mouse‐tail lesion model of VV demonstrated a protective effect of VIGIV (Shearer 2005). Clinical trials of VIGIV in healthy volunteers attenuated the endogenous immune response to vaccinia vaccination, consistent with the hypothesis of in vivo neutralization of VV by VIGIV (Wittek 2006). Data are not available on the effectiveness of VIGIV against mpox virus or in mpox viral infection.
Why it is important to do this review
To date (January 2023), 85,158 cases of human mpox have been reported across 110 locations, including 103 locations which have not previously recorded cases (CDC 2022a). In some countries, people experiencing severe disease or at risk for severe disease may be offered treatment with investigational therapeutics. In the USA, tecovirimat is offered to some mpox patients through an expanded access investigational new drug (EA‐IND) protocol. In the UK, brincidofovir and tecovirimat may be offered to people with severe disease.
A review on the safety and efficacy of cidofovir, brincidofovir, and tecovirimat (Yu 2022) summarized data from all study types, including in vitro and in vivo animal studies, human clinical trials, and human case reports of orthopoxvirus infection. The review included nine trials in humans that investigated the safety of tecovirimat or brincidofovir in healthy humans, humans with other viral infections, or recipients of haematopoietic cell transplant (HCT). This review demonstrated that both drugs were generally safe and well‐tolerated in both the healthy and immunocompromised populations. This evidence of safety was limited in that it did not investigate drugs specifically in participants with mpox. Similarly, this review summarized data on the efficacy of drugs for treating viral infections, which may be applicable to mpox, but there were no randomized controlled trials (RCTs) on participants with mpox infection available for inclusion.
The most recent outbreak of mpox has permitted the development of trials on the safety and efficacy of therapeutics for mpox in infected human populations, which will provide an improved evidence base for recommendations on appropriate treatment options.
Non‐randomized studies review
Since we are aware that trials investigating therapeutics for mpox are mostly ongoing, and we anticipated a small number of included studies, we also conducted a review of non‐randomized studies (presented in an appendix). Non‐randomized studies (NRS) may provide additional evidence regarding the effects of therapeutics for mpox, particularly when used to treat individuals who are commonly excluded from RCTs, such as those with pre‐existing medical conditions. The non‐randomized studies review is described in full detail in Appendix 1.
Objectives
-
To systematically review the existing evidence on the effectiveness of therapeutics for mpox infection in humans compared to:
another different therapeutic for mpox, or
placebo, or
supportive care, defined as the treatment of physical and psychological symptoms arising from the disease.
To assess the safety of therapeutics for mpox infection from non‐randomized studies (NRS).
Methods
Criteria for considering studies for this review
Types of studies
There are two parts to this review; the main review concerning evidence from randomized trials, and a narrative review of safety data from non‐randomized studies. The full methods for the narrative review of safety data from non‐randomized studies is contained within Appendix 1, and summarized briefly at the end of the Methods section. The methods detailed below relate to the review of randomized controlled trials (RCTs).
Studies were eligible if they were of randomized controlled design. Cluster‐randomized controlled trials would have been eligible; however, none were identified. Trials with a cross‐over design were not eligible.
Types of participants
Humans with mpox virus infection confirmed by reverse transcription polymerase chain reaction (RT‐PCR), inclusive of participants with confirmed mpox infection as a subset population within a study, were eligible for inclusion.
Studies conducted in any country or setting were eligible for this review, including inpatient and outpatient settings.
Types of interventions
All therapeutics used for the treatment of human mpox were eligible for this review. This included, but was not limited to, tecovirimat, brincidofovir, cidofovir, NIOCH‐14, immunomodulators, and vaccine immune globulin.
Control
Placebo, or
Supportive care, or
Placebo and supportive care, or
A different intervention therapeutic and supportive care
Supportive care refers to symptomatic relief measures and procedures required for specific symptoms or complications arising from the disease.
Types of outcome measures
The main review addresses patient‐important outcomes as determined by the WHO Clinical Management and Infection Prevention and Control guideline development group (GDG), described below. These are outcomes of interest, and we did not use them as criteria for study inclusion.
Critical outcomes
Serious adverse events (defined as any adverse event that may be life‐threatening or could result in death, up to 28 days)
Development of disease‐related complications (proctitis, urethritis, ophthalmic complications, up to 28 days or the longest period as reported by study authors)
Admission to hospital for non‐hospitalized participants (up to 28 days)
Pain, as judged by any visual or numerical pain scale (up to 28 days)
Level of virus detected in clinical samples (as an indicator of infectiousness) (up to 28 days or the longest period as reported by study authors)
Time to healing of all skin lesions (up to 28 days or the longest period as reported by study authors)
Mortality (up to 28 days)
Important outcomes
Scarring, as judged by an appropriate scale such as the Vancouver Scar Scale (VSS) (up to 28 days)
Clinical disease progression for hospitalized participants, as judged by study investigators (up to 28 days)
Duration of illness defined as time to resolution of all signs and symptoms (days since symptom onset, up to 28 days, or the longest period as reported by study authors)
Adverse maternal outcomes (up to 28 days)
Adverse perinatal outcomes (up to 28 days)
Mental health outcomes (any mental health condition reported during study period, up to 28 days, or the longest period as reported by study authors)
Search methods for identification of studies
Electronic searches
We searched the following databases using the search terms detailed in Appendix 2: MEDLINE (OVID; 1946 to 25 January 2023), Embase (OVID; 1947 to 25 January 2023), Biosis previews (Web of Science; 1926 to 25 January 2023), CAB Abstracts (Web of Science; 1910 to 25 January 2023), and Cochrane Central Register of Controlled Trials (CENTRAL) (Issue 1 of 12, January 2023). There were no date or language limits placed on the search.
We also searched ClinicalTrials.gov and the WHO ICTRP for trials in progress, on 25 January 2023 using the terms shown in Appendix 2.
Searching other resources
We asked experts from the WHO Clinical Management and Infection Prevention and Control guideline development group for details of potentially relevant studies or ongoing trials to be considered for inclusion in the review.
Data collection and analysis
Selection of studies
Three review authors (Tilly Fox, Rebecca Kuehn, Naveena Princy) independently screened the titles and abstracts of the search results for potentially relevant articles using the inclusion criteria. If there was any doubt over the potential for inclusion of a study, it was included for full‐text screening.
Two review authors (TF, RK) independently reviewed the full text of studies identified through title and abstract screening; any disagreements were resolved by discussion with all authors.
Data extraction and management
Two review authors (TF, RK) independently extracted data using a pre‐piloted standardized data extraction form. We contacted the study authors to obtain missing data if applicable. At each step of data extraction, we resolved any discrepancies through discussion between all review authors.
We extracted the following information.
General information: author, title, publication date, country, date(s) of study, funding details, conflict of interest statement
Study characteristics: study setting (inpatient or outpatient); study design; dates of recruitment; eligibility criteria; length of follow‐up; loss to follow‐up; and adherence to assigned treatment.
Participant characteristics: number of participants (recruited, allocated, and evaluated); source of participants; age; sex; disease severity; vaccination status; concurrent treatments; pregnancy; and comorbidities (e.g. skin disease, immunosuppression).
Interventions: type; route of administration; dosage; timing; frequency; duration of treatment; and duration of follow‐up.
Control: type (placebo/active treatment); route of administration; dosage; timing; frequency; duration of treatment; and duration of follow‐up.
Outcomes: data on the prespecified outcomes in both the intervention and control arms as follows: for dichotomous outcomes, number of events and participants; for continuous outcomes, mean, standard deviation (SD), and total number of participants; and for time‐to‐event outcomes, hazard ratios (HRs).
Assessment of risk of bias in included studies
Two review authors (TF, RK) planned to independently assess the risk of bias using the Cochrane risk of bias 2 tool (RoB 2) (Cochrane 2022; Higgins 2022; Sterne 2019). Any disagreements would be settled by a third review author. We planned to justify judgements made in the risk of bias tables. The effect of interest was to be the effect of assignment to the intervention at baseline, regardless of whether the interventions were received as intended (the ‘intention‐to‐treat effect’). We intended to manage the assessments using the RoB 2 Excel tool for randomized trials and assess risk of bias for the critically important outcomes included in the analyses. We planned to use the following domains to assess bias:
bias arising from the randomization process;
bias due to deviations from intended interventions;
bias due to missing outcome data;
bias in measurement of the outcome; and
bias in selection of the reported result.
For trials that had randomized clusters, we planned to assess an additional component, namely bias arising from identification or recruitment of individual participants within clusters.
We planned to answer signalling questions as either yes; probably yes; probably no; no; or no information. We would then use these to determine the overall risk of bias for each domain (high, some concerns, low) and later, the overall risk of bias for each primary outcome from the included studies (high, some concerns, low). We planned to judge study outcomes to have an overall low risk of bias if all domains were at low risk, some concerns if any domain had some concerns, and high if we assessed any domain to be at high risk of bias.
Measures of treatment effect
For dichotomous outcomes (serious adverse events, development of disease‐related complications, admission to hospital, detection of virus, mortality, scarring, adverse maternal outcomes, adverse perinatal outcomes, and mental health outcomes), we planned to use the risk ratio (RR) with the corresponding 95% interval (CI) as the effect measure.
For continuous outcomes (pain, time to healing of all skin lesions, clinical disease progression, and duration of illness) when outcomes were measured in the same way between trials, we planned to use mean differences (MD) with the corresponding 95% interval (CI) as the effect measure. We planned to use the standardized mean difference (SMD) to combine trials that measured the same outcome with different methods.
For time‐to‐event data we planned to use the hazard ratio (HR) with the corresponding 95% interval (CI).
Unit of analysis issues
The unit of analysis for this review was planned to be the individual randomized participant. If multi‐arm trials were identified, to enable individual pairwise comparisons, we planned to either select relevant arms for inclusion in our analyses, or if more than two arms were relevant to the review, we planned to combine intervention arms to allow a single comparison if appropriate (e.g. study arms with different doses of the same therapeutic). If it would not be reasonable to combine intervention groups, we planned to split the 'shared' comparator group to avoid double‐counting of participants.
We did not anticipate that any cluster‐randomized studies would meet the inclusion criteria for this review. However, if any studies that met the inclusion criteria were identified, we planned to undertake analyses at the individual level while accounting for the clustering in the data.
Dealing with missing data
We planned to contact study authors to obtain missing study characteristics, missing outcomes, missing summary data, and missing individual data.
We planned to assess the risk of reporting bias due to missing studies and missing outcomes as described in the Assessment of reporting biases section.
If we were unable to obtain missing summary data, we planned to calculate or estimate the required data from other reported statistics using the formulae specified in the Cochrane Handbook for Systematic Reviews of Interventions.
If we were unable to obtain missing individual data, we planned to assess the risk of bias using the RoB 2 tool (Higgins 2022; Sterne 2019). In the first instance, we planned to conduct a complete‐case analysis, and we could have performed analyses to investigate the impact of missing data. For example, we could have varied the event rate within missing individuals from intervention and control groups within plausible limits as suggested by clinical experts, or we could have excluded studies thought to be at risk of bias due to missing data (rated as some concerns or high risk of bias in the 'Bias due to missing outcome data' for RoB2) from our meta‐analyses.
Assessment of heterogeneity
We planned to present results of the included studies in forest plots, which we would have inspected visually to assess heterogeneity (that is, non‐overlapping CIs generally signify statistical heterogeneity). We also planned to use the Chi² test with a P value of less than 0.1 to indicate statistical heterogeneity. Heterogeneity would have been quantified using the I² statistic, which describes the percentage of the variability in effect estimates that is due to heterogeneity rather than sampling error. We would have interpreted this statistic using the following guidance from the Cochrane Handbook:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity*;
50% to 90%: may represent substantial heterogeneity*;
75% to 100%: considerable heterogeneity*.
*The importance of the observed value of I2 depends on (1) magnitude and direction of effects, and (2) strength of evidence for heterogeneity (e.g. P value from the Chi2 test, or a confidence interval for I2: uncertainty in the value of I2 is substantial when the number of studies is small).
Assessment of reporting biases
We searched for ongoing trials that met our eligibility criteria and classified them as ‘ongoing’ until their publication.
If we had included 10 studies in a meta‐analysis, we planned to explore the possibility of small study biases (a tendency for estimates of the intervention effect to be more beneficial in smaller studies) for the primary outcomes using funnel plots. In the case of asymmetry, we planned to consider various explanations such as publication bias, poor study design and the effect of study size.
Data synthesis
We planned to perform all meta‐analyses using random‐effects models. Where performing a meta‐analysis would be considered to be inappropriate due to important clinical or methodological heterogeneity, or if study results differed to the extent that combining them in a pooled analysis would not make sense, we planned to summarize data using other methods outlined in the Cochrane Handbook (McKenzie 2022).
