

ABSTRACT
NPPA/atrial natriuretic peptide (natriuretic peptide type A) exerts critical pleiotropic effects in the cardiovascular system, limiting cardiomyocyte hypertrophy and death, reducing cardiac fibrosis and promoting vascular integrity. However, the molecular mechanisms underlying these beneficial effects still need to be clarified. We demonstrated for the first time that macroautophagy/autophagy is involved in the local protective effects of NPPA in cardiomyocytes (CMs), both in vitro and in vivo. Exogenous NPPA rapidly activates autophagy in CMs through NPR1/type A natriuretic peptide receptor and PRKG/protein kinase G signaling and also increases cardiac autophagy in mice. Remarkably, endogenous NPPA is secreted by CMs in response to glucose deprivation or hypoxia, thereby stimulating autophagy through autocrine/paracrine mechanisms. NPPA preserves cell viability and reduces hypertrophy in response to stress through autophagy activation. In vivo, we found that Nppa knockout mice undergoing ischemia-reperfusion (I/R) show increased infarct size and reduced autophagy. Reactivation of autophagy by Tat-Beclin D11 limits I/R injury. We also found that the protective effects of NPPA in reducing infarct size are abrogated in the presence of autophagy inhibition. Mechanistically, we found that NPPA stimulates autophagy through the activation of TFEB (transcription factor EB). Our data suggest that NPPA is a novel extracellular regulator of autophagy in the heart.
KEYWORDS: Atrial natriuretic peptide, autophagy, cardiomyocytes, ischemia-reperfusion, transcription factor EB
Introduction
NPPA/atrial natriuretic peptide (natriuretic peptide type A) is a cardiac hormone belonging to the family of natriuretic peptides (NPs) [1]. It is mostly secreted by atrial cardiomyocytes in response to mechanical stress, such as pressure or volume overload [2,3]. Once secreted, NPPA exerts diuretic, natriuretic, and vasodilatory effects, which together regulate water and salt balance and blood pressure levels [2,3]. These effects of NPPA are mostly mediated through the interaction with NPR1/type A natriuretic peptide receptor (natriuretic peptide receptor 1), a guanylate cyclase receptor widely distributed in the body, which promotes the elevation of intracellular levels of cyclic guanosine monophosphate (cGMP) and the activation of PRKG/protein kinase G (protein kinase, cGMP-dependent) [1].
Besides its systemic effects, NPPA plays critical pleiotropic effects in the cardiovascular system, through autocrine and paracrine mechanisms [4]. For example, NPPA is rapidly secreted by cardiomyocytes in response to mechanical stress, thereby limiting cardiac hypertrophy, fibrosis, remodeling, and dysfunction. Genetic ablation of both Nppa and Npr1 genes in mice exacerbated cardiac hypertrophy and maladaptive remodeling in response to mechanical overload [5–10]. In accordance, NPPA administration reduced cardiac damage in pre-clinical models of pressure overload and ischemia-reperfusion [11,12]. In addition, NPPA was also shown to be secreted by endothelial cells [13,14] and to exert pro-angiogenic and anti-inflammatory effects in the vascular system [15]. This evidence suggests that NPPA is an important regulator of cardiovascular remodeling. However, the molecular mechanisms underlying the pleiotropic effects of NPPA still need to be clarified.
Accumulating lines of evidence demonstrated that macroautophagy/autophagy is a critical regulator of cardiovascular homeostasis and response to stress. Autophagy is an intracellular mechanism devoted to the digestion of old, damaged, or unused cytoplasmic elements, such as dysfunctional organelles, misfolded or senescent proteins, through the use of double-membrane vesicles called autophagosomes, which finally fuse with lysosomes [16]. Amino acids, lipids, and nucleotides are then reused for other cellular purposes. Previous work showed that activation of autophagy limits cardiac remodeling and dysfunction in response to mechanical stress and myocardial infarction, reduces acute ischemic injury, attenuates genetic cardiomyopathies, and ameliorates metabolic cardiomyopathy [17–19]. In addition, autophagy preserves vascular integrity and promotes angiogenesis [20,21]. However, the mechanisms regulating autophagy in the cardiovascular system need to be fully elucidated, particularly the extracellular regulators. In this regard, the possible role of NPPA in the regulation of autophagy is completely unknown.
In our study, we tested for the first time the hypothesis that NPPA may protect cardiac cells through autophagy activation. To this aim, we evaluated the role of NPPA in the promotion of cardiac autophagy both in vitro and in vivo, and we also dissected the molecular mechanisms involved in this process. Importantly, we investigated the biological significance of autophagy activation by endogenous NPPA in response to stress stimuli, such as glucose deprivation, hypoxia, and cardiac ischemia/reperfusion.
Results
NPPA increased autophagy and autophagic flux in cardiomyocytes through an NPR1-PRKG signaling pathway
First of all, we assessed the level of autophagy in primary neonatal rat ventricular cardiomyocytes (CMs) exposed to physiological concentrations of NPPA (10−11 M) at different time points (Figure 1 A,B). NPPA rapidly increased MAP1LC3/LC3-II levels, a well-known marker of autophagy. It also increased the expression levels of ATG7 and BECN1, two autophagy-related proteins (Figure 1C,D). NPPA also improved autophagic flux in CMs, as indicated by increased levels of LC3-II with respect to control treatment either in the presence or in the absence of bafilomycin A1, an inhibitor of lysosomal digestion (Figure 1 E,F). We further corroborated these results by observing that in cells with adenoviral-mediated overexpression of mRFP-GFP-LC3, NPPA significantly increased the number of both autophagosomes (yellow dots) and autolysosomes (red only dots) (Figure 1G,H). The number of autophagosomes was higher in CMs treated with NPPA in the presence of bafilomycin A1. Overall, these data indicated that physiological levels of NPPA stimulate autophagy and autophagic flux in cardiomyocytes.
Figure 1.
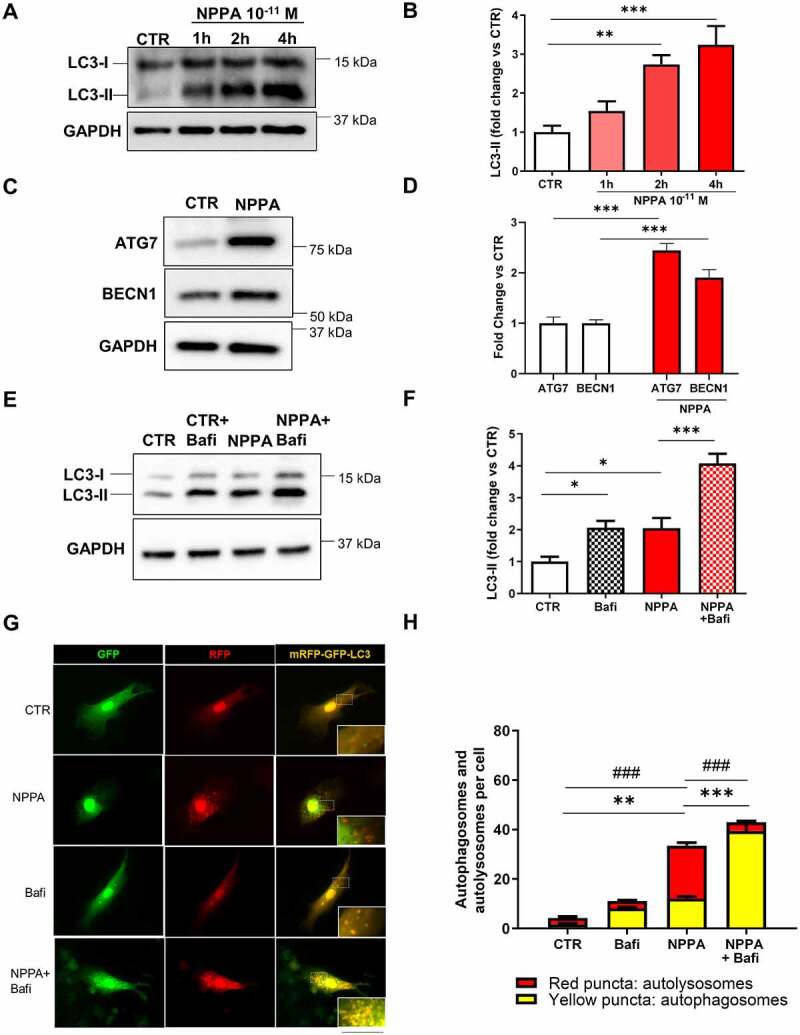
NPPA stimulated autophagy in rat neonatal cardiomyocytes (CMs). (A–B) CMs were treated with NPPA (10−11 M) for the indicated times in order to evaluate the autophagic response. Representative western blot for LC3-II (A) and corresponding quantification (B) (N=5). (C–D) Representative western blots for BECN1 and ATG7 (C) and corresponding densitometric analyses (D) of CMs treated with 10−11 M NPPA for 4 h (N=5). (E–F) CMs were treated with bafilomycin A1 (100 nM for 4 h) to evaluate autophagic flux in the presence or absence of 10−11 M NPPA for 4 h. Representative western blot for LC3-II (E) and corresponding quantification (F) (N=8). (G–H) CMs were transduced with an adenovirus overexpressing mRFP-GFP-LC3 for 48 h and then treated with 10−11 M NPPA for 4 h to evaluate LC3 puncta, in the presence or absence of bafilomycin A1. Yellow dots (merged red and green) indicate autophagosomes whereas red dots indicate autolysosomes. Scale bar: 100 μm (N=5–11). Values are expressed as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 obtained by using the one-way ANOVA followed by Bonferroni’s multiple comparison test (B, F, H) and ***p<0.001 obtained by using the student T test (D). (H) **p<0.01, ***p<0.001, comparison between yellow dots and ###p<0.001, comparison between red dots.
