

Abstract
Cannabidiol (CBD) is approved for treatment of seizures associated with two forms of epilepsy that become apparent in infancy or early childhood. To consider an adult physiologically‐based pharmacokinetic (PBPK) model for pediatric scaling, we assessed in vitro‐derived cytochrome P450 (CYP) and uridine 5′‐diphospho‐glucuronosyltransferase (UGT) enzyme contributions to CBD clearance in human. An i.v. PBPK model was constructed using CBD physicochemical properties and knowledge of disposition. The i.v. datasets were used for model building and evaluation. Oral PBPK models for CBD administered in fasted and fed states were developed using single dose oral datasets and parameters optimized from the i.v. model and evaluated with multiple dose datasets. Relative contributions of CBD metabolizing enzymes were partitioned according to in vitro studies. Clinical drug–drug interaction (DDI) studies were simulated using CBD fed state, itraconazole, fluconazole, and rifampicin PBPK models. Linear mixed effect modeling was used to estimate area under the concentration‐time curve from zero to infinity (AUC0–∞) perpetrator + CBD versus CBD alone. The i.v. and oral datasets used in model evaluation produced acceptable average fold error (AFE) of 1.28 and absolute AFE of 1.65. Relative contributions of drug‐metabolizing enzymes to CBD clearance were proposed from in vitro data: UGT1A7 4%, UGT1A9 16%, UGT2B7 10%, CYP3A4 38%, CYP2C19 21%, and CYP2C9 11%. The simulated DDI studies using the in vitro‐derived values produced AUC0–∞ treatment ratios comparable to observed: itraconazole 1.24 versus 1.07, fluconazole 1.45 versus 1.22, and rifampicin 0.49 versus 0.69. The constructed CBD PBPK models can predict adult exposures and have potential for use in pediatrics where exposure estimates are limited.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Cannabidiol (CBD) treatment for seizures in children could benefit from improved understanding of CBD exposure in pediatrics. To use physiologically‐based pharmacokinetic (PBPK) models for CBD exposure predictions in pediatrics, we need to attain metabolizing enzyme contributions to CBD clearance in adults.
WHAT QUESTION DID THIS STUDY ADDRESS?
The appropriateness of a CBD PBPK model utilizing in vitro‐determined CBD clearance enzyme contributions to predict exposures in adults was verified.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Results of simulated drug–drug interaction clinical studies using PBPK models verified use of in vitro‐derived enzyme contributions of CBD clearance in exposure predictions. The in vitro‐derived values produced area under the concentration‐time curve from zero to infinity (AUC0–∞) geometric mean treatment ratios comparable to observed.
HOW MIGHT THIS CHANGE DRUG DISCOVERY, DEVELOPMENT, AND/OR THERAPEUTICS?
Understanding metabolizing enzyme contributions to CBD clearance can be used to develop pediatric PBPK models to improve prediction of exposure estimates in this population. Our study provides an example of the bottom‐up approach, lending support to in vitro data use for in vivo estimates in drugs with sparse clinical data.
INTRODUCTION
The Cannabis sativa plant, commonly known as cannabis, is widely recognized for its pharmaceutical effects in humans. These effects have been mainly studied in a group of cannabis extracted chemicals referred to as cannabinoids. 1 The most widely known cannabinoid is delta‐9‐tetrahydrocannabinol (THC), which is mainly responsible for the psychoactive effects of cannabis. Cannabidiol (CBD), a cannabinoid that is an isomer of THC, is less known but has been gaining attention for its therapeutic potential without having any psychotoxic effects. In 2018, the US Food and Drug Administration (FDA) approved an oral solution of CBD, Epidiolex, to treat Lennox–Gastaut and Dravet syndrome. These forms of epilepsy manifest in infancy and early childhood.
A study in subjects given an i.v. administration of CBD characterized its pharmacokinetics (PKs) with an average half‐life of 24 h, a volume of distribution of 32.7 ± 8.6 L/kg, and a clearance of 74 ± 14 L/h. 2 , 3 Additionally, 16% and 33% of the total dose were found in urine (fraction unchanged not reported) and feces (12% unchanged), respectively. 3 The plasma concentration versus time profiles of these subjects also suggested no significant enterohepatic circulation nor reabsorption once CBD is cleared through the biliary route. 3 Although a significant presence of CBD in feces revealed the occurrence of biliary clearance, in vitro studies to date have not uncovered the mechanism of transport into the bile. These studies reported CBD unlikely to be a substrate of P‐glycoprotein and breast cancer resistance protein. 4 Furthermore, CBD was not a substrate for a number of human renal and hepatic uptake transporters. 4 Following single oral administration doses of 1500–6000 mg in fasted adults, CBD exhibits nonlinear kinetics with less than a proportional increase in exposure with dose. 5
CBD is a Biopharmaceutics Classification System (BCS) class II drug with high permeability and low solubility. It is highly lipophilic with an in silico predicted logP of ~6 log units. 6 CBD has a low absolute oral bioavailability of 6% in humans 7 and it increases to 14–25% with the administration of food. 8 The low bioavailability of CBD is likely due to incomplete absorption and significant presystemic elimination and 70–75% of an oral dose is estimated to be removed by hepatic metabolism before reaching systemic circulation. 7 The extensive metabolism of CBD by cytochrome P450 (CYP) and uridine 5′‐diphospho‐glucuronosyltransferase (UGT) enzymes appear to support this estimation. Yet to receive consensus in the scientific community is the identity of CYP and UGT enzymes responsible for CBD metabolism and their extent of contribution to CBD clearance. In a drug–drug interaction (DDI) study where subjects were given a THC/CBD oromucosal spray, metabolism of CBD was attributed to CYP3A4 and not CYP2C19. 9 Conversely, a DDI study with subjects administered an oral solution of CBD demonstrated the significance of CYP2C19 rather than CYP3A4 in CBD clearance. 10 Neither of these studies assessed the potential contribution of other CYP enzymes on CBD metabolism. However, in vitro studies have shown no significant contribution by CYP2C9 11 and potential contributions by UGT1A7, UGT1A9, and UGT2B7 to phase II metabolism of CBD. 12 Most recently, Beers et al. 13 performed a thorough in vitro investigation of enzyme contributions to CBD clearance. The authors showed a larger influence of CYP enzymes compared to UGT enzymes, and contributions from three CYP enzymes, CYP3A4, CYP2C19, and CYP2C9.