Subgroup analysis and investigation of heterogeneity
We planned to conduct subgroup analysis to explore the following sources of heterogeneity as determined by the WHO Clinical Management and Infection Prevention and Control GDG.
Disease severity – severe disease versus non‐severe disease. We planned to use the Centre for Disease Control guidance for a severe case: when a patient has conditions such as hemorrhagic disease; a large number of lesions such that they are confluent; necrotic lesions; severe lymphadenopathy that can be necrotizing or obstructing (such as in airways); involvement of multiple organ systems and associated comorbidities (for example, pulmonary involvement with nodular lesions; sepsis; encephalitis; myocarditis; ocular or periorbital infections); or other conditions requiring hospitalization (CDC 2022b). The therapeutic effect of an intervention may differ according to disease severity.
Days from symptom onset to therapeutic administration – less than seven days versus seven or more days. The effect of an intervention may differ depending on time to therapeutic administration.
We also intend to explore the following subgroups of absolute effects.
Immunosuppression – immunosuppressed versus non‐immunosuppressed. Immunosuppression for the purposes of this review is defined as a participant having any comorbid condition or taking any medication associated with a reduction of the activation or efficacy of the immune system, as reported by study authors. The therapeutic effect of an intervention may differ according to level of participant immune function.
Age – less than 60 years old versus 60 years old or more. The therapeutic effect of an intervention may differ due to altered physiological changes in older persons compared to younger persons.
Skin disease – pre‐existing skin disease versus no pre‐existing skin disease. Pre‐existing skin disease is defined as any skin condition diagnosed in a participant before their diagnosis with mpox, as reported by study authors. Persons with pre‐existing skin disease may modify the effect of a therapeutic, including for the outcomes of time to healing of skin lesions, pain, and scarring, compared to persons with no pre‐existing skin disease.
Sensitivity analysis
We planned to perform sensitivity analyses to investigate the impact of missing data. For example, we may have assessed varying the event rate within missing patients from intervention and control groups within plausible limits, or we may exclude studies thought to be at high risk of attrition bias from our meta‐analyses.
Summary of findings and assessment of the certainty of the evidence
We planned to present the main results of the review in summary of findings tables including our rating of the certainty of evidence based on the GRADE approach. An example summary of findings table is displayed in Appendix 3. We planned to follow current GRADE guidance as recommended in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2022).
Two review authors (TF, RK) planned to assess the certainty of the evidence, considering the domains of risk of bias, inconsistency, imprecision, indirectness, and publication bias.
We planned to construct one summary of findings table for each therapeutic comparison. The summary of findings tables were planned to include all critical outcomes, outlined below.
Serious adverse events (up to 28 days)
Development of disease‐related complications (up to 28 days or the longest period as reported by study authors)
Admission to hospital for non‐hospitalized participants (up to 28 days)
Pain, as judged by any visual or numerical pain scale (up to 28 days)
Level of virus detected in clinical samples (as an indicator of infectiousness) (up to 28 days)
Time to healing of all skin lesions (up to 28 days, or the longest period as reported by study authors)
Mortality (up to 28 days)
Methods for future updates
We will manually check trials platforms and use our network of contacts to stay informed regarding new trials. The review will be updated once data (results and information to allow assessment of risk of bias) of one or more trial(s) become available to enable a meta‐analysis. We will monitor trial completion and publication closely and follow the guidance in Garner 2016 for review updates.
Non‐randomized studies review
Full methods for the NRS review are detailed in Appendix 1.
Criteria for considering studies for this review
Types of studies
Prospective and retrospective cohort studies, including single‐arm and comparative studies with at least two participants, were eligible.
Participants
All participants, of any age or gender, were eligible.
We included studies from any country or setting, including inpatient and outpatient settings.
Interventions
Any therapeutics for treating mpox were eligible, including but not limited to tecovirimat, brincidofovir, cidofovir, NIOCH‐14, immunomodulators, and vaccine immune globulin.
Outcomes
Critical
Serious adverse events (defined as any serious event that is not reported under any other outcome) (up to 28 days)
Development of complications (proctitis, urethritis, ophthalmic complications)
Mortality (up to 28 days)
Important
Mental health outcomes (up to 28 days)
Adverse maternal outcomes (up to 28 days)
Adverse perinatal outcomes (up to 28 days)
Search methods
Vittoria Lutje (Cochrane Infectious Diseases Group Information Specialist) searched the following databases on 22 September 2022: Cochrane Central Register of Controlled Trials (CENTRAL; Issue 9 of 12, 2022), published in the Cochrane Library; MEDLINE (Ovid); Embase (Ovid); and Scopus (Elsevier). She also searched the WHO International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/en/) and ClinicalTrials.gov (clinicaltrials.gov) for trials in progress. Further details of the search strategy are provided in Annex 1 of Appendix 1. We undertook a call to experts in the field for relevant studies to be considered for inclusion in the review.
Study selection
Two review authors (TF and RK) independently screened all titles and abstracts. Two authors (TF and RK) independently reviewed the full text of studies identified through title and abstract screening; any disagreements were resolved by discussion with all authors.
Data collection and analysis
Full details of the methods used for the data collection and analysis are provided in Appendix 1. We assessed risk of bias for all critical outcomes reported in controlled non‐randomized studies using the ROBINS‐I tool (Sterne 2019). We assigned uncontrolled non‐randomized studies a critical overall risk of bias due to the inherent biases associated with the study design. We extracted data on study characteristics and data relevant to the study population, intervention, comparator, and outcomes. We summarized the data on the frequency of adverse events in each study and described this narratively.
Results
Description of studies
This Results section chiefly concerns results for the main review of RCTs investigating the effectiveness and safety of therapeutics in mpox. The full results from the narrative review of non‐randomized studies concerning the safety of therapeutics is in Appendix 1, and is presented briefly in the Effects of interventions section below.
Results of the search
The literature searches and appeal to experts identified 368 potentially relevant studies (366 from searches and two from communications with our expert group). After removing 190 duplicates, we screened 178 records, from which we considered seven for full‐text screening. Five of these were ongoing RCTs eligible for our review. Two studies were excluded due to having an ineligible study design (Excluded studies).
Overall, we did not identify any completed RCTs that met our inclusion criteria (Figure 1). We identified five ongoing placebo‐controlled RCTs; all ongoing trials on tecovirimat for the treatment of human mpox virus (ISRCTN17461766; NCT05534165; NCT05534984; NCT05559099; NCT05597735).
1.

We have described the five ongoing trials in this main review.
Ongoing studies
The complete details of the identified ongoing RCTs investigating tecovirimat compared to placebo are described in the Characteristics of included studies section. These ongoing trials are summarized below.
NCT05534165
Trial design and location
PLATINUM‐CAN is a multicentre, randomized, placebo‐controlled trial of tecovirimat in non‐hospitalized patients with presumptive or PCR‐confirmed mpox infection, which is being conducted in Canada.
Participants
Participants will be aged over 18 years with laboratory‐confirmed mpox (determined by PCR, culture, or antigen test obtained from a sample collected from blood, oropharynx, anal or skin lesion within four days of randomization) or presumptive mpox (skin lesion(s), mucosal lesion(s) or proctitis consistent with a high probability of mpox infection in the opinion of the site investigator and sexual contact with one or more persons in the 21 days prior to symptom onset, or any person with known close exposure to another person known to be infected with mpox infection). The trial will randomize 120 participants (1:1) into the control and intervention groups.
Interventions
The study plans to administer 600 mg tecovirimat as three 200 mg oral capsules taken twice daily for 14 days to individuals in the intervention group. In the control group, participants will receive a placebo oral capsule to be administered in the same pattern.
Outcomes
The planned primary outcomes for this study are: as follows.
Time to active lesion resolution (up to 28 days)
Feasibility and acceptability of conducting a pragmatic phase 3 interventional trial for outpatients with mpox in Canada (up to four months).
The planned secondary outcomes are as follows.
Time to complete lesion resolution (up to 28 days)
Time to negative throat swab viral culture (up to 28 days)
Time to negative skin or mucosa swab viral culture (up to 28 days)
Secondary feasibility outcomes (up to four months).
Estimated primary completion date
January 2023
NCT05534984
Trial design and location
STOMP is a randomized, placebo‐controlled, double‐blind, multicentre study being conducted in the USA to establish the efficacy of tecovirimat for the treatment of people with laboratory‐confirmed or presumptive mpox disease.
Participants
Participants of all ages are eligible for this study providing they have laboratory confirmed or presumptive mpox infection. 530 participants will be randomized 2:1 to receive either tecovirimat or placebo, respectively. Participants with any of the following conditions or characteristics will be placed in an open‐label tecovirimat category: severe disease, significant skin conditions, severe immune suppression, pregnant or breastfeeding, aged less than 18 years, receiving a potent inducing concomitant medication.
Interventions
The study plans to administer participants in arm 1 with tecovirimat oral capsules twice daily for 14 days, dosed according to weight. Participants in the control group (arm 2) will receive an identical oral placebo capsule twice daily for 14 days. Participants in the open‐label tecovirimat group (arm 3) will receive oral tecovirimat capsules dosed and scheduled according to weight and age (less than seven days old, more than seven days old).
Outcomes
The planned primary outcome for this study is time to clinical resolution, defined as the first day on which all skin lesions are scabbed, desquamated or healed, and visible mucosal lesions are healed (up to 29 days).
The planned secondary outcomes are as follows.
Pain assessed by an 11‐point numerical rating scale for pain (up to 29 days)
Time to development of severe mpox in those without severe mpox at baseline (up to 57 days)
Level of mpox virus in blood (up to 57 days)
Level of mpox virus in skin lesions (up to 57 days)
Level of mpox virus in oropharynx (up to 57 days)
Level of mpox virus in rectum (up to 57 days)
Level of mpox virus in genital secretions (up to 57 days)
Time to complete lesion healing defined as all lesions being re‐epithelialized (up to 29 days)
Participant‐reported adherence (up to 15 days)
Participant‐reported quality of life as measured by EQ‐5D‐5L (up to 29 days)
Occurrence of Grade 3 or greater adverse event (up to 57 days)
All‐cause mortality (up to 57 days)
Tecovirimat concentrations in blood in children less than 18 years of age (up to 15 days)
Estimated primary completion date
April 2023
NCT05559099
Trial design and location
PALM007 is a randomized, placebo‐controlled, double‐blind study being conducted in the Democratic Republic of Congo to establish the efficacy of tecovirimat for the treatment of people with laboratory‐confirmed mpox who have at least one active lesion that is not yet scabbed.
Participants
Participants of all ages are eligible for this study, providing they have laboratory‐confirmed infection. 450 participants will be randomized 1:1 to receive either tecovirimat or placebo.
Interventions
The study plans to administer participants in the intervention arm with 200 mg tecovirimat oral capsules for 14 days, dosed and scheduled according to weight. Participants in the control group will receive an identical oral placebo capsule twice daily for 14 days. All participants will also receive standard care.
Outcomes
The planned primary outcome for this study is time to lesion resolution defined as the number of days to the first day on which all lesions on the total body are scabbed or desquamated or a new layer of epidermis has formed (up to day 28).
The planned secondary outcomes are as follows.
Number of days to the first of two consecutive negative mpox test results (up to day 28)
Mortality (up to 28 days post‐randomization)
Number of days to the first negative mpox PCR result (up to day 28)
Number of days to participant death (up to day 28)
Frequency of solicited clinical symptoms (up to day 59)
Clinical symptoms defined as: nausea, vomiting, abdominal pain, diarrhoea, anorexia, cough, lymphadenopathy, dysphagia, sore throat, muscle aches, fatigue/lack of energy, fever, chills, night sweats, headache, ocular lesions, eye pain, change in vision, buccal ulcers, nasal congestion, cough, joint pain, pain with urination, painful skin lesions, pruritic skin lesions.
Duration of solicited clinical symptoms (up to day 59)
Incidence of serious adverse events requiring drug discontinuation (up to day 14)
Incidence of adverse events requiring drug discontinuation (up to day 14)
Incidence of other adverse events (up to day 28)
Incidence of bacterial infections (up to day 28)
Number of clinically defined bacterial infections (up to day 28)
Estimated primary completion date
August 2024
NCT05597735
Trial design and location
UNITY is a randomized, placebo‐controlled, double‐blinded trial being conducted in Brazil and Switzerland to investigate the safety and efficacy of tecovirimat for the treatment of adults and adolescents with laboratory‐confirmed mpox virus infection.
Participants
Included participants must be 14 years or older, have at least one visible active skin or mucosal lesion, and be reachable by smartphone if they are an outpatient. 150 participants will be randomized 1:1 to receive either tecovirimat or placebo.
Interventions
The trial aims to administer participants in the intervention arm with tecovirimat, available as immediate‐release oral capsules containing tecovirimat monohydrate, equivalent to 200 mg of tecovirimat. It will be dosed according to international recommendations. Participants in the control group will receive an identical placebo.
