

Abstract
Inflammatory bowel diseases (IBD) arise from a convergence of genetic risk, environmental factors, and gut microbiota, where each is necessary but not sufficient alone to cause disease. Emerging evidence supports a bidirectional relationship between disease progression and changes in microbiota membership and function. Thus, the study of the gut microbiome and host-microbe interactions should provide critical insights into disease pathogenesis as well as leads for developing microbiome-based diagnostics and interventions for IBD. In this article, we review the most recent advancement of the relationship between gut microbiota and IBD and highlight the importance of going beyond establishing description and association to gain mechanistic insights into causes and consequences of the disease. The review aims at contextualizing recent findings to form conceptional frameworks for understanding the etiopathogenesis of IBD and for the future development of microbiome-based diagnostics and interventions.
Keywords: inflammatory bowel diseases, gut microbiota, inflammation, host-microbial interactions, dysbiosis, pathobionts
1. Introduction
Inflammatory bowel diseases (IBD) are a heterogeneous collection of chronic inflammatory disorders that generally include two separate conditions: ulcerative colitis (UC) and Crohn’s disease (CD) (1, 2). The first IBD case which resembled what we call UC today was recorded in the late 1700s, and the term “ulcerative colitis” was first coined in 1859 (3). Later in 1932, CD was recognized as an entity separate from UC due to its transmural and often patchy pattern of inflammation that can affect any part of the GI tract (4). In contrast, UC is restricted to the colon, typically starting from the rectum and spreading proximally to the cecum in many patients (5).
Despite the many advances in technologies and experimental models, the etiology of IBD is still unknown. The greater incidence of IBD among identical versus fraternal twins and certain families supports a heritable component for IBD. Moreover, the identification of hundreds of genetic polymorphisms and mutations through Genome-Wide Association Studies (GWAS) of IBD provides further support of a genetic basis for IBD (6, 7). However, genetics alone is rarely sufficient to cause disease. The incidence and prevalence of IBD have also been rising concomitantly with the industrialization, lifestyle changes, and urbanization of modern societies in a time frame too short to be explained by genetic drift or natural selection (8). In this regard, non-inheritable factors, such as environmental factors, shifts away from plant-based to animal-based processed diets, smoking, antibiotic administration, etc., must be considered in the etiopathogenesis of these diseases (9).
The idea that transmissible bacterial agents may be responsible for IBD was first proposed in the early 1900s (4). However, after decades of searching, no pathogens in the traditional sense have been found for causing IBD. In this regard, there has been an increased focus on the concept that indigenous gut microbes, when given the opportunity, can transform, trigger, and contribute to the etiopathogenesis of IBD. Major shifts in gut microbiota have been reported in association with active disease, e.g., increases in the major phylotype Proteobacteria and decreases in Firmicutes (10, 11). In experimental IBD models, several genetically-susceptible mouse strains only develop colitis in the presence of gut microbiota, and the frequency and severity of spontaneous colitis is largely determined by the composition of the microbiota (12).
IBD arises from a convergence of genetic risk, environmental, and microbial factors each necessary but insufficient alone to cause disease (13, 14). We refer readers to a prior systematic review of the gut microbiome and IBD that summarizes past findings that support these relationships (15). Here, we highlight more recent advances that may potentially shift paradigms of IBD risk, etiopathogenesis, and eventual best practices relevant to the prevention, management, and better clinical outcomes of IBD.
2. Changes in Gut Microbiota of IBD Patients: Cause or Effect?
Major shifts in the gut microbiota membership and function that promote potential disease states (dysbiosis) are commonly observed in IBD patients, but the significance of these changes is unclear. A key question remains: is dysbiosis a cause or consequence of immune activation and inflammation in IBD or both? While the gut microbiota is fairly stable throughout life, it can be perturbed by dietary changes, environmental shifts, infectious pathogens, lifestyle factors, medications, etc. The development of IBD dysbiosis in turn has repercussions on host responses (e.g. immune and metabolic) that attempt to restore the balance in host-microbe relationships(Figure 1). Host response includes the release of antimicrobial peptides (AMP), reactive oxygen species, immune mediators, mucus, and other changes to impact the composition and functions of the gut microbial community, the latter through modifications of the regional ecosystems of the gut. Several studies have shown that the IBD-associated microbiota exhibit reduced diversity and enrichment of less abundant phyla such as Gammaproteobacteria (16, 17). However, few mechanistic insights have emerged from these associations because important clinical metadata to help contextualize the findings are usually lacking. In addition, there has been a heavy reliance on low-resolution 16S rRNA amplicon sequence analytics that provide compositional information limited to the family or genus level which is insufficient to resolve inter-individual variations among individuals with IBD. Moreover, 16S rRNA amplicon sequence variants (ASVs) provides no functional information, which is needed to better understand host-microbe interactions relevant to states of health and disease. Increasing numbers of studies now employ metagenomics, metatranscriptomics, and metabolomics that when viewed in the context of host metadata (e.g. clinical course, medications, mucosal gene expression, histology, immune indices) can be much more informative. The application of open-source, community-driven analysis and visualization, and machine-learning platforms can further facilitate the analysis and integration of these complex and large datasets (18–21). For example, Miyoshi et al. accurately predicted which IL-10 gene deficient (IL10-KO) mice would develop colitis based on the metagenomic signatures of their antibiotic-induced dysbiosis (22).
Figure 1. Hypothetical model for IBD development and progression.
