

ABSTRACT
Macroautophagy/autophagy is a multistep degradative process that is essential for maintaining cellular homeostasis and is often dysregulated during disease. Systematically quantifying flux through this pathway is critical for gaining fundamental insights and effectively modulating this process. Established methods to quantify flux use steady-state measurements, which provide limited information about the perturbation and the cellular response. We present a theoretical and experimental framework to measure autophagic steps in the form of rates under non-steady-state conditions. We use this approach to measure temporal responses to rapamycin and wortmannin treatments, two commonly used autophagy modulators. We quantified changes in autophagy rates in as little as 10 min, which can establish direct mechanisms for autophagy perturbation before feedback begins. We identified concentration-dependent effects of rapamycin on the initial and temporal progression of autophagy rates. We also found variable recovery time from wortmannin’s inhibition of autophagy, which is further accelerated by rapamycin. Furthermore, we applied this approach to study the effect of serum and glutamine starvation on autophagy. Serum starvation led to a rapid and transient increase in all the rates. Glutamine starvation led to a decrease in the rates on a longer timescale. In summary, this new approach enables the quantification of autophagy flux with high sensitivity and temporal resolution and facilitates a comprehensive understanding of this process.
KEYWORDS: Autophagy flux, live cell fluorescence microscopy, rapamycin, starvation, temporal dynamics, wortmannin
Introduction
Macroautophagy (hereafter referred to as autophagy) is an intracellular recycling process that breaks down misfolded proteins and damaged organelles into their primary building blocks. This dynamic process involves autophagosome formation, the fusion of autophagosomes with lysosomes, and the turnover of autolysosomes. Constitutive autophagy is important for cellular homeostasis and is modulated during many extrinsic stresses such as nutrient deprivation or pathogen infection [1,2]. This process is also dysregulated during chronic diseases associated with aging, neurodegeneration, and cancer [3,4]. Autophagy can be modulated using pharmacological agents and is a major drug development target for treating cancer, neurodegeneration, and pathogen infection [5,6]. Along with medical applications, autophagy modulation has also shown potential to enhance biomanufacturing by increasing cell longevity [7]. Thus, quantifying autophagy dynamics will be critical in any application that involves modulating this process.
A major challenge in determining how autophagy is modulated is the limited tools that allow for direct and absolute quantification of each step in the process. Western blot is commonly used to estimate autophagy; however, it is typically less quantitative and less sensitive. Western blot also does not allow measurement of all the autophagic steps. Fluorescent reporter systems have enabled the quantification of autophagosomes in live cells. Green fluorescent protein (GFP) labeling of MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3), a protein associated with autophagosomes and autolysosomes, allows for quantification of autophagosome accumulation. GFP is pH-sensitive and is bleached in the acidic environment of the autolysosome in this system [8,9]. Tandem green and red (GFP:RFP) labeling of LC3 has also been used to identify acidic autolysosomes since RFPs are acid-stable [10,11]. Although these systems allow direct monitoring of autophagosomes and autolysosomes in real time, they do not provide direct quantification of each step, also known as “autophagy flux”. For instance, the accumulation of autophagosomes could be a result of an increase in the formation or decrease in the clearance of autophagosomes. The same principle applies to autolysosomes.
To quantitatively measure autophagy flux, the inputs and outputs of autophagosome and autolysosome accumulation must be dissected. Measuring autophagosome accumulation after inhibiting clearance with small molecules such as bafilomycin A1 is a commonly used approach for measuring autophagic flux [12–14]. Nevertheless, this approach does not provide a direct quantification of autophagosome and autolysosome clearance steps. A new approach using a novel fluorescent probe was described to quantify autophagic flux by measuring the GFP:RFP signal ratio without adding lysosomal inhibitors [15]. However, this method does not provide a direct quantification of autolysosomes. It is also not sensitive to identify differences in the autophagic flux if the changes are relatively similar. For example, inhibition of autophagosome formation could lead to similar changes in both GFP and RFP signals. Thus, a true change in flux may still result in a similar GFP:RFP ratio as basal, leading to ambiguity. Another case could be equal and simultaneous initiation and inhibition of autophagosome formation and clearance (resulting in a constant GFP signal), which could lead to no observable difference in the fluorescence levels. Finally, most measurements made using the methods discussed above are made long after perturbation, when the system reaches a new steady-state where the rates of all the steps are equal. Although this provides very useful information about the final autophagy state, it is incapable of informing the nature of the perturbation and the dynamic response of the cells to the perturbation. To illustrate, an autophagosome formation inhibitor and an autophagosome formation inducer might reach the same final steady-state even though they perturb autophagy very differently. Therefore, it is essential to temporally quantify all the steps involved in autophagy to gain a better understanding about the perturbation as well as the regulatory mechanisms of this dynamic process.
Here, we present an approach to quantify the rates of each step of the autophagy pathway under non-steady-state conditions. This approach directly builds upon previous advances that provided a quantitative framework to measure autophagy rates at a steady-state [12,13] and is enabled by high-throughput live cell imaging and measurement of instantaneous autophagy rates. With this approach, we study the effects of well-characterized autophagy regulators rapamycin and wortmannin. Through our non-steady-state analysis, we show that rapamycin dynamically regulated autophagy flux to reach an elevated state that decreases back to basal levels over time. We use the non-steady-state rate approach to reveal two modes of regulation that cause this dynamic behavior. We further show that rapamycin concentrations can be used to precisely modulate autophagy flux. Conversely, we show that wortmannin initially inhibits autophagy flux, which recovers over time in a concentration-dependent manner. Additionally, we performed a similar analysis during serum and glutamine starvation. Measuring rates after starvation indicated that autophagy rates are rapidly and transiently induced by serum starvation, while glutamine starvation inhibited autophagy rates on a longer timescale. Taken together, this innovative approach has the potential to provide novel insights related to autophagy and mechanisms driving autophagy-regulating perturbations through quantitative measurements.
Results
Non-steady-state measurement of autophagy rates
Measurement of steady-state autophagy flux has long been performed [14,16]. Loos and colleagues established a formal framework to quantify autophagy flux or the rate of autophagosome formation under steady-state conditions [12,13]. The model considers the whole autophagic process as a multistep process governed by three steps: 1) the rate of formation of autophagosomes (R1), 2) the rate of autolysosome formation via fusion of autophagosomes with lysosomes (R2) and 3) the rate of degradation of autolysosomes (R3) (Figure 1A). This method relies on quantifying autophagosomes and their accumulation over time in live cells by using GFP-labeled LC3 as described above. Performing a mass balance on the autophagosomes (AP) yields an expression for the rate of change of autophagosomes:
| (1) |
Figure 1.
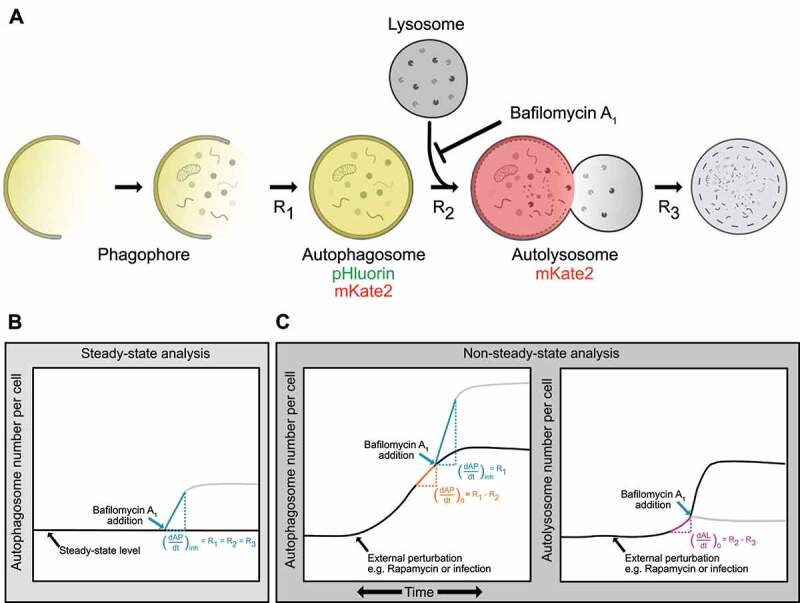
Conceptual framework of non-steady-state analysis of autophagy rates. (A) Phagophores expand to form autophagosomes. Autophagosomes fuse with lysosomes to form autolysosomes. Contents are degraded in autolysosomes. The rates of each of these steps (R1, R2, and R3) can be measured using a mass action model and live-cell imaging. Fluorescently tagged LC3 (pHluorin-mKate2-LC3) can be used to quantify autophagosomes (pHluorin- and mKate2-positive) and autolysosomes (mKate2-positive, pHluorin is quenched at low pH). (B) Measurement of autophagosome numbers following inhibition of autophagosome-lysosome fusion using bafilomycin A1 allows for measurement of R1, the rate of autophagosome formation. When performed at a steady-state, this rate is equal to the other rates in the pathway. (C) When changes in autophagosome and autolysosome numbers are measured using an instantaneous rate approach, all rates in the autophagy pathway (R1, R2, and R3), which may not be equal under dynamic conditions, can be measured.
Similarly, the rate of change of autolysosomes (AL) can be written as:
| (2) |
Under steady-state, no change in autophagosomes or autolysosomes over time is observed because R1, R2, and R3 are equal. Using bafilomycin A1 to inhibit the fusion of the autophagosome with lysosomes sets and results in autophagosome accumulation. Immediately post-inhibition:
| (3) |
where is the accumulation rate of autophagosomes following inhibition of R2 by bafilomycin A1 [12,13] (Figure 1B).
Despite the success of quantitative autophagy flux measurements, non-steady-state measurements (dynamic conditions in which R1, R2, and R3 may not be equal) remain out of reach. We expanded on this previous work to develop a non-steady-state rate approach that enables the evaluation of all three rates under dynamic conditions (Figure 1C). Measuring the change in the number of autophagosomes with time just before the chemical inhibition would provide the net rate of change of autophagosomes at that time point . R2 can then be evaluated using Equations (1) and (3):
| (4) |
We can extend this analysis to also quantify the instantaneous net rate of change of autolysosomes prior to chemical inhibition. R3 can be evaluated using Equations (2) and (4):
| (5) |
Thus, we can recover the absolute formation and fusion rates of autophagosomes along with the degradation rate of autolysosomes as a function of time by carrying out this approach at multiple time points.
Experimental system to monitor autophagy dynamics
Accurately quantifying autophagy rates over time requires a method to distinguish autophagosomes from autolysosomes and to track them in live cells simultaneously. Several tandem reporter systems that can distinguish autophagosomes from autolysosomes have been previously described [17–19], though none have been used to extract rate data for all steps in the autophagy pathway. We used the previously developed Super-Ecliptic, pHluorin-mKate2-LC3 system [11]. pHluorin is an acid-sensitive GFP, while mKate2 is an acid-stable RFP. Thus, autophagosomes are green and red, while the acidic autolysosomes are only red (Figure 1A).
We first confirmed that the accumulation of pHluorin-mKate2-labeled puncta is specific to autophagosomes using the tandem reporter with wild-type (WT) LC3 and a LC3 mutant (LC3 ΔG). LC3 ΔG lacks the glycine at the carboxyl-terminus, which is essential for proper lipidation and association with autophagosomes [8,20]. The addition of bafilomycin A1 and rapamycin, two well-established modulators of autophagy [21,22], led to the expected accumulation of pHluorin-mKate2-labeled puncta in cells expressing WT LC3, but not in LC3 ΔG-expressing cells (Figure 2A).
Figure 2.
