

Abstract
Inhibitors of Bruton tyrosine kinase (BTK), a kinase downstream of BCR, display remarkable activity in a subset of mantle cell lymphoma (MCL) patients, but the drug resistance remains a considerable challenge. In this study, we demonstrate that aberrant expression of ROR1 (receptor tyrosine kinase-like orphan receptor 1), seen in a large subset of MCL, results in BCR/BTK–independent signaling and growth of MCL cells. ROR1 forms a functional complex with CD19 to persistently activate the key cell signaling pathways PI3K–AKT and MEK–ERK in the BCR/BTK–independent manner. This study demonstrates that ROR1/CD19 complex effectively substitutes for BCR–BTK signaling to promote activation and growth of MCL cells. Therefore, ROR1 expression and activation may represent a novel mechanism of resistance to inhibition of BCR/BTK signaling in MCL. Our results provide a rationale to screen MCL patients for ROR1 expression and to consider new therapies targeting ROR1 and/or CD19 or their downstream signaling pathways for MCL-expressing ROR1.
Introduction
Mantle cell lymphoma (MCL) is a clinically aggressive B cell lymphoma (1). The BCR–Bruton tyrosine kinase (BTK) signaling pathway and its inhibitors play a pivotal role in, respectively, pathogenesis and therapy of B cell lymphomas, including MCL (2–4). Most MCL patients treated with the BTK inhibitor ibrutinib display clinical responses (3,4), but resistance to the drug almost inevitably develops (5,6). Although mutations of genes encoding BTK or signaling proteins downstream of BTK can lead to the inhibitor resistance (7–9), cell reprogramming represents a potential alternative mechanism to the resistance (10). Identification of these mechanisms is of uttermost importance in designing an appropriate therapeutic strategy to counteract the reprogramming.
ROR1 belongs to the receptor tyrosine kinase-like orphan receptor (ROR) family and displays very restricted expression in normal tissues (11,12). ROR1 is aberrantly expressed in various malignancies, including small lymphocytic lymphoma/chronic lymphocytic leukemia (SLL/CLL) and MCL. ROR1 activates signaling molecules, such as RAC-1 and contractin (13,14), to promote cell proliferation, survival, and migration (13–15). CD19 is a B cell–specific receptor capable of stimulating the growth of malignant B cells (16). In normal B lymphocytes, it is activated by SRC family kinases and SYK, both downstream of BCR (17). Once activated, CD19 cosignals with BCR to activate PI3K–AKT and other signaling pathways. In this study, we demonstrate that ROR1 associates in MCL cells with CD19. The ROR1/CD19 complex activates PI3K–AKT and MEK–ERK signaling pathways and promotes cell growth independently of BCR and BTK.
Materials and Methods
The MCL-RL line was developed in our laboratory; its identity as MCL has been confirmed by the CD19+CD5+ B cell phenotype and IgH–Cyclin D1 translocation and expression of the Cyclin D1 protein. JeKo-1 and REC-1 were from the American Type Culture Collection; Mino and SP49 cells were obtained from Dr. R. Lai, University of Alberta; Maver and JVM were from Dr. L. Wang, University of Chicago, Granta was from the German Cell Culture Collection (Braunschweig, Germany); in all of these cell lines, translocation and protein expression of Cyclin D1 has been confirmed. MCL patient samples were obtained from the Stem Cell and Xenograft Core (University of Pennsylvania, Philadelphia, PA). Cells were grown in RPMI 1640 with 10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin.
Flow cytometry
Cell surface expression of ROR1, ROR2, and CD19 was analyzed using PE-conjugated anti-human ROR1 and ROR2 and FITC-conjugated anti-human CD19 Abs or control IgG Abs (BioLegend, San Diego, CA). Flow analyses were done on an LSR II instrument (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software v9.0.2 (TreeStar, Ashland, OR).
Western blot and immunoprecipitation analysis
Western blotting was performed using Abs against ROR1, CD19, p-BTK, p-PI3K, p-AKT, p-ERK1/2 (Cell Signaling Technology, Danvers, MA), and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), according to standard protocols. For immunoprecipitation analysis, cellular lysates were precleared with Protein G Sepharose, incubated with 10 μl ROR1 Ab (Cell Signaling Technology), 10 μl CD19 Ab (Cell Signaling Technology, ), 3.3 μl nonspecific rabbit IgG (Cell Signaling Technology) or 3.3 μl nonspecific mouse IgG (Cell Signaling Technology) overnight at 4°C. Proteins were precipitated by using Protein A Sepharose beads (20 μl) at 4°C for 30 min. The beads were washed five times with 500 μl of 1× lysis buffer (Cell Signaling Technology), followed by Western immunoblotting.
