

Abstract
The off-label use of racemic ketamine and the FDA approval of (S)-ketamine are promising developments for the treatment of depression. Nevertheless, racemic ketamine and (S)-ketamine are controlled substances with known abuse potential and their use is associated with undesirable side effects. For these reasons, research efforts have focused on identifying alternatives. One candidate is (2R,6R)-hydroxynorketamine ((2R,6R)-HNK), a ketamine metabolite that in preclinical models lacks the dissociative and abuse properties of ketamine while retaining its antidepressant-like behavioral efficacy. (2R,6R)-HNK’s mechanism of action however is unclear. The main goals of this study were to perform an in-depth pharmacological characterization of (2R,6R)-HNK at known ketamine targets, to use target deconvolution approaches to discover novel proteins that bind to (2R,6R)-HNK, and to characterize the biodistribution and behavioral effects of (2R,6R)-HNK across several procedures related to substance use disorder liability. We found that unlike (S)- or (R)-ketamine, (2R,6R)-HNK did not directly bind to any known or proposed ketamine targets. Extensive screening and target deconvolution experiments at thousands of human proteins did not identify any other direct (2R,6R)-HNK-protein interactions. Biodistribution studies using radiolabeled (2R,6R)-HNK revealed non-selective brain regional enrichment, and no specific binding in any organ other than the liver. (2R,6R)-HNK was inactive in conditioned place preference, open-field locomotor activity, and intravenous self-administration procedures. Despite these negative findings, (2R,6R)-HNK produced a reduction in immobility time in the forced swim test and a small but significant increase in metabolic activity across a network of brain regions, and this metabolic signature differed from the brain metabolic profile induced by ketamine enantiomers. In sum, our results indicate that (2R,6R)-HNK does not share pharmacological or behavioral profile similarities with ketamine or its enantiomers. However, it could still be possible that both ketamine and (2R,6R)-HNK exert antidepressant-like efficacy through a common and previously unidentified mechanism. Given its pharmacological profile, we predict that (2R,6R)-HNK will exhibit a favorable safety profile in clinical trials, and we must wait for clinical studies to determine its antidepressant efficacy.
Keywords: depression, ketamine, drug development, antidepressant
Introduction
The off-label use of racemic, (R,S)-ketamine (ketamine) and the FDA approval of its (S)-ketamine enantiomer (i.e, esketamine or Spravato®) are promising developments for the treatment of depression and chronic pain[1, 2]. However, ketamine and (S)-ketamine are controlled substances with abuse potential and their use is associated with undesirable side effects, including dissociation[3, 4]. This may be especially problematic for the treatment of depression and chronic pain, which shares high comorbidity with substance use disorders (SUDs) [5, 6].
Recent research efforts have focused on identifying alternatives to ketamine and (S)-ketamine. One candidate currently in clinical trials is (R)-ketamine[7, 8]. (R)-ketamine shows greater antidepressant-like efficacy and exhibits a weaker abuse liability profile than (S)-ketamine in preclinical models[8–12]. A recent small open-label (R)-ketamine clinical trial reported rapid and long-lasting antidepressant effects with no major dissociation or other side-effects[13]. However, double-blind, randomized controlled trials are necessary to establish (R)-ketamine’s antidepressant efficacy. Another promising candidate is (2R,6R)-hydroxynorketamine ((2R,6R)-HNK), a ketamine metabolite reported in preclinical studies to lack the dissociative and abuse properties of ketamine and its enantiomers while retaining antidepressant-like behavioral efficacy and robust synaptic potentiation effects at excitatory synapses [12, 14, 23, 15–22]. An extensive number of studies have identified antidepressant-relevant effects of (2R,6R)-HNK, across diverse behavioral tasks including decreasing immobility time in the forced swimming[11, 12, 27–33, 14, 16–18, 23–26] and tail suspension tests[32, 33], reducing time to feed in the novelty-suppressed feeding test[12, 14, 18, 23, 33], reversal of anhedonia deficits[11, 12, 17, 25, 30, 32, 34] recovery of social interaction deficits[12], modifying fear conditioned responses[16, 24] and reversing learned helplessness[11, 12, 19, 27, 30]. (2R,6R)-HNK also reverses behavioral learned helplessness when administered directly to the ventricles of mice[11]. Additionally, metabolic conversion of ketamine or (R)-ketamine to (2R,6R)-HNK has been identified to contribute to ketamine’s preclinical antidepressant-like behavioral effects in rodents[11, 12]. These extensive behavioral data are further supported by ex vivo and in vitro findings that (2R,6R)-HNK exerts biochemical and synaptic effects consistent with antidepressant-like activity, including the rapid potentiation of excitatory synapses as observed in hippocampal slices[12, 14, 22, 26, 28, 35–38]. This is in addition to therapeutic effects in rodent models of chronic pain[39]. However, these findings are potentially in contrast with recent clinical studies where higher plasma levels of (2R,6R)-HNK and (2S,6S;2R,6R)-HNK correlated with less clinical improvement of depression[40, 41]. (2R,6R)-HNK is currently in Phase 1 clinical trials[42] and therefore, like (R)-ketamine, its clinical antidepressant efficacy is still unknown. Furthermore, (2R,6R)-HNK exerts an analgesic effect in several preclinical pain models [39].
Although ketamine is a non-competitive antagonist at N-methyl-D-aspartate receptors (NMDARs)[20], it is not clear to what extent NMDARs are the mediators of the antidepressant effects of ketamine, since more selective NMDAR antagonists do not recapitulate ketamine’s antidepressant-like[43, 44] or clinical antidepressant profile[45]. Moreover, (R)-ketamine is a weaker NMDAR antagonist than (S)-ketamine, but it exhibits greater antidepressant-like efficacy than (S)-ketamine in rodents[8]. Remarkably, (2R,6R)-HNK exhibits negligible affinity for NMDARs compared to ketamine and its enantiomers[9, 12].
We recently performed an in-depth pharmacological characterization of ketamine’s enantiomers and found that in addition to their action at NMDARs, (S)-ketamine and (R)-ketamine differentially engage opioid and other receptors and produce divergent behavioral and metabolic brain activity profiles[9]. However, the profile of (2R,6R)-HNK in these areas has not been thoroughly reported. A parsimonious explanation would conclude that since ketamine and (2R,6R)-HNK are structurally related they would share, at least partially, a common mechanism of action. As such, the main goal of this study was to perform an in-depth pharmacological characterization of (2R,6R)-HNK at known ketamine targets, to use high-content target deconvolution approaches to discover novel (2R,6R)-HNK-binding proteins, and to characterize the biodistribution, brain metabolic activity profile, and behavioral effects of (2R,6R)-HNK across several procedures related to SUDs.
Results
(2R,6R)-HNK neither binds to nor activates known ketamine targets
First, we tested (2R,6R)-HNK’s capacity to bind to known ketamine targets. Unlike ketamine and its enantiomers, (2R,6R)-HNK failed to displace [3H]MK801 binding in rat brain membranes (Fig. 1A), confirming its lack of affinity for NMDARs at this site as previously reported[12, 46, 47]. Also unlike (S)- and (R)-ketamine, which bind to mu (MOR) and kappa (KOR) opioid receptors[9], (2R,6R)-HNK showed no MOR or KOR binding as it failed to displace [3H]DAMGO and [3H]U69,593 from rat brain membranes (Fig. 1B, C).
Figure 1. (2R,6R)-HNK does not bind nor activate known ketamine binding sites.
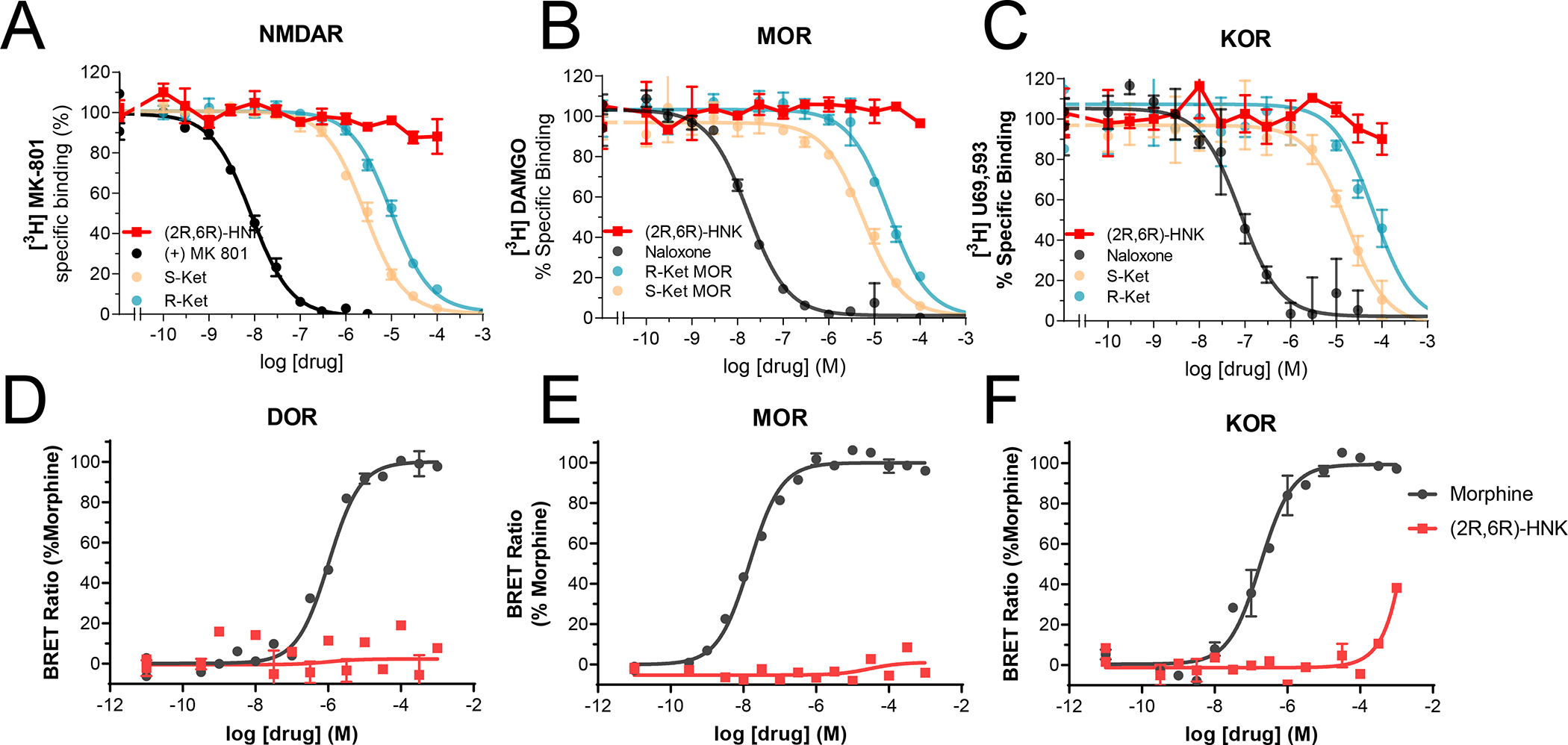
Competition binding assays of (2R,6R)-HNK (red) or reference ligands (MK-801 or naloxone, black) versus radioligands ([3H]-MK801, [3H]-DAMGO or [3H]-U69,593) labeling NMDA receptors (A), mu-opioid receptors, MOR, (B) or kappa-opioid receptors, KOR (C), respectively. The curves corresponding to (S)- and (R)-ketamine were published previously[9] and are displayed for reference. All binding assays were performed at the same time in rat whole brain (except cerebellum) membrane suspensions. In vitro signaling elicited by morphine or (2R,6R)-HNK in HEK-293 cells transiently transfected with MOR or KOR (D–F) and cAMP BRET-based biosensor. All data points are mean ± SEM of representative experiments performed in triplicate (experiments were performed 3–6 times to estimate the parameters (Ki, EC50 and Emax) or for statistical evaluation of the main effects of each drug reported in the main text.Figure 2.
