

ABSTRACT
How macroautophagy/autophagy influences neurofilament (NF) proteins in neurons, a frequent target in neurodegenerative diseases and injury, is not known. NFs in axons have exceptionally long half-lives in vivo enabling formation of large stable supporting networks, but they can be rapidly degraded during Wallerian degeneration initiated by a limited calpain cleavage. Here, we identify autophagy as a previously unrecognized pathway for NF subunit protein degradation that modulates constitutive and inducible NF turnover in vivo. Levels of NEFL/NF-L, NEFM/NF-M, and NEFH/NF-H subunits rise substantially in neuroblastoma (N2a) cells after blocking autophagy either with the phosphatidylinositol 3-kinase (PtdIns3K) inhibitor 3-methyladenine (3-MA), by depleting ATG5 expression with shRNA, or by using both treatments. In contrast, activating autophagy with rapamycin significantly lowers NF levels in N2a cells. In the mouse brain, NF subunit levels increase in vivo after intracerebroventricular infusion of 3-MA. Furthermore, using tomographic confocal microscopy, immunoelectron microscopy, and biochemical fractionation, we demonstrate the presence of NF proteins intra-lumenally within autophagosomes (APs), autolysosomes (ALs), and lysosomes (LYs). Our findings establish a prominent role for autophagy in NF proteolysis. Autophagy may regulate axon cytoskeleton size and responses of the NF cytoskeleton to injury and disease.
KEYWORDS: 3-MA, calpeptin, Con A, MG115, proteasome, rapamycin
Introduction
Neurofilaments (NFs), composed of four subunits NEFL, NEFM, NEFH, and INA/α-internexin in the central nervous system or peripherin in the peripheral nervous system, form a highly stable lattice within the axonal cytoskeleton where they regulate caliber and nerve conduction velocities [1] as well as act as a reversible docking platform for vesicular organelles to coordinate their distributions and dynamic behaviors in axons [2]. NF subunit assemblies are also integral components of synapses where they modulate synaptic plasticity and specific in vivo behaviors by regulating Dopamine D1 and N-methyl D-aspartate receptor activity and stability at synaptic surfaces [3,4]. NF accumulations are pathological hallmarks of amyotrophic lateral sclerosis (ALS) and contribute to the neurofibrillary lesions in Alzheimer's disease (AD) [5]. Cerebrospinal fluid (CSF) levels of NEFH and NEFL are useful biomarkers of axonal injury in neurological diseases [6,7], and NEFL detection in blood (and CSF) has recently emerged as an index of disease onset and progression in AD, albeit not a disease-specific index [8].
Calpains initiate the proteolysis of NEFH and NEFM subunits by cleaving off their long carboxyl-terminal tail domains. Phosphorylation of these domains by multiple kinases confers resistance to calpain proteolysis [9,10], which contributes to the exceptionally long half-lives of NFs comprising the axonal cytoskeleton [11]. NF proteins nevertheless undergo slow constitutive turnover in part via calpains [12] and the degradation initiated by calpains is greatly accelerated in states of calcium injury to axons [12,13] or axotomy resulting in Wallerian degeneration [13]. In addition to calpains, the Gigaxonin-mediated ubiquitin-proteasome system [14–16] and lysosomal CTSD (cathepsin D) [17] have been implicated in NF degradation, although the possible role of autophagy in NF proteolysis is not known.
Macroautophagy, the major autophagic route to lysosomes, can sequester and degrade most cellular constituents, including protein aggregates and organelles [18,19]. This suggests that the sterically complex NF subunits and even the heteropolymeric protofilaments that might predominate in perikarya, and synapses are candidate autophagic substrates. Neuronal macroautophagy (hereafter referred to as autophagy) is constitutive and inducible in response to cellular stress and injury in physiological and disease contexts [20,21]. Autophagy is initiated by inhibiting MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1), or by activating AMP-activated protein kinase (AMPK) and ULK1 kinase [21]. Formation of the autophagosome (AP), a compartment that contains substrates sequestered by a phagophore, is initiated at the ER by the activation of a ULK1 (unc-51 like kinase 1)-PIK3C3/VPS34 complex and coordination by multiple protein complexes. These structures fuse with multivesicular bodies (MVBs) or late endosomes to form amphisomes and they later fuse with lysosomes (LYs) to form autolysosomes (ALs). APs can also directly fuse with LYs to form ALs [21] to degrade the contents and regenerate new LYs. AP Formation, maturation, and complete degradation of substrates in ALs is termed autophagic flux [22].
In this report, we demonstrate that NF subunit protein levels are increased when NF-transfected N2a cells are either exposed to an autophagy inhibitor (3-MA), by depletion of autophagy protein ATG5 using shRNA, or a combination of ATG5 depletion and 3-MA exposure, or by blockade at the lysosomal proteolytic stage. Conversely, activation of autophagy with rapamycin decreased NF protein levels. In vivo, NF protein levels in the mouse brain rose upon inhibiting autophagy via intracerebroventricular infusion of 3-MA. Furthermore, in the neocortex of wild-type mice, NF proteins were immunolocalized within the lumens of autophagic vesicles (AVs) containing MAP1LC3/LC3 (APs) or CTSD (ALs), and were enriched in APs, ALs, and LYs fractions isolated from the brain. Based on these observations, we conclude that autophagy is a previously unrecognized degradative pathway for NF subunit proteins. These results provide new insights into neuronal cytoskeleton regulation and into possible pathogenic mechanisms in neurofibrillary diseases, where autophagy is frequently implicated [21].
Results
NF formation and degradation in N2a cells
Undifferentiated N2a cells express only NEFH protein. NEFM and NEFL proteins are absent in these cells since their genes are transcriptionally repressed [23]. To avoid confounding effects of differentiation agents, such as retinoic acid and dibutyryl cyclic AMP on autophagy in our culture system, we used undifferentiated N2a cells transfected with NF genes for our in vitro analyses. To examine NF formation, we transfected NF genes (Nefh, Nefm, Nefl) into N2a cells as previously described [24] and immunostained cells 42 h later with NF antibodies (NEFL, red and NEFM, green). Only NF-transfected cells expressed NEFL and NEFM (Figure 1A) while these subunits were undetected in untransfected (Mock) cells (Figure 1A). NF-transfected cells formed filaments (Figure 1C, purple arrows) running parallel to each other with extensive crosslinking (Figure 1C, see red arrows) resembling the NFs seen in axons in vivo (see Figure 6A,F). Double-label immunoelectron microscopy (IEM) of NF-transfected cells with antibodies to NEFL (10 nm) and NEFH (6 nm) (Figure 1D), and NEFM (10 nm) and NEFL (6 nm) (Figure 1E) gold particles decorated filaments confirming these filaments to be NFs.
Figure 1.

NF formation and degradation in N2a cells. Untransfected (Mock, A) and NF-gene-transfected (NFs, B) N2a cells were immunolabeled with NEFL and NEFM antibodies (A and B). Scale bar: 10 µm. Electron microscopy of NF containing cells show formation of parallel NF filaments (purple arrows) that are interconnected with cross-bridges (C, see red arrows). Immunolabeling of these filamentous cells with NF antibodies (D, NEFL, 10 nm and NEFHCOOH, 6 nm) (E, NEFM, 10 nm and NEFL, 6 nm gold particles) show labeling of filaments with all 3 NF antibodies (NEFL, NEFM and NEFH). Scale bar: 50 nm. NF-transfected cells treated with cycloheximide (CHX) show reduction in NF proteins at 1.5 h (F and G, compared to 100% at 0 h time point, NEFL 70%, n = 4, p = 0.0115; NEFM 79%, n = 4, p = 0.0439; NEFH-MYC-79%, n = 4, p = 0.0240), and at 3 h time point (F and G, compared to 100% at 0 h time point, NEFL 53%, n = 5, p = 0.0001; NEFM 69%, n = 5, p = 0.0113; NEFH-MYC 77%, n = 8, p = 0.0007; n = 8). *p < 0.05; ***p < 0.001, one-way ANOVA.
Figure 6.
