

ABSTRACT
High blood glucose is one of the risk factors for metabolic disease and INS (insulin) is the key regulatory hormone for glucose homeostasis. Hypoinsulinemia accompanied with hyperglycemia was diagnosed in mice with pancreatic β-cells exhibiting autophagy deficiency; however, the underlying mechanism remains elusive. The role of secretory autophagy in the regulation of metabolic syndrome is gaining more attention. Our data demonstrated that increased macroautophagic/autophagic activity leads to induction of insulin secretion in β-cells both in vivo and in vitro under high-glucose conditions. Moreover, proteomic analysis of purified autophagosomes from β-cells identified a group of vesicular transport proteins participating in insulin secretion, implying that secretory autophagy regulates insulin exocytosis. RAB37, a small GTPase, regulates vesicle biogenesis, trafficking, and cargo release. We demonstrated that the active form of RAB37 increased MAP1LC3/LC3 lipidation (LC3-II) and is essential for the promotion of insulin secretion by autophagy, but these phenomena were not observed in rab37 knockout (rab37-/-) cells and mice. Unbalanced insulin and glucose concentration in the blood was improved by manipulating autophagic activity using a novel autophagy inducer niclosamide (an antihelminthic drug) in a high-fat diet (HFD)-obesity mouse model. In summary, we reveal that secretory autophagy promotes RAB37-mediated insulin secretion to maintain the homeostasis of insulin and glucose both in vitro and in vivo.
KEYWORDS: Glucose-stimulated insulin secretion, insulin, LC3, RAB37, secretory autophagy
Introduction
Autophagy regulates cellular metabolism via the conventional degradation autophagic pathway to properly protect the cell from stresses that cause damage. Accumulating evidence shows that autophagosomes selectively recruit cargos (aggregated proteins, damaged organelles, pathogens, and microRNA) in the cytosol, followed by fusion with the lysosome for degradation [1–3]. Recently, a novel function of autophagy, namely secretory autophagy (also known as autosecretion or type III secretion), has gained more attention. In contrast to traditional degradative autophagy, the recruited cargos are carried by the double-membrane autophagosome and exported to the extracellular environment (exocytosis) [4,5]. This unconventional secretory autophagy is involved in vesicle trafficking and cytokine secretion (IL1B/IL-1β, IL6, IL18, HMGB1, and TNF/TNF-α) for innate and adaptive immune responses [6]. Furthermore, autophagosomes fuse with the plasma membrane to release engulfed cargos, including pathogens (viruses and bacteria) [7,8]. In addition, Kraya et al. demonstrated that autophagy-related secretion affects the extracellular environment through intracellular autophagy dynamics in cells [9]. There is growing evidence that autophagy plays pleiotropic roles within the cell involving both degradative and secretory pathways. However, the function of secretory autophagy in normal physiologic and pathogenic conditions are largely unknown.
Autophagy is known to play a protective role in pancreatic β-cell homeostasis [10–12]. Increasing evidence shows that the reduction of autophagic activity is accompanied by the pathogenesis of type-II diabetes mellitus (DM) and related complications [13–15]. Sun et al. reported that endoplasmic reticulum stress-induced diabetes can be improved by autophagic degradation of misfolded proinsulin molecules and damaged organelles [16]. Several antidiabetic drugs, including metformin, exendin-4 (a GLP-1 analog), and MK-626 (a DPP4 [dipeptidylpeptidase 4] inhibitor), have been proved to promote β-cell autophagy and augment insulin secretion [17–20]. Furthermore, autophagy deficiency (deletion of the Atg7 gene) in mouse β-cells resulted in hyperglycemia, glucose-intolerance, reduced β-cell mass, decreased insulin production, and decreased insulin secretion function [21–24]. These findings imply that autophagy plays a pivotal role in the regulation of insulin and β-cell homeostasis. However, the underlying molecular mechanism of autophagy-regulated insulin secretion in β-cell and its role in DM remains unclear.
RAB proteins belonging to the RAB small GTPase subfamily contribute to cellular endocytosis, vesicle transition, and exocytosis pathways [25]. RAB proteins cycling between GDP-bound inactive and GTP-bound active forms are responsible for the initiation and termination of vesicle trafficking [26]. Diverse RAB members (RAB1, RAB5, RAB7, RAB8B, RAB9, RAB24, and RAB33) participating in the canonical degradative autophagy machinery have been reported [25,27]. Moreover, RAB8A is known to be involved in secretory autophagy [28,29]. These findings imply that RAB proteins may play a critical role in the fate of cargoes undergoing either degradation or secretion. In this study, we confirmed that high glucose-induced active-form RAB37 accompanied by increased autophagic activity promotes insulin secretion from β-cells. RAB37 functions as the determining factor shifting conventional insulin secretion to unconventional secretory autophagy to promote the translocation of insulin granules from the cytosol to the plasma membrane. In HFD-obese mouse and ob/ob mouse models, increased autophagic activity induces more insulin secretion accompanied by improved glucose consumption, which implies that this mechanism may have therapeutic potential for hypoinsulinemia and hyperglycemia-related metabolic diseases.
Results
High glucose (HG) stimulation increases autophagic activity and promotes the colocalization of insulin and LC3 in vitro and in vivo
Initially, we used a mouse model to investigate the kinetics of autophagic activity, insulin secretion, and glucose concentration in the blood of mice receiving an intraperitoneal glucose tolerance test (IPGTT) for various time intervals. Our data showed that high glucose (HG) treatment for 90 min significantly increased LC3-II level (a marker of autophagosomes) (Figure 1A) accompanied by increased kinetics of glucose (Fig. S1A) and insulin (Fig. S1B) with a peak at 30 min in the blood of the mice after IPGTT. Moreover, the coexistence of LC3 (red) and insulin (green) was increased in the β-cells of the murine pancreatic tissues after HG treatment for 90 min (Figure 1B, yellow). In summary, insulin was associated with increased autophagic activity in the IPGTT mouse model.
Figure 1.
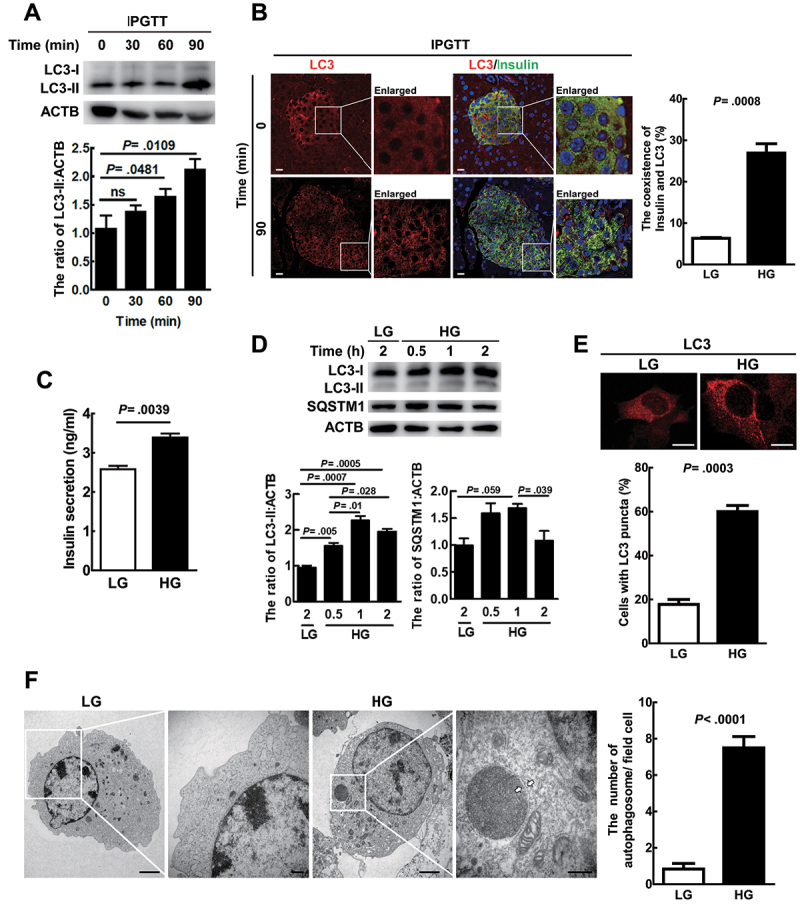
HG-Induced autophagic activity was associated with increased insulin colocalization with LC3 and release both in vitro and in vivo. (A) Mice (N = 5 9 mice per group) pancreatic tissues were collected, protein was extracted, and LC3 protein level was evaluated by immunoblotting at various times post-IPGTT. ACTB/β-actin was used as the internal control. (B) the coexistence of LC3 (red) and insulin (green) in mice pancreatic islet tissue sections using antibodies against LC3 and insulin conjugated with FITC or rhodamine at 0 min and 90 min post-IPGTT under a confocal microscope, N = 200. Scale bars: 10 μm. (C) MIN6 cells were treated with LG (5 mM) or HG (25 mM) for 1 h. The levels of secreted insulin in the cultured media were measured by ELISA. The bars represent mean ± SEM of three independent experiments in triplicate. (D) MIN6 cells were treated with LG or HG for the time as indicated. The protein levels of LC3 and SQSTM1 were determined by immunoblotting using specific antibodies. (E) MIN6 cells were treated with LG or HG for 1 h, and the endogenous LC3 puncta, which were labeled by anti-LC3 antibody followed by Alexa Fluor 568 secondary antibody, were investigated under a confocal microscope, N = 60. Scale bars: 10 μm. (F) MIN6 cells treated with LG or HG for 1 h and autophagosomes were observed by transmission electron microscopy. The white arrows show the double-membrane structure. Scale bar: 10 μm in (F); scale bar: 250 nm in enlarged images.
We treated immortalized mouse β-cells (MIN6) with either HG (25 mM) or low glucose (5 mM, LG) for various time intervals. Our data show that HG treatment of MIN6 cells for 1 h significantly increased insulin secretion compared to LG treatment cells (Figure 1C). Under the same treatment, LC3-II expression was gradually increased and SQSTM1/p62 accumulation decreased from 0.5 to 2 h in HG-treated cells compared to LG-treated cells (Figure 1D). Moreover, increased numbers of LC3 puncta (Figure 1E) and double-membrane autophagosome-like vesicles (Figure 1F) were detected in HG-treated MIN6 cells compared to LG-treated cells for 1 h. Altogether, HG was shown to increase autophagic activity and insulin secretion both in vitro and in vivo.
LC3 and insulin migrated toward the plasma membrane, and insulin secretion is enhanced by secretory autophagy under HG conditions
Increasing evidence indicates that secretory autophagy is responsible for the trafficking of specific cargos from the cytoplasm to the extracellular environment [7,30,31]. Herein, we attempted to determine whether secretory autophagy is involved in insulin exocytosis. Initially, we observed that HG treatment increased colocalization of LC3 (red) and insulin (green) in the cytoplasm compared with LG-treatment in MIN6 cells (Figure 2A, yellow). We then isolated immature insulin secretory granules (iISGs) and mature ISGs (mISGs) by subcellular fractionation of MIN6 cells [32,33]. Our data showed that insulin was highly expressed in both iISGs and mISGs fractions compared to whole cell lysate (WC) and post-nuclear supernatant (PNS). STX6 (syntaxin 6; a marker of immature insulin granules) was highly expressed in the iISGs fraction, and CALR (calreticulin; a marker of the endoplasmic reticulum) was mainly expressed in the WC and PNS. Notably, LC3-II protein (lipidated form of LC3) was greatly increased in the fraction of mISGs compared to that of iISGs under high glucose conditions (Figure 2B; the quantification was shown as bar diagrams in Fig. S2A). These data imply that autophagic vesicles may associate with mature ISGs while they move toward the plasma membrane for secretion.
Figure 2.