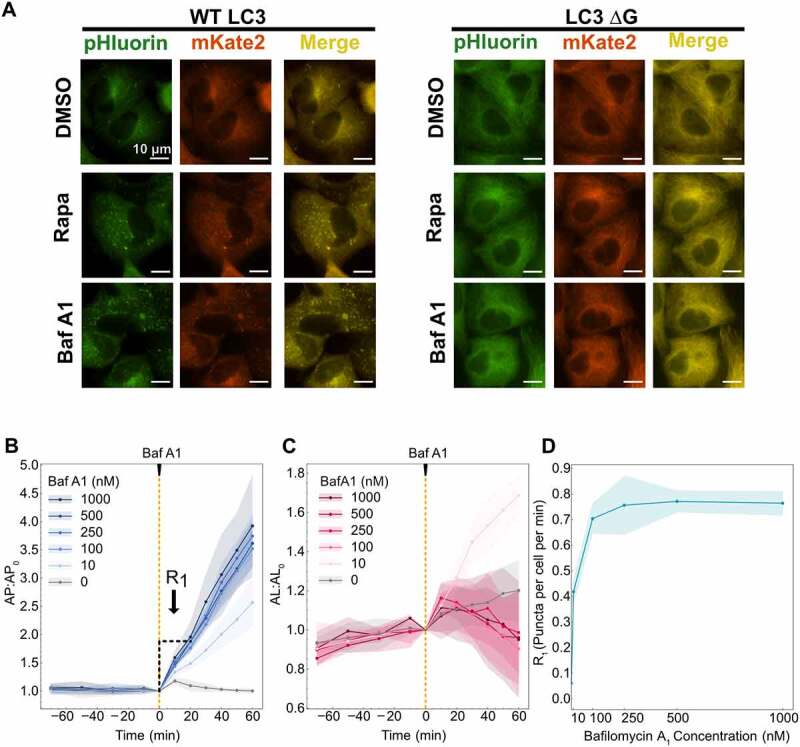
Calibration of system conditions for data collection. (A) Images of cells expressing the pHluorin-mKate2-LC3 tandem fluorescent reporter following DMSO, 100 nM rapamycin (Rapa) and 500 nM bafilomycin A1 (Baf A1) treatment. WT LC3 represent wild type and LC3 ΔG was used as a negative control because this mutant cannot be lipidated for phagophore association. (B) Autophagosomes and (C) autolysosomes were quantified over 90 min before addition of bafilomycin A1 and 60 min after. R1 was calculated using the first 20 min of data following bafilomycin A1 treatment. (D) R1 rates are plotted as a function of bafilomycin A1 concentration. At least 150–200 cells were imaged for all experiments.
We then calibrated our experimental system to determine the optimal concentration of bafilomycin A1 to inhibit R2 completely. We monitored autophagosome and autolysosome dynamics over time before and after the addition of bafilomycin A1 at various concentrations (Figure 2B,C). Autophagosome and autolysosome numbers were confirmed to be at a steady-state prior to the addition of bafilomycin A1. We observed a constant increase in autophagosomes over time following the addition of bafilomycin A1 for all concentrations tested, with more dramatic increases at higher concentrations. For autolysosomes, we did not observe any considerable changes for higher concentrations (100 nM and above) but for 10 nM bafilomycin A1 we observed an increase in the autolysosome numbers. This could be due to complete inhibition of autolysosome clearance by bafilomycin A1 [14,21] but only partial inhibition of the fusion step as indicated by the lower slope of autophagosome increase, leading to continuous autolysosome production but no clearance.
We used autophagosome data to determine R1 as a function of bafilomycin A1 concentration. R1 was measured as the slope using 20 min of data immediately following the addition of bafilomycin A1 (Figure 2B). The ability to measure rates within 20 min is a major advantage of this system as it allows the measurement of instantaneous R1 with minimal feedback from bafilomycin A1 addition. A saturation of R1 was observed starting at 100 nM bafilomycin A1 (Figure 2D). However, to ensure complete inhibition of R2 even during induced conditions (e.g., higher autophagosome and lysosome numbers), a concentration of 500 nM bafilomycin A1 was used for all subsequent experiments. We also acknowledge that it may not be possible to differentiate between autophagosomes and autolysosomes after bafilomycin A1 treatment. This is due to the unclear mechanism of bafilomycin A1 of either inhibiting fusion of autophagosomes with lysosomes or inhibition of acidification of autolysosomes [23]. Therefore, the measured number of autophagosomes could be accumulated either in the form of unfused autophagosomes or unacidified autolysosomes. Nevertheless, this does not affect our analysis as the approach only relies on measuring autophagosome accumulation after inhibition and we can measure the accumulation in either form. A mathematical explanation is provided in the supplemental information.
Rapamycin-induced autophagosome and autolysosome dynamics are concentration-dependent
We next tested the ability to monitor autophagosome and autolysosome temporal dynamics using rapamycin, which induces autophagosome formation through the inhibition of MTORC1 (mechanistic target of rapamycin kinase complex 1) [24–26]. We tested seven concentrations of rapamycin (Figures 3, S1A,B). Autophagosomes increased following rapamycin treatment, with higher rapamycin concentrations resulting in a more rapid increase (Figures 3A, S1A). For the highest concentrations of rapamycin (>10 nM), this rapid increase peaked at 30 min post-treatment, followed by a gradual decrease. We observed a saturation behavior for concentrations above 10 nM. A mid-range concentration of rapamycin (1 nM) resulted in a more gradual increase in autophagosome numbers, followed by a slight decrease. The lowest concentration of rapamycin tested (0.1 nM) had no effect. Autolysosome dynamics followed similar concentration-dependent trends, with slightly delayed peaks at 1.5–2 h post-treatment for higher concentrations (Figures 3B, S1B). Interestingly, there was no difference in autophagosome and autolysosome peak time (3.5–4 h) for the mid-range concentrations of rapamycin (0.5 and 1 nM). The raw autophagosome and autolysosome data are provided in the supplemental (Figure S1C,D). We also confirmed rapamycin inhibition of MTORC1 activity by monitoring change in the phosphorylation status of the RPS6/S6 ribosomal protein at the Ser 240/244 site, which is a downstream substrate of MTORC1 (Figure S1E).
Figure 3.
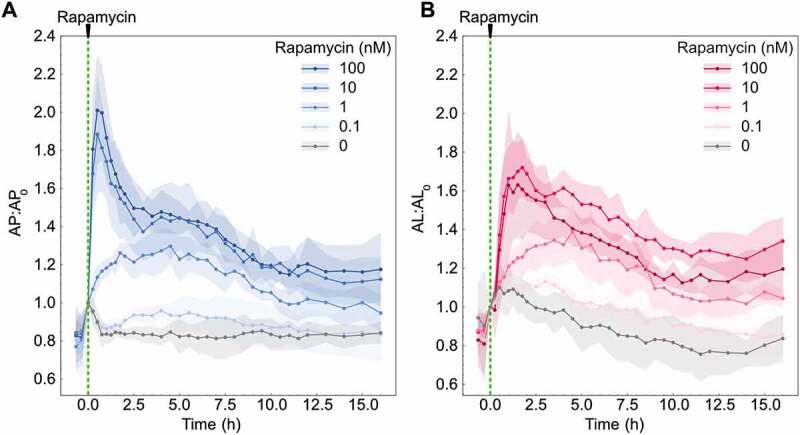
Autophagosome and autolysosome dynamics are a function of rapamycin concentration. (A) Autophagosome and (B) autolysosome number dynamics after rapamycin treatment. The indicated concentration of rapamycin was added at 0 min. The number of autophagosomes and autolysosomes at 0 min was used as the normalization factor. Data points represent mean while shaded area represents ± standard deviation. Four independent replicates were performed. At least 150–200 cells were imaged for all experiments.
Time evolution of autophagy rates reveals initial rate-limiting steps
The autophagosome dynamics for high concentrations of rapamycin (>10 nM) suggested two possible models of cellular response to rapamycin treatment. The sudden increase in autophagosome numbers followed by a decrease could be driven by a rapid increase in the rate of autophagosome formation (R1), while the rates of autolysosome formation and degradation (R2 and R3, respectively) lag due to latency in the pathway response. Alternatively, R1 could increase and then decrease due to feedback mechanisms induced by sustained rapamycin treatment. We next sought to distinguish between these two possible cellular response models using the non-steady-state rate approach described earlier.
To understand which mode of response cells were operating, we measured R1, R2, and R3 over time following rapamycin treatment. Initially, we focused on cells treated with a high concentration of rapamycin (100 nM) compared to untreated cells (DMSO). Raw autophagosomes and autolysosomes data used for rate measurements at 30 min are shown as an example to illustrate the procedure followed (Figure 4A,B). Cells were at a steady-state before rapamycin treatment, with no changes in either autophagosome or autolysosome numbers. Following rapamycin addition, we observed an increase in autophagosome and autolysosome numbers, similar to our previous experiments. Bafilomycin A1 was then added to measure rates. This overall procedure was repeated to collect rate data from 10 min to 15 h post-treatment. Importantly, the rates of untreated cells (basal autophagy rates) were at a steady-state, meaning all three rates were equal and did not vary over time (Figure S2).
Figure 4.
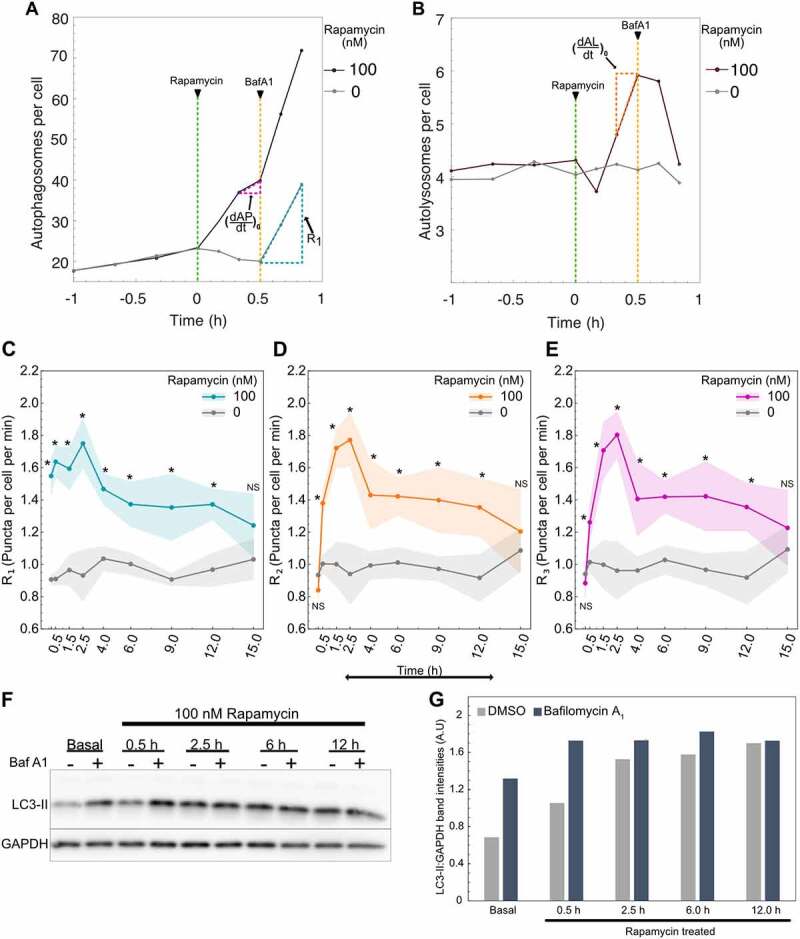
Autophagy rates change over time following rapamycin treatment. (A) Raw autophagosome and (B) raw autolysosome dynamics for rate measurement at 30 min post-rapamycin treatment. R1 was calculated using the autophagosome data 20 min post-bafilomycin A1 addition. and were calculated using the autophagosome and autolysosome data respectively 10 min before bafilomycin A1 addition. (C) Change in R1 (D) R2 and (E) R3 over time. Data points represent mean while shaded area represents ± standard deviation. Four independent replicates were performed. (*) indicates p-value <0.05 and NS indicates not significant. P-values were calculated using an independent two-tail t-test. (F) LC3-II and GAPDH protein quantification using western blot. Cells were treated with 100 nM rapamycin for different time points followed by 500 nM bafilomycin A1 (Baf A1) treatment. Basal samples represent DMSO-treated cells. (G) Quantification of the western blot shown in Fig 4F using densitometry. LC3-II band intensity is normalized with the respective GAPDH band intensity in the same lane. At least 150–200 cells were imaged for all imaging experiments.