In consideration of the conflicting results from these existing in vivo and in vitro studies, further investigation of CBD metabolism pathways is warranted. Appropriately characterizing the role of metabolism in CBD clearance is especially important in the pediatric context. Prediction of CBD exposure in pediatrics relies on an understanding of accurate relative enzyme contributions to account for the maturation of these metabolic enzyme pathways. The active metabolite of CBD formation and exposure in pediatrics are also of interest. Over 30 CBD metabolites have been identified by Harvey and Mechoulam. 14 Of the known metabolites, 7‐hydroxy‐CBD (7‐OH‐CBD) has similar activity as CBD and exhibits a little more than half of its exposure. 8 Prediction of 7‐OH‐CBD exposure in pediatrics also requires properly partitioned enzyme contributions to CBD clearance.
To address these gaps, this current study applies physiologically‐based pharmacokinetic (PBPK) modeling. PBPK models can provide in silico estimates of drug exposure given the proper parameterization with host physiology and drug properties. 15 These models can be used to leverage existing in vitro data to confirm enzyme contributions to CBD clearance in humans. Thus, the first objective was to develop and validate an adult oral CBD PBPK model that incorporates in vitro‐determined enzyme contributions to CBD clearance. The second objective was to assess whether the in vitro estimates accurately predict enzyme contributions observed in CBD DDI studies in human. Appropriately partitioned CBD clearance by enzyme metabolism pathways demonstrated by this study would give confidence to scale the adult PBPK model for pediatric use. As a result, predicted CBD and 7‐OH‐CBD exposures in infants and children using the scaled PBPK model can address a gap where PK data are currently limited in these populations.
METHODS
Software
PBPK modeling was performed using the open‐source PBPK modeling platform, PK‐Sim version 9.1 (Open Systems Pharmacology Suite). Published PK profiles were digitized with Plot Digitizer version 2.6.8 (by Joseph Huwaldt) to obtain concentration‐time data. Analyses of the clinical DDI study simulations, including linear mixed effect modeling, were conducted using R (R Core Team, 2019, Vienna, Austria).
i.v. PBPK model construction and evaluation
An i.v. model was constructed using CBD physicochemical properties and knowledge of absorption, distribution, metabolism, and excretion (ADME), and an i.v. dataset. Prediction methods for cellular permeability (PK‐Sim Standard) and partition coefficients (Rodgers and Rowland, 16 , 17 , 18 Schmitt, 19 Berezkhovskiy, 20 and PK‐Sim Standard) were evaluated. Local optimization was carried out in PK‐Sim using a Monte Carlo approach for exploring the parameter space of influential model variables. The optimized model was evaluated using a different i.v. dataset than used for optimization. Model evaluation was quantitatively assessed by calculating the average fold error (AFE; bias) and absolute AFE (AAFE; precision) of drug plasma concentrations. Two‐fold error was deemed reasonable.
Oral PBPK model construction and evaluation
Leveraging knowledge of the systemic disposition from the i.v. model, an oral PBPK model was built for CBD administered in the fasted and fed states. Absorption‐specific parameters (specific intestinal permeability and dissolution model) were adjusted to account for absorption‐related PK nonlinearity. 21 Specific intestinal permeability was set based on the BCS class II drug status of CBD. Model evaluation was quantitatively assessed by calculating AFE and AAFE as described for the i.v. PBPK model evaluation. Additionally, the oral model predicted area under the plasma concentration‐time curve from time point 0 to the end of the dosing interval (AUC0‐τ) were compared to observed values (development and evaluation datasets) by calculating the percent difference, attained by (observed – predicted)/observed × 100%.
Population models construction and evaluation
To assess the ability of the models to reproduce PK variability following their respective i.v. and oral administrations, adult virtual populations of 100 individuals were created. Virtual populations in PK‐Sim are created based on the methods described by Willmann et al. 22 Briefly, virtual populations were built based on sex, age, and weight distributions of clinical studies used to evaluate PK variability where PK variability was therefore a function of anatomic and physiological interindividual variability for relevant model parameters.
Metabolite model construction and evaluation
An initial evaluation of the in vitro study‐informed clearance partitioning was performed using knowledge that CBD to 7‐OH‐CBD metabolite formation is mainly attributed to CYP2C19 and CYP2C9 metabolism. 13 An oral model was constructed using 7‐OH‐CBD physicochemical properties and its known ADME. The metabolite model was evaluated by assessing AFE and AAFE with published observed 7‐OH‐CBD plasma concentration‐time profiles.
Clinical DDI studies simulation
In order to evaluate clearance partitioning, completed using in vitro information, clinical DDI studies reported in Patsalos et al. 10 were simulated. CBD was the victim drug and modeled using the fed state oral CBD PBPK model. In this model, metabolic clearance was partitioned according to the relative contributions of CBD metabolizing enzymes as defined by in vitro studies. 12 , 13 Three perpetrator drugs were used in the clinical DDI studies of Patsalos et al. 10 The inhibitor itraconazole and inducer rifampicin, and inhibitor fluconazole, were included for their strong effects on CYP3A4 and CYP2C19, respectively. Compound properties of these perpetrator drugs were extracted from existing PBPK models. 23 , 24 , 25 These models were modified to incorporate the main inhibition or induction processes affecting CBD clearance and relevant formulations of the perpetrator drug. Although rifampicin and fluconazole additionally induces CYP2C19 and CYP2C9, 26 and inhibits CYP3A4 and CYP2C9, 27 , 28 respectively, only activity on CYP3A4 for both perpetrators and CYP2C19 for fluconazole were included in their PBPK models to reflect dominant metabolism pathways.
To create a population of subjects who were administered CBD in the fed state, 100 individuals were built based on the sex, age, and weight distributions reported by Patsalos et al. 10 The virtual subjects were given a single dose of 750 mg CBD oral solution after 30 min of a high‐fat breakfast for the CBD alone arm. These subjects were subsequently administered the perpetrator drug according to their respective protocol. 10 On the last day of perpetrator drug treatment, 750 mg CBD oral solution was co‐administered 1 h after itraconazole or fluconazole and within 30 min of starting a high‐fat breakfast. The rifampicin group received 750 mg CBD concomitantly and within 30 min of starting a high‐fat breakfast on day 16 of treatment. The predicted CBD AUC from zero to infinity (AUC0–∞) of each subject from CBD alone and CBD co‐administered with perpetrator were exported from PK‐Sim and into R.