Outcomes
The planned primary outcome for this study is time to all visible lesion resolution (up to day 28).
The planned secondary outcomes are as follows.
All‐cause mortality within the first 28 days (applies to all participants) (up to day 28)
All‐cause unplanned admission to hospital within first 28 days (applies to outpatients)
Occurrence of patients with a complication within first 28 days (applies to all participants who did not already have a complication at baseline)
Time to resolution of symptoms and signs within first 28 days (applies to all participants)
Viral clearance up to 28 days after randomization
Frequency of adverse events and serious adverse events for specific therapeutics (applies to all participants) (up to day 60)
Estimated primary completion date
January 2024
ISRCTN17461766
Trial design and location
PLATINUM is a placebo‐controlled randomized trial of tecovirimat in non‐hospitalized people with mpox.
Participants
Included participants can be any age, with laboratory confirmed mpox infection and at least one active skin or mucosal lesion. 500 participants will be randomized 1:1 to receive either tecovirimat or placebo.
Interventions
Participants in the intervention arm will be administered tecovirimat as an oral capsule, dosed at 1200 mg per day for participants over 18 years, and according to body weight for children and adolescents.
Outcomes
The planned primary outcome for this study is time to active lesion resolution.
The planned secondary outcomes are as follows.
Time to complete lesion resolution (up to day 28)
Time to negative throat swab viral culture (at days 7, 14, 21, and 28)
Time to negative skin or mucosa swab viral culture (at days 7, 14, 21, and 28)
Estimated primary completion date
June 2023
Excluded studies
We excluded two studies at full‐text screening level. Neither were RCTs so were ineligible for the main review. The reasons for exclusion are outlined in Excluded studies. One study was a non‐randomized trial of tecovirimat (jRCTs031220169). The other study was a non‐randomized expanded access protocol (ISRCTN43307947), which was not eligible for this section of the review, but we included in the review of non‐randomized studies in Appendix 1 (Mbrenga 2022).
Risk of bias in included studies
We did not identify any completed studies. When studies publish data that can be included in our review, we will assess the risk of bias.
Effects of interventions
Randomized controlled trials review
We found no completed RCTs meeting the inclusion criteria for the review.
Non‐randomized studies review
The effects of interventions of non‐randomized studies are summarized briefly below and provided in full detail in Appendix 1.
Tecovirimat
Serious adverse events
One study reported serious adverse events meeting our definition in Appendix 1. They were reported by one out of 14 (7%) participants (Mbrenga 2022). They reported one case of life‐threatening anaemia on day 9 of treatment. This participant was also positive for HIV and malaria and received a blood transfusion to treat the anaemia, recovering without sequelae on day 13. The medical monitor for the study did not consider this adverse event to be related to the treatment with tecovirimat.
Liver function tests
O'Laughlin 2022 also reported asymptomatic elevated liver function tests for one participant out of 340 (0.3%) treated with tecovirimat. The authors did not provide detail on which liver enzyme tests were performed (i.e. which enzymes were tested) and whether these were performed routinely or due to other clinical indicators of drug‐induced liver injury (DILI); therefore, we cannot determine the severity of this result.
One participant in Adler 2022 had a haematological, renal, and liver profile that remained within normal limits during the first week of therapy, and they reported no adverse effects.
Brincidofovir
Liver function tests
Adler 2022 reported changes in alanine transaminase (ALT) from baseline in participants throughout the treatment period. In 100% of participants (three of three participants), abnormal increases in ALT were reported. The level peaked at > 500 U/L (day 21), > 300 U/L (day 8) and > 100 U/L (day 14) for the three participants, and treatment was discontinued as a result. For participant 1, ALT peaked 7 days after the first dose, resulting in discontinuation of treatment. Participants 2 and 3 received two doses before elevation of ALT, which peaked on day 21 and day 14, respectively. This is represented graphically in Figure 2 (see details in Appendix 1).
2.
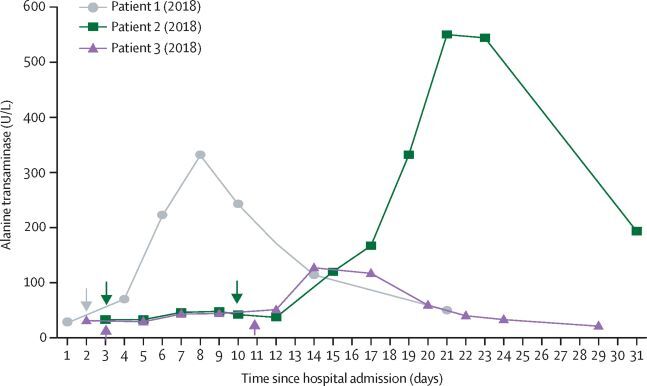
Alanine transaminase values of the three participants who received therapy with brincidofovir (reproduced with permission from Adler 2022)
These cases may meet the criteria of at least five‐fold elevation above the upper limit of normal (ULN) for ALT. In the UK, the normal range for ALT is considered to be 0 to 50 U/L (NHS 2022). Based on this range and the criteria for drug‐induced liver injury (Table 2 in Appendix 1), at least two of three reported changes in ALT in this study may suggest mild drug‐induced liver injury. It is unknown whether these participants would have developed moderate or severe DILI due to discontinuation of brincidofovir. Authors stated that no other significant biochemical or haematological disturbances were observed, and they did not state if further investigations (such as liver ultrasound) were undertaken. All participants made a full recovery.
Discussion
Summary of main results
Randomized controlled trials review
We did not identify any published RCTs investigating the safety and efficacy of therapeutics for mpox. It is encouraging to have identified five ongoing trials that are in progress in the USA, Canada, Brazil, Switzerland, UK, and Democratic Republic of Congo to investigate tecovirimat for the treatment of mpox. We will update this review as evidence becomes available.
Non‐randomized studies review
Included studies investigated the safety of tecovirimat and brincidofovir. No other therapeutics were investigated.
Tecovirimat
Tecovirimat appears to be safe and well‐tolerated based on very low‐certainty evidence from two studies (Mbrenga 2022; O'Laughlin 2022). The frequency of adverse events reported was low: 11/340 reported mild adverse events; 2/341 reported mental health outcomes; 1/341 reported increased liver enzymes; 1/14 reported death, which was not thought to be related to the study drug; and 1/14 reported a serious adverse event (anaemia) that was not thought to be related to the study drug (See Results Table 1 in Appendix 1).
Brincidofovir
Brincidofovir was investigated in three participants, all of whom reported increases in ALT, which indicated mild drug‐induced liver injury and resulted in discontinuation of the study drug (See Results Table 2 in Appendix 1). We judged that two of these cases (whose ALT peaked at > 500U/L and > 300U/L) met the criteria for mild drug‐induced liver injury (at least five‐fold elevation above the upper limit of normal for ALT, described in full detail in the NRS review in Appendix 1)(Aithal 2011). There are no data to inform whether continuation of treatment with brincidofovir would have resulted in a higher grade of drug‐induced liver injury; however, the guidance for the use of brincidofovir evidences severe hepatobiliary adverse events in 1% of 392 people receiving treatment, demonstrating the potential for more serious hepatic outcomes (Chimerix 2022). Based on the very low‐certainty evidence for adverse effects, caution and close clinical monitoring in the event of the use of brincidofovir for the treatment of mpox is recommended.
Overall completeness and applicability of evidence
Randomized controlled trials review
No completed RCTs were included in this review. Five ongoing trials were identified that intend to include populations with, or at greater risk of, severe disease, including paediatric populations and people with immunosuppression and skin diseases. This will allow the safety and efficacy of tecovirimat to be investigated in the general population, as well as in more vulnerable populations.
Non‐randomized studies review
Three non‐randomized studies contributed to the outcomes of safety in this review (Appendix 1). Two studies included very small populations. The safety of brincidofovir was only investigated in three participants, and tecovirimat was investigated in 355 participants. We were unable to disaggregate results based on age, days from symptom onset to initiation of treatment, pre‐existing skin disease, immunosuppression, or disease severity to determine if safety profiles differed between subgroups. We also found no studies comparing one therapeutic to another, so we are unable to compare safety between different therapeutic options. We did not find any evidence on other potential therapeutic options as outlined in How the intervention might work.
Certainty of the evidence
Randomized controlled trials review
Since no completed RCTs were identified, we could not assess the quality of the evidence. In the update of this review, we will assess the certainty of the evidence from RCTs and cluster‐RCTs using the GRADE approach.
Non‐randomized studies review
We assigned uncontrolled non‐randomized studies a critical overall risk of bias due to the inherent biases associated with the study design. One study did not clearly report methods for measuring or reporting safety‐related outcomes in their study (Adler 2022).
Potential biases in the review process
We attempted to minimize bias at all stages in the review process and performed study screening, data extraction, and risk of bias assessments in duplicate. We only included studies available in the English language, therefore we may have missed studies published in other languages.
Agreements and disagreements with other studies or reviews
The review of non‐randomized studies in Appendix 1 demonstrates that, in a small population of participants with mpox, tecovirimat is associated with a low frequency of adverse events, which is in agreement with studies conducted in healthy and immunocompromised populations (Yu 2022).
For brincidofovir, the data in Appendix 1 demonstrate adverse effects in three out of three (100%) participants with mpox. In comparison, the company producing brincidofovir reported a frequency of ALT elevation > 3 x ULN in 27 of 392 (7%) participants; however, these individuals did not have mpox (Chimerix 2022).
We agree with the conclusions of a recent scoping review on antiviral therapies for mpox, that further studies are needed to elucidate the full safety profile of therapeutics for treating mpox (Kuroda 2023).
Authors' conclusions
Implications for practice.
Randomized controlled trials review
This review found no evidence from randomized trials from which to draw certain conclusions concerning the efficacy of therapeutics in humans with mpox.
Non‐randomized studies review
Very low‐certainty evidence from non‐randomized studies indicates no serious safety signals emerging for the use of tecovirimat in people with mpox infection (Appendix 1). Our data suggest that there is a risk of elevated liver enzymes and subsequent drug‐induced liver injury associated with the use of brincidofovir; however, the extent of this risk is unclear due to the small population size in which this has been investigated. There is a need for further studies investigating the safety and efficacy of therapeutics for mpox, which should aim to carefully monitor the effect of the drug on hepatic outcomes to further elucidate this risk. We were unable to compare the safety and efficacy of one therapeutic option for treating mpox with another, so no recommendations can be made for one therapeutic over another. We also do not know the effectiveness and safety profile of different therapeutics in different subpopulations.
Future updates
This review will contribute to the development of World Health Organization guidelines on the treatment of mpox, and will be updated once evidence from the ongoing trials becomes available. This will provide evidence on patient‐centred outcomes of treatment and should assist policymakers, health professionals and consumers in making appropriate decisions for treatment of mpox.
Implications for research.
The 2022 mpox outbreak has been characterized by human‐to‐human transmission outside endemic areas. The significant number of cases that have occurred in the current outbreak presents a unique opportunity to investigate the effectiveness of therapeutics for mpox that have not yet been studied in human efficacy trials. Well‐designed trials currently in progress have been identified and will allow further evaluation of this evidence base. We will update the review once the data (results and information to allow assessment of risk of bias) of one or more than one trial become available to enable us to perform a meta‐analysis.
What's new
| Date | Event | Description |
|---|---|---|
| 30 March 2023 | Amended | The published review had RoB 2 tables showing although it is an empty review, with no included studies. RoB 2 integration has now been disabled, and set to RoB 1, to address this issue. |
History
Review first published: Issue 3, 2023
Acknowledgements
The Cochrane Infectious Diseases Group (CIDG) editorial base is funded by UK aid from the UK government for the benefit of low‐ and middle‐income countries (project number 300342‐104). The views expressed herein do not necessarily reflect the UK government’s official policies.
Tilly Fox, Vittoria Lutje, and Rebecca Kuehn are supported by, and Susan Gould is partly supported by, the Research, Evidence and Development Initiative (READ‐It). READ‐It (project number 300342‐104) is funded by UK aid from the UK government; however, the views expressed herein do not necessarily reflect the UK government’s official policies.
Editorial and peer‐reviewer contributions
The following people conducted the editorial process for this article.
Sign‐off Editor (final editorial decision): Dr Paul Hine, CIDG
Managing Editor (selected peer reviewers, collated peer‐reviewer comments, provided editorial guidance to authors, edited the article): Deirdre Walshe, CIDG
Copy Editor (copy editing and production): Andrea Takeda, Cochrane Central Production Service
-
Peer‐reviewers (provided comments and recommended an editorial decision):
Stephen Spencer, Liverpool School of Tropical Medicine (clinical/content review); one additional peer reviewer provided clinical/content peer review, but chose not to be publicly acknowledged
Rachel Richardson, Cochrane Evidence Production and Methods Directorate (methods review)
Jo Platt, Information Specialist, Cochrane GNOC (search review)
Tilly Fox, Vittoria Lutje, and Rebecca Kuehn are members of the CIDG editorial team, but were not involved in the editorial process of this review.