For rapamycin-treated cells, we observed a nearly immediate increase in R1, with significant changes in R1 measured as soon as 10 min post-treatment compared to untreated cells (Figure 4C). This significantly elevated rate was maintained until 12 h post-treatment. This result is consistent with the known mechanism of rapamycin inducing autophagy upstream of phagophore expansion and thus validates the proposed non-steady-state approach to characterize the effects of external perturbation on autophagy. Interestingly, R2 and R3 were slower to increase, with significantly increased rates starting at 30 min post-treatment (Figure 4D,E). Similar to R1, R2 and R3 maintained significantly elevated rates until 12 h post-treatment. At 15 h post-treatment, all rapamycin rates were statistically indistinguishable from the basal rates of untreated cells. The dynamics we observed suggest that R2 and R3 may represent rate-limiting steps initially, after which there is a general decrease in all rates.
To confirm the puncta and rate dynamics observed using the new method are consistent with the traditionally used method, we measured LC3-II protein levels using western blot. We treated cells with 100 nM rapamycin for different time points followed by the addition of 500 nM bafilomycin A1 to measure R1 (Figures 4F,G and S3A,B). Cells were treated with bafilomycin A1 for 2 h to ensure a consistent and detectable change in the LC3-II levels. Here, the higher sensitivity of the new method is noteworthy, as it can detect changes as soon as 20 min after bafilomycin A1 treatment compared to 2 h for western blot. We confirmed the increase in LC3-II levels for DMSO samples after treating with bafilomycin A1, indicating the inhibition of the fusion step. For just rapamycin treatment, we observed an increase in LC3-II levels starting at 30 min followed by constant maintenance of LC3-II levels until 12 h post-treatment. This is contrary to the observed autophagosome and autolysosome puncta dynamics where there is an initial increase followed by a decrease (Figure 3A,B). We hypothesize that the variation between puncta dynamics measurements and western blot measurements is caused by the intrinsic nature of each measurement. LC3-II protein levels measured using western blot indicate the summation of LC3-II protein on autophagosomes and autolysosomes. Both autophagosome and autolysosome puncta remained higher than the initial state even at 15 h, indicating higher levels of LC3-II protein at those time points. Moreover, the number of LC3-II molecules bound to each autophagosome could be dynamic and is challenging to measure. Nonetheless, the initial accumulation of LC3-II was consistent between the two methods. To validate the observed R1 dynamics, bafilomycin A1 was added at multiple time points after treating with rapamycin (Figures 4F,G and S3A,B). We observed a clear increase in LC3-II accumulation at 30 min, while for the later time points, it only caused a modest increase. This indicates R1 is higher initially and declines over time. These results were also consistent with measurements made using the non-steady-state method, where R1 increased over 2.5 h post-treatment, followed by a gradual decrease.
Latency and feedback contribute to rapamycin-driven autophagy rate dynamics
We next set out to test these temporal differences in rates by comparing rates at different time points for rapamycin-treated cells. At 10 and 30 min post-treatment, R1 was significantly greater than R2 and R3 (Figure 5A), consistent with the rapid increase in the autophagosome numbers until 30 min post-treatment. But this difference was eliminated by 1.5 h post-treatment because of increases in R2 and R3 (Figure 5A), which is also consistent with the peak time of autolysosome numbers (Figure 3B). We focused our detailed temporal analysis on R1 since R2 and R3 reached the same level as R1 and followed the same trend from 1.5 h onward. Interestingly, we observed a constant R1 until 2.5 h post-treatment, at which point there was a gradual decrease until 15 h (Figure 5B). Thus, an increase in the overall flux through the pathway in response to rapamycin is initially limited by latency in R2 and R3, but not R1. This is followed by a general decrease in autophagy rates after 2.5 h. Thus, both models of regulation we initially hypothesized to exist are playing a role in the autophagy dynamics we observed.
Figure 5.
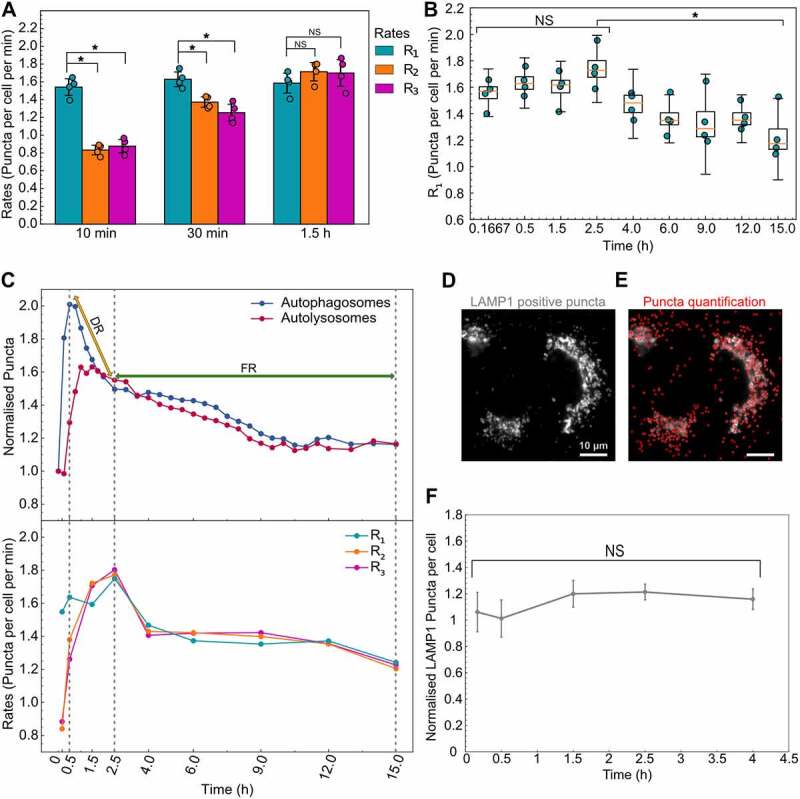
Autophagy rates indicate a hybrid model of cellular response to high concentrations of rapamycin. (A) Statistical comparison of autophagy rates for cells treated with 100 nM rapamycin at three different time points. (B) Temporal change in R1 for cells treated with 100 nM rapamycin. (*) indicates p-value <0.05 and NS indicates not significant. Statistical significance for the first four points was calculated using a one-way ANOVA test. The P-value for the statistical test between 2.5 and 15 h is calculated using paired two-tail t-test. (C) Normalized mean values of autophagosome (AP:AP0) and autolysosome numbers (AL:AL0) along with mean values of autophagic rates (R1, R2, R3) are compared to visualize the two regimes of cellular response. DR and FR represent degradative and feedback regimes, respectively. (D) A549 cells stained with LAMP1 antibody. (E) LAMP1 positive puncta detection (shown in red). Scale bar: 10 µm. (F) Temporal change in the normalized LAMP1 puncta/cell (rapamycin treated to DMSO treated) after treatment with 100 nM rapamycin. Error bars represent standard error for four independent replicates. NS indicates not significant, statistical significance was calculated using a one-way ANOVA test. At least 150–200 cells were imaged for all experiments.
To illustrate this hybrid model of cellular response and regulation of autophagy rates, we juxtaposed the autophagosome and autolysosome dynamics with rate data (Figure 5C). We used 30 min as a reference point to compare the temporal changes in R1 since autophagosome numbers peak at 30 min. The immediate spike in R1 but lag for R2 and R3 caused the initial accumulation of autophagosomes in the first 30 min. From 30 min to 2.5 h, the decrease in autophagosomes was caused by an increase in R2 and R3 to the same level as R1, leading to the degradation of accumulated autophagosomes, which we named the degradative regime (DR). However, after 2.5 h, the decrease in autophagosome numbers was a result of the decrease in R1 along with R2 and R3, which we named the feedback regime (FR). These results underscore the overall consistency of temporal rate data with the autophagosome and autolysosome dynamics.
We hypothesized that the initial lag in R2 and R3 in degradative regime is due to a lack of lysosomes to fuse with the newly formed autophagosomes. To test this hypothesis, we performed immunofluorescence staining for LAMP1 (lysosomal-associated membrane protein 1) after treating cells with 100 nM rapamycin for 4 h (Figure 5D). We chose 4 h, as R2 and R3 reach R1 by 1.5 h and remain the same thereafter. We quantified the LAMP1-positive puncta for basal and 100 nM rapamycin-treated cells (Figure 5E). Contrary to our hypothesis, there was no significant difference in the normalized puncta (rapamycin-treated relative to basal) between 10 min and 4 h of rapamycin treatment (Figure 5F). This indicates that the number of lysosomes is not a limiting factor for the fusion step, and another aspect of autophagosome-lysosome fusion is limiting.
Initial autophagy rates and time evolution of rates depend on rapamycin concentration
Given the concentration-dependent effects of rapamycin on autophagosome and autolysosome dynamics, we hypothesized that autophagy rates might also exhibit concentration-dependent effects. We thus measured all the rates for a range of rapamycin concentrations over 15 h (Figures 6A and S4). Mid-range concentrations of rapamycin (0.5–1.0 nM) resulted in a more gradual increase in R1 compared to high rapamycin concentrations (10–100 nM). To quantify rapamycin’s ability to induce autophagy, we modeled R1 using the Hill equation [27] (Figure 6B). R1 at 10 min was used to model rapamycin induction kinetics because this time point represents the effect of rapamycin on autophagy with minimal time for feedback mechanisms from the cells. In the Hill equation, (R1) Basal represents the basal rate of autophagosome formation in the absence of any perturbation. This basal rate was estimated to be 0.90 puncta per cell per minute. Vm and Km represent the rapamycin-induced maximal autophagy level and half-maximal rapamycin concentration, respectively. Vm and Km were estimated to be 0.685 puncta per cell per minute and 1.1 nM. The exponent n represents the observed Hill coefficient and was estimated to be 1.9. This model can be used to predict rapamycin’s ability to induce R1 at early stages and will be useful in developing a complete temporal model. This method may be extended to various autophagy perturbations, which may relate to the mechanism of action based on the perturbed initial response and enable modeling of the response.
Figure 6.