In order to demonstrate the involvement of the NPR1-PRKG signaling in the activation of autophagy by NPPA, we checked levels of LC3-II in cells with Npr1 or Prkg1 knockdown (Figure S1A–D) and treated with NPPA for 4 h (Figure 2A–F). As expected, NPPA failed to increase autophagy in CMs with Npr1 (Figure 2A,B,E,F) or Prkg1 (Figure 2C,D) knockdown. This evidence demonstrated that NPPA induces autophagy through the NPR1-PRKG pathway.
Figure 2.
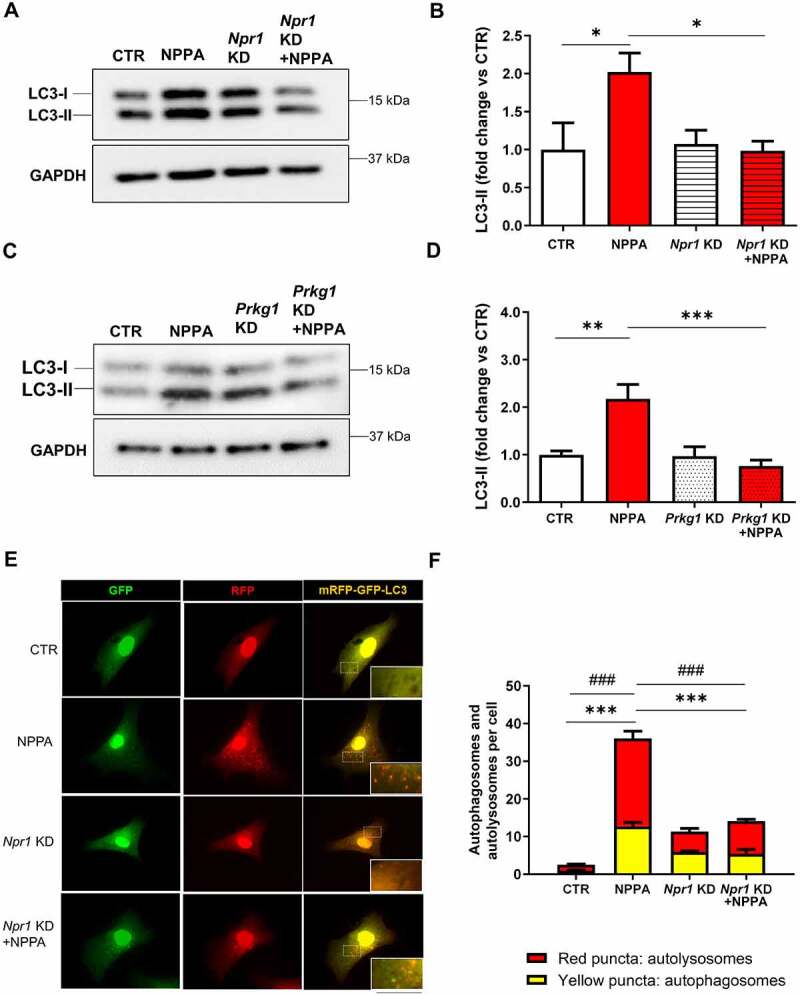
NPPA stimulated autophagy in CMs through NPR1and PRKG signaling. (A–B) CMs were silenced for Npr1 and treated with 10−11 M NPPA for 4 h to evaluate autophagy. (C–D) CMs were silenced for Prkg1 and treated with 10−11 M NPPA for 4 h to evaluate autophagy. Representative western blots for LC3-II and corresponding densitometric analyses are reported. (E–F) CMs were silenced for Npr1 and transduced with an adenovirus overexpressing mRFP-GFP-LC3 for 48 h and then treated with 10−11 M NPPA for 4 h to evaluate LC3 puncta. Yellow dots (merged red and green) indicate autophagosomes whereas red dots indicate autolysosomes. Scale bar: 100 μm (N=4). Values are expressed as mean ± SEM; (N=4–5) *p<0.05, **p<0.01, ***p<0.001 obtained by using the one-way ANOVA followed by Bonferroni’s multiple comparison test. (F) ***p<0.001, comparison between yellow dots and ###p<0.001, comparison between red dots.
Endogenous NPPA secretion was required for autophagy activation in response to stress stimuli
Then, we assessed whether endogenous NPPA production is needed for autophagy activation in response to stress stimuli. To this aim, we exposed CMs to glucose deprivation or to hypoxia. In these conditions, we observed a rapid and progressive accumulation of NT-proNPPA, the amino terminal fragment of NPPA prohormone, in conditioned media over time (Figure 3A and Figure S2A). Notably, Npr1 or Prkg1 knockdown blunted autophagy activation in response to 8 h of glucose deprivation as indicated by lower LC3-II levels and mRFP-GFP-LC3 dots in cells with knockdown (Figure 3B–G). On the other hand, Npr1 or Prkg1 knockdown did not affect autophagy activation at earlier time points of stress (data not shown). We also observed that CMs subjected to 8 h of hypoxia were unable to activate autophagy in the presence of Npr1 knockdown. The number of autophagosomes and autolysosomes in response to hypoxia decreased when compared to non-silenced cells (Figure S2B–C). These findings demonstrated that endogenous NPPA secretion is required for autophagy activation in a later phase of stress (glucose deprivation and hypoxia), through autocrine/paracrine effects.
Figure 3.
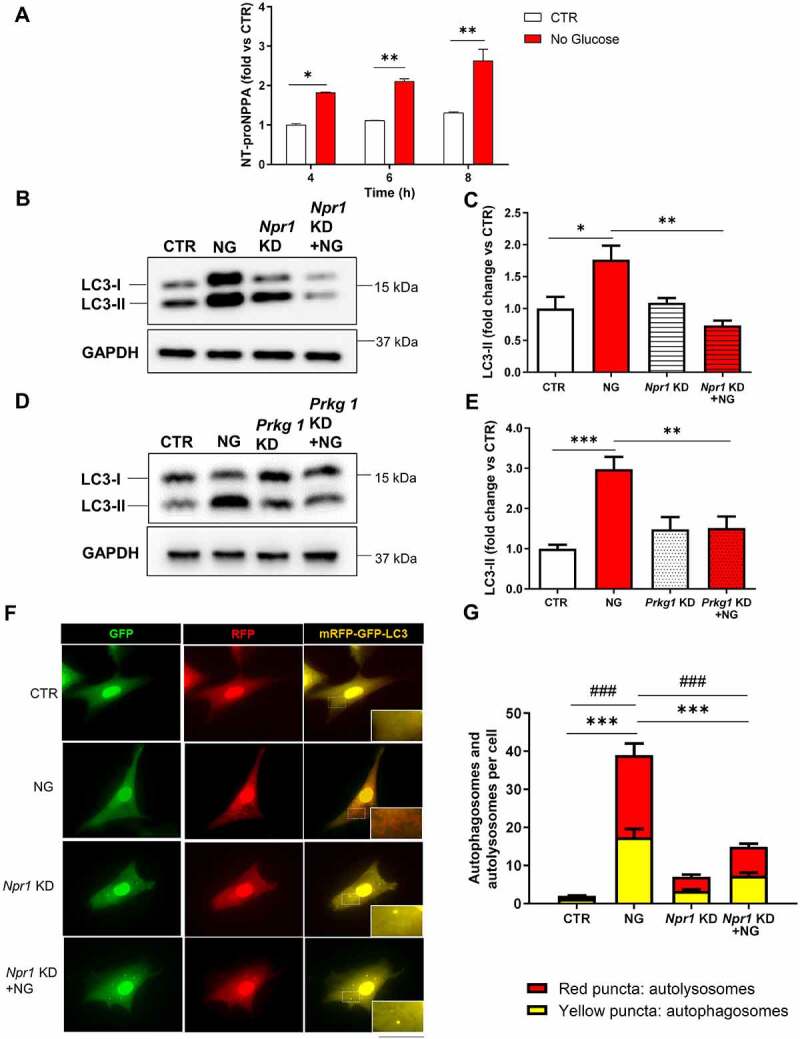
NPPA was released by cardiomyocytes in response to stress and regulated stress-induced autophagy. (A) NT-proNPPA secretion in CMs exposed to no glucose medium (NG) (A) for the indicated time points. Values are expressed as mean ± SEM; (N=3) *p<0.05, **p<0.01, obtained by using the two-way ANOVA followed by Bonferroni’s multiple comparison test. (B–E) CMs were silenced for Npr1 (B–C) or Prkg1 (D–E) and treated for 8 h with NG for autophagy evaluation. Representative western blots for LC3-II and corresponding densitometric analyses are shown (N=4–5). (F–G) CMs were silenced for Npr1 and transduced with an adenovirus overexpressing mRFP-GFP-LC3 for 48 h and then exposed to NG to evaluate LC3 puncta. Yellow dots (merged red and green) indicate autophagosomes whereas red dots indicate autolysosomes. Scale bar: 100 μm (N=4). CTR indicates untreated and not silenced cells. Values are expressed as mean ± SEM; *p<0.05, **p<0.01, ***p<0.001 obtained by using one-way ANOVA followed by Bonferroni’s multiple comparison test. (G) ***p<0.001, comparison between yellow dots and ###p<0.001, comparison between red dots.