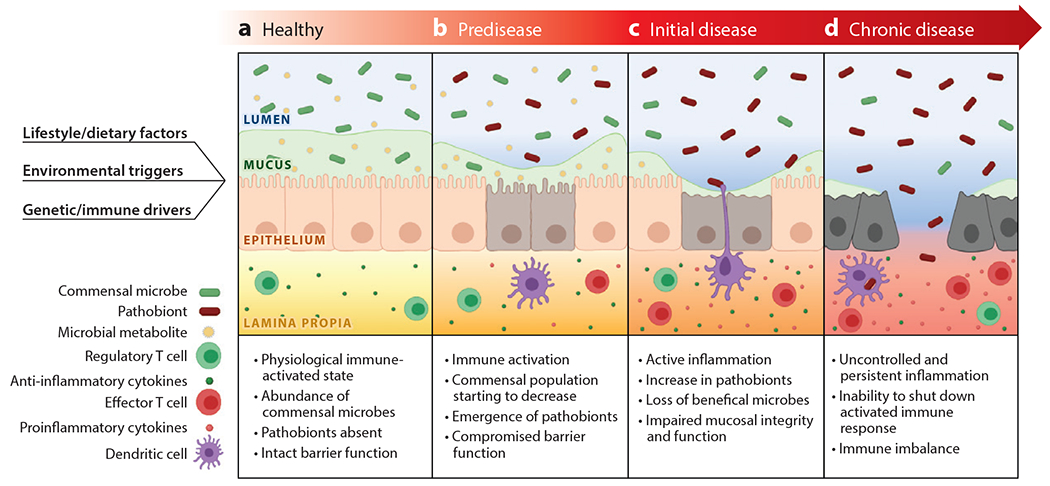
Under healthy conditions, commensal microbes produce beneficial metabolites to help maintain an impermeable barrier comprised of intact mucosal and epithelial layers (Panel A). IBD starts at a pre-disease state where the disease happens at a sub-clinical level. Genetic/immune drivers, environmental triggers, as well as lifestyle/diet can all contribute to the occurrence of pre-disease (Panel B). At this stage, certain commensal microbes transition to pathobionts that are more fit in an ecosystem where immune activation, the compromised barrier, mucus depletion, and other conditional factors come into play. As the disease progress, patients enter the initial stage of the disease, where patients display active inflammation mucosal damage (Panel C). The abundance of commensal bacteria significantly decreases as blooming pathobionts emerge and bloom. As a result, the microbiota produce fewer beneficial metabolites. If the condition persists for a long time, patients will reach a chronic stage of disease with persistent inflammation. The persistent inflammation and long-term dysbiosis lead to immune imbalance and the inability for mucosal healing which maintains the inflammatory and dysbiotic state creating a chronic inflammatory cycle (Panel D).
Study designs have also improved to incorporate longitudinal observations, time sequence sample collections, better controls, stratification of patients (e.g. CD vs UC), and associated clinical metadata (medications, regional involvement, active vs inactive disease). As one example, UC patients with ileal pouch-anal anastomosis were followed for up to 2 years with serial endoscopy and sampling of both host mucosa and pouch microbiomes. By performing both 16S rRNA and metagenomic analysis of luminal aspirates, mucosal brushings, and pinch biopsies, the emergence of potential pathobionts, i.e. commensal microbes that can transform into disease-promoting states, could be observed before the appearance of endoscopic changes or clinical symptoms (23, 24). The study also found that UC, but not FAP (familial adenomatous polyposis) pouch patients exhibited anomalous mucosal gene responses, potentially rendering them susceptible to developing of pouchitis. The Integrative Human Microbiome Project prospectively examined the dynamic changes of the microbiome of individual IBD patients over one year through combined metagenomic with meta-transcriptomic analyses (19, 20). Certain microbial populations detected by metagenomic analysis appeared dormant, whereas others that were not detectable by metagenomics appeared functionally active based on metatranscriptomic profiling. Jacobs et al. reported shifts in microbial composition as well as in fecal metabolites (fecal bile acids, taurine, and tryptophan) that were associated with the development of IBD in pediatric patients compared with healthy siblings and parents (25). These observations lead us to propose the hypothetical sequence of events illustrated in Figure 1, where a variety of environmental, dietary, xenobiotic, immunological, and other factors create conditions that promote the transformation of certain commensal microbes to pathobionts that then trigger and/or contribute to the onset of disease in genetically susceptible individuals.
The majority of clinical studies of intestinal microbiomes have relied on fecal samples which are an admixture of distal intestinal microbiota and not representative of region-specific gut microbiota. Thus, more studies are turning to endoscopic approaches to acquire region-specific samples. Hirano et al. , for example, performed a paired analysis of mucosal biopsies of both inflamed and non-inflamed sites of individual patients (26), finding increases in the relative abundances of Cloacibacterium and Tissierellaceae and decreases in the relative abundance of Neisseria in the former (26). Libertucci et al. analyzed mucosa-associated microbiota from site-matched colonic mucosal biopsies from CD patients with and without injury (27), finding altered microbial communities in the latter compared with the former or with non-IBD controls (27). Nishino et al. collected mucosal-associated samples by brushing mucosal surfaces, finding significant differences in several genera between CD and UC mucosal surfaces (28). Collectively, these studies highlight the complex biogeography and diversity of mucosal microbiota that may underpin states of health and disease, but they still come up short in determining if these associations are cause or consequence.
Few studies have examined the role of other microbial kingdoms of the gut microbiome such as fungi and viruses. Fungi have been implicated in some types of IBD, e.g. patients with CD who are positive for the serum biomarker ASCA (anti-Saccharomyces cerevisiae antibody) (29). Three groups have reported an increase in Candida albicans in IBD patients, especially among CD patients (30–32). Another fungal species, Malassezia restricta, which is highly abundant in some CD patients, appears to elicit inflammatory responses through the CARD9 (33) which is involved in anti-fungal defense and downstream from Dectin-1, an innate fungal β-glucan receptor (34). Risk polymorphisms of CARD9 and Dectin-1 have both been associated with IBD (33). In an animal study, Dectin-1, mannose receptors, and macrophage C-type lectin receptors have all shown to be involved in intestinal inflammation (35).
Changes in the viral community of the gut microbiota of IBD patients have also been described. Bacteriophages are viruses that parasitize and replicate within a bacterium, and their incorporation into bacterial genomes can affect bacterial gene expression and function. Increases of certain bacteriophage species of both luminal and mucosal samples of IBD patients have been reported with deep metagenomic sequencing (36, 37). However, the functional significance of altered bacteriophage profiles in IBD remains unclear.
3. Host Factors that Shape Gut Microbiota Composition and Functions
IBD-associated immune dysfunction and mucosal inflammation can greatly alter regional ecosystems, affecting intestinal permeability, mucus content and composition, mucosal gene expression, function, and cellular content, and immune signals in the gut. These changes also affect gut microbial assemblage, function, and the dynamics and connectivity among microbial members in ways that impact healthy host-microbe interactions to promote immune activation and inflammation in susceptible individuals. In this section, we review recent evidence that show the impact of host factors on the gut microbiota.