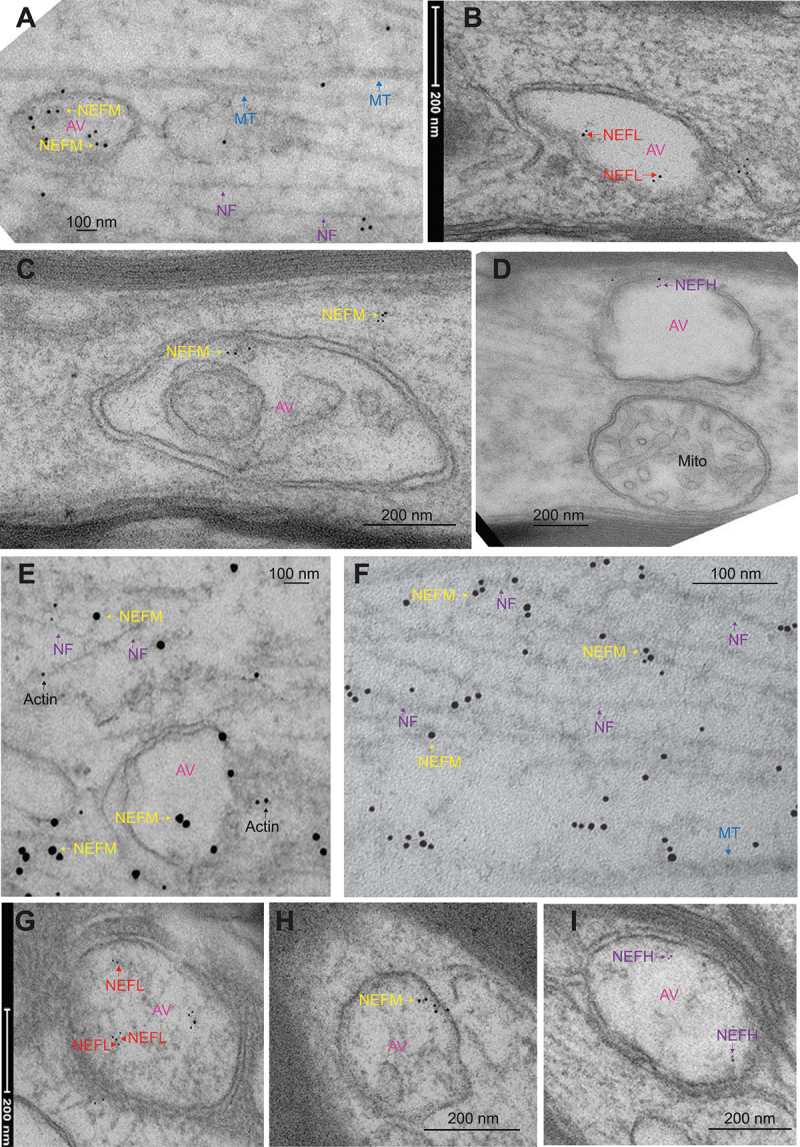
Immunoelectron microscopy (IEM) indicates NF proteins are present in autophagic vesicles in optic axons (A-I). Mouse optic axons were subjected to IEM with NEFM (RMO44; A,C,E,F, and H), NEFL (B and G), and NEFHCOOH (D and I) antibodies. NF protein labeling in lumens of double membrane AVs in optic axons (B, 646 nm; C, 1075 nm; D, 711 nm), and cytoplasmic labeling with NEFM (A, C, E and F, see yellow arrows) and NEFL (D, red arrows) antibodies is also seen in axons while microtubules (MT, blue arrows in A and F) and mitochondria (Mito. Figure D) are not stained. AVs of 260-600 nm were also stained with NEFL (G, 409 nm, see arrow heads for NEFL staining on short filaments), NEFM (A, 644 nm; E, 606 nm; H, 260 nm, yellow arrows), and NEFHCOOH (I, 377 nm, purple arrows) antibodies. Panel E double labeled with NEFM (15 nm gold particles, yellow arrows) and actin (6 nm gold particles, black arrows). The size of the gold particles in images from A-D, and G-I, 6 nm and in F, 10 nm. AV-autophagic vesicle, NF-neurofilament.
To measure NF protein degradation in N2a cells, NF-gene-transfected cells (after 42 h) were treated with cycloheximide (CHX, 10 µM) at 0–180 min to block new protein synthesis, cell extracts were made, and immunoblotted with NF antibodies. Immunoblot data from CHX-treated cells indicated time-dependent loss of NF proteins. NF subunit protein levels were significantly reduced at 1.5 h (Figure 1F,G), and further reduced at the 3 h time point (Figure 1F,G).
NF subunit proteins are degraded by macroautophagy in an in vitro cell model
To test the role of autophagy in NF protein degradation, NF-gene-transfected N2a cells were treated with the autophagy inhibitor, 3-MA (5 mM) [25] for 42 h, and the immunoblot analyses of the cell extracts with NF antibodies demonstrate that levels of all three NF proteins were significantly increased (Figure 2A). Similarly, NF-gene-transfected 3-MA-treated cells displayed significantly more immunostaining for NEFL (Figure 2B,C) and NEFM (Figure 2B,C) compared to NF-gene-transfected untreated cells (NF) (Figure 2B-D).
Figure 2.

3-MA, and Con A treatments inhibited NF protein degradation. (A) Treatment of NF-gene-transfected N2a cells with 3-MA (5 mM, 42 h) and measurement of levels of NF proteins (NEFL, 3-fold, p = 0.0003; NEFM, 2.3-fold, p < 0.0015 and NEFH-MYC, 1.4-fold, p < 0.0042; n = 3 for all subunits). NF-gene-transfected N2a cells (NFs) were treated with 3-MA (NF + 3-MA), stained with NEFL and NEFM antibodies (B, lower and higher mag. Scale bars: 10-µm) and quantification of NEFL (C, n = 115 cells for NF and n = 122 cells for NF + 3-MA, p < 0.0001) and NEFM (D, n = 119 cells for NF and n = 110 for NF + 3-MA, p < 0.0001, unpaired t-test) staining in cells. Con A treatment of NF-gene-transfected cells and measurement of levels of NF proteins (E, NEFL, 2-fold, n = 4, p < 0.0051; NEFM, 2-fold, n = 4, p < 0.0188; NEFH-MYC, 3-fold, n = 3, p < 0.0116), MAP1LC3-I (E, n = 4, p < 0.0054), MAP1LC3-II (E, n = 4, p < 0.0003), the ratio of MAP1LC3-II:MAP1LC3-I (E, n = 4, p < 0.0001), and the formation of mature-CTSD (E, n = 4, p < 0.0091). Panels A-E, unpaired t-test; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To further show that NFs are degraded by the autolysosomal pathway, NF-gene-transfected cells were treated with an inhibitor of the lysosomal vacuolar-type H+-ATPase, concanamycin A (Con A; 0.5 µM, 24 h), which resulted in significant increases in the levels of all three NF proteins (Figure 2E). Con A treatment of the cells significantly inhibited the pH-dependent formation of mature CTSD (Figure 2E) and greatly inhibited clearance of MAP1LC3-II (Figure 2E), which established the efficacy of Con A. These observations indicate that inhibiting autolysosomal function prevents NF degradation and raises NF protein levels in N2a cells.
Depletion of ATG5 (autophagy related 5) protein in cells is known to block AP formation and suppress autophagy flux [26]. We tested whether depleting ATG5 levels in N2a cells with Atg5-shRNA would increase NF protein levels. Atg5-shRNA transduction significantly decreased ATG5 protein in untransfected (Figure 3A,C) as well as in NF-transfected N2a cells (Figure 3A,C). ATG5-depletion with Atg5-shRNA significantly increased levels of all three NF proteins in NF-transfected N2a cells (Figure 3A,D). Importantly, Atg5-shRNA transduction increased endogenous NEFH levels in un-transfected N2a cells by 3.6-fold compared to non-transduced cells (Figure 3A,D). ATG5 protein depletion in these cells also had a significant reduction in MAP1LC3-I, MAP1LC3-II, and the MAP1LC3-II:MAP1LC3-I ratio (Figure 3A,C). Transduction of scrambled shRNA (SC-shRNA) into N2a cells did not alter the levels of ATG5, MAP1LC3-I, MAP1LC3-II, the ratio of MAP1LC3-II:MAP1LC3-I and all three NF proteins (Figure 3B,E,F). The lowered levels of MAP1LC3-II and the ratio of MAP1LC3:MAP1LC3-I in ATG5-depleted N2a cells is consistent with the blockage of AP formation and slowdown of autophagy [26] in N2a cells, thus implicating autophagy in NF protein degradation.
Figure 3.

ATG5-depletion inhibited NF protein degradation. Transduction of N2a cells with Atg5-shRNA, measurement of ATG5 protein in transduced cells (Atg5-shRNA) (A and C, n=4, p<0.0001), in NF-transfected cells (A and C, n=4, p<0.0001), measurement of levels of NF proteins (D, NEFL, n=5, p<0.0003; NEFM, n=8, p<0.0001; NEFH, n=8, p<0.0001; endogenous-NEFH (NEFHCOOH) in Atg5-shRNA cells, n=4, p<0.0147), MAP1LC3-I(C, n=8, p<0.0005), MAP1LC3-II (C, n=8, p<0.0004) and MAP1LC3-II:MAP1LC3-I ratio (C, n=8, p<0.0012) in Atg5-shRNA+NF cells. Transduction of SC-shRNA into NF-transfected N2a cells and measurement of the levels of ATG5, MAP1LC3, MAP1LC3-II, the ratio of MAP1LC3-II:MAP1LC3-I, transfected NF proteins and the endogenous NEFH protein (B, E-F). ATG5-depletion (G, NF vs NF+Atg5-shRNA, n=6, p<0.0001; NF+3-MA vs NF+Atg5-shRNA+3-MA, n=6, p<0.0001) coupled with 3-MA treatment, and measurement of NF subunit levels (G, NEFL, n=4-6, 8.2-fold increase, p<0.0001; NEFM, n=6, 70% increase, p<0.0001; NEFH-MYC, n=4-6, 1.6-fold increase, p<0.0001), MAP1LC3-I (G, NF+3-MA vs NF+Atg5-shRNA+3-MA, n=5-6, p<0.0001), MAP1LC3-II (G, NF+3-MA vs NF+Atg5-shRNA+3-MA, n=5-6, p<0.0001) and the ratio of MAP1LC3-II: MAP1LC3-I (G, NF+3-MA vs NF+Atg5-shRNA+3-MA, n=5-6, p<0.0008). eGFP overexpression in Atg5-shRNA transduced cells as well as in NF-transfected cells and treatment of these cells with 3-MA and measurement of eGFP protein in cells (C, n=4, p=0.6863; G, n=4, p=0.5441). 3-MA treatment and measurement of the levels of MAP1LC3-I (H, Mock vs 3-MA, n=3, p<0.0001; NF vs NF+3-MA, n=3, p=0.0001), MAP1LC3-II (H, Mock vs 3-MA, n=3, p=0.0001; NF vs NF+3-MA, n=3, p=0.0001), the ratio of MAP1LC3-II:MAP1LC3-I (H, Mock vs 3-MA, n=3, p=0.0001; NF vs NF+3-MA, n=3, p<0.0001), and SQSTM1/p62 (H, Mock vs 3-MA, n=3, p<0.0001; NF vs NF+3-MA, n=3, p=0.0001). Treatment of NF-transfected N2a cells with rapamycin (10 nM, 24 h) and measurement of levels of NF proteins (I, NEFL, n=9, 30% reduction, p=0.0077; NEFM, n=9, 50% reduction, p=0.0062; NEFH-MYC, n=7, 20% reduction, p=0.0272), MAP1LC3-I (I, n=6, p<0.0036), MAP1LC3-II (I, n=6, p<0.0081), the ratio of MAP1LC3-II: MAP1LC3-I (I, n=6, p<0.0130), and calpain activity (I, n=6, p<0.9795). Panels C-G,I, unpaired t-test; Panels C,E,G,H, one-way ANOVA; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Combined ATG5 depletion and 3-MA exposure further elevates NF protein levels in N2a cells
Blockage of autophagy with ATG5 depletion significantly increased all three NF protein levels in N2a cells (Figure 3A,C,D). We tested whether treatment of ATG5-depleted cells with 3-MA to further block autophagy would increase NF protein levels even more over ATG5-depleted cells. Our data from ATG5-depleted cells treated with 3-MA significantly further increased all three NF protein levels over the ATG5-depleted cells (Figure 3G). The above results prove that blockage of autophagy either genetically or pharmacologically or by combining both the treatments increase NF levels, as predicted.