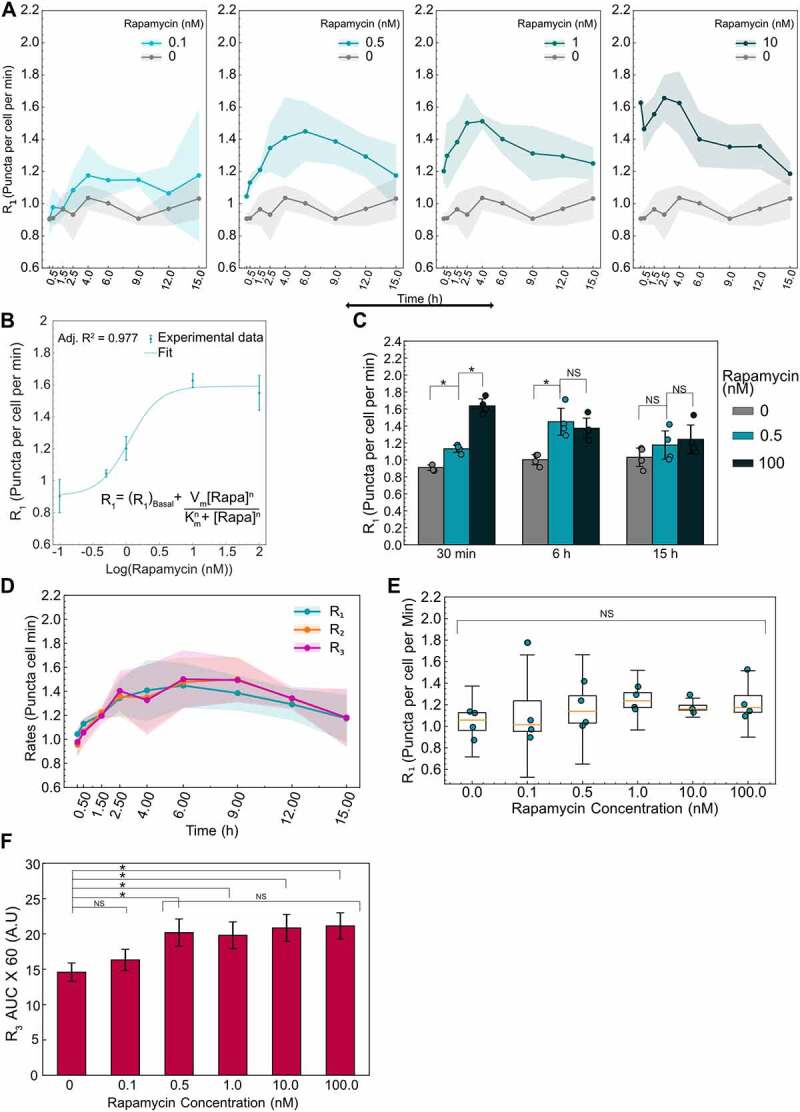
Initial and time evolution of autophagy rates depend on rapamycin concentration. (A) Temporal dynamics of R1 for different concentrations of rapamycin. Data points represent the mean while shaded area represents ± standard deviation. Four independent replicates were performed. (B) R1 at 10 min after rapamycin addition is plotted as a function of rapamycin concentration. Individual data points represent experimental data while the dotted line represents the model fit. The model used for fitting the data along with the adjusted R2 value is also shown. (C) Statistical comparison of R1 for three different rapamycin concentrations at three different time points. (*) indicates p-value <0.05 and NS indicates not significant. P-values were calculated using an independent two-tail t-test. (D) Temporal evolution of all autophagic rates (R1, R2, R3) for 0.5 nM rapamycin-treated cells. Data points represent the mean while the shaded area represents ± standard deviation. (E) R1 at 15 h as a function of rapamycin concentration. NS indicates not significant. P-values were calculated using a one-way ANOVA test. (F) AUC for R3 data for different concentrations of rapamycin. Data represent mean ± standard deviation. P-values were calculated using a one-way ANOVA test followed by Tukey’s post hoc test for pairwise comparison. At least 150–200 cells were imaged for all experiments.
In addition to concentration-dependent effects on initial R1, we also observed concentration-dependent effects on the time evolution of R1. For a mid-range concentration of rapamycin (0.5 nM), R1 gradually increased and reached the same level as R1 of higher concentrations (100 nM) over 6 h (Figure 6C). This was surprising given the very low accumulation of autophagosomes and autolysosomes for 0.5 nM treatment (Figure S1A,B). Consequently, we explored the temporal nature of all autophagy rates (R1, R2, and R3) for this mid-range concentration of rapamycin. We hypothesized that the slower response time for R1 at mid-range concentrations of rapamycin (Figure 6A) might allow adequate time for R2 and R3 to adjust in sync with R1, even at early time points, compared to the rapid response for high concentrations of rapamycin (100 nM). Measuring R2 and R3 over time showed a similar trend, equal to R1 over the entire 15 h time course, thus resulting in a low accumulation of the autophagic vesicles (Figure 6D). Interestingly, rates decreased at longer time points. R1 was indistinguishable from basal levels by 15 h post-treatment for 0.5 nM rapamycin (Figure 6C), similar to the high concentration of rapamycin (Figure 4C). This led us to look at long-term impacts on R1 for all concentrations of rapamycin. For all concentrations tested, R1 was not significantly different from basal levels by 15 h post-treatment (Figure 6E), suggesting adaptation of the autophagy response to long-term inhibition of MTOR complexes. Taken together, rapamycin treatment results in early responses that are concentration-dependent, with mid-range concentrations resulting in slower responses that evolve as steady-state flux through the pathway. These results further signify the importance of measuring all the autophagy rates temporally to capture the complete response.
Given the temporal and concentration-dependent behavior of autophagy, we wanted to develop a simple metric for measuring the total amount of autophagy processed until steady-state conditions are reached. We measured the area under the curve (AUC) for R3. Assuming the average cargo captured and degraded are the same for each condition, this measure would indicate the total amount of cargo completely degraded through the autophagic pathway. For example, perturbations such as rapamycin that induce autophagic flux would have higher AUC while perturbations that inhibit autophagy initiation or clearance would have low AUC. Using the R3 temporal data spanning 15 h, we calculated the AUC for basal and all rapamycin treatments (Figure 6F). While 0.1 nM rapamycin was indistinguishable from the basal condition, all other rapamycin concentrations lead to significantly higher AUC, indicating higher autophagic flux. Intriguingly, all rapamycin concentrations above 0.1 nM degraded similar amounts of cargo. These results were consistent with the observed slow response for the mid-range concentration (0.5 and 1 nM) and a faster but shorter response for higher concentrations (10 and 100 nM) as discussed earlier. Henceforth, this measurement can be used as an additional metric to track the total amount of autophagy perturbed.
Wortmannin temporarily inhibits basal and rapamycin-induced autophagosome formation
After validating our non-steady-state approach using an autophagy inducer, we next set out to test the method using wortmannin, a commonly used inhibitor of autophagosome formation [28,29]. We used 1 µM wortmannin to test its ability to inhibit basal and rapamycin-induced autophagy. First, we measured the autophagosome and autolysosome temporal dynamics after treating with wortmannin and/or rapamycin (Figure 7A,B). We observed an immediate decrease in the autophagosome numbers for wortmannin, as well as wortmannin with rapamycin-treated conditions, while the autolysosomes numbers remained constant. Surprisingly, after 30 min, we observed an increase in the autophagosome numbers in cells treated with wortmannin only and a combination of wortmannin with rapamycin. Moreover, the rate of increase in autophagosome number for wortmannin with rapamycin-treated cells was faster than just wortmannin-treated cells. The autophagosomes for wortmannin with rapamycin reached initial levels by 1 h and kept increasing to saturate at a higher level than basal (1.8-fold) by 6 h, followed by a slight downward trend by 15 h. Wortmannin-only treatment took 3.5–4 h for autophagosome numbers to reach the initial level and saturated at a slightly higher level (1.3-fold). For autolysosomes, we observed a higher accumulation for wortmannin with rapamycin treatment (1.5-fold) compared to just wortmannin (1.1-fold), similar to the autophagosome behavior. We used 100 nM rapamycin and basal (DMSO) as controls and their behavior remained the same as earlier experiments. From the known inhibitor mechanism of wortmannin, we hypothesized the initial drop in autophagosomes is a result of inhibition of R1. However, the increase after 30 min could either be a result of an increase in R1 or a much lower decrease in R2 and R3 compared to R1 or a combination of both. Rates for each step are needed to uncover the dynamics involved.
Figure 7.
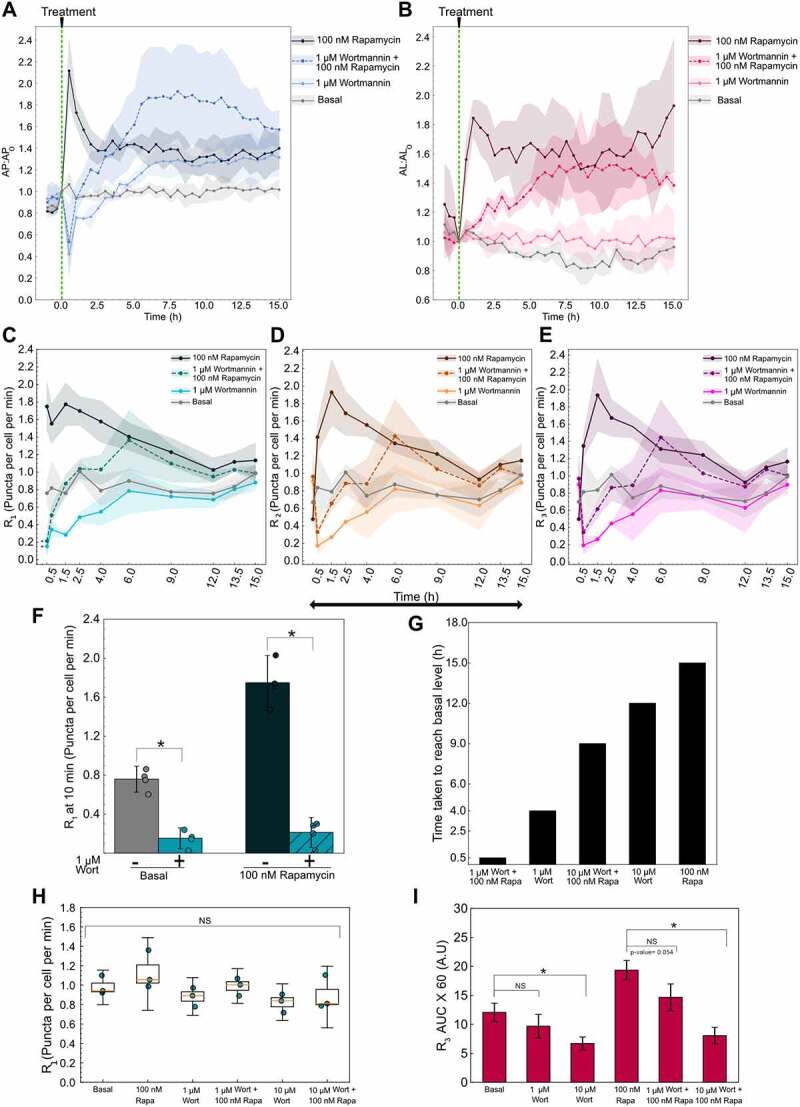
Variable autophagy recovery time from wortmannin’s inhibition. (A) Autophagosome and (B) autolysosome number dynamics after treatment. The indicated concentration of small molecule was added at 0 min. The number of autophagosomes and autolysosomes at 0 min was used as the normalization factor. Data points represent the mean while the shaded area represents ± standard deviation. Three independent replicates were performed. (C) R1 (D) R2 (E) R3 temporal dynamics for basal, 1 µm wortmannin (Wort) with and without 100 nM rapamycin (Rapa), and 100 nM rapamycin alone. Data points represent the mean while the shaded area represents ± standard deviation. Three independent replicates were performed. (*) indicates p-value <0.05, p-values were calculated using an independent two-tail t-test. (F) Statistical comparison of R1 at 10 min for different treatments. (*) indicates p-value <0.05, p-values were calculated using an independent two-tail t-test. (G) Time taken for R1 of each treatment condition to reach the basal level. (H) R1 at 15 h plotted as a function of treatment condition. NS indicates not significant. p-values were calculated using a one-way ANOVA test. (I) AUC for R3 for different treatments. P-values were calculated using a one-way ANOVA test followed by Tukey’s post hoc test for pairwise comparison. (*) indicates p-value <0.05 and NS indicates not significant. At least 150–200 cells were imaged for all experiments.