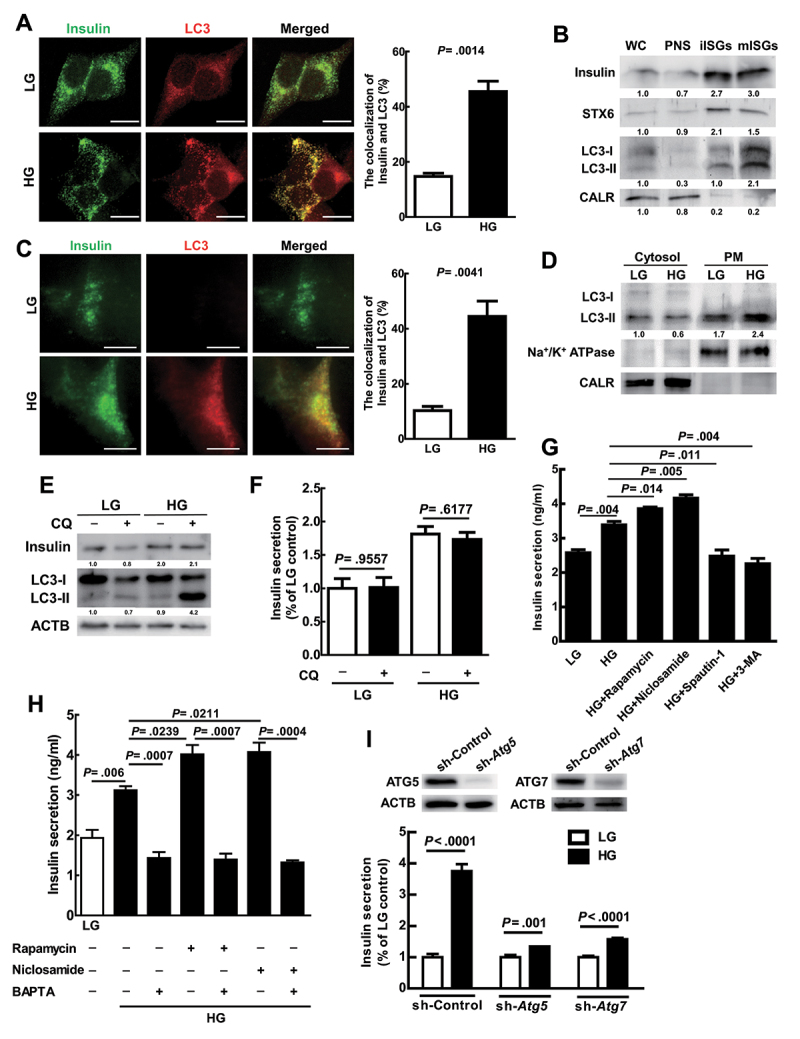
LC3 and insulin migrated toward the plasma membrane, and insulin secretion was enhanced by secretory autophagy under HG conditions. (A) Colocalization of LC3 (red) and insulin (green) in MIN6 cells after LG or HG treatment for 1 h was observed under confocal microscopy (N = 60). Scale bars: 10 μm. (B) After HG treatment for 1 h in MIN6 cells, immature insulin secretory granules (iIsgs), and mature ISGs (mIsgs) were purified through OptiPrep gradient. Western blot analyses of whole cell lysate (WC), postnuclear supernatant (PNS), iIsgs, and mIsgs using antibodies against insulin, STX6, LC3, and CALR. (C) Colocalization of LC3 (red) and insulin (green) in MIN6 cells after LG or HG treatment for 1 h was observed under TIRF microscopy (N = 30). Scale bars: 10 μm. (D) Cells were treated with LG or HG for 1 h and plasma membrane proteins were isolated by plasma membrane protein extraction kit. Protein levels of LC3, sodium-potassium ATPase (Na+/K+-ATPase), and CALR in the cytosol and plasma membrane fractions were determined by specific antibodies. (E) MIN6 cells were treated with LG or HG in the presence or absence of CQ (50 μM) for 1 h, and the levels of LC3 and insulin proteins were determined by immunoblotting using specific antibodies. ACTB/β-actin was used as the internal control. (F) the same treatment as in (E) and the secreted insulin in the cultured media were determined by ELISA. (G) MIN6 cells were treated with LG, HG, or HG together with pharmacological regulators of autophagy including rapamycin (100 nM), niclosamide (100 nM), spautin-1 (10 μM), or 3-MA (0.5 mM) for 1 h. The levels of secreted insulin in the cultured media were measured by ELISA. This experiment was conducted in quintuplicate and repeated 3 times. (H) MIN6 cells were pretreated with low glucose medium (LG, 5 mM) for 1.5 h, and the solutions were replaced with fresh LG medium with or without 10 μM BAPTA-AM for 30 min incubation. After LG (with or without BAPTA) treatment, MIN6 cells were followed by treated high glucose (HG, 25 mM) and/or autophagy inducers including rapamycin (100 nM) and niclosamide (100 nM) for 1 h. The levels of secreted insulin in the cultured media were measured by ELISA. This experiment was conducted in quintuplicate and repeated 3 times. (I) MIN6 cells harboring sh-Atg5, sh-Atg7, and sh-Control genes were established by lenti-virus shRNA system. The protein levels of ATG5 and ATG7 in sh-Atg5, sh-Atg7, and sh-Control cell lines were determined using specific antibodies. These cell lines were treated with LG or HG for 1 h followed by measurement of secreted insulin using ELISA. Data are shown as mean ± SEM of three independent experiments in triplicate. Student’s t-test was used for statistical analysis.
We further confirmed increased colocalization of LC3 (red) and insulin (green) proximal to the plasma membrane of MIN6 cells under HG conditions under Total internal reflection fluorescence (TIRF) microscopy (Figure 2C, merged yellow). TIRF microscope is particularly useful for investigating the molecule proximal to the specific membrane (within 200 nanometers) [34]. This result implies that high glucose increases the colocalization of autophagic vesicles and insulin granules, which are very close to the plasma membrane. To confirm that vesicles harboring LC3 migrate to the plasma membrane under HG conditions, the cytosol and plasma membrane fractions from MIN6 cells under LG and HG treatments were extracted and compared. Our result showed that the LC3-II level was increased in the plasma membrane fraction in MIN6 cells of the HG treatment compared to the LG treatment. Accordingly, Na+/K+ ATPase, the marker of the plasma membrane, was detected in the plasma membrane fraction (PM) and CALR was detected in the cytosol fraction. Our data indicate that HG treatment promotes the translocation of autophagic vesicles to the plasma membrane (Figure 2D; the quantification was shown as bar diagrams in Fig. S2B).
Riahi et al. reported that autophagy regulates proinsulin degradation [35]. Similarly, HG treatment triggered the autophagic degradation machinery, as demonstrated by the increased LC3-II accumulation in the presence of chloroquine (CQ) for 1 h compared to the LG group (Figure 2E, lane 2 vs. lane 4; the quantification was shown as bar diagrams in Fig. S2C). CQ is a fusion blocker of autophagosome and lysosome, an inhibitor of degradative autophagic progression. The intracellular insulin level was increased in the HG-treated cells compared to the LG-treated cells with or without CQ treatment (Figure 2E); however, HG-induced intracellular insulin expression (Figure 2E, lane 3 vs. lane 4) or insulin secretion was not affected by CQ treatment (Figure 2F). Our data indicate that HG-induced degradative autophagic progression does not affect the level of intracellular and extracellular insulin.
Moreover, we transiently transfected ptfLC3 plasmid DNA into MIN6 cells to monitor autophagic progression from autophagosome (yellow puncta) to autolysosome (red puncta) under HG conditions [36]. We detected higher numbers of yellow (autophagosome) and red puncta (autolysosome) in the HG-treated cells compared to the LG-treated cells under fluorescent microscopy (Fig. S2D). Furthermore, we detected more yellow puncta close to the plasma membrane of HG-treated cells, but not LG-treated cells under TIRF microscopy (Fig. S2E). These data suggest that HG treatment increased the translocation of autophagosome-like, but not autolysosome-like vesicles to the plasma membrane of MIN6 cells. Taken together, our findings reveal that autophagy regulates insulin secretion in a degradative autophagy-independent manner.
To explore the role of secretory autophagy in insulin secretion, we used autophagy inducers and inhibitors to manipulate autophagic activity and determine insulin secretion. We demonstrated that the autophagy inducer, either rapamycin or niclosamide (an anthelmintics drug and the inhibitor of MTOR [37]) increased HG-induced insulin secretion in MIN6 cells (Figure 2G, columns 3 and 4 vs. 2). In contrast, the autophagy inhibitor, either spautin-1 or 3-MA, decreased HG-induced insulin secretion in MIN6 cells (Figure 2G, columns 5 and 6 vs. 2). To compare autophagy-related insulin secretion under HG with the conventional insulin secretion under GSIS, we investigated the role of systemic calcium (Ca2+) signaling in Min-6 cells using BAPTA-AM (intracellular calcium chelator). We reveal that HG-induced insulin secretion was decreased by BAPTA-AM (10 μM) with or without autophagy inducers (rapamycin and niclosamide) in Min-6 cells (Figure 2H). Our data indicate that HG-induced autophagy-mediated insulin secretion was affected by calcium flux, which is similar to the conventional insulin secretion. Notably, the secreted insulin was no significant difference either by autophagy inducers or inhibitors under low glucose conditions (Fig. S2F). These findings imply that autophagy promotes insulin secretion only under HG treatment. Consistently, HG-induced insulin secretion was significantly suppressed by genetic silencing of Atg5 or Atg7 genes compared with the sh-Control group in MIN6 cells (Figure 2I; the quantification was shown as bar diagram in Fig. S2 G).
Runwal et al. reported that LC3-I, rather than LC3-II puncta, associated with SQSTM1-positive aggregates were detected when autophagy function was impaired [38]. Similarly, high glucose stimulation did not increase LC3-II puncta as well as colocalization with insulin in Atg5 knockdown MIN6 cells (Fig. S2 H), indicating that LC3-phosphatidylethanolamine (PE) conjugation is essential for autophagosome formation and insulin secretion. We further measured the basal level of proinsulin and insulin proteins in wild-type Atg5 and knockdown MIN6 cells under LG treatment. Our data showed no difference in proinsulin and insulin level between these cell lines (Fig. S2I), indicating that both proinsulin and insulin expression was not affected by autophagy without stimulation.
Altogether, under HG conditions, manipulation of autophagic activity affected insulin secretion in β-cells, which confirms that secretory autophagy participates in insulin secretion.
RAB37 is activated and colocalized with insulin and autophagosomes in MIN6 cells under HG treatment
RAB3A, RAB27A, and RAB37 are known to regulate insulin secretion [25]. We purified autophagosomes of MIN6 cells after HG treatment by discontinuous gradient centrifugation, and proteins within the autophagosome were analyzed by Mass Spectrometry (MS)-based quantitative proteomics analysis to clarify what possible RAB family proteins are involved in autophagy-related insulin secretion. Gene Ontology Biological Processes analysis revealed the proteins participating in insulin secretory in the purified autophagosomes of MIN6 cells (Figure 3A). Furthermore, insulin granule proteins as well as three RAB family proteins (RAB3A, RAB27A, and RAB37) were identified in the proteomics analysis (Table 1). Kiral et al. reported that RAB proteins regulate intracellular membrane trafficking in a GTP-dependent manner [39]. Notably, among the three RAB proteins identified in the autophagosome, only active-form RAB37 (GTP-bound) was increased after HG treatment for 1 h in MIN6 cells (Figure 3B; the quantification was shown as bar diagrams in Fig. S3A). This indicates that active-form RAB37 may play a pivotal role in autophagosome-mediated recruitment and secretion of insulin.
Figure 3.