Atg5-shRNA containing replication-deficient viral vector also contains an eGFP gene and transduction of this vector alone or into NF-transfected cells or treatment of Atg5-shRNA-transduced cells with 3-MA did not alter the levels of eGFP significantly in N2a cells (Figure 3A,C,G). These results indicate that the 3-MA effect of raising neurofilament levels is specific.
3-MA inhibits clearance of known autophagic substrates in N2a cells
To test whether 3-MA inhibits clearance of known autophagic substrates in N2a cells, cell extracts treated with 3-MA were immunoblotted with MAP1LC3 and SQSTM1/p62 (sequestosome 1) antibodies. Our immunoblot data demonstrate that the levels of MAP1LC3-I (Figure 3H), MAP1LC3-II (Figure 3H), the ratio of MAP1LC3-II: MAP1LC3-I (Figure 3H), and SQSTM1/p62 (sequestosome 1) (Figure 3H) were significantly elevated while the levels of TUBB/β-tubulin (Figure 3H) were unaltered in 3-MA-treated N2a cells.
Activation of autophagy with rapamycin enhances NF protein degradation in N2a cells
Since blockage of autophagy with 3-MA increases NF protein levels, we tested whether activation of autophagy with rapamycin would increase NF protein degradation in N2a cells. As expected, NF-transfected N2a cells treated with rapamycin (10 nM, 24 h) significantly increased NF protein degradation and reduced the levels of all three NF proteins (Figure 3I). Rapamycin significantly increased the levels of MAP1LC3-I, MAP1LC3-II, and the ratio of MAP1LC3-II:MAP1LC3-I (Figure 3I) while it did not affect the levels of calpain activity as measured by the ratio of calpain-cleaved 150-kDa-SPTA1/α-Spectrin fragment to full-length (FL)-SPTA1 (Figure 3I).
We also tested whether 3-MA influences the other two proteolytic systems, namely calpain and proteasome in N2a cells. 3-MA-treated cell extracts from untransfected cells were immunoblotted with SPTA1 and ubiquitin antibodies, which demonstrated a significantly reduced ratio of calpain- cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 compared to untreated extracts (Fig. S1A–B) indicating an inhibition of calpain activity. The same extracts have unaltered ubiquitin levels (Fig. S1A and S1C) indicating that 3-MA did not influence proteasome activity in N2a cells.
NF proteins are also degraded by calpain and proteasome systems in N2a cells
Since NF proteins are degraded by autophagy, we tested whether other proteolytic systems, such as calpains and the proteosome also degrade NF proteins in N2a cells. To test these two possibilities, we first treated NF-gene-transfected cells with a synthetic calpain inhibitor, calpeptin (40 µM, 24 h), which decreased the ratio of 150-kDa calpain cleaved SPTA1 fragment:FL-SPTA1 (Figure 4A), as expected. Calpain inhibition led to a significant increase in the levels of all three NF proteins (Figure 4A) while TUBB levels remained unaltered (Figure 4A). The calpeptin effect on NF levels was slightly less compared to that induced by blocking autophagy with 3-MA or inhibiting lysosomal proteases by elevating lysosomal pH with Con A (Table 1).
Figure 4.

NF proteins are degraded by calpain and proteasome systems in N2a cells. NF-gene-transfected N2a cells were treated with Calpeptin (40 µm, 24 h) and measurement of levels of NF proteins (A, NEFL, 2.2-fold, n = 3, p < 0.0345; NEFM, 1.6-fold, n = 4, p < 0.0111; NEFH-MYC, 1.4-fold, n = 3–4, p < 0.0377) and the ratio of calpain-cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 (A, n = 4, p < 0.0010). MG-132 treatment (0.5 µm, 24 h) of NF-transfected cells and measurement of proteasome activity (B, n = 4, p < 0.0001) and the levels of NF subunits (B, NEFL, 2.1-fold, n = 3, p < 0.0314; NEFM, 1.5-fold, n = 3, p < 0.0260; NEFH-MYC, 1.5-fold, n = 3–4, p < 0.0168). Panels A and B, unpaired t-test; *p < 0.05; **p < 0.01; ****p < 0.0001.
Table 1.
Effect of inhibitors on transfected NF proteins in N2a cells.
| Inhibitor effect on NF Proteins in N2a cells (fold increase) | ||||
|---|---|---|---|---|
| Protein | 3-MA | Con A | Calpeptin | MG-132 |
| NEFH | 1.340 ± 0.051 (**) | 2.947 ± 0.528 (*) | 1.441 ± 0.241 (*) | 1.526 ± 0.0165 (*) |
| NEFM | 2.302 ± 0.130 (**) | 2.036 ± 0.437 (*) | 1.649 ± 0.063 (*) | 1.504 ± 0.048 (*) |
| NEFL | 3.025 ± 0.154 (***) | 1.952 ± 0.217 (**) | 2.167 ± 0.082 (*) | 2.167 ± 0.079 (*) |
Note: Effect of autophagy (3-MA), lysosomal vacuolar-type H+-ATPase (Con A), calpain (calpeptin) and proteasome (MG132) inhibitors on NF protein levels in N2a cells. The fold increases are over untreated samples (one-fold increase equals to 100% increase). Significance is indicated in parenthesis. *p < 0.05, **p < 0.01, ***p < 0.001. The data from Figure 2A, Figure 2E, Figure 4A, and Figure 4 are compared in Table 1.
To test the role of the proteasome in NF proteolysis in N2a cells, we treated NF-gene-transfected cells with MG-132 (an inhibitor of the proteasome, 0.5 µM, 24 h). Increased levels of total ubiquitin (Figure 4B) with MG-132 treatment confirmed the inhibition of proteasome activity. MG-132 treatment of NF-transfected cells showed a significant increase in levels of all three NF proteins (Figure 4B), while the levels of TUBB (Figure 4B) were unaltered. The increases for each NF subunit due to proteasome inhibition were slightly less compared to those using autophagy/lysosomal inhibition (Table 1).
NF proteins are sequestered within compartments of the autophagic pathway in vivo
To determine whether NF proteins undergo autophagic degradation pathway in vivo, we performed double fluorescence immunocytochemistry on sections of the mouse brain to detect NEFM protein and its possible colocalization with MAP1LC3-positive and CTSD-positive vesicular compartments. Generally, NEFM antibody immunolabeled a fibrous network in cortical neuronal cell bodies, proximal dendrites (Fig. S2A, see arrows) and axons (Fig. S2A, and B, for NEFM panel). To visualize NEFM label in vesicular structures in neuronal cell bodies, we intentionally reduced the gain of the green fluorescence (NEFM label) channel on the microscope, and images were captured (Figure 5 panel A and B for NEFM panels representing unmodified images). We then adjusted all the images in Adobe Photoshop CC 2015 (see Materials and Methods for details) to visualize NEFM puncta in cortical cell bodies (modified images in Figure 5C, Ci, D, and Di are shown). NEFM immunolabel was colocalized with MAP1LC3 (Figure 5C) or CTSD (Figure 5D) positive vesicles. To determine the amount of NEFM label in vesicular structures colocalized with MAP1LC3 and CTSD positive vesicles, we quantified the extent of colocalization within cortical neuronal cell bodies. Our colocalization quantitative data demonstrate that one-fifth of the NEFM vesicular immunolabel was associated with MAP1LC3 (Figure 5E) and with CTSD (Figure 5F) vesicles. We unequivocally established that NEFM localization within the lumens of MAP1LC3-positive (AP and AL) and CTSD-positive lysosomal/autolysosomal (LY/AL) compartments by analyzing Z-stack series of images through entire vesicles immunopositive for NEFM-MAP1LC3 (Figure 5Ci) and NEFM-CTSD (Figure 5Di). These results establish that NEFM protein is present in APs and AL/LY vesicles of the autophagic pathway in mouse cortex in vivo.
Figure 5.

NEFM is present in AP and AL compartments of mouse cortical neurons. Cortices were double immunolabeled with NEFM and MAP1LC3 (A, unmodified images) or NEFM and CTSD antibodies (B, unmodified images). Images in C and D are modified in Photoshop (see Materials and Methods for details) to visualize NEFM-label in vesicular structures that is colocalized with MAP1LC3 (an AP marker, C, see arrows for colocalization) and CTSD (an AL/LY marker, Rudy-4, D, see arrows for colocalization) puncta. Colocalized puncta (yellow) were imaged through Z-stacks to show NEFM in the lumen of APs (see arrows in Ci to follow the same vesicles) and AL vesicles (see arrows in Di to follow the same vesicles) in mouse cortex layer III. Scale bars A-D, Ci and Di: 2 µm. Quantification of NEFM label in vesicular structures indicates 1/5 of NEFM label is colocalized with MAP1LC3 (E, n = 93 neurons), and CTSD (F, n = 109 neurons) vesicles in cortical neuronal cell bodies.