3.1. Genetic Drivers and Intestinal Permeability
Over two hundred risk variants have been identified by GWAS, and more are likely to be identified from studies of other ethnic groups or newly industrialized societies with rising incidence and prevalence of IBD (8). Not surprisingly, many IBD-gene variants appear to be associated with microbial sensing and clearance, T-cell differentiation and maintenance, and regulation of inflammatory mediators. Among these risk variants, mutations of the immunomodulatory interleukin 10 (IL-10) cytokine promote very early onset IBD in pediatric patients which may be dominant, as patients can present at birth with disease before acquisition of gut microbiota (38). Similarly, the Samp1/Yit mouse line displays a CD-like disease of the terminal ileum that is largely driven by genetic factors, as offspring can exhibit this disease at birth. Furthermore, the severity increases when gut microbiota are present (39). In contrast, IL-10KO mice develop frank colitis as adults, and like most human IBD, the penetrance of disease is variable depending on institutional housing conditions, diet, and the presence of disease-promoting pathobionts (40).
A healthy mucus layer plays a crucial role in maintaining the symbiotic relationship between the host and gut microbiota for intestinal homeostasis. The mucus layer creates spatial separation between microbes and intestinal epithelium but also serves as a selective filter to allow critical host-microbe interactions. Intestinal microbes produce many important small molecules and food-derived nutrients for proper host immune, intestinal, and metabolic functions. Concomitantly, the host produces mucus, antimicrobial peptides (AMPs), and immunoglobulins that not only keep microbes at bay, but help select, nurture, and shape regional gut microbiomes. Both clinical and experimental studies have implicated impairment of intestinal mucosal barrier function as an important contributing factor to the risk and development of IBD. A thinner mucus layer leaves intestinal epithelium exposed to the luminal microbes and induces immune activation to generate an inflammatory response. For example, mutations in mucin synthesis genes such as MUC2 are associated with the development of colitis (41). In MDR1A-deficient mice, intestinal mucosal dysbiosis associated with a significantly thinner mucus layer can be detected before the development of fecal dysbiosis and colitis (42).
Deficiency in host defense against microbes also leads to increased susceptibility to inflammation. Consistently, some gene variants associated with IBD (e.g. NOD2, ATG16L1, CYBB, etc.) are involved in autophagy and pathogen clearance (7). NOD2, for example, is an innate receptor for microbe-derived muramyl dipeptide and plays an essential role in microbial sensing. Combined deficiency of NOD2 and CYBB leads to the accumulation of Mucispirillum scheaedleri, a mucolytic anaerobe which in turn could trigger the onset of IBD (43).
3.2. Mucosal Immunoglobulins and Antimicrobial Peptides
In recent years, mucosal immunoglobulins have been linked to the development of IBD (44). Mucosal immunoglobulins are antibodies produced by B-cell-rich mucosal inductive sites. Among different classes of mucosal immunoglobins, the role of immunoglobin A (IgA) in IBD is most well-studied (44). Selectively enriched IgA-binding fecal bacteria from IBD patients cause more severe colitis in an animal model (45). Another study showed that “natural”, low-affinity, polyreactive antibodies that are T-cell and antigen-independent appear to contribute to the shaping of the gut microbiome, particularly of the small intestine (46). Other studies have reported that specific monoclonal IgAs activate the polysaccharide utilization loci of Bacteroides thetaiotaomicron and promote epithelial adherence of Bacteroides fragilis (47, 48).
In addition to IgA, mucosal IgM, IgG, and IgD may contribute to intestinal homeostasis (44). Mucosal IgG, for example, exhibits selectivity in microbial binding (49). Furthermore, mucosal IgG of UC patients appears to engage gut-resident macrophages and induce intestinal inflammation (50). The effects of immunoglobulin binding to gut microbes remain poorly understood.
AMPs secreted by specialized intestinal epithelial cells, including Paneth cells, have also been linked to intestinal homeostasis, regulation of gut microbiota, and IBD etiopathogenesis. Several CD-associated polymorphic genes, including NOD2, ATG16L, IRGM, XBP-1, TCF7L2, and LRP6 have been associated with Paneth cell dysfunction. For example, reduced α-defensin has been detected in ileal CD but not with UC or colonic (51). Recently, Pierre et al. reported that peptide YY (PYY), previously known as an endocrine peptide involved in satiety control, is expressed in Paneth cells, suggesting an alternate role as an AMP. PYY was shown to have antifungal activity, selectively targeting the hyphae or virulent form of C. albicans (32). Intriguingly, an increased abundance of C. albicans has been observed in CD patients, possibly arising from Paneth cell dysfunction and compromised PYY production or secretion (30, 31).
3.3. Diet, Lifestyle, and Environmental Factors
Westernized diets high in saturated fats, simple carbohydrates, and low in dietary fiber and other lifestyle changes have been suspected of promoting the rapid rise of IBD cases among newly industrialized countries (9, 52). Diet is an important and dominant driver of gut microbial assembly and function. One animal study showed that a low-fiber diet over multiple generations results in progressive and irreversible changes in gut microbiota, including decreased diversity and loss of polysaccharide-digesting microbes (53). In addition to diet, a link between smoking, breast feeding, urban living, pollutants, and xenobiotic exposure have been implicated as risk factors in a meta-analysis of observational studies of IBD (52). Several studies have suggested that circadian disruptions can increase intestinal inflammation and more aggressive CD (54). Furthermore, diurnal oscillation disruptions of certain members of the gut microbiota potentially promote imbalances in host-microbe interactions and contribute to IBD risk and progression (55).
3.4. The Early Life Microbiome and Immune Development
The development of immune tolerance to the commensal microbiota happens early in life, during which the immune system develops antigen-specific tolerance to microbes and microbial products (56–58). The early life microbiome is critical for the proper development of immune tolerance by presenting a diverse repertoire of antigens and/or induction of regulatory T cells (Treg) and natural killer T cells (NKT) (59–62). IL-10 KO mice with antibiotic-induced dysbiosis in early life are more prone to the development of spontaneous colitis if the immune system fails to develop tolerance to key members of the gut microbiota (60). In the early stage of life, goblet cell-associated antigen passages also form in the colon, facilitating the transportation of a variety of bacterial antigens from the lumen to lamina propria (57). Perturbations of the early life microbiome with antibiotics exposure, C-sections, formula feeding, diets, etc., can limit the diversity and maturation of the early life microbiome to compromise the development of immune tolerance to important commensal microbiota, thereby increasing long term risk for complex immune disorders like IBD (22, 60, 62). The implications of these findings are significant, as they suggest a rethinking of best practices to interventions aimed at restoring the “health” of the early life microbiome to promote long-term health and reduce the risk for individuals genetically prone to certain disorders associated with gut dysbiosis.