For each perpetrator, a linear mixed effect model was estimated with log‐transformed AUC0–∞ values with treatment as fixed effect and subject as random effect. The estimated treatment effect was obtained, representing the log‐mean difference of AUC0–∞ of CBD and co‐administration of the perpetrator, and CBD alone. The estimate was then back‐transformed to obtain the ratio of treatment medians, also known as the AUC0 ‐ ∞ geometric mean treatment ratio. The difference between the predicted and observed treatment ratios were quantified by percent error. The percent errors were calculated by taking the absolute of the observed value subtracted by the predicted value divided by the observed value and multiplying by 100%.
RESULTS
i.v. PBPK model
The drug‐specific parameters of CBD and the values used for the i.v. model prior to optimization, termed the naïve i.v. model, are presented in Table 1. Table 2 presents the CBD dataset used to construct the i.v. model.
TABLE 1.
Physicochemical properties and ADME of CBD for i.v. model construction
| Used in naïve model | Used in optimized model | |
|---|---|---|
| Physicochemical properties | ||
| Lipophilicity (logP) | 6.1 log units (ALOGPS), 6.3 log units (ChemAxon) 6 | 2.43 log units |
| Fraction unbound in plasma (f u) | 0.06–0.07, 44 , 45 0.18 43 | 0.18 |
| Fraction excreted in urine, feces | 0.16, 0.12 (unchanged) 3 | 0.16, 0.12 (unchanged) |
| Molecular weight | 314.5 g/mol 8 | 314.5 g/mol |
| pKa | 9.7 (acid) 46 | 9.7 (acid) |
| Solubility | 0.0126 mg/mL (water, ALOGPS), 6 34 ± 7.5 μM (FaSSIF buffer), 47 40 ± 2.5 μM (FeSSIF buffer) 47 | 1.2 × 10−6 mg/ml (water, fasted state), 1.88 mg/ml (water, fed state) |
| ADME | ||
| Partition coefficient | Rodgers and Rowland, Schmitt, Berezkhovskiy, PK‐Sim Standard | Schmitt |
| Cellular permeability | PK‐Sim Standard | PK‐Sim Standard |
| CYP3A4 reference concentration, a CLspec, CL contribution | 4.32 μM, 0 1/min, 38% 13 | 4.32 μM, 0.34 1/min, 38% |
| CYP2C19 reference concentration, a CLspec, CL contribution | 0.76 μM, 0.1/min, 21% 13 | 0.76 μM, 1.06 1/min, 21% |
| CYP2C9 reference concentration, a CLspec, CL contribution | 3.84 μM, 0.1/min, 11% 13 | 3.84 μM, 0.10 1/min, 11% |
| UGT1A7 reference concentration, a CLspec, CL contribution | 1 μM, 0.1/min, 4% 12 | 1 μM, 0.21 1/min, 4% |
| UGT1A9 reference concentration, a CLspec, CL contribution | 1 μM, 0.1/min, 16% 12 | 1 μM, 5.3 1/min, 16% |
| UGT2B7 reference concentration, a CLspec, CL contribution | 1 μM, 0.1/min, 10% 12 | 1 μM, 0.37 1/min, 10% |
| Efflux biliary K m, Vmax | 0 μM, 0 μM/L/min | 2000 μM, 1742 μM/L/min |
| GFR fraction b | 1.0 | 1.0 |
Abbreviations: ADME, absorption, distribution, metabolism, and excretion; CBD, cannabidiol; CL, clearance; CLspec, specific clearance; FaSSIF, fasted state simulated intestinal fluid; FeSSIF, fed state simulated intestinal fluid; GFR, glomerular filtration rate; Vmax, maximal rate of metabolism.
Reference concentrations are 100% for the organ with the most abundant enzyme (e.g., liver); it is a fraction thereof for all other relevant organs.
GFR fraction of 1.0 indicates renal clearance calculated as GFR*fu with no reabsorption or tubular secretion.
TABLE 2.
Pharmacokinetic datasets for i.v. and oral model construction and evaluation
| Study | Dose and administration | Cohort | N | Age (years) a | Weight (kg) a |
|---|---|---|---|---|---|
| i.v. PBPK model construction | |||||
| Ohlsson et al. 48 , c | 20 mg i.v. infusion over 2 min | European men | 5 | 26.4 ± 5.7 | 78.6 ± 10.9 |
| i.v. PBPK model evaluation | |||||
| Wall et al. 3 | 20 mg i.v. bolus | White American men | 5 | 25.0 b | 76.9 b |
| Oral PBPK model construction – fasted state | |||||
| Tayo et al. 49 , d | 200 mg oral solution | European men (63%) and women | 8 | 60.4 ± 11.5 | 75.0 b |
| Taylor et al. 45 , d | 200 mg oral solution | European men (50%) and women | 8 | 55.0 ± 10.0 | 89.4 ± 11.6 |
| Crockett et al. 35 , c | 750 mg oral solution | European men (41%) and women | 29 | 36.6 ± 14.3 | 74.7 ± 11.0 |
| Schoedel et al. 50 , c | 750 mg oral solution | American men (72%) and women | 41 | 37.7 ± 8.9 | 81.0 b |
| Center for Drug Evaluation and Research 51 , d | 750 mg oral solution | American men (44%) and women | 49 | 33.0 | 66.0 b |
| Schoedel et al. 50 , c | 1500 mg oral solution | American men (72%) and women | 41 | 37.7 ± 8.9 | 81.0 b |
| Taylor et al. 5 , d | 1500 mg oral solution | European men (17%) and women | 6 | 26.0 ± 3.2 | 62.0 b |
| Taylor et al. 5 , d | 3000 mg oral solution | European men (50%) and women | 6 | 25.0 ± 4.7 | 70.0 b |
| Schoedel et al. 50 , c | 4500 mg oral solution | American men (72%) and women | 41 | 37.7 ± 8.9 | 81.0 b |
| Taylor et al. 5 , d | 4500 mg oral solution | European women | 6 | 25.8 ± 7.9 | 57.0 b |
| Center for Drug Evaluation and Research 51 , d | 4500 mg oral solution | American men (44%) and women | 48 | 33 | 66.0 b |
| Taylor et al. 5 , d | 6000 mg oral solution | European men (33%) and women | 6 | 22.8 ± 3.2 | 62.0 b |
| Oral PBPK model construction – fed state | |||||
| Crockett et al. 35 , c | 750 mg oral solution with high‐fat meal | European men (60%) and women | 15 | 41.1 ± 12.4 | 71.6 ± 13.0 |
| Taylor et al. 5 | 1500 mg oral solution with high‐fat meal | European men (33%) and women | 12 | 25.1 ± 6.2 | 59.0 b |
| Oral PBPK model evaluation – fasted and fed state | |||||
| Morrison et al. 36 , e | 750 mg oral solution b.i.d. fed state | European men (60%) and women | 15 | 27.7 ± 8.2 | 74.8 ± 13.0 |
| Morrison et al. 36 e | 750 mg oral solution b.i.d. fed state | European men (67%) and women | 12 | 35.1 ± 12.9 | 81.1 ± 14.4 |
| Morrison et al. 36 ,e | 750 mg oral solution b.i.d. fed state | European men (64%) and women | 14 | 29.9 ± 10.5 | 74.5 ± 12.3 |
| Taylor et al. 5 | 750 mg oral solution b.i.d. fasted and fed states | European men (22%) and women | 9 | 28.6 ± 8.5 | 59.0 b |
| Taylor et al. 5 | 1500 mg oral solution b.i.d. fasted and fed states | European men (56%) and women | 9 | 25.1 ± 4.8 | 69.0 b |
Abbreviation: PBPK, physiologically‐based pharmacokinetic.