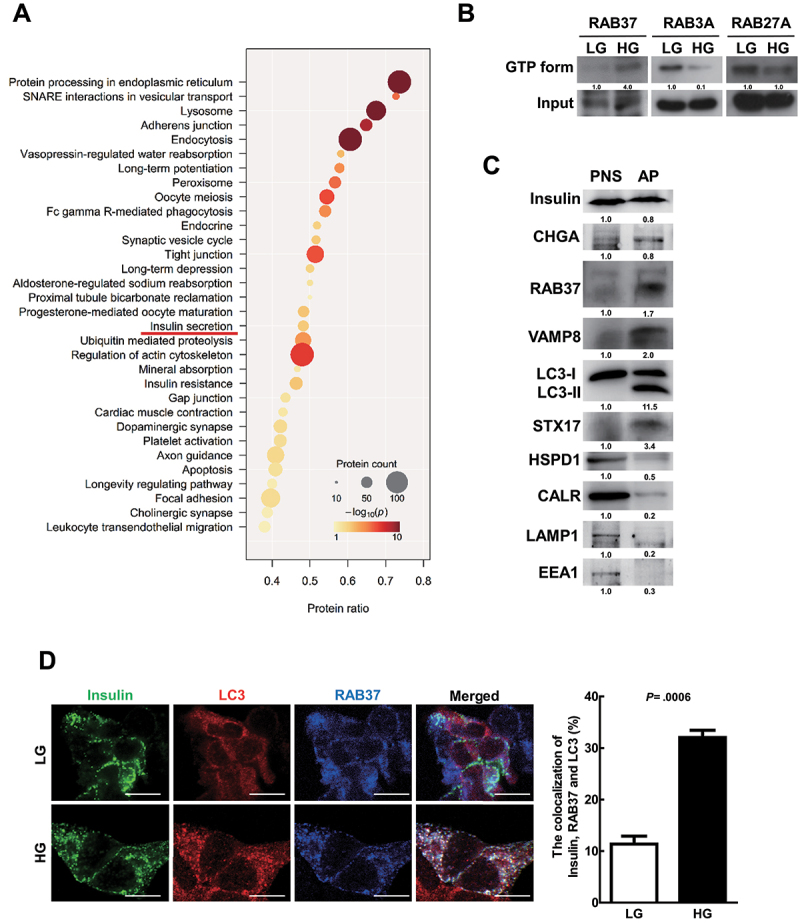
RAB37 was activated and colocalized with insulin and autophagosomes in MIN6 cells under HG treatment. (A) MIN6 cells were treated with HG (25 mM) for 1 h followed by purification of autophagosomes (AP) using density gradient centrifugation. The proteins in the purified autophagosome were identified by HPLC/MS/MS followed by the analysis of Gene Ontology Biological Processes (GOBP). Circular size is proportional to the fold enrichment and the color is proportional to significance. (B) MIN6 cells were treated with LG or HG for 1 h. The levels of active-forms of RAB3A, RAB27A, and RAB37 proteins were determined using extraction beads of active-form RAB proteins or total cellular input followed by immunoblotting. (C) the protein levels of various vesicle markers in the postnuclear supernatant (PNS) and purified autophagosomes (AP) were evaluated by immunoblotting using specific antibodies. (D) Colocalization of insulin (green) with LC3 (red) and RAB37 (blue) in MIN6 cells under LG or HG treatment was investigated under confocal microscopy, N = 120. Scale bars: 10 μm. Student’s t-test was used for statistical analysis.
Table 1.
Insulin secretion-related proteins identified in purified autophagosomes of MIN6 cells.
| Cargos |
| INS (insulin) IAPP (islet amyloid polypeptide) CHGA (chromogranin A) CHGB/secretogranin-1 (chromogranin B) SCG2/CHGC(secretogranin II)) SCG3 (secretogranin III) SCG5 (secretogranin V) IGF2 (insulin-like growth factor 2) MIF (macrophage migration inhibitory factor (glycosylation-inhibiting factor)) |
| Metabolic enzymes |
| PCSK1 (proprotein convertase subtilisin/kexin type 1) PCSK2 (proprotein convertase subtilisin/kexin type 2) CPE (carboxypeptidase E) |
| Transmembrane proteins |
| PTPRN (protein tyrosine phosphatase, receptor type, N) PTPRN2 (protein tyrosine phosphatase, receptor type, N polypeptide 2) SYT5 (synaptotagmin V) VAMP2 (vesicle-associated membrane protein 2) VAMP3 (vesicle-associated membrane protein 3) VAMP7 (vesicle-associated membrane protein 7) VAMP8 (vesicle-associated membrane protein 8) |
| RAB proteins |
| RAB3A (RAB3A, member RAS oncogene family) RAB27A (RAB27A, member RAS oncogene family) RAB37 (RAB37, member RAS oncogene family) |
| Ion channel proteins |
| CLCN3 (chloride channel, voltage-sensitive 3) SLC30A8/ZnT8 (solute carrier family 30 (zinc transporter), member 8) ATP6AP1 (ATPase, H+ transporting, lysosomal accessory protein 1) |
The autophagosomes were purified from mouse MIN6 cells after HG treatment for 1 h.
Many studies have reported that LC3 protein may anchor on diverse single and double-membrane vesicles designated as autophagic vesicles including phagosomes, macropinosomes, amphisomes, multiple vesicle bodies (MVB), and endosomes with degradative functions by fusing with lysosomes [31,40]. To confirm that the AP fraction represents autophagosomes, we investigated the markers of other vesicles including endosome (EEA1), lysosome (LAMP1), mitochondria (HSPD1/Hsp60), and endoplasmic reticulum (CALR) in the AP fraction of MIN6 cells after high glucose treatment. Our data showed very low levels of other vesicle markers except LC3-II in the AP fraction compared to the PNS fraction (Figure 3C; the quantification was shown as bar diagrams in Fig. S3B). Moreover, we detected five highly expressed proteins, including insulin, chromogranin A (major cargo in insulin secretory vesicles), RAB37, VAMP8, and STX17 in the AP fraction, which are possibly involved in insulin secretion (Figure 3C). Furthermore, the size of the purified insulin granule is about 200 nm in diameter under the transmission electron microscope (TEM) [41], but the size of the autophagosome is between 200 and 1,500 nm in diameter in mammals [42]. In our TEM data from the purified AP, the average size was about 681.9 ± 21 nm in diameter (mean ± SEM; n = 100; Fig. S3C). The dramatic difference in size rules out the possibility of insulin granules contamination in AP fraction. We further detected increased colocalization of insulin (green) with LC3 (red) and RAB37 (blue) proteins in MIN6 cells after HG treatment compared to LG-treated cells (Figure 3D, white color). In summary, HG treatment of mouse β-cells induces active-form RAB37 accompanied by autophagic activity, which promotes insulin secretion.
RAB37 is required for autophagy to promote insulin secretion under HG treatment
We established rab37 knockout stable clones (rab37-/-) derived from MIN6 cells using the CRISPR-Cas9 system (Fig. S4A). The protein levels of RAB37 were dramatically decreased in clone 3 (Figure 4A). Accordingly, the LC3-II levels were greatly decreased in clone 3 compared to the wild-type (WT) Rab37+/+ cells (Figure 4A; the quantification was shown as bar diagrams in Fig. S4B). We determined the insulin status in Rab37+/+ and rab37-/- cells to clarify whether RAB37 is required for insulin production. We analyzed the levels of insulin mRNA and protein in rab37-/- cells, and found that the status of insulin of rab37-/- cells were similar to that of Rab37+/+ cells (Fig. S4C and S4D). In Rab37+/+ cells, HG-induced insulin secretion was further boosted by niclosamide (the autophagy inducer). In contrast, in rab37-/- cells, insulin secretion was greatly decreased regardless of treatment with LG, HG, or HG plus the autophagy inducer (Figure 4B). In addition, in rab37-/- MIN6 cells, colocalization of insulin and LC3 was significantly decreased compared to the Rab37+/+ under confocal microscopy and TIRF microscopy (Figure 4c,d, yellow). The following plasmids of wild-type (WT), active-form (Q89 L) RAB37, and inactive-form (T43N) RAB37, as well as vector plasmid control were transiently transfected into MIN6- rab37-/- cells to clarify which forms of RAB37 protein affects insulin secretion (Figure 4E). Our data showed that in the cells harboring active-form RAB37 plasmid, there was far greater induction of insulin secretion compared with the other groups under HG conditions (Figure 4F). In summary, RAB37 was indispensable for the translocation of autophagic vesicles toward the plasma membrane, and active-form RAB37 plays an essential role in autophagy-mediated insulin secretion.
Figure 4.
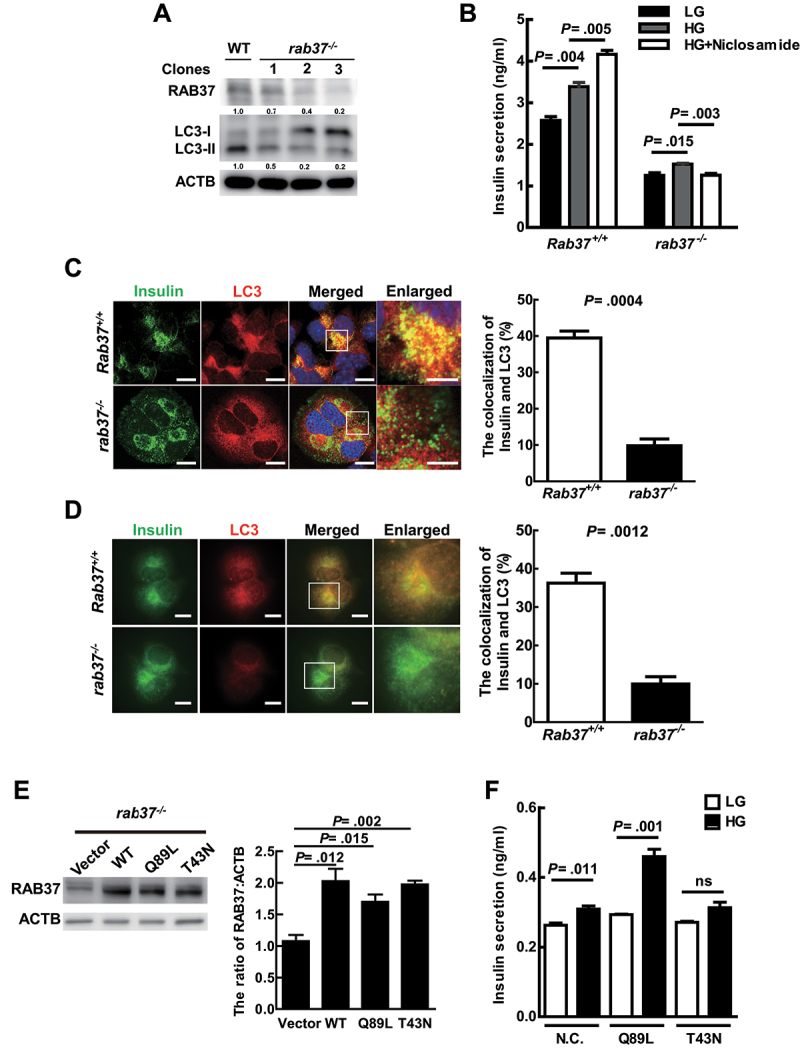
Active-Form RAB37 is required for autophagy to promote insulin secretion under HG treatment. (A) MIN6 rab37-/- cell lines (clone 1–3) were established using the CRISPR-Cas9 system. The protein levels of RAB37 and LC3 were determined by immunoblotting using specific antibodies. ACTB/β-actin was used as the internal control. WT: Wild-type MIN6 cells. (B) Rab37+/+ and rab37-/- MIN6 cells were treated with LG, HG, or HG+niclosamide (100 nM) for 1 h. The levels of secreted insulin in the cultured media were determined by ELISA. Data are shown as mean ± SEM of three independent experiments in triplicate. (C and D) Rab37+/+ and rab37-/- cells were treated with HG for 1 h. Colocalization of insulin (green) and LC3 (red) proteins in the cell were investigated under a confocal microscope (N = 120) in (C) and a TIRF microscope (N = 45) in (D). (E) Wild-type RAB37 (WT), active-form RAB37 (Q89 L), inactive-form RAB37 (T43N), or vector plasmid DNA was transiently transfected into the MIN6-rab37-/- cells. The expression level of RAB37 protein in these transfected cells was measured using anti-RAB37 antibody by immunoblotting. ACTB/β-actin was the internal control. (F) the transfected cells (Vector, Q89 L, and T43N) were treated with either LG or HG for 1 h, and secreted insulin in the media of these three cell lines was determined by ELISA. Data are shown as mean ± SEM of three independent experiments in triplicate. Scale bars: 10 μm in (C) and (D); Scale bars: 5 μm in enlarged images. Student’s t-test was used for statistical analysis.