In addition to immune development, maternal antibodies from placental transfer or breast milk may also play a role in regulating immune homeostasis during early life. NOD2/CYBB-deficient mice develop spontaneous colitis after weaning, as maternal antibodies appear to mitigate inflammatory responses of offspring to microbiota before weaning, albeit the mechanisms underlying this action remain poorly understood.
4. Impact of Gut Microbiota on Host Health
The composition and functional impact of gut microbiota, partially shaped by the host environment, can in turn affect states of intestinal health and disease. In healthy individuals, gut microbiota perform important functions including nutrient absorption, immune regulation, and barrier maintenance. Perturbations of gut microbiota, particularly in genetically susceptible individuals, can contribute to the development of disease. This notion is supported by the observation that genetically susceptible animals rarely develop spontaneous colitis under germ-free conditions. One study showed reduced Treg cells and B-cell class switching in the colon when transferring IBD-associated microbiota to germ-free mice (16). Recent studies further identified microbial-derived metabolites that directly interact with host immune signaling.
4.1. Pathobionts: Commensal Microbes to Opportunistic Pathogens
For many years, grouping microbes into those that were commensal and health-promoting versus those that were thought to be disease-promoting was common practice. However, it has become more apparent that commensal microorganisms, given the opportunity, can cause an imbalance in host-microbiome interactions to promote disease. These microbes are defined as “pathobionts”, as their fitness and functional properties can transition between states that differentially impact the host.
In a longitudinal study of UC pouch patients, certain strains of B. fragilis, normally found in low abundance in the commensal microbiota, were found to bloom before and during pouchitis suggesting they may have triggered the onset of the disease (23). Another Bacteroides was also shown to increase the incidence of colitis in mice with antibiotic-altered microbiota (60). In a time-series study, a transient increase of Ruminococcus gnavus was detected during active diseases (63). The group further identified a specific polysaccharide in R. gnavus that induced the production of TNFα, a pro-inflammatory cytokine (64). Bilophila wadsworthia, a bile-tolerant, sulfite-reducing Proteobacteria, increases in abundance with consumption of a high saturated fat diet to promote colitis in IL-10KO mice (40). B. wadsworthia has also been implicated in human IBD despite being often found in the gut microbiota of healthy individuals (65). Thus, the state of pathobionts and their impact in promoting disease are highly context dependent.
4.2. Production of Short-Chain Fatty Acids (SCFA)
SCFAs are products of bacterial fermentation and play an important role in intestinal homeostasis (66). Propionate and acetate are mainly produced by Bacteroidetes, whereas butyrate is predominantly produced by Firmicutes. SCFAs can be utilized as a carbon source by intestinal epithelial cells, but also bind to G-protein coupled receptors (GPCR). Thus, SCFAs play an important role in mucosal healing, histone deacetylation, and gene expression. In addition, SCFAs exhibit immune-modulatory activity, including Treg development, cytokine production, and anti-inflammatory effects (67). SCFAs also regulate the rest of the microbial population by modifying the ecological milieu to select for specific microbial community members.
Among different SCFAs, butyrate function is the most well-understood. Microbial-derived butyrate promotes IL-10 independent epithelial barrier function through repression of Claudin-2, a channel forming tight junction protein that normally disrupts gut barrier function (68). Butyrate was also shown to induce macrophage differentiation and AMP production (69). Due to its many beneficial functions, reduced butyrate production often signals dysbiosis, e.g. decreased butyrate-synthetic capacity of the gut microbiota has been reported in patients with active IBD (70).
4.3. Secondary Bile Acids
Bile acids are important bile components to facilitate lipid digestion and absorption. Primary bile acids (PBAs) enter the intestinal tract from the duodenum and are converted to secondary bile acids (SBAs) in the colon by microbes (71). In IBD, many factors can perturb the bile acid pool and composition, including malabsorption and decreased conversion of PBA to SBA (72, 73). Inflammation-induced changes in intestinal epithelium can further impair the reabsorption of PBAs and SBAs. Such changes pose a higher risk of infection by Clostridioides difficile, as these conditions promote de-sporulation of C. difficile and activation of virulence properties (73).
Recent studies suggest that SBAs produced by microbes play an important role in T cell differentiation (74, 75). One study found that SBAs modulate a population of RORγ expressing Treg cells, and deletion of microbial bile acid metabolic pathways decreased this Treg population (75). Another study screened a library of bile acid metabolites and identified two bile acid derivatives with distinct functions: 3-xoxLCA that inhibits the differentiation of Th17 cells, whereas isoalloLCA promotes the differentiation of Treg cells (74). The observation was further confirmed in vivo (74). These findings suggest an important role of microbial metabolites in modulating the homeostasis of host immune response.
A recent study of UC and FAP patients with ileal pouches reported a decreased abundance of microbial bile acid conversion genes and reduced levels of lithocholic and deoxycholic acids (76). Using an animal model, they found that the administration of these bile acids mitigated DSS-induced colitis via the activation of the TGR5 receptor (76).
4.4. Aromatic Amino Acid Catabolites
Microbe-derived aromatic amino acids may play an important role in immune activation, intestinal epithelial barrier function, and in promoting anti-inflammatory or anti-oxidative conditions (77). These metabolites, often indole-derived compounds, are produced by different microbial species during the breakdown of tryptophan or other aromatic amino acids. The microbial-derived amino acid catabolites reach the systemic circulation to interact with host receptors.
The serum levels of tryptophan and tryptophan metabolites are reduced in IBD, especially among CD patients (78, 79) and IBD patients exhibit a diminished capacity to utilize tryptophan (80). Microbial-derived tryptophan catabolites serve as a host signaling molecule through the activation of the aryl hydrocarbon receptor (AHR) or pregnane X receptor (77). AHR is highly expressed in intestinal epithelium and appears to promote gene expression involved in maintaining barrier integrity and immune cell differentiation (81). The activating ligands of AHR include a variety of tryptophan metabolites: indoleacrylic acid, indole-3-propionic acid, indole-3-ethanol, indole-3-pyruvate, and indole-3-aldehyde (80, 82–84). In an animal model, transplanting microbiota with impaired tryptophan catabolism increased susceptibility to colitis and inflammation in the recipient (85). Finally, administration of tryptophan and some of the metabolites appears to reduce the colitis severity in a murine model (79), raising the possibility that they may have therapeutic potential in IBD (86).