NPPA exerted beneficial cellular effects in response to stress through autophagy activation in vitro
In order to evaluate the biological relevance of NPPA-induced autophagy, we also tested the impact of autophagy inhibition on the cellular effects of NPPA (Figure S3). For this purpose, we studied the effects of NPPA on cell viability and hypertrophy in stressed cells in the presence of Atg7 gene knockdown (Figure S1E-H). The latter encodes a key protein required for autophagy activation. We observed that lack of ATG7 does not allow NPPA to preserve cell viability (Figure S3A) and hypertrophy (Figure S3B) in CMs exposed to glucose deprivation or phenylephrine, respectively. These results indicated that autophagy inhibition blunts the protective effects of NPPA.
NPPA was required for autophagy activation in response to stress in vivo
Next, we sought to corroborate the in vitro evidence with in vivo findings. To this aim, we subjected heterozygous Nppa knockout mice (Nppa+/-) to cardiac ischemia-reperfusion (I/R) (Figure 4A). We used heterozygous Nppa+/- mice because our intention was to mainly attenuate NPPA secretion in response to stress, without a significant interference with baseline cardiovascular homeostasis, as previously shown [22]. In fact, previous work demonstrated that homozygous KO mice have high blood pressure levels which may have a significant impact on cardiac mass and adaptation to stress [5,6]. On the other hand, Nppa+/- mice have normal blood pressure [5], as also confirmed in our experiments (systolic blood pressure, wild-type: 112 mmHg ±9.61 S.D.; Nppa+/-: 117.6 mmHg ±3.64 S.D.; N = 5, p = 0.26).
Figure 4.
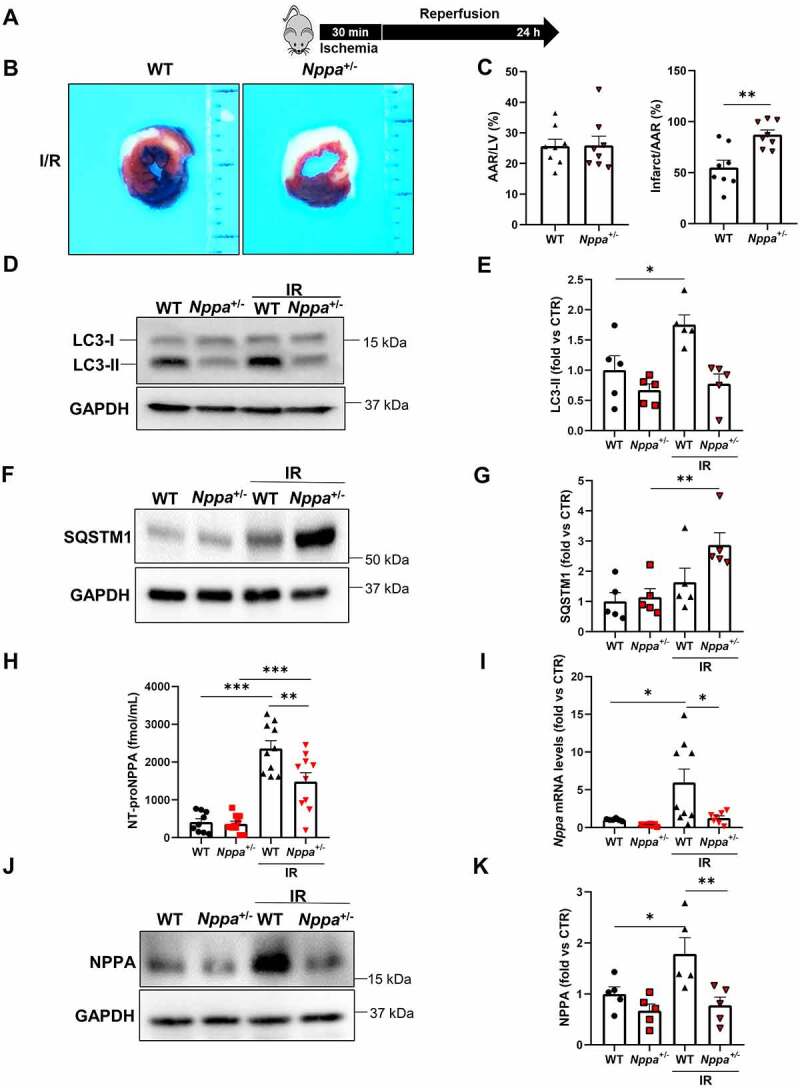
Nppa knockout mice showed increased infarct size and impaired cardiac autophagy in response to ischemia/reperfusion. (A) Wild-type and Nppa knockout mice (Nppa+/-) were subjected to 30 min of ischemia with 24 h of reperfusion. (B-C) Representative images of LV myocardial sections after Alcian blue and TTC staining (B) and corresponding quantification of area at risk (AAR) and infarction area (C) (scale bar: 1mm) (N=8 for each group). (D-G) Evaluation of autophagy in ischemic border region of wild-type and Nppa+/- mice after I/R. Representative western blot for LC3 and SQSTM1 (D, F) and corresponding quantification (E, G) are reported (N=5 for each group). (H) Serum levels of NT-proNPPA in WT and Nppa+/- mice undergoing I/R (N=9–10) (I) Nppa mRNA levels in ischemic border region of I/R wild-type and Nppa+/- mice (N=6–9). (J-K) Representative western blot for NPPA (J) and corresponding quantification (N=5 for each group) are reported (K). Values are expressed as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 obtained by using student T test (C) and one-way ANOVA followed by Bonferroni’s multiple comparison test (E,G, H, I, K).
Nppa+/- mice undergoing I/R showed an increased infarct size as compared to wild-type mice (Figure 4B,C). We also observed that Nppa+/- mice fail to activate cardiac autophagy, as indicated by lower LC3-II levels and increased levels of SQSTM1 (Figure 4D–G). The lack of autophagy activation in Nppa+/- mice was associated with significantly reduced NPPA serum levels in response to I/R as compared to wild-type mice (Figure 4H). Remarkably, we observed that Nppa ventricular gene expression and NPPA protein levels are reduced in Nppa+/- mice undergoing I/R, compared to wild-type mice (Figure 4I–K). These results suggested that a defective NPPA secretion abrogates the activation of autophagy upon stress. We also detected a significant reduction of autophagy at baseline in homozygous nppa mice (nppa−/−), compared to wild-type mice (Figure S4A-D). The latter suggested that NPPA is required to maintain physiological levels of cardiac autophagy in unstressed conditions.
To further understand the role of NPPA-induced autophagy in cardioprotection during I/R, we performed rescue experiments by using Tat-Beclin D11, a potent synthetic inducer of autophagy, administered 6 h before ischemia (Figure 5A). We observed that Tat-Beclin D11 administration to Nppa+/- mice reduces infarct size (Figure 5B,C) and increases cardiac autophagy, evaluated by increased levels of LC3-II and decreased levels of SQSTM1 (Figure 5D–G). The reduction of infarct size by Tat-Beclin D11 was more pronounced in Nppa+/- mice compared to wild-type (Figure 5B,C). Thus, NPPA secretion in the heart was required for autophagy activation and cardioprotection in response to I/R.
Figure 5.