We used the non-steady-state method discussed earlier to measure the individual rates following wortmannin treatment. At 10 min, we observed an immediate decrease in the R1 while R2 and R3 remained at the basal level for wortmannin-treated cells, confirming the known inhibitory mechanism of action of wortmannin (Figure 7C–E). Moreover, R1 for wortmannin with rapamycin was also significantly lower at 10 min compared to basal and rapamycin treatments (Figure 7C,F). Therefore, wortmannin initially inhibits basal as well as rapamycin-induced autophagosome formation. R2 and R3 decreased by 30 min due to the lack of autophagosomes to degrade because of decreased R1 (Figure 7D,E). Interestingly, R1 increased over time following initial inhibition by wortmannin. Wortmannin is less stable in cell culture media and could be the major reason for recovery after wortmannin treatment [30]. However, the possibility of feedback cannot be completely ruled out. R2 and R3 follow a similar trend as R1 after 30 min with a slight delay (Figure S5A,B). This indicates that the behavior is mainly driven by R1 and the downstream flow of autophagosomes through the pathway is unperturbed. Rapamycin and basal rate behaviors were consistent with the previous results (Figure 7C–E).
We tested a higher wortmannin concentration (10 µM) to probe if it plays a role in the recovery of R1 (Figure S5C–E). We analyzed the time taken for R1 to reach back to a statistically insignificant level as basal and potentially exceed it (Figure 7G). A treatment of 1 µM wortmannin reached basal level by 4 h while 10 µM wortmannin took approximately 12 h, suggesting wortmannin’s concentration is a governing factor. Moreover, we observed a faster recovery of wortmannin with rapamycin-treated cells. For 1 µM wortmannin, cells also treated with rapamycin reached basal level by 30 min compared to 4 h for wortmannin alone. Similarly, for 10 µM wortmannin, the cells with rapamycin took 9 h compared to 12 h with wortmannin alone. The accelerated recovery of rapamycin-treated cells could be due to the additional autophagosome induction capacity of rapamycin.
We next analyzed the final steady-state rates and the total cargo degraded in terms of R3 AUC over 15 h. At 15 h, the rates of all treatment conditions were indistinguishable from each other as well as the basal condition (Figure 7H). This result reemphasizes the importance of temporal monitoring of autophagy, as rapamycin and wortmannin, which have opposing effects on autophagosome formation, reach the same final steady-state. Finally, using the R3 data collected over 15 h, we calculated the AUC for 1 µM and 10 µM wortmannin treatment conditions (Figure 7I). We anticipated a decrease in the overall cargo degraded as wortmannin inhibited the initiation of autophagosome formation and thus reduced the overall flux through the pathway. We did not observe a significant decrease in the overall cargo degraded for 1 µM wortmannin treatment compared to basal but did for 10 µM wortmannin treatment. For cells treated with wortmannin along with rapamycin, 1 µM wortmannin caused a clear decrease in cargo degraded compared to the rapamycin sample even though it did not meet our statistical criteria (p-value = 0.0542). Conversely, rapamycin with 10 µM wortmannin treatment significantly decreased the cargo degraded compared to rapamycin-induced conditions. These results are consistent with the faster recovery of autophagy under 1 µM wortmannin treatment compared to 10 µM. These measurements can be utilized to further guide the precise tuning of autophagy.
As these observations were made using a single A549- pHluorin-mKate2-LC3 clone, we tested the behavior of bulk sorted A549-pHluorin-mKate2-LC3 cells. We measured the change in autophagosome and autolysosome dynamics as well as the rates for 100 nM rapamycin and 1 µM Wortmannin treatment. The overall behavior of the bulk sorted cells was similar to that of the individual clone which was used for all the measurements (Figure S6A–E), indicating the clone’s behavior is representative of the general A549 cell line. To further confirm that the method is expandable to other cell lines, we developed a bulk sorted U2OS-pHluorin-mKate2-LC3 cell line. A similar analysis was performed on U2OS cells and parallel behaviors were observed between U2OS and A549 cell lines under rapamycin and wortmannin treatment (Figure S6F–J). There was substantial variation in the amount of LC3 expressed among the bulk sorted cells, which could be the main reason for the variation observed in the data. Finer optimization of the sorting process for selecting similar expressing populations could mitigate such variability.
Serum and glutamine starvation regulate autophagosome dynamics on different timescales
Serum starvation and nutrient deprivation regulate autophagy in many biological systems such as cancer and bioproduction [31,32]. We tested the dynamic autophagy response to serum starvation, glutamine starvation, and a combination of both in our clonal A549 cell line. Under normal conditions, cells were stably cultured at 8% fetal bovine serum (FBS) and 4 mM glutamine concentration. Cells were imaged for 1 h before diluting the FBS and glutamine concentration to 0.64% and 0.32 mM, respectively, by serial dilution via repeated partial media replacements. A serial dilution approach was used to minimize the stress induced by replacing the media in the wells and to prevent cells from being completely dry. Nevertheless, due to the high sensitivity of the system, we noticed differences between unperturbed conditions, in which cells were completely undisturbed, compared to the basal control, which involved removing and adding complete media to the cells (Figure S7A).
Serum starvation caused an immediate increase in autophagosome and autolysosome numbers, peaking within 2 h followed by a decrease (Figure 8A,B). Thereafter, the autophagosome numbers stabilized at the initial steady-state number, while the autolysosomes saturated at an appreciably lower steady-state number (Figure 8A,B). Conversely, glutamine starvation led to a modest increase in autophagosome number compared to basal fluctuations (Figure 8A). As discussed previously, the fluctuations in basal can be associated due to the stress induced by the removal and addition of media to cells (Figure S7A). By 20 h we observed a downward trend in the autophagosome numbers for glutamine-starved conditions compared to basal (Figure 8A). Autolysosomes overall followed similar trends as autophagosomes, with a modest increase followed by a modest decrease (Figure 8B). For combined serum and glutamine starvation, there was a rapid increase and decrease in autophagosomes and autolysosomes in the first 8 h, similar to serum starvation (Figure 8A,B). We then observed a continuing downward trend in autophagosomes, similar to glutamine starvation (Figure 8A,B). This suggests that serum starvation may dominate at the initial stages while glutamine starvation comes into effect at a later stage.
Figure 8.
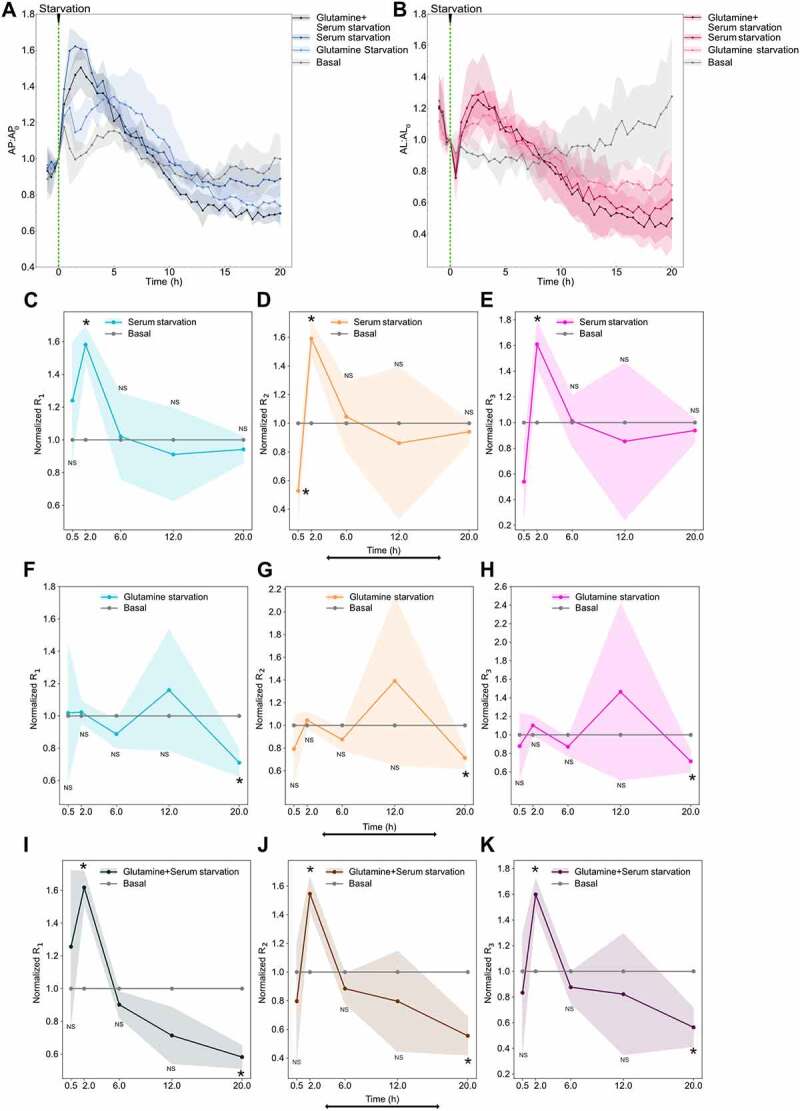
Serum starvation induces autophagy flux while glutamine starvation inhibits autophagy flux. (A) Autophagosome and (B) autolysosome number dynamics under starvation conditions. Normal media was removed partially and media with no serum or no glutamine or both were added at 0 min to dilute the concentration. The number of autophagosomes and autolysosomes at 0 min was used as the normalization factor. Data points represent the mean while shaded area represents ± standard deviation. Three independent replicates were performed. Normalized (C) R1 (D) R2 (E) R3 temporal dynamics of serum-starved with respect to basal. Basal line at one is provided for reference. Normalized (F) R1 (G) R2 (H) R3 temporal dynamics for glutamine-starved with respect to basal. Normalized (I) R1 (J) R2 (K) R3 temporal dynamics for glutamine + serum-starved with respect to basal. Data points represent the mean while the shaded area represents ± standard deviation. Three technical replicates were performed. Statistical comparison between various starvation conditions and basal rates at every time point were made using an independent two-tail t-test. (*) indicates p-value <0.05 and NS indicated not significant. At least 150–200 cells were imaged for all experiments.
Serum and glutamine starvation have opposing impacts on autophagy rates
To decipher the rate dynamics leading to the observed autophagosomes and autolysosome dynamics, we measured the rates using the non-steady-state approach described earlier. Rates were measured from 30 min to 20 h after starvation. An early timepoint was not chosen because the unstarved cells were also stressed initially due to the mixing involved during the change of media (Figure S7B–D). Therefore, all the rates were normalized with the basal condition to account for the stress induced by the exchange of media (Figure 8C–K). We only observed a significant difference between serum starvation and basal rates at 2 h post starvation, where all rates from serum starvation were higher (Figure 8C–E). This was consistent with the observed maximum autophagosomes and autolysosome accumulation during that time period. We also observed that the differences among the three rates were minor, indicating that the overall flux was increased at 2 h post starvation with minimal lag. After 2 h, all three rates returned to basal levels.
For glutamine starvation, we did not detect a significant difference between basal and glutamine-starved rates until 20 h post starvation (Figure 8F–H). This was consistent with the observed late response of autophagosome and autolysosome dynamics for glutamine starvation (Figure 8A,B). We also observed no significant differences between the three rates, indicating that the overall flux decreased at 20 h post starvation.