Opioid receptor activation is associated with inhibition of cyclic adenosine monophosphate (cAMP) accumulation and/or recruitment of β-arrestins [9]. (S)- and (R)-ketamine differentially activate MOR and KOR signaling pathways, acting as partial agonists to inhibit cAMP accumulation but do not activate β-arrestin signaling[9]. A recent paper used computational modeling to report that (2R,6R)-HNK binds to opioid receptors with high (i.e. in the nanomolar range) affinity but this finding was not tested experimentally[48]. Furthermore, the authors claimed that (2R,6R)-HNK exhibited inverse agonist properties at opioid receptors[48]. These findings are inconsistent with our binding data which reveal that (2R,6R)-HNK does not bind to opioid receptors. To test whether (2R,6R)-HNK affects opioid receptor signaling, we measured its ability to inhibit cAMP accumulation in HEK-293 cells transiently transfected with either human delta-opioid receptor (DOR; OPRD1), MOR (OPRM1) or KOR (OPRK1) cDNA. We found that (2R,6R)-HNK did not induce any cAMP response up to a concentration of 1 mM, far beyond the concentration that (2R,6R)-HNK achieves (~10 μM) in the brain after administration of antidepressant-like doses[14] (Fig. 1D–F). In sum, these results reveal that (2R,6R)-HNK lacks the target engagement profile of ketamine and its enantiomers at their most commonly attributed targets as it does not bind to NMDA or opioid receptors. Furthermore, unlike ketamine and its enantiomers, (2R,6R)-HNK does not activate opioid receptors at physiological concentrations and therefore it is unlikely that it is involved in opioid-mediated antidepressant effects of ketamine[49, 50].
(2R,6R)-HNK exhibits an inert pharmacological profile
Given the lack of interactions that we observed between (2R,6R)-HNK and the most commonly attributed ketamine enantiomer targets, we directed our efforts towards the discovery of novel proteins that may interact with (2R,6R)-HNK. First, we performed a competitive screen using two concentrations of (2R,6R)-HNK (100 nM and 10 μM) at >80 receptor and enzymatic binding sites which are known targets for several neuropsychiatric medications. No hits were identified using 100 nM of (2R,6R)-HNK (Fig. 2A). Using 10 μM, we found that (2R,6R)-HNK increases binding of Substance P to the Neurokinin receptor (NK1), suggesting a potential allosteric interaction between (2R,6R)-HNK and Substance P at this receptor. The screen did not identify any other hits. To confirm the NK1R finding, we performed radioligand binding experiments using [3H]-Substance P and (2R,6R)-HNK in rat whole brain membrane homogenates. We found that (2R,6R)-HNK did not alter [3H]-Substance P binding (Fig. 2B), indicating a lack of interaction between (2R,6R)-HNK and NK1R in the rat brain.
Figure 2. Target deconvolution assays and other suggested targets.
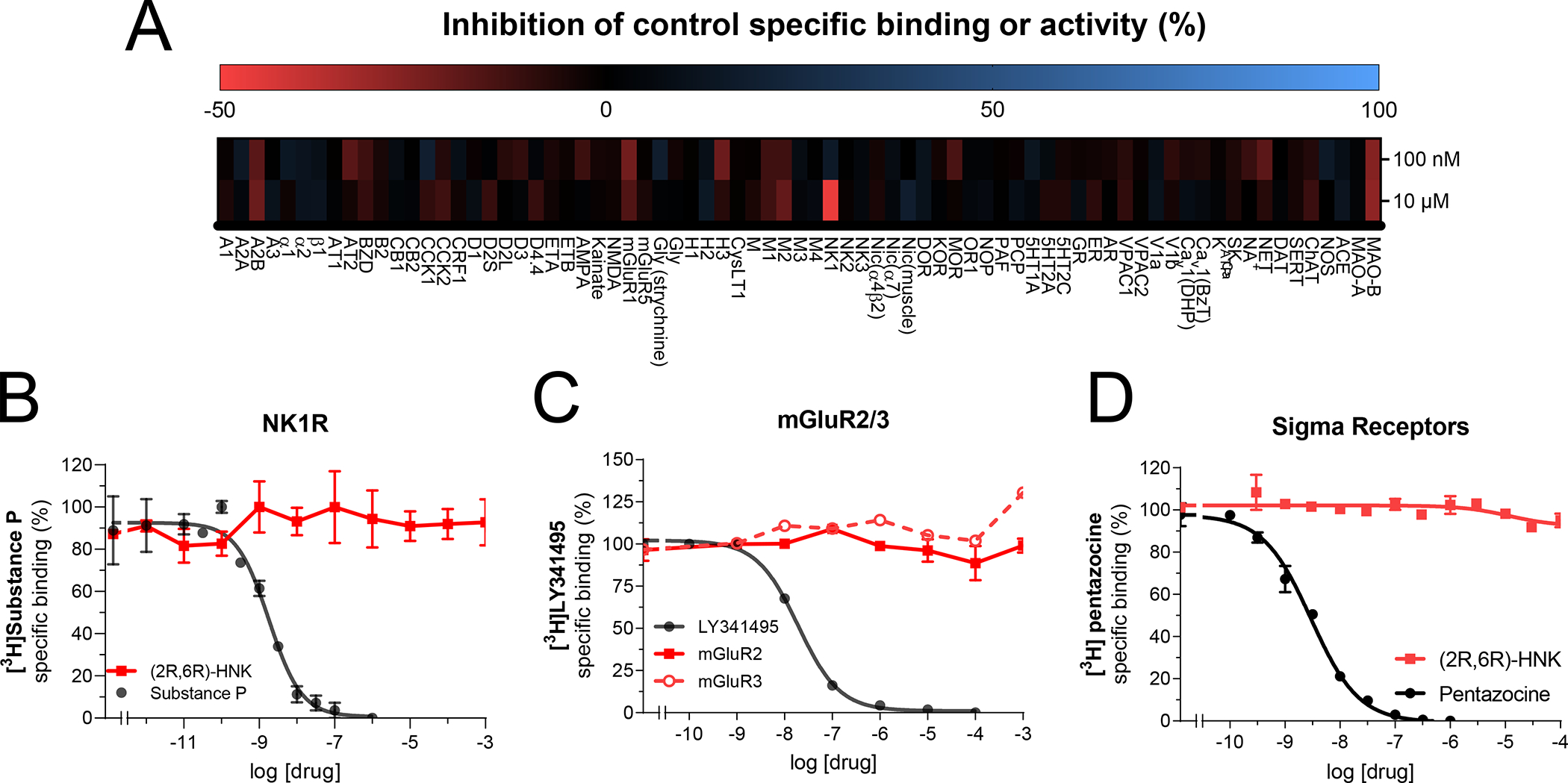
Receptor and enzyme competitive screen at two concentrations (100 nM and 10 μM) of (2R,6R)-HNK (A). Competition binding assays of (2R,6R)-HNK (red) or reference ligands (Substance P, LY341495 (on mGluR2) or pentazocine, black) versus radioligands ([3H]-Substance P, [3H]-LY341495 or [3H]-pentazocine) labeling Tachykinin receptors, NK1R (B), metabotropic glutamate receptors types 2 and 3, mGluR2/3 (C) or sigma-1 receptors (D), respectively. All binding assays were performed in rat whole brain (except cerebellum) membrane suspensions. All data points are mean ± SEM of representative experiments performed in triplicate (experiments were performed 3–6 times to estimate the parameters (Ki, EC50 and Emax) or for statistical evaluation of the main effects of each drug reported in the main text.Figure 3.
Previous research has implicated effects of (2R,6R)-HNK on metabotropic glutamate receptor (mGluR) types 2 or 3 signaling as being involved in the antidepressant-like mechanism of action [15]. To examine whether (2R,6R)-HNK binds to mGluR2/3 receptors, we performed radioligand competition experiments using the mGluR2/3 antagonist [3H]LY341475 in membranes expressing human recombinant mGluR2 or mGluR3 receptors. We found that (2R,6R)-HNK failed to displace [3H]LY341475 at either receptor (Fig. 2C). In agreement with these results, (2R,6R)-HNK did not affect functional responses at mGluR2/3 (Supplemental Figure S1). (2R,6R)-HNK’s potential interaction was also tested in functional assays at mGluR1, mGluR4, mGluR5, mGluR6, and mGluR8 but no effects of (2R,6R)-HNK were observed (Supplemental Figure S1).
Ketamine has been reported to inhibit HCN1 channels [51]. We found that neither (2R,6R)-HNK nor ketamine interacted with the HCN1 channel (Supplemental Fig. 1) indicating that the effects of (2R,6R)-HNK are not mediated by direct interactions with this channel. Finally, unlike ketamine and its enantiomers which exhibit weak binding to sigma receptors[9], we found that (2R,6R)-HNK did not bind to this site (Fig. 2D).
The above in vitro assays failed to identify direct interactions between (2R,6R)-HNK and discrete pharmacological targets using target-specific pharmacological probes. To expand the range of targets tested, we radiolabeled (2R,6R)-HNK with tritium [3H] to use it as a direct probe. First, we tested [3H]-(2R,6R)-HNK for binding to the high-density protein microarrays HuProt™ (>16,000 human gene products; ~81% of the human proteome) and Protoarray® (>9000 human proteins) but found that neither assay yielded any high-affinity specific binding hits (Supplemental Figs. 2, 3). Notably, high-density purified protein arrays can be susceptible to improper protein folding, especially for membrane proteins. To address this issue, we leveraged Retrogenix™ target deconvolution technology to test [3H]-(2R,6R)-HNK binding to >6000 cell surface and secreted human proteins transiently transfected in HEK-293 cells and tested in their cellular environment. No high-affinity specific binding to any of the tested proteins was observed (Supplemental Fig. 4).
Since the above screening approaches did not result in any positive target identification, we used an alternative target deconvolution strategy, a yeast 3-hybrid screen, which affords potentially greater sensitivity and readout amplification as well as an increase in the number of targets profiled[52]. Two (2R,6R)-HNK probes were synthesized which each contained the (2R,6R)-HNK motif, a short polyethyleneglycol (PEG) linker and a methotrexate anchor. The two probes differed in the placement of the linker, with probe one linking through the (2R,6R)-HNK aryl group, while probe 2 linked through the amine functional group. Both probes were initially tested for toxicity and permeability assessment in yeast, and both probes were found to have no toxicity or permeability issues in the assay. The probes were then screened against a cDNA library derived from adult mouse brain with more than 45,000 unique cDNAs. Despite the wide range of targets contained in this library, the screen did not identify any positive high-confidence probe-protein interactions, other than known false positives routinely detected in this specific type of screen or that are known to be hardly-detectable due to low mRNA library representation, or prey folding and prey toxicity. Following this, screens were performed against cDNA libraries derived from human adult brain, human lung cancer cells, and human placenta, but again despite the wide range of targets contained in these libraries, the screen did not identify any positive high-confidence (2R,6R)-HNK-protein interactions (Supplemental Fig. 5).
Collectively, we profiled direct interactions between (2R,6R)-HNK and >30,000 proteins but failed to identify any (2R,6R)-HNK-protein interactions, indicating that (2R,6R)-HNK exhibits an inert pharmacological profile.
(2R,6R)-HNK exhibits low uptake and no specific binding in brain
An alternative approach for understanding a drug’s mechanism of action is to characterize its biodistribution profile. Prior studies have reported (2R,6R)-HNK’s pharmacokinetics in brain tissue and in blood using analytical methods[14, 53]. To extend these observations to other organs in the body and in particular, to examine (2R,6R)-HNK’s region-specific accumulation in the brain, we administered a trace dose of [3H]-(2R,6R)-HNK (2 μCi/g, intravenous (i.v.)) to adult naïve male and female rats (n = 6) and analyzed their tissue biodistribution ex vivo. We found that [3H]-(2R,6R)-HNK showed a serum clearance rate of 50% in 30 min (Fig. 3A). In an in vitro saturation assay, we did not detect any specific binding of [3H]-(2R,6R)-HNK in rat serum (Fig. 3B). Accordingly, both serum and brain [3H]-(2R,6R)-HNK uptake were not blocked via pretreatment with a pharmacological dose of (2R,6R)-HNK (10 mg/kg, intraperitoneal (i.p.)) (Fig. 3C). In fact, we could not detect [3H]-(2R,6R)-HNK specific binding in any organ other than the liver (Fig. 3C). We also found that [3H]-(2R,6R)-HNK showed very low brain enrichment, with no region-specific accumulation in the brain (Fig. 3D, E). These findings indicate that (2R,6R)-HNK’s accumulation and binding in the brain or other organs, with the exception of the liver, are neither saturable nor specific and hence cannot be attributed to interactions with any given protein. The high and non-specific accumulation of [3H]-(2R,6R)-HNK in the kidneys is indicative of renal excretion. The only specific binding observed, in the liver, likely reflects [3H]-(2R,6R)-HNK’s saturable and specific binding to metabolic enzymes, as it is known that (2R,6R)-HNK undergoes metabolization through P450 liver enzymes[54, 55].
Figure 3. Fast clearance and no brain regional specificity of (2R,6R)-HNK uptake.