Mean and SD (if reported).
Estimated values are presented since demographics were not reported by the study.
Studies were used population models development.
Studies were also used for 7‐OH‐CBD oral model evaluation.
Data are from separate clobazam, stiripentol, and valproate drug‐drug interaction studies.
A naïve model was set up for a mean male individual weighing 78.6 kg. Of the four partition coefficient calculation methods (Table 1), Schmitt was selected based on visual model performance for curve shape. Lipophilicity, as a scalar for tissue‐to‐plasma partition coefficients (K p), was set 2.5‐fold lower than predicted in silico to be in alignment with experimental values. The experimental values were attained from a study by Gronewold and Skopp, 29 which measured CBD concentrations in body fluids and tissues from human cadavers. Gronewold and Skopp 29 performed single dose rather than continuous infusion studies, thus potentially underestimating actual steady‐state K p values. Nevertheless, the study by Gronewold and Skopp 29 justified that volume of distribution was well captured with a lipophilicity of 2.43 resulting in predicted K p values (median: 3.7; range: 0.18, 43.4), which were similar to K p values measured in humans (median: 3.3; range: 1.8, 21.3). 29
Clearance was partitioned as biliary and metabolic. The i.v. plasma profiles from the study by Wall et al. 3 did not present a distinct second peak. Therefore, absence of significant enterohepatic circulation was suggested and therefore not modeled. The study also suggested significant biliary clearance occurring. To model this process, a transporter was added to the apical side of the liver and its properties optimized to reach a fraction excreted unchanged in feces of 12%. Glomerular filtration rate times fraction unbound in plasma accounted for renal clearance.
CYPs and UGTs contributed 70% and 30% of total metabolic clearance, respectively. 13 Clearance was further partitioned according to the relative contributions of metabolizing enzymes, CYP3A4, CYP2C19, CYP2C9, UGT1A7, UGT1A9, and UGT2B7, based on in vitro studies by Beers et al. 13 and Mazur et al. 12 (Table 1). The organ‐specific expression of UGT1A7 was informed by the Human Protein Atlas (https://www.proteinatlas.org/) and Strassburg et al. 30 The remaining enzymes were populated by the PK‐Sim expression database reverse transcription polymerase chain reaction profiles. 31 , 32 , 33
The optimized values for the i.v. PBPK model are presented in Table 1. The outcomes of i.v. model optimization and evaluation are presented in Figure S1 and Figure 1, respectively.
FIGURE 1.
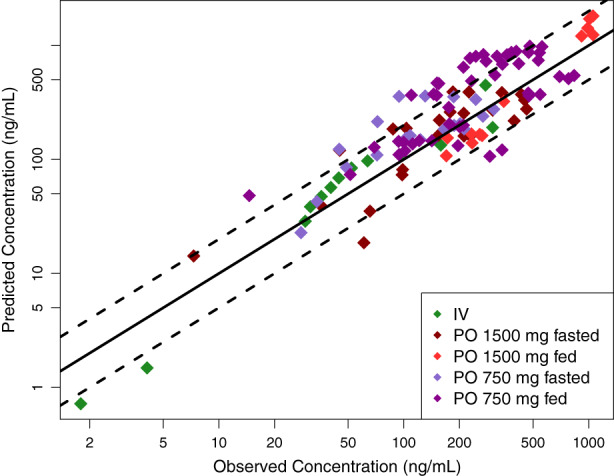
The i.v. and oral model predicted vs observed mean CBD plasma concentrations for evaluation (R 2 = 0.68). The solid and dashed lines represent the identity line and two‐fold differences, respectively. CBD, cannabidiol.
Oral PBPK models
The systemic parameters developed for the mean male i.v. PBPK model were used for the model defining oral administration. Drug/formulation‐specific parameters that were defined in the oral model included CBD solubility, formulation dissolution, and specific intestinal permeability. The single dose administration PK datasets used for model building are shown in Table 2. CBD as an oral solution is virtually insoluble in water. 34 As a result, solubility was expected to rate limit absorption and therefore specific intestinal permeability was set to a high and non‐rate‐limiting value. The expected dissolution‐precipitation‐dissolution cycle was modeled assuming overall dissolution followed a Weibull function. To account for absorption‐related PK nonlinearity, 21 dissolution shape and half‐time were optimized for each dose, and solubility was optimized globally. To account for the significant change in CBD bioavailability due to the food effect, 5 , 35 dissolution parameters and solubility were optimized using studies with fed state subjects (Table 2). The results of the optimization of oral absorption parameters in both the fasted and fed states are shown in Table 3.
TABLE 3.
Oral absorption parameters for fasted and fed state oral models construction
| Used in naïve model | Used in optimized model | |||
|---|---|---|---|---|
| Fasted state | Fed state | Fasted state | Fed state | |
| Dissolution half‐time, shape | ||||
| 200 mg | 10 min, 0.92 | NA | 790 min, 1.0 | NA |
| 750 mg | 10 min, 0.92 | NA | 460 min, 2.2 | NA |
| 1500 mg | 10 min, 0.92 | 10 min, 0.92 | 350 min, 3.3 | 95 min, 1.6 |
| 3000 mg | 10 min, 0.92 | 10 min, 0.92 | 650 min, 2.4 | 120 min, 2.0 |
| 4500 mg | 10 min, 0.92 | NA | 350 min, 3.3 | NA |
| 6000 mg | 10 min, 0.92 | NA | 840 min, 2.4 | NA |
| Water solubility | 0.0107 mg/ml6 | 1.20 × 10−6 mg/ml | 1.20 × 10−6 mg/ml | 1.88 mg/ml |
| Specific intestinal permeability | 2.47 × 10−5 cm/min | 2.47 × 10−5 cm/min | 1 cm/min | 1 cm/min |
Note: NA, no available fed state data to inform this parameter.