For both glutamine and serum starvation, we observed a significant increase in all the rates at 2 h, similar to serum starvation alone, while at 20 h we observed a significant decrease in all three rates, similar to glutamine starvation alone (Figure 8I–K). Although significant, we only observed minor differences between the three rates at 2 h, suggesting an increase in overall flux with minor lag. At 20 h, there was no significant difference among the three rates, indicating that the overall flux decreased relative to the basal flux at a longer time period. As hypothesized earlier, this behavior corroborates the observed autophagosome and autolysosome dynamics and highlights the different timescales of glutamine and serum starvation responses. In summary, these results suggest that serum starvation transiently induces autophagy flux on a short timescale while glutamine starvation inhibits autophagy flux on a longer timescale.
Serum addition to serum-starved cells transiently inhibits autophagy flux
Given the faster response to serum starvation, we added back serum to the serum-starved cells at 20 h post-starvation to observe the dynamics. Serum-starved cells that were replenished with serum immediately decreased autophagosome numbers within 30 min while the continuously serum-starved cells remained at the same level (Figure 9A). Lower levels of autophagosomes were observed for 3–4 h after replenishment followed by an increase of autophagosomes to the basal level (Figure 9A). The autolysosome numbers had a slower response to serum replenishment (Figure 9B). These observations suggest that serum is an inhibitor of autophagy flux, which could be confirmed by rate measurements.
Figure 9.
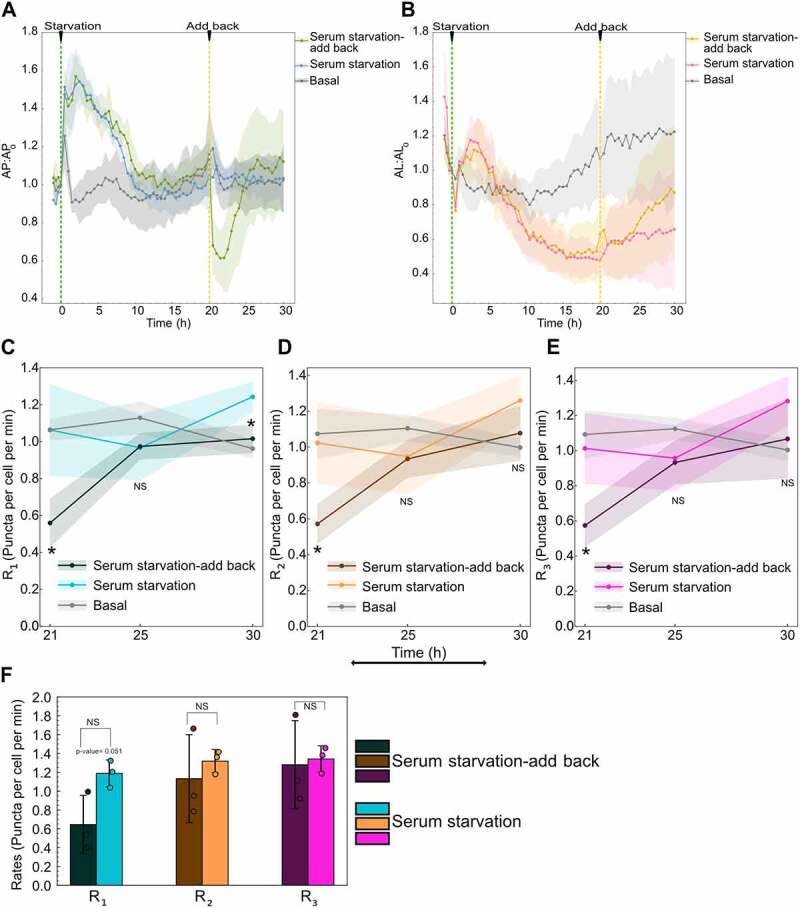
Serum addback to serum-starved cells transiently inhibits autophagy flux. (A) Autophagosome and (B) autolysosome number dynamics under continued serum-starved, serum-starved with serum replenishment at 20 h, and basal. The number of autophagosomes and autolysosomes at 0 min was used as the normalization factor. Data points represent the mean while the shaded area represents ± standard deviation. Three independent replicates were performed. Combined serum and glutamine starvation data are plotted on all the plots to facilitate comparison (E) R1 (F) R2 (G) R3 temporal dynamics for basal, serum-starved, and serum-starved after serum replenishment at 20 h. Data points represent mean while the shaded area represents ± standard deviation. Three independent replicates were performed. Independent two-tail t-test was used to compare statistical significance between serum-starved with no replenishment and serum-starved with replenishment. (*) indicates p-value <0.05. (H) Comparison of R1 R2, and R3 after 10 min of serum addback to serum-starved cells compared to continued serum-starved cells. Bar graphs represent the mean while the error bars represent ± standard deviation. Three technical replicates were performed. (*) indicates p-value <0.05, p-values were calculated using an independent two-tail t-test. At least 150–200 cells were imaged for all experiments.
We measured the rates after serum addition at 21, 25, and 30 h (1, 5, and 10 h post-serum addback, respectively). We observe lower rates for serum addback cells compared to serum-starved cells at 21 h. After 21 h, the rates recover to reach the basal level at 25 and 30 h (Figure 9C–E). As all three rates reach the same level by 21 h, we compared the rates immediately (10 min) after adding back serum to capture the latency in decrease among R1, R2, and R3. Even though not significant, we observe a considerable decrease in R1 immediately compared to R2 and R3 after the serum is added back compared to cells that continue in serum-starved conditions (Figure 9F). Overall, these results indicate serum transiently inhibits autophagy flux, with an initial latency in R2 and R3 responses.
Discussion
Quantitatively measuring all the autophagic steps remains a significant challenge and is key to developing better autophagy-based applications. This is especially critical for developing autophagy-based therapies, where dysfunction of cellular pathways is disease- and environment-specific, leading to variable response to the same treatment. Therefore, it is crucial to systematically characterize the disease state, kind of perturbation (for example, inducer or inhibitor) as well as the cellular response to gain a comprehensive understanding. Moreover, since autophagy is a dynamic process, it is pivotal to temporally monitor the process to capture the complete dynamic response until a steady-state is reached. These measurements will be essential in informing the overall change in the autophagic state after a perturbation, the feedback mechanisms involved, and their timescales. For example, this information will assist in developing combinatorial therapies for effectively modulating autophagy to treat diseases with finer control and minimal side effects [33,34].
We present a method to quantify autophagy rates in live cells. Previous studies have quantified the rate of autophagosome production under steady-state conditions [12,13,16]. We expand on these studies by creating a theoretical and experimental framework to measure autophagy rates for all three steps in the autophagy pathway under non-steady-state conditions. We do so by monitoring autophagosome and autolysosome numbers before and after inhibition of autophagosome-lysosome fusion. When combined with the instantaneous rate approach it enables measurement of all three autophagy rates without the requirement of them being equal.
By measuring autophagy rates under non-steady-state conditions for rapamycin, we were able to validate our system using a well-characterized inducer of autophagy. We observed concentration-dependent increases in initial rates of autophagosome formation. These results were consistent with previous studies measuring autophagy flux [35,36] and rapamycin’s well-established mode of action upstream of phagophore formation. We also observed an overall return to basal autophagy rates, consistent with previous indirect observations [36]. These results are indicative of long-term feedback mechanisms at play. Importantly, our approach enables measuring initial rates with high time resolution (10 min), which can uncover the direct mode of action of an autophagy perturbation, before long-term feedback mechanisms convolute measurements.
The non-steady-state approach also revealed novel insights into the mechanisms regulating the cellular response to rapamycin. We uncovered temporal responses to high concentrations of rapamycin that could be explained by a hybrid model of regulation of autophagy rates. Latency in the rates of autolysosome formation and degradation revealed rate-limiting steps leading to autophagosome accumulation at very early time points. Moreover, we have also shown the latency in the fusion step is not due to a limited number of lysosomes, and future efforts to dissect mechanisms of latency could be leveraged to overcome them. Probing for the fusion governing proteins that may be rate limiting can be valuable for elucidating the fundamental mechanism involved as well as for developing new targets to inhibit the fusion step. At later time points, feedback mechanisms lowered the overall flux through the pathway. Understanding the timeline of feedback mechanisms for additional perturbations could help refine control over autophagy. Conversely, low concentrations of rapamycin treatment led to a slower but steady response in rates. We hypothesize this behavior is due to the complex interplay between multiple feedback mechanisms of MTORC1 [37,38]. Using these measurements in conjunction with fluorescent protein activity reporters at a single cell level can elucidate the complex dynamics involved [39,40] and will be important to dissect in the future. The temporal nature of this new approach also enabled the development of a new metric in the form of overall cargo degraded (AUC for R3) which can be used as an additional property to characterize the system. This result underlines the dynamic nature of the pathway and the significance of this metric to fine-tune the flux through the pathway. In the future, it will also be interesting to determine if autophagy-associated diseases are due to a general reduction in degradative capacity (AUC for R3), or defects in the degradation of specific cargo.
We also measured rates for wortmannin to demonstrate the universality of this method to different types of perturbations. We observed concentration-dependent effects as well as a differential rate of recovery from wortmannin inhibition. This information can be used for modeling the system and extracting parameters such as half-maximal concentration, degradation constants, and maximal induction capacity. Additionally, this information can also be used to probe for the feedback mechanisms involved and their specific pathways. For example, if there is a MTORC1 independent feedback mechanism involved in the increase of R1 after wortmannin inhibition, the addition of rapamycin after recovery would lead to a higher R1 value and vice versa. Future work can be focused on extracting system parameters to develop a predictive model using the current data and some additional experimentation.
To validate the behavior observed beyond a single clone and cell line, we tested the dynamics of bulk A549 and U2OS cells for rapamycin and wortmannin treatment. The overall behavior of bulk cells was similar to that of the single clone and consistent across cell lines. Therefore, this method can also be used to characterize different cell lines about the inherent basal state and their response to perturbations and makes measurements in unique cancer and neurodegenerative disease models a possibility. However, various parameters, such as the variability in the amount of fluorescent LC3 protein expressed in each line need to be carefully assessed before drawing such comparisons.
We expanded our analysis to physiological conditions such as serum and glutamine starvation. Serum starvation transiently upregulated autophagy flux while adding back serum almost immediately inhibited autophagy flux, indicating, that serum inhibits autophagy. Conversely, glutamine starvation inhibited autophagy flux on a longer timescale indicating glutamine is required for autophagy. Our observations were consistent with previously reported findings [41,42]. However, the measurements were noisier compared to chemical perturbations. One important reason for this could be the significant cell-to-cell variability observed under these starvation conditions, especially glutamine starvation (data not shown). Single-cell measurements could alleviate these limitations and could provide interesting new findings.
While our method enables novel measurements of autophagy rates, expanding its use will require improvements in autophagy-related tools. For example, our current approach uses fluorescent proteins to monitor autophagosome and autolysosome numbers and is thus limited to engineered cells. Performing similar experiments with live-cell organelle dyes could overcome this limitation. This would enable autophagy rate measurements in difficult-to-engineer cells, and open the door to measurements in patient-derived cells [43,44]. Possible applications include precision medicine for autophagy-related diseases such as cancer and neurodegeneration [45,46]. Moreover, expanding such measurements to in vivo systems is vital for clinical translation [47].
In conclusion, our work demonstrates quantitative measurement of rates for all three steps in the autophagy pathway under non-steady-state conditions. This study revealed novel mechanisms of regulation for rapamycin induction of autophagy and differential temporal kinetics of wortmannin’s inhibition. In the future, these approaches could be applied to uncover mechanisms of action for novel autophagy-regulating compounds, develop predictive models, and characterize unique responses based on cellular genetic background. Moreover, integration with other live cell measurements would create a quantitative and holistic picture of autophagy as it connects to other cellular pathways.