Two recent studies found that metabolites derived from gut microbiota function as human GPCR ligands (87, 88). GPCRs are critical signaling molecules for immune and inflammatory responses and the maintenance of intestinal barrier function, and their dysfunction appears linked with the pathogenesis of IBD (89).
4.5. Sphingolipids
Sphingolipids, a class of molecules produced by both host and microbes, are often different between IBD and non-IBD subjects (90). Host-derived sphingolipids are important signaling molecules that regulate inflammation, immunity, and have been implicated in the IBD pathogenesis (91). However, sphingolipids can also be produced by gut microbes that can modulate host immune responses. For example, sphingolipids produced by Bacteroides inhibit the proliferation of invariant NKT cells and protect against chemically-induced colitis (92). IBD patients display diminished bacterial-derived sphingolipid production, potentially compensated by an increase in host-derived sphingolipids (90). The lack of microbial-derived sphingolipids resulted in intestinal inflammation and altered host ceramide pools in a murine model (93).
5. Advances in Microbiome-Based IBD Therapeutics and Diagnostics
Despite the rapid advances in enabling technologies, conceptual and mechanistic insights into the microbial basis of IBD lag behind, thus presenting a challenge to the development of novel microbiome-based biotherapeutics. Current pharmacological treatments for both UC and CD primarily aim at inhibiting host inflammation: aminosalicylates, corticosteroids, Janus kinase inhibitors, and monoclonal antibodies to TNFα, IL-12/23, or integrins (94). A major limitation of these treatments is the low response rate to the initial treatment and further loss of response over time among the initial responders (94). Microbiome-based interventions are being actively explored for IBD, but so far their promise remains unrealized (95). Proposed microbial therapies for IBD include fecal microbial transplantation (FMT), probiotics, synbiotics, prebiotics, and postbiotics. The underline principles of these methods are to either introduce or promote potential beneficial microbes in IBD patients. FMT appears to be effective for UC patients in several clinical trials, however, the long-term effect remains unknown (96, 97). So far, no commercial probiotics have shown evidence of efficacy in the treatment of IBD in clinical trials (98). In addition, several clinical trials of dietary intervention for IBD were conducted but appear to be ineffective (99). Although microbial-derived therapeutics are still in the early stages, ongoing research and clinical trials seem to suggest that following the microbial route may be the most fruitful for developing an effective IBD therapeutic.
6. Future Issues
Despite the recent advancement in the understanding the role of microbiome in IBD, several obstacles remain for the development of effective microbiome-based therapies. Researchers must first understand if engraftment of microbes is needed for sustained benefit, and if so, what microbes are best suited for the purpose? Given the large inter-individual differences in gut microbiota, it is likely that a highly personalized treatment would be required for microbiome-based interventions. The timing of the intervention is also important. If disease-promoting gut microbiota emerge prior to the onset of IBD, it would be important to intervene at this early stage to rebalance the gut microbiome and reduce the risk for disease or to achieve sustained remission. On the other hand, experimental studies have shown that introducing keystone microbiota of the early life microbiome after the immune developmental window cannot lower risk for development of colitis. Thus, the timing and precision of microbiome-based interventions will be critical issues to resolve in developing next generation biotherapeutics.
Acknowledgments:
This research is supported by the following grants from the National Institute of Diabetes and Digestive and Kidney Diseases: RC2DK122394, R01DK47722, R01DK113788, and NIH T32 DK07074; and the Center for Interdisciplinary Study of Inflammatory Intestinal Diseases (P30 DK42086). Additional support has been provided by the Gastrointestinal Research Foundation of Chicago, the David and Ellen Horing Research Fund, and the Helmsley Charitable Trust. Figure 1 was created using Biorender.com
Footnotes
Disclosure Statement:
Eugene B. Chang is the co-founder and Chief Medical Officer for AVnovum Therapeutics. Yue Shan and Mirae Lee have no conflicts of interest.
References:
- 1.Roda G, Chien Ng S, Kotze PG, Argollo M, Panaccione R, et al. 2020. Crohn’s disease. Nat. Rev. Dis. Prim 6(1):22. [DOI] [PubMed] [Google Scholar]
- 2.Kobayashi T, Siegmund B, Le Berre C, Wei SC, Ferrante M, et al. 2020. Ulcerative colitis. Nat. Rev. Dis. Prim 6(1):74. [DOI] [PubMed] [Google Scholar]
- 3.Actis GC, Pellicano R, Fagoonee S, Ribaldone DG. 2019. History of Inflammatory Bowel Diseases. J. Clin. Med 8(11):1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mulder DJ, Noble AJ, Justinich CJ, Duffin JM. 2014. A tale of two diseases: The history of inflammatory bowel disease. J. Crohn’s Colitis 8(5):341–48 [DOI] [PubMed] [Google Scholar]
- 5.Flynn S, Eisenstein S. 2019. Inflammatory Bowel Disease Presentation and Diagnosis. Surg. Clin. North Am 99(6):1051–62 [DOI] [PubMed] [Google Scholar]
- 6.Kaplan GG, Ng SC. 2017. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology. 152(2):313–321.e2 [DOI] [PubMed] [Google Scholar]
- 7.Graham DB, Xavier RJ. 2020. Pathway paradigms revealed from the genetics of inflammatory bowel disease. Nature. 578(7796):527–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, et al. 2017. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 390(10114):2769–78 [DOI] [PubMed] [Google Scholar]
- 9.Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, et al. 2018. Environmental triggers in IBD: A review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol 15(1):39–49 [DOI] [PubMed] [Google Scholar]
- 10.Hurst RD, Molinari M, Chung TP, Rubin M, Michelassi F. 1996. Prospective study of the incidence, timing and treatment of pouchitis in 104 consecutive patients after restorative proctocolectomy. Arch Surg. 131(5):497–500 [DOI] [PubMed] [Google Scholar]
- 11.Simchuk EJ, Thirlby RC. 2000. Risk factors and true incidence of pouchitis in patients after ileal pouch-anal anastomoses. World J Surg. 24(7):851–56 [DOI] [PubMed] [Google Scholar]