RAB37 induces LC3 lipidation through interaction with LC3
Lipidated LC3 (LC3-II) covalently binds to phosphatidylethanolamine (PE) and attaches on the phagophore membrane [43]. Our data showed that the levels of exogenous RAB37 protein were associated with LC3-II and LC3-I to LC3-II conversion in a dose-dependent manner in MIN6 cells (Figure 5A). Accordingly, knockout of the endogenous RAB37 gene in MIN6 cells led to decreased LC3-II protein expression and LC3-I to LC3-II conversion (Figure 4A, lane 1 vs. lanes 2 to 4). These data imply that RAB37 may affect LC3 lipidation (LC3-II). To confirm this speculation, we added purified RAB37 protein and the required ingredients (including E1-like Atg7, E2-like Atg3, synthetic liposomes, and ATP) to reconstitute LC3 lipidation reaction in an in vitro system, following Nath et al.’s protocol [44]. Initially, we confirmed that the purified RAB37 protein had much higher GTPase activity in the presence of GTP compared to other treatments in the in vitro system (Figure 5B). We further detected increased LC3 lipidation (LC3B-PE) in the reaction mixture containing the purified RAB37 protein (RAB37) compared to the treatment without RAB37 (N.T.) (Figure 5C; the quantification was shown as the bar diagram in Fig. S5A). The RAB proteins anchored on the vesicle membrane are known to be responsible for vesicle trafficking [45]. To clarify whether the RAB37 anchored vesicle is involved in autophagosome formation, we constructed RAB37-bound liposomes at various concentrations using a headgroup-modified lipid chelator DOGS-NTA 1,2-dioleoyl-sn-glycero-3-(N-[5-amino-1-carboxypentyl] iminodiacetic acid)-succinyl, which was membrane-anchored by the C-terminal polyhistidine-tag of RAB37 (RAB37-His12) [46] (Fig. S5B and S5C, PE-NTA vs. PE). Using this system, we confirmed that the RAB37-liposome promoted LC3 lipidation (LC3B – PE) in a dose-dependent manner (Figure 5D the quantification was shown as the bar diagram in Fig. S5D). This finding implies that RAB37-anchored vesicle promotes autophagosome formation through increased LC3 lipidation. We also demonstrated that the interaction between RAB37 and LC3 was slightly increased in MIN6 cells after HG stimulation compared to LG treatment by immunoprecipitation analysis (Figure 5E; the quantification was shown as bar diagrams in Fig. S5E). RAB37 protein contains three LC3-interacting region (LIR) motifs. To determine which motif is essential for the interaction of RAB37 and LC3 protein, we generated three LIR motif mutants RAB37-mLIR1 (FRSV, 92 95), RAB37-mLIR2 (FDNI, 118 121), and RAB37-mLIR3 (FLAI, 184 187) (Figure 5F; the quantification was shown as bar diagrams in Fig. S5F). Compared to wild-type RAB37 (WT), the interaction between LC3 and RAB37 was notably decreased when the LIR1 or LIR3 motif was mutated by immunoprecipitation analysis (Figure 5F, WT vs. M1 or M3). In summary, active-form RAB37 increased LC3 lipidation to form LC3-II and triggered autophagosome formation possibly through interaction with LC3 protein via LIR motifs.
Figure 5.
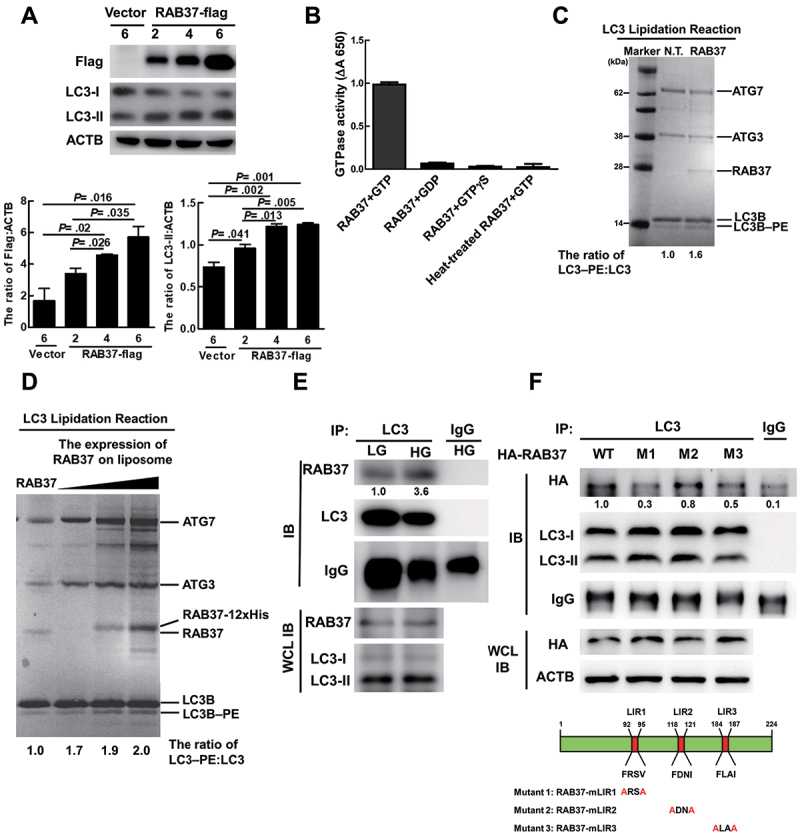
RAB37 induces LC3 lipidation through interaction with LC3. (A) MIN6 cells were transfected with Vector or different amounts (μg) of pLp’s-RAB37-Flag plasmid DNA. The expression levels of Flag and LC3 were determined by immunoblotting analysis using specific antibodies. ACTB/β-actin was used as the internal control. (B) the recombinant or heat-treated RAB37 protein (100°C for 15 min, 4 μM) was incubated with GTP (1 mM), GDP (1 mM), or GTPγS (analog of GTP, 1 mM) at 30°C for 2 h. The solution was mixed with BIOMOL GREEN™, and then incubated at 30°C for 30 min. The produced phosphate was detected using a spectrophotometric multi-well plate reader. (C) LC3 lipidation reaction. The reaction mixture containing 2 μM ATG7, 2 μM ATG3, 10 μM LC3B, 2 mM lipid (400-nm liposomes containing 10 mol% bl-PI, 30 mol% DOPE, 58 mol% POPC, 1 mol% Rhod, and 1 mol% NBD), 1 mm dithiothreitol, 1 mM ATP with or without 4 μM RAB37 protein, and 1 mM GTP was incubated at 37°C for 90 min followed by SDS PAGE analysis of the mixture solution. NT: The reaction solution without RAB37 protein. (D) the PE-NTA lipidation reaction mixture (400 nm liposomes containing POPE:POPC:Rhod:DOGS-NTA = molar ratio 20:73:1:6) plus 4 μM RAB37 protein (RAB37) and various concentrations of RAB37-12xhis-bound liposomes (20, 100, 300 μM RAB37-12xhis with 1.5 mM liposome plus 1 mm GTP) were incubated at 37°C for 90 min. (C and D) the reaction mixtures were analyzed in 12% Bis-Tris PAGE, and total protein levels were determined by Coomassie Brilliant Blue staining. (E) MIN6 cells were treated with LG or HG for 1 h and total protein extraction was immunoprecipitated by anti-LC3 antibody. (F) Human embryonic kidney 293T cells were transfected with plasmid DNA (24 μg) of pCMV-HA-RAB37-WT, pCMV-HA-RAB37-mLIR1, pCMV-HA-RAB37-mLIR2, or pCMV-HA-RAB37-mLIR3, and the total protein extraction was immunoprecipitated by anti-LC3 antibody. The expression levels of HA in the precipitates and whole-cell lysates were determined using anti-HA antibody by immunoblotting. Schematic diagrams of RAB37 mutants in the LIR motif are shown.
Autophagy-Mediated insulin secretion requires RAB37 in mice
Ljubicic et al. reported that RAB37 participates in insulin secretion of pancreatic β-cells in vitro [47]. However, the mechanism remains unclear. Herein, we used a RAB37 knockout rab37-/- mouse model to clarify whether autophagy-mediated insulin secretion is RAB37-dependent. There were no significant differences between rab37-/- mice and their wild-type littermates in body weight, body temperature, longevity, or reproductive rate under general housing conditions. Our data showed that insulin expression level in islets of Langerhans in the pancreas was no different between Rab37+/+ and rab37-/- mice (Fig. S4E). However, significant low insulin secretion accompanied with high blood glucose was detected in rab37-/- mice compared to Rab37+/+ mice after IPGTT treatment for 30 and 60 min (Figure 6A, white column), indicating that RAB37 affects both of insulin secretion and glucose consumption in the blood of the mice. To clarify whether RAB37 is required for autophagy-mediated insulin secretion in mice, both Rab37+/+ and rab37-/- mice were fed a chow diet (CD, normal diet) in the presence or absence of the autophagy inducer niclosamide (1500 ppm) for two weeks. In the islets of Langerhans of the Rab37+/+ mice, niclosamide treatment greatly increased the coexistence of insulin (green) and LC3 (red) (Fig. S6A, yellow), and many autophagosome-like vesicles engulfing or fusing with insulin granules were detected using a TEM (Fig. S6B). We also detected increased coexistence of insulin (green) and LC3 (red) in the pancreatic β-cells of Rab37+/+ mice, but not in rab37-/- mice fed with CD (Figure 6B, yellow). Furthermore, insulin secretion was significantly increased accompanied with a dramatic decrease of blood glucose in Rab37+/+ mice after IPGTT treatment in the presence of niclosamide for 60 min (Figure 6C). However, this phenomenon was not seen in rab37-/- mice (Figure 6D). In addition, we isolate the islets from Rab37+/+ and rab37-/- mice. The isolated islets were incubated with low glucose (LG, 2.8 mM), high glucose (HG, 16.7 mM) or HG with the autophagy inducer rapamycin, niclosamide or the inhibitor 3-MA for 1 h. Our results demonstrated that HG treatment increased insulin secretion in islets of both Rab37+/+ and rab37-/- mice. But the level of HG-induced insulin secretion was slightly impaired in islets of rab37-/- mice (Fig. S4F and S4 G; bar 1 vs. bar 2). Notably, in islets of Rab37+/+ mice, the trend of insulin secretion was further boosted by two autophagy inducers under HG conditions. Accordingly, HG-induced insulin secretion was suppressed by 3-MA. However, autophagy-related insulin secretion by autophagy regulators was not observed in islets of rab37-/- mice. In conclusion, our in vivo and ex vivo studies were consistent with the results of Figure 4B, showing that autophagy has no effect on insulin secretion in rab37-/- MIN6 cells. Altogether, the aforementioned findings demonstrate that autophagy-mediated insulin secretion requires the RAB37 pathway both in vitro and in vivo (Figure 7).
Figure 6.
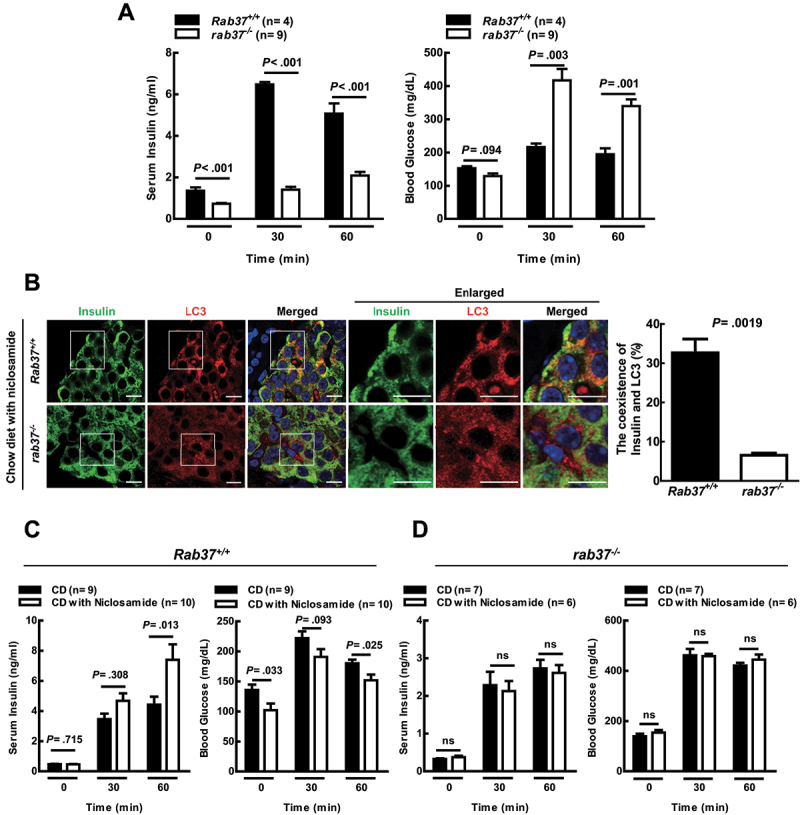
Promotion of insulin secretion by autophagy requires RAB37 in mice. (A) After IPGTT, the concentrations of insulin and glucose in the blood of Rab37+/+ and rab37-/- male mice were evaluated by ELISA. (B) the coexistence of insulin (green) and LC3 (red) in the tissue sections of islets of Langerhans from Rab37+/+ and rab37-/- mice after feeding with chow diet (CD) together with niclosamide (1500 p.p.m.) for 14 days under a confocal microscope, N = 200. (C) Rab37+/+ and (D) rab37-/- mice were fed with CD in the presence or absence of niclosamide for 14 days followed by IPGTT. The concentrations of insulin and glucose in the blood of these mice were measured at the times as indicated. Data were presented as mean ± SEM. N = 4 9 mice per group. Student’s t-test was used for statistical analysis. ns: no significance. Scale bars: 10 μm.