We acknowledge the WHO Clinical Management and Infection Prevention and Control Guideline Development Group (GDG) 2022 for their role in the development of the research (Population, Intervention, Comparator and Outcomes ‐ PICO) question.
Appendices
Appendix 1. Review of non‐randomized studies
Results tables
Results table 1: Safety of tecovirimat for treating mpox infection in children and adults
|
Patient or population: children and adults infected with mpox Setting: UK, USA, Central African Republic Intervention: tecovirimata | |||
| Outcomes |
Number of events with tecovirimat n (%) |
No. of participants (studies) | Comment |
| Serious adverse events | 2 (0.6%) | 355 (3) | Two out of 355 participants treated with tecovirimat across three studies reported serious adverse events, including one case of elevated liver enzyme levels and one case of severe anaemia. |
| Other adverse events | 11 (3.2%) | 340 (1) | Eleven out of 340 participants treated with tecovirimat across one study reported other adverse events. |
| Mortality | 1 (7%) | 14 (1) | One participant out of 14 treated with tecovirimat across one study died. |
| Mental health outcomes | 2 (0.6%) | 341 (2) | Two participants out of 341 treated with tecovirimat across two studies reported mental health outcomes, including low mood and hospitalization for psychiatric reasons. |
| Footnotes aThe included studies do not include control populations; therefore, there are no comparison statistics included in the results table and no GRADE assessment has been performed. We judged these studies to be at critical risk of bias due to an observational, non‐comparative study design. | |||
Results table 2: Safety of brincidofovir for treating mpox infection in adults
|
Patient or population: adults infected with mpox Setting: UK Intervention: bricidofoviraa | |||
| Outcomes |
Number of events with brincidofovir n (%) |
No. of participants (studies) | Comment |
| Serious adverse events | 3 (100%) | 3 (1) | Three out of three participants treated with brincidofovir across one study reported elevated alanine transaminase levels. |
| Mental health outcomes | 2 (67%) | 3 (1) | Two out of three participants treated with brincidofovir across one study reported mental health outcomes, including low mood and emotional lability. |
| Footnotes aThe included studies do not include control populations; therefore, there are no comparison statistics included in the results table and no GRADE assessment has been performed. We judged these studies to be at critical risk of bias due to an observational, non‐comparative study design. | |||
Background
We conducted this systematic review to present the safety of therapeutics for mpox from non‐randomized, observational studies. Safety data on therapeutics for treating mpox from RCTs are presented in the main review.
Methods
Criteria for considering studies for this review
Types of studies
Prospective and retrospective cohort studies (including single‐arm and comparative studies) that included at least two participants were eligible.
Participants
All participants, of any age or gender, were eligible. We included any country or setting, including in patient and outpatient settings.
Interventions
We included any therapeutics for treating mpox, including but not limited to tecovirimat, cidofovir, brincidofovir, NIOCH‐14, immunomodulators, and vaccine immune globulin.
Outcomes
Critical
Serious adverse events (defined as any serious event that is not reported under any other outcome) (up to 28 days)
Development of complications (proctitis, urethritis, ophthalmic complications)
Mortality (up to 28 days)
Important
Mental health outcomes (up to 28 days)
Adverse maternal outcomes (up to 28 days)
Adverse perinatal outcomes (up to 28 days)
Search methods
Vittoria Lutje (Cochrane Infectious Diseases Group Information Specialist) searched the following databases on 22 September 2022: Cochrane Central Register of Controlled Trials (CENTRAL; Issue 9 of 12, 2022), published in the Cochrane Library; MEDLINE (Ovid); Embase (Ovid); and Scopus (Elsevier). She also searched the WHO International Clinical Trials Registry Platform (ICTRP; www.who.int/ictrp/en/) and ClinicalTrials.gov (clinicaltrials.gov) for trials in progress. Further details of the search strategy are provided in Annex 1 of this appendix.
We undertook a call to experts in the field for relevant studies to be considered for inclusion in the review.
Study selection
Two review authors (TF and RK) independently screened all titles and abstracts. Two authors (TF and RK) independently reviewed the full text of studies identified through title and abstract screening; any disagreements were resolved by discussion with all authors.
Data extraction
One review author (TF) extracted all data. We aimed to extract the following information.
General information: author, title, publication date, country, date(s) of study, funding details, conflict of interest statement
Study characteristics: study setting (inpatient or outpatient); study design; dates of recruitment; eligibility criteria; length of follow‐up; loss to follow‐up; and adherence to assigned treatment.
Participant characteristics: number of participants (recruited, allocated, and evaluated); source of participants; age; sex; disease severity; vaccination status; concurrent treatments; pregnancy; and comorbidities (e.g. skin disease, immunosuppression).
Interventions: type; route of administration; dosage; timing; frequency; duration of treatment; and duration of follow‐up.
Control: type (placebo/active treatment); route of administration; dosage; timing; frequency; duration of treatment; and duration of follow‐up.
Outcomes: frequency of participants experiencing events in the control and intervention groups.
Risk of bias assessment
Two review authors (TF, RK) planned to independently conduct a risk of bias assessment for all critical outcomes reported in controlled non‐randomized studies using the ROBINS‐I tool. We planned to resolve any disagreements with a third review author (SG). We would have assessed the domains outlined in Sterne 2019: confounding, selection of participants, classification of interventions, deviations from intended interventions, missing data, measurement of outcomes, and selection of reported results. These domains would have been compared against a theoretical target RCT. We identified potential confounders as comorbidities, use of adjunctive treatments, age, or disease complications at baseline.
We assigned uncontrolled non‐randomized studies a critical overall risk of bias due to the inherent biases associated with the study design.
Results of the search
We identified 506 potentially relevant studies through our search strategy. After removing duplicates, we screened 380 records, from which we considered 37 for full‐text screening. We excluded 34 studies after full‐text screening due to ineligible study type (5), ineligible population (8), ineligible intervention (20), or no relevant outcomes (1).
Included studies
Three studies met our inclusion criteria and were included in this report (Adler 2022; Mbrenga 2022; O'Laughlin 2022). These studies are summarized in Table 1 of Appendix 1, and further study details are outlined in the section 'Characteristics of included studies' in Appendix 1.
Table 1: study characteristics
| Author, year | Study design | Participants (n) | Therapeutic administered (n) | Safety outcomes |
| Adler 2022 | Retrospective observational study | 4 receiving therapeutic treatment | Brincidofovir (3), tecovirimat (1) | Serious adverse events, development of complications, mental health outcomes |
| Mbrenga 2022 | Open access programme | 14 | Tecovirimat (14) | Serious adverse events, mortality |
| O'Laughlin 2022 | Retrospective observational study | 340 with safety information | Tecovirimat (340) | Serious adverse events, mental health outcomes |
Trial design and location
Two of the included studies were retrospective observational studies (Adler 2022; O'Laughlin 2022), and one was an open access programme (Mbrenga 2022). One study was conducted in the Central African Republic (Mbrenga 2022), one in the UK (Adler 2022), and one in the USA (O'Laughlin 2022).
Interventions
In two studies, tecovirimat was the only therapeutic administered for the treatment of mpox infection (Mbrenga 2022; O'Laughlin 2022), and in one study both brincidofovir and tecovirimat were independently administered (Adler 2022).
Participants
Most participants across all studies were male (96.6%). Only one study reported the safety of therapeutics for treating mpox infection in children (0.9% of participants, aged 0 to 18 years) (Mbrenga 2022). No female participants in any studies were pregnant.
One participant was confirmed to have HIV (Mbrenga 2022). Other comorbidities were also reported in this study, including 11 participants positive for malaria. The earliest time that any participant began treatment after the onset of symptoms was five days, and the latest was 45 days.
Outcomes
Serious adverse events
One study reported serious adverse events in partipants treated with tecovirimat under an expanded access program (Mbrenga 2022). These outcomes were not predefined.
Liver function tests
Two studies reported elevated liver enzyme levels, which we categorized as a serious adverse event (Adler 2022; O'Laughlin 2022). Adler 2022 reported changes in alanine transaminase (ALT) from baseline throughout the study period. O'Laughlin 2022 did not specify methods for reporting liver enzyme levels. Neither study indicated whether participants were excluded from treatment based on baseline liver enzyme levels.
Increases in liver enzyme levels, including ALT, AST (aspartate transaminase), bilirubin, and albumin, may indicate drug‐induced liver injury (DILI) and should be carefully monitored in patients. Guidance on DILI has been produced by the European Association for the Study of the Liver (EASL) which outline a variety of host‐dependent (such as age, sex) and drug‐dependent risk factors (such as dose and hepatic drug metabolism) for DILI (EASL 2019). The guidance explains that most liver alterations associated with drugs are acute (meaning they occur suddenly) and can be identified by elevations in liver enzymes, as outlined above. Liver biochemical tests can detect these elevations, but some forms of DILI presentation will not have distinct features (EASL 2019). Criteria for DILI, including a DILI severity index (see Table 2), was developed by an Expert Working Group and published in Aithal 2011. The criteria are described in Annex 2 of Appendix 1.
Table 2: Drug‐induced liver injury severity index
| Category | Severity | Description |
| 1 | Mild | Elevated ALT/ALP concentration reaching criteria for drug‐induced liver injury, but bilirubin concentration < 2 × ULN |
| 2 | Moderate | Elevated ALT/ALP concentration reaching criteria for drug‐induced liver injury, and bilirubin concentration ≥ 2 × ULN, or symptomatic hepatitis |
| 3 | Severe | Elevated ALT/ALP concentration reaching criteria for drug‐induced liver injury, bilirubin concentration ≥ 2 × ULN, and 1 of the following.
Other organ failure considered to be due to drug‐induced liver injury |
| 4 | Fatal or transplantation | Death or transplantation due to drug‐induced liver injury |
Abbreviations: ALP: alkaline phosphatase; ALT: alanine aminotransferase; ULN: upper limit of normal
Development of complications
One study reported the development of complications related to the illness and outlined the management needed for these (Adler 2022).
Mortality
One study reported mortality rates (Mbrenga 2022).
Mental health outcomes
Two studies reported mental health outcomes experienced by participants during the study period (Adler 2022; O'Laughlin 2022). Neither study specified whether these were related to the illness or to the therapeutic administered.
Adverse maternal or perinatal outcomes
No studies included maternal or perinatal participants; therefore, these outcomes were not reported.
Excluded studies
We identified one retrospective observational analysis study that we have excluded from this review (Girometti 2022). This study included one participant who was treated with tecovirimat for mpox, but the study did not provide any relevant safety outcomes. We have contacted the study authors to determine if they can provide any outcomes of interest, and we will include this study in future if possible.
Risk of bias assessment
We planned to use the ROBINS‐I risk of bias tool to assess the risk of bias of controlled non‐randomized studies for the two primary outcomes, serious adverse events (Adler 2022; Mbrenga 2022; O'Laughlin 2022) and mortality (Mbrenga 2022). None of the included studies included a control arm, so we assessed these studies to have an overall critical risk of bias due to the inherent biases associated with the study design.
Overall risk of bias
Overall, we judged all studies to be at critical risk of bias due to inherent biases associated with the uncontrolled study design (Table 3).
Table 3: Risk of bias assessment
| Study ID | Overall risk of bias assessment |
| Adler 2022 | Critical risk of bias due to observational, non‐comparative study designs. |
| Mbrenga 2022 | |
| O'Laughlin 2022 |
Effects of interventions
Tecovirimat
See Results table 1.
Serious adverse events
In one study reporting serious adverse events, they were reported by one out of 14 (7%) participants (Mbrenga 2022; Table 4). They reported one case of life‐threatening anaemia on day 9 of treatment. This participant was also positive for HIV and malaria and received a blood transfusion to treat the anaemia, recovering without sequelae on day 13. The medical monitor for the study did not consider this adverse event to be related to the treatment with tecovirimat.
Table 4: Reporting of serious adverse events in participants treated with tecovirimat
| Study | Number of serious adverse events reported n/N (%) | Description |
| Mbrenga 2022 | 1/14 (7%) | Case 1: life‐threatening anaemia at day 9. Also positive for malaria and HIV. |
| Total | 1/14 (7%) |
Liver function tests
O'Laughlin 2022 also reported asymptomatic elevated liver function tests for one participant out of 340 (0.3%) treated with tecovirimat. The authors did not provide detail on which liver enzyme tests were performed (i.e. which enzymes were tested) and whether these were performed routinely or due to other clinical indicators of DILI; therefore, we cannot determine the severity of this result.