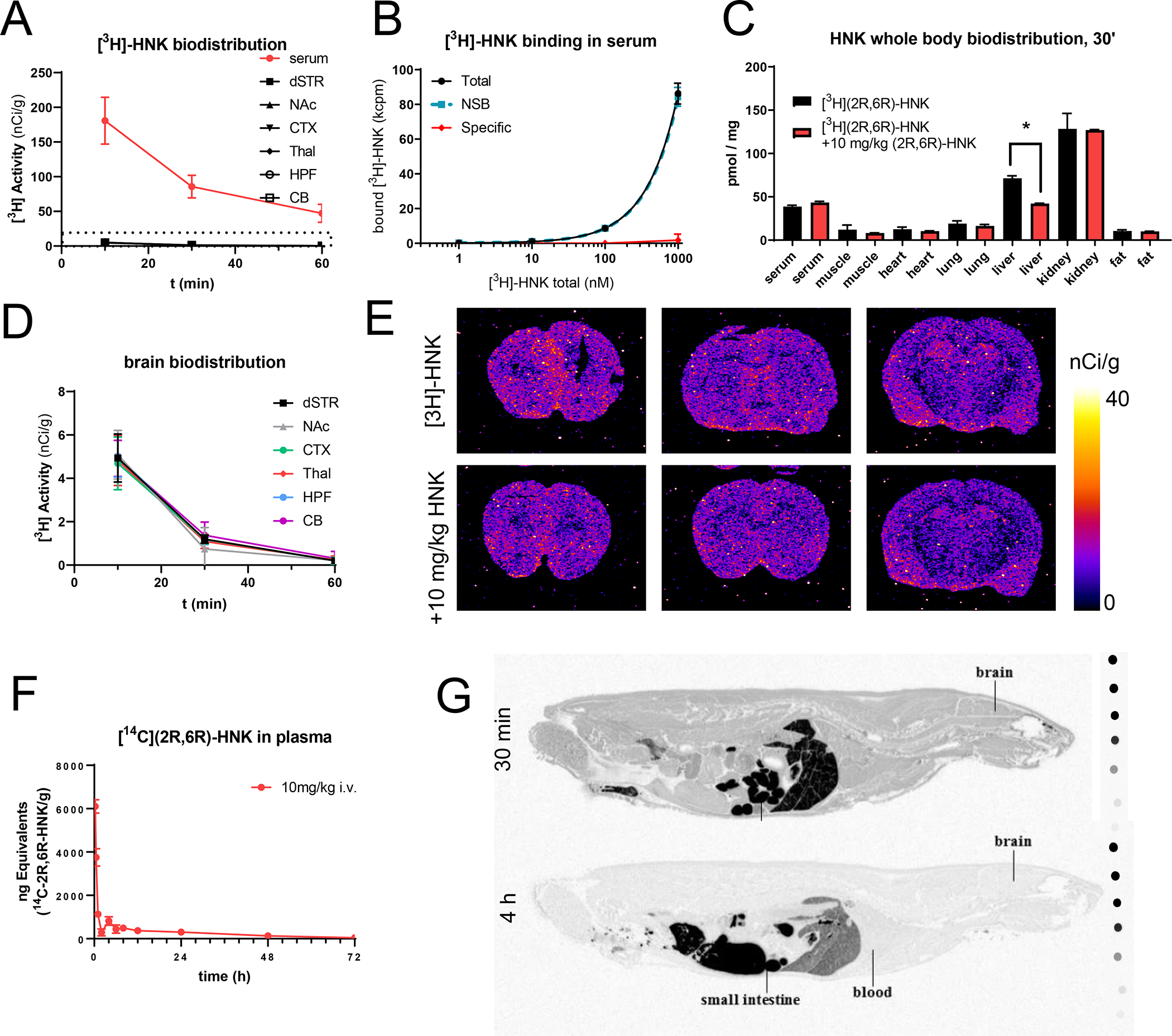
Activity detected in serum and brain after bolus i.v. (1 μCi/g) administration of [3H](2R,6R)-HNK in rats (A). Saturation binding experiments using rat serum indicate the lack of high-affinity specific binding in serum proteins (B). Biodistribution of [3H](2R,6R)-HNK 30 min after IV administration, uptake was not blocked in any organ, except the liver, when pretreating the animals with 10 mg/kg of (2R,6R)-HNK (IP) (C). Lack of enrichment in any selected brain region of rats injected (i.v., 1μCi/g) with radiolabeled [3H](2R,6R)-HNK (D). Representative autoradiograms of coronal brain sections of rats injected (IV, 1μCi/g) with radiolabeled[3H](2R,6R)-HNK 30 min after i.v. administration with or without pretreatment with 10 mg/kg of (2R,6R)-HNK (IP) (E). Activity detected in plasma after bolus i.v. (100 μCi/g) administration of [3H](2R,6R)-HNK in rats (F). Whole body autoradiograms of rats injected with radiolabeled [14C](2R,6R)-HNK 30 min and 4h after bolus i.v. administration (10 mg/kg). All data points are mean ± SEM.
The studies using [3H]-(2R,6R)-HNK are limited to administering trace doses of the radiopharmaceutical. In order to establish the biodistribution of a pharmacological dose of (2R,6R)-HNK, and to test safety of 14C exposure for future human pharmacokinetic studies, we labeled the molecule with [14C] and performed a mass balance and quantitative whole-body autoradiography (QWBA) study in rats (Fig. 3F, G. Supplemental Fig. 6). The pharmacokinetics of [14C]-(2R,6R)-HNK (100 μCi, 10 mg/kg, i.v.) were comparable to its tritiated analog and radioactivity was widely distributed through the entire body at 30 min (without increased brain accumulation or regional brain enrichment) (Fig. 3G). Furthermore, [14C]-(2R,6R)-HNK was rapidly metabolized in the liver by glucuronidation and was primarily excreted in urine and bile (Supplemental Fig 6). Specifically in males, the maximum mean blood and plasma concentrations (Cmax) of radioactivity were 4570 and 5240 ng equivalents [14C]-(2R,6R)-HNK/g, respectively, observed at the first collection time point (15 min post injection). The mean concentrations of radioactivity in blood and plasma then declined through 120 hours. In females the maximum mean blood and plasma concentrations (Cmax) of radioactivity were 6100 and 7200 ng equivalents [14C]-(2R,6R)-HNK/g, respectively, also observed at the first collection time point (15 min post injection). The mean concentrations of radioactivity in blood and plasma then also declined through 72 hours post injection. Comparison of area under curve (AUC) values of radioactivity in plasma from males and females showed similar exposure suggesting no sex differences. (2R,6R)-HNK-glucuronide-1 was the major circulating component in male and female rats after an IV dose. Plasma exposures AUC 0–72h of (2R,6R)-HNK-glucuronide-1 were 12800 ng eq·h/g (51.9% of the total radioactivity exposure through 72 hours) and 13500 ng eq·h/g (51.1% of the total radioactivity exposure through 72 hours) in males and females, respectively, after an intravenous dose. (2R,6R)-HNK was a major circulating component in intact male and female rats, with plasma exposures of 5330 ng eq·h/g (21.6% of the total radioactivity exposure through 72 hours) and 7090 ng eq·h/g (26.7% of the radioactivity exposure through 72 hours), respectively. Hydroxy-(2R,6R)-HNK-2 and hydroxy-(2R,6R)-HNK-3 were minor metabolites and individually contributed to <5% of the total radioactivity exposure through 72 hours.
(2R,6R)-HNK leads to changes in brain activity distinct from those induced by ketamine enantiomers
The pharmacological characterization studies we performed did not identify molecular targets or regional substrates for (2R,6R)-HNK. However, it could be possible that both ketamine and (2R,6R)-HNK exert their pharmacological actions through a common and previously unidentified mechanism. To examine this, we evaluated changes in brain metabolic activity induced by a prolonged (2R,6R)-HNK i.v. infusion in rats. We used a dose (10 mg/kg in 40 min) that is known to elicit antidepressant-like effects in rodents[12, 15] and that is expected to have a human equivalent dose of ~1.5 mg/kg over 40 min[56], which falls in the middle of the dose range of (2R,6R)-HNK tested in the current Phase 1 trial[57]. We injected [18F]-fluorodeoxyglucose (FDG) (~400 μCi, IP) to naïve awake rats at 10 min after starting the (2R,6R)-HNK IV infusion and then anesthetized them 30 min later and scanned them using positron emission tomography (PET) to visualize whole brain [18F]-FDG uptake as a proxy for brain metabolic activity. Using a voxel-wise analysis, we found that the prolonged (2R,6R)-HNK infusion led to a mild but significant increase in metabolic activity in a brain network comprising regions such as the insula, nucleus accumbens, globus pallidus, dorsomedial thalamus, motor, sensory, and visual cortices, and midline cerebellum (Fig. 4A–C). A secondary analysis using a region of interest (ROI)-based approach which confers lower spatial resolution due to predefined regional brain parcellations, only showed a significant increase in FDG uptake in the nucleus accumbens (~10% increase) (Fig. 4D). The ANOVA, which included the between-subjects factor of treatment (saline or (2R,6R)-HNK) and the within-subjects factor brain ROI (58 predefined brain regions), did reveal significant effects of the treatment (F(1, 14) = 5.45, p = 0.035), brain ROI (F(57, 798) = 134, p < 0.0001), and interaction between the two factors (F(57, 798) = 1.887, p = 0.0002). A post-hoc Šídák’s multiple comparisons test showed significant differences in the left nucleus accumbens (adjusted p = 0.0077). Overall, these effects were weaker and differed substantially in their regional makeup from those elicited by ketamine enantiomers as previously reported[9]. Moreover, the regional metabolic connectivity profile associated with (2R,6R)-HNK exposure also markedly differed from the regional metabolic connectivity profile of (S)-ketamine (Fig. 4E, Supplemental Fig. 6) and closely resembled the saline condition. These findings indicate that (2R,6R)-HNK leads to subtle but significant increases in brain metabolic activity in a region-specific manner, which markedly differs from the regional brain metabolic activity profiles induced by ketamine enantiomers, further supporting the notion that (2R,6R)-HNK and ketamine enantiomers differ in their target engagement profiles.
Figure 4. Changes in metabolic activity induced by (2R,6R)-HNK differ from those produced by (S)-ketamine.
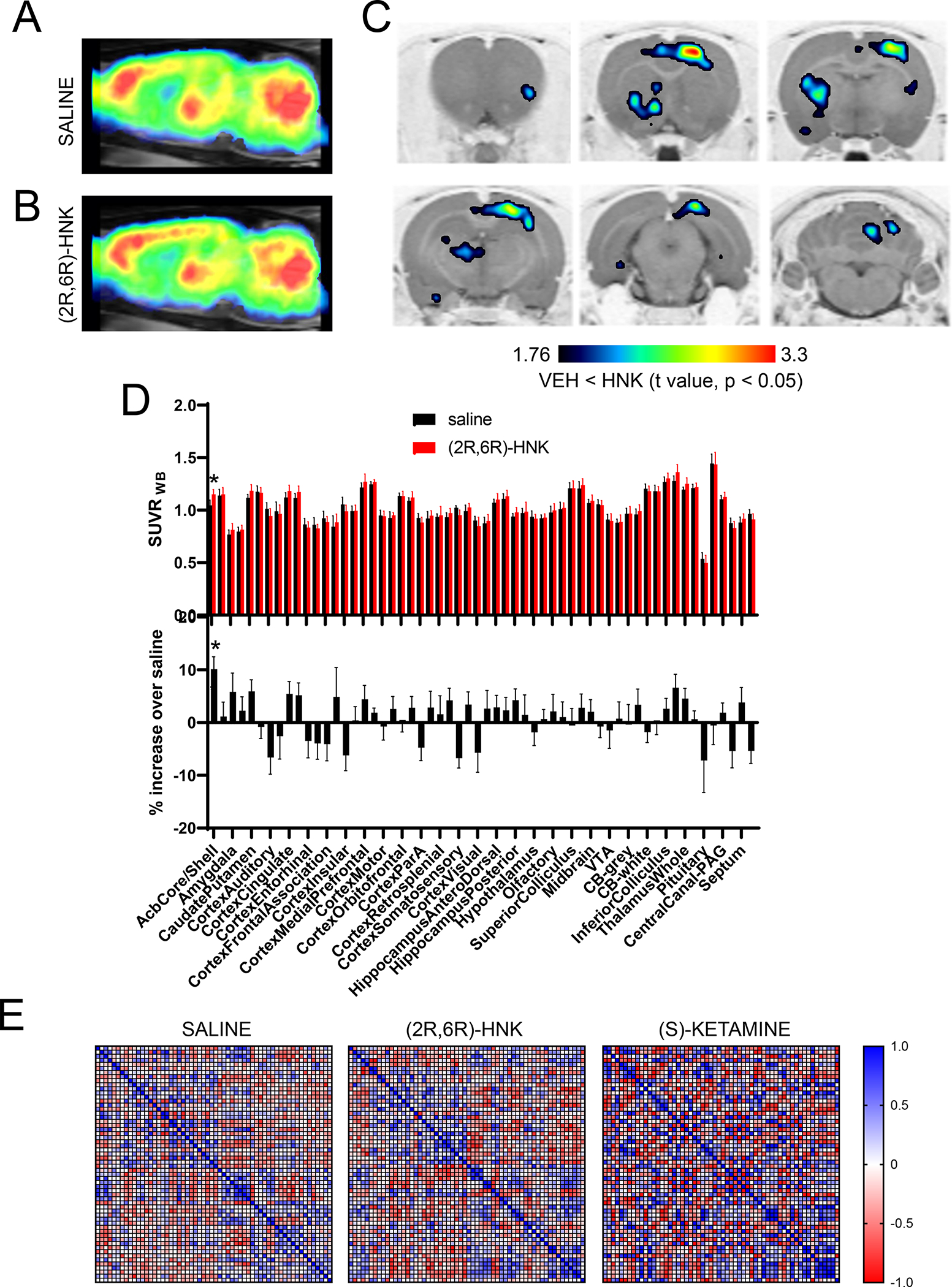
Brain metabolic mapping using [18F]-FDG PET scanning in rats. [18F]-FDG uptake images obtained after administration of saline (baseline, n = 4) or (2R,6R)-HNK (10 mg/kg over 40 min, n = 5). They show the average SUVRWB (standardized uptake value ratio) calculated using the whole brain as a reference region (A,B). Voxel-based parametric mapping analyses revealed significantly increased metabolic activity from baseline values in areas such as the insula, nucleus accumbens, globus pallidus, dorsomedial thalamus, motor, sensory, and visual cortices, and midline cerebellum. Statistical parametric maps of significant decreases of [18F]-FDG uptake (P < 0.05, paired t test) (C). Regional [18F]-FDG uptake (SUVRWB) in the brain regions of animals infused with saline or (2R,6R)-HNK (D). Correlation matrices of the metabolic activity across brain regions of animals infused with saline, (2R,6R)-HNK or (S)-ketamine. The data corresponding to the (S) ketamine dataset was reported in [9]. Full data sets are available in Supplemental Information 6 (E).