Figure 7.
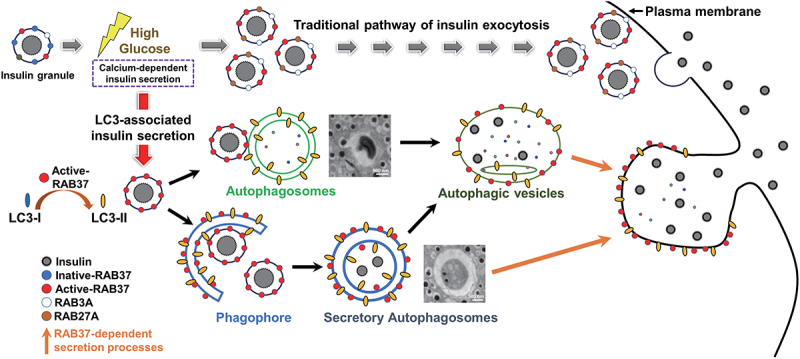
A schematic hypothesis of RAB37-related autophagy promotes insulin secretion in β-cell under high glucose conditions. RAB3A, RAB27A, and RAB37 participate in exocytosis of the traditional insulin secretion under high glucose treatment. Furthermore, RAB37 is also involved in the unconventional insulin secretion through secretory autophagy. High glucose activates RAB37 accompanied by increased autophagic activity in β-cells. We proposed two pathways that autophagy participates in insulin secretion. First, activated-RAB37 interacts with LC3 to promote lipidation of LC3 (LC3-II) followed by recruitment of insulin granule by phagophore expansion to form the mature autophagosome. Second, active RAB37-anchored insulin granules move to autophagosomes followed by fusion. These two pathways provide the possible explanation how RAB37 is anchored on the autophagosome and how insulin is recruited into the autophagosome. Moreover, the trafficking of LC3-anchored insulin vesicles was through a RAB37-dependent secretion process under GSIS. In summary, in addition to the traditional insulin secretion pathway, activated-RAB37, together with increased autophagy, increases insulin exocytosis to maintain glucose homeostasis in the blood.
Discussion
Autophagic activity is induced to modulate unbalanced metabolic stresses, including deprivation of nutrients or starvation both in vitro and in vivo. In contrast, autophagy has been shown to be suppressed in β-cells under starvation conditions through a protein kinase D-dependent regulation pathway [48]. Increasing evidence indicates the involvement of autophagic molecular machinery in cellular secretion, which has been confirmed by either induction or inhibition of autophagy mediators followed by dramatic alterations in the cellular secretory profile [7]. Secretory autophagy affects the secretion of a plethora of factors ranging from cytokines to granule contents, and viral particles [49]. Encompassing a wide range of secretory factors, autophagy-dependent secretion is implicated in diseases ranging from cancer to neurodegeneration [50].
Under long-term HG incubation, autophagy is induced through modulation of the 5’-AMP-activated protein kinase axis and plays a protective role in promoting the survival of pancreatic β-cells [51,52]. In contrast, under short-term HG conditions, β‐cells sense extracellular glucose and secrete insulin in a biphasic manner. The first phase culminates 5–6 min after stimulation, and a gradual increase in insulin release over 60 min ensues, which is called the second phase [53,54]. In this study, we reveal that HG stimulation induces autophagic activity within 1–2 h and promotes insulin secretion both in vitro and in vivo (Figure 1), indicating that secretory autophagy-mediated insulin secretion participates in the second phase of insulin secretion. We further confirmed that HG stimulation also activates RAB37 and participates in insulin secretion. In summary, autophagy plays a secretory function for insulin release from β cells in a RAB37-dependent fashion.
Because the precise mechanism of interaction between insulin granules and LC3-anchored vesicles remains unclear, we investigated two potential RAB37-autophagy-related insulin secretion pathways under HG conditions based on our observation under TEM (Figure 7). The results revealed the following: (a) insulin granules fused with autophagosomes (Fig. S6Bi), and (b) insulin granules were engulfed by the phagophore and proceeded to autophagosome formation (Fig. S6Bii). However, the following pathways related to the autophagy-mediated insulin secretion can not be excluded. 1: In TMED10-channeled unconventional protein secretion, a set of secretory cargoes containing a common motif to bind with the C-terminal tail of TMED10 enter into vesicles, which are precursors of phagophores. They further expand to form autophagosomes for unconventional secretion [55,56]. Insulin is stored in large dense-core vesicles (LDCV) awaiting release on demand [57], indicating that it is present within secretory vesicles (insulin granules). These insulin granules may participate in autophagosomal membrane generation by fusion with the autophagosome under HG stimulation. 2: LC3 decoration occurs on single-membrane vesicles, which are bound for the unconventional secretion of viruses and extracellular vesicle cargoes [31,58]. Increasing evidence supports the role of LC3 protein in viral exocytosis, and several viruses acquire LC3-conjugated membranes for their exocytosis [59,60]. Leidal et al. also reported that LC3-anchored multivesicular bodies (MVB) are responsible for the trafficking of RNA binding proteins and incorporate small non-coding RNAs toward extracellular vesicles (EVs) for secretion in an autophagosome-independent manner [61]. Our detection of a high level of LC3-II in the fraction of mISGs (Figure 2B) implies that active-form RAB37 may increase LC3 lipidation on the insulin granules under HG treatment. Taken together, the evidence shows RAB37-anchored insulin granules proceed to LC3-related vesicles for secretion through either phagophore elongation or LC3-II directly decorating insulin granules. Further research is warranted to more precisely elucidate the underlying mechanisms.
Dupont et al. reported that during IL1B/IL-1β secretion, autophagosomes containing IL1B fuse with the lysosome, and lysosomal hydrolase CTSB (cathepsin B) promotes IL1B secretion driven by the autophagosome [62]. We demonstrated that HG increases autophagic vesicles (Fig. S2D), which are transported toward the plasma membrane for secretion without fusing with the lysosome in β cells (Fig. S2E). The STX17 protein, localized on the outer membrane of the mature autophagosome, interacts with VAMP8 (the effector of RAB family members) on the outer membrane of the lysosome through an adaptor protein SNAP29 followed by fusion with autophagosomes to form autolysosomes [63]. It has been reported that VAMP8 on the membrane of insulin granules is responsible for insulin exocytosis [64]. It is probable that VAMP8 on the membrane of insulin granules and lysosome competes to bind to STX17 on the autophagosome membrane, and once the insulin granule fuses with the autophagosome, it is bound for secretion.
RAB family proteins are essential for vesicle formation and trafficking at different stages [65]. RAB2A localized on perinuclear immature granules regulates the early stage of insulin granule biogenesis [66]. RAB37 is mainly located on insulin secretory granules and responds to docking and priming for insulin secretion [25]. The levels of several RAB proteins including pancreatic RAB3A, RAB27, and RAB37 are decreased under exposure to physiopathological conditions that favor the development of diabetes [47,67]. These reports indicate that RAB proteins are critical in maintaining glucose homeostasis acting in the pancreas and involved in diabetes mellitus development. We demonstrated that RAB37 expression did not affect insulin levels both in vitro and in vivo (Figure 4C and Fig. S4). These findings imply that RAB37 modulates insulin expression through the regulation of the secretion process. Despite active-form RAB37 increased lipidation of LC3, LC3 puncta still can be detected in both rab37-/- cells and rab37-/- mice under the stimulation of either HG (Figure 4C) or niclosamide (Figure 6B), implying that RAB37 is not essential for autophagosome formation. Moreover, the number of LC3 puncta proximal to the plasma membrane was greatly decreased in rab37-/- cells compared to Rab37+/+ cells under HG treatment by TIRF (Figure 4D), suggesting that RAB37 is required for LC3-anchored vesicles moving toward the plasma membrane. Taken together, active-form RAB37 not only increases autophagosome formation through LC3 lipidation, but also plays a pivotal role in facilitating autophagic vesicles-mediated insulin secretion. RAB GTPase protein family are key regulators responsible for intracellular trafficking of vesicles, including exosomes, MVBs, amphisomes and lysosomes. Various RAB proteins are responsible for regulation of autophagy progression such as phagophore formation (RAB33), autophagosome formation (RAB30), and autophagosome-lysosome fusion (RAB7) [68]. Moreover, RAB8A and RAB8B are involved in secretory and degradative autophagy, respectively. RAB8A is required for autophagy-mediated IL1B secretion pathway under starvation and inflammasome activation. In contrast, RAB8B is responsible for autophagosome maturation that fuses with lysosomes in degradative autophagy [30]. In our study, we reveal that high glucose-induced active-form RAB37 is responsible for insulin release through secretory autophagy. Similar to our findings, Schroeder et al. reported that starvation-induced active-form RAB7 is required for lysosomes and multi-vesicular fusion and migration to the surface of lipid droplets during lipophagic progression [69]. Above findings imply that different stimulus-induced autophagy may activate different RAB family proteins to execute either the degradative or secretory autophagy machinery for specific functions. Altogether, our novel findings warrant further exploration of the role of pancreatic RAB37-mediated secretory autophagy in obesity/diabetes and the possible involvements of other exocytosis-related RAB proteins, including RAB3A, RAB27A, and RAB8A in insulin secretion through secretory autophagy.
Diabetes mellitus (DM) refers to multiple metabolic diseases that are characterized by a high level of blood sugar (hyperglycemia) over a prolonged period. About 90% of DM patients have type-II DM, and typical symptoms include overweight and insulin resistance. Recently, Zhang et al. showed that obesity causes impaired insulin transcription and secretion by upregulating MIR802, leading to hyperglycemia [70]. Prolonged hyperglycemia in type-II DM patients leads to damage of multiple organs, including the heart and blood vessels, as well as the eyes, kidneys, and nervous system [71]. Therefore, the conventional strategies to treat type-II DM include stimulating β-cells to increase insulin secretion (sulfonylureas and meglitinides) or improving the cellular sensitivity to insulin (metformin and thiazolidinediones) [72]. It may be advantageous to clarify whether regulating autophagic activity can ameliorate the pathophysiological symptoms of hyperglycemia by increasing insulin secretion and decreasing glucose level in the blood. In this study, both the expression and coexistence of insulin and LC3 proteins in pancreatic β-cells increased in HFD-fed mice, as well as ob/ob obese mice, compared to CD-treated mice under a confocal microscope (Fig. S7A). The HFD-fed mice were further treated with niclosamide to induce autophagic activity, and our data show that the coexistence of insulin and LC3 was increased in the niclosamide treatment group (HFD+niclosamide) compared to the non-treatment (HFD) group under HFD conditions (Fig. S7B). Furthermore, a significant increase in insulin secretion associated with a decrease of blood glucose level was detected at 60 min after IPGTT treatment in the niclosamide-treated mice (Fig. S7C). It has been reported that in the HFD-induced obese mouse model, compensatory hyperplasia of β-cell in the tissue mass of the islet was induced, resulting in increased insulin secretion [73]. We also did a morphometric analysis of the islets of Langerhans and found no difference in islet cell mass (Fig. S7D) and β-cell proliferation (Fig. S7E) compared to mice with or without niclosamide treatment. This indicates that autophagy did not enhance the compensatory hyperplasia of β-cells. Our results confirmed that induction of autophagy could improve the pathophysiological symptom of hyperglycemia in the HFD-induced obese mouse model by promoting insulin secretion in a RAB37-dependent manner. However, both dysregulation of insulin secretion and insulin resistance are characteristic features in the HFD-induced obese mouse model [70,74]. Therefore, whether inducing autophagic activity to increase insulin secretion could remedy the disease symptoms in HFD-mice warrants further exploration. Nevertheless, our findings that secretory autophagic machinery promotes insulin release via an active-RAB37-dependent process may open a new avenue for DM therapy.
Niclosamide is a traditional FDA-approved anthelmintic drug used for treating intestinal tapeworm infection. Niclosamide is also a mitochondrial uncoupler that improves blood glycemic control and reduces hepatic steatosis in mice by increasing energy expenditure and lipid metabolism [75]. Several safe chemical uncouplers have been used as potential antidiabetic agents [76,77]. Niclosamide is also an autophagy inducer that inhibits the MTORC1 signaling pathway [37]. To support our finding that induction of autophagy indeed promotes insulin secretion, we also used the well-known autophagy inducer, rapamycin. Similarly, Liu et al. reported that rapamycin promotes insulin secretion in isolated islets from obese mice [18]. Moreover, Tat-BECN1 (a small peptide autophagy inducer) was also shown to increase insulin secretion in ex vivo fasted human islets [48]. Taken together, the evidence indicates that increased autophagy promotes insulin secretion.