One participant in Adler 2022 had a haematological, renal, and liver profile that remained within normal limits during the first week of therapy, and they reported no adverse effects.
Other adverse events
One study reported other adverse events for 11 out of 340 (3.2%) participants (O'Laughlin 2022). The events included headache (three), nausea (two), visual disturbance (two), weakness (two), vomiting (one), depression with suicidality (one), rash (one), hives (one), numbness (one), fatigue (one), and dizziness (one).
Mortality
One death was reported in the study by Mbrenga 2022. The death was reported on day 17 after treatment initiation, but there was no official report on the cause of death. The participant had been discharged on day 14, at which time they were doing well, with all lesions resolved, and tested PCR negative.
Mental health outcomes
Mental health outcomes have been associated with previous outbreaks of mpox and attributed to stigmatization and social isolation (Ahmed 2022). Regrettably, during the 2017 outbreak, one case of suicide was reported in a man admitted to an isolation facility (Ogoina 2022).
Mental health outcomes were reported for two out of 341 participants (Adler 2022; O'Laughlin 2022). One participant experienced low mood, and the other participant was hospitalized for psychiatric reasons. No further detail was provided.
Brincidofovir
See Results table 2.
Serious adverse events
For treatment with brincidofovir, the only serious adverse events reported were those relating to liver function tests.
Liver function tests
Adler 2022 reported changes in alanine transaminase (ALT) from baseline in participants throughout the treatment period. In 75% of participants (three out of four), abnormal increases in ALT were reported. The level peaked at > 500 U/L (day 21), > 300 U/L (day 8) and > 100 U/L (day 14) for the three participants, and treatment was discontinued as a result. Figure 2, reproduced with permission from Adler 2022, shows the changes in ALT from the three participants throughout the treatment period, with arrows noting doses of brincidofovir. For patient 1, ALT peaked seven days after the first dose, resulting in discontinuation of treatment. Patients 2 and 3 received two doses before elevation of ALT.
The criteria for drug‐induced liver injury from Aithal 2011 are presented in Table 1. Two cases had at least five‐fold elevation above the ULN for ALT (> 500U/L and > 300U/L). In the UK, the normal range for ALT is considered to be 0 to 50 U/L (NHS 2022). Based on this range and the criteria for drug‐induced liver injury (Table 1), two of three participants (patients 1 and 2) met the criteria for mild drug‐induced liver injury. There were insufficient data to determine whether patient 3 met the criteria (unable to determine their exact ALT value at baseline from Figure 2).
It is unknown whether these participants would have developed moderate or severe DILI due to discontinuation of brincidofovir. Authors stated that no other significant biochemical or haematological disturbances were observed, and all participants made a full recovery (Adler 2022).
Development of complications
Adler 2022 reported the development of complications throughout the study period. Three participants in this study experienced complications, including ulcerated inguinal lesion with delayed healing, deep tissue abscesses, conjunctivitis, and painful disruption of thumbnail from subungual lesion. The full details of the complications reported are detailed in Table 5.
Table 5: Development of complications
| Study | Case number | Therapeutic | Complications |
| Adler 2022 | 1 | Brincidofovir | Ulcerated inguinal lesion with delayed healing |
| 2 | Brincidofovir | Deep tissue abscesses, severe pain. | |
| 3 | Brincidofovir | Conjunctivitis, painful disruption of thumbnail from subungual lesion | |
| 4 | Tecovirimat | None reported |
Mental health outcomes
Two participants in the study by Adler 2022 reported experiencing low mood, and one also reported emotional lability that required clinical psychology input. The third participant did not report any mental health outcomes.
Subgroup analysis
We had planned to perform subgroup analysis to investigate the effect of disease severity and the time between symptom onset and administration of therapeutic. This was not possible due to no control groups being included in the studies, and the limited reporting of these characteristics.
Discussion
In the three studies identified, the study design did not include a comparator (control) population. The issue with this study design is that we do not know whether the outcome or effect seen relates to the intervention used, or to another factor such as the disease itself. Further, in studies with no comparator, we do not know if the intervention under assessment performs worse or better than another intervention. As a result, we judged these studies to be at critical risk of bias.
Tecovirimat
Overall, a low number of adverse events was reported (16 in 355 participants), the majority of which were not serious (headache, nausea, visual disturbance, weakness, vomiting, rash, hives, numbness, fatigue, and dizziness). Elevated liver enzymes were reported in one participant (1/341), but the authors did not provide any detail on which liver enzymes they measured, so we cannot judge the severity of this outcome. Based on very low‐certainty data from non‐randomized studies, tecovirimat appears to be safe and well‐tolerated.
Brincidofovir
In the three participants treated with brincidofovir, all participants reported abnormal elevations in ALT. In one participant this increase began within two days of the first dose. For the other two participants it began within two to three days of receiving the second dose. For all participants, this resulted in discontinuation of treatment.
Based on criteria outlined in Table 2, we judged two of these cases to have mild drug‐induced liver injury. Guidance for the administration of brincidofovir recommends monitoring liver laboratory parameters due to previous observations of elevated ALT, aspartate aminotransferase (AST), and total bilirubin in people treated with brincidofovir (Chimerix 2022).
Evidence from phase 2/3 clinical trials that included 392 participants treated with brincidofovir demonstrated ALT elevations > 3 x the ULN in 7% of participants, and the occurence of severe hepatobiliary adverse events (such as hyperbilirubinemia, acute hepatitis, hepatic steatosis, and venoocclusive liver disease) in less than 1% of participants (Chimerix 2022). It is difficult to compare the results from our review with these data as the participants did not have mpox, and their characteristics have not been described. It is also unclear what effect would be seen in people with raised liver enzymes at baseline, or those at higher risk of DILI, as the participants in our review were all reported to have normal liver enzyme levels at baseline.
Overall, the evidence base will be considerably enriched by evidence from RCTs, particularly those which include participants with comorbidities that place them at higher risk of drug‐induced liver injury.
Overall conclusion
Due to the critical risk of bias identified and the low the number of studies and participants, we consider the current evidence on the safety of tecovirimat and brincidofivir for mpox to be of very low certainty. The evidence from non‐randomized studies indicates no serious safety signals emerging for the use of tecovirimat in people with mpox infection.
Our data suggest that there is a risk of elevated liver enzymes and subsequent drug‐induced liver injury associated with the use of brincidofovir; however, the extent of this risk is unclear due to the small population size in which this has been investigated.
Characteristics of included studies
Adler 2022
General information
Authors: Adler H, Gould S, Hine P, Snell LB, Wong W, Houlihan CF, et al.
Title: Clinical features and management of human monkeypox: a retrospective observational study in the UK
Country: UK
Date(s) of study: 15 August 2018 to 10 September 2021
Funding details: none
Study characteristics
Study setting: high‐consequence infectious diseases (HCID) centres in the UK
Study design: retrospective observational study
Dates of recruitment: 15 August 2018 to 10 September 2021
Eligibility criteria: people admitted to any HCID centre in the UK with confirmed mpox (defined as a compatible clinical illness with positive mpox viral polymerase chain reaction (PCR) from any anatomical site) between 15 August 2018 and 10 September 2021.
Length of follow‐up: duration of hospitalization (between 10 and 35 days).
Adherence to assigned treatment: full adherence
Participant characteristics
Number of participants: 4
Source of participants: HCID hospital inpatient
Source of infection (setting, mode of transmission)
Cases 1 and 2: acquired in Nigeria
Case 3: healthcare worker exposed to participant 2 18 days prior to rash development
Case 4: mother of a child infected after travel to Nigeria (child suspected to have caught infection from father)
Age: 30 to 40 years
Sex: 50% male
Disease severity and complications
Case 1: lymphadenopathy, maximum 150 concurrent lesions on face, scalp, trunk, limbs, palms, glans penis, and scrotum. Low mood and emotional ability. Ulcerated inguinal lesion with delayed healing. Clinical psychology input required.
Case 2: lymphadenopathy, maximum 100 concurrent lesions on face, trunk, limbs, palms, soles, and scrotum. Deep tissue abscesses, severe pain, and low mood.
Case 3: maximum 32 concurrent lesions on face, trunk, hands (including nail bed), and labia majora. Conjunctivitis, painful disruption of thumbnail from subungual lesion.
Case 4: maximum 10 concurrent lesions on face, trunk, arms, and hands. Low mood.
Vaccination status: 75% unvaccinated (3), 25% Modified Vaccinia Ankara (MVA) six days post‐exposure or 12 days pre‐illness (1)
Concurrent treatments
Case 1: clinical psychology input
Case 2: Empiric broad‐spectrum antibiotics, abscess drainage, and analgesia
Case 3: Antibacterial eye drops
Case 4: None
Pregnancy: none
Comorbidities: negative for HIV, hepatitis B, and hepatitis C status
Interventions
Therapeutic: cases 1 to 3: brincidofovir 200 mg orally, case 4: tecovirimat 600 mg orally
Route of administration: oral capsule
Dosage, timing, and duration of treatment
Case 1: brincidofovir 200 mg one dose once a week for one week
Cases 2 and 3: brincidofovir 200 mg two doses once a week for 2 weeks
Case 4: tecovirimat 600 mg twice daily for 14 days
Duration of follow‐up: duration of hospitalization: Case 1: 26 days, Case 2: 27 days, Case 3: 35 days, Case 4: 10 days.
Day that therapeutic was administered post symptom onset (PSO): Case 1: day 7, Case 2: day 6, Case 3: day 7, Case 4: day 5
Outcomes
Outcomes: changes in alanine transaminase levels; discontinuation of treatment.
Mbrenga 2022
General information
Authors: Mbrenga F, Nakoune E, Malaka C, Bourner J, Dunning J, Vernet G, et al.
Title: Monkeypox treatment with tecovirimat in the Central African Republic under an Expanded Access Programme
Country: Central African Republic
Date(s) of study: December 2021 to February 2022
Funding details: SIGA donated 100 treatments for use under the Expanded Access Programme (EAP). The EAP was financed with core funds of IPB, financial support to IPB from SIGA, the University of Oxford under the UK Foreign, Commonwealth and Development Office and Wellcome (215091/Z/18/Z) and the Bill & Melinda Gates Foundation (OPP1209135).
Study characteristics
Study setting: Lobaye district of CAR, Mbaïki hospital – the district primary and referral health care facility
Study design: expanded access protocol (preprint)
Dates of recruitment: December 2021 to February 2022
Eligibility criteria: eligible participants received laboratory confirmation of mpox infection and weighed ≥ 13 kg. People receiving repaglinide were ineligible due to risk of interactions with tecovirimat.
Length of follow‐up: 28 days
Adherence to assigned treatment: full adherence
Participant characteristics
Number of participants: 14
Source of participants: intensified active surveillance system: suspected cases were identified through referrals from community health workers, other health facilities and community leaders, and through contact tracing in villages of the district by the EAP team.
Source of infection (setting, mode of transmission): not specified but stated that cases typically peak around the ‘caterpillar season’ between July and December when people are more exposed to wildlife. Cases tend to occur in small clusters of < 10 cases within households or communities, indicating human‐to‐human transmission.
Age: Range 4 to 38 years
Sex: 4 males, 10 females
Disease severity and complications: 14 (100%) participants presented with muscle pain, headache, lymphadenopathy, and lesions of any type. Fever (64%), back pain (79%), upper respiratory symptoms (79%), cough (36%), lower respiratory symptoms (29%), clear sputum (21%) and keratitis (14%). By day 4, all (100%) participants reported lymphadenopathy and lesions (any type). At day 8, all (100%) participants continued to present with lymphadenopathy and lesions (any type).
Vaccination status: not stated
Concurrent treatments: not stated
Pregnancy: none
Comorbidities: 11 participants tested positive for malaria on rapid diagnostic test (RDT), and one participant subsequently tested positive for HIV
Interventions
Therapeutic: tecovirimat
Route of administration: oral capsule
Dosage, timing, and duration of treatment: adult participants received 600 mg of oral tecovirimat (3 x 200 mg capsules) twice daily for 14 days. Dosing for paediatric participants was based on weight: children weighing 13 kg to < 25 kg received 200 mg of tecovirimat (1 capsule) twice daily for 14 days; children weighing 25 kg to < 40 kg received 400 mg of tecovirimat (2 capsules) twice daily for 14 days; and children weighing ≥ 40 kg received 600 mg of tecovirimat (three capsules) of tecovirimat twice daily for 14 days.
Duration of follow‐up: 28 days. Due to logistical constraints, as participants lived in remote areas that are difficult to access, the day 28 visit was conducted between days 19 and 55.
Day that therapeutic was administered (pso): median 21 days (range 5 to 45 days)
Outcomes
Serious adverse events; mortality.
O’Laughlin 2022
General information
O’Laughlin K, Tobolowsky FA, Elmor R, Overton R, O’Connor S, Damon IK, et al.