(2R,6R)-HNK is inert in behavioral procedures related to substance use disorder liability
Regardless of its efficacy as an antidepressant, a significant limitation of ketamine is its abuse liability. We recently reported that (S)-ketamine is the enantiomer implicated in the abuse liability of ketamine, and while (S)-ketamine is self-administered in rats, (R)-ketamine is not, even at doses equipotent for NMDAR antagonism[9]. The data reported in this manuscript (lack of binding to opioid receptors and different brain metabolic activity profile) would predict that (2R,6R)-HNK does not share the same limitation as ketamine in regard to SUD liability.
To test (2R,6R)-HNK’s behavioral actions in rodents we ran a battery of behavioral paradigms relevant to depression and SUDs. In the forced swimming test in mice (n= 8 per condition) a single administration of (2R,6R)-HNK (3 or 10 mg/kg, IP) reduced immobility time 24 hours after drug administration (Fig. 5A). The ANOVA which considered a between-subject factor of drug dose (0, 1, 3 or 10 mg/kg) revealed significant effects of drug dose ((F3, 28) =12.9, p > 0.001) and a Dunnett’s post-hoc multiple comparisons test detected significant differences of 3 and 10 mg/kg (p < 0.001) doses but not 1 mg/kg (p = 0.86), compared to vehicle-treated mice. Nonetheless, in male mice (n = 8 per condition) a single acute administration of (2R,6R)-HNK (10, 30, 65 mg/kg, IP) did not alter locomotor activity (Fig. 5B). The mixed ANOVA, which included the between-subjects factor of drug dose (0, 10, 30 or 65 mg/kg) and the within-subjects factor of time bin (5-min bins, from 0 to 30), did not reveal significant effects of the drug (F(3, 28) = 2.2, p = 0.11), nor an interaction between the two factors (F(15, 140) = 1.33, p = 0.19).
Figure 5. Lack of effects of (2R,6R)-HNK on locomotor activity, CPP and self-administration.
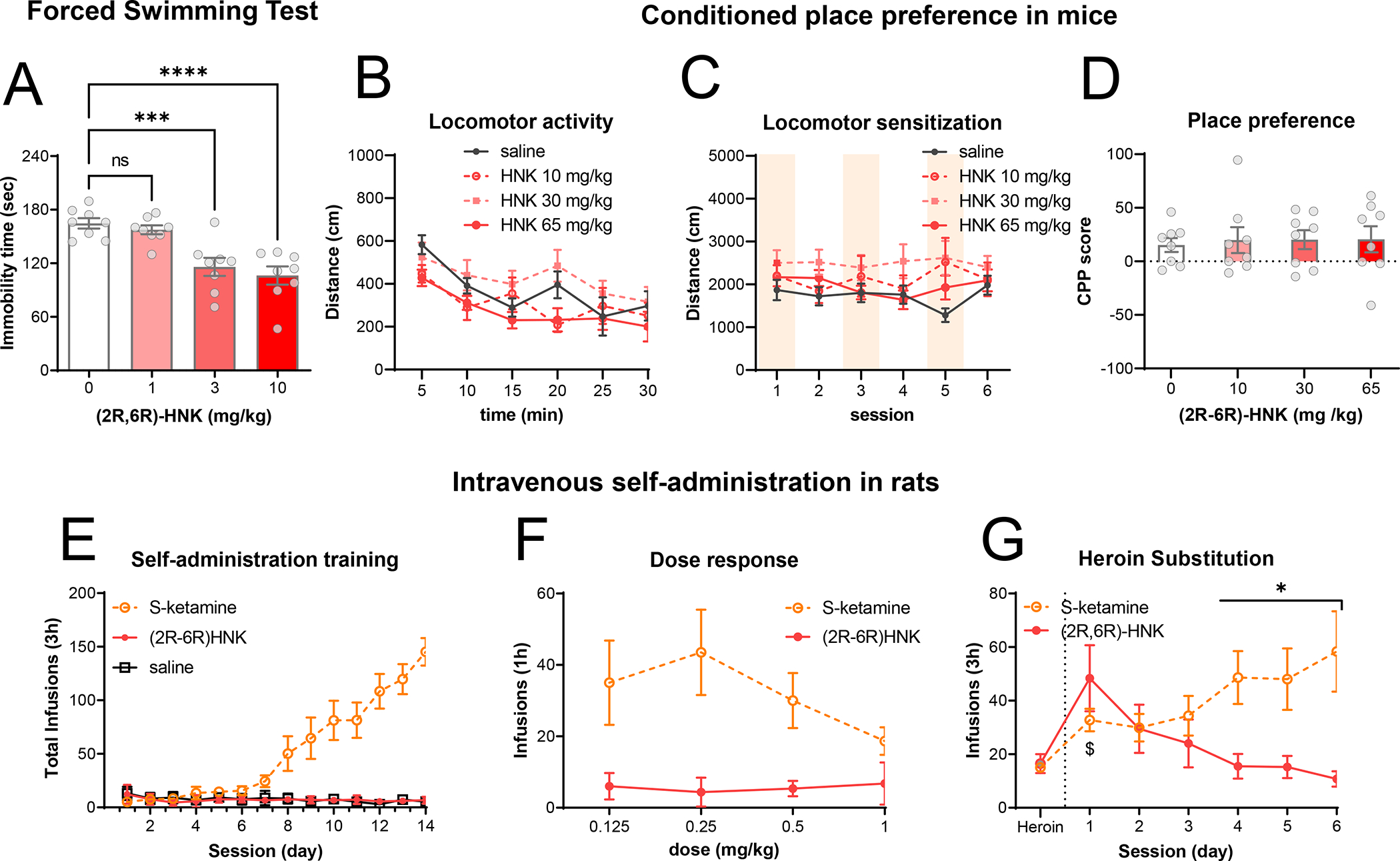
Forced Swimming Test (A): male mice were tested in the forced swimming test 24 hours after a single administration of (2R,6R)-HNK (1, 3 and 10 mg/kg, IP). Plots display immobility time and are representing individual data points with bars indicating mean ± SEM. *** and **** denote statistical significance between groups (P < 0.001 and P < 0.0001, respectively). Acute locomotor activity (B): male mice were and injected with saline or (2R,6R)-HNK (10, 30, and 65 mg/kg, IP) and their locomotor activity was measured. Plots display distance traveled in open field arenas in 5min time bins. CPP: Male mice were injected with (2R,6R)-HNK (0, 10, 30 or 65 mg/kg) or saline on either side of the CPP arena for 6 days and were allowed to explore both sides. Plots in (C) display the distance traveled during the consecutive conditioning sessions. At the end of the conditioning phase the preference for the drug-paired side was quantified as a CPP score (D). In all cases, data points are displayed as mean ± SD of 8 mice per condition or individual data points with bars displaying mean ± SEM. Self-administration: Self-administration training: mean ± SEM number of infusions of saline, (2R,6R)-HNK or (S)-ketamine under the FR1 20-s timeout reinforcement schedule (E). Dose–response curve: mean ± SEM number of infusions for different unit doses of (2R,6R)-HNK or (S)-ketamine under the FR1 20-s timeout reinforcement schedule (F). Data from the (S)-ketamine animals was previously published in [9]. In (G), rats (n=16) were trained to press for heroin and after steady responding it was substituted by either (2R,6R)-HNK or (S)-ketamine. $ denotes statistical significance with respect to heroin infusions within the same group (P < 0.05) and * denotes statistical significance between groups (P < 0.01). In all cases data points represent mean ± SEM of number of infusions.
Next, we tested for rewarding effects of (2R,6R)-HNK in mice using the conditioned place preference (CPP) procedure. We injected male mice (n = 8 per group) with either saline or (2R,6R)-HNK (0, 10, 30 or 65 m/kg, IP) and placed them in one of two sides of a two-compartment chamber for 15 min. On alternate days, we injected the same mice with vehicle injections and placed them on the other side for 15 min. We performed the drug or vehicle pairing over 6 days and counterbalanced the vehicle and drug injections during these days. During the conditioning phases of the procedure, we monitored the locomotor activity of the mice in order to detect sensitization or other locomotor effects derived from repeated exposure. Repeated administration of (2R,6R)-HNK did not produce any changes in locomotor activity and did not induce sensitization (Fig. 5C). The mixed ANOVA, which included the between-subjects factor of drug dose (0, 10, 0 or 65 mg/kg) and the within-subjects factor of session (1 to 6), did not show significant effects of drug dose (F(3, 28) = 2.05, p = 0.13), session (F(5, 140) = 0.55, p = 0.73), nor an interaction between the two factors (F(15, 140) = 1.04, p = 0.41). In addition, repeated administration of (2R,6R)-HNK (10, 30, 65 mg/kg, IP) did not induce CPP at any of the tested doses (Fig. 5D). The one-way ANOVA, which included the between-subjects factor of drug dose (0, 10, 30 or 65 mg/kg), did not show significant effects of drug dose (F(3,28) = 0.06, p = 0.98) on the CPP score.
Based on our prior studies with ketamine enantiomers[9], we tried to train male rats (n = 14) to self-administer (2R,6R)-HNK using a unit dose known to promote (S)-ketamine and ketamine self-administration, [9, 58] (0.5 mg/kg/inf, IV) for 16 days under a fixed ratio 1 (FR1) reinforcement schedule with a 20-s timeout. During this period, rats were primed to press the lever to ensure that they experienced the drug and the visual cues associated with it. However, rats did not learn the task, indicating that (2R,6R)-HNK was not a reliable reinforcer in the self-administration procedure (Fig. 5E). The mixed ANOVA, which included the between-subjects factor of drug (saline or (2R,6R)-HNK) and the within-subjects factor of session (14 training sessions), did not show significant effects of drug (F(1, 140) = 0.54, p = 0.46), nor an interaction between the two factors (F(13, 140) = 0.59, p = 0.85). Next, we conducted a dose-response experiment to examine whether animals would respond to other unit doses (0.125, 0.25, or 1 mg/kg/inf) but rats did not respond to any of the (2R,6R)-HNK doses tested (Fig. 5F). A one-way ANOVA, which included the between-subjects factor of (2R,6R)-HNK unit dose (0.125, 0.25, or 1 mg/kg/inf), did not reveal significant effects of drug unit dose. To address a possibility that (2R,6R)-HNK infusions could be rewarding but not salient enough to drive naïve rats to learn to self-administer it, we tested its reinforcing properties in rats previously exposed to heroin self-administration. We trained male rats (n=16) to self-administer heroin (0.05 mg/kg/inf, IV) for 10 days under a FR1 schedule with a 20-s timeout. Once rats achieved steady lever responding (15 ± 3 infusions per session), heroin infusions were substituted by either (S)-ketamine (0.5 mg/kg/inf, IV) or (2R,6R)-HNK (0.5 mg/kg/inf, IV). On the first session, rats increased their pressing for (S)-ketamine but pressed significantly more for (2R,6R)-HNK (p= 0.001, according to a Sidak’s post-hoc test, see below), indicating increased drug seeking in the (2R,6R)-HNK group due to the lack of the expected rewarding effects via the drug substitution. The mixed ANOVA, which included the between-subjects factor of drug (S-ketamine or (2R,6R)-HNK) and the within-subjects factor of session, revealed significant effects of drug (F(1, 14) = 4.81, p = 0.045), session (F(6, 84) = 5.21, p = 0.0001) and an interaction between the two factors (F(6, 84) = 12.0, p < 0.0001). On successive sessions, rats showed escalation of self-administration for S-ketamine but rapid extinction of self-administration for (2R,6R)-HNK (Fig. 5G). Overall, these results indicate that rats trained to self-administer heroin generalized their training to (S)-ketamine and continued to self-administer the new drug but did not generalize to (2R,6R)-HNK, indicating that (2R,6R)-HNK lacks reinforcing properties and suggesting that it would have a weak SUD liability profile.