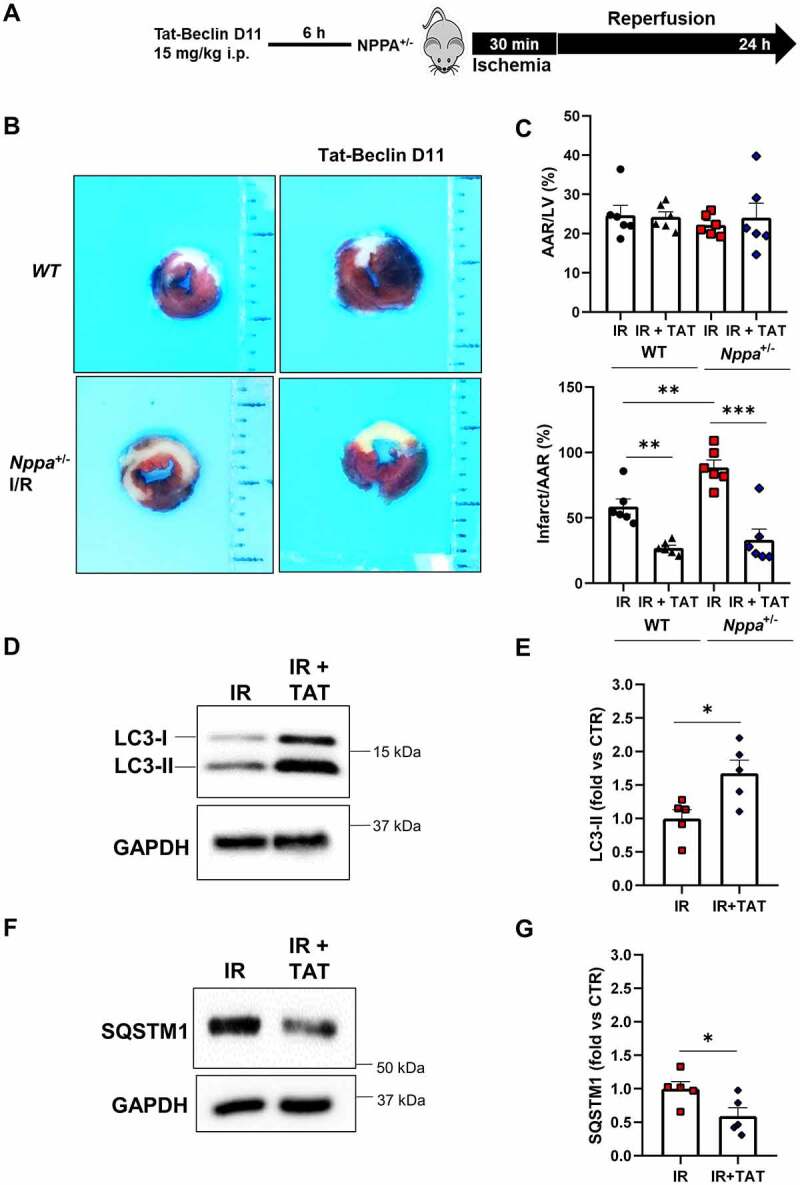
Tat-Beclin D11 decreased infarct size in Nppa knockout mice and rescued cardiac autophagy in response to I/R. (A). Nppa+/- and wild-type mice were injected with 15 mg/kg of Tat-beclin D11 and then subjected to 30 min of ischemia and 24 h of reperfusion. (B-C). Representative images of LV myocardial sections after alcian blue and TTC staining (B) and corresponding quantification of area at risk (AAR) and infarction area (C) (scale bar: 1mm) (N=6 for each group). (D-G). Evaluation of autophagy in ischemic border region of Nppa+/- mice receiving or not Tat-beclin D11 and then subjected to I/R. Representative western blot for LC3 and SQSTM1 (D, F) and corresponding quantification (E, G) are reported (N=5 for each group). Values are expressed as mean ± SEM. *p<0.05, **p<0.01, ***p<0.001 obtained by using one-way ANOVA followed by Bonferroni’s multiple comparison test (C) and student T test (E,G).
The protective effect of NPPA toward I/R injury was mediated by autophagy activation in vivo
Next, we tested whether the therapeutic potential of NPPA administration in vivo was mediated by autophagy activation in the heart. First of all, we observed a significant increase of LC3-II levels in response to NPPA treatment, as compared to mice receiving saline solution (Figure 6A,B). Then, we tested the effects of NPPA in mice undergoing I/R in the presence or in the absence of pre-treatment with chloroquine, an autophagy inhibitor (Figure 6C). We found that NPPA reduces infarct size and this effect was abrogated by autophagy inhibition (Figure 6D,E). These results suggested that NPPA reduces I/R injury through autophagy-dependent mechanism also in vivo.
Figure 6.
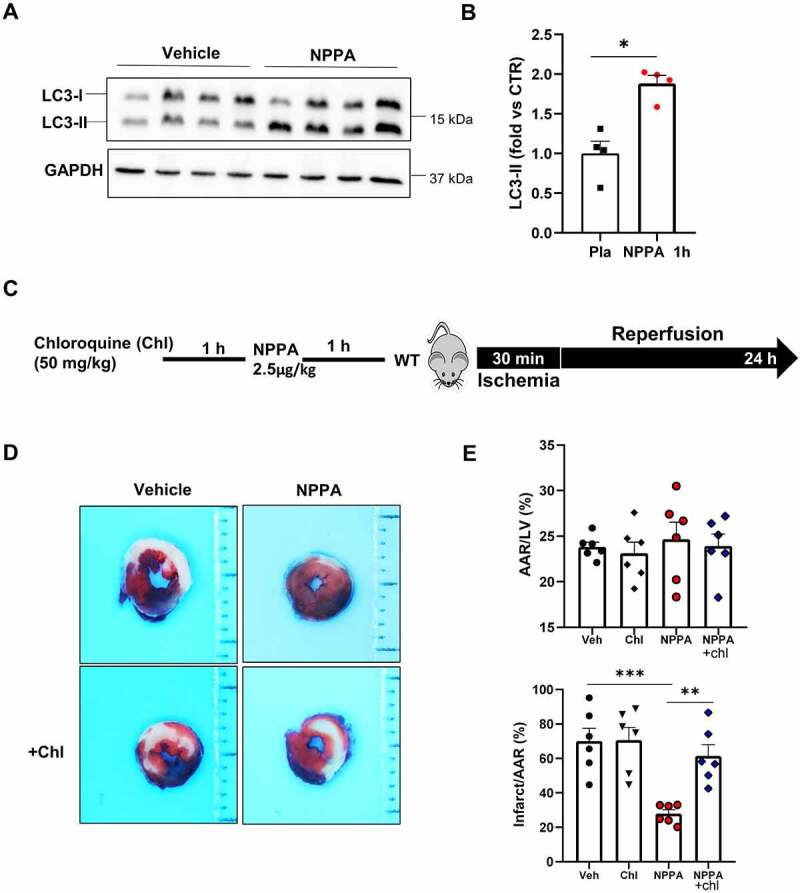
NPPA stimulated cardiac autophagy in mice. (A–B). Evaluation of autophagy in the heart of wild-type mice receiving or not NPPA (2.5 μg/kg). Representative western blot for LC3 after 1 h of NPPA injection and corresponding quantification are reported (N=4 for each group). C. Wild-type mice were injected with 2.5 μg/kg NPPA for 1 h and then subjected to 30 min of ischemia and 24 h of reperfusion, in the presence or absence of chloroquine pre-treatment (50 mg/kg i.p.). D-E. Representative images of LV myocardial sections after Alcian blue and TTC staining (D) and corresponding quantification of area at risk (AAR) and infarction area (E) (scale bar: 1 mm) (N=6 for each group). *p<0.05, **p<0.01, ***p<0.001 by using one-way ANOVA followed by Bonferroni’s Multiple comparison test (E) and student T test (B).
NPPA stimulated autophagy through TFEB nuclear translocation
Finally, we sought to investigate the mechanism involved in NPPA-induced autophagy. Complex 1 of MTOR (MTORC1) is a master regulator of autophagy, inhibiting the process [23]. Interestingly, we found that NPPA significantly activates MTORC1, as indicated by the increased phosphorylation of its downstream targets RPS6KB/p70S6K and EIF4EBP1/4E-BP1 [24] (Figure S5A-D). This result suggested that NPPA regulates autophagy through MTORC1-independent mechanisms. TFEB (transcription factor EB) has also recently emerged as a critical transcriptional regulator of autophagy in cells [25]. Since we found that NPPA secretion during stress is required for autophagy activation in a later phase, it seems likely that NPPA regulates autophagy by interfering with transcriptional mechanisms. Therefore, we evaluated the possible involvement of TFEB in the proautophagic effects of NPPA. Western blot analysis revealed that NPPA induces a significant increase in TFEB nuclear localization, indicating its activation (Figure 7A,B) [25,26]. The dynamic changes of intracellular TFEB localization associated with NPPA treatment were also evaluated through immunofluorescence quantitative analysis. Upon NPPA treatment, the nuclear translocation of TFEB was documented by the TFEB colocalization with DAPI nuclear signal in the merged images (Figure 7C) and highlighted by a significant increase of TFEB/DAPI colocalization coefficient and TFEB nuclear:cytoplasmic ratio (Figure 7D). On the other hand, NPPA treatment did not affect the expression of phospho-TFEB levels at Ser142, a phosphorylation induced by MTORC1 and retaining TFEB in the cytosol [27] (Figure S5E-F). This result further indicated that MTORC1 is not involved in these mechanisms. We also performed ChIP experiments in H9c2 cardiac cells to investigate the binding of TFEB to the promoter regions of autophagic genes, such as Becn1, Sqstm1, and Wipi1 which are known to be direct targets of TFEB [25,28,29]. We found that NPPA promotes the binding of TFEB to promoter regions of all these genes, indicating that NPPA promotes the transcriptional activity of TFEB (Figure 7E). We also observed a reduced TFEB nuclear translocation in Nppa+/- mice undergoing I/R injury (Figure S6A-D), compared to wild-type.