In conclusion, we reveal herein that secretory autophagy promotes insulin secretion in a RAB37-dependent manner under high glucose conditions both in vitro and in vivo (Figure 7). We further showed that niclosamide, an off-label agent (also an autophagy inducer), may have the potential to improve glucose homeostasis in DM patients with hyperglycemia or hypoinsulinemia.
Materials and methods
Cell lines and reagents
The MIN6 insulinoma cell line was a kind gift from Dr. Jun-ichi Miyazaki (Osaka University, Osaka, Japan) [78]. MIN6 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, 12,100,061). Cells were cultured with 450 mg/dl glucose, 10% fetal bovine serum (Biological Industries, 04-0001-1A), penicillin-streptomycin (Sigma, P4333), and 50 µM β-mercaptoethanol (Sigma, M6250) at 37° in a 5% CO2 incubator. Rapamycin (Sigma R0395), niclosamide (Sigma, N3510), spautin-1, 3-MA (Sigma, M9281), BAPTA-AM (Sigma, A1076), and chloroquine (Sigma, C6628) were purchased from Sigma.
Insulin detection
Insulin was measured according to a previously described procedure [79]. Basal DMEM was serum-free with no glucose. Low (5 mM; Gibco, 11,885,084) or high (25 mM; Gibco, 12,100,061) concentration of glucose was added into the medium. MIN6 cells were seeded into the 12-well plates with LG medium for 2 h. The medium was replaced with LG or HG media for 1 h. The supernatant medium was collected for measurement of insulin concentration by Mercodia Mouse Insulin ELISA kit (Mercodia AB, 10-1247-01). The insulin secretion capacity was determined after normalization with total protein.
Western blot analysis
Total protein was collected from the cells after treatment. Immunoblotting was performed according to a previously described procedure [80]. The following antibodies were used: monoclonal antibodies for ACTB/β-actin (Sigma, A5441), LC3 (Medical and Biological Laboratories, PM036), SQSTM1/p62 (Medical and Biological Laboratories, PM045), ATG5 (Abcam, ab228668), ATG7 (Abcam, ab133528), insulin (Proteintech, 15,848-1-AP), RAB37 (MyBioSouce, MBS9133732), STX17 (GeneTex, GTX13012), VAMP8 (GeneTex, GTX132181), STX 6 (GeneTex, GTX115375), CHGA/chromogranin A (GeneTex, GTX113165), HSPD1/Hsp-60 (GeneTex, FTX110089), CALR/calreticulin (GeneTex, GTX111627), and Na+/K+ ATPase (GeneTex, GTX113390).
Autophagosome extraction
The MIN6 cells were suspended in 0.4 ml 10% sucrose (Sigma, S0389) and mixed with 0.5 ml 1 M HEPES, pH 7.3, 0.1 M EDTA and homogenized using a Dounce homogenizer (Merck, DWK885300–0001). This homogenate was diluted with a homogenization buffer (HB; 0.25 M sucrose, 10 mM HEPES, 1 mM EDTA, pH 7.3) containing 1.5 mM glycyl-l-phenylalanine 2-naphthylamide (Sigma, D9628) and 1% DMSO. After incubation for 7 min at 37°C to destroy the lysosomes, the homogenate was cooled to 4°C. The extraction was performed as previously reported [81].
Isolation of iIsgs and mIsgs
The iISGs and mISGs were purified following the previous protocol with minor modifications [33,82]: Briefly, MIN6 cells were treated with HG for 1 h, and then homogenized in homogenization buffer [0.3 mol/L sucrose (Sigma, SLCH3216), 1 mmol/L EDTA (Merck, 654,833), 1 mmol/L MgSO4 (Macron avantor, 6066–04), 10 mmol/L MES-KOH (Sigma, M8250), 1 mmol/L PMSF (Thermo Scientific, 36,978), pH 6.5]. The nuclear debris was removed by centrifugation at 1000 g for 5 min. The supernatant was loaded on top of a discontinuous Optiprep (Sigma, D15561) gradient, prepared by 5 layers of 30%, 23.4%, 17.6%, 13.2%, and 8.8% of Optiprep. The Optiprep gradient was centrifuged for 75 min at 100,000 g in an SW41 rotor (Beckman), and the gradients were divided into twelve fractions. Both fraction 6 (interface between 13.2% and 17.6%) and fraction 8 (interface between 17.6% and 23.4%) containing insulin were iISGs and mISGs, respectively.
In solution digestion and LC-MS/MS analyses
The protein was extracted from the autophagosomes of MIN6 cells. Protein extracts were denatured and then alkylated in dithiothreitol (7 mM) and iodoacetamine (21 mM), respectively, at 37°C for 30 min. Proteins were digested by trypsin (Sigma, T4049) at 37°C for 16 h. The peptide mixtures were separated on a 3 × 150 mm C18 column (Gemini, Phenomenex, 00F-4435-E0) coupled to a high-performance liquid chromatography system (Beckman Coulter, CA, USA) using an acetonitrile gradient in 0.1% ammonium hydroxide solution, with approximately 30 fractions. The peptide fractions were analyzed on a nanoLC-Q ExactiveTM HF mass spectrometer (Thermo Fisher, San Jose, USA) equipped with an HPLC system (M Class, Waters, MA, USA). MS raw files were uploaded into Proteome Discoverer (version 2.1, Thermo Fisher, MA, USA) with the default setting to generate peak lists for protein identification using the MASCOT search engine (version 2.5, Matrix Science, MA, USA) against the Swiss-Prot Mus musculus protein database (released in Jan, 2016). The peptides sharing an identical sequence among multiple proteins were assigned to the one with the highest protein score. The identification of peptides and proteins with a false discovery rate of less than 1% was considered acceptable.
Transmission electron microscope (TEM)
MIN6 cells and pancreas tissues were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4 containing 4% sucrose, 1 mM MgCl2, and 1 mM CaCl2, and post-fixed in 1% osmium tetroxide. Tissues and cells were embedded with LR White (Agar Scientific, AGR1281) after dehydration with ethanol. Ultrathin sections were stained with saturated uranyl acetate and lead citrate at room temperature (RT) for 1 h and investigated under TEM (HITACHI-7000, Tokyo, Japan) [83].
Fluorescent immunohistochemistry (FIHC) staining
FIHC staining of paraffin section slides was performed as previously described [84]. Anti-RAB37 (Proteintech, 13,051-AP), anti-insulin (Dako, A0564), and anti-LC3 (Medical and Biological Laboratories, M153–3) antibodies were added to the slide and incubated for 1 h at RT. After washing, cells were dyed with secondary antibodies with Alexa Fluor 408, 488, and 568 (Invitrogen, A48254, SA5– 10,094, and A11004) and incubated for 1 h with/without Hoechst staining for 30 min. Fluorescent changes of the cells were investigated either under a fluorescent microscope (DP 70; Olympus, Tokyo, Japan) or a confocal microscope (FV-1000; Olympus). Representative images are shown in Figures, and colocalization was determined by counting 200–250 cells from four independent mice.
Plasma membrane protein extraction
A plasma membrane protein extraction kit (Abcam, ab65400) was used to purify the cytosol and plasma membrane fraction from MIN6 cells after treatment of LG or HG. Briefly, we separated the cell membrane fraction from the total membrane component. The cytosol and plasma membrane fractions were investigated for the markers and LC3 by immunoblotting using specific antibodies. Sodium-potassium ATPase was the marker for cell membrane, and CALR was the marker for cytosol.
Cell transfection
The cells (2 × 105/well) were seeded into a six-well plate were transfected with pLP’s-Flag vector (Clontech, 11,703), pLP’s-RAB37-WT-Flag, pLP’s-RAB37Q89 L-Flag, pLP’s-RAB37T43N-Flag, pCMV-HA-RAB37, pCMV-HA-RAB37[FAVA] or pCMV-HA-RAB37[FAIA] or ptfLC3 (4 μg) by Lipofectamine 2000™ following the manufacturer’s instructions (Invitrogen, 11,668–019).
IFA and TIRF imaging
MIN6-related cells were seeded on a 29-mm glass-bottom dish. After 24 h, cells were pretreated with LG medium for 2 h. The medium was replaced with LG or HG media for 1 h. Furthermore, we investigated the localization of endogenous LC3, insulin, and RAB37 using immunofluorescence staining followed by confocal (FV-1000; Olympus) or TIRF microscopy investigation. The TIRF system was built on an inverted microscope (Olympus I×81, Japan). TIRF microscopy delivers images with an outstandingly high axial resolution below 100 nm. This allows the observation of membrane-associated processes. Representative images are shown in Figures and colocalization was quantified by counting 100–120 cells from three independent experiments.
Small hairpin RNA (shRNA) lentiviral infection system
MIN6 cells (1 × 106/well) were seeded into six-well plates. After 24 h, cells were infected with lentivirus sh-GFP (control), sh-Atg5, or sh-Atg7 and incubated at 37° in 5% CO2 overnight. Cells were selected by puromycin (2 μg/ml) for 2 weeks. The lentiviral shRNA Atg5 target sequence was AGCCGAAGCCTTTGCTCAATG and the shRNA Atg7 target sequence was GCCAACATCCCTGGATACAAG. All lentivirus shRNA were purchased from National RNAi Core Facility, Academia Sinica, Taiwan.
CRISPR-Cas9 system
MIN6 cells were seeded onto 10-cm culture dishes. After 24 h, cells were transfected with expression vectors for Rab37 guide RNA (19 μg) (U6-gRNA-Rab37), Cas9 gene (4 μg) (CMV-p-Cas9), and pCMV-BSD (1 μg). Cells were selected by blasticidin (1 μg/ml) for 2 weeks. The monoclonal colonies were identified for mutant/KO by genotyping and immunoblotting. The guide RNA for targeting Rab37 was sg-RNA-Rab37: CGTTGGGAAGACATCTCTACTGG. U6-gRNA-Rab37 and CMV-p-Cas9 were purchased from ToolGen (ATC-0050).
(Proteo-)Liposome preparation
The following lipids were used: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC; Avanti Polar Lipids, 850457C), 1,2-dioleoyl-sn-glycero-3-phosphoethanol-amine (DOPE; Avanti Polar Lipids, 850725C), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE; Avanti Polar Lipids, 850757P), 1,2-dioleoyl-sn-glycero-3-([N-{5-amino-1-carboxypentyl}iminodiacetic acid]succinyl) (DOGS-NTA; Avanti Polar Lipids, 790,404), and L-phosphatidylinositol from bovine liver (bl-PI; Sigma, P8443). Furthermore, 1 2 mol% N-(lissamina rodamina B sulfonil)-1,2-dioleoil-sn-3-fosfatidiletanolamina (Rhod-DOPE) or N-(7nitrobenz-2-oxa-1,3-diazol-4-il)-1,2-dioleoil-sn-glicero-3-fosfoetanolamina (NBD-DOPE) (Avanti Polar Lipids, 810,158 and 810141P) were used as lipid dyes. The lipid ratios were bl-PI:DOPE:POPC:Rhod:NBD (molar ratio 10:30:58:1:1), POPE:POPC:Rhod:DOGS-NTA (molar ratio 20:73:1:6), or POPE:POPC:Rhod:NBD (molar ratio 20:78:1:1), and then the solution was solubilized in chloroform and dried to form a lipid film by N2 and vacuum in glass tubes. The dried lipid film was resuspended in SNARE buffer (20 mM HEPES, 100 mM KCl, pH 7.4). After five freeze – thaw cycles, unilamellar proteoliposomes were extruded through polycarbonate filters (50 nm pore size, Structure Probe, F0058-MB) at least 21 times.
RAB37 GTPase activity assay
GTP-hydrolysis activity of recombinant RAB37 was determined by the Malachite Green-based reagent Biomol Green (Enzo Life Sciences, BML-AK111). Purified RAB37 protein (4 μM) or heat-treated RAB37 (100°C for 15 min, 4 μM) were incubated at 30°C for 2 h in buffer containing HEPES (20 mM), pH 7.2, KCl (100 mM), and GTP (1 mM; Sigma, G8877), GDP (1 mM; Sigma, G7127), or GTPγS (1 mM; Sigma, 20–176) where indicated. The mixtures were mixed with Biomol Green reagent (Enzo Life Sciences, BML-AK111) and then incubated at 30°C for 30 min. The absorbance of samples at 650 nm were detected using a SmartSpecTM Plus spectrophotometer (BIO-RAD). Means and standard deviations of the corrected values (DA650: subtracting the absorbance value of the control reaction assayed in the absence of RAB37) were determined from three independent experiments [46].