Discussion
Here we show that unlike ketamine and its enantiomers, (2R,6R)-HNK did not displace binding of [3H]MK-801 to NMDAR which replicates prior findings[12, 47, 59]. Specifically, both our findings presented here, and those from prior reports tested (2R,6R)-HNK for NMDAR binding at concentrations up to 100 μM and observed no displacement of [3H]MK-801. Similarly, another study reported that 10 μM (2R,6R)-HNK tested on cultured hippocampal neurons had no effect on NMDAR function[60]. This finding is also consistent with a previous report that tested effects of (2R,6R)-HNK on NMDAR current activity, on NMDA-induced lethality in mice, NMDAR-mediated field excitatory postsynaptic potentials (fEPSPs) in the CA1 field of mouse hippocampal slices, NMDAR-mediated miniature excitatory postsynaptic currents (mEPSCs) and NMDA-evoked currents in CA1 pyramidal neurons of rat hippocampal slices, and recombinant NMDARs expressed in Xenopus oocytes [12]. However, Suzuki et al. also tested (2R,6R)-HNK at a higher concentration (50 μM) which led to a ~40% reduction in synaptic NMDAR currents[60]. This finding is at odds with our [3H]MK-801 binding data as well as the [3H]MK-801 binding data published previously. Notably, a 50 μM concentration of (2R,6R)-HNK is unlikely to be relevant for its antidepressant-like effects, as (2R,6R)-HNK reaches a maximum extracellular concentration of ~8 μM in the hippocampus after i.p. injection of antidepressant-relevant doses in mice[14]. Whereas it is possible that (2R,6R)-HNK injected i.v. may bypass metabolic degradation and lead to higher brain concentrations, our [3H](2R,6R)-HNK uptake findings showing that i.v.-injected (2R,6R)-HNK has low brain uptake and retention, and recent findings showing that (2R,6R)-HNK, even at i.v. doses up to 160 mg/kg does not produce NMDAR-associated neurotoxicity[53] further support the conclusion that (2R,6R)-HNK does not interact with NMDARs at antidepressant-like relevant doses. These findings are also consistent with our QWBA study that revealed minimal brain uptake, no region-specific enrichment, and rapid clearance of a high dose (10 mg/kg, i.v. bolus) of [14C]-(2R,6R)-HNK.
We also found that unlike ketamine and its enantiomers, (2R,6R)-HNK did not displace binding of [3H]DAMGO to MOR, [3H]U69,593 to KOR, or [3H]Pentazocine to sigma receptors. Also, unlike ketamine and its enantiomers, (2R,6R)-HNK did not induce any functional response through the MOR, KOR or DOR (up to concentrations of 1 mM). Notably, a recent study reported that (2R,6R)-HNK binds to opioid receptors with nM affinity but this conclusion was derived by computational modeling and was not tested experimentally[48]. This finding is inconsistent with our experimental data which show that (2R,6R)-HNK does not bind to opioid receptors at concentrations up to 100 μM. Furthermore, this same study also claimed that (2R,6R)-HNK exhibited inverse agonist properties at opioid receptors[48]. However, the experimental results supporting this claim do not appear to provide convincing evidence to conclude that (2R,6R)-HNK affects functional responses of opioid receptors, as the effect sizes reported were small. Overall, the findings from Joseph et al. are in stark contrast to our findings as described here as well as other findings which revealed that unlike ketamine, (2S,6S;2R,6R)-HNK had no effect on acute morphine nociception or tolerance in rats[61] and that (2R,6R)-HNK-mediated pain reduction in mice was not blocked by naloxone pretreatment[39]. Collectively, our findings reveal that (2R,6R)-HNK has a distinct pharmacological profile from racemic ketamine and its enantiomers, and that it does not interact with any known ketamine targets such as NMDA, opioid, or sigma receptors at pharmacologically-relevant concentrations.
In addition to these known ketamine targets, we performed detailed characterizations of (2R,6R)-HNK’s pharmacological profile at other receptors previously implicated in either (2R,6R)-HNK’s or ketamine’s actions. (2R,6R)-HNK was recently reported to exert antidepressant-relevant effects in mice convergent with those of mGluR2-inhibition[15] but whether this occurs via direct effects of (2R,6R)-HNK at mGluR2 receptors has not been previously reported. We found that (2R,6R)-HNK did not affect binding of [3H]LY341495 to mGlu2 or mGlu3 receptors. In agreement with these binding results, (2R,6R)-HNK did not affect functional responses at mGluR2/3 or to other mGluRs indicating that mGluR2-dependent antidepressant-relevant effects of (2R,6R)-HNK are not mediated via direct interactions with mGluR2 or other mGluRs. Ketamine has been reported to inhibit HCN1 channels[51] and HCN1 was reported necessary for ketamine to exert antidepressant-like actions in mice[62]. Furthermore, HCN1 inactivity leads to stress resilience in mice[63–66]. We found that (2R,6R)-HNK did not interact functionally with the HCN1 channel indicating that the effects of (2R,6R)-HNK are not mediated by direct interactions with this channel. Finally, our study adds to existing literature that neither ketamine’s enantiomers nor its metabolites had affinity or functional agonist or antagonist actions at dopamine receptors (D1-D5) or binding affinity or function effects on monoamine transporters (up to 10 μM)[67].
In addition to examining interactions between (2R,6R)-HNK and known targets of ketamine previously implicated in its putative mechanism of action, we also examined the extent to which (2R,6R)-HNK might exert its actions through an unidentified or unsuspected target. Hence, we undertook extensive screening and target deconvolution experiments, specifically, four different screening strategies which collectively profiled (2R,6R)-HNK at >30,000 human proteins, including mouse and human tissues. These efforts did not reveal any significant interactions between (2R,6R)-HNK and the numerous proteins tested. Notably, among the many proteins that we profiled in these experiments, (2R,6R)-HNK specifically failed to interact with tropomyosin receptor kinase B (TrkB), estrogen receptor alpha (ERα), and the glucocorticoid receptor (GR) which were previously reported to bind or interact with (2R,6R)-HNK as well as ketamine[16, 26, 68]. Therefore, our results show that (2R,6R)-HNK has a largely inert in vitro pharmacological profile, and that like ketamine and its enantiomers, which we previously profiled using similar screening assays[9], does not bind to TrkB, ERα or GR. The discrepancy between our findings related to TrkB, ERα, and GR and those published previously[16, 26, 68] may be due to technical differences in the respective assays employed. However, the report describing (2R,6R)-HNK binding to TrkB used an indirect methodology[16], evaluating binding of several biotin-conjugated antidepressants- of different classes and with distinct known binding sites- for their ability to displace a biotin-conjugated fluoxetine analog bound to TrkB. Notably, fluoxetine, an extensively investigated compound, is a known selective serotonin reuptake inhibitor and its binding to TrkB has not been previously described[69]. In regard to the GR, the effect of (2R,6R)-HNK on dexamethasone-induced GR activation was modest and significantly decreased the GR response only at a single dexamethasone concentration (1 nM) along the dose response curve[26]. Given the fact that i) we could not detect any interaction between ketamine enantiomers and TrkB, ERα, or GR in prior experiments[9], ii) we did not detect any direct interactions between (2R,6R)-HNK and TrkB, ERα, or GR using the different unbiased screening approaches employed here, and iii) that ketamine/(2R,6R)-HNK binding or interactions to TrkB, ERα, and GR have not yet been replicated, indicate that the previously reported binding/interactions between ketamine/(2R,6R)-HNK and these receptors may not comprise robust phenomena, or may be artefactual or non-specific.
Although prior pharmacokinetic studies using (2R,6R)-HNK demonstrated reasonable brain permeability[12, 14, 38], our results from ex vivo autoradiography and uptake experiments using radiolabeled (2R,6R)-HNK preclude the existence of any active metabolic byproducts and show that it has non-specific brain uptake (it was not blocked by administration of a pharmacological dose of non-radiolabeled (2R,6R)-HNK) and uniform brain regional distribution. Notably, our [3H]-(2R,6R)-HNK experiments revealed that specific binding was observed only in the liver. Taken together with the above findings, these results suggest that (2R,6R)-HNK exhibits a largely inert profile at common ketamine targets and that it lacks high-affinity specific binding in the brain and in the other organs we examined, with the specific binding observed in the liver most likely attributed to (2R,6R)-HNK’s interaction with metabolizing liver cytochrome P450 enzymes[54, 55, 70–72]. Overall, these findings are consistent with prior findings where (2R,6R)-HNK, even at i.v. doses up to 160 mg/kg, did not demonstrate any neurotoxicological effects in rats[53].
Compared to saline-infused rats, a prolonged (2R,6R)-HNK i.v. infusion (10 mg/kg over 40 min) in rats led to a mild but significant increase in metabolic activity in a brain network comprising regions such as the insula, globus pallidus, dorsomedial thalamus, motor, sensory, and visual cortices, and midline cerebellum, with the strongest effect observed in the nucleus accumbens (~10% increase). Moreover, this effect was different from the brain regional metabolic profile elicited by (S)-ketamine infusion. The effect of (2R,6R)-HNK i.v. infusion on brain activity is intriguing given its largely inert pharmacological profile and is supported by recent studies which reported that antidepressant-like doses of (2R,6R)-HNK in mice are sufficient to modulate cortical and thalamic activity[15, 73]. Whereas the magnitude of these effects was small compared to effects produced by (S)-ketamine[9], this is not surprising, given the differences in potency and overall pharmacological profile between (2R,6R)-HNK and (S)-ketamine. Indeed, a prior study showed that racemic ketamine and (2R,6R)-HNK markedly differ in their capacity to induce calcium activity in the prefrontal cortex (PFC)[54]. In agreement with this prior study that injected rats with 30 mg/kg (2R,6R)-HNK, we did not see any metabolic activity in the PFC after (2R,6R)-HNK. Finally, it is important to note that the increase in brain metabolic activity observed with (2R,6R)-HNK was unlikely to be due to metabolic byproducts or variation in anesthesia since the rats were exposed to FDG uptake during awake and freely moving conditions.
Finally, we evaluated (2R,6R)-HNK across several behavioral procedures relevant to SUDs. Consistent with prior findings[11, 21], (2R,6R)-HNK significantly reduced immobility in the forced swim test and did not modify locomotion in mice. Finally, we found that rats did not self-administer (2R,6R)-HNK using the i.v. drug self-administration procedure which was also previously observed in mice[12]. Overall, and with the exception if the forced swim test data, these negative results are consistent with (2R,6R)-HNK’s low in vivo toxicity and wide putative therapeutic window[21, 53] but also with the inert pharmacological profile we report in this study. Notably, these behavioral procedures were performed in naïve rodents and not in anxiety or depression models typically exposed to stress or anxiogenic manipulations. It may be possible that (2R,6R)-HNK may have a different pharmacological profile or behavioral efficacy in such models, where the underlying neurobiology can be altered due to the exposure of a stressful or anxiogenic environment. As summarized in the introduction, many studies have identified antidepressant-like effects of (2R,6R)-HNK[11, 14–19]. Nevertheless, it is important to also point out that several studies from a single laboratory have reported negative findings with respect to (2R,6R)-HNK’s behavioral efficacy as an antidepressant[70, 74–76]. Accordingly, such discrepancies may be explained by its largely inert profile or by specific behavioral adaptations associated with stress exposure in laboratory rodents. Indeed, it has also been reported that (2R,6R)-HNK exerts an analgesic effect in several preclinical pain models[39]. Finally, the inert behavioral effects we observed here and those observed in prior studies may also be explained by seasonal variability in rodent behavior, lab-to-lab variability, or experimenter-specific effects[77, 78], which have in fact been recently described for (2R,6R)-HNK as well as ketamine[79].