Title: Clinical Use of Tecovirimat (Tpoxx) for Treatment of Monkeypox Under an Investigational New Drug Protocol
Country: USA
Date(s) of study: May to August 2022
Funding details: Centers for Disease Control and Prevention (CDC)
Study characteristics
Study setting: US healthcare settings
Study design: retrospective observational study
Dates of recruitment: retrospective, participants treated May to August 2022
Eligibility criteria: treated with tecovirimat and followed up for one visit during treatment and one post treatment.
Length of follow‐up: to post‐treatment.
Adherence to assigned treatment: yes
Participant characteristics
Number of participants: 495 (results for 340)
Source of participants: US healthcare settings
Source of infection (setting, mode of transmission): not stated
Age: 36.5 years (31.4 to 43.9)
Sex: 97.7% male, 2.3% female
Disease severity and complications: two thirds (232, 64.6%) of tecovirimat recipients had lesions affecting < 10% of their body; 17 (4.7%) had lesions affecting 75% to 100% of their body. 191 (79.6%) lesions in anatomic areas that might result in serious sequelae. 74 (30.8%) at risk for severe disease.
Vaccination status: 88.9% not vaccinated
Concurrent treatments: not stated
Pregnancy: none
Comorbidities: 46.3% HIV positive. 1.3% other immunocompromising condition.
Interventions
Therapeutic: tecovirimat
Route of administration: oral capsule 494 (99.8%); intravenous 1 (0.2%)
Dosage, timing, and duration of treatment: not stated
Duration of follow‐up: not stated
Day that therapeutic was administered (pso): median (interquartile range) 7.0 (5 to 10)
Outcomes
Adverse effects
Annex 1 – Search strategy for Appendix 1
Ovid MEDLINE(R) and In‐Process, In‐Data‐Review & Other Non‐Indexed Citations <1946 to 22 September 2022>
1 Monkeypox virus/ or Monkeypox/ or monkeypox.mp.
2 (treatment or therapeu* or drug*).mp.
3 (tecovirimat or (cidofovir or brincidofovir) or NIOCH* or "vaccinia immune globulin*" or immunomodulat*).mp. [mp=title, book title, abstract, original title, name of substance word, subject heading word, floating sub‐heading word, keyword heading word, organism supplementary concept word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms]
4 2 or 3
5 1 and 4
6 (safe* or complication* or adverse* or side effect* or harm* or risk* or tolerability or teratogen* or toxic* or pharmacotox* or neurotox* or cardiotox* or nephrotox* or immunotox* or hepatox* or cytotox* or immunocytotox*).ti,ab.
7 adverse event*.mp. or "Drug‐Related Side Effects and Adverse Reactions"/
8 Safety/
9 6 or 7 or 8
10 5 and 9
11 limit 5 to adverse effects
12 10 or 11
Embase 1947‐Present, updated daily
1 Monkeypox virus/ or Monkeypox/ or monkeypox.mp.
2 (treatment or therapeu* or drug*).mp.
3 (tecovirimat or (cidofovir or brincidofovir) or NIOCH* or "vaccinia immune globulin*" or immunomodulat*).mp. [mp=title, abstract, heading word, drug trade name, original title, device manufacturer, drug manufacturer, device trade name, keyword heading word, floating subheading word, candidate term word]
4 2 or 3
5 1 and 4
6 (safe* or complication* or adverse* or side effect* or harm* or risk* or tolerability or teratogen* or toxic* or pharmacotox* or neurotox* or cardiotox* or nephrotox* or immunotox* or hepatox* or cytotox* or immunocytotox*).ti,ab.
7 adverse event/
8 drug safety/
9 6 or 7 or 8
10 5 and 9
11 limit 10 to human
12 limit 5 to adverse effects
13 limit 12 to human
14 11 or 13
Scopus
65 document results
( ( TITLE‐ABS‐KEY ( monkeypox ) ) AND ( TITLE‐ABS‐KEY ( tecovirimat OR cidofovir OR brincidofovir OR nioch* OR "vaccinia immune globulin*" OR immunomodulat* OR treatment ) ) AND ( TITLE‐ABS‐KEY ( ( safe* OR complication* OR adverse* OR side AND effect* OR harm* OR risk* OR tolerability OR teratogen* OR toxic* ) ) ) ) OR ( ( TITLE‐ABS‐KEY ( monkeypox ) ) AND ( TITLE‐ABS‐KEY ( tecovirimat OR cidofovir OR brincidofovir OR nioch* OR "vaccinia immune globulin*" OR immunomodulat* OR treatment ) ) AND ( TITLE‐ABS‐KEY ( safety OR "side effect*" OR complications OR "adverse event*" OR "adverse effect*" ) ) )
Annex 2 ‐ Criteria for drug‐induced liver injury
Clinical chemistry criteria for drug‐induced liver injury
Any one of the following.
At least five‐fold elevation above the ULN for ALT.
At least two‐fold elevation above the ULN for ALP (particularly with accompanying elevations in concentrations of 5′‐nucleotidase or γ‐glutamyl transpeptidase in the absence of known bone pathology driving the rise in ALP level).
At least three‐fold elevation in ALT concentration and simultaneous elevation of bilirubin concentration exceeding 2 × ULN.
Appendix 2. Search strategy
Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process, In‐Data‐Review & Other Non‐Indexed Citations, Daily and Versions <1946 to 20 January 2023>
1 Monkeypox virus/ or Monkeypox/
2 monkeypox*.mp.
3 (Mpox or Mpx or Hmpxv or Hmpx).mp.
4 1 or 2 or 3
5 tecovirimat.mp.
6 ST‐246.mp.
7 TPOXX.mp.
8 Cidofovir.mp. or Cidofovir/
9 Vistide.mp.
10 Brincidofovir.mp.
11 Tembexa.tw.
12 NIOCH‐14.mp.
13 Immunomodulator*.mp. or Immunologic Factors/
14 Vaccinia immune globulin.mp.
15 VIGIV.mp.
16 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15
17 4 and 16
18 (treatment or therapeu* or drug*).mp.
19 4 and 18
20 17 or 19
21 randomized controlled trial.pt.
22 controlled clinical trial.pt.
23 randomized.ab.
24 (placebo or randomly or trial or groups).ti,ab.
25 dt.fs.
26 21 or 22 or 23 or 24 or 25
27 Animals/
28 humans/
29 27 and 28
30 27 not 29
31 26 not 30
32 20 and 31
Embase 1947‐Present, updated daily
1 Monkeypox virus/ or Monkeypox/
2 monkeypox*.mp.
3 (Mpox or Mpx or Hmpxv or Hmpx).mp.
4 1 or 2 or 3
5 tecovirimat.mp.
6 ST‐246.mp.
7 TPOXX.mp.
8 Cidofovir.mp. or Cidofovir/
9 Vistide.mp.
10 Brincidofovir.mp.
11 Tembexa.tw.
12 NIOCH‐14.mp.
13 Immunomodulator*.mp. or immunomodulating agent/
14 Vaccinia immune globulin.mp.
15 VIGIV.mp.
16 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15
17 4 and 16
18 (treatment or therapeu* or drug*).mp.
19 4 and 18
20 17 or 19
21 Randomized Controlled Trial/ or randomization/ or controlled clinical trial/ or Double Blind Procedure/ or Single Blind Procedure/ or Crossover Procedure/
22 (clinica* adj3 trial*).tw.
23 ((singl* or doubl* or trebl* or tripl*) adj3 (mask* or blind* or method*)).tw.
24 exp Placebo/
25 (placebo* or random*).ti,ab.
26 ((control* or prospectiv*) adj3 (trial* or method* or stud*)).tw.
27 (crossover* or cross‐over*).ti,ab.
28 21 or 22 or 23 or 24 or 25 or 26 or 27
29 exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/
30 human/ or normal human/ or human cell/
31 29 and 30
32 29 not 31
33 28 not 32
34 20 and 33
BIOSIS Previews and CABI: CAB Abstracts® (Web of Science)
#5 #4 AND #1
#4 #2 OR #3
#3 tecovirimat or ST‐246 or TPOXX or Cidofovir (Abstract) or Vistide or Brincidofovir or Tembexa or NIOCH‐14. (Abstract) or Immunomodulator* or Vaccinia immune globulin or VIGIV (Abstract)
#2 tecovirimat or ST‐246 or TPOXX or Cidofovir (Title) or Vistide or Brincidofovir or Tembexa or NIOCH‐14. (Title) or Immunomodulator* or Vaccinia immune globulin or VIGIV (Title)
#1 monkeypox (Topic) or Mpox or Mpx or Hmpxv or Hmpx (Title) or Mpox or Mpx or Hmpxv or Hmpx (Abstract)
Cochrane Central Register of Controlled Trials
Issue 1 of 12, January 2023
#1 MeSH descriptor: [Monkeypox] explode all trees
#2 MeSH descriptor: [Monkeypox virus] explode all trees
#3 (Mpox or Mpx or Hmpxv or Hmpx):ti,ab,kw
#4 #1 or #2 or #3
#5 tecovirimat or ST‐246 or TPOXX or Cidofovir (Title) or Vistide or Brincidofovir or Tembexa or NIOCH‐14
#6 immunomodulator*
#7 MeSH descriptor: [Immunologic Factors] explode all trees
#8 Vaccinia immune globulin or VIGIV
#9 #5 or #6 or #7 or #8
#10 #4 and #9
ClinicalTrials.gov.
Recruiting, Not yet recruiting, Active, not recruiting, Enrolling by invitation Studies | Interventional Studies | Monkeypox
WHO ICTRP
monkeypox and (tecovirimat or ST‐246 or TPOXX or Cidofovir or Vistide or Brincidofovir or Tembexa or NIOCH‐14)
Appendix 3. Draft summary of findings table
|
Patient or population: people infected with mpox Setting: USA, Canada, Democratic Republic of Congo, Brazil, Switzerland Intervention: tecovirimat Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects (95% CI) | Relative effect (95% CI) | No. of participants (studies) | Certainty of the evidence (GRADE) | Comment | |
| Risk with placebo | Risk with tecovirimat | |||||
| Serious adverse events | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Development of complications (proctitis, urethritis, ophthalmic complications) | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Admission to hospital | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Pain | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Risk of onward transmission | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Time to healing of all skin lesions “re‐epithelialization” | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
| Mortality | ‐ | ‐ | ‐ | ‐ | ‐ | ‐ |
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ISRCTN43307947 | Ineligible study design |
| jRCTs031220169 | Ineligible study design: this is an ongoing non‐randomized trial |
Characteristics of ongoing studies [ordered by study ID]
ISRCTN17461766.
| Study name | PLATINUM |
| Methods | Study setting: community ‐ non‐hospitalized patients who are well enough to remain at home Study design: interventional double‐blind randomized parallel‐group placebo‐controlled trial Dates of recruitment: 18 August 2022 Eligibility criteria: people with laboratory‐confirmed mpox who are well enough to be at home Inclusion criteria
Exclusion criteria
Length of follow‐up: 28 days Loss to follow‐up: not applicable, ongoing Adherence to assigned treatment: not applicable, ongoing |
| Participants | Number of participants: 500 Source of participants: enroled by doctors Age: any Sex: any Disease severity: not stated but non‐hospitalized patients so unlikely to be severe disease. Inclusion criteria states 'well enough to be at home.' Vaccination status: not stated Concurrent treatments: not stated Pregnancy: none Comorbidities: not stated |
| Interventions |
Intervention Therapeutic: tecovirimat Route of administration: oral capsule Dosage, timing, frequency and duration
Control Type: identical placebo Route of administration: oral capsule Dosage, timing, frequency and duration: Identical to intervention |
| Outcomes |
Primary Time to active lesion resolution, defined as the first day on which all skin lesions are scabbed or desquamated (and mucosal lesions healed) up to 28 days after randomization Secondary
|
| Starting date | 18 August 2022 |
| Contact information | platinumtrial@ndph.ox.ac.uk |
| Notes | Author: Prof. Sir Peter Horby, platinumtrial@ndph.ox.ac.uk Title: Antiviral treatment with tecovirimat for patients managed at home with monkeypox (PLATINUM) Publication date: ongoing, estimated completion date June 2023 Country: UK Funding details: National Institute for Health and Care Research (UK) CoI statement: not stated |
NCT05534165.