Plasmid construction and recombinant protein preparation
For recombinant protein manipulation, HsRAB37-WT was cloned into the pGEX-4T1 vector (Sigma, GE28-9545-49) by PCR amplification, and restriction enzymes, SalI and NotI (New England Biolabs Inc., R3138 and R3189), were used for digestion and ligation, respectively. pCMV-HA-RAB37[FAVA] and -RAB37[FAIA] were established by performing mutagenesis twice using QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, 200,523). For establishing RAB37 with a 12xHis tag with C terminal, pET-28a (Sigma, 69,864) was used to construct pET-28a-12His. The pET-28a contains the C-terminal 6xHis tag for protein reconstitution, and we attempted to create another 6xHis prior to using this tag. Another 4xHis sequence with two restriction enzyme cutting sequences, SacI and XhoI, were designed with sense and antisense primers for annealing. This annealing DNA was constructed such that pET-28a was transformed into pET-28a-4His-XhoI-6His. pET-28a-4His-XhoI-6His was created using another 2xHis through mutagenesis on XhoI sequences, yielding pET-28a-12His. The hRAB37-WT was cloned into pET-28a-12His vector by PCR amplification and restriction enzymes, NdeI and SacI (New England Biolabs Inc) for digestion and ligation. These plasmids were transformed into the competent cells, BL-21 (Yeastern Biotech Co. Ltd, Taipei, Taiwan), and were cultured in LB medium with Isopropyl β-D-1-thiogalactopyranoside (IPTG) 0.2 mM at 37°C for 3 h to induce protein expression. Bacteria cultures were centrifuged at 1,700 x g. at 4°C for 15 min, and then 50 ml cold SNH buffer (50 mM Tris, pH 8, 100 mM NaCl, 1 mM MgCl2) with 1:100 bacteria protease inhibitor (G-bioscences, 786–330) and DTT (1 mM) were added and mixed by vortex. Bacteria lysates were exploded by sonication and centrifuged at 15,300 x g for 30 min. The supernatant was added to a column containing glutathione beads (Millipore, G4510) and washed with PBS (Sigma, D8537) three times. Finally, recombinant proteins were eluted by thrombinase (Sigma, GE27-0846-01) or 500 mM imidazole buffer in Tris-HCl, pH 8.0. Protein quality and purification were analyzed by Coomassie Brilliant Blue staining and WB examination.
Flotation assay
Nycodenz liposome flotation assay was performed following a previously described procedure [85]. Briefly, lipidation reaction products were prepared with 40% Nycodenz (ProteinGenix, 1,002,424) by mixing with 100% (w:v) Nycodenz in SNH buffer (50 mM Tris, pH 8, 100 mM NaCl, 1 mM MgCl2). Reaction mixture (1.5 mM liposome, 1 mM DTT, and 20, 100, or 300 μM RAB37-12xHis) was incubated at 30°C for 120 min. In ultraclear ultracentrifuge tubes (Beckmann, 343,778), Nycodenz gradients were assembled with 300 μl of reaction mixture (40% Nycodenz) at the bottom, followed by a 250 μl middle layer (30% Nycodenz), and a 50 μl top layer (SNH buffer). Each sample underwent centrifugation at 200,000 g in a SW55Ti rotor (Beckman) for 4 h. The liposomes and lipidated proteins were collected from the top 75 μl of the gradient.
Construction of rab37 knockout (KO) mice
The mouse Rab37 (MmRab37) gene contains 9 exons (NM_021411). The specific Mmrab37 KO target vector was constructed using the Transgenic Mouse Models Core (National Taiwan University). The target vector of MmRab37 was deleted for exon 1 and exon 2 with neomycin resistance gene to generate the Mmrab37 KO mice (C57BL/6J strain). This KO construct was injected into JM8A3 embryonic stem cells (The KOMP Repository, UCDAVIS, Knockout Mouse Project). Cells were screened for neomycin and candidate cell lines were genotyped by Southern blotting. The selected cell lines were injected into blastocysts to generate chimeric mice in the C57BL/6J strain. The Mmrab37 KO mice were maintained in a Specific Pathogen-Free control feeding room at the animal care center of National Cheng Kung University. The founders and their offspring mice were genotyped by RT-PCR or/and Southern blot analysis, and the expression of RAB37 was confirmed by both qPCR and immunohistochemistry.
Animals and treatment
Four-week-old male C57BL/6J mice were obtained from the Laboratory Animal Center of National Cheng Kung University and the B6.V-Lepob/JNarl (ob/ob) mice (LEP-deficient mice representing severe obesity with the symptom of hyperinsulinemia) were purchased from the National Laboratory Animal Center (Tainan, Taiwan). The animals were maintained in a pathogen-free facility under isothermal conditions with regular photoperiods. The experimental protocol adhered to the regulations of Taiwan’s Animal Protection Act and was approved by the Laboratory Animal Care and Use Committee of the university. Five-week-old mice were randomized into different groups and fed with various diets. For the C57BL/6J mice, the animals were fed with CD, HFD (60% fat calories; Research Diet, D12492), or one of the two diets (CD or HFD) with the addition of 1,500 p.p.m. niclosamide (Sigma, N3510). Blood glucose and insulin tolerance concentrations were determined in mice after an 8-h fast followed by intraperitoneal injection of 1.0 g/kg glucose (intraperitoneal glucose tolerance test [IPGTT]). Blood samples were collected from the retro-orbital sinus of the mice at various time points after injection of glucose for blood glucose and insulin determination using a commercial glucose assay kit (Biosystems, 11,538) and the Mercodia Mouse Insulin ELISA kit (Mercodia AB, 10-1247-01), respectively.
Quantification and statistical analysis
The western blot assay results were quantified by ImageJ (https://imagej.nih.gov/ij/). Summary data were presented as means ± SEM (standard deviation) and analyzed using the Student’s t-test in Prism 8 version 1.0 (GraphPad Software, Inc., San Diego, CA). P < 0.05 was considered significant. ns: no significance.
Supplementary Material
Acknowledgments
We thank Dr. Thomas Melia (Yale University, USA) for providing the in vitro system for the LC3 and GL1 lipidation reactions. We thank the technical services provided by the “Transgenic Mouse Model Core Facility of the National Core Facility Program for Biotechnology, Ministry of Science and Technology, Taiwan” and the “Gene Knockout Mouse Core Laboratory of National Taiwan University Center of Genomic Medicine”. This work was financially supported by the Cancer Progression Research Center of National Yang Ming Chiao Tung University, which runs the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project (Ministry of Education, Taiwan), Ministry of Science and Technology (MOST 109-2320-B-010-020 and MOST 109-2314-B-038-119-MY2), Taipei, Taiwan, and Kaohsiung Medical University Research Center (KMUTC108A04-0 and KMU-TC108A04-2), Kaohsiung, Taiwan. Taipei Medical University (TMU108-AE1-B39), Taipei, Taiwan.
Correction Statement
This article has been republished with minor changes. These changes do not impact the academic content of the article.
Funding Statement
The work was supported by the Ministry of Science and Technology, Taiwan [MOST 109-2314-B-038-119-MY2]; Ministry of Science and Technology, Taiwan [MOST 109-2320-B-010-020]; Kaohsiung Medical University Research Center [KMUTC108A04-0]; Kaohsiung Medical University Research Center [KMU-TC108A04-2]; Taipei Medical University [TMU108-AE1-B39]
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2123098
References
- [1].Lan SH, Wu SY, Zuchini R, et al. Autophagy suppresses tumorigenesis of hepatitis B virus-associated hepatocellular carcinoma through degradation of microRNA-224. Hepatology. 2014. Feb;59(2):505–517. DOI: 10.1002/hep.26659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014 Jan 23 53;(2)167–178. 10.1016/j.molcel.2013.12.014 [DOI] [PubMed] [Google Scholar]
- [3].Wu SY, Lan SH, Liu HS.. Degradative autophagy selectively regulates CCND1 (cyclin D1) and MIR224, two oncogenic factors involved in hepatocellular carcinoma tumorigenesis. Autophagy. 2019. Apr;15(4):729–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Padmanabhan S, Manjithaya R. Facets of autophagy based unconventional protein secretion-the road less traveled. Front Mol Biosci. 2020;7:586483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Martinelli S, Anderzhanova EA, Bajaj T, et al. Stress-Primed secretory autophagy promotes extracellular BDNF maturation by enhancing MMP9 secretion. Nat Commun. 2021 Jul 30 12;(1)4643. 10.1038/s41467-021-24810-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bustos SO, Antunes F, Rangel MC, et al. Emerging autophagy functions shape the tumor microenvironment and play a role in cancer progression - implications for cancer therapy. Front Oncol. 2020;10:606436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].New J, Thomas SM. Autophagy-Dependent secretion: mechanism, factors secreted, and disease implications. Autophagy. 2019. Oct;15(10):1682–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Li MY, Naik TS, Siu LYL, et al. Lyn kinase regulates egress of flaviviruses in autophagosome-derived organelles. Nat Commun. 2020 Oct 15 11;(1)5189. 10.1038/s41467-020-19028-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kraya AA, Piao S, Xu X, et al. Identification of secreted proteins that reflect autophagy dynamics within tumor cells. Autophagy. 2015;11(1):60–74. DOI: 10.4161/15548627.2014.984273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Mazza S, Maffucci T. Autophagy and pancreatic beta-cells. Vitam Horm. 2014;95:145–164. [DOI] [PubMed] [Google Scholar]
- [11].Marasco MR, Linnemann AK. Beta-Cell autophagy in diabetes pathogenesis. Endocrinology. 2018 May 1;159(5):2127–2141. DOI: 10.1210/en.2017-03273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vivot K, Pasquier A, Goginashvili A, et al. Breaking bad and breaking good: beta-cell autophagy pathways in diabetes. J Mol Biol. 2020 Mar 6 432;(5)1494–1513. 10.1016/j.jmb.2019.07.030 [DOI] [PubMed] [Google Scholar]
- [13].Wang Y, Li YB, Yin JJ, et al. Autophagy regulates inflammation following oxidative injury in diabetes. Autophagy. 2013. Mar;9(3):272–277. DOI: 10.4161/auto.23628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hc K, Ed A. Heart failure in type 2 diabetes mellitus. Circ Res. 2019 Jan 4;124(1):121–141. DOI: 10.1161/CIRCRESAHA.118.311371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mukherjee A, Morales-Scheihing D, Butler PC, et al. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015. Jul;21(7):439–449. DOI: 10.1016/j.molmed.2015.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun J, Cui J, He Q, et al. Proinsulin misfolding and endoplasmic reticulum stress during the development and progression of diabetes. Mol Aspects Med. 2015. Apr;42:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li Q, Jia S, Xu L, et al. Metformin-Induced autophagy and irisin improves INS-1 cell function and survival in high-glucose environment via AMPK/SIRT1/PGC-1alpha signal pathway. Food Sci Nutr. 2019. May;7(5):1695–1703. DOI: 10.1002/fsn3.1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu L, Liu J, Yu X. Dipeptidyl peptidase-4 inhibitor MK-626 restores insulin secretion through enhancing autophagy in high fat diet-induced mice. Biochem Biophys Res Commun. 2016 Feb 12;470(3):516–520. DOI: 10.1016/j.bbrc.2016.01.116 [DOI] [PubMed] [Google Scholar]
- [19].Zummo FP, Krishnanda SI, Georgiou M, et al. Exendin-4 stimulates autophagy in pancreatic beta-cells via the RAPGEF/EPAC-Ca(2+)-PPP3/calcineurin-TFEB axis. Autophagy. 2021;1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Arakawa M, Ebato C, Mita T, et al. Effects of exendin-4 on glucose tolerance, insulin secretion, and beta-cell proliferation depend on treatment dose, treatment duration and meal contents. Biochem Biophys Res Commun. 2009 Dec 18 390;(3)809–814. 10.1016/j.bbrc.2009.10.054 [DOI] [PubMed] [Google Scholar]
- [21].Jung HS, Chung KW, Won Kim J, et al. Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab. 2008. Oct;8(4):318–324. DOI: 10.1016/j.cmet.2008.08.013 [DOI] [PubMed] [Google Scholar]
- [22].Quan W, Lim YM, Lee MS. Role of autophagy in diabetes and endoplasmic reticulum stress of pancreatic beta-cells. Exp Mol Med. 2012 Feb 29;44(2):81–88. DOI: 10.3858/emm.2012.44.2.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ebato C, Uchida T, Arakawa M, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008. Oct;8(4):325–332. DOI: 10.1016/j.cmet.2008.08.009 [DOI] [PubMed] [Google Scholar]