- 12.Mizoguchi A 2012. Animal models of inflammatory bowel disease. Prog. Mol. Biol. Transl. Sci 105:263–320 [DOI] [PubMed] [Google Scholar]
- 13.Wu H, Shen B. 2009. Pouchitis: lessons for inflammatory bowel disease. Curr Opin Gastroenterol. 25(4):314–22 [DOI] [PubMed] [Google Scholar]
- 14.Cleynen I, Boucher G, Jostins L, Schumm LP, Zeissig S, et al. 2016. Inherited determinants of Crohn’s disease and ulcerative colitis phenotypes: A genetic association study. Lancet. 387(10014):156–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee M, Chang EB. 2021. Inflammatory Bowel Diseases (IBD) and the Microbiome-Searching the Crime Scene for Clues. Gastroenterology. 160(2):524–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Torres J, Hu J, Seki A, Eisele C, Nair N, et al. 2020. Infants born to mothers with IBD present with altered gut microbiome that transfers abnormalities of the adaptive immune system to germ-free mice. Gut. 69(1):42–51 [DOI] [PubMed] [Google Scholar]
- 17.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, et al. 2014. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 15(3):382–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vich Vila A, Imhann F, Collij V, Jankipersadsing SA, Gurry T, et al. 2018. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med 10(472):eaap8914. [DOI] [PubMed] [Google Scholar]
- 19.Schirmer M, Franzosa EA, Lloyd-Price J, McIver LJ, Schwager R, et al. 2018. Dynamics of metatranscription in the inflammatory bowel disease gut microbiome. Nat. Microbiol 3:337–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lloyd-Price J, Arze C, Ananthakrishnan AN, Schirmer M, Avila-Pacheco J, et al. 2019. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature. 569:655–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yilmaz B, Juillerat P, Øyås O, Ramon C, Bravo FD, et al. 2019. Microbial network disturbances in relapsing refractory Crohn’s disease. Nat. Med 25(2):323–36 [DOI] [PubMed] [Google Scholar]
- 22.Miyoshi J, Lee STMM, Kennedy M, Puertolas M, Frith M, et al. 2020. Metagenomic Alterations in Gut Microbiota Precede and Predict Onset of Colitis in the IL10 Gene-Deficient Murine Model. Cell. Mol. Gastroenterol. Hepatol 11(2):491–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vineis JH, Ringus DL, Morrison HG, Delmont TO, Dalal S, et al. 2016. Patient-specific Bacteroides genome variants in pouchitis. MBio. 7(6):e01713–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang Y, Dalal S, Antonopoulos D, Hubert N, Raffals LH, et al. 2017. Early Transcriptomic Changes in the Ileal Pouch Provide Insight into the Molecular Pathogenesis of Pouchitis and Ulcerative Colitis. Inflamm. Bowel Dis 23(3):366–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jacobs JP, Goudarzi M, Singh N, Tong M, McHardy IH, et al. 2016. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. CMGH. 2(6):750–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirano A, Umeno J, Okamoto Y, Shibata H, Ogura Y, et al. 2018. Comparison of the microbial community structure between inflamed and non-inflamed sites in patients with ulcerative colitis. J. Gastroenterol. Hepatol 33(9):1590–97 [DOI] [PubMed] [Google Scholar]
- 27.Libertucci J, Dutta U, Kaur S, Jury J, Rossi L, et al. 2018. Inflammation-related differences in mucosa-associated microbiota and intestinal barrier function in colonic Crohn’s disease. Am. J. Physiol.-Gastrointest. Liver Physiol 315(3):G420–31 [DOI] [PubMed] [Google Scholar]
- 28.Nishino K, Nishida A, Inoue R, Kawada Y, Ohno M, et al. 2018. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol 53(1):95–106 [DOI] [PubMed] [Google Scholar]
- 29.Richard ML, Lamas B, Liguori G, Hoffmann TW, Sokol H. 2015. Gut fungal microbiota: The Yin and Yang of inflammatory bowel disease. Inflamm. Bowel Dis 21(3):656–65 [DOI] [PubMed] [Google Scholar]
- 30.Sokol H, Leducq V, Aschard H, Pham HP, Jegou S, et al. 2017. Fungal microbiota dysbiosis in IBD. Gut [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai T, Inoue R, Kawada Y, Morita Y, Inatomi O, et al. 2019. Characterization of fungal dysbiosis in Japanese patients with inflammatory bowel disease. J. Gastroenterol 54(2):149–59 [DOI] [PubMed] [Google Scholar]
- 32.Pierre JF, La Torre D, Sidebottom A, Kambal A, Zhu X, et al. 2020. Peptide YY: a novel Paneth cell antimicrobial peptide that maintains fungal commensalism. bioRxiv. 2020.05.15.096875 [Google Scholar]
- 33.Limon JJ, Tang J, Li D, Wolf AJ, Michelsen KS, et al. 2019. Malassezia Is Associated with Crohn’s Disease and Exacerbates Colitis in Mouse Models. Cell Host Microbe. 25(3):377–88.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, et al. 2012. Interactions between commensal fungi and the C-type lectin receptor dectin-1 influence colitis. Science (80-. ). 336(6086):1314–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rahabi M, Jacquemin G, Prat M, Meunier E, AlaEddine M, et al. 2020. Divergent Roles for Macrophage C-type Lectin Receptors, Dectin-1 and Mannose Receptors, in the Intestinal Inflammatory Response. Cell Rep. 30(13):4386–98.e5 [DOI] [PubMed] [Google Scholar]
- 36.Zuo T, Lu XJ, Zhang Y, Cheung CP, Lam S, et al. 2019. Gut mucosal virome alterations in ulcerative colitis. Gut. 68(7):1169–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duerkop BA, Kleiner M, Paez-Espino D, Zhu W, Bushnell B, et al. 2018. Murine colitis reveals a disease-associated bacteriophage community. Nat. Microbiol 3(9):1023–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shim JO. 2019. Recent advance in very early onset inflammatory bowel disease. Pediatr. Gastroenterol. Hepatol. Nutr 22(1):41–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pizarro TT, Pastorelli L, Bamias G, Garg RR, Reuter BK, et al. 2011. SAMP1/YitFc mouse strain: A spontaneous model of Crohn’s disease-like ileitis. Inflamm. Bowel Dis 17(12):2566–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, et al. 2012. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/−mice. Nature. 487(7405):104–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liso M, De Santis S, Verna G, Dicarlo M, Calasso M, et al. 2020. A Specific Mutation in Muc2 Determines Early Dysbiosis in Colitis-Prone Winnie Mice. Inflamm. Bowel Dis 26(4):546–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Glymenaki M, Singh G, Brass A, Warhurst G, McBain AJ, et al. 2017. Compositional Changes in the Gut Mucus Microbiota Precede the Onset of Colitis-Induced Inflammation. Inflamm. Bowel Dis 23(6):912–22 [DOI] [PubMed] [Google Scholar]