| Study name | PLATINUM‐CAN |
| Methods | Study setting: community ‐ non‐hospitalized patients (including hospital emergency rooms, outpatient HIV and infectious diseases clinics, community sexual health clinics, primary care, or through public health services) Study design: multicentre, randomized (1:1), placebo‐controlled trial. Quadruple masking: participant, care provider, investigator, outcomes assessor Dates of recruitment: not yet recruiting (as of 29 September 2022) Eligibility criteria Inclusion criteria
Exclusion criteria
Length of follow‐up: 28 days Loss to follow‐up: not applicable, ongoing Adherence to assigned treatment: not applicable, ongoing |
| Participants | Number of participants: 120 Source of participants: not stated Age: 18 years and older Sex: any Disease severity: not stated but non‐hospitalized patients so unlikely to be severe disease. Inclusion criteria state "able to be managed without hospitalisation". Vaccination status: not stated Concurrent treatments: not stated Pregnancy: not stated Comorbidities: not stated |
| Interventions |
Intervention Therapeutic: tecovirimat Route of administration: oral capsule Dosage: 600 mg (administered as three 200 mg capsules) Timing: every 12 hours, 30 min after a meal Frequency: twice daily Duration of treatment: 14 days Duration of follow‐up: 28 days Control Type: identical placebo Route of administration: oral capsule Dosage: 600 mg (administered as three 200 mg capsules) Timing: every 12 hours, 30 min after a meal Frequency: twice daily Duration of treatment: 14 days Duration of follow‐up: 28 days |
| Outcomes |
Primary
Secondary
Other outcomes
|
| Starting date | Registered 9 September 2022 |
| Contact information | |
| Notes | Authors: Marina Klein, McGill University Title: Tecovirimat in Non‐hospitalized Patients with Monkeypox (PLATINUM‐CAN). Official titile: Placebo‐controlled Randomized Trial of Tecovirimat in Non‐hospitalized Patients with Monkeypox: Canadian Feasibility Study (PLATINUM‐CAN) Publication date: Ongoing, estimated completion date January 2023 Country: Canada Funding details: sponsors and collaborators: McGill University Health Centre/Research Institute of the McGill University Health Centre, University Health Network Toronto, Unity Health Toronto, University of British Columbia, CIHR Canadian HIV Trials Network CoI statement: not stated |
NCT05534984.
| Study name | STOMP |
| Methods | Double‐blinded, placebo‐controlled, randomized trial with 3 arms Arm A: tecovirimat oral capsule Arm B: placebo Arm C: tecovirimat open‐label. Randomized 2:1 Dates of recruitment: recruitment started 12 September 2022 Inclusion criteria (all participants; Arms A, B, and C)
Additional Inclusion Criteria for Arms A and B
Additional Inclusion Criteria for Arm C Participants who meet the above entry criteria who also meet any of the following criteria will be registered to Arm C.
Those with or without severe disease and with one or more of the following will also be enrolled into Arm C
Exclusion criteria (all participants; Arms A, B, and C)
Length of follow‐up: 57 days. Loss to follow‐up: not applicable, ongoing trial. Adherence to assigned treatment: not applicable, ongoing trial. |
| Participants | Number of participants: 530 Source of participants: hospitals and health centres at 64 sites in the USA Age: any Sex: any Disease severity: any Vaccination status: not stated Concurrent treatments: not stated Pregnancy: participants who are pregnant or breastfeeding will receive open‐label tecovirimat after discussion of the potential risks and benefits. Comorbidities: participants with significant skin conditions or severe immune suppression will receive open label tecovirimat. |
| Interventions |
Intervention Therapeutic: tecovirimat Route of administration: oral capsule Dosage Arm A
Arm C
Timing, frequency and duration of treatment Arm A
Arm C
Duration of follow‐up: 57 days. As per the different outcomes in length of follow‐up box. Control Type: placebo Route of administration: oral capsule Dosage, timing and frequency: participants weighing 25 kg to < 40 kg: placebo 400 mg every 12 hours for 14 days Participants weighing 40 kg to < 120 kg: placebo 600 mg every 12 hours for 14 days Participants weighing 120 kg and over: placebo 600 mg every 8 hours for 14 days Duration of treatment: 14 days Duration of follow‐up: 57 days. As per the different outcomes in length of follow‐up box. |
| Outcomes | Primary
Secondary
|
| Starting date | Registered 10 September 2022 |
| Contact information | |
| Notes | Authors: Timothy Wilkin, Cornell Official title: A Randomized, Placebo‐Controlled, Double‐Blinded Trial of the Safety and Efficacy of Tecovirimat for the Treatment of Human Monkeypox Virus Disease Publication date: ongoing, estimated completion date: 30 September 2023 Country: USA, 64 locations, including Puerto Rico. Funding details: sponsor: National Institute of Allergy and Infectious Diseases (NIAID) Collaborator: SIGA technologies CoI statement: not stated |
NCT05559099.
| Study name | PALM007 |
| Methods | Study setting: Not stated Study design: randomized (1:1), placebo‐controlled trial Dates of recruitment: recruiting (29 September 2022) Eligibility criteria Inclusion criteria
Exclusion criteria
Length of follow‐up: 28 days, long term assessment at 59 days Loss to follow‐up: not applicable, ongoing Adherence to assigned treatment: not applicable, ongoing |
| Participants | Number of participants: 450 Source of participants: not stated Age: any Sex: any Disease severity: not stated but must have at least one active, not yet scabbed, lesion Vaccination status: not stated Concurrent treatments: standard of care Pregnancy: not eligible to participate Comorbidities: not stated |
| Interventions |
Intervention Therapeutic: tecovirimat Route of administration: oral capsule Dosage: 200 mg capsules Timing and frequency: number of capsules and frequency of dosage will be based on participant weight:
Duration of treatment: 14 days Duration of follow‐up: 28 days Control Type: identical placebo Route of administration: oral capsule Dosage: 200 mg Timing: every 12 hours Frequency: twice daily Duration of treatment: 14 days Duration of follow‐up: 28 days |
| Outcomes | Primary Time to lesion resolution (up to day 28) defined as the number of days to the first day on which all lesions on the total body are scabbed or desquamated or a new layer of epidermis has formed. Secondary
|
| Starting date | Registered 29 September 2022 |
| Contact information | |
| Notes | Authors: Principal investigator Jean‐Jacques Muyembe‐Tamfum, Kinshasa University Title: A Randomized, Placebo‐controlled, Double‐blinded Trial of the Safety and Efficacy of Tecovirimat for the Treatment of Adult and Pediatric Patients With Monkeypox Virus Disease Publication date: ongoing, estimated completion date August 2024 Country: Democratic Republic of Congo Funding details: Sponsors: National Institute of Allergy and Infectious Diseases (NIAID), Institut National de Recherche Biomédicale, Kinshasa, République Démocratique du Congo CoI statement: not stated |
NCT05597735.
| Study name | UNITY |
| Methods | Study setting: mixed, most participants will be managed at home or in the community. Study design: randomized, placebo‐controlled trial Dates of recruitment: not yet recruiting Inclusion criteria
Exclusion criteria
Length of follow‐up: 60 days Loss to follow‐up: not stated, ongoing Adherence to assigned treatment: not stated, ongoing |
| Participants | Number of participants: 150 Sex: male and female Disease severity: any severity, must have at least one active lesion Vaccination status: not stated, ongoing Concurrent treatments: not stated, ongoing Pregnancy: not stated, ongoing Comorbidities: not stated, ongoing |
| Interventions |
Intervention Therapeutic: tecovirimat Route of administration: immediate‐release oral capsules containing tecovirimat monohydrate Dosage, frequency and timing: 200 mg, dosed on weight:
Duration of treatment: 14 days Duration of follow‐up: 28 days follow‐up and long‐term assessment at 60 days Control Route of administration: oral capsule Dosage, frequency and timing: to match intervention group Duration of treatment: 14 days Duration of follow‐up: 28 days follow‐up and long‐term assessment at 60 days |
| Outcomes | Primary
For skin lesions, typically this means the lesion has scabbed, desquamated and new layer of skin has been formed. For mucosal lesions, the phase of scabbing and desquamation is absent, and healing with new layer of skin ensues. Secondary
|
| Starting date | Registered 1 November 2022 |
| Contact information | |
| Notes | Authors: Study director Yazdan Yazdapanah, Pr. ANRS, Emerging Infectious Diseases Alexandra Calmy alexandra.calmy@hcuge.ch Olivier Segeral olivier.segeral@hcuge.ch Title: Assessment of the Efficacy and Safety of Tecovirimat in Patients with Monkeypox Virus Disease (UNITY) Country: Brazil, Switzerland Date(s) of study: estimated primary completion January 2024 Estimated actual completion: January 2025 Funding details: Calmy Alexandra, Oswaldo Cruz Foundation, ANRS, Emerging Infectious Diseases CoI statement: not stated |
CoI: conflict of interest; PCR: polymerase chain reaction
Differences between protocol and review
The methods used in the review are consistent with the planned methods published in the protocol (Fox 2022). We have provided some further detail for our outcomes, including specifying the time point of interest (up to 28 days) and provided an example scale for the scarring outcome (Vancouver Scar Scale (VSS)).
Due to the lack of completed RCTs identified, we were unable to perform the risk of bias assessment, planned analyses, or GRADE assessment; hence we have no summary of findings table.
Vittoria Lutje (VL) joined the author team at review stage.
Contributions of authors
Tilly Fox (TF), Rebecca Kuehn (RK), and Naveena Princy (NP) selected studies. TF and RK extracted study data and characteristics, and assessed the risk of bias. Susan Gould (SG) and Tim Rowland (TR) contributed to the development of the review. Vittoria Lutje (VL) developed the search strategy and performed the literature search. All authors contributed to review design and approved the final version.
Sources of support
Internal sources
Liverpool School of Tropical Medicine, UK
External sources
-
Foreign, Commonwealth, and Development Office (FCDO), UK
Project number 300342‐104
Declarations of interest
TF is a Cochrane Infectious Diseases Group (CIDG) Research Associate, and was not involved in the editorial process. She has no known conflicts of interest to declare.
SG is a co‐author on one included study in this review. SG was not involved in any inclusion decisions, data extraction, or risk of bias assessment for this study.
NP has no known conflicts of interest to declare.
TR has no known conflicts of interest to declare.
VL is the CIDG Information Specialist, and was not involved in the editorial process. She has no known conflicts of interest to declare.
RK is a CIDG Research Associate, and was not involved in the editorial process. She has no known conflicts of interest to declare.
Edited (no change to conclusions)
References
References to studies excluded from this review
ISRCTN43307947 {published data only}
- ISRCTN43307947. Expanded access protocol for the use of tecovirimat for the treatment of monkeypox infection. www.isrctn.com/ISRCTN43307947 (first received 4 June 2021).
jRCTs031220169 {published data only}
- jRCTs031220169. A multicenter, open-label, double-arm trial to evaluate the efficacy and safety of oral tecovirimat therapy for patients with smallpox or monkeypox. rctportal.niph.go.jp/en/detail?trial_id=jRCTs031220169 (first received 28 June 2022).
References to ongoing studies
ISRCTN17461766 {published data only}
- ISRCTN17461766. Antiviral treatment with tecovirimat for patients managed at home with monkeypox (PLATINUM) [Placebo-controlled randomised trial of tecovirimat in non-hospitalised monkeypox patients (PLATINUM)]. isrctn.com/ISRCTN17461766 (first received 17 August 2022).
NCT05534165 {published data only}
- NCT05534165. Tecovirimat in non-hospitalized patients with monkeypox (PLATINUM-CAN) [Placebo-controlled randomized trial of tecovirimat in non-hospitalized patients with monkeypox: Canadian feasibility study (PLATINUM-CAN)]. clinicaltrials.gov/ct2/show/NCT05534165 (first received 9 September 2022).
NCT05534984 {published data only}
- NCT05534984. Study of tecovirimat for human monkeypox virus (STOMP) [A randomized, placebo-controlled, double-blinded trial of the safety and efficacy of tecovirimat for the treatment of human monkeypox virus disease]. clinicaltrials.gov/ct2/show/NCT05534984 (first received 10 September 2022).
NCT05559099 {published data only}
- NCT05559099. Tecovirimat for treatment of monkeypox virus (PALM 007) [A randomized, placebo-controlled, double-blinded trial of the safety and efficacy of tecovirimat for the treatment of adult and pediatric patients with monkeypox virus disease]. clinicaltrials.gov/ct2/show/NCT05559099 (first received 29 September 2022).
NCT05597735 {published data only}
- NCT05597735. Assessment of the efficacy and safety of tecovirimat in patients with monkeypox virus disease (UNITY) [A Phase III, multi-country, randomized, placebo-controlled, double-blinded trial to assess the efficacy and safety of tecovirimat antiviral treatment for patients with monkeypox virus disease]. clinicaltrials.gov/ct2/show/NCT05597735 (first received 28 October 2022).