Figure 7.
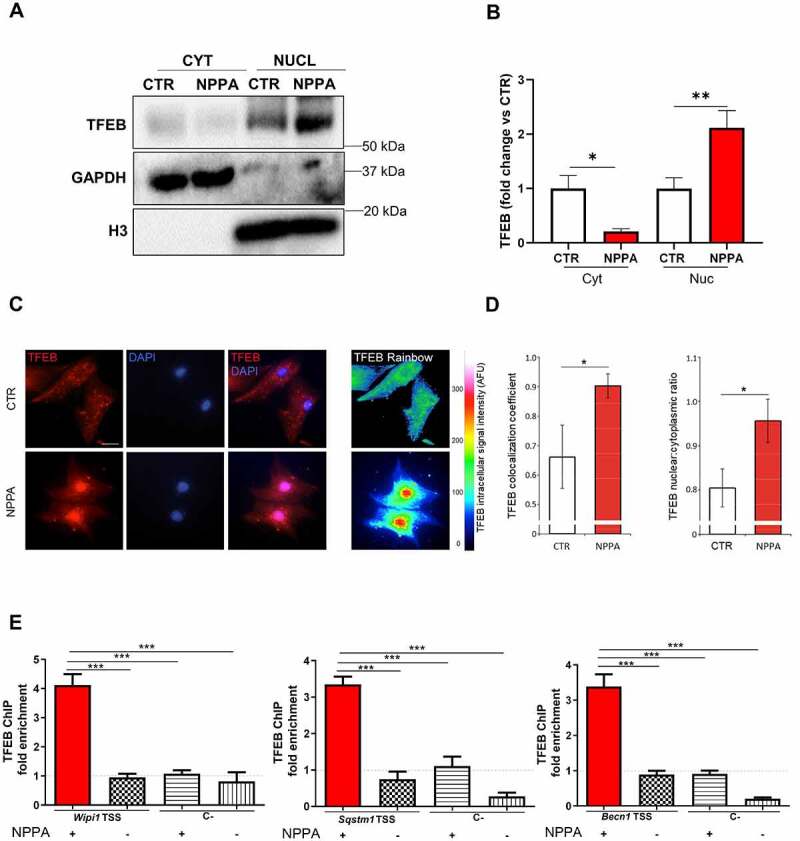
NPPA induced the nuclear translocation of transcription factor EB (TFEB) in cardiomyocytes. (A–E) CMs were treated with 10−11 M NPPA for 4 h to evaluate cytoplasm (cyt) and nuclear (nuc) expression of TFEB. (A–B) Representative western blot and corresponding densitometric analysis. Cytoplasm and nuclear expression of TFEB were normalized with GAPDH and histone 3 (H3) levels, respectively. Values are expressed as mean ±SEM (N=5) *p<0.05; **p<0.01 obtained by using the one-way ANOVA followed by Bonferroni’s Multiple Comparison Test. (C) Representative images show the colocalization of TFEB (red) and DAPI (blue) in NPPA-treated CMs and the intracellular intensity of TFEB signal in untreated and treated cells (Rainbow scale of pseudocolor). Scale bar: 20 μm. (D) the columns represent the correlation coefficient and the nuclear:cytoplasmic ratio of TFEB. Values are expressed as mean ±SEM (N=4). *p<0.0001 was obtained by Student T test. (E) Quantification of chromatin immunoprecipitation for TFEB binding to the transcriptional starting site (TSS) of Wipi1, Sqstm1 and Becn1 genes, using the anti-TFEB antibody, in H9c2 cells treated without or with NPPA. Regions 2.5–4 Kb upstream to the TSS of Wipi1, Sqstm1 and Becn1 were used as negative controls of TFEB binding (C-). Data are mean fold increases ± SEM (N=4). ***p < 0.001 was obtained by Student T test.
Finally, in order to test whether TFEB activation directly contributes to autophagy activation in cells treated with NPPA, we checked the effects of NPPA on autophagy in cardiomyocytes in the absence and presence of Tfeb knockdown (Figure S1I). We found that NPPA fails to activate autophagy in the presence of Tfeb knockdown (Figure 8A–D), as observed by the analysis of LC3-II level (Figure 8A,B) and by mRFP-GFP-LC3 dots (Figure 8C,D). These data indicated that TFEB is needed for NPPA-dependent activation of autophagy.
Figure 8.
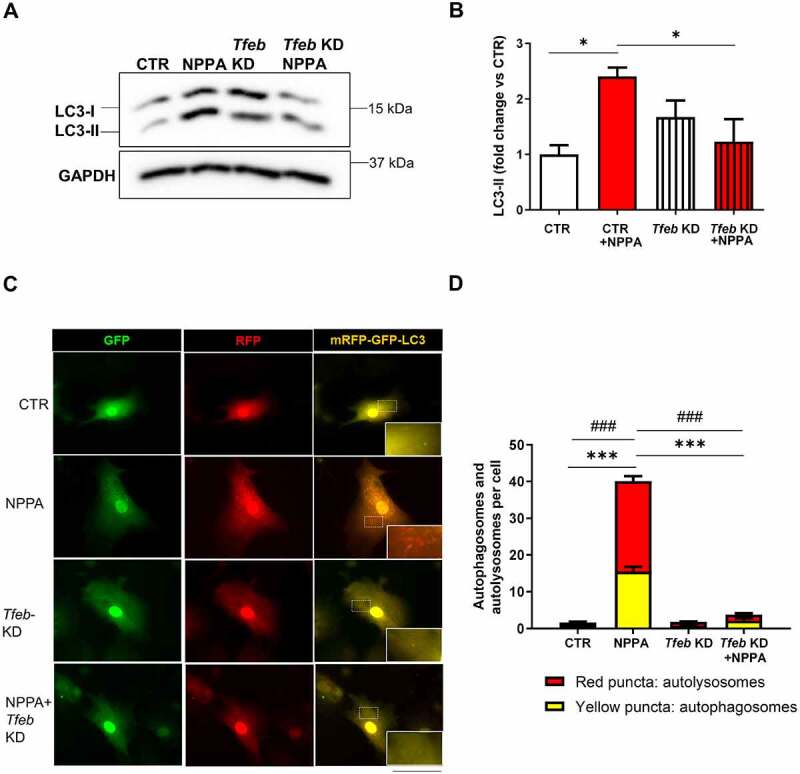
TFEB-Mediated NPPA-induced autophagy in cardiomyocytes. (A–B) CMs were silenced for Tfeb and treated for 4 h with 10−11 M NPPA for autophagy evaluation. Representative western blot for LC3-II and corresponding densitometric analysis are shown (N=4). (C–D) CMs were silenced for Tfeb and transduced with an adenovirus overexpressing mRFP-GFP-LC3 for 48 h and then exposed and treated with 10−11 M NPPA for 4 h to evaluate LC3 puncta. Yellow dots (merged red and green) indicate autophagosomes whereas red dots indicate autolysosomes. Scale bar: 100 μm (N=4). Controls: cells treated with control adenovirus (Ad-LACZ). Values are expressed as mean ± SEM. *p<0.05 obtained by using the one-way ANOVA followed by Bonferroni’s multiple comparison test. ***p<0.001, comparison between yellow dots and ###p<0.001, comparison between red dots.
Discussion
In this study, we demonstrated for the first time that: 1) physiological concentrations of NPPA promote autophagy in CMs through the NPR1-PRKG pathway; 2) endogenous NPPA is required for stress-induced autophagy activation; 3) autophagy activation mediates the beneficial effects of NPPA on CMs in vitro and in vivo and 4) NPPA stimulates autophagy through TFEB activation (Figure S7).