The developed oral PBPK models in the fasted and fed states were evaluated in five datasets with multiple dosing presented in Table 2. The study by Morrison et al. 36 was used to evaluate the fed state model with 750 mg oral solution administered every other day. In the studies by Taylor et al., 5 an oral solution dose was administered in the morning after 10 h of fasting and administered again in the evening 2 h after the end of a meal. This regimen was repeated for a total of 7 days. Model performance for the evaluation is presented in Figure 1 and Figures S2‐S4. The i.v. and oral evaluations produced acceptable AFE and AAFE values of 1.28 and 1.65, respectively. The oral model predicted AUC0‐τ compared to those observed were well captured, resulting in a mean [range] percent difference of −8.54% [−90 to 28%].
Metabolite model
The oral metabolite model was parameterized with 7‐OH‐CBD physicochemical and ADME properties presented in Table 4. Uncertain parameters, such as fraction unbound in plasma and partition coefficient prediction method, were informed by the optimized CBD model. As with CBD, lipophilicity was set 2.5‐fold lower than predicted in silico. CBD single dose administration studies with measured 7‐OH‐CBD plasma concentrations used to evaluate the metabolite model are presented in Table 2. The results of metabolite model evaluation are shown in Figure 2. The sum of CBD intrinsic clearance by CYP2C19 and CYP2C9 was used to define 7‐OH‐CBD intrinsic clearance. Further increases in 7‐OH‐CBD clearance retained the parallel elimination rates of 7‐OH‐CBD and CBD while reducing the first phase of the former compound. Therefore, the set 7‐OH‐CBD intrinsic clearance value supported a formation rate limited process and led to a reasonable representation of the observed data. Acceptable AFE and AAFE values of 1.09 and 1.71, respectively, were produced.
TABLE 4.
Physicochemical properties and ADME of 7‐OH‐CBD for model construction
| Used in naïve model | Used in optimized model | |
|---|---|---|
| Physicochemical properties | ||
| Lipophilicity (logP) | 5.3 log units (XLogP3 3.0), 52 5.0 log units (ChemAxon) | 1.94 log units a |
| Fraction unbound in plasma (f u) | 0.06–0.07, 44 , 45 0.18 43 | 0.18 a |
| Molecular weight | 330.5 g/mol 52 | 330.5 g/mol |
| pKa | 9.7 (acid) 46 | 9.7 (acid) a |
| Solubility | 4 × 10−3 mg/ml (water, ChemAxon), 0.26 mg/ml (water, ChemSpider) 52 | 4 × 10−3 mg/ml |
| ADME | ||
| Partition coefficient | Schmitt | Schmitt a |
| Cellular permeability | PK‐Sim Standard | PK‐Sim Standard a |
| P450 concentration, CLspec b | 1 μM, 0 1/min | 1 μM, 1.16 1/min |
| Efflux biliary K m, V max | 0 μM, 0 μM/L/min | 2000 μM, 1742 μM/L/min a |
| GFR fraction c | 1.0 | 1.0 a |
Abbreviations: ADME, absorption, distribution, metabolism, and excretion; CBD, cannabidiol; CLspec, specific clearance; FaSSIF, fasted state simulated intestinal fluid; FeSSIF, fed state simulated intestinal fluid; GFR, glomerular filtration rate; Vmax, maximal rate of metabolism.
Uncertain parameters were assumed from CBD physicochemical properties and ADME processes.
The identity of enzymes involved in 7‐OH‐CBD metabolism are yet to be elucidated.
GFR fraction of 1.0 indicates renal clearance calculated as GFR*fu with no reabsorption or tubular secretion.
FIGURE 2.
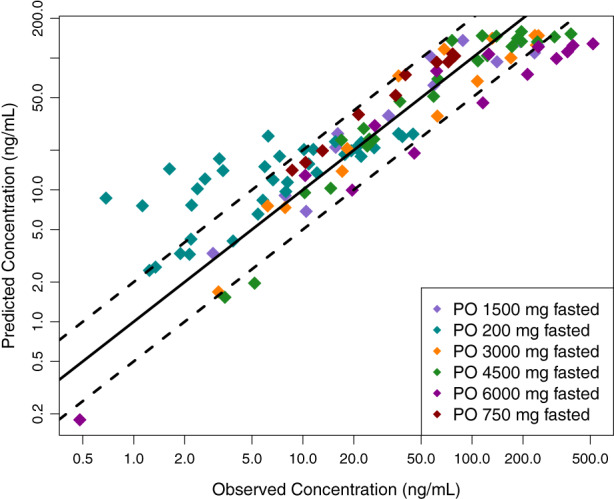
Oral model predicted vs observed mean 7‐OH‐CBD plasma concentration for evaluation (R 2 = 0.65). The solid and dashed lines represent the identity line and two‐fold differences, respectively. CBD, cannabidiol.
Population models
To assess the accuracy of predicted PK variability, virtual populations were constructed as in the respective clinical study presented in Table 2. Whereas anatomy and physiology variability are captured in PK‐Sim, the interindividual variability in enzyme concentration is not included for every enzyme and must therefore be user‐defined. User‐defined proteins included UGT1A7 and UGT1A9. The reference concentrations of these enzymes were previously defined in Table 1 and represent the most abundant organ concentration of the enzyme with all other relevant organs as a fraction of the concentration. For UGT1A7 and UGT1A9, a geometric SD of 1.5 was applied, based on the following assessment.
The variability of UGT1A9 concentration was based on the in vitro studies by Badée et al. 37 and Miyagi et al. 38 In these studies, enzyme activity was attained by measuring UGT1A9 glucuronidation in human liver microsomes of 0–78 year old subjects. Activity was not found to be age dependent, and thus a linear function with a geometric mean of 1 and geometric SD of 1.5 was used to describe UGT1A9 activity. To address the lack of variability information on UGT1A7, variability was assumed to be similar to UGT1A9.
Observed variability was well‐captured with the i.v. population model (Figure S5) but tended to be underestimated following oral administration. This finding reflected the status of CBD as a poorly soluble and highly permeable molecule with low bioavailability. Therefore, a SD of 50% of the mean dissolution half‐time was added per dose and the final oral population models are presented in Figures S6–S10.