To further demonstrate that NF proteins are present in AVs in vivo, we performed IEM of mouse (n = 4, 6 months) optic axons (Figure 6), cortical neuronal cell bodies (Fig. S3), and mouse primary cortical neurons (Fig. S4–S6) with NF antibodies. The double-membrane AVs have a very short half-life (5–10 min) in mammalian cells [27], and their sizes and shapes significantly vary based on the state of the cell and depending on the cargo [28]. Our IEM results with NF antibodies demonstrated that NEFM antibody (RMO44) labels axonal neurofilaments (indicated with purple arrows in Figure 6A,E,F), and all three NF antibodies labeled double membrane-limited AVs (yellow arrows in Figure 6C for NEFM; red arrows in Figure 6B for NEFL, and purple arrows in Figure 6D for NEFH) in optic axons in the size range of 600–1000 nm. We also found NF protein labeling in lumens of much smaller AVs (260–600 nm) (Figure 6E,G,I) that do not have clear double membrane or appear as a single membrane (Figure 6A,H) may represent ALs. Similarly, labeling of cortical neuronal cell bodies with NEFH, NEFM, and NEFL antibodies demonstrate that NF proteins are present in the lumens of single membrane AVs (Fig. S3A–F, NF label is indicated with arrows in each AV) in the size range of 600–960 nm. Immunolabeling of NFs in these AVs is specific since the antibodies did not label the microtubules (MT, blue arrows in Figure 6A,F) or mitochondria (Mito., Figure 6D), and there was no labeling of AVs that were incubated with only mouse (Fig. S3 G) or rabbit secondary antibodies (Fig. S3 H).
To examine the interactions between NF proteins and autophagic vesicles in primary cultures, mouse primary cortical neurons from mouse 17-day embryos (E17) with 7–10 DIV were treated with Con A (10 nM) for 6 h, immuno-colocalized with the AP marker MAP1LC3 and NF antibodies. Our colocalization data demonstrate that all three NF proteins were colocalized with MAP1LC3 in neuronal processes of mouse cortical neurons (Fig. S4A–C) although the occurrence of these interactions were very rare (similar colocalization in untreated cells was not observed). To further examine NFs and AVs interaction in these cultures, we performed IEM with NF antibodies and our results show that NF antibodies decorated various sizes of single membrane AVs (300–700 nm) in untreated mouse primary cortical neurons (Fig. S5A–F). Con A-treated neurons contained much bigger AVs (600–1078 nm) with NF labeling within the lumen of the vesicles (see arrows for NF labeling within AVs, Fig. S5G–L) and the same vesicles also contained multiple membrane structures (Fig. S5G–L). We further examined the neurites of mouse primary cortical neurons for the presence of NF proteins by performing IEM with NF antibodies. We found all the three NF antibodies decorating the filamentous structures in the neurites of untreated (Fig. S6A,C,E) and Con A-treated (Fig. S6B,D,F) cultures. Con A-treated neurites with NEFL antibody (NR-4) identified an AV with membranous structure within the lumen showed NEFL labeling (Fig. S6F, see the arrow within the AV).
Our IEM analyses of NF proteins with mouse optic axons, and mouse primary cortical neurons identified short filamentous structures within the lumen of AVs that were labeled with NF antibodies (see arrow heads for string of gold particles in Figure 6G, in optic axon and in mouse primary cortical neuron cell bodies in Fig. S5C–D). Most of the NF labeling detected within the lumen of AVs do not have filamentous structures (Figure 6A–E; Fig. S3A–F; Fig. S5A–B,E,G–L), which indicated non-filamentous oligomers and/or monomers of NF subunits are present in these AVs. Based on these data, we propose that, consistent with the bulk of sequestration capability of macroautophagy, AVs contain neurofilament proteins in various assembly forms, including short filament/protofilament assemblies, oligomers, or subunits.
To further demonstrate that NFs are degraded through autophagosomal-autolysosomal compartments in vivo, we isolated subcellular fractions from mouse brain enriched in APs, ALs, LYs, and mitochondria. Immunoblot analyses revealed the presence of NF subunits in AP and AL fractions (Figure 7A) while smaller amounts of NF proteins were present in LYs (Figure 7A), presumably due to degradation by cathepsin CTSD [17]. By contrast, NF proteins were absent in a mitochondrial fraction (Figure 7A) that was enriched in TOMM20 (translocase of outer mitochondria membrane 20) [29].
Figure 7.
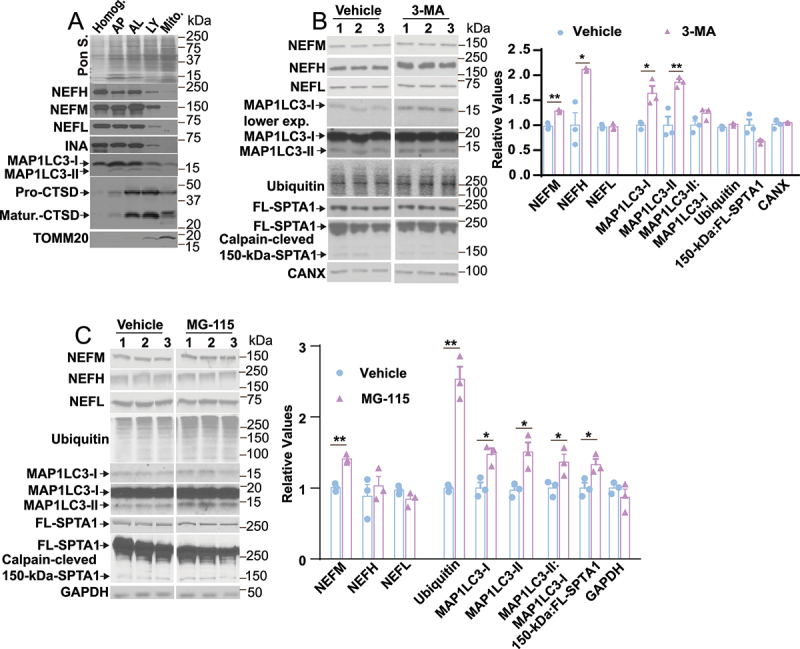
Nfs are enriched in AP, and AL fractions, and infusion of 3-MA and MG-115 into mouse brain increases NF protein levels in vivo. (A) Organelle fractions (20 µg) from mouse brain except for total homogenate (Homog. 7 µg) were immunoblotted with NEFM, NEFHCOOH, NEFL, INA, MAP1LC3, CTSD and TOMM20 antibodies. Pon. S- Ponceau S stain. ICV infusion of 3-MA (9 mg/ml) into the right ventricle of mice and measurement of NEFM (B, 1.3-fold, n = 3, p < 0.0017), NEFH (B, NEFHCOOH, 2.1-fold, n = 3, p < 0.0114), NEFL (B, n = 3, p = 0.9066), MAP1LC3-I (B, n = 3, p = 0.0142), MAP1LC3-II (B, n = 3, p = 0.0090), the ratio of MAP1LC3-II:MAP1LC3-I (B, n = 3, p = 0.0967), ubiquitin (B, n = 3, p = 0.0874), the ratio of calpain-cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 (B, n = 3, p = 0.0508) and CANX (B, n = 3, p = 0.5436) proteins. ICV Infusion of MG-115 (0.1 mg/ml) into the right ventricle and measurement of NEFM (C, 1.4-fold, n = 3, p < 0.0011), NEFL (C, n = 3, p = 0.1349), NEFH (C, NEFHCOOH, n = 3, p = 0.5212), Ubiquitin (C, n = 3, p < 0.0010), MAP1LC3-I (C, n = 3, p < 0.0145), MAP1LC3-II (C, n = 3, p < 0.0220), the ratio of MAP1LC3-II:MAP1LC3-I (C, n = 3, p < 0.0489), the ratio of calpain-cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 (C, n = 3, p < 0.0349) and GAPDH (C, n = 3, p = 0.3512) proteins. Veh: Vehicle. Panels B and C, unpaired t-test; *p < 0.05; **p < 0.01.
Intraventricular infusion of 3-MA in mice in vivo prevents autophagic degradation of NF proteins
To assess whether autophagy is constitutively active in the degradation of NFs in vivo in the brain, we infused 3-MA (9 mg/ml) with ALZET osmotic pumps into the lateral ventricle of mice for four weeks. This concentration was previously shown not to cause detectable neurotoxicity. Immunoblots of cortex/HP from 3-MA infused mice demonstrated elevated levels of NEFM and NEFH (Figure 7B) but not NEFL (Figure 7B) compared to vehicle treatment. 3-MA treatment significantly increased MAP1LC3-I (Figure 7B), MAP1LC3-II (Figure 7B) levels, while it did not alter the ratio of MAP1LC3-II:MAP1LC3-I (Figure 7B). Importantly, the unaltered ratio of calpain-cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 (Figure 7B) and unaltered levels of ubiquitin upon 3-MA treatment indicate unchanged calpain and proteasome activities in mouse cortex/HP.
MG-115 infusion slows only NEFM proteolysis in mouse brain
To determine the contribution of proteasome activity to NF proteolysis in vivo, mice were infused with MG-115 (0.1 mg/ml, 4 weeks duration) into the right ventricle of C57BL/6J mice and NF protein levels were measured in cortex/hippocampus by immunoblot analyses. An elevated level of total ubiquitin (Figure 7C) [30] after MG115 treatment confirmed the effectiveness of the inhibitor. MG115 significantly prevented the degradation of NEFM (Figure 7C) but had no effect on the degradation of NEFL, NEFH, and GAPDH (Figure 7C). MG-115 infusion also significantly increased MAP1LC3-I (Figure 7C), MAP1LC3-II (Figure 7C), the ratio of MAP1LC3-II:MAP1LC3-I (Figure 7C), and the ratio of calpain-cleaved 150-kDa-SPTA1 fragment:FL-SPTA1 (Figure 7C); however, despite statistical significance, the changes of MAP1LC3 and SPTA1 are very small and unlikely to influence NF levels.