MTT enzymatic conversion assay
Cells were plated in 96-well plates (3–5 × 103 cells per well), incubated with drugs, drug vehicle, short hairpin RNA (shRNA), or empty vector, labeled with MTT (Promega, Madison, WI), and evaluated using a Titertek Multiskan reader.
RNA sequence analysis
RNA sequence analysis (RNA-Seq) was performed using Illumina’s TruSeq Ribo-Zero Stranded Library Prep Kit. The libraries were sequenced on an Illumina HiSeq 2500, using V4. Reads were trimmed to remove low-quality sequences, then aligned to the genome (Human hg19) and quantified on RefSeq transcripts using the RNA-Seq Unified Mapper. Length-normalized counts (fragments per kb of transcript per million counts) were extracted from the RNA-Seq Unified Mapper output. Pairwise comparisons between groups were carried using a custom script that implemented Bioconductor software package edgeR to compute a p value and fold change for each transcript.
Short interfering RNA assay
A mixture of four short interfering RNAs (siRNAs) specific for human ROR1 or control siRNAs (Dharmacon, Lafayette, CO) were introduced into cells for 86 h using Lipofectamine 2000 Transfection Reagent (Life Technologies, Carlsbad, CA).
shRNA assay
The oligonucleotides targeting CD79B (5′-TTTCTCAGGACACACCGTT-3′), ROR1 (5′-CATGCAATCCCTCTGTATG-3′), and CD19 (5′-TCGGGCCTGACTTCCATGG-3′) were cloned into pELNSAvr vector obtained from the Milone laboratory (University of Pennsylvania, Philadelphia, PA). To generate the viral particles, HEK-293T producer cells were cotransfected with the lenti vectors and the packaging plasmids. Viral supernatants were harvested after 24 and 48 h, filtered, and used to transduce MCL-RL and Jeko-1 cells.
Fluorescence staining and image analysis by Bright Detail Similarity and Amnis ImageStream
Cells were washed twice and incubated with either PE-conjugated ROR1 Ab or FITC-conjugated CD19 Ab (BioLegend). Cells were then analyzed by imaging flow cytometry using the Amnis ImageStream X (Amnis, Seattle, WA), with 488-nm lasers used for excitation of FITC and PE and 785-nm laser for the side scatter. For each sample, 10,000 events were recorded at original magnification ×60. The data analysis for both Bright Detail Similarity (BDS) and ImageStream was performed on the IDEAS software (Amnis) to determine the colocalization between ROR1–PE and CD19–FITC. Templates of the software settings were created in IDEAS to standardize analysis. For BDS, pixel location was calculated and presented as a BDS score reflecting colocalization of the PE and FITC probes.
Results and Discussion
ROR1 is highly expressed in BTK inhibitor-insensitive MCL
As shown in Fig. 1A, MCL cell lines differ in their sensitivity to the BTK inhibitor ibrutinib. Whereas MCL-RL cells are highly sensitive to the drug, JeKo-1 cells are rather resistant, with the other two cell lines, SP49 and Mino, showing an intermediate sensitivity. Evaluation of an additional four MCL cell lines showed that one, REC-1, displayed intermediate sensitivity, whereas the remaining three (MAVER, JVM, and GRANTA) were drug-resistant (Supplemental Fig. 1A). The mechanisms underlying resistance to ibrutinib are still poorly understood but include mutations of BTK or its substrate PLCγ2 (7–8). Because we did not see these mutations in JeKo-1 cells (data not presented), we searched for alternative mechanisms potentially responsible for the BTK- and, thus, BCR-independent growth of JeKo-1 and, possibly, other MCL cells.
Figure 1: ROR1 is highly expressed in MCL cells poorly responsive to BTK inhibitor.