DDI simulations
The PBPK model for fluconazole 24 did not include inhibition on CYP2C19, a main effect of the perpetrator drug. 26 A competitive inhibition process (Ki = 2.1 μM) 39 was added and the model was qualified in humans 40 with results presented in Figure S11. Furthermore, the rifampicin PBPK model 25 was updated to include a capsule formulation using typical BCS class II properties of 200 min dissolution half‐time for a Weibull function. The model was evaluated using data from subjects administered 600 mg of rifampicin capsule daily (Figure S12). 41 The fed state oral CBD, itraconazole, and updated fluconazole and rifampicin PBPK models produced treatment ratios presented in Table 5. The calculated percent error comparing the predicted and observed AUC0–∞ geometric mean treatment ratios were 16%, 19%, and 29% for itraconazole, fluconazole, and rifampicin, respectively.
TABLE 5.
Model‐predicted and observed AUC0–∞ geometric mean treatment ratios
| Study | Itraconazole | Fluconazole | Rifampicin |
|---|---|---|---|
| Patsalos et al. 10 | 1.07 | 1.22 | 0.69 |
| This study | 1.24 | 1.45 | 0.49 |
Note: AUC0–∞, area under the concentration‐time curve from zero to infinity.
DISCUSSION
This study assessed the appropriateness of using in vitro estimates of enzyme contributions to CBD clearance for predicting exposures in humans. The developed i.v. and oral CBD PBPK models demonstrated acceptable AFE and AAFE values in model evaluation. Although clinical data on CBD metabolism pathways are sparse, we were able to propose the enzymes involved and their contributions to CBD clearance through in vitro studies. According to our assessment of the in vitro data, CYPs provided a 2.3‐fold greater contribution than UGTs, and the individual enzymes involved by order of decreasing contributions included: CYP3A4, CYP2C19, UGT1A9, CYP2C9, UGT2B7, and UGT1A7. Finally, model‐predicted treatment ratios were reasonable with a percent error ranging from 16–30%. Thus, the partitioning of clearance was deemed reasonable, reflecting the observed degree of influence of the perpetrator drugs on CBD clearance.
The methods used in this study present several advantages. To our knowledge, only two other groups have reported developed CBD PBPK models to date. 4 , 42 The first model was constructed by the sponsors for the main purposes of predicting DDIs with CBD as a perpetrator in adult and pediatric populations (≥2 years old). However, a limitation to the model was that the major metabolic enzyme of CBD, CYP2C19, was not incorporated. 4 The second model was developed by Qian and Markowitz, 42 with one of the aims to study interactions between CBD and methylphenidate. The model mainly differs from the model in this current study by incorporating a lower fraction unbound value and only the CYP class of enzymes. Our study provides an improved adult PBPK model that incorporates all potentially clinically important metabolic pathways of CBD via both CYP and UGT enzyme classes. Additionally, our work uses an updated fraction unbound value determined by the three‐solvent extraction technique, which provides higher cannabinoid recovery and assay reproducibility. 43 Moreover, our study provides a robust evaluation step by validating the models developed with single dose administration data in subjects who were administered multiple dosing. The limited number of published CBD PBPK models may be related to the difficulty in modeling this BCS class II drug. Our modeling methods addressed a highly permeable and poorly soluble compound through increasing permeability to a non‐rate‐limiting value, setting timing of dissolution changes as a function of dose to capture nonlinear absorption, and modeling the fed state to confirm the sensitivity of dissolution half‐time and solubility in affecting CBD bioavailability. Although we used these methods, the variability in absorption was still underestimated in most model predictions. With the inability for all oral PK variability to be adequately captured, variability of treatment effect ratios (i.e., as 90% confidence intervals) were not examined in this study. Nevertheless, the assessment with treatment effect ratios was sufficient to verify the appropriateness of in vitro‐determined metabolic enzyme contributions to CBD clearance. Furthermore, based on the use cases of the adult CBD PBPK model to predict exposures and subsequently scale to pediatric populations for exposure estimations, the model was deemed appropriate with AUC0–∞ being similar to observed. Ultimately, with absorption, distribution, and excretion parameters solidified, we had increased confidence that most of the model uncertainty belonged to the partitioning of clearance through metabolism.
In relation to previous work, Jiang et al 11 also attempted to classify the relative CBD clearance contributions by CYP enzymes. In their studies, the effect of anti‐CYP3A4 antibody on 6α‐OH‐CBD, 6β‐OH‐CBD, and 4′´‐OH‐CBD metabolite hydrolase activities were examined in human liver microsomes. Results showed that all activities measured were inhibited to ~50% of the control level when the antibody was added. Due to the potential activity of CYP3A4 and CYP2C19 in forming these metabolites, it may be inferred that each enzyme produced even contributions of CBD clearance in the liver. Additionally, chemical inhibition studies and correlation analysis by Jiang et al. 11 suggested CYP2C19 to play a role in 7‐OH‐CBD metabolite formation. These results by Jiang et al. 11 provided an alternative to the percent contributions determined through the studies by Beers et al. 13 and Mazur et al. 12 Our study used the results of the latter publications to explore the contributions of a greater range of CYP and UGT enzymes.
In vitro studies were used to determine the relative enzyme CBD clearance contributions. There are limitations to use of these data particularly for the partitioning of clearance by CYP and UGT enzymes, and among UGT enzymes. For distinguishing the contributions provided by each enzyme superfamily, we used results from substrate‐depletion studies in human liver microsomes. 13 Specifically, we based the relative contributions from their depletion rate constants. To further support this decision, an in vitro study measuring total metabolite formation by each superfamily to derive intrinsic clearance values and their estimated percent contributions would be necessary. Moreover, specific UGT enzyme contributions were derived from an in vitro study that measured UGT isoform activity from incubated microsomal protein that contained recombinant UGT and human liver microsomes. 12 Activity toward CBD was limited and UGT1A9, UGT2B7, and UGT2B17 only formed minimal amounts of glucuronidated CBD product. 12 Further substrate depletion and metabolite formation studies would be needed to confirm the relative contributions of UGT enzymes using human liver microsomes. These two main sources of in vitro‐derived enzyme partitioning of clearance uncertainties may have affected the 16–30% difference seen across the three perpetrator drugs. A sensitivity analysis revealed that the CYP versus UGT enzymes relative contributions to total CBD clearance would be influential to the AUC0–∞ geometric mean treatment ratio predictions. As examples, when CYPs provide nine‐fold and 0.43‐fold difference in contribution compared to UGTs, predicted treatment ratios are 1.36 and 1.08 with itraconazole, 1.74 and 1.13 with fluconazole, and 0.44 and 0.69 with rifampicin, respectively. Consequently, our study may have incorporated an overestimation of activity from CYP enzymes and thus greater magnitudes of treatment ratios than expected. It may also be possible that the contribution by CYP3A4 was overestimated. The potential overpartitioning of CYP3A4 is relevant for fluconazole and rifampicin because they are known to act on several enzymes that also metabolize CBD, whereas itraconazole is thought to mainly inhibit CYP3A4. Therefore, decreasing the relative contribution of CYP3A4 by increasing the contributions of one or more of CYP2C19, CYP2C9, or UGT enzymes may result in a decreased percent error. Due to the large uncertainty on the exact source of percent error deviation, we based our study on in vitro data. Further exploration of potential clinical DDIs in CYP2C9, UGT1A7, UGT1A9, and UGT2B7 would assist in assessing their roles on CBD exposure in human participants.