Discussion
This study is the first demonstration that the subunits of neurofilaments are substrates for autophagy and that steady-state levels of NFs in vivo are modulated in part by autophagic activity. At least five lines of evidence from the study support an autophagic turnover of NF proteins: (1) levels of exogenously expressed NF proteins are elevated by inhibiting autophagy with 3-MA or depletion of autophagy protein ATG5 with its shRNA in vitro, depletion of ATG5 combined with 3-MA exposure, or by inhibiting lysosomal proteolysis with Con A in N2a cells (Table 1). (2) In vivo ICV infusion of 3-MA in mice increases NF levels in the mouse brain (Table 2). (3) NF proteins are unequivocally localized within neuronal APs and ALs/LYs by double immunofluorescence cytochemistry, immunoelectron microscopy, and by biochemical analyses of subcellular fractions. (4) Moreover, autophagy inhibitor 3-MA protected NEFL and NEFM proteins more effectively compared to the proteasome inhibitor, MG-132, and the calpain inhibitor, calpeptin in N2a cells (Table 1). Additionally, 3-MA was much more effective in protecting NEFM and NEFH than MG-115 in mice (Table 2). (5) Finally, activation of autophagy with rapamycin reduced neurofilament protein levels in N2a cells.
Table 2.
Effect of inhibitors on NF protein levels in mouse brain.
| Inhibitor effect in mouse brain on NF proteins (fold increase) | |||
|---|---|---|---|
| Proteins | 3-MA | MG-115 | CAST Tg |
| NEFH | 2.114 ± 0.020 (*) | NC | NC |
| NEFM | 1.312 ± 0.017 (*) | 1.408 ± 0.035 (*) | 0.14 (Ref. 41) |
| NEFL | NC | NC | 0.07 (Ref. 41) |
Note: Effect of autophagy (3-MA), proteasome (MG115) and calpain (CAST, calpastatin) inhibitors in brains on NF protein levels in mice. The fold increases are over vehicle treated or untreated samples (one-fold increase equals to 100% increase). NC, no change. Significance is indicated in parenthesis. *p < 0.05. The data from Figure 7B, Figure 7C and from Reference 41 are compared in Table 2.
Our findings in N2a cells and in vivo in the brain further document that NF proteins are substrates for all three major proteolytic systems (calpain, proteasome, and autolysosomal) and that the different subunits vary in their relative susceptibility to a given proteolytic system. The basis for the observed differential susceptibility to a given protease system in a given cell or in the brain likely reflects multiple factors, including the different structural properties of the subunits and their post-translational modifications, their assembly state in different cell models, which affects accessibility to proteases, the varying composition of proteolytic activities in different cell types, and the variable effectiveness of proteolysis inhibitors at the concentrations used. Although it is well established that calpains execute limited proteolysis on native proteins, they can also act on selected cleavage products that they, or other proteases generate. It is likely, however, which the complete digestion of a given NF protein involves the participation of all three proteolytic systems studied here and possibly other cellular proteases. Evidence suggests that the major proteolytic systems cross talk, Gigaxonin protein involved in NF protein degradation through the ubiquitin protease system (UPS) also controls autophagy by the turnover of ATG16L1 [31,32], calpains with autophagy [33], and calpains with the UPS [34,35]. We speculate that the incomplete protection of NF degradation by individually inhibiting activity of each of the three studied proteolytic systems may in part reflect a need to block more than one proteolytic system at a time. Increased protection seen with 3-MA in NF protein levels compared to other inhibitors may be due to 3-MA partial inhibition of calpain (N2a cells) in NF-transfected cells and, for NEFM specifically, proteasome activities in the mouse brain. Also, given the exceptional metabolic stability of the neuronal NF cytoskeleton in adult mouse brain (half-life 2 months) [11,24,36] and the multiple factors (phosphorylation, interactions with cross-linking proteins, differential turnover rates of the NF subunits), a month of partial inhibition of a single proteolytic system in vivo may be insufficient time to allow reassembly of the axonal NF cytoskeleton, which requires months to build during development of the mouse brain. Other intermediate filament proteins, such as vimentin in cells [37], and keratin in mice have been shown to undergo autophagy [38].
The NEFL gene produces two alternately spliced mRNAs [39] and both these mRNAs could produce different size proteins. The change in levels of NEFL species in response to the PtdIns3K inhibitor, 3-MA, may be due to inhibition of some of the proteases that degrade higher molecular weight NEFL, while the lower molecular weight NEFL may be degraded by Con A-sensitive lysosomal proteases.
We demonstrate that autophagy degrades NF subunit proteins constitutively in normal neurons (Figure 8). ATG5-depletion prevents AP formation. 3-MA treatment inhibits PtdIns3K, which prevents AP formation and NF sequestration, thereby blocking its autophagy (Figure 8).
Figure 8.

A model depicting NF subunit protein degradation through autolysosomal pathway in neurons. NF subunit proteins are sequestered into APs and degraded in ALs after fusion with LYs. Inhibition of autophagy initiation with ATG5-depletion or with 3-MA at initiation (by inhibiting PtdIns3K) as well as clearance in AL may prevent NF degradation through autolysosomal pathway in neurons.
Calpains can initiate the rapid degradation of the assembled NF cytoskeleton in pathological states and injury models by cleaving the long C-terminal tails of NEFM and NEFH, a process that is opposed by the extensive phosphorylation of the tails as the NF cytoskeleton matures in developing axons [9,10,40]. Calpains, however, only carry out limited proteolysis of substrates and the proteases responsible for the complete degradation of NF subunits are unknown. NFs are rapidly degraded by calpain under experimental conditions of axotomy, traumatic brain injury, and neurodegenerative disorders [13,41,42]. NF cytoskeleton is rapidly dismantled under conditions of Wallerian degeneration or retraction of axons during development. Conversely, NFs accumulate as a signature pathological feature of ALS [43–45] and related neuroaxonal dystrophies [46–48], and are components of the abnormal inclusions in other neurofibrillary diseases, such as AD [5] and Pick disease [49]. We hypothesize that autophagy normally degrades NF subunit proteins constitutively in neurons, but in neurons with defective autophagy, NFs buildup in cell bodies and proximal axons to become the pathological hallmarks of ALS and AD. We propose that the autophagic pathway may modulate NF subunit protein levels in axons and dendrites and thereby influence axonal diameters and nerve conduction velocity in neurons in normal and disease states.
Materials and methods
All the animal protocols described in the paper were approved by the Animal Care and Use Committees of the Nathan Kline Institute and NYU Langone Medical Center.
Mice
Both male and female mice of C57BL/6J (6–9 months) were utilized for our studies.
DNA constructs
Full-length mouse genomic Nefh (a kind gift from Dr. Don Cleveland, University of California, San Diego, La Jolla, CA, USA), mouse genomic Nefm and Nefl clones were kindly provided by Dr. Jean-Pierre-Julien (University of Laval, Canada) were cloned into pcDNA 3.1 expression vector was described previously [24].
Antibodies and Fine chemicals
The following monoclonal antibody (mAb) and polyclonal antibodies (pAb) are used in the study: mAb RMO44 for NEFM subunit (ThermoFisher Scientific, 13–0500), pAb to NEFH (NEFHCOOH) [39,50]; The following antibodies and reagents are from Sigma-Millipore: NEFL mAb NR-4 (N5134) and polyclonal Ab (AB9568), NEFM polyclonal Ab (AB1987), mAbs to TUBA/α-tubulin (05–829), TUBB (T8328), INA (I0282), SPTA1 (MAB1622), ATG5 (MAB2605), calpeptin (117,591-20-5), GAPDH (CB1001), 3-methyladenine (3-MA, M9281), (Con A, 344-085), rapamycin (37,094), and cycloheximide (C7698); the MYC-tagged NEFH with mAb 9E10 (Santa Cruz Biotechnology, SC-40), SQSTM1/p62 (sequestosome 1; Cell Signaling Technology, 23,214), Rabbit MAP1LC3 (Novous Biologicals,100–2220), mouse MAP1LC3 (MBL International Corporation, M152–3), CTSD (Rudy-4 [51]; D-2-3 [52]), Ad5-hAtg5-shRNA (shADV-201,644), and Ad5-SC-shRNA (1122N) (Vector Biolabs), ubiquitin (DAKO, Z0458), CANX/calnexin (Assay Designs, SPA-860), MG-115 (474,780), and MG-132 (474,791) (Calbiochem).