In summary, here we performed a comprehensive investigation into the pharmacological and behavioral properties of (2R,6R)-HNK. Our findings indicate that (2R,6R)-HNK is a largely inert molecule that does not share the pharmacological profile similarities of ketamine and that it lacks high-affinity specific binding in the brain. Consequently, the mechanism of action of (2R,6R)-HNK remains elusive. The inertness of (2R,6R)-HNK, as we describe here, has important implications for the mechanistic basis of its reported antidepressant-like effects and to what extent the in vitro and in vivo testing we performed here can be used to demonstrate such mechanisms or predict clinical performance. The approach and findings we report converge on the notions that (2R,6R)-HNK’s putative antidepressant-like mechanism of action is distinct from that of ketamine’s or that it may involve a common yet unidentified mechanism. Our findings indicate that if such a mechanism exists, it is unlikely to involve high affinity interactions between (2R,6R)-HNK and specific molecular targets. Instead, our findings indicate that (2R,6R)-HNK may produce its antidepressant-like effects indirectly, by interacting with chemical or other physical processes, or via weak, low affinity interactions at multiple molecular targets that converge upon one or more biological systems. In any case, the in vitro and in vivo testing we performed here does not allow us to predict clinical antidepressant efficacy. However, we do predict that if clinical trials testing the efficacy of (2R,6R)-HNK for depression, chronic pain, and other indications are successful, (2R,6R)-HNK will constitute a promising pharmacotherapy devoid of the abuse liability and other negative side-effects associated with ketamine and (S)-ketamine, which are currently used for the treatment of refractory depression.
Materials and methods
Animals
Male wild-type mice (C57BL/6J or CD-1) were ordered from the Jackson or Charles River Laboratory, weighing 20 to 25 g or ~35 grams respectively. Mice were maintained under a normal 12-hour light/dark cycle with food and water freely available. Male Sprague-Dawley rats (strain code #400) were ordered from Charles River Laboratories, weighing 200 to 250 g. Rats were maintained under a reverse 12-hour light/dark cycle with ad libitum access to food and water. We housed two rats per cage before surgery and individually after surgery. Rats feeding regimen was restricted to 18 to 20 g per day throughout the experiment task in order to maintain a stable weight of the rats and facilitate acquisition of drug self-administration. All experiments and procedures complied with all relevant ethical regulations for animal testing and research and followed the National Institutes of Health (NIH) guidelines and were approved by each institute’s animal care and use committees. The experimenters were blind to the group allocation during data acquisition.
Cell culture and transfection
Human embryonic kidney (HEK-293, ATCC) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, USA) supplemented with 2 mM L-glutamine, 0.1 mM nonessential amino acids, antibiotic/antimycotic (all supplements from Gibco) and 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Inc. Flowery Branch, GA, USA) and kept in an incubator at 37°C and 5% CO2. Cells were routinely tested for myclopasma contamination (MycoAlert® Mycoplasma Detection Kit, Lonza). Cells were seeded on 60 cm2 dishes at 4 × 106 cells/dish 24 hours before transfection. The indicated amount of cDNA was transfected into HEK-293 cells using polyethylenimine (PEI; Sigma-Aldrich) in a 1 to 2 DNA:PEI ratio. Cell harvesting for radioligand binding experiments or signaling assays were performed approximately 48 hrs after transfection.
Radioligand binding assays
Transfected HEK-293 cells were and harvested 48 hrs after transfection. Cells were suspended in Tris-HCl 50 mM pH 7.4 supplemented with protease inhibitor cocktail (1:100, Sigma-Aldrich, St Louis, MO, USA). The dissected brain tissue was diluted in Tris-HCl 50 mM buffer supplemented with protease inhibitor cocktail (1:1000). HEK-293 cells or brain tissue were disrupted with a Polytron homogenizer (Kinematica, Basel, Switzerland). Homogenates were centrifuged at 48,000 g (50 min, 4 °C) and washed 2 times in the same conditions to isolate the membrane fraction. Protein was quantified by the bicinchoninic acid method (Pierce). For competition experiments, membrane suspensions (50 μg of protein/ml) were incubated during 2hr at RT in 50 mM Tris-HCl (pH 7.4) containing 10 mM MgCl2, and [3H]MK-801 (30 Ci/mmol, Novandi Chemistry AB, Södertälje, Sweden), [3H]DAMGO (NIDA Drug Supply), [3H]U69,593 (36 Ci/mmol, Novandi Chemistry AB, Södertälje, Sweden), [3H]LY341495 (54 Ci/mmol, Novandi Chemistry AB, Södertälje, Sweden), [3H](+)Pentazocine (28.3 Ci/mmol, Perkin Elmer), or [3H]Substance P (28 Ci/mmol, Perkin Elmer), and increasing concentrations of (2R,6R)-HNK (0.1 nM to 1 mM, NCATS) or the reference compound. Non-specific binding was determined in the presence of 10 μM of each reference ligand. In all cases, free and membrane-bound radioligand were separated by rapid filtration of 500-μl aliquots in a 96-well plate harvester (Brandel, Gaithersburg, MD, USA) and washed with 2 ml of ice-cold Tris-HCl buffer. Microscint-20 scintillation liquid (65 μl/well, PerkinElmer) was added to the filter plates, plates were incubated overnight at RT and radioactivity counts were determined in a MicroBeta2 plate counter (PerkinElmer, Boston, MA, USA) with an efficiency of 41%. One-site competition curves were fitted using Prism 9 (GraphPad Software, La Jolla, CA, USA). Ki values were calculated using the Cheng-Prusoff equation.
In vitro functional assays
BRET assays were performed to detect inhibition of cAMP accumulation by employing the CAMYEL (yellow fluorescence protein-Epac-Rluc) biosensor[80]. HEK cells were transfected with 5 μg of untagged receptor (MOR, KOR or DOR) and 5 μg CAMYEL biosensor using the PEI method described above. BRET experiments were performed 48 hours after transfection. On experiment day, cells were harvested, washed, and resuspended in DPBS containing 100 μM sodium metabisulfite and 5.5 mM glucose. Cells were plated in 96-well white, solid bottom-plates (Greiner Bio-One) and incubated in the dark for 45 min. Then, cells were pre-treated for 5 minutes with 10 μM Forskolin to stimulate cAMP accumulation followed by incubation for 5 minutes with 5 μM Coelenterazine H (substrate for luciferase). Finally, cells were stimulated with indicated compounds. BRET signal was determined 5 minutes after ligand addition by calculating the ratio of the light emitted by mVenus (535/30 nm) over that emitted by Rluc (475/30 nm) using a Pherastar plate reader (BMG Labtech, Cary, NC). The net BRET values were obtained by subtracting the background ratio from untreated cells (cells with only Forskolin). Agonist-promoted BRET changes were expressed as a percent of the maximum response for each receptor. Fluorescence levels were also monitored to control for expression across experiments by plating cells in 96-well black solid bottom plates (Greiner Bio-One) and measuring mVenus emission (excitation at 480 nm, emission at 530 nm) for 1 second recordings.
Binding and enzyme profile screen
These experiments were performed by an outside vendor (Eurofins, France). Briefly, membrane homogenates from stable cell lines expressing each receptor/enzyme were incubated with the respective radioligand in the absence or presence of (2R,6R)-HNK or reference control compounds in a buffer. In each experiment, the respective reference compound was tested concurrently with the test compound to assess the assay reliability. Nonspecific binding was determined in the presence of a specific agonist or antagonist at the target. Following incubation, the samples were filtered rapidly under vacuum through glass fiber filters presoaked in a buffer and rinsed several times with an ice-cold buffer using a 48-sample or 96-sample cell harvester. The filters were counted for radioactivity in a scintillation counter using a scintillation cocktail.
Ex-vivo biodistribution of [3H](2R,6R)-HNK
Rats received an injection (IP) of [3H](2R,6R)-HNK (2 μCi/g), at the indicated time point and brain, blood, and tissues were collected for radiometric analyses. The brains were flash frozen in isopentane (Sigma-Aldrich) and stored at −80°C until use. The blood was collected and after clotting centrifuged (13,000 rpm, 10 min at RT) and serum was collected. The tissues were solubilized with Solvable™ (PerkinElmer) and bleached with hydrogen peroxide (Sigma-Aldrich). Serum and tissue samples were dissolved in scintillation cocktail and radioactivity counts were determined in a Beckman LS 60000TA scintillation counter (BeckmanCoulter, Indianapolis, IN, USA). The brains were flash frozen in isopentane. Then the brain tissue was sectioned (10 μm) on a cryostat (Leica, Germany) and thaw mounted onto ethanol-washed glass slides. Slides were air dried and placed in a Hypercassette™ (Amersham Biosciences) and covered with a BAS-TR2025 Storage Phosphor Screen (FujiFilm, Japan). The slides were exposed to the screen for 10–14 days and imaged using a phosphorimager (Typhoon FLA 7000; GE Healthcare).
Ex-vivo biodistribution of [14C](2R,6R)-HNK
This study was performed by Covance laboratories Inc. The detailed methods and additional results are included in Supplemental Figure 6.
[18F]-Fluorodeoxyglucose PET
This procedure was based on previous studies [9, 81, 82]. Rats were habituated to experimenter handling and the open field arena. Rats were fasted 16 h before the experiment. On the day of the experiment, rats received a continuous i.v. infusion of vehicle (buffered saline), (S)-ketamine (10 mg/kg), or HNK (10 mg/kg) over 40 min in an open field arena. Ten minutes after start of infusion, rats were injected (IP) with 13 MBq of 2-deoxy-2-[18F]-fluoro-D-glucose (FDG; Cardinal Health). After 30 min, rats were anesthetized with 1.5% isoflurane, placed on a custom-made bed of a nanoScan small animal PET/CT scanner (Mediso Medical Imaging Systems) and scanned for 20 min on a static acquisition protocol, followed by a CT scan. The PET data were reconstructed and coregistered to an MRI template using PMOD4.2 software environment (PMOD Technologies, Switzerland). The standardized uptake value ratio (SUVRWB) images were calculated using the whole brain as a reference region. All statistical parametric mapping analyses were performed using Matlab R2016 (Mathworks) and SPM12 (University College London). The ROI-based data was obtained using PMOD and the Schiffer rat VOI-atlas[83] and the statistical analyses were performed using GraphPad’s Prism 9.
Forced Swimming Test
Male wild-type mice (CD-1, 35–40 g, N=32) were tested for immobility time. The forced swim test was performed in normal light conditions (800 Lux). (2R,6R)-HNK, or vehicle (saline), was administered 24 hours prior to testing. Mice were placed in clear Plexiglass cylinders (30 cm height x 20 cm diameter) filled with 15 cm of water (23 ± 1°C) for 6 minutes, which were recorded using a digital video camera. Immobility time, defined as passive floating with no additional activity other than that necessary to keep the animal’s head above water, was subsequently scored during the last 4 minutes of the 6-minute test by a trained observer blinded to the treatment assignments.
Locomotor activity in mice
Male wild-type mice (C57BL/6J, 20–25 g, N=32) were tested for drug-induced locomotor activity. The mice (N = 8 per group) received an injection (IP, 10 ml/kg) of (2R,6R)-HNK (10, 30 or 65 mg/kg) or saline. After the injections, the mice were placed immediately in a 20 by 20 cm arena with equipped infrared beam sensors to track locomotor activity (Opto-varimex ATM3, Columbus Instruments). Locomotor activity was tracked for 30 min as infrared beam crossing and traveled distance were converted to cm and binned into 5 min time-bins.
Conditioned place preference in mice
Male wild-type mice (C57BL/6J, 20–25 g, N=32) were tested for CPP. The task consisted of 8 sessions, 1 per day, in chambers with two visually distinct sides, one with clear walls and white floor and one with checkered walls and black floor. The sides were separated by a wall with a center pass. Locomotor activity was measured by way of time spent in each chamber as well as total distance traveled (Opto-varimex ATM3, Columbus Instruments) as described above. In the first session, the mice could freely explore both sides of a conditioning box for 20 min to determine inherent side preference, designated as the Pre-Test. Using this data, the drug-paired side was assigned as their less preferred side. In an alternating fashion for 6-days, mice were injected (IP) with either saline or (2R,6R)-HNK (0, 10, 30 or 65 mg/kg) and placed in the predetermined drug/no drug side of the chamber for 30 min. In the last session (designated as Post-Test) the animals had access to both sides during 20 min, and their CPP score was quantified as: (Tpost – Tpre) / Tpre, where Tpost and Tpre are the times spent in the drug paired chamber during the post-test and pre-test sessions, respectively.