We attempted to simulate the clinical DDI study by Stott et al., 9 where THC/CBD was administered as an oromucosal spray and subsequently with ketoconazole or omeprazole. Because ketoconazole and omeprazole as perpetrator drugs mainly target CYP3A4 and CYP2C19, respectively, uncovering similar model‐predicted and observed treatment effect ratios may reinforce our study findings. However, there was substantial uncertainty in the dose directly absorbed by the nasal versus oral passages, which was an essential input for a drug with very low bioavailability (F = 6%). As a result, we were unable to draw conclusions from the simulation exercise using the study by Stott et al. 9 Despite this limitation, contributions to CBD clearance by CYP2C19 and CYP2C9 were adequately supported by our developed metabolite model presented in this study.
Finally, a limitation of the model is use of describing the dissolution‐precipitation process with Weibull equations that were dose‐specific. Therefore, the absorption model was fit to describe the data and we were able to meet the study objective to verify in vitro‐derived enzyme contributions to CBD clearance for exposure predictions in adults. However, as an outcome of fitting our absorption model, extrapolating to a new population such as pediatrics will require careful consideration.
Our study provides a basis for an understanding of the metabolizing enzymes involved and their potential relative contributions to CBD clearance. The resulting increased confidence in the relative enzyme contributions to CBD clearance allows for the investigation of PK predictions in the pediatric population. Currently, CBD and active metabolite PK data are limited in pediatrics and thus exposure predictions in this populations would be valuable.
AUTHOR CONTRIBUTIONS
C.H.T.Y., J.L.B., K.D.J., and A.N.E. wrote the manuscript. C.H.T.Y. and A.N.E. designed the research. C.H.T.Y. and J.L.B. performed the research. C.H.T.Y. analyzed the data.
FUNDING INFORMATION
C.H.T.Y. is supported by the Canadian Institutes of Health Research (CIHR) Frederick Banting and Charles Best Canada Graduate Scholarships Doctoral Award (CGS‐D), a Canada Graduate Scholarship to Honor Nelson Mandela; Award Number: DF2‐171445. J.L.B. is supported by the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH) under award T32GM086330.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
Supporting information
Appendix S1:
ACKNOWLEDGMENTS
The authors thank Dagmar Hadjucek (PhD, University of Waterloo) for guidance and assistance in our statistical analyses.
Yeung CHT, Beers JL, Jackson KD, Edginton AN. Verifying in vitro‐determined enzyme contributions to cannabidiol clearance for exposure predictions in human through physiologically‐based pharmacokinetic modeling. CPT Pharmacometrics Syst Pharmacol. 2023;12:320‐332. doi: 10.1002/psp4.12908
REFERENCES
- 1. Hanuš LO, Meyer SM, Muñoz E, Taglialatela‐Scafati O, Appendino G. Phytocannabinoids: a unified critical inventory. Nat Prod Rep. 2016;33:1357‐1392. [DOI] [PubMed] [Google Scholar]
- 2. Millar SA, Stone NL, Yates AS, O'Sullivan SE. A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharmacol. 2018;9:1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wall ME, Brine DR, Perez‐Reyes M. Metabolism of Cannabinoids in Man. Raven Press; 1976. [Google Scholar]
- 4. Center for Drug Evaluation and Research . Clinical Pharmacology and Biopharmaceutics Review(s): Epidiolex. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000ClinPharmR.pdf. [Accessed March 31, 2022].
- 5. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double‐blind, placebo‐controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32:1053‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cannabidiol [Internet]. Available from: https://go.drugbank.com/drugs/DB09061. [Accessed October 8, 2021].
- 7. Perucca E, Bialer M. Critical aspects affecting cannabidiol oral bioavailability and metabolic elimination, and related clinical implications. CNS Drugs. 2020;34:795‐800. [DOI] [PubMed] [Google Scholar]
- 8. European Medicines Agency. Assessment Report: Epidyolex . Committee for Medicinal Products for Human Use (CHMP). 2019. Available from: https://www.ema.europa.eu/en/documents/assessment‐report/epidyolex‐epar‐public‐assessment‐report_en.pdf. [Accessed March 31, 2022].