Growth of Neura-2A (N2a) cells, transfections, transductions, and inhibitor treatments Undifferentiated N2a cells were transfected with NF-subunit genes (Nefl, Nefm, and Nefh) as previously described to form NF subunits into organized NF networks [24]. We can detect only NEFH with NEFH polyclonal antibodies [47] in undifferentiated and untransfected (UT) N2a cells but fail to detect NEFM (with RM044) or NEFL (NR-4) on immunoblots since these genes are repressed [23]. Undifferentiated N2a cells were grown in Dulbecco’s Modified Eagle Medium (Gibco, 11,955–065) with 10% fetal bovine serum (Gibco, 16-000-069) with antibiotics (Penicillin/Streptomycin, Gibco, 05140–122) at 0.5 x 106 cells/well were plated on 6 well plates, 1.5-µg of DNA for each construct was transfected with Lipofectamine 2000 (ThermoFisher Scientific, 11,688,019) (in each experiment all 3 WT-NF constructs were used at equal concentrations) for 4 h, fresh medium was replenished, and after 24 h, cells were grown in presence or absence of an autophagy inhibitor, 3-methyladenine (3-MA, 5 mM, 42 h); proteasome inhibitor, MG115 (0.5 µM, 24 h); lysosomal proteolysis inhibitor, Con A (25 nM, 24 h); or calpain inhibitor, calpeptin (40 µM, 24 h) or rapamycin (10 nM, 24 h) were added in each experiment. Protein synthesis was inhibited in NF-transfected cells by treating with cycloheximide (10-µg/ml) for 0–180 min to examine proteolysis. For Atg5-shRNA (Ad5-eGFP-U6-h/m-Atg5-shRNA; Vector Biolabs, 201,644) transduction, cells were first transfected with NF genes for 4 h, after 24 h cells were transduced with (100 Genome copies/cell, stock 2.4x1010 GC/ml) Atg5-shRNA or SC-shRNA (Ad5-eGFP-U6-scrambled-shRNA; Vector Biolabs, 1122; 100 Genome copies/cell, stock 5 × 1010 GC/ml) for 24 h, medium changed, cells were harvested after 48 h of transduction. For combined treatment of Atg5-shRNA transduction with 3-MA, first cells were transfected with NF subunit genes for 4 h, medium changed, after 24 h cells were transduced with Atg5-shRNA as indicated above for 24 h, then fresh medium replenished with or without 3-MA (5 mM) for 42 h. Cells were washed in 1X phosphate-buffered saline (PBS, pH 7.4; Gibco, 10,010–023), scraped in 1X PBS, centrifuged, and lysed in a mammalian protein extraction reagent (Pierce, 78,501) with protease and phosphatase inhibitors. Protein concentration was determined using bicinchoninic acid assay kit (ThermoFisher Science, 23,225). Equal amounts of protein extracts were immunoblotted with the antibodies indicated above.
SDS-Polyacrylamide gel electrophoresis and immunoblot analyses
Mouse tissue extracts were made from 6–9-month-old animals (male and female) as described previously [12], and protein concentrations were determined as indicated earlier, and equal amounts of protein extract were separated on SDS-polyacrylamide gels, transferred to nitrocellulose membranes, and incubated with primary antibodies. Blots were then washed, incubated with appropriate secondary antibodies conjugated with HRP (Southern Biotech, goat anti-mouse HRP, 1030–05; goat anti-rabbit HRP, 4030–05), and developed with ECL reagent and immunoreactive bands were quantified using Multi Gauge V3.0 (Fuji Film, Japan).
Intracerebroventricular (ICV) infusion of 3-MA and MG-115
3-methyladenine (3-MA, 9 mg/ml in 50% DMSO containing 20 mM HEPES, pH 7.4) or MG-115 (0.1 mg/ml in 25% DMSO containing 20 mM HEPES, pH 7.4) or the vehicle was infused into the lateral ventricle [51] of 9-month-old mice (n = 3 for each treatment) for a period of 4 weeks using an ALZET osmotic pump brain infusion kit (ALZET Osmatic Pumps, Model 2004 mini-osmotic pump) (0.25 µl/h delivery rate). ALZET pumps were loaded with testing drug or the vehicle and connected to the brain infusion assembly with polyethylene tubing and incubated in sterile saline at 37ºC for 48 h prior to implant. Mice were anesthetized with a mixture (0.01 ml/g body weight, i.p., 26 G needle) of ketamine (10 mg/ml) and xylazine (1 mg/ml) and placed in a stereotaxic apparatus with a mouse adapter (David Kopf Instruments). The scalp was shaved, and a midline incision made starting slightly behind the eyes, exposing the skull area. Through this opening, a hemostat was used to open a pocket on the back of the mouse to house the pump. The coordinates for the cannula placement in the lateral ventricle were AP −0.3 mm to Bregma, ML 1.0 mm to Bregma, and DV 2.5 mm to cranium. Cannula length was adjusted with spacers. A hole was drilled in the skull, the cannula was glued to the cleaned and scraped skull with Loctite 454, and the incision closed over the assembly. After infusion, cortex/hippocampal areas were dissected from mice, homogenized in THB buffer (tissue homogenization buffer: 0.32 M sucrose, 20 mM Tris-HCl, pH 7.4, 1 mM each of EDTA, EGTA, and DTT; 10% homogenates) and the protein estimation was done using the BCA method. Immunoblots using antibodies indicated above were performed with equal protein loading.
Subcellular fractionation.
Fresh cerebral cortices (n = 4 mice, both sexes, 6 months old) were homogenized and centrifuged to remove nuclei and unbroken cells. Post-nuclear supernatants were subjected to differential centrifugation and by flotation in a discontinuous gradient of Nycodenz® (Accurate chemical and scientific corporation, AN1002424; 50%, 26%, 24%, 20%, and 10%) to isolate fractions enriched mainly in APs at an interface of 10–20%, ALs at 20–24%, LY at 24–26% and mitochondria at 26–50% interface [29,53]. These fractions were washed in 0.25 M sucrose (Sigma-Millipore, S0389), centrifuged, and the pellets were resuspended in 1X PBS, protein assayed and subjected to immunoblots (20-µg of protein) with NF and organelle-specific antibodies.
Immunohistochemistry.
WT mice at 9 months of age (n = 4, male and female mice) were perfused with 4% paraformaldehyde, sagittal brain sections of 40 µm were immunostained with NEFM mAb (RMO44) or RM044 + CTSD (Rudy-4) [51], or RMO44+ MAP1LC3 (pAb, Novus) as described previously [12]. After washes, sections were incubated with appropriate secondary antibodies, washed, and sections were mounted onto cover slips and images from layer III and V of the cortex were captured with Zeiss LSM880 confocal microscope with a pan Apochromat 40x/1.4 oil objective lens, and optically zoomed to 100x (2.5 times) using the following steps: Alexa Fluor 488 (ex:488, em490–560 with MBS 488), and Alexa Fluor 568 (ex: 561, em:582–640 with MBS 458/561). As NEFM antibody strongly labels NF networks in neuronal cell bodies and proximal dendrites (Fig. S2A,B). To visualize NEFM vesicular structures in neuronal cell bodies, we intentionally attenuated contributions of signal from background labeling and the fibrous neurofilaments. We color balanced the images before using the filter for noise reduction in Adobe Photoshop CC 2015. More specifically, we increased the highlights of the green and red channels by 100 with the “color balance” function and subsequently reduced the shadows of these channels by 30 and 25, respectively. We then removed the background with “Dust and Scratch” filter using the radius of 2 and threshold of 25 on all images. These NEFM-positive vesicular structures in cortical cell bodies were colocalized with MAP1LC3 and CTSD vesicles. To follow the colocalization of NEFM-MAP1LC3 and NEFM-CTSD puncta, Z-stack image series were captured at 0.1 µm thickness and colocalized puncta in images were shown with arrows. CTSD and MAP1LC3 puncta colocalizations were determined using the redirect function of the NIH Image J program.
Preparation of mouse cortical primary neurons
Mouse primary cortical neurons were prepared from E17 embryos of C57BL/6J timed pregnant mice as described previously [54]. Briefly, cerebral cortices were dissected from E17 pups on ice, minced, incubated in Hibernate E medium (Brainbits, HE) containing 10 U/ml of papain (Worthington Biochemical, LK003176) and 0.2 mg/ml of DNase (Sigma-Aldrich, DN25) at 37°C for 15 min, then equal volume of Hibernate E medium with 10% FBS was added, and centrifuged in Sorvall ST 16 R at 184x g for 5 min. Pellets containing the cells were resuspended in plating medium (DMEM/F12, GlutaMax supplement; ThermoFisher, 10,565–018) containing 5% horse serum (Gibco, 10,650,122) and 5% FBS (Gibco, 16,000,069) filtered (through 40-µm nylon membrane filter), neurons in filtrate were plated on poly-D-lysine-coated glass cover slips at a density of 0.2 x 106 cells/cm2 in plating medium or on 6 well plates at a density of 3 × 106 cell/ml. The medium on the cells was replaced with Neurobasal medium (ThermoFisher, 12,348–017) supplemented with B27 (ThermoFisher, 17,504–044) and GlutaMax (ThermoFisher, 35,050–061) after 1.5 h. Neurons in the culture was maintained for 7–10 DIV at 37°C in 95% air and 5% CO2 with half of the medium replenished every 3 days until cells were used. Some of the cells on coverslips were treated with Con A (10 nm) for 6 h and processed for ICC and immunoelectron microscopy (IEM).
Immunocytochemistry and Immuno-EM of mouse primary cortical neurons
After 7 days of differentiation, neurons on cover slips were treated with Con A (10 µM) for 6 h, fixed in 4% paraformaldehyde in cacodylate buffer for 30 min, and the neurons on 6-well plates were fixed in 4% PFA +2.5% glutaraldehyde at RT for 30 min and then plates were stored at 4°C overnight. Cells on cover slips were subjected to immunocytochemistry with neurofilament and MAP1LC3 antibodies as described above, and the primary neurons in 6-well plates were immunostained with neurofilament antibodies for IEM as described below.