- 43.Caruso R, Mathes T, Martens EC, Kamada N, Nusrat A, et al. 2019. A specific gene-microbe interaction drives the development of Crohn’s disease–like colitis in mice. Sci. Immunol 4(34):eaaw4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen K, Magri G, Grasset EK, Cerutti A. 2020. Rethinking mucosal antibody responses: IgM, IgG and IgD join IgA. Nat. Rev. Immunol 20(7):427–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, et al. 2014. Immunoglobulin A Coating Identifies Colitogenic Bacteria in Inflammatory Bowel Disease. Cell. 158(5):1000–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bunker JJ, Erickson SA, Flynn TM, Henry C, Koval JC, et al. 2017. Natural polyreactive IgA antibodies coat the intestinal microbiota. Science (80-. ). 358(6361):eaan6619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nakajima A, Vogelzang A, Maruya M, Miyajima M, Murata M, et al. 2018. IgA regulates the composition and metabolic function of gut microbiota by promoting symbiosis between bacteria. J. Exp. Med 215(8):2019–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Donaldson GP, Ladinsky MS, Yu KB, Sanders JG, Yoo BB, et al. 2018. Gut microbiota utilize immunoglobulin a for mucosal colonization. Science (80-. ). 360(6390):795–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Armstrong H, Alipour M, Valcheva R, Bording-Jorgensen M, Jovel J, et al. 2019. Host immunoglobulin G selectively identifies pathobionts in pediatric inflammatory bowel diseases. Microbiome. 7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Castro-Dopico T, Dennison TW, Ferdinand JR, Mathews RJ, Fleming A, et al. 2019. Anti-commensal IgG Drives Intestinal Inflammation and Type 17 Immunity in Ulcerative Colitis. Immunity. 50(4):1099–1114.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bevins CL. 2017. The Immune System in IBD: Antimicrobial Peptides. In Crohn’s Disease and Ulcerative Colitis: From Epidemiology and Immunobiology to a Rational Diagnostic and Therapeutic Approach, ed Baumgart DC, pp. 75–86. Cham: Springer International Publishing [Google Scholar]
- 52.Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos GK, Lytras T, Bonovas S. 2019. Environmental Risk Factors for Inflammatory Bowel Diseases: An Umbrella Review of Meta-analyses. Gastroenterology. 157(3):647–59.e4 [DOI] [PubMed] [Google Scholar]
- 53.Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. 2016. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 529(7585):212–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canakis A, Qazi T. 2020. Sleep and Fatigue in IBD: an Unrecognized but Important Extra-intestinal Manifestation. Curr. Gastroenterol. Rep 22(2):8. [DOI] [PubMed] [Google Scholar]
- 55.Bishehsari F, Voigt RM, Keshavarzian A. 2020. Circadian rhythms and the gut microbiota: from the metabolic syndrome to cancer. Nat. Rev. Endocrinol 16(12):731–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milani C, Duranti S, Bottacini F, Casey E, Turroni F, et al. 2017. The First Microbial Colonizers of the Human Gut: Composition, Activities, and Health Implications of the Infant Gut Microbiota. Microbiol. Mol. Biol. Rev 81(4):e00036–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knoop KA, Gustafsson JK, McDonald KG, Kulkarni DH, Coughlin PE, et al. 2017. Microbial antigen encounter during a preweaning interval is critical for tolerance to gut bacteria. Sci. Immunol 2(18):eaao1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gensollen T, Iyer SS, Kasper DL, Blumberg RS. 2016. How colonization by microbiota in early life shapes the immune system. Science (80-. ). 352(6285):539–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Al Nabhani Z, Dulauroy S, Marques R, Cousu C, Al Bounny S, et al. 2019. A Weaning Reaction to Microbiota Is Required for Resistance to Immunopathologies in the Adult. Immunity. 50(5):1276–88.e5 [DOI] [PubMed] [Google Scholar]
- 60.Miyoshi J, Bobe AM, Miyoshi S, Huang Y, Hubert N, et al. 2017. Peripartum Antibiotics Promote Gut Dysbiosis, Loss of Immune Tolerance, and Inflammatory Bowel Disease in Genetically Prone Offspring. Cell Rep. 20(2):491–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olszak T, An D, Zeissig S, Vera MP, Richter J, et al. 2012. Microbial exposure during early life has persistent effects on natural killer T cell function. Science (80-. ). 336(6080):489–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schulfer AF, Battaglia T, Alvarez Y, Bijnens L, Ruiz VE, et al. 2018. Intergenerational transfer of antibiotic-perturbed microbiota enhances colitis in susceptible mice. Nat. Microbiol 3(2):234–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hall AB, Yassour M, Sauk J, Garner A, Jiang X, et al. 2017. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9(1):103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. 2019. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. U. S. A 116(26):12672–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, et al. 2014. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 505(7484):559–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belzer C, Chia LW, Aalvink S, Chamlagain B, Piironen V, et al. 2017. Microbial metabolic networks at the mucus layer lead to diet-independent butyrate and vitamin B12 production by intestinal symbionts. MBio. 8(5):e00770–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Russo E, Giudici F, Fiorindi C, Ficari F, Scaringi S, Amedei A. 2019. Immunomodulating Activity and Therapeutic Effects of Short Chain Fatty Acids and Tryptophan Post-biotics in Inflammatory Bowel Disease. Front. Immunol 10:2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zheng L, Kelly CJ, Battista KD, Schaefer R, Lanis JM, et al. 2017. Microbial-Derived Butyrate Promotes Epithelial Barrier Function through IL-10 Receptor–Dependent Repression of Claudin-2. J. Immunol 199(8):2976–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, et al. 2019. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity. 50(2):432–45.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laserna-Mendieta EJ, Clooney AG, Carretero-Gomez JF, Moran C, Sheehan D, et al. 2018. Determinants of reduced genetic capacity for butyrate synthesis by the gut microbiome in Crohn’s disease and ulcerative colitis. J. Crohn’s Colitis 12(2):204–16 [DOI] [PubMed] [Google Scholar]