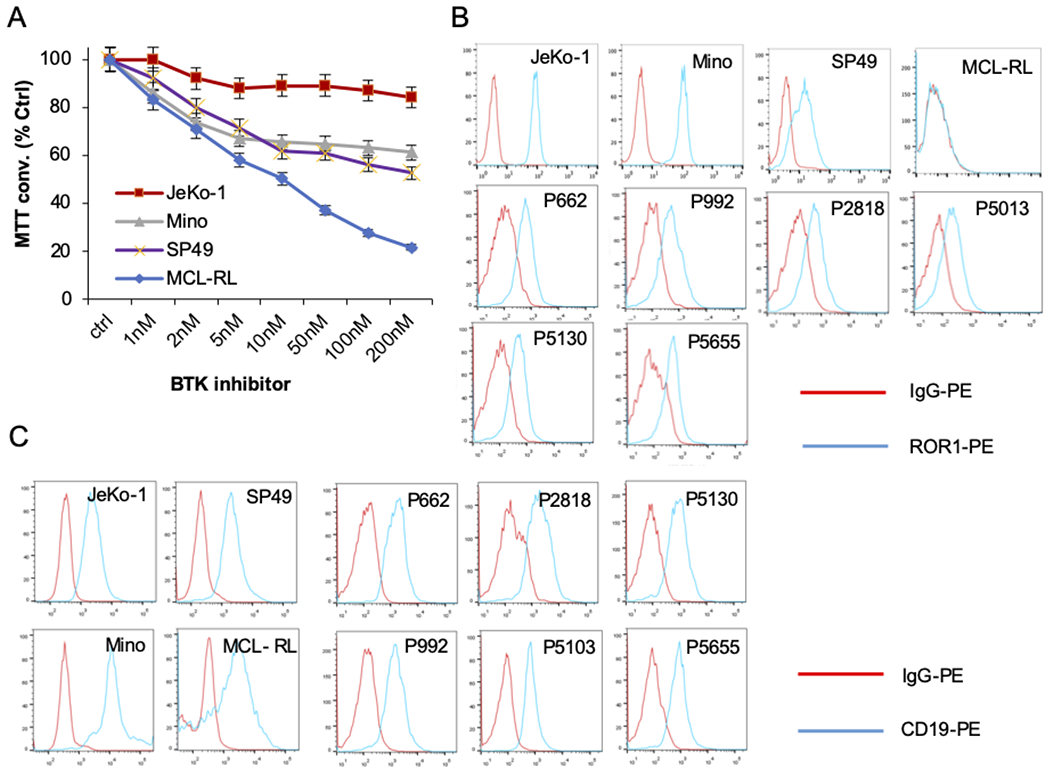
(A) Impact of BTK inhibitor Ibrutinib on growth MCL cell lines (JeKo-1, Mino, Mino, SP49, and MCL-RL) evaluated using MTT conversion assay at 48 h. The results are representative of three experiments; the bars reflect the SD values of experimental triplicates. (B) Expression of ROR1 by MCL cell lines and primary cells detected by flow cytometry. (C) Expression of CD19 in the MCL cell lines and patient samples evaluated by flow cytometry.
Because of the emerging role of ROR1 in pathogenesis and therapy of SLL/CLL (13,14) and results of our RNA sequencing screen indicating expression of the ROR1 gene in JeKo-1 but not MCL-RL cells (data not shown), we assessed by flow cytometry the expression of ROR1 protein in the MCL cell lines and primary cells from six patients (Fig. 1B, Supplemental Fig. 1B). ROR1 was variably expressed by the cell lines, but JeKo-1 and some of the other lines, fully or partially resistant to ibrutinib, expressed the receptor, whereas MCL-RL did not (Fig. 1B, Supplemental Fig. 1B). All six primary cell samples examined displayed ROR1 expression (Fig. 1B), suggesting that expression of this receptor is frequent in MCL. This contrasted with the expression of another member of the ROR family, ROR2, which could not be detected on either cultured or primary MCL cells (Supplemental Fig. 1C), indicating that this receptor does not play an important role in MCL, as opposed to SLL/CLL, in which it forms a functional complex with ROR1 (15). Given the lack of ROR2 expression, we focused next on CD19, a key receptor normally activated with BCR to trigger the intracellular signaling cascade (17), as a potential cell signaling partner of ROR1.