N2a Cells or mouse primary cortical neurons grown in 6 well plates were fixed as indicated above and processed the next day with 1% osmium tetroxide for 30 min followed by washes and then incubation with 1% uranyl acetate at 4°C in the dark overnight. The next day, the cells were washed and dehydrated with increasing concentrations of ethanol followed by infiltration with increasing concentrations of Spurr’s resin. Excess resin was drained off the plate and resin-filled capsules were inverted over the infiltrated cell layer before being incubated at 65°C overnight. The next day, the capsules were snapped off and block face trimmed before being sectioned on a Leica Ultramicrotome using a diamond knife. Sections were cut at 70-nm thickness and collected on 75-mesh carbon/formvar coated nickel grids (Electron Microscopy Sciences). Sections were then etched with 4% sodium metaperiodate for 10 min, washed twice in water, and blocked for 1 h. Grids were incubated with a combination of either NEFL and NEFM or NEFL and NEFH antibodies at 4°C overnight. Some of the grids were also incubated separately with NEFL, NEFM, and NEFH antibodies. The next day the grids were washed seven times in 1X PBS, and were then incubated with either mouse (6 nm; Electron Microscopy Sciences, 25,124) or rabbit (6 nm; Electron Microscopy Sciences, 25,104) or both anti-mouse 10 nm (Electron Microscopy Sciences, 25,129) and anti-rabbit 6 nm (Electron Microscopy Sciences, 25,104) gold secondary antibodies for 1 h. Then the grids were washed seven times in 1X PBS, and twice in distilled water. Grids were finally post stained in 1% uranyl acetate for 5 min followed by two washes in water and then stained with lead citrate for 5 min followed by final two washes in distilled water. The grids were then imaged with a Ceta Camera on a Thermo Fisher Talos L120C operating at 120 kV.
Immuno-EM of mouse cortical neurons and optic axons
C57BL/6J mice of 6-month-old (both sexes, n = 4 for each genotype) were perfused transcardially with 4% paraformaldehyde, 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.2, brains and optic nerves (OPN) were post fixed for 12 h, brain sections (500 µm) and OPN were treated with 2% osmium tetroxide, washed, dehydrated, and embedded in Durcupan (Sigma-Aldrich, 44,611). NF-gene-transfected N2a cells after 42 h were washed in 1X PBS and fixed in EM fixative as indicated above. Ultra-thin sections (60 nm) of Hippocampus, OPN, and N2a cells were stained with NEFM (RMO44 mAb) or NEFM (Millipore-Sigma, polyclonal, AB1987) + NEFL (mAb, NR-4) or NEFL (mAb, NR-4) + NEFH antibodies (NEFHCOOH), and followed by either with 10-nm gold conjugated with goat-anti mouse secondary Ab (Electron Microscopy Sciences, 25,129) or with 6-nm gold conjugated with goat-anti mouse secondary Ab (Electron Microscopy sciences, 25,124) to detect NEFL and 15-nm gold conjugated with goat-anti rabbit secondary Ab (Electron Microscopy Sciences, 25,112) to detect NEFM. The images were captured with a Jeol 100 CX Transmission electron microscope at 80 KV with Eagle 4k x 4k camera as described previously [55]. NF labeled AVs (double-membrane APs, and single-membrane ALs) were identified by previously published morphological criteria [29,56] in cortical neuronal cell bodies and optic axons.
Statistical analyses.
We performed two-tailed unpaired t-test for single comparison, and One-way ANOVA analyses (with Tukey’s post-hoc) for multiple comparison tests in GraphPad Prism 6 software to obtain quantitative data. Most of the data are presented as bar graphs with individual data points and mean ±SEM (fewer than 8 repeats). Statistical significance is indicated by asterisks *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. The number and ages of the animals used for each experiment is indicated in each method.
Supplementary Material
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Funding Statement
This work was funded by the National Institutes of Health/National Institute on Aging AG005604 (R.A.N. and M.V.R). We thank Dr. Kuldeep Sachdeva for organizing the figures in Adobe Illustrator.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2124500
References
- [1].Waxman SG. Determinants of conduction velocity in myelinated nerve fibers. Muscle & nerve. PubMed PMID: 6245357. 1980;3(2):141–150. DOI: 10.1002/mus.880030207 [DOI] [PubMed] [Google Scholar]
- [2].Rao MV, Mohan PS, Kumar A, et al. The myosin Va head domain binds to the neurofilament-L rod and modulates endoplasmic reticulum (ER) content and distribution within axons. PLoS One. 2011;6(2):e17087. PubMed PMID: 21359212; PubMed Central PMCID: PMC3040190: 10.1371/journal.pone.0017087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yuan A, Sershen H, Veeranna, et al. Neurofilament subunits are integral components of synapses and modulate neurotransmission and behavior in vivo. Mol Psychiatry. 2015;20(8):986–994. PubMed PMID: 25869803: 10.1038/mp.2015.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Yuan A, Veeranna, Sershen H, et al. Neurofilament light interaction with GluN1 modulates neurotransmission and schizophrenia-associated behaviors. Transl Psychiatry. 2018;8(1):167. PubMed PMID: 30143609; PubMed Central PMCID: PMC6109052: 10.1038/s41398-018-0194-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Rudrabhatla P, Jaffe H, Hc P. Direct evidence of phosphorylated neuronal intermediate filament proteins in neurofibrillary tangles (NFTs): phosphoproteomics of Alzheimer’s NFTs. Faseb J. 2011;25(11):3896–3905. PubMed PMID: 21828286; PubMed Central PMCID: PMC3205835: DOI: 10.1096/fj.11-181297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shaw G, Yang C, Ellis R, et al. Hyperphosphorylated neurofilament NF-H is a serum biomarker of axonal injury. Biochem Biophys Res Commun. 2005;336(4):1268–1277. PubMed PMID: 16176808: 10.1016/j.bbrc.2005.08.252. [DOI] [PubMed] [Google Scholar]
- [7].Takahashi H, Aoki Y, Nakajima A, et al. Phosphorylated neurofilament subunit NF-H becomes elevated in the cerebrospinal fluid of patients with acutely worsening symptoms of compression myelopathy. J Clin Neurosci. 2014;21(12):2175–2178. PubMed PMID: 25065845: 10.1016/j.jocn.2014.04.021. [DOI] [PubMed] [Google Scholar]
- [8].Bacioglu M, Maia LF, Preische O, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron. 2016;91(2):494–496. PubMed PMID: 27477021: 10.1016/j.neuron.2016.07.007. [DOI] [PubMed] [Google Scholar]
- [9].Hall GF, Lee VM. Neurofilament sidearm proteolysis is a prominent early effect of axotomy in lamprey giant central neurons. J Comp Neurol. 1995;353(1):38–49. PubMed PMID: 7714248: DOI: 10.1002/cne.903530106. [DOI] [PubMed] [Google Scholar]
- [10].Pant HC. Dephosphorylation of neurofilament proteins enhances their susceptibility to degradation by calpain. Biochem J. 1988;256(2):665–668. PubMed PMID: 2851997; PubMed Central PMCID: PMC1135461: DOI: 10.1042/bj2560665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nixon RA, Logvinenko KB. Multiple fates of newly synthesized neurofilament proteins: evidence for a stationary neurofilament network distributed nonuniformly along axons of retinal ganglion cell neurons. J Cell Biol. 1986;102(2):647–659. PubMed PMID: 2418034; PubMed Central PMCID: PMC2114090: DOI: 10.1083/jcb.102.2.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Rao MV, McBrayer MK, Campbell J, et al. Specific calpain inhibition by calpastatin prevents tauopathy and neurodegeneration and restores normal lifespan in tau P301L mice. J Neurosci. 2014;34(28):9222–9234. PubMed PMID: 25009256; PubMed Central PMCID: PMC4087203: 10.1523/JNEUROSCI.1132-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ma M, Ferguson TA, Schoch KM, et al. Calpains mediate axonal cytoskeleton disintegration during Wallerian degeneration. Neurobiol Dis. PubMed PMID: 23542511; PubMed Central PMCID: PMC3721029 2013;56: 34–46.doi: 10.1016/j.nbd.2013.03.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chen PC, Qin LN, Li XM, et al. The proteasome-associated deubiquitinating enzyme Usp14 is essential for the maintenance of synaptic ubiquitin levels and the development of neuromuscular junctions. J Neurosci. 2009;29(35):10909–10919. PubMed PMID: 19726649; PubMed Central PMCID: PMC2766780: 10.1523/JNEUROSCI.2635-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Israeli E, Dryanovski DI, Schumacker PT, et al. Intermediate filament aggregates cause mitochondrial dysmotility and increase energy demands in giant axonal neuropathy. Hum Mol Genet. 2016;25(11):2143–2157. PubMed PMID: 27000625; PubMed Central PMCID: PMC5081048: 10.1093/hmg/ddw081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mahammad S, Murthy SN, Didonna A, et al. Giant axonal neuropathy-associated gigaxonin mutations impair intermediate filament protein degradation. J Clin Invest. 2013;123(5):1964–1975. PubMed PMID: 23585478; PubMed Central PMCID: PMC3635735: 10.1172/JCI66387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nixon RA, Marotta CA. Degradation of neurofilament proteins by purified human brain cathepsin D. J Neurochem. 1984;43(2):507–516. PubMed PMID: 6429280: DOI: 10.1111/j.1471-4159.1984.tb00928.x. [DOI] [PubMed] [Google Scholar]
- [18].Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889. PubMed PMID: 16625204: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- [19].Yao J, Jia L, Feathers K, et al. Autophagy-Mediated catabolism of visual transduction proteins prevents retinal degeneration. Autophagy. 2016;12(12):2439–2450. PubMed PMID: 27753525; PubMed Central PMCID: PMC5173283: 10.1080/15548627.2016.1238553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Moreno ML, Merida S, Bosch-Morell F, et al. Autophagy dysfunction and oxidative stress, two related mechanisms implicated in retinitis pigmentosa. Front Physiol. 2018;9:1008. PubMed PMID: 30093867; PubMed Central PMCID: PMC6070619: DOI: 10.3389/fphys.2018.01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. PubMed PMID: 23921753: DOI: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- [22].Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. PubMed PMID: 16874052: DOI: 10.4161/auto.1.2.1697. [DOI] [PubMed] [Google Scholar]