- 71.Wahlström A, Sayin SI, Marschall HU, Bäckhed F. 2016. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism [DOI] [PubMed] [Google Scholar]
- 72.Fitzpatrick LR, Jenabzadeh P. 2020. IBD and Bile Acid Absorption: Focus on Pre-clinical and Clinical Observations. Front. Physiol 8(11):1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Allegretti JR, Kearney S, Li N, Bogart E, Bullock K, et al. 2016. Recurrent Clostridium difficile infection associates with distinct bile acid and microbiome profiles. Aliment. Pharmacol. Ther 43(11):1142–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hang S, Paik D, Yao L, Kim E, Jamma T, et al. 2019. Bile acid metabolites control TH17 and Treg cell differentiation. Nature. 576(7785):143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Song X, Sun X, Oh SF, Wu M, Zhang Y, et al. 2020. Microbial bile acid metabolites modulate gut RORγ+ regulatory T cell homeostasis. Nature. 577(7790):410–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sinha SR, Haileselassie Y, Nguyen LP, Tropini C, Wang M, et al. 2020. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe. 27(4):659–70.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Roager HM, Licht TR. 2018. Microbial tryptophan catabolites in health and disease. Nat. Commun 9(1):3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lai Y, Xue J, Liu CW, Gao B, Chi L, et al. 2019. Serum metabolomics identifies altered bioenergetics, signaling cascades in parallel with exposome markers in Crohn’s disease. Molecules. 24(3):449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nikolaus S, Schulte B, Al-Massad N, Thieme F, Schulte DM, et al. 2017. Increased Tryptophan Metabolism Is Associated With Activity of Inflammatory Bowel Diseases. Gastroenterology. 153(6):1504–16.e2 [DOI] [PubMed] [Google Scholar]
- 80.Wlodarska M, Luo C, Kolde R, D’Hennezel E, Annand JW, et al. 2017. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe. 22(1):25–37.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Neavin DR, Liu D, Ray B, Weinshilboum RM. 2018. The role of the aryl hydrocarbon receptor (AHR) in immune and inflammatory diseases. Int. J. Mol. Sci 19(12):3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Alexeev EE, Lanis JM, Kao DJ, Campbell EL, Kelly CJ, et al. 2018. Microbiota-Derived Indole Metabolites Promote Human and Murine Intestinal Homeostasis through Regulation of Interleukin-10 Receptor. Am. J. Pathol 188(5):1183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scott SA, Fu J, Chang PV. 2020. Microbial tryptophan metabolites regulate gut barrier function via the aryl hydrocarbon receptor. Proc. Natl. Acad. Sci. U. S. A 117(32):19376–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, et al. 2017. A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature. 551(7682):648–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lamas B, Richard ML, Leducq V, Pham HP, Michel ML, et al. 2016. CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med 22(6):598–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhang J, Zhu S, Ma N, Johnston LJ, Wu C, Ma X. 2020. Metabolites of microbiota response to tryptophan and intestinal mucosal immunity: A therapeutic target to control intestinal inflammation. Med. Res. Rev 41(2):1061–88 [DOI] [PubMed] [Google Scholar]
- 87.Cohen LJ, Esterhazy D, Kim SH, Lemetre C, Aguilar RR, et al. 2017. Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature. 549(7670):48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen H, Nwe PK, Yang Y, Rosen CE, Bielecka AA, et al. 2019. A Forward Chemical Genetic Screen Reveals Gut Microbiota Metabolites That Modulate Host Physiology. Cell. 177(5):1217–31.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zeng Z, Mukherjee A, Varghese AP, Yang X-LL, Chen S, Zhang H. 2020. Roles of G protein-coupled receptors in inflammatory bowel disease. World J. Gastroenterol 26(12):1242–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Franzosa EA, Sirota-Madi A, Avila-Pacheco J, Fornelos N, Haiser HJ, et al. 2019. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol 4(2):293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bryan PF, Karla C, Edgar Alejandro MT, Sara Elva EP, Gemma F, Luz C. 2016. Sphingolipids as Mediators in the Crosstalk between Microbiota and Intestinal Cells: Implications for Inflammatory Bowel Disease. Mediators Inflamm. 2016:9890141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.An D, Oh SF, Olszak T, Neves JF, Avci FY, et al. 2014. Sphingolipids from a symbiotic microbe regulate homeostasis of host intestinal natural killer T cells. Cell. 156(1–2):123–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brown EM, Ke X, Hitchcock D, Jeanfavre S, Avila-Pacheco J, et al. 2019. Bacteroides-Derived Sphingolipids Are Critical for Maintaining Intestinal Homeostasis and Symbiosis. Cell Host Microbe. 25(5):668–680.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Plichta DR, Graham DB, Subramanian S, Xavier RJ. 2019. Therapeutic Opportunities in Inflammatory Bowel Disease: Mechanistic Dissection of Host-Microbiome Relationships. Cell. 178(5):1041–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shan Y, Segre JA, Chang EB. 2019. Responsible stewardship for communicating microbiome research to the press and public. Nat. Med 25(6):872–74 [DOI] [PubMed] [Google Scholar]
- 96.Paramsothy S, Kamm MA, Kaakoush NO, Walsh AJ, van den Bogaerde J, et al. 2017. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: a randomised placebo-controlled trial. Lancet (London, England). 389(10075):1218–28 [DOI] [PubMed] [Google Scholar]
- 97.Paramsothy S, Nielsen S, Kamm MA, Deshpande NP, Faith JJ, et al. 2019. Specific Bacteria and Metabolites Associated With Response to Fecal Microbiota Transplantation in Patients With Ulcerative Colitis. Gastroenterology. 156(5):1440–54.e2 [DOI] [PubMed] [Google Scholar]
- 98.Preidis GA, Weizman AV, Kashyap PC, Morgan RL. 2020. AGA Technical Review on the Role of Probiotics in the Management of Gastrointestinal Disorders. Gastroenterology. 159(2):708–38.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Limketkai BN, Iheozor-Ejiofor Z, Gjuladin-Hellon T, Parian A, Matarese LE, et al. 2019. Dietary interventions for induction and maintenance of remission in inflammatory bowel disease. Cochrane database Syst. Rev 2(2):CD012839. [DOI] [PMC free article] [PubMed] [Google Scholar]