Additional references
Adler 2022
- Adler H, Gould S, Hine P, Snell L, Wong W, Houlihan C, et al. Clinical features and management of human monkeypox: a retrospective observational study in the UK. Lancet Infectious Diseases 2022;22(8):1153-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ahmed 2022
- Ahmed SK, Abdulqadir SO, Hussein SH, Omar RM, Ahmed NA, Essa RA, et al. The impact of monkeypox outbreak on mental health and counteracting strategies: a call to action. International Journal of Surgery 2022;106:106943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Aithal 2011
- Aithal GP, Watkins PB, Andrade RJ, Larrey D, Molokhia M, Takikawa H, et al. Case definition and phenotype standardization in drug-induced liver injury. Clinical Pharmacology & Therapeutics 2011;89(6):806-15. [DOI] [PubMed] [Google Scholar]
Americo 2022
- Americo J, Earl P, Moss B. Virulence differences of monkeypox virus clades 1, 2a and 2b.1 in a small animal model. bioRxiv 2022;Preprint:12.01.518711. [DOI: 10.1101/2022.12.01.518711] [DOI] [Google Scholar]
Andrei 2010
- Andrei G, Snoeck R. Cidofovir activity against poxvirus infections. Viruses 2010;2(12):2803-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Beer 2019
- Beer EM, Rao VB. A systematic review of the epidemiology of human monkeypox outbreaks and implications for outbreak strategy. PLOS Neglected Tropical Diseases 2019;13(10):e0007791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cangene 2010
- Cangene. Highlights of prescribing information Vaccinia Immune Globulin Intravenous (Human). www.fda.gov/media/77004/download (accessed 14 December 2022).
CDC 2022a
- Centers for Disease Control and Prevention. Monkeypox outbreak global map. www.cdc.gov/poxvirus/monkeypox/response/2022/world-map.html (accessed 15 December 2022).
CDC 2022b
- Centers for Disease Control and Prevention. Treatment information for healthcare professionals. Updated 31 October 2022. Interim clinical guidance for the treatment of monkeypox. www.cdc.gov/poxvirus/monkeypox/clinicians/treatment.html (accessed 27 January 2023).
CDC 2023a
- Centers for Disease Control and Prevention. Science brief: detection and transmission of mpox (formerly monkeypox) virus during the 2022 Clade IIb outbreak. www.cdc.gov/poxvirus/monkeypox/about/science-behind-transmission (accessed 9 February 2023).
CDC 2023b
- Centers for Disease Control and Prevention. Clinician FAQs. www.cdc.gov/poxvirus/mpox/clinicians/faq.html (accessed 6 February 2023).
Chimerix 2022
- Chimerix. TEMBEXA Highlights of prescribing information. www.accessdata.fda.gov/drugsatfda_docs/label/2021/214460s000,214461s000lbl.pdf (accessed 2 November 2022).
Cochrane 2022
- Cochrane. Cochrane Risk of bias 2 tool. www.riskofbias.info/welcome/rob-2-0-tool/current-version-of-rob-2 (accessed 6 December 2022).
De Clerq 2002
- De Clerq E. Cidofovir in the treatment of poxvirus infections. Antiviral Research 2002;55(1):1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
EASL 2019
- European Association for the Study of the Liver. EASL clinical practice guidelines: drug-induced liver injury. Journal of Hepatology 2019;70(6):1222-61. [DOI] [PubMed] [Google Scholar]
Falendysz 2017
- Falendysz E, Lopera J, Doty J, Nakazawa Y, Matzke C, Lorenzsonn F, et al. Characterization of Monkeypox virus infection in African rope squirrels (Funisciurus sp.). PLOS Neglected Tropical Diseases 2017;11:e0005809. [DOI] [PMC free article] [PubMed] [Google Scholar]
Foster 2017
- Foster S, Parker S, Lanier R. The role of brincidofovir in preparation for a potential smallpox outbreak. Viruses 2017;9(11):320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Frenois‐Veyrat 2022
- Frenois-Veyrat G, Gallardo F, Gorgé O, Marcheteau E, Ferraris O, Baidaliuk A, et al. Tecovirimat is highly efficient on the Monkeypox virus lineage responsible for the international 2022 outbreak. bioRxiv 2022;Pre-print:07.19.500484. [DOI: 10.1101/2022.07.19.500484] [DOI] [Google Scholar]
Garner 2016
- Garner P, Hopewell S, Chandler J, MacLehose H, Akl E A, Beyene J, et al. When and how to update systematic reviews: consensus and checklist. BMJ 2016;354:i3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
Girometti 2022
- Girometti N, Byrne R, Bracchi M, Heskin J, McOwan A, Tittle V, et al. Demographic and clinical characteristics of confirmed human monkeypox virus cases in individuals attending a sexual health centre in London, UK: an observational analysis. Lancet Infectious Diseases 2022;22(9):1321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grosenbach 2018
- Grosenbach DW, Honeychurch K, Rose EA, Chinsangaram J, Frimm A, Maiti B, et al. Oral tecovirimat for the treatment of smallpox. New England Journal of Medicine 2018;379(1):44-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Grossi 2017
- Grossi IM, Foster SA, Gainey MR, Krile RT, Dunn JA, Brundage T, et al. Efficacy of delayed brincidofovir treatment against a lethal rabbitpox virus challenge in New Zealand white rabbits. Antiviral Research 2017;143:278-86. [DOI] [PubMed] [Google Scholar]
Higgins 2022
- Higgins JP, Savović J, Page MJ, Elbers RG, Sterne JA. Chapter 8: Assessing risk of bias in a randomized trial. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Hoy 2018
- Hoy S. Tecovirimat: first global approval. Drugs 2018;78(13):1377-82. [DOI] [PubMed] [Google Scholar]
Hutson 2021
- Hutson CL, Kondas AV, Mauldin MR, Doty JB, Grossi IM, Morgan CN, et al. Pharmacokinetics and efficacy of a potential smallpox therapeutic, brincidofovir, in a lethal monkeypox virus animal model. mSphere 2021;6(1):e00927-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Jordan 2009
- Jordan R, Goff A, Frimm A, Corrado ML, Hensley LE, Byrd CM, et al. ST-246 antiviral efficacy in a nonhuman primate monkeypox model: determination of the minimal effective dose and human dose justification. Antimicrobial Agents and Chemotherapy 2009;53(5):1817-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kern 2003
- Kern E. In vitro activity of potential anti-poxvirus agents. Antiviral Research 2003;57:35-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kuroda 2023
- Kuroda N, Shimizu T, Hirano D, Ishikane M, Kataoka Y. Lack of clinical evidence of antiviral therapy for human monkeypox: a scoping review. Journal of Infection and Chemotherapy 2023;29(2):228-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
LiverTox 2022
- National Institute of Diabetes and Digestive and Kidney Diseases. LiverTox: clinical and research information on drug-induced liver injury. Available from www.ncbi.nlm.nih.gov/books/NBK585270/. [PubMed]
Mazurkov 2016
- Mazurkov OY, Kabanov AS, Shishkina LN, Sergeev AA, Skarnovich MO, Bormotov NI, et al. New effective chemically synthesized anti-smallpox compound NIOCH-14. Journal of General Virology 2016;97(5):1229-39. [DOI] [PubMed] [Google Scholar]
Mbrenga 2022
- Mbrenga F, Nakouné E, Malaka C, Bourner J, Dunning J, Vernet G, et al. Monkeypox treatment with tecovirimat in the Central African Republic under an Expanded Access Programme. medRxiv 2022;Pre-print:2022.08.24.22279177. [DOI: 10.1101/2022.08.24.22279177] [DOI] [PMC free article] [PubMed] [Google Scholar]
McKenzie 2022
- McKenzie JE, Brennan SE. Chapter 12: Synthesizing and presenting findings using other methods. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 6.3 (updated February 2022). Available from training.cochrane.org/handbook.
NHS 2022
- NHS Medway. FOI 5839 response. www.medway.nhs.uk/downloads/FOI%205839%20Response.pdf (accessed 15 December 2022).
NHS inform 2023
- Public Health Scotland. Mpox (monkeypox). www.nhsinform.scot/illnesses-and-conditions/infections-and-poisoning/mpox-monkeypox/ (accessed 9 February 2023).
NIAID 2022
- National Institute of Allergy and Infectious Diseases. Mpox (formerly monkeypox) treatment. www.niaid.nih.gov/diseases-conditions/mpox-treatment (accessed 6 February 2023).
O'Laughlin 2022
- O’Laughlin K, Tobolowsky FA, Elmor R, Overton R, O’Connor SM, Damon IK, et al. Clinical use of Tecovirimat (Tpoxx) for treatment of monkeypox under an investigational new drug protocol — United States, May-August 2022. Morbidity and Mortality Weekly Report 2022;71(37):1190-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ogoina 2022
- Ogoina D, Mohammed A, Yinka-Ogunleye A, Ihekweazu C. A case of suicide during the 2017 monkeypox outbreak in Nigeria. IJID Regions 2022;3:226-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Patel 2022
- Patel A, Tam J, Mason C, Murphy J, Sundramoorthi R, Naidu V, et al. Clinical features and novel presentations of human monkeypox in a central London centre during the 2022 outbreak: descriptive case series. BMJ 2022;378:e072410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rimoin 2010
- Rimoin A, Mulembakani P, Johnston S, Lloyd Smith J, Kisalu N, Kinkela T, et al. Major increase in human monkeypox incidence 30 years after smallpox vaccination campaigns cease in the Democratic Republic of Congo. PNAS 2010;107(37):16262-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sbrana 2007
- Sbrana E, Jordan R, Hruby DE, Mateo RI, Xiao SY, Siirin M, et al. Efficacy of the antipoxvirus compound ST-246 for treatment of severe orthopoxvirus infection. American Journal of Tropical Medicine and Hygiene 2007;76(4):768-73. [PubMed] [Google Scholar]
Schünemann 2022
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 6.3 (updated February 2022). Cochrane, 2022. Available from training.cochrane.org/handbook.
Seley‐Radtke 2021
- Seley-Radtke K, Thames J, Waters C. Broad spectrum antiviral nucleosides—our best hope for the future. Elsevier Public Health Emergency Collection 2021;57:109-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shearer 2005
- Shearer J, Siemann L, Gerkovich M, House R. Biological activity of an intravenous preparation of human vaccinia immune globulin in mouse models of vaccinia virus infection. Antimicrobial Agents and Chemotherapy 2005;49(7):2634-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Siegrist 2022
- Siegrist EA, Sassine J. Antivirals with activity against mpox: a clinically oriented review. Clinical Infectious Diseases 2023;76(1):155-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Smith 2011
- Smith SK, Self J, Weiss S, Carroll D, Braden Z, Regnery RL, et al. Effective antiviral treatment of systemic orthopoxvirus disease: ST-246 treatment of prairie dogs infected with monkeypox virus. Journal of Virology 2011;85(17):9176-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI] [PubMed] [Google Scholar]
Trost 2015
- Trost LC, Rose ML, Khouri J, Keilholz L, Long J, Godin SJ, et al. The efficacy and pharmacokinetics of brincidofovir for the treatment of lethal rabbitpox virus infection: a model of smallpox disease. Antiviral Research 2015;117:115-21. [DOI] [PubMed] [Google Scholar]
WHO 2022a
- World Health Organization. Monkeypox: experts give virus variants new names. www.who.int/news/item/12-08-2022-monkeypox--experts-give-virus-variants-new-names (accessed 9 February 2023).
WHO 2022b
- World Health Organization. WHO Director-General declares the ongoing monkeypox outbreak a Public Health Emergency of International Concern. www.who.int/europe/news/item/23-07-2022-who-director-general-declares-the-ongoing-monkeypox-outbreak-a-public-health-event-of-international-concern (accessed 6 February 2023).
WHO 2022c
- World Health Organization. Monkeypox factsheet. www.who.int/news-room/fact-sheets/detail/monkeypox (accessed 6 February 2023).
WHO 2023
- World Health Organization. Fourth meeting of the International Health Regulations (2005) (IHR) Emergency Committee on the multi-country outbreak of monkeypox (mpox). who.int/news/item/15-02-2023-fourth-meeting-of-the-international-health-regulations-(2005)-(ihr)-emergency-committee-on-the-multi-country-outbreak-of-monkeypox-(mpox) (accessed 02 March 2023).
Wittek 2006
- Wittek R. Vaccinia immune globulin: current policies, preparedness, and product safety and efficacy. International Journal of Infectious Diseases 2006;10(3):193-201. [DOI] [PubMed] [Google Scholar]
Yu 2022
- Yu J, Raj SM. Efficacy of three key antiviral drugs used to treat orthopoxvirus infections: a systematic review. Global Biosecurity 2022;1:1. [DOI: 10.31646/gbio.12] [DOI] [Google Scholar]
References to other published versions of this review
Fox 2022
- Fox T, Gould S, Princy N, Rowland T, Kuehn R. Therapeutics for treating mpox in humans. PROSPERO 2022. CRD42022382675. www.crd.york.ac.uk/prospero/display_record.php?RecordID=382675 (accessed 9 December 2022).