In recent years, NPPA has emerged as a critical regulator of cardiovascular homeostasis, exerting important pleiotropic effects in the heart and in the vascular system through autocrine and paracrine mechanisms [3,4,30]. Mice with systemic Nppa gene deletion or with cardiac-specific Npr1 gene deletion develop massive cardiac hypertrophy, dilation, and dysfunction in response to pressure or volume overload [2,3,7,22,31]. NPPA reduces cardiac remodeling in mice with chronic myocardial infarction induced by permanent coronary artery ligation [11]. NPPA activates PRKG signaling by increasing intracellular cGMP levels in cardiomyocytes [2,3]. Our data suggested that autophagy may be a critical mechanism underlying the beneficial actions of NPPA. We found that NPPA activates autophagy through NPR1-PRKG axis. Remarkably, autophagy inhibition by Atg7 knockdown abrogated the beneficial effects of NPPA on cell viability and hypertrophy in response to stress. Our in vitro data were also confirmed by in vivo evidence, evaluating the phenotype of systemic Nppa gene knockout mice subjected to I/R stress. First of all, we found that ventricular NPPA secretion is increased in response to I/R injury. This mechanism appeared to be required for limiting myocardial injury through the activation of autophagy. In fact, we found increased I/R injury and reduced cardiac autophagy in Nppa+/- mice, as compared to wild-type mice. On the other hand, autophagy reactivation with Tat-Beclin D11 rescued infarct size in Nppa+/- mice. This result was in line with recent data demonstrating that stimulation of autophagy with Tat-Beclin 1 before I/R reduces infarct size, whereas autophagy may be maladaptive when stimulated exclusively during the late phase of I/R [18]. We also observed that NPPA stimulates cardiac autophagy in vivo and limits infarct size. The protective effects of NPPA in models of I/R were already demonstrated in previous studies [32–34]. Here, we reported for the first time that autophagy inhibition through chloroquine reduces the protective effects of exogenous NPPA administration, suggesting that NPPA exerts cardiac beneficial effects through autophagy stimulation.
Of note, we did not test whether NPPB/brain natriuretic peptide (natriuretic peptide B), another member of NP family, also regulates autophagy. We decided to focus only on NPPA since NPPB does not share all the cardioprotective functions of NPPA, whereas it mostly limits cardiac fibrosis [35].
Notably, our study dissected a new extracellular mechanism of regulation of autophagy in the cardiovascular system. We found that NPPA is rapidly released by cardiac cells upon stress stimuli, such as glucose deprivation or hypoxia. Knockdown of NPPA receptor, Npr1, and its main downstream target, Prkg1, attenuated autophagy activation during stress, indicating that NPPA release by cells is required for autophagy activation through autocrine/paracrine effects. Of note, Npr1-Prkg1 knockdown attenuated autophagy activation in CMs after 8 h of stress, whereas no effects were observed at earlier time points (4 h). This result indicated that NPPA is dispensable for autophagy activation in the early phase of stress, whereas it is fundamental in a later phase.
Our study also clarified a new signaling pathway regulating autophagy, the NPR1-PRKG-TFEB pathway. Recent work demonstrated that cardiac TFEB is activated in mice undergoing I/R injury, in line with our results [36]. Our study extends this evidence demonstrating that NPPA represents an endogenous activator of TFEB and autophagy in the heart during I/R. Future studies are warranted to test whether this pathway is important for autophagy regulation also in different cell types and systems. Of note, a recent elegant study showed that PRKG activates autophagy in cardiomyocytes through the inhibition of MTORC1 [37]. MTORC1 is a negative regulator of autophagy and TFEB [23]. We observed that NPPA activates MTORC1, suggesting that MTORC1 may not be involved in TFEB activation induced by NPPA. It will be important to test in the future whether PRKG directly phosphorylates and activates TFEB in response to NPPA exposure.
Materials and Methods
Cell cultures
Neonatal rat ventricle cardiomyocytes (CMs) were isolated from 1 to 2 days neonatal Wistar Kyoto rats through an enzymatic digestion followed by a magnetic separation, by using the Miltenyi technology (Miltenyi Biotec, 130-098-373). CMs were cultured as previously described [26].
NPPA treatment
NPPA synthetic peptide was purchased from Sigma-Aldrich (A8208), dissolved in PBS (Euroclone, ECB4004 L) and diluted in complete medium at the final concentration of 10−11 M. For the evaluation of autophagy, cells were treated with NPPA for different times of incubation (from 1 to 4 h). The effects of NPPA on autophagy levels were also tested in Npr1 and Prkg1 knockdown cells. Control cells were treated with vehicle (PBS 0.001%).
Autophagic flux
Autophagic flux was evaluated by treating cells with NPPA 10−11 M for 4 h in the presence or absence of bafilomycin A1 (100 nM for 4 h; Sigma Aldrich, B1793).
Autophagic flux was also assessed in CMs by using the mRFP-GFP-LC3 probe, as reported elsewhere [38,39]. Cells were plated in 8-well chamber slides and transduced with adenovirus overexpressing mRFP-GFP-LC3 (kindly provided by Junichi Sadoshima, Rutgers New Jersey Medical School, USA) for 48 h, in the presence or in the absence of 10−11 M NPPA for 4 h and with or without bafilomycin A1. GFP is degraded by lysosomal acids, whereas mRFP is resistant. Thus, autophagosomes were detected both in red (mRFP) and green fluorescence (GFP), whereas autolysosomes were detected only in red fluorescence. Images were acquired with an epifluorescence microscope. mRFP-GFP-LC3 experiments were performed also in the presence of Npr1 or Tfeb knockdown, and in CMs undergoing hypoxia or glucose deprivation.
Glucose deprivation and hypoxia
In order to evaluate levels of autophagy under stress conditions in CMs, cells were subjected to glucose deprivation (NG) for 8 h using a DMEM no glucose medium (Thermo Fisher Scientific, 11966–025).
Control cells were treated with a complete medium used for the routine cultures. The effects of NG on autophagy were also tested in cells with Npr1 or Prkg1 knockdown.
In a separate set of experiments, the exposure to NG was performed for 24 h, in the presence or absence of NPPA 10−11 M (24 h) in order to evaluate the effects on cell viability. The same effects were evaluated in cells with Atg7 knockdown. Cell viability was assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma Aldrich, M5655) following the manufacturer’s instructions.
For hypoxia experiments, CMs were plated in a 6-well plate for the evaluation of NPPA secretion or in 8-chamber slide for mRFP-GFP-LC3 experiments. CMs were then incubated with the hypoxic solution and transferred to a hypoxia chamber (Bugbox Plus; Baker Ruskinn) for 4 h and 8 h. Hypoxic stimulation was maintained at 37°C with 5% CO2 and 1% O2.
Cell hypertrophy
Cell hypertrophy was assessed in phenylephrine (Sigma Aldrich, P-6126; 200 μM for 24 h)-treated CMs in the presence or absence of NPPA 10−11 M (24 h). Control cells were silenced for Atg7. Hypertrophy was assessed by immunofluorescence for ACTB/β-actin (Santa Cruz Biotechnology, sc -69879) and cell size was quantified in randomly chosen fields by ImageJ (National Institutes of Health, USA).
Gene silencing
Gene silencing was performed using species-specific commercially available siRNAs (Npr1, Prkg1 and Atg7 [Silencer Pre-designed siRNAs, Ambion, Thermo Fisher Scientific, Npr1 (s219559) Prkg1 (s132550), Atg7 (s161901)] and following the manufacturer’s protocol. Briefly, a specific amount of siRNAs was incubated with a transfection agent, Lipofectamine RNAi max, (Thermo Fisher Scientific, 13778150) in OPTIMEM-reduced serum medium (Thermo Fisher Scientific, 31985062) as previously reported [40,41]. After 5 h, OPTIMEM was replaced by a complete medium and the treatments (NPPA, NG) were started after 48 h. Control cells were incubated with only lipofectamine. The efficiency of gene silencing was assessed by RT-PCR analysis and western blot. RT-PCR was performed as previously reported [42]. Primer sequences are listed in Table S1.
TFEB localization
CMs, grown on multiwell plate and incubated with or without NPPA as described above, were fixed with 4% paraformaldehyde (Electron Microscopy Sciences, 50-259-99) in PBS for 30 min at 25°C and subsequently treated with 0.1 M glycine (Sigma-Aldrich, G8898) for 20 min at 25°C and with 0.1% Triton X-100 (Sigma-Aldrich, 93,418) for an additional 5 min at 25°C to allow permeabilization. Cells were then incubated for 1 h at 25°C with rabbit polyclonal anti-TFEB (1:100 in PBS; MyBioSource, MBS7049264). The primary antibody was visualized using goat anti-rabbit IgG-TXred (1:400 in PBS; Jackson ImmunoResearch Laboratories, 111-075-144) for 30 min at 25°C. Nuclei were stained with DAPI (1:1000 in PBS; Sigma-Aldrich, D9542). Fluorescence signals were analyzed by ApoTome System connected with an Axiovert 200 inverted microscope (Zeiss). Image analysis was performed by the Axiovision software (Zeiss) equipped with colocalization module.