Materials and methods
Cell culture and media
A549 cells (ATCC, CCL-185) and U2OS cells (ATCC, HTB-96) were used for autophagy experiments. HEK 293T cells (ATCC, CRL-11268) were used for lentivirus packaging. Cells were cultured in a humidified incubator at 37°C and 5% CO2. All cell lines were maintained in DMEM (Gibco, 11965118) supplemented with 8% fetal bovine serum (FBS [Gibco, 10438-026]). U2OS cells were also cultured with 1% penicillin-streptomycin (Gibco, 15070063). For live-cell imaging, A549 cells were cultured in FluoroBrite DMEM (Gibco, A1896701) supplemented with 8% FBS and 4 mM of GlutaMAX (Gibco, 35050061). U2OS reporter cells were cultured in the same imaging media along with 1% penicillin-streptomycin (Gibco, 15070063).
Reporter cell line construction
The FUGW-PK-hLC3 lentivirus was used to develop A549 and U2OS reporter cell lines. Lentivirus was packaged in HEK 293Ts in 6-well format as previously described [48]. The harvested lentivirus media was stored at −80°C until further use. A549 and U2OS cells were plated overnight at a density of 0.1 million cells per well in a 24-well plate. The media was replaced with lentivirus-containing media. After an hour, the lentivirus media was replaced with fresh media, and the cells were scaled up upon reaching confluency. For bulk populations, A549 and U2OS cells positive for pHluorin and mKate2 signal were sorted using a Beckman Coulter “Astrios EQ”:18-Color cell sorter. For clonal selection, transduced A549 cells were sorted into individual cells into a 96-well plate using a Beckman Coulter “Astrios EQ”:18-Color cell sorter. Each clone population was scaled up upon reaching confluency. A single clone population was used for most experiments to decrease noise arising from different integration sites. FUGW-PK-hLC3 ΔG reporter cell line was also developed using the same approach. FUGW-PK-hLC3 and FUGW-PK-hLC3 ΔG were gifts from Isei Tanida (Addgene, 61460; http://n2t.net/addgene:61460; RRID: Addgene_61460; Addgene, 61461; http://n2t.net/addgene:61461; RRID: Addgene_61461, respectively).
Chemical treatments
Bafilomycin A1 (Selleck Chemicals, S1413), rapamycin (Selleck Chemicals, S1039), and wortmannin (Selleck Chemicals, S2758) were used for treating the cells. For measuring rates, 500 nM bafilomycin A1 was added along with a final concentration of 0.2 µg/mL Hoechst 33342 trihydrochloride solution (Hoechst 33342 [Invitrogen, H3570]) to ensure proper mixing. All basal conditions were treated with DMSO (Sigma Aldrich, 472301).
Starvation conditions
A549 reporter cells were stably cultured in FluoroBrite DMEM (Gibco, A1896701) supplemented with 8% FBS and 4 mM of GlutaMAX (Gibco, 35050061) for 12–16 h before the experiment in a 96 well plate. Each well contained a culture volume of 100 µL. To initiate starvation conditions, 60 µL of media were removed and 160 µL of FluoroBrite DMEM containing either containing 8% FBS or 4 mM GlutaMAX or neither was added to the wells. After mixing, 160 µL of the media were removed from the wells and an additional 60 µL of FluoroBrite DMEM containing either 8% FBS or 4 mM GlutaMAX or neither was added to the wells. Therefore, leading to a 12.5-fold dilution. For addback experiments, FBS was added back to wells to bring back the serum levels to 8%. The same volume of FBS starved media and full media was added to continued serum-starved and basal wells, respectively.
Live cell microscopy
All reporter cell lines were seeded in 96-well glass-bottom plates with #1.5 cover glass (Cellvis, P96-1.5H-N). A549 cells were directly plated while U2OS cells were plated after treating the 96-well plate with collagen solution (Gibco, A1048301) to increase cell adherence. Live cell imaging was performed using Nikon Ti2 inverted microscope with an okolab stage top incubator to maintain 37°C and 5% CO2. Cells were plated at approximately 1.7 X 104 cells per well for 12–18 h prior to performing the experiment. A total of 4–5 positions were imaged in each well at the indicated time using the NIS-Elements AR software. A549 reporter cell line and A549 bulk sorted cells for all chemical treatment experiments were imaged at 25% LED intensity and 200 ms exposure for GFP channel and at 30% LED intensity and 350 ms for TRITC channel images. U2OS reporter cells and A549 reporter clonal cell line for starvation experiments were imaged at 35% LED intensity and 200 ms exposure for GFP channel and at 45% LED intensity and 350 ms for TRITC channel images. Images were acquired using CFI PLAN APO LAMBDA 40X CF160 Plan Apochromat Lambda 40X objective lens, N.A. 0.95, W.D. 0.17–0.25 mm, F.O.V. 25 mm, DIC, Correction collar 0.11–0.23 mm, Spring Loaded, and using Andor Zyla VSC-08688 camera.
Immunofluorescence
A549s were seeded in a 96-well glass-bottom plate. Cells were treated with either DMSO or 100 nM rapamycin for indicated times. After treatment, 100% ice-cold methanol (Fisher Scientific, A412–4) was added to the cells and were incubated for 20 min at −20°C. After aspirating methanol, cells were rinsed thrice with 1X DPBS (Gibco, 21600069) solution for 5 min each. Following DPBS wash, cells were incubated with 5% goat serum (Sigma Aldrich, G9023) in 1X DPBS with 0.3% TritonTM X-100 (Fischer Scientific, BP151–100) for an hour. After aspirating the serum solution, cells were incubated with 1:600 anti-LAMP1 primary antibody solution (LAMP1 [D2D11] XP rabbit mAb [Cell Signaling Technology, 9091], 1X DPBS, 1% bovine serum albumin [BSA; Sigma Aldrich, 126609], 0.3% TritonTM X-100) overnight. After removing the primary antibody solution, cells were rinsed thrice with 1X DPBS solution. After rinsing, cells were incubated with secondary antibody solution (1:1000 goat anti-rabbit IgG H+L] Alexa flour 488 [InvitrogenTM, A-11008], 1:5000 Hoechst 33342 in 1% BSA in 1X DPBS with 0.3% TritonTM X-100) for an hour in the dark. Finally, cells were washed thrice with 1X DPBS solution for 5 min each before imaging. Each condition at each time point had three replicates. The average number of LAMP1 positive puncta from the triplicates was used as one biological replicate value. The experiment was repeated four times independently.
Immunofluorescence microscopy
After fixation, cells were imaged using Nikon Ti2 inverted microscope. LAMP1 positive puncta and nuclear staining were imaged using the green channel (GFP) and blue channel (DAPI), respectively. GFP images were acquired at 25% LED intensity and 200 ms exposure. DAPI images were acquired at 15% LED intensity and 75 ms exposure settings. Images were acquired using CFI PLAN APO LAMBDA 40X CF160 Plan Apochromat Lambda 40X objective lens, N.A. 0.95, W.D. 0.17–0.25 mm, F.O.V. 25 mm, DIC, Correction collar 0.11–0.23 mm, Spring Loaded, and using Andor Zyla VSC-08688 camera.
Western blot
Cells (80,000 per well) were plated in a 12-well plate overnight. The next day, cells were treated with DMSO or 100 nM rapamycin for different time points. Bafilomycin A1 (500 nM) was added to the cells for 2 h at different timepoints for measuring LC3 accumulation. At the specific time point, the media in the wells was aspirated and the cells are quickly rinsed using 1X DPBS. Cells were then lysed with RIPA buffer (150 mM sodium chloride [NaCl; Fischer Scientific, S271], 50 mM Tris, pH 8 [Fischer Scientific, BP152], 1% Triton X-100 [Fisher Scientific, BP151-100], 0.1% sodium dodecyl sulfate [Fischer Scientific, BP166-500], 0.5% sodium deoxycholate [Sigma Aldrich, D6750]) containing protease inhibitors (Thermo ScientificTM, A32955). The lysed cells in RIPA buffer were incubated on ice for 30 min. After 30 min, the samples were centrifuged at 16,128 x g for 20 min, after which the supernatant of the samples was collected and stored at −20°C. Sample protein content was normalized using the PierceTM BCA protein assay kit (Thermo ScientificTM, 23225). LDS (InvitrogenTM, NP0007) and TCEP (Thermo ScientificTM, 77720) in 4:1 ratio was then added to the normalized samples and were heated in a thermocycler for 10 min at 95°C. The samples were then run on an SDS gel containing 4% stacking and 15% resolving gel compartments. The proteins were resolved at 115 volts for 15 min initially followed by 150 volts for an hour. The proteins were then transferred onto methanol-activated Amersham Hybond P 0.2 PVDF membrane (Cytiva, 10600021) at 150 volts for an hour. The membrane is then reactivated using methanol and quickly rinsed in distilled water. After reactivation, the membrane is blocked using 5% milk in TBS-T buffer (Tris-Buffered Saline pH 7.6 [TBS; 20 mM Tris, pH 8, 150 mM NaCl, hydrochloric acid [Sigma Aldrich, 320331] with 0.1% Tween- 20 [Fischer Scientific, BP337-100]) solution for an hour. The membrane slices were then incubated in their respective primary antibody diluted in 2.5% BSA in TBS-T solution overnight with gentle agitation. The antibody dilution for each antibody is as follows, 1: 1000 anti- phopho-RPS6/S6 ribosomal protein (Ser240/244; D68F8) rabbit mAb (Cell Signaling Technology, 5364), 1:1000 anti-RPS6/S6 ribosomal protein (5G10) rabbit mAb (Cell Signaling Technology, 2217), 1:1000 anti- LC3B (D11) XP rabbit mAb (Cell Signaling Technology, 3868), and 1:1000 anti-GAPDH (14C10) rabbit mAb (Cell Signaling Technology, 2118). Following primary antibody incubation, the membrane was rinsed thrice with TBS-T solution for 5 min each. The membrane was then incubated with 1:5000 goat anti-rabbit IgG-HRP (SouthernBiotech, 4030-05) secondary antibody in 5% Milk TBS-T solution. The membrane was washed twice with TBS-T followed by a TBS wash. Finally, the membrane was incubated with ECL western blotting substrate (Thermo ScientificTM, 32109) for 5 min before acquiring images. Amersham Imager 600 system (GE Healthcare) was used for imaging.
Image processing for live cell imaging
NIS-Elements AR software was used for extracting autophagosomes and autolysosome puncta numbers. GFP and TRITC channel images were processed and analyzed using the General analysis job functionality in the NIS-Elements AR software. Both GFP and TRITC channel images were background corrected using the rolling ball correction method with a radius of 1.95 µm. Following background correction, Spot Detection functionality was used for thresholding and detecting puncta in both channels. GFP channel images were used for estimating autophagosome puncta numbers as the green signal is only detected in autophagosomes. Bright-clustered detection method in the Spot Detection tool was used for detecting circular areas in the GFP channel with a typical spot diameter of 0.8 µm and a minimum contrast value of 5. The contrast value acts as a thresholding parameter to only detect puncta whose difference between mean intensity inside and mean intensity outside the spot is higher than the contrast value provided. Similarly, for the TRITC channel, 0.8 m was used as the typical spot diameter and 7.5 was used as the contrast value. A higher contrast value was used for detecting puncta in the TRITC channel because of the lower signal-to-noise ratio and thus to minimize false-positive puncta. However, the puncta from the TRITC channel includes both autophagosomes as well as autolysosomes count as both have a red signal. Therefore, to extract autolysosome-only count, we compared the colocalization of puncta in GFP and TRITC channels using the AND binary operation. The number of colocalized puncta (representing autophagosomes) were then subtracted from the total TRITC puncta, thus providing the autolysosome count. All the other parameters in the spot detection tool were left as default. The puncta detection accuracy was confirmed through manual inspection of multiple images under various conditions (untreated, rapamycin and bafilomycin A1 treatment). Post analysis, the autolysosome and autophagosome count for each image were exported as a spreadsheet.