Self-administration in rats
Surgery
The procedure is based on previous studies[9]. Rats were anesthetized with isoflurane (5% induction; 2–3% maintenance). Catheters were made from Silastic tubing attached to a modified 22-gauge cannula (Plastics One, Cat# C313G-5up) and cemented to polypropylene mesh (Elko Filtering Co., Cat# 05–1000/45). The catheter was inserted into the jugular vein, and the mesh was fixed to the mid-scapular region of the rat. Rats were injected with carprofen (2.5 mg/kg, SC, Norbrook) after surgery and on the following day to relieve pain and decrease inflammation. Rats recovered for 6–8 days before drug self-administration training. During all experimental phases, catheters were flushed daily with gentamicin (4.25 mg/mL, Fresenius Kabi, Cat# 1002) dissolved in sterile saline. If we suspected catheter failure, we tested patency with a short-acting barbiturate anesthetic Brevital (methohexital sodium, 10 mg/mL in buffered saline, 0.1–0.2 ml injection volume, i.v.).
Apparatus
All rats were trained and tested in standard Med Associates self-administration chambers. Each chamber had either one or two levers located 7.5–8 cm above the grid floor. Lever presses on the active, retractable lever activated the infusion pump, whereas lever presses on the inactive, retractable lever had no consequences. Each session began with the illumination of a house light that remained on for the entire session. The active and inactive lever was inserted into the chamber 10 s after the house light was illuminated. During the self-administration sessions, a fixed-ratio-1 (FR1) reinforcement schedule (each lever press is reinforced) was used. Each infusion was paired with a 20-s white-light cue and there was a 20-s timeout before responses resulted in another infusion. At the end of the session, the house light was turned off and levers were retracted.
Self-administration training
Several cohorts of male and female rats were trained to self-administer (S)-ketamine (Toronto Research Chemicals, Cat# K165310), (2R,6R)-HNK or heroin (NIDA Pharmacy) during 3 h/day (three 1 h sessions separated by 10 min) or 1 h/day for 16 days. In all cases, (S)-ketamine or (2R,6R)-HNK were infused over 3.5 s at a dose of 0.5 mg/kg/infusion, while heroin was infused over 3.5 s at a dose of 0.05 mg/kg/infusion. Responses on the active lever during the timeout period were recorded but did not result in drug infusions. In all cases, rats were considered trained if there was a significant difference between their active and inactive lever presses and there was less than 20% variation in their infusions per session during consecutive daily sessions. The data from rats that did not meet this criterion or that did not pass the catheter patency test were excluded from the study.
Dose response
One cohort (n=8/condition) was placed on a multiple-dose schedule to observe dose-dependent (S)-ketamine or (2R,6R)-HNK self-administration according to a randomized unit dose sequence (0.125, 0.25, 0.5, or 1.0 mg/kg/infusion) for 1 h/day for 8 days. All parameters (reinforcement schedule, light cues, etc.) remained the same as training, except for drug dose. The number of infusions earned, active, and inactive lever responses were recorded for each session.
Heroin substitution
After 8 days of training, the cohort of rats trained to press for heroin had their infusions substituted for (S)-ketamine or (2R,6R)-HNK (0.5 mg/kg/infusion) (N=8 per group) for 6 days. Their responding was evaluated as described above.
Statistics
Sample sizes were chosen based on our results from previous experiments. Depending on experiment, we used paired/two-sample t-tests or single factor and multifactor ANOVAs with Sidak or Tukey post-hoc tests, taking repeated measures into account where appropriate. All statistical tests were evaluated at the P≤0.05 level.
Supplementary Material
Acknowledgements
Some Ki determinations, and agonist and/or antagonist functional data were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program, Contract # HHSN-271–2018-00023-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA. This work was supported by the NIDA Intramural Research Program (ZIA000069), the NIA, NIMH and NCATS Intramural Research Programs and by Grants RYC-2019–027371-I (JB) and PID2020–117989RA-I00 (JB) funded by MCIN/AEI /10.13039/501100011033 and by “ESF Investing in your future”. TDG was supported by NIH R01-MH107615 and RAI145211A, and VA Merit Awards 1I01BX004062 and 101BX003631–01A1.
Footnotes
Conflict of interest.
CZ is listed as a co-inventor on a patent for the use of ketamine in major depression and suicidal ideation. CZ and RM are co-inventors on a patent for the use of (2R,6R)-hydroxynorketamine, (S)-dehydronorketamine, and other stereoisomeric dehydroxylated and hydroxylated metabolites of (R,S)-ketamine metabolites in the treatment of depression and neuropathic pain. PZ, RM, PM, CJT, CAZ and TDG are co-inventors on a patent application for the use of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine in the treatment of depression, anxiety, anhedonia, suicidal ideation, and post-traumatic stress disorders, and on a patent on the crystal forms and methods of synthesis of (2R,6R)-hydroxynorketamine and (2S,6S)-hydroxynorketamine. PM and CJT are co-inventors on a patent application for the salts of (2R,6R)-hydroxynorketamine, their crystal forms, and methods of making the same and the process for synthesis and purification of (2R,6R)-hydroxynorketamine. RM, PM, CAZ, and CT have assigned their patent rights to the U.S. government but will share a percentage of any royalties that may be received by the government. PZ and TDG have assigned their patent rights to the University of Maryland Baltimore but will share a percentage of any royalties that may be received by the University of Maryland Baltimore. MM has received research funding from AstraZeneca, Redpin Therapeutics and Attune Neuroscience. All other authors declare no conflicts of interest.
References
- 1.Krystal JH, Abdallah CG, Sanacora G, Charney DS, Duman RS. Ketamine: A Paradigm Shift for Depression Research and Treatment. Neuron. 2019;101:774–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bahji A, Vazquez GH, Zarate CA. Comparative efficacy of racemic ketamine and esketamine for depression: A systematic review and meta-analysis. J Affect Disord. 2021;278:542–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schatzberg AF. A word to the wise about intranasal esketamine. Am J Psychiatry. 2019;176:422–424. [DOI] [PubMed] [Google Scholar]
- 4.Schatzberg AF. A word to the wise about ketamine. Am J Psychiatry. 2014;171:262–264. [DOI] [PubMed] [Google Scholar]
- 5.Davis L, Uezato A, Newell JM, Frazier E. Major depression and comorbid substance use disorders. Curr Opin Psychiatry. 2008;21:14–18. [DOI] [PubMed] [Google Scholar]
- 6.Compton WM, Thomas YF, Stinson FS, Grant BF. Prevalence, correlates, disability, and comorbidity of DSM-IV drug abuse and dependence in the United States: Results from the national epidemiologic survey on alcohol and related conditions. Arch Gen Psychiatry. 2007;64:566–576. [DOI] [PubMed] [Google Scholar]
- 7.Hashimoto K Rapid-acting antidepressant ketamine, its metabolites and other candidates: A historical overview and future perspective. Psychiatry Clin Neurosci. 2019;73:613–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hashimoto K Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem Pharmacol. 2020;177. [DOI] [PubMed] [Google Scholar]
- 9.Bonaventura J, Lam S, Carlton M, Boehm MA, Gomez JL, Solís O, et al. Pharmacological and behavioral divergence of ketamine enantiomers:implications for abuse liability. Mol Psychiatry. 2021. 2021. 10.1038/s41380-021-01093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jelen LA, Young AH, Stone JM. Ketamine: A tale of two enantiomers. J Psychopharmacol. 2021;35:109–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zanos P, Highland JN, Liu X, Troppoli TA, Georgiou P, Lovett J, et al. (R)-Ketamine exerts antidepressant actions partly via conversion to (2R,6R)-hydroxynorketamine, while causing adverse effects at sub-anaesthetic doses. Br J Pharmacol. 2019;176:2573–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zanos P, Moaddel R, Morris PJ, Georgiou P, Fischell J, Elmer GI, et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature. 2016;533:481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leal GC, Bandeira ID, Correia-Melo FS, Telles M, Mello RP, Vieira F, et al. Intravenous arketamine for treatment-resistant depression: open-label pilot study. Eur Arch Psychiatry Clin Neurosci. 2021;271:577–582. [DOI] [PubMed] [Google Scholar]
- 14.Lumsden EW, Troppoli TA, Myers SJ, Zanos P, Aracava Y, Kehr J, et al. Antidepressant-relevant concentrations of the ketamine metabolite (2R,6R)-hydroxynorketamine do not block NMDA receptor function. Proc Natl Acad Sci U S A. 2019;116:5160–5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, et al. (2R,6R)-hydroxynorketamine exerts mGlu2 receptordependent antidepressant actions. Proc Natl Acad Sci U S A. 2019;116:6441–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Casarotto PC, Girych M, Fred SM, Kovaleva V, Moliner R, Enkavi G, et al. Antidepressant drugs act by directly binding to TRKB neurotrophin receptors. Cell. 2021;184:1299–1313.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou D, Peng HY, Bin Lin T, Lai CY, Hsieh MC, Wen YC, et al. (2R,6R)-hydroxynorketamine rescues chronic stress-induced depression-like behavior through its actions in the midbrain periaqueductal gray. Neuropharmacology. 2018;139:1–12. [DOI] [PubMed] [Google Scholar]
- 18.Aguilar-Valles A, De Gregorio D, Matta-Camacho E, Eslamizade MJ, Khlaifia A, Skaleka A, et al. Antidepressant actions of ketamine engage cell-specific translation via eIF4E. Nature. 2021;590:315–319. [DOI] [PubMed] [Google Scholar]
- 19.Elmer GI, Tapocik JD, Mayo CL, Zanos P, Gould TD. Ketamine metabolite (2R,6R)-hydroxynorketamine reverses behavioral despair produced by adolescent trauma. Pharmacol Biochem Behav. 2020;196:172973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zanos P, Moaddel R, Morris PJ, Riggs LM, Highland JN, Georgiou P, et al. Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol Rev. 2018;70:621–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Highland JN, Zanos P, Riggs LM, Georgiou P, Clark SM, Morris PJ, et al. Hydroxynorketamines: Pharmacology and potential therapeutic applications. Pharmacol Rev. 2021;73:763–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Riggs LM, Aracava Y, Zanos P, Fischell J, Albuquerque EX, Pereira EFR, et al. (2R,6R)-hydroxynorketamine rapidly potentiates hippocampal glutamatergic transmission through a synapse-specific presynaptic mechanism. Neuropsychopharmacol 2019 452. 2019;45:426–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukumoto K, Fogaca MV., Liu RJ, Duman C, Kato T, Li XY, et al. Activity-dependent brain-derived neurotrophic factor signaling is required for the antidepressant actions of (2R,6R)-hydroxynorketamine. Proc Natl Acad Sci U S A. 2019;116:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen BK, Luna VM, LaGamma CT, Xu X, Deng SX, Suckow RF, et al. Sex-specific neurobiological actions of prophylactic (R,S)-ketamine, (2R,6R)-hydroxynorketamine, and (2S,6S)-hydroxynorketamine. Neuropsychopharmacology. 2020;45:1545–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou D Brain-derived neurotrophic factor in the ventrolateral periaqueductal gray contributes to (2R,6R)-hydroxynorketamine-mediated actions. Neuropharmacology. 2020;170. [DOI] [PubMed] [Google Scholar]
- 26.Herzog DP, Perumal N, Manicam C, Treccani G, Nadig J, Rossmanith M, et al. Longitudinal CSF proteome profiling in mice to uncover the acute and sustained mechanisms of action of rapid acting antidepressant (2R,6R)-hydroxynorketamine (HNK). Neurobiol Stress. 2021;15:100404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Highland JN, Morris PJ, Zanos P, Lovett J, Ghosh S, Wang AQ, et al. Mouse, rat, and dog bioavailability and mouse oral antidepressant efficacy of ( 2R,6R)-hydroxynorketamine. J Psychopharmacol. 2019;33:12–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pham TH, Defaix C, Xu X, Deng SX, Fabresse N, Alvarez JC, et al. Common Neurotransmission Recruited in (R,S)-Ketamine and (2R,6R)-Hydroxynorketamine-Induced Sustained Antidepressant-like Effects. Biol Psychiatry. 2018;84:e3–e6. [DOI] [PubMed] [Google Scholar]
- 29.Wulf HA, Browne CA, Zarate CA, Lucki I. Mediation of the behavioral effects of ketamine and (2R,6R)-hydroxynorketamine in mice by kappa opioid receptors. Psychopharmacology (Berl). 2022. 23 April 2022. 10.1007/S00213-022-06118-4. [DOI] [PubMed] [Google Scholar]
- 30.Zanos P, Highland JN, Stewart BW, Georgiou P, Jenne CE, Lovett J, et al. (2R,6R)-hydroxynorketamine exerts mGlu2 receptordependent antidepressant actions. Proc Natl Acad Sci U S A. 2019;116:6441–6450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aleksandrova LR, Wang YT, Phillips AG. Ketamine and its metabolite, (2R,6R)-HNK, restore hippocampal LTP and long-term spatial memory in the Wistar-Kyoto rat model of depression. Mol Brain. 2020;13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peng WH, Kan HW, Ho YC. Periaqueductal gray is required for controlling chronic stress-induced depression-like behavior. Biochem Biophys Res Commun. 2022;593:28–34. [DOI] [PubMed] [Google Scholar]
- 33.Ju L, Yang J, Zhu T, Liu P, Yang J. BDNF-TrkB signaling-mediated upregulation of Narp is involved in the antidepressant-like effects of (2R,6R)-hydroxynorketamine in a chronic restraint stress mouse model. BMC Psychiatry. 2022;22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong X, Ouyang C, Liang W, Dai C, Zhang W. (2R,6R)-Hydroxynorketamine Alleviates Electroconvulsive Shock-Induced Learning Impairment by Inhibiting Autophagy. Neuropsychiatr Dis Treat. 2021;17:297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wray NH, Schappi JM, Singh H, Senese NB, Rasenick MM. NMDAR-independent, cAMP-dependent antidepressant actions of ketamine. Mol Psychiatry. 2019;24:1833–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collo G, Cavalleri L, Chiamulera C, Merlo Pich E. (2R,6R)-Hydroxynorketamine promotes dendrite outgrowth in human inducible pluripotent stem cell-derived neurons through AMPA receptor with timing and exposure compatible with ketamine infusion pharmacokinetics in humans. Neuroreport. 2018;29:1425–1430. [DOI] [PubMed] [Google Scholar]
- 37.Yao N, Skiteva O, Zhang X, Svenningsson P, Chergui K. Ketamine and its metabolite (2R,6R)-hydroxynorketamine induce lasting alterations in glutamatergic synaptic plasticity in the mesolimbic circuit. Mol Psychiatry. 2018;23:2066–2077. [DOI] [PubMed] [Google Scholar]
- 38.Shaffer CL, Dutra JK, Tseng WC, Weber ML, Bogart LJ, Hales K, et al. Pharmacological evaluation of clinically relevant concentrations of (2R,6R)-hydroxynorketamine. Neuropharmacology. 2019;153:73–81. [DOI] [PubMed] [Google Scholar]
- 39.Kroin JS, Das V, Moric M, Buvanendran A. Efficacy of the ketamine metabolite (2R,6R)-hydroxynorketamine in mice models of pain. Reg Anesth Pain Med. 2019;44:111–117. [DOI] [PubMed] [Google Scholar]
- 40.Farmer CA, Gilbert JR, Moaddel R, George J, Adeojo L, Lovett J, et al. Ketamine metabolites, clinical response, and gamma power in a randomized, placebo-controlled, crossover trial for treatment-resistant major depression. Neuropsychopharmacology. 2020;45:1398–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grunebaum MF, Galfalvy HC, Choo TH, Parris MS, Burke AK, Suckow RF, et al. Ketamine metabolite pilot study in a suicidal depression trial. J Psychiatr Res. 2019;117:129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phase 1 Evaluation of (2R,6R)-Hydroxynorketamine - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT04711005?term=%28R%29-ketamine&draw=3&rank=40. Accessed 18 November 2021.