As shown in Fig. 1C and Supplemental Fig. 1B, CD19 was almost universally expressed in our set of MCL cell lines and primary cells as detected by flow cytometry. Its expression, as well as expression of ROR1, was also evaluated in the selected MCL cell lines by Western blot (Fig. 2A), confirming for both the expression pattern seen in the flow cytometry analysis.
Figure 2: ROR1 forms a complex with CD19.

(A) Detection of ROR1 and CD19 protein expression in MCL cell lines by Western blot. (B) Double staining of ROR1 and CD19 in MCL lines and primary cells analyzed by flow cytometry. (C) Association of ROR1 and CD19 as detected by co-immunoprecipitation (IP). (D) Extent of ROR1-CD19 co-localization evaluated using BDS score. (E) Co-localization (yellow) of CD19 (green) with ROR1 (red) in MCL cell lines and patient samples detected by imaging flow cytometry.
ROR1 associates with the native CD19 receptor
ROR1 and CD19 were coexpressed in the same cells in the MCL cell lines and primary cells, as documented by double-staining (Fig. 2B, Supplemental Fig. 1B). ROR1 and CD19 also physically associated with each other, as documented by bidirectional coimmunoprecipitation studies (Fig. 2C), strongly suggesting that they form a functional complex. To demonstrate that ROR1 and CD19 colocalize within the cell membrane, further indicating that these cell surface receptors may interact with each other, we performed image analysis of the double-stained MCL cells. Evaluation of the signal overlaps between ROR1 and CD19 stains using BDS analysis indicated that these receptors indeed colocalize, with a BDS score correlating in the MCL cell lines with their ROR1 expression level, as schematically depicted in Fig. 2D. The CD19–ROR1 colocalization was present not only in cell lines but MCL primary cells, as detected in the ImageStream analysis (Fig. 2E, Supplemental Fig. 2).
ROR1/CD19 activates key signaling pathways in BTK inhibitor-resistant MCL cells
To determine whether the ROR1/CD19 receptor complex plays a pathogenic role in MCL cells, in particular, the ones growing independently of BCR–BTK signaling, such as JeKo-1 cells, we depleted in these cells components of the BCR complex as well as ROR1 and CD19. Although JeKo-1 cells expressed CD79A and CD79B members of the BCR complex at the concentrations similar to MCL-RL cells (Fig. 3A), shRNA-mediated depletion in JeKo-1 cells of CD79B impaired the high basal phosphorylation of BTK but had no effect on the high basal phosphorylation of PI3K, AKT, and ERK (Fig. 3B, left panel), indicating that PI3K–AKT and MEK–ERK, the key intracellular pathways, are activated in JeKo-1 cells independently of BCR–BTK signaling. In contrast, CD79B depletion in the ibrutinib-responsive MCL-RL cells inhibited phosphorylation of not only BTK but also of PI3K, AKT, and ERK (Fig. 3B, right panel), confirming the dependence of these cells on BCR-induced cell signaling. Strikingly, shRNA-mediated depletion in JeKo-1 cells of either ROR1 or CD19 profoundly suppressed phosphorylation of PI3K and ERK (Fig. 3C); similar results were obtained using also ROR1 siRNA in these cells (Supplemental Fig. 3A). Similar to JeKo-1, shRNA-mediated depletion of either ROR1 or CD19 suppressed PI3K phosphorylation in three additional MCL cell lines: Mino, REC-1, and MAVER; CD79B depletion exerted no effect on the PI3K phosphorylation status (Supplemental Fig. 3B). The ROR1 or CD19 but not CD79A depletion also inhibited ERK phosphorylation, as seen in Mino; the basal p-ERK signal in REC-1 and MAVER was too weak for reliable evaluation. These results indicate that the ROR1/CD19 complex activates PI3K–AKT and MEK–ERK pathways in these MCL cells, and it does so independently of BCR.
Figure 3: ROR1/CD19 activates key intracellular signaling pathways and promotes growth of MCL cells independently of BCR complex.

(A) Expression of CD79A and CD79B in MCL cell lines detected by flow cytometry. (B) Effect of shRNA-mediated CD79B depletion at day 5 on activation of key cell signaling pathways evaluated by Western blot using the depicted phospho-specific Abs. (C) Impact of shRNA-mediated ROR1 and CD19 depletion at day 5 in BTK inhibitor resistant MCL cells (JeKo-1) on key cell signaling pathway examined by Western blot and the phospho-specific Abs. (D) Effect of shRNA-mediated CD19 and ROR1 depletion (upper panel) or PI3K inhibitor (BEZ235) on growth of JeKo-1 cells determined by MTT conversion assay.