Cellpose was used for counting cells in each image [49]. The ND2 files were converted to RGB tif files and the GFP channel images were used for segmenting and extracting the cell count. Cellpose was implemented in Python 3.7 using a custom script and 120 was used as the diameter input for segmenting individual cells. The segmentation accuracy was confirmed by manual inspection as well as by comparing with Hoechst-based nucleus count.
After extracting the cell, autophagosome and autolysosome count from each position imaged in a well. The total number of autophagosomes and autolysosomes in all the positions imaged were added and was divided by the total number of cells providing a population level autophagosome and autolysosome count per cell. This analysis was done using a custom script in MATLAB.
Image processing for immunofluorescence microscopy
LAMP1-positive puncta in fixed samples were estimated using spot detection tool in NIS-Elements AR software. A radius of 0.8 µm and a contrast value of 10 was used for detecting LAMP1-positive puncta. Similarly, the number of cells was estimated using the spot detection tool on the nuclear stain with a typical diameter of 15 µm and a contrast value of 1.5 as parameters.
Data fitting and area under the curve estimation
Curve fitting toolbox in MATLAB was used to fit the data. Custom equations were provided for fitting the data and the Nonlinear least-squares method was used for the fit. Trapz function in MATLAB was used for calculating the area under the curve and a custom script was used for propagating the error.
Statistical analysis
At least 150–200 cells were imaged for all experiments. A minimum of three experimental replicates was performed for all the quantitative experiments. Independent t-test, paired t-test, and one-way ANOVA were used as indicated for comparing statistical significance for various experiments. All statistical tests were performed in Python 3.8 using the SciPy package. ANOVA along with Tukey post hoc test for AUC calculations was performed using a webpage (https://statpages.info/anova1sm.html). Box and whisker plots indicate the median value as an orange line, interquartile range (IQR) as a box, and range [ as whiskers.
Supplementary Material
Acknowledgments
We thank members of the Shah and Albeck groups for feedback on project development and the manuscript. We also want to thank Shivaani Krishna from the Nunnari lab for providing U2OS cells.
Funding Statement
The work was supported by the W. M. Keck Foundation [].
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2022.2117515
References
- [1].Green DR, Levine B.. To be or not to be? How selective autophagy and cell death govern cell fate. Cell. 2014;157(1):65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Choi Y, Bowman JW, Jung JU. Autophagy during viral infection — a double-edged sword. Nat Rev Microbiol. 2018;16(6):341–354. DOI: 10.1038/s41579-018-0003-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Hansen M, Rubinsztein DC, Walker DW. Autophagy as a promoter of longevity: insights from model organisms. Nat Rev Mol Cell Biol. 2018;19(9):579–593. Available from: https://www.nature.com/articles/s41580-018-0033-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nixon RA The role of autophagy in neurodegenerative disease. Nat Med. 2013(19):8 :983–997. Available from: https://www.nature.com/articles/nm.3232. [DOI] [PubMed] [Google Scholar]
- [5].Mulcahy Levy JM, Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020;27(3):843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Galluzzi L, Pedro JMB, Levine B, et al. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov. 2017;16:487–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kim YJ, Baek E, Lee JS, et al. Autophagy and its implication in Chinese hamster ovary cell culture. Biotechnol Lett. 2013;35(11):1753–1763. DOI: 10.1007/s10529-013-1276-5 [DOI] [PubMed] [Google Scholar]
- [8].Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19(21):5720–5728. DOI: 10.1093/emboj/19.21.5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bampton ETW, Goemans CG, Niranjan D, et al. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2005;1(1):23–36. DOI: 10.4161/auto.1.1.1495 [DOI] [PubMed] [Google Scholar]
- [10].Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3(5):452–460. [DOI] [PubMed] [Google Scholar]
- [11].Tanida I, Ueno T, Uchiyama Y. A super-ecliptic, pHluorin-mKate2, tandem fluorescent protein-tagged human LC3 for the monitoring of mammalian autophagy. PLOS ONE. 2014;9:3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Loos B, Du Toit A, Hofmeyr JHS. Defining and measuring autophagosome flux—concept and reality. Autophagy. 2014;10(11):2087–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].du Toit A, Hofmeyr JHS, Gniadek TJ, et al. Measuring autophagosome flux. Autophagy. 2018;14(6):1060–1071. DOI: 10.1080/15548627.2018.1469590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fass E, Shvets E, Degani I, et al. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281(47):36303–36316. DOI: 10.1074/jbc.M607031200 [DOI] [PubMed] [Google Scholar]
- [15].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. DOI: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- [16].Klionsky DJ, Abdel-Aziz AK, Abdelfatah S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy. 2021;117:1–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhou C, Zhong W, Zhou J, et al. Monitoring autophagic flux by an improved tandem fluorescent-tagged LC3 (mTagrfp-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy. 2012;8(8):1215–1226. Available from: https://www.tandfonline.com/action/journalInformation?journalCode=kaup20. [DOI] [PubMed] [Google Scholar]
- [18].Pankiv S, Clausen TH, Lamark T, et al. P62/sqstm1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. DOI: 10.1074/jbc.M702824200 [DOI] [PubMed] [Google Scholar]
- [19].Tanida I, Ueno T, Uchiyama Y. Use of pHlurorin-mKate2-human LC3 to Monitor Autophagic Responses. Methods Enzymol. 2017. DOI: 10.1016/bs.mie.2016.09.054. [DOI] [PubMed]
- [20].Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279(46):47704–47710. [DOI] [PubMed] [Google Scholar]
- [21].Yamamoto1 A, Tagawa1 Y, Yoshimori2 T, et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct. 1998;23(1):33–42. DOI: 10.1247/csf.23.33 [DOI] [PubMed] [Google Scholar]
- [22].Rubinsztein DC, Gestwicki JE, Murphy LO, et al. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6(4):304–312. DOI: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- [23].Klionsky DJ, Elazar Z, Seglen PO, et al. Does bafilomycin a 1 block the fusion of autophagosomes with lysosomes? Autophagy. 2008;4(7):849–850. DOI: 10.4161/auto.6845 [DOI] [PubMed] [Google Scholar]
- [24].Brown EJ, Albers MW, Bum Shin T, et al. A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature. 1994;369:(6483):756–758. DOI: 10.1038/369756a0. [DOI] [PubMed] [Google Scholar]
- [25].Sabatini DM, Erdjument-Bromage H, Lui M, et al. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78:35–43. http://www.cell.com/article/0092867494905703/fulltext [DOI] [PubMed] [Google Scholar]
- [26].Sabersu CJ, Martinl1 MM, Brunnj GJ, et al. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270(2):815–822. [DOI] [PubMed] [Google Scholar]
- [27].Goutelle S, Maurin M, Rougier F, et al. The hill equation: a review of its capabilities in pharmacological modelling. Fundam. Clin. Pharmacol. 2008;22(6):633–648. https://onlinelibrary.wiley.com/doi/full/10.1111/j.1472-8206.2008.00633.x. [DOI] [PubMed] [Google Scholar]
- [28].Blommaart EFC, Krause P, Schellens JPM, et al. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243(1–2):240–246. DOI: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- [29].Petiot A, Ogier-Denis E, Blommaart EFC, et al. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–998. DOI: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- [30].Ihle NT, Williams R, Chow S, et al. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3(7):763–772. DOI: 10.1158/1535-7163.763.3.7. [DOI] [PubMed] [Google Scholar]
- [31].Altman BJ, Stine ZE, Dang CV. From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer. 2016;16:619–634. https://www.nature.com/articles/nrc.2016.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Patel DB, Santoro M, Born LJ, et al. Towards rationally designed biomanufacturing of therapeutic extracellular vesicles: impact of the bioproduction microenvironment. Biotechnol Adv. 2018;36(8):2051–2059. DOI: 10.1016/j.biotechadv.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Egan DF, Chun MGH, Vamos M, et al. Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell. 2015;59(2):285–297. DOI: 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Liu T, Zhang J, Li K, et al. Combination of an autophagy inducer and an autophagy inhibitor: a smarter strategy emerging in cancer therapy. Front Pharmacol. 2020;11:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Decuypere J, Kindt D, Luyten T, et al. mTOR-controlled autophagy requires intracellular Ca 2 + Signaling. PLOS ONE. 2013;8(4)e61020. DOI: 10.1371/journal.pone.0061020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].de Wet S, Du Toit A, Loos B. Spermidine and rapamycin reveal distinct autophagy flux response and cargo receptor clearance profile. Cells. 2021;10(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Gonzá Lez A, Hall MN, Lin S-C, et al. Cell metab review AMPK and TOR: the Yin and Yang of cellular nutrient sensing and growth control. Cell Metab. 2020;31(3):472–492. DOI: 10.1016/j.cmet.2020.01.015. [DOI] [PubMed] [Google Scholar]
- [38].Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans. 2013;41(4):939–943. DOI: 10.1042/BST20130030. [DOI] [PubMed] [Google Scholar]
- [39].Zhou X, Clister TL, Lowry PR, et al. Dynamic visualization of mTORC1 activity in living cells. Cell Rep . 2015;10(10):1767–1777. DOI: 10.1016/j.celrep.2015.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Konagaya Y, Terai K, Hirao Y, et al. A highly sensitive FRET biosensor for AMPK exhibits heterogeneous AMPK responses among cells and organs. Cell Rep. 2017;21(9):2628–2638. DOI: 10.1016/j.celrep.2017.10.113. [DOI] [PubMed] [Google Scholar]
- [41].Sakiyama T, Musch MW, Ropeleski MJ, et al. Glutamine increases autophagy under basal and stressed conditions in intestinal epithelial cells. Gastroenterology. 2009;136(3):924–932.e2. DOI: 10.1053/j.gastro.2008.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chen R, Zou Y, Mao D, et al. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014;206(2):173. DOI: 10.1083/jcb.201403009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Martínez-Pizarro A, Desviat LR, Ugarte M, et al. Endoplasmic reticulum stress and autophagy in homocystinuria patients with remethylation defects. PLoS One. 2016;11(3):e0150357. DOI: 10.1371/journal.pone.0150357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mohammadi A, Kelly OB, Filice M, et al. Differential expression of microRnas in peripheral blood mononuclear cells identifies autophagy and TGF-beta-related signatures aberrantly expressed in inflammatory bowel disease. J Crohn’s Colitis. 2018;12(5):568–581. DOI: 10.1093/ecco-jcc/jjy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer (Nature Publishing Group). 2017;17(9):528–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Scrivo A, Bourdenx M, Pampliega O, et al. Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol. 2018;17(9):802–815. DOI: 10.1016/S1474-4422(18)30238-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Loos B, Klionsky DJ, Du TA, et al. On the relevance of precision autophagy flux control in vivo – points of departure for clinical translation for clinical translation. Autophagy. 2020;16(4):750–762. DOI: 10.1080/15548627.2019.1687211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Minami SA, Shah PS. Transient light-activated gene expression in Chinese hamster ovary cells. BMC Biotechnol. 2021;21(1)13. https://link.springer.com/articles/10.1186/s12896-021-00670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Stringer C, Wang T, Michaelos M, et al. Cellpose: a generalist algorithm for cellular segmentation. Nat Methods. 2021;18(1):100–106. DOI: 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.