- 43.Hillhouse TM, Porter JH. Ketamine, but not MK-801, produces antidepressant-like effects in rats responding on a differential-reinforcement-of-low-rate operant schedule. Behav Pharmacol. 2014;25:80–91. [DOI] [PubMed] [Google Scholar]
- 44.Hillhouse TM, Porte JH, Negus SS. Comparison of antidepressant-like and abuse-related effects of phencyclidine in rats. Drug Dev Res. 2014;75:479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Henter ID, Park LT, Zarate CA. Novel Glutamatergic Modulators for the Treatment of Mood Disorders: Current Status. CNS Drugs. 2021;35:527–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Eur J Pharmacol. 2013;698:228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Morris PJ, Moaddel R, Zanos P, Moore CE, Gould T, Zarate CA, et al. Synthesis and N-Methyl- d -aspartate (NMDA) Receptor Activity of Ketamine Metabolites. Org Lett. 2017;19:4572–4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Joseph TT, Bu W, Lin W, Zoubak L, Yeliseev A, Liu R, et al. Ketamine Metabolite (2 R,6 R)-Hydroxynorketamine Interacts with μ and κ Opioid Receptors. ACS Chem Neurosci. 2021;12:1487–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams NR, Heifets BD, Blasey C, Sudheimer K, Pannu J, Pankow H, et al. Attenuation of Antidepressant Effects of Ketamine by Opioid Receptor Antagonism. Am J Psychiatry. 2018;175:1205–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Williams NR, Heifets BD, Bentzley BS, Blasey C, Sudheimer KD, Hawkins J, et al. Attenuation of antidepressant and antisuicidal effects of ketamine by opioid receptor antagonism. Mol Psychiatry. 2019;24:1779–1786. [DOI] [PubMed] [Google Scholar]
- 51.Chen X, Shu S, Bayliss DA. HCN1 channel subunits are a molecular substrate for hypnotic actions of ketamine. J Neurosci. 2009;29:600–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shepard AR, Conrow RE, Pang IH, Jacobson N, Rezwan M, Rutschmann K, et al. Identification of PDE6D as a molecular target of anecortave acetate via a methotrexate-anchored yeast three-hybrid screen. ACS Chem Biol. 2013;8:549–558. [DOI] [PubMed] [Google Scholar]
- 53.Morris PJ, Burke RD, Sharma AK, Lynch DC, Lemke-Boutcher LE, Mathew S, et al. A comparison of the pharmacokinetics and NMDAR antagonism-associated neurotoxicity of ketamine, (2R,6R)-hydroxynorketamine and MK-801. Neurotoxicol Teratol. 2021;87:106993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adams JD, Baillie TA, Trevor AJ, Castagnoli N. Studies on the biotransformation of ketamine 1—Identification of metabolites produced in vitro from rat liver microsomal preparations. Biol Mass Spectrom. 1981;8:527–538. [DOI] [PubMed] [Google Scholar]
- 55.Hijazi Y, Boulieu R. Contribution of CYP3A4, CYP2B6, and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–858. [DOI] [PubMed] [Google Scholar]
- 56.Nair A, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Phase 1 Safety Testing of SAR405838 - Full Text View - ClinicalTrials.gov. https://clinicaltrials.gov/ct2/show/NCT01636479?term=SAR405838&draw=2&rank=1. Accessed 1 October 2021.
- 58.De Luca MT, Badiani A. Ketamine self-administration in the rat: Evidence for a critical role of setting. Psychopharmacology (Berl). 2011;214:549–556. [DOI] [PubMed] [Google Scholar]
- 59.Moaddel R, Abdrakhmanova G, Kozak J, Jozwiak K, Toll L, Jimenez L, et al. Sub-anesthetic concentrations of (R,S)-ketamine metabolites inhibit acetylcholine-evoked currents in α7 nicotinic acetylcholine receptors. Eur J Pharmacol. 2013;698:228–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET, Monteggia LM. Effects of a ketamine metabolite on synaptic NMDAR function. Nature. 2017;546:E1–E3. [DOI] [PubMed] [Google Scholar]
- 61.Lilius TO, Viisanen H, Jokinen V, Niemi M, Kalso EA, Rauhala PV. Interactions of (2S,6S;2R,6R)-Hydroxynorketamine, a Secondary Metabolite of (R,S)-Ketamine, with Morphine. Basic Clin Pharmacol Toxicol. 2018;122:481–488. [DOI] [PubMed] [Google Scholar]
- 62.Li J, Chen FF, Chen XD, Zhou C. Inhibition of HCN1 channels by ketamine accounts for its antidepressant actions. J Sichuan Univ (Medical Sci Ed. 2014;45. [PubMed] [Google Scholar]
- 63.Lewis AS, Vaidya SP, Blaiss CA, Liu Z, Stoub TR, Brager DH, et al. Deletion of the hyperpolarization-activated cyclic nucleotide-gated channel auxiliary subunit TRIP8b impairs hippocampal Ih localization and function and promotes antidepressant behavior in mice. J Neurosci. 2011;31:7424–7440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim CS, Chang PY, Johnston D. Enhancement of dorsal hippocampal activity by knockdown of hcn1 channels leads to anxiolytic- and antidepressant-like behaviors. Neuron. 2012;75:503–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Han Y, Heuermann RJ, Lyman KA, Fisher D, Ismail QA, Chetkovich DM. HCN-channel dendritic targeting requires bipartite interaction with TRIP8b and regulates antidepressant-like behavioral effects. Mol Psychiatry. 2017;22:458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Knoll AT, Halladay LR, Holmes A, Levitt P. Quantitative Trait Loci and a Novel Genetic Candidate for Fear Learning. J Neurosci. 2016;36:6258–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Can A, Zanos P, Moaddel R, Kang HJ, Dossou KSS, Wainer IW, et al. Effects of Ketamine and Ketamine Metabolites on Evoked Striatal Dopamine Release, Dopamine Receptors, and Monoamine Transporters. J Pharmacol Exp Ther. 2016;359:159–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ho MF, Zhang C, Zhang L, Li H, Weinshilboum RM. Ketamine and active ketamine metabolites regulate STAT3 and the Type i interferon pathway in human microglia: Molecular mechanisms linked to the antidepressant effects of ketamine. Front Pharmacol. 2019;10:1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sommi RW, Crismon ML, Bowden CL. Fluoxetine: A Serotonin-specific, Second-generation Antidepressant. Pharmacother J Hum Pharmacol Drug Ther. 1987;7:1–14. [DOI] [PubMed] [Google Scholar]
- 70.Yamaguchi J, Toki H, Qu Y, Yang C, Koike H, Hashimoto K, et al. (2R,6R)-Hydroxynorketamine is not essential for the antidepressant actions of (R)-ketamine in mice. Neuropsychopharmacology. 2018;43:1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zarate CA, Brutsche N, Laje G, Luckenbaugh DA, Venkata SLV, Ramamoorthy A, et al. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Desta Z, Moaddel R, Ogburn ET, Xu C, Ramamoorthy A, Venkata SLV, et al. Stereoselective and regiospecific hydroxylation of ketamine and norketamine. Xenobiotica. 2012;42:1076–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Goswamee P, Rice R, Leggett E, Zhang F, Manicka S, Porter JH, et al. Effects of subanesthetic ketamine and (2R,6R) hydroxynorketamine on working memory and synaptic transmission in the nucleus reuniens in mice. Neuropharmacology. 2022;208:108965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang K, Fujita Y, Hashimoto K. Lack of metabolism in (R)-ketamine’s antidepressant actions in a chronic social defeat stress model. Sci Reports 2018 81. 2018;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hashimoto K, Shirayama Y. What Are the Causes for Discrepancies of Antidepressant Actions of (2R,6R)-Hydroxynorketamine? Biol Psychiatry. 2018;84:e7–e8. [DOI] [PubMed] [Google Scholar]
- 76.Xiong Z, Fujita Y, Zhang K, Pu Y, Chang L, Ma M, et al. Beneficial effects of (R)-ketamine, but not its metabolite (2R,6R)-hydroxynorketamine, in the depression-like phenotype, inflammatory bone markers, and bone mineral density in a chronic social defeat stress model. Behav Brain Res. 2019;368:111904. [DOI] [PubMed] [Google Scholar]
- 77.Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, et al. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat Methods. 2014;11:629–632. [DOI] [PubMed] [Google Scholar]
- 78.Mogil JS. Laboratory environmental factors and pain behavior: The relevance of unknown unknowns to reproducibility and translation. Lab Anim (NY). 2017;46:136–141. [DOI] [PubMed] [Google Scholar]
- 79.Georgiou P, Zanos P, Mou T-CM, An X, Gerhard DM, Dryanovski DI, et al. Experimenter sex modulates mouse biobehavioural and pharmacological responses. BioRxiv. 2022:2022.01.09.475572. [Google Scholar]
- 80.Jiang LI, Collins J, Davis R, Lin K-M, DeCamp D, Roach T, et al. Use of a cAMP BRET Sensor to Characterize a Novel Regulation of cAMP by the Sphingosine 1-Phosphate/G13 Pathway. J Biol Chem. 2007;282:10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cai N-S, Quiroz C, Bonaventura J, Bonifazi A, Cole TO, Purks J, et al. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J Clin Invest. 2019;129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bonaventura J, Eldridge MAG, Hu F, Gomez JL, Sanchez-Soto M, Abramyan AM, et al. High-potency ligands for DREADD imaging and activation in rodents and monkeys. Nat Commun. 2019;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schiffer WK, Mirrione MM, Biegon A, Alexoff DL, Patel V, Dewey SL. Serial microPET measures of the metabolic reaction to a microdialysis probe implant. J Neurosci Methods. 2006;155:272–284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.