Depletion of ROR1 and CD19 inhibits growth of MCL cells
To gain further insight into the kind of dependence of MCL cells on the ROR1/CD19 complex, we examined the impact of shRNA-mediated depletion of ROR1 and CD19 on cell growth. After confirming the high efficiency of transduction of JeKo-1 cells with the ROR1 and CD19 shRNA constructs (Supplemental Fig. 4A), we examined these cells in the MTT conversion assay. Depletion of either CD19 or ROR1 profoundly inhibited cell growth in a time-dependent manner of not only JeKo-1 cells (Fig. 3D; upper panel) but also the three other MCL cell lines examined (Supplemental Fig. 4C). Cell growth-suppressing effects were also achieved using a small molecule PI3K inhibitor of PI3K BEZ235 (Fig. 3D), supporting the notion that PI3K–AKT signaling pathway is the key effector of the growth-promoting signals generated by the ROR1/CD19 complex. Interestingly, forced expression of ROR1 in MCL-RL cells did not confer on these cells ibrutinib independence (data not shown), suggesting that ROR1 expression is but one step in reprogramming from the BCR-to ROR1-dependent cell growth.
In summary, whereas ibrutinib-responsive MCL cells relay on the BCR–BTK pathway to activate the key downstream signaling pathways to sustain cell growth, ibrutinib-resistant MCL may aberrantly express ROR1 that associates with the endogenous CD19 to form a functional complex capable of activating similar key downstream cell signaling pathways and support cell growth in the BCR–BTK–independent manner (Fig. 4). These findings have translational implications, foremost by describing a mechanism of escape from BTK inhibition by ectopic expression of ROR1. It is likely that this type of cell reprogramming is prevalent among lymphomas and other malignancies [e.g., signaling through IGF-R1 can result in resistance to EZH2 inhibition (18)]. Therefore, evaluation of patients treated with inhibitors of the BCR–BTK signaling pathway for ROR1 expression and activation as a potential mechanism of inhibitor resistance may be considered. Importantly, reprogramming of this kind does not make malignant cells therapy-resistant in general terms. In contrast, it creates new therapeutic targets while leaving some other pre-existing targets intact. ROR1 has already been identified as a therapeutic target in SLL/CLL, MCL, and other malignancies (19–22). CD19 is successfully targeted by immunotherapy with CART19 cells (23–25), suggesting that this type of therapy may be warranted in lymphoma patients who developed resistance to BCR–BTK pathway inhibition via activation of the ROR1/CD19 complex. Finally, because the key downstream signaling pathways are active in the reprogrammed cells, they also could be targeted in the BCR–BTK inhibitor-resistant cells.
Figure 4:
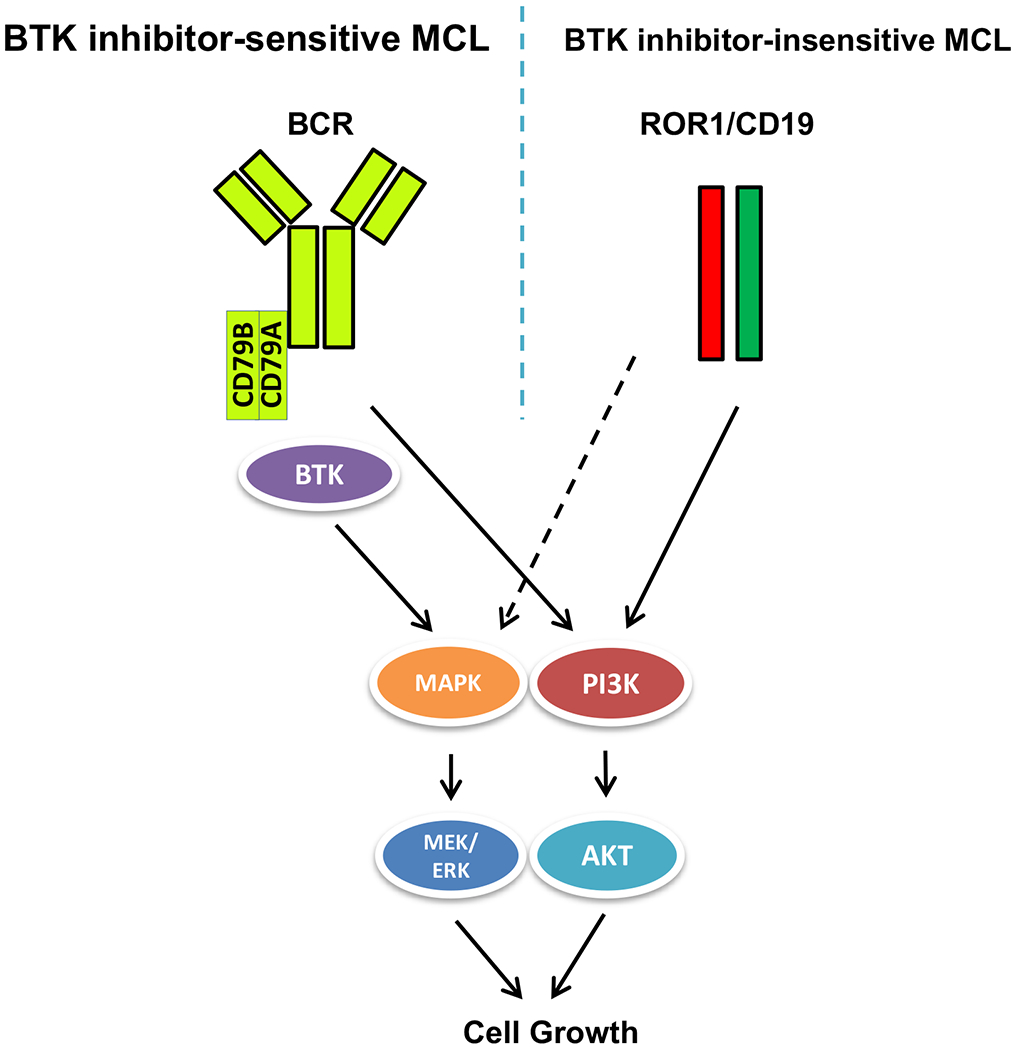
Schematic diagram of BCR/BTK- and ROR1/CD19-activated cell signaling pathways in BTK-sensitive and –resistant MCL cells.
Supplementary Material
Acknowledgments