TFEB knockdown
Tfeb knockdown was performed by incubating CMs for 72 h with an adenovirus expressing a short hairpin sequence targeting Tfeb (Ad-Tfeb KD), by Vector Biolabs (shADV-294304). In order to evaluate autophagy in the presence of Tfeb knockdown, NPPA 10−11 M was administered during the last 4 h of adenoviral infection. Control cells were incubated with an empty adenovirus (LACZ).
Animal procedures
8- to 10-week-old wild-type C57BL6/J and heterozygous Nppa knockout mice (Nppa+/-) (Jackson Laboratory, B6.129P2-Nppatm1Unc/J, stock n° 002685) were subjected to cardiac ischemia-reperfusion (I/R). Animals were anesthetized by intraperitoneal injection of ketamine (100 mg/kg i.p.) and xylazine (5 mg/kg i.p.). Ischemia was achieved by ligating the anterior descending branch of the left anterior descending artery (LAD) by 8–0 prolene suture 2 mm below the border between left atrium and LV, with silicon tubing (1 mm OD) placed on top of LAD. After occlusion for 30 min, the silicon tubing was removed to achieve reperfusion. After 24 h, Alcian blue dye (1%; Sigma-Aldrich, A5268) was used to demarcate the ischemic area at risk (AAR) in anesthetized animals. Then, animals were sacrificed, hearts were removed and 1 mm left ventricle (LV) sections were incubated with 1% TTC (Sigma-Aldrich, T8877) solution at 37°C for 15 min. The infarct size and AAR were calculated by ImageJ software as reported elsewhere [43]. For rescue experiments, Nppa+/- mice undergoing I/R were injected with Tat-Beclin D11 (Novus biological, NBP2–49888) (15 mg/kg, i.p.) 6 h before ischemia. Systolic blood pressure was evaluated by the tail-cuff sphygmomanometer. Mice were adapted to the procedure a few days before performing the measurement. In different experiments, I/R and control mice were used for the evaluation of cardiac autophagy (LC3-II and SQSTM1), cytoplasm, and nuclear level of TFEB, Nppa gene expression and NPPA secretion. Cardiac autophagy was also evaluated in 8- to 10-week-old wild-type homozygous nppa knockout mice (nppa−/−).
NPPA treatment in vivo
A bolus injection of exogenous NPPA (Sigma-Aldrich, A8208; 2.5 μg/kg) was given by tail vein in wild-type mice and cardiac autophagy was evaluated after 1 h post-injection. In a separate set of experiments, NPPA was injected to wild-type mice 1 h before I/R protocol in the presence or absence of chloroquine (Sigma Aldrich, C6628) pre-treatment for 1 h (50 mg/kg i.p.). In these experimental groups, infarct size was determined as described above.
Ethics
All procedures involving animals were performed in accordance with the Italian and European Community (Directive 2010/63/EU) for animal experiments, and the protocol was approved by Italian Ministry of Health (protocol number: 1014/2020-PR).
Assessment of NT-proNPPA levels and of NPPA gene expression
NT-proNPPA levels were assayed in CM supernatants (glucose deprivation and hypoxia) and in serum from wild type and Nppa+/- mice by using a colorimetric commercially available ELISA kit (Biomedica Gruppe-Pantec, BI-20892). CMs were plated on a 6-multiwell plate and, after reaching >95% confluence, they were treated for different times (4, 6, 8 h) with NG or subjected to hypoxia (4 and 8 h). Mice undergoing I/R were sacrificed and 200 μL of serum were collected and used for the analyses. Values were acquired by a microplate reader and the levels of NT-proNPPA were calculated by following the manufacturer’s protocol. Nppa mRNA expression was evaluated in cardiac ventricle by RT-PCR. Briefly, ventricular RNA was extracted by trizol (Invitrogen, 15596026) and 1 μg retrotranscribed into cDNA by using Superscript VILO (Invitrogen, 11756050). Nppa mRNA levels were normalized with Gapdh as a housekeeping gene (Table S1)
Chromatin immunoprecipitation (ChIP)
All the buffers used for chromatin immunoprecipitation were supplemented with protease inhibitors (Roche, 11697498001). ChIPs were performed as previously described, with minor changes [44]. Briefly, 6 × 106 H9c2 cells (ATCC®, CRL-1446™) were treated with NPPA 10−11 M for 4 h, and then, fixed with 1% formaldehyde for 10 min at room temperature (RT). Subsequently, chromatin was sonicated in ice-cold lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1), to obtain fragments of ~ 300 bp in length.
The sheared chromatin was diluted 10-fold in dilution buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1) and precleared with protein A-Sepharose (GE-Healthcare, GE17-0780-01). The 5% of the supernatant was saved as the input. Appropriate amounts of chromatin were incubated with 5 μg goat anti-TFEB antibody (abcam, ab2636), or with 5 μg normal goat IgG antibodies (Millipore, NI02).
The immunoprecipitated complexes were recovered with protein A-Sepharose and washed with ice-cold buffers as it follows: one wash in buffer 1 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1), four washes in buffer 2 (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl, 20 mM Tris-HCl, pH 8.1), one wash in buffer 3 (250 mM LiCl, 1% NP-40 [Santa Cruz Biotechnology, 9016-45-9], 1% sodium deoxycholate [Sigma Aldrich, D6750], 1 mM EDTA, 10 mM Tris-HCl, pH 8.1) and three washes in TE buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA). The immuno-complexes were then reverse crosslinked, and the DNA was purified and used for qPCR, which was performed with SsoAdvance Universal SYBR Green supermix (Bio-Rad, 1725270) on a Real-Time PCR system (CFX96; Bio-Rad), according to the manufacturer’s protocols.
Wipi1 Fw and Rev, Sqstm1 Fw and Rev, Becn1 Fw and Rev were used as primers to amplify Wipi1, Sqstm1 and Becn1 promoters (Table S2). Regions 2.5–4 kb upstream the TSS of Wipi1, Sqstm1, and Becn1, used as negative controls of TFEB binding, were amplified using primers listed in Table S2.
Data are expressed as fold enrichment over background (IgG), which were calculated in terms of , as described elsewhere [44].
Western blot analysis
Proteins were extracted from CMs and hearts with RIPA buffer (Santa Cruz Biotechnology, sc -24948) and western blot analysis was performed as previously reported [42] using the following primary antibodies: anti-LC3 (MBL International, M186–3), anti-ATG7 (Cell Signaling Technology, 8538), anti-BECN1 (Cell Signaling Technology, 3738), anti-GAPDH (Santa Cruz Biotechnology, sc -32233), anti-histone 3 H3 (Cell Signaling Technology, 9715), anti-TFEB (Bethyl, A303-673A), anti-phospho-TFEB (Ser142; Millipore, ABE1971-I) anti-phospho-RPS6KB/S6K (Thr389; Cell Signaling Technology, 9205), anti-RPS6KB/S6K (Cell Signaling Technology, 9202), anti-phospho-EIF4EBP1/4E-BP1 (Thr37/46; Cell Signaling Technology, 2855), anti-EIF4EBP1/4E-BP1 (Abcam, ab2606), anti-NPR1/NPR-A (abcam, ab14356), anti-PRKG/PKG (Mybiosource, MBS565721), anti-VCL/VINCULIN (Santa Cruz Biotechnology, sc-73614); anti-SQSTM1 (abcam, ab109012), anti-NPPA/ANP (abcam, ab225844). Secondary antibodies were anti-rabbit and anti-mouse (Millipore, anti-rabbit, 401315; anti-mouse, 401215).
Statistical analysis
All continuous variables are presented as mean ± SEM. Statistical analysis between the two groups was performed by the Student t-test. Statistical analysis between multiple groups was evaluated using one- or two-way ANOVA followed by Bonferroni post-hoc test. Graphs and statistical analyses were performed by Graph Pad Prism (GraphPad Software, Inc. La Jolla, CA, USA) and statistical significance was set at P < 0.05 level. The n of experiment is reported in figure legends.