- [24].Sheng Q, Xiao X, Prasadan K, et al. Autophagy protects pancreatic beta cell mass and function in the setting of a high-fat and high-glucose diet. Sci Rep. 2017 Nov 27 7;(1)16348. 10.1038/s41598-017-16485-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Veluthakal R, Thurmond DC. Emerging roles of small GTPases in Islet β-Cell function. Cells. 2021 Jun 15;10(6):1503. DOI: 10.3390/cells10061503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Li G, Marlin MC. Rab family of GTPases. Methods Mol Biol. 2015;1298:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014. Mar;21(3):348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Szatmari Z, Sass M. The autophagic roles of Rab small GTPases and their upstream regulators: a review. Autophagy. 2014. Jul;10(7):1154–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Noh SH, Gee HY, Kim Y, et al. Specific autophagy and ESCRT components participate in the unconventional secretion of CFTR. Autophagy. 2018;14(10):1761–1778. DOI: 10.1080/15548627.2018.1489479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jiang S, Dupont N, Castillo EF, et al. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun. 2013;5(5):471–479. DOI: 10.1159/000346707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nieto-Torres JL, Leidal AM, Debnath J, et al. Beyond autophagy: the expanding roles of ATG8 proteins. Trends Biochem Sci. 2021. Aug;46(8):673–686. DOI: 10.1016/j.tibs.2021.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Norris N, Yau B, Kebede MA. Isolation and proteomics of the insulin secretory granule. Metabolites. 2021 Apr 30;11(5):288. DOI: 10.3390/metabo11050288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cao M, Mao Z, Kam C, et al. PICK1 and ICA69 control insulin granule trafficking and their deficiencies lead to impaired glucose tolerance. PLoS Biol. 2013;11(4):e1001541. DOI: 10.1371/journal.pbio.1001541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sharifi MN, Mowers EE, Drake LE, et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 2016 May 24 15;(8)1660–1672. 10.1016/j.celrep.2016.04.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Riahi Y, Wikstrom JD, Bachar-Wikstrom E, et al. Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia. 2016. Jul;59(7):1480–1491. DOI: 10.1007/s00125-016-3868-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010 Feb 5;140(3):313–326. DOI: 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Balgi AD, Fonseca BD, Donohue E, et al. Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009 Sep 22 4;(9)e7124. 10.1371/journal.pone.0007124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Runwal G, Stamatakou E, Siddiqi FH, et al. LC3-Positive structures are prominent in autophagy-deficient cells. Sci Rep. 2019 Jul 12 9;(1)10147. 10.1038/s41598-019-46657-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kiral FR, Kohrs FE, Jin EJ, et al. Rab GTPases and membrane trafficking in neurodegeneration. Curr Biol. 2018 Apr 23 28;(8)R471–R486. 10.1016/j.cub.2018.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Heckmann BL, Teubner BJW, Tummers B, et al. LC3-Associated endocytosis facilitates beta-amyloid clearance and mitigates neurodegeneration in murine alzheimer’s disease. Cell. 2019 Jul 25 178;(3)536–551 e14. 10.1016/j.cell.2019.05.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kreutzberger AJB, Kiessling V, Doyle CA, et al. Distinct insulin granule subpopulations implicated in the secretory pathology of diabetes types 1 and 2. Elife. 2020 Nov 9;9: 10.7554/eLife.62506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res. 2014. Jan;24(1):58–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thukral L, Sengupta D, Ramkumar A, et al. The molecular mechanism underlying recruitment and insertion of lipid-anchored LC3 protein into membranes. Biophys J. 2015 Nov 17 109;(10)2067–2078. 10.1016/j.bpj.2015.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nath S, Dancourt J, Shteyn V, et al. Lipidation of the LC3/GABARAP family of autophagy proteins relies on a membrane-curvature-sensing domain in Atg3. Nat Cell Biol. 2014. May;16(5):415–424. DOI: 10.1038/ncb2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011. Jan;91(1):119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tamura N, Mima J. Membrane-Anchored human Rab GTPases directly mediate membrane tethering in vitro. Biol Open. 2014 Oct 31;3(11):1108–1115. DOI: 10.1242/bio.20149340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Ljubicic S, Bezzi P, Brajkovic S, et al. The GTPase Rab37 participates in the control of insulin exocytosis. PLoS One. 2013;8(6):e68255. DOI: 10.1371/journal.pone.0068255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Goginashvili A, Zhang Z, Erbs E, et al. Insulin granules. Insulin secretory granules control autophagy in pancreatic beta cells. Science. 2015 Feb 20 347;(6224)878–882. 10.1126/science.aaa2628 [DOI] [PubMed] [Google Scholar]
- [49].Buratta S, Tancini B, Sagini K, et al. Lysosomal exocytosis, exosome release and secretory autophagy: the autophagic- and endo-lysosomal systems go extracellular. Int J Mol Sci. 2020 Apr 8 21;(7)2576. 10.3390/ijms21072576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Gonzalez CD, Resnik R, Vaccaro MI. Secretory autophagy and its relevance in metabolic and degenerative disease. Front Endocrinol (Lausanne). 2020;11:266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Bugliani M, Mossuto S, Grano F, et al. Modulation of autophagy influences the function and survival of human pancreatic beta cells under endoplasmic reticulum stress conditions and in type 2 Diabetes. Front Endocrinol (Lausanne). 2019;10:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Han D, Yang B, Olson LK, et al. Activation of autophagy through modulation of 5’-AMP-activated protein kinase protects pancreatic beta-cells from high glucose. Biochem J. 2010 Jan 15 425;(3)541–551. 10.1042/BJ20090429 [DOI] [PubMed] [Google Scholar]
- [53].Komatsu M, Takei M, Ishii H, et al. Glucose-Stimulated insulin secretion: a newer perspective. J Diabetes Investig. 2013 Nov 27 4;(6)511–516. 10.1111/jdi.12094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest. 2011. Jun;121(6):2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang M, Liu L, Lin X, et al. A translocation pathway for vesicle-mediated unconventional protein secretion. Cell. 2020 Apr 30 181;(3)637–652 e15. 10.1016/j.cell.2020.03.031 [DOI] [PubMed] [Google Scholar]
- [56].Zhang M, Kenny SJ, Ge L, et al. Translocation of interleukin-1beta into a vesicle intermediate in autophagy-mediated secretion. Elife. 2015 Nov 2;4: 10.7554/eLife.11205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Weiss M, Steiner DF, Philipson LH, et al. In: In: Feingold K, Anawalt BBoyce A, editors. Insulin biosynthesis, secretion, structure, and structure-activity relationships. South Dartmouth (MA): Endotext;2000. [PubMed] [Google Scholar]
- [58].Leidal AM, Debnath J. LC3-Dependent extracellular vesicle loading and secretion (LDELS). Autophagy. 2020. Jun;16(6):1162–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Buckingham EM, Jarosinski KW, Jackson W, et al. Exocytosis of varicella-zoster virus virions involves a convergence of endosomal and autophagy pathways. J Virol. 2016 Oct 1 90;(19)8673–8685. 10.1128/JVI.00915-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Taisne C, Lussignol M, Hernandez E, et al. Human cytomegalovirus hijacks the autophagic machinery and LC3 homologs in order to optimize cytoplasmic envelopment of mature infectious particles. Sci Rep. 2019 Mar 14 9;(1)4560. 10.1038/s41598-019-41029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Leidal AM, Huang HH, Marsh T, et al. The LC3-conjugation machinery specifies the loading of RNA-binding proteins into extracellular vesicles. Nat Cell Biol. 2020. Feb;22(2):187–199. DOI: 10.1038/s41556-019-0450-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Dupont N, Jiang S, Pilli M, et al. Autophagy-Based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J. 2011 Nov 8 30;(23)4701–4711. 10.1038/emboj.2011.398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012 Dec 7;151(6):1256–1269. DOI: 10.1016/j.cell.2012.11.001 [DOI] [PubMed] [Google Scholar]
- [64].Zhu D, Zhang Y, Lam PP, et al. Dual role of VAMP8 in regulating insulin exocytosis and islet beta cell growth. Cell Metab. 2012 Aug 8 16;(2)238–249. 10.1016/j.cmet.2012.07.001 [DOI] [PubMed] [Google Scholar]
- [65].Xiong QY, Yu C, Zhang Y, et al. Key proteins involved in insulin vesicle exocytosis and secretion. Biomed Rep. 2017. Feb;6(2):134–139. DOI: 10.3892/br.2017.839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Matsunaga K, Taoka M, Isobe T, et al. Rab2a and Rab27a cooperatively regulate the transition from granule maturation to exocytosis through the dual effector Noc2. J Cell Sci. 2017 Feb 1 130;(3)541–550. 10.1242/jcs.195479 [DOI] [PubMed] [Google Scholar]
- [67].Gendaszewska-Darmach E, Garstka MA, Blazewska KM. Targeting small gtpases and their prenylation in diabetes mellitus. J Med Chem. 2021 Jul 22;64(14):9677–9710. DOI: 10.1021/acs.jmedchem.1c00410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Lu Q, Wang PS, Yang L. Golgi-Associated Rab GTPases implicated in autophagy. Cell Biosci. 2021 Feb 8;11(1):35. DOI: 10.1186/s13578-021-00543-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Schroeder B, Schulze RJ, Weller SG, et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015. Jun;61(6):1896–1907. DOI: 10.1002/hep.27667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Zhang F, Ma D, Zhao W, et al. Obesity-Induced overexpression of miR-802 impairs insulin transcription and secretion. Nat Commun. 2020 Apr 14 11;(1)1822. 10.1038/s41467-020-15529-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Mrugacz M, Bryl A, Zorena K. Retinal vascular endothelial cell dysfunction and neuroretinal degeneration in diabetic patients. J Clin Med. 2021 Jan 25;10(3):458. DOI: 10.3390/jcm10030458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Padhi S, Nayak AK, Behera A. Type II diabetes mellitus: a review on recent drug based therapeutics. Biomed Pharmacother. 2020. Nov;131:110708. [DOI] [PubMed] [Google Scholar]
- [73].Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. 2010. Mar;21(3):151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Prentki M, Nolan CJ. Islet beta cell failure in type 2 diabetes. J Clin Invest. 2006. Jul;116(7):1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Tao H, Zhang Y, Zeng X, et al. Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med. 2014. Nov;20(11):1263–1269. DOI: 10.1038/nm.3699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kanemoto N, Okamoto T, Tanabe K, et al. Antidiabetic and cardiovascular beneficial effects of a liver-localized mitochondrial uncoupler. Nat Commun. 2019 May 15 10;(1)2172. 10.1038/s41467-019-09911-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Perry RJ, Zhang D, Zhang XM, et al. Controlled-Release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science. 2015 Mar 13 347;(6227)1253–1256. 10.1126/science.aaa0672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Miyazaki J, Araki K, Yamato E, et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990. Jul;127(1):126–132. DOI: 10.1210/endo-127-1-126 [DOI] [PubMed] [Google Scholar]
- [79].Cheng K, Delghingaro-Augusto V, Nolan CJ, et al. High passage MIN6 cells have impaired insulin secretion with impaired glucose and lipid oxidation. PLoS One. 2012;7(7):e40868. DOI: 10.1371/journal.pone.0040868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yeh HH, Wu CH, Giri R, et al. Oncogenic Ras-induced morphologic change is through MEK/ERK signaling pathway to downregulate Stat3 at a posttranslational level in NIH3T3 cells. Neoplasia. 2008. Jan;10(1):52–60. DOI: 10.1593/neo.07691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Seglen PO, Brinchmann MF. Purification of autophagosomes from rat hepatocytes. Autophagy. 2010. May;6(4):542–547. [DOI] [PubMed] [Google Scholar]
- [82].Li M, Du W, Zhou M, et al. Proteomic analysis of insulin secretory granules in INS-1 cells by protein correlation profiling. Biophys Rep. 2018;4(6):329–338. DOI: 10.1007/s41048-018-0061-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Lee YR, Lei HY, Liu MT, et al. Autophagic machinery activated by dengue virus enhances virus replication. Virology. 2008 May 10 374;(2)240–248. 10.1016/j.virol.2008.02.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Wu SY, Lan SH, Wu SR, et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Hepatology. 2018. Jul;68(1):141–154. DOI: 10.1002/hep.29781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Choy A, Dancourt J, Mugo B, et al. The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science. 2012 Nov 23 338;(6110)1072–1076. 10.1126/science.1227026 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.