This work was supported by grants from the Lymphoma Research Foundation, the Berman Family Fund, the Daniel B. Allanoff Foundation, the Abramson Cancer Center Translational Center of Excellence in Lymphoma, and the Donald E. and Shirley C. Morel, Stanley and Stella Bayster Endowed Chair funds of Fox Chase Cancer Center.
Footnotes
Disclosures
The authors declare no conflict of interest related to this study.
References
- 1.Cheah CY, Seymour JF, and Wang ML. 2016. Mantle Cell Lymphoma. J Clin Oncol 34: 1256–1269. [DOI] [PubMed] [Google Scholar]
- 2.Young RM, and Staudt LM. 2013. Targeting pathological B cell receptor signalling in lymphoid malignancies. Nat Rev Drug Discov 12: 229–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, and O’Brien S. 2013. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med 369: 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Inamdar AA, Goy A, Ayoub NM, Attia C, Oton L, Taruvai V, Costales M, Lin YT, Pecora A, and Suh KS. 2016. Mantle cell lymphoma in the era of precision medicine-diagnosis, biomarkers and therapeutic agents. Oncotarget 7: 48692–48731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, and Blum KA. 2013. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med 369: 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang ML, Blum KA, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Zhang L, Baher L, Cheng M, Lee D, Beaupre DM, and Rule S. 2015. Long-term follow-up of MCL patients treated with single-agent ibrutinib: updated safety and efficacy results. Blood 126: 739–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woyach JA, Furman RR, Liu TM, Ozer HG, Zapatka M, Ruppert AS, Xue L, Li DH, Steggerda SM, Versele M, Dave SS, Zhang J, Yilmaz AS, Jaglowski SM, Blum KA, Lozanski A, Lozanski G, James DF, Barrientos JC, Lichter P, Stilgenbauer S, Buggy JJ, Chang BY, Johnson AJ, and Byrd JC. 2014. Resistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N Engl J Med 370: 2286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP, and Chen-Kiang S. 2014. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov 4: 1022–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma J, Lu P, Guo A, Cheng S, Zong H, Martin P, Coleman M, and Wang YL. 2014. Characterization of ibrutinib-sensitive and-resistant mantle lymphoma cells. Br J Haematol 166: 849–861. [DOI] [PubMed] [Google Scholar]
- 10.Zhao X, Lwin T, Silva A, Shah B, Tao J, Fang B, Zhang L, Fu K, Bi C, Li J, et al. Sotomayor. 2017. Unification of de novo and acquired ibrutinib resistance in mantle cell lymphoma. Nat Commun 8: 14920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Al-Shawi R, Ashton SV, Underwood C, and Simons JP. 2001. Expression of the Ror1 and Ror2 receptor tyrosine kinase genes during mouse development. Dev Genes Evol 211: 161–171. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S, Chen L, Wang-Rodriguez J, Zhang L, Cui B, Frankel W, Wu R, and Kipps TJ. 2012. The onco-embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol 181: 1903–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hasan MK, Yu J, WidhopfII GF, Rassenti LZ, Chen L, Shen Z, Briggs SP, Neuberg DS, Kipps TJ. 2018. Wnt5a induces ROR1 to recruit DOCK2 to activate Rac1/2 in chronic lymphocytic leukemia. Blood 132: 170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hasan MK, Rassenti L, WidhopfII GF, Yu J, Kipps TJ. 2019. Wnt5a causes ROR1 to complex and activate cortactin to enhance migration of chronic lymphocytic leukemia cells. Leukemia 33: 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J, Chen L, Cui B, Widhopf GF, Shen Z, Wu R, Zhang L, Zhang S, Briggs SP, and Kipps TJ. 2016. Wnt5a induces ROR1/ROR2 heterooligomerization to enhance leukemia chemotaxis and proliferation. J Clin Invest 126: 585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung EY, Psathas JN, Yu D, Li Y, Weiss MJ, and Thomas-Tikhonenko A. 2012. CD19 is a major B cell receptor-independent activator of MYC-driven B-lymphomagenesis. J Clin Invest 122: 2257–2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schweighoffer E, Tybulewicz VL. 2018. Signalling for B cell survival. Curr. Opin. Cell Biol . 51: 8–14. [DOI] [PubMed] [Google Scholar]
- 18.Bisserier M, and Wajapeyee N. 2018. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 131: 2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi MY, Widhopf GF, Wu CC, Cui B, Lao F, Sadarangani A, Cavagnaro J, Prussak C, Carson DA, Jamieson C, and Kipps TJ. 2015. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clin Lymphoma Myeloma Leuk 15 Suppl: S167–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang S, Cui B, Lai H, Liu G, Ghia EM, Widhopf GF, Zhang Z, Wu CC, Chen L, Wu R, Schwab R, Carson DA, and Kipps TJ. 2014. Ovarian cancer stem cells express ROR1, which can be targeted for anti-cancer-stem-cell therapy. Proc Natl Acad Sci U S A 111: 17266–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karvonen H, Chiron D, Niininen W, Ek S, Jerkeman M, Moradi E, Nykter M, Heckman CA, Kallioniemi O, Murumägi A, and Ungureanu D. 2017. Crosstalk between ROR1 and BCR pathways defines novel treatment strategies in mantle cell lymphoma. Blood Adv 1: 2257–2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu J, Chen Y, Chen L, Zhang L, Rassenti LZ, Widhopf GF, and Kipps TJ. 2018. Cirmtuzumab inhibits ibrutinib-resistant, Wnt5a-induced Rac1 activation and proliferation in mantle cell lymphoma. Oncotarget 9: 24731–24736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Porter DL, Levine BL, Kalos M, Bagg A, and June CH. 2011. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365: 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sadelain M 2017. CD19 CAR T Cells. Cell 171: 1471. [DOI] [PubMed] [Google Scholar]
- 25.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, Brogdon JL, Pruteanu-Malinici I, Bhoj V, Landsburg D, Wasik M, Levine BL, Lacey SF, Melenhorst JJ, Porter DL, and June CH. 2017. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med 377: 2545–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.