- [23].Bruce J, Schwartz ML, Shneidman PS, et al. Methylation and expression of neurofilament genes in tissues and in cell lines of the mouse. Brain Res Mol Brain Res. 1993;17(3–4):269–278. PubMed PMID: 8510499: DOI: 10.1016/0169-328x(93)90011-d. [DOI] [PubMed] [Google Scholar]
- [24].Yuan A, Sasaki T, Rao MV, et al. Neurofilaments form a highly stable stationary cytoskeleton after reaching a critical level in axons. J Neurosci. 2009;29(36):11316–11329. PubMed PMID: 19741138; PubMed Central PMCID: PMC2788791: 10.1523/JNEUROSCI.1942-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lee JA, Gao FB. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J Neurosci. 2009;29(26):8506–8511. PubMed PMID: 19571141; PubMed Central PMCID: PMC2726650: DOI: 10.1523/JNEUROSCI.0924-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cassidy LD, Young AR, Perez-Mancera PA, et al. A novel Atg5-shRNA mouse model enables temporal control of Autophagy in vivo. Autophagy. 2018;14(7):1256–1266. Epub 2018/07/13PubMed PMID: 29999454; PubMed Central PMCID: PMCPMC6103714: 10.1080/15548627.2018.1458172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annual review of nutrition. PubMed PMID: 17311494. 2007;27: 19–40. DOI: 10.1146/annurev.nutr.27.061406.093749 [DOI] [PubMed] [Google Scholar]
- [28].Jin M, Klionsky DJ. Regulation of autophagy: modulation of the size and number of autophagosomes. FEBS Lett. 2014;588(15):2457–2463. PubMed PMID: 24928445; PubMed Central PMCID: PMC4118767: DOI: 10.1016/j.febslet.2014.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yang DS, Stavrides P, Saito M, et al. Defective macroautophagic turnover of brain lipids in the TgCRND8 Alzheimer mouse model: prevention by correcting lysosomal proteolytic deficits. Brain. 2014;137(Pt 12):3300–3318. PubMed PMID: 25270989; PubMed Central PMCID: PMC4240291: 10.1093/brain/awu278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ang E, Pavlos NJ, Rea SL, et al. Proteasome inhibitors impair RANKL-induced NF-kappaB activity in osteoclast-like cells via disruption of p62, TRAF6, CYLD, and IkappaBalpha signaling cascades. J Cell Physiol. 2009;220(2):450–459. Epub 2009/04/15PubMed PMID: 19365810: 10.1002/jcp.21787. [DOI] [PubMed] [Google Scholar]
- [31].Vi K, Fm M, Dc R. Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 2010;584(7):1393–1398. PubMed PMID: 20040365: DOI: 10.1016/j.febslet.2009.12.047. [DOI] [PubMed] [Google Scholar]
- [32].Scrivo A, Codogno P, Bomont P. Gigaxonin E3 ligase governs ATG16L1 turnover to control autophagosome production. Nat Commun. 2019;10(1):780. PubMed PMID: 30770803; PubMed Central PMCID: PMC6377711: DOI: 10.1038/s41467-019-08331-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Menzies FM, Garcia-Arencibia M, Imarisio S, et al. Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 2015;22(3):433–444. PubMed PMID: 25257175; PubMed Central PMCID: PMC4326573: 10.1038/cdd.2014.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Demarchi F, Bertoli C, Copetti T, et al. Calpain is required for macroautophagy in mammalian cells. J Cell Biol. 2006;175(4):595–605. PubMed PMID: 17101693; PubMed Central PMCID: PMC2064596: 10.1083/jcb.200601024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Park C, Cuervo AM. Selective autophagy: talking with the UPS. Cell biochemistry and biophysics. PubMed PMID: 23709310; PubMed Central PMCID: PMC3758803. 2013;67(1):3–13. DOI: 10.1007/s12013-013-9623-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Millecamps S, Gowing G, Corti O, et al. Conditional NF-L transgene expression in mice for in vivo analysis of turnover and transport rate of neurofilaments. J Neurosci. 2007;27(18):4947–4956. PubMed PMID: 17475803: DOI: 10.1523/JNEUROSCI.5299-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang RC, Wei Y, An Z, et al. Akt-Mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959. PubMed PMID: 23112296; PubMed Central PMCID: PMC3507442: 10.1126/science.1225967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vijayaraj P, Kroger C, Reuter U, et al. Keratins regulate protein biosynthesis through localization of GLUT1 and -3 upstream of AMP kinase and Raptor. J Cell Biol. 2009;187(2):175–184. PubMed PMID: 19841136; PubMed Central PMCID: PMC2768834: DOI: 10.1083/jcb.200906094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rao MV, Houseweart MK, Williamson TL, et al. Neurofilament-Dependent radial growth of motor axons and axonal organization of neurofilaments does not require the neurofilament heavy subunit (NF-H) or its phosphorylation. J Cell Biol. 1998;143(1):171–181. PubMed PMID: 9763429; PubMed Central PMCID: PMCPMC2132801: DOI: 10.1083/jcb.143.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Greenwood JA, Troncoso JC, Costello AC, et al. Phosphorylation modulates calpain-mediated proteolysis and calmodulin binding of the 200-kDa and 160-kDa neurofilament proteins. J Neurochem. 1993;61(1):191–199. PubMed PMID: 8515266: DOI: 10.1111/j.1471-4159.1993.tb03555.x. [DOI] [PubMed] [Google Scholar]
- [41].Rao MV, Campbell J, Palaniappan A, et al. Calpastatin inhibits motor neuron death and increases survival of hSOD1(G93A) mice. J Neurochem. 2016;137(2):253–265. PubMed PMID: 26756888; PubMed Central PMCID: PMC4828294: DOI: 10.1111/jnc.13536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Saatman KE, Creed J, Raghupathi R. Calpain as a therapeutic target in traumatic brain injury. Neurotherapeutics. 2010;7(1):31–42. PubMed PMID: 20129495; PubMed Central PMCID: PMC2842949: DOI: 10.1016/j.nurt.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Matsumoto S, Kusaka H, Murakami N, et al. Basophilic inclusions in sporadic juvenile amyotrophic lateral sclerosis: an immunocytochemical and ultrastructural study. Acta Neuropathol. 1992;83(6):579–583. PubMed PMID: 1636375: DOI: 10.1007/BF00299405. [DOI] [PubMed] [Google Scholar]
- [44].Sasaki S, Warita H, Abe K, et al. Impairment of axonal transport in the axon hillock and the initial segment of anterior horn neurons in transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2005;110(1):48–56. PubMed PMID: 15920660: DOI: 10.1007/s00401-005-1021-9. [DOI] [PubMed] [Google Scholar]
- [45].Xu Z, Cork LC, Griffin JW, et al. Increased expression of neurofilament subunit NF-L produces morphological alterations that resemble the pathology of human motor neuron disease. Cell. 1993;73(1):23–33. PubMed PMID: 8462100: DOI: 10.1016/0092-8674(93)90157-L. [DOI] [PubMed] [Google Scholar]
- [46].Adebola AA, Di Castri T, He CZ, et al. Neurofilament light polypeptide gene N98S mutation in mice leads to neurofilament network abnormalities and a Charcot-Marie-Tooth Type 2E phenotype. Hum Mol Genet. 2015;24(8):2163–2174. PubMed PMID: 25552649; PubMed Central PMCID: PMC4380066: 10.1093/hmg/ddu736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cifuentes-Diaz C, Nicole S, Velasco ME, et al. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11(12):1439–1447. PubMed PMID: 12023986: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- [48].Samantaray S, Knaryan VH, Shields DC, et al. Critical role of calpain in spinal cord degeneration in Parkinson’s disease. J Neurochem. 2013;127(6):880–890. PubMed PMID: 23875735; PubMed Central PMCID: PMC3859802: DOI: 10.1111/jnc.12374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bu B, Klunemann H, Suzuki K, et al. Niemann-Pick disease type C yields possible clue for why cerebellar neurons do not form neurofibrillary tangles. Neurobiol Dis. 2002;11(2):285–297. PubMed PMID: 12505421: 10.1006/nbdi.2002.0551. [DOI] [PubMed] [Google Scholar]
- [50].Rao MV, Garcia ML, Miyazaki Y, et al. Gene replacement in mice reveals that the heavily phosphorylated tail of neurofilament heavy subunit does not affect axonal caliber or the transit of cargoes in slow axonal transport. J Cell Biol. 2002;158(4):681–693. PubMed PMID: 12186852; PubMed Central PMCID: PMC2174004: 10.1083/jcb.200202037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yang DS, Kumar A, Stavrides P, et al. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol. 2008;173(3):665–681. PubMed PMID: 18688038; PubMed Central PMCID: PMC2527090: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cataldo AM, Nixon RA. Enzymatically active lysosomal proteases are associated with amyloid deposits in Alzheimer brain. Proc Natl Acad Sci U S A. 1990;87(10):3861–3865. PubMed PMID: 1692625; PubMed Central PMCID: PMC54003: DOI: 10.1073/pnas.87.10.3861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. PubMed PMID: 19339967; PubMed Central PMCID: PMC2676208: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28(27):6926–6937. PubMed PMID: 18596167; PubMed Central PMCID: PMC2676733: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Lugli G, Larson J, Martone ME, et al. Dicer and eIf2c are enriched at postsynaptic densities in adult mouse brain and are modified by neuronal activity in a calpain-dependent manner. J Neurochem. 2005;94(4):896–905. PubMed PMID: 16092937: DOI: 10.1111/j.1471-4159.2005.03224.x. [DOI] [PubMed] [Google Scholar]
- [56].Yang DS, Stavrides P, Kumar A, et al. Cyclodextrin has conflicting actions on autophagy flux in vivo in brains of normal and Alzheimer model mice. Hum Mol Genet. 2017;26(5):843–859. PubMed PMID: 28062666; PubMed Central PMCID: PMC6075207: 10.1093/hmg/ddx001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.