Supplementary Material
Funding Statement
This work was supported by grants from the Italian Ministry of Health to S.Ru., M.V., and S.S. (Ricerca corrente; GR-2013-02355401), Italian Ministry of Research to SS (PRIN_2017N8K7S2_002; PRIN_20202020YRETTX), Pasteur Institute-Cenci Bolognetti Foundation to SS, University Sapienza to S.Ru. (project number RM1181641BF8C865). We thank “Leducq Transatlantic Networks of Excellence: Modulating autophagy to treat cardiovascular disease (15CBD04)” for the partial support and all its members for the advices and critical discussion.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2115675
References
- [1].Suga S, Nakao K, Hosoda K, et al. Receptor selectivity of natriuretic peptide family, atrial natriuretic peptide, brain natriuretic peptide, and C-type natriuretic peptide. Endocrinol. 1992. Jan;130(1):229–239. DOI: 10.1210/endo.130.1.1309330 [DOI] [PubMed] [Google Scholar]
- [2].Rubattu S, Sciarretta S, Valenti V, et al. Natriuretic peptides: an update on bioactivity, potential therapeutic use, and implication in cardiovascular diseases. Am J Hypertens. 2008. Jul;21(7):733–741. [DOI] [PubMed] [Google Scholar]
- [3].Volpe M, Rubattu S, Burnett JsJr.. Natriuretic peptides in cardiovascular diseases: current use and perspectives. Eur Heart J. 2014. Feb;35(7):419–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Forte M, Madonna M, Schiavon S, et al. Cardiovascular pleiotropic effects of natriuretic peptides. Int J Mol Sci. 2019. Aug 8;20(16):3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].John SW, Krege JH, Oliver PM, et al. Genetic decreases in atrial natriuretic peptide and salt-sensitive hypertension. Sci. 1995. Feb 3;267(5198):679–681. [DOI] [PubMed] [Google Scholar]
- [6].Feng JA, Perry G, Mori T, et al. Pressure-independent enhancement of cardiac hypertrophy in atrial natriuretic peptide-deficient mice. Clin Exp Pharmacol Physiol. 2003. May-Jun;30(5–6):343–349. [DOI] [PubMed] [Google Scholar]
- [7].Mori T, Chen YF, Feng JA, et al. Volume overload results in exaggerated cardiac hypertrophy in the atrial natriuretic peptide knockout mouse. Cardiovasc Res. 2004. Mar 1;61(4):771–779. [DOI] [PubMed] [Google Scholar]
- [8].Oliver PM, Fox JE, Kim R, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor a. Proc Natl Acad Sci U S A. 1997. Dec 23;94(26):14730–14735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Knowles JW, Esposito G, Mao L, et al. Pressure-independent enhancement of cardiac hypertrophy in natriuretic peptide receptor a-deficient mice. J Clin Invest. 2001. Apr;107(8):975–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kishimoto I, Rossi K, Garbers DL.. A genetic model provides evidence that the receptor for atrial natriuretic peptide (guanylyl cyclase-A) inhibits cardiac ventricular myocyte hypertrophy. Proc Natl Acad Sci U S A. 2001. Feb 27;98(5):2703–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kasama S, Furuya M, Toyama T, et al. Effect of atrial natriuretic peptide on left ventricular remodelling in patients with acute myocardial infarction. Eur Heart J. 2008. Jun;29(12):1485–1494. [DOI] [PubMed] [Google Scholar]
- [12].Hayashi M, Tsutamoto T, Wada A, et al. Intravenous atrial natriuretic peptide prevents left ventricular remodeling in patients with first anterior acute myocardial infarction. J Am Coll Cardiol. 2001. Jun 1;37(7):1820–1826. [DOI] [PubMed] [Google Scholar]
- [13].Brandt RR, Heublein DM, Mattingly MT, et al. Presence and secretion of atrial natriuretic peptide from cultured human aortic endothelial cells. Am J Physiol. 1995. Feb;268(2 Pt 2):H921–5. [DOI] [PubMed] [Google Scholar]
- [14].Potter LR, Yoder AR, Flora DR, et al. Natriuretic peptides: their structures, receptors, physiologic functions and therapeutic applications. Handb Exp Pharmacol. 2009. ;191:341–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kuhn M, Volker K, Schwarz K, et al. The natriuretic peptide/guanylyl cyclase–a system functions as a stress-responsive regulator of angiogenesis in mice. J Clin Invest. 2009. Jul;119(7):2019–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008. Jan 11;132(1):27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Sciarretta S, Zhai P, Volpe M, et al. Pharmacological modulation of autophagy during cardiac stress. J Cardiovasc Pharmacol. 2012. Sep;60(3):235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Sciarretta S, Maejima Y, Zablocki D, et al. The role of autophagy in the heart. Annu Rev Physiol. 2018. Feb 10;80(1):1–26. [DOI] [PubMed] [Google Scholar]
- [19].Nakai A, Yamaguchi O, Takeda T, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007. May;13(5):619–624. [DOI] [PubMed] [Google Scholar]
- [20].Torisu T, Torisu K, Lee IH, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von willebrand factor. Nat Med. 2013. Oct;19(10):1281–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nussenzweig SC, Verma S, Finkel T. The role of autophagy in vascular biology. Circ Res. 2015. Jan 30; 116(3): 480–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Franco V, Chen YF, Oparil S, et al. Atrial natriuretic peptide dose-dependently inhibits pressure overload-induced cardiac remodeling. Hypertens. 2004. Nov;44(5):746–750. [DOI] [PubMed] [Google Scholar]
- [23].Sciarretta S, Forte M, Frati G, et al. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. 2018. Feb 2;122(3):489–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017Apr 6;169(2):361–371. [DOI] [PubMed] [Google Scholar]
- [25].Settembre C, Di Malta C, Polito VA, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011. Jun 17;332(6036):1429–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sciarretta S, Yee D, Nagarajan N, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018. May 8;71(18):1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Martina JA, Chen Y, Gucek M, et al. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012. Jun;8(6):903–914. DOI: 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Palmieri M, Impey S, Kang H, et al. Characterization of the clear network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011. Oct 1;20(19):3852–3866. [DOI] [PubMed] [Google Scholar]
- [29].Sardiello M, Palmieri M, di Ronza A, et al. A gene network regulating Iysosomal biogenesis and function. Sci. 2009Jul 24;325(5939):473–477. [DOI] [PubMed] [Google Scholar]
- [30].Rubattu S, Volpe M. Natriuretic peptides in the cardiovascular system: multifaceted roles in physiology, pathology and therapeutics. Int J Mol Sci. 2019. Aug 16;20(16): 3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Holtwick R, van Eickels M, Skryabin BV, et al. Pressure-Independent cardiac hypertrophy in mice with cardiomyocyte-restricted inactivation of the atrial natriuretic peptide receptor guanylyl cyclase-A. J Clin Invest. 2003. May;111(9):1399–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Okawa H, Horimoto H, Mieno S, et al. Preischemic infusion of alpha-human atrial natriuretic peptide elicits myoprotective effects against ischemia reperfusion in isolated rat hearts. Mol Cell Biochem. 2003. Jun;248(1–2):171–177. [DOI] [PubMed] [Google Scholar]
- [33].Takagi G, Kiuchi K, Endo T, et al. Alpha-human atrial natriuretic peptide, carperitide, reduces infarct size but not arrhythmias after coronary occlusion/reperfusion in dogs. J Cardiovasc Pharmacol. 2000. Jul;36(1):22–30. [DOI] [PubMed] [Google Scholar]
- [34].Zhang WW, Hasaniya NW, Premaratne S, et al. Atrial natriuretic peptide protects against ischemia-reperfusion injury in rabbit hearts in vivo. Vasc Endovascular Surg. 2008. Jun-Jul;42(3):263–267. [DOI] [PubMed] [Google Scholar]
- [35].Tamura N, Ogawa Y, Chusho H, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A. 2000. Apr 11;97(8):4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Nah J, Sung EA, Zhai P, et al. Tfeb-mediated transcriptional regulation of autophagy induces autosis during Ischemia/reperfusion in the heart. Cells. 2022. Jan 13;11(2):258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ranek MJ, Kokkonen-Simon KM, Chen A, et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature. 2019. Feb;566(7743):264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Sciarretta S, Zhai P, Shao D, et al. Activation of NADPH oxidase 4 in the endoplasmic reticulum promotes cardiomyocyte autophagy and survival during energy stress through the protein kinase RNA-activated-like endoplasmic reticulum kinase/eukaryotic initiation factor 2alpha/activating transcription factor 4 pathway. Circ Res. 2013. Nov 8;113(11):1253–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Cai W, Fujita T, Hidaka Y, et al. Translationally controlled tumor protein (TCTP) plays a pivotal role in cardiomyocyte survival through a Bnip3-dependent mechanism. Cell Death Dis. 2019. Jul 18;10(8):549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ramirez-Sagredo A, Quiroga C, Garrido-Moreno V, et al. Polycystin-1 regulates cardiomyocyte mitophagy. Faseb J. 2021. Aug;35(8):e21796. [DOI] [PubMed] [Google Scholar]
- [42].Forte M, Bianchi F, Cotugno M, et al. Pharmacological restoration of autophagy reduces hypertension-related stroke occurrence. Autophagy. 2019. Nov;12:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nah J, Zhai P, Huang CY, et al. Upregulation of rubicon promotes autosis during myocardial ischemia/reperfusion injury. J Clin Invest. 2020. Jun 1;130(6):2978–2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Fioriniello S, Csukonyi E, Marano D, et al. MeCP2 and major satellite forward RNA cooperate for pericentric heterochromatin organization. Stem Cell Rep. 2020. Dec 8;15(6):1317–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.