- 9. Stott C, White L, Wright S, Wilbraham D, Guy G. A phase I, open‐label, randomized, crossover study in three parallel groups to evaluate the effect of rifampicin, ketoconazole, and omeprazole on the pharmacokinetics of THC/CBD oromucosal spray in healthy volunteers. Springerplus. 2013;2:236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Patsalos PN, Szaflarski JP, Gidal B, VanLandingham K, Critchley D, Morrison G. Clinical implications of trials investigating drug‐drug interactions between cannabidiol and enzyme inducers or inhibitors or common antiseizure drugs. Epilepsia. 2020;61:1854‐1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89:165‐170. [DOI] [PubMed] [Google Scholar]
- 12. Mazur A, Lichti CF, Prather PL, et al. Characterization of human hepatic and extrahepatic UDP‐glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37:1496‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beers JL, Fu D, Jackson KD. Cytochrome P450‐catalyzed metabolism of cannabidiol to the active metabolite 7‐hydroxy‐cannabidiol. Drug Metab Dispos. 2021;49:882‐891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harvey DJ, Mechoulam R. Metabolites of cannabidiol identified in human urine. Xenobiotica. 1990;20:303‐320. [DOI] [PubMed] [Google Scholar]
- 15. Maharaj AR, Barrett JS, Edginton AN. A workflow example of PBPK modeling to support pediatric research and development: case study with lorazepam. AAPS J. 2013;15:455‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rodgers T, Leahy D, Rowland M. Physiologically based pharmacokinetic modeling 1: predicting the tissue distribution of moderate‐to‐strong bases. J Pharm Sci. 2005;94:1259‐1276. [DOI] [PubMed] [Google Scholar]
- 17. Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95:1238‐1257. [DOI] [PubMed] [Google Scholar]
- 18. Rodgers T, Rowland M. Mechanistic approaches to volume of distribution predictions: understanding the processes. Pharm Res. 2007;24:918‐933. [DOI] [PubMed] [Google Scholar]
- 19. Schmitt W. General approach for the calculation of tissue to plasma partition coefficients. Toxicol In Vitro. 2008;22:457‐467. [DOI] [PubMed] [Google Scholar]
- 20. Berezhkovskiy LM. Volume of distribution at steady state for a linear pharmacokinetic system with peripheral elimination. J Pharm Sci. 2004;93:1628‐1640. [DOI] [PubMed] [Google Scholar]
- 21. Lim SY, Sharan S, Woo S. Model‐based analysis of cannabidiol dose‐exposure relationship and bioavailability. Pharmacotherapy. 2020;40:291‐300. [DOI] [PubMed] [Google Scholar]
- 22. Willmann S, Höhn K, Edginton A, et al. Development of a physiology‐based whole‐body population model for assessing the influence of individual variability on the pharmacokinetics of drugs. J Pharmacokinet Pharmacodyn. 2007;34:401‐431. [DOI] [PubMed] [Google Scholar]
- 23. Open Systems Pharmacology ‐ PBPK Model Library ‐ Itraconazole [Internet] . 2020. Available from: https://github.com/Open‐Systems‐Pharmacology/OSP‐PBPK‐Model‐Library/tree/v9.1.1/Itraconazole. [Accessed October 24, 2021].
- 24. Open Systems Pharmacology ‐ PBPK Model Library ‐ Fluconazole [Internet] . 2021. Available from: https://github.com/Open‐Systems‐Pharmacology/Fluconazole‐Model. [Accessed October 13, 2021].
- 25. Open Systems Pharmacology ‐ PBPK Model Library ‐ Rifampicin [Internet] . 2021. Available from: https://github.com/Open‐Systems‐Pharmacology/OSP‐PBPK‐Model‐Library/tree/master/Rifampicin. [Accessed February 16, 2022].
- 26. U.S. Food and Drug Administration . Drug Development and Drug Interactions: Table of Substrates, Inhibitors and Inducers. 03/10/2020. Available from: https://www.fda.gov/drugs/drug‐interactions‐labeling/drug‐development‐and‐drug‐interactions‐table‐substrates‐inhibitors‐and‐inducers. [Accessed March 31, 2022].
- 27. Olkkola KT, Ahonen J, Neuvonen PJ. The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82:511‐516. [DOI] [PubMed] [Google Scholar]
- 28. Kunze KL, Wienkers LC, Thummel KE, Trager WF. Warfarin‐fluconazole. I. Inhibition of the human cytochrome P450‐dependent metabolism of warfarin by fluconazole: in vitro studies. Drug Metab Dispos. 1996;24:414‐421. [PubMed] [Google Scholar]
- 29. Gronewold A, Skopp G. A preliminary investigation on the distribution of cannabinoids in man. Forensic Sci Int. 2011;210:e7‐e11. [DOI] [PubMed] [Google Scholar]
- 30. Strassburg CP, Manns MP, Tukey RH. Expression of the UDP‐glucuronosyltransferase 1A locus in human colon. Identification and characterization of the novel extrahepatic UGT1A8. J Biol Chem. 1998;273:8719‐8726. [DOI] [PubMed] [Google Scholar]
- 31. Nishimura M, Yaguti H, Yoshitsugu H, Naito S, Satoh T. Tissue distribution of mRNA expression of human cytochrome P450 isoforms assessed by high‐sensitivity real‐time reverse transcription PCR. Yakugaku Zasshi. 2003;123:369‐375. [DOI] [PubMed] [Google Scholar]
- 32. Nishimura M, Naito S. Tissue‐specific mRNA expression profiles of human ATP‐binding cassette and solute carrier transporter superfamilies. Drug Metab Pharmacokinet. 2005;20:452‐477. [DOI] [PubMed] [Google Scholar]
- 33. Nishimura M, Naito S. Tissue‐specific mRNA expression profiles of human phase I metabolizing enzymes except for cytochrome P450 and phase II metabolizing enzymes. Drug Metab Pharmacokinet. 2006;21:357‐374. [DOI] [PubMed] [Google Scholar]
- 34. Greenwich Biosciences Inc . EPIDIOLEX (Cannabidiol) Oral Solution Label; 6/2018. Report No.: 4282447.
- 35. Crockett J, Critchley D, Tayo B, Berwaerts J, Morrison G. A phase 1, randomized, pharmacokinetic trial of the effect of different meal compositions, whole milk, and alcohol on cannabidiol exposure and safety in healthy subjects. Epilepsia. 2020;61:267‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Morrison G, Crockett J, Blakey G, Sommerville K. A phase 1, open‐label, pharmacokinetic trial to investigate possible drug‐drug interactions between clobazam, stiripentol, or valproate and cannabidiol in healthy subjects. Clin Pharmacol Drug Dev. 2019;8:1009‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Badée J, Qiu N, Collier AC, et al. Characterization of the ontogeny of hepatic UDP‐glucuronosyltransferase enzymes based on glucuronidation activity measured in human liver microsomes. J Clin Pharmacol. 2019;59(Suppl 1):S42‐s55. [DOI] [PubMed] [Google Scholar]
- 38. Miyagi SJ, Milne AM, Coughtrie MW, Collier AC. Neonatal development of hepatic UGT1A9: implications of pediatric pharmacokinetics. Drug Metab Dispos. 2012;40:1321‐1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wienkers LC, Wurden CJ, Storch E, Kunze KL, Rettie AE, Trager WF. Formation of (R)‐8‐hydroxywarfarin in human liver microsomes. A new metabolic marker for the (S)‐mephenytoin hydroxylase, P4502C19. Drug Metab Dispos. 1996;24:610‐614. [PubMed] [Google Scholar]
- 40. Kang BC, Yang CQ, Cho HK, Suh OK, Shin WG. Influence of fluconazole on the pharmacokinetics of omeprazole in healthy volunteers. Biopharm Drug Dispos. 2002;23:77‐81. [DOI] [PubMed] [Google Scholar]
- 41. Aarnoutse RE, Kibiki GS, Reither K, et al. Pharmacokinetics, tolerability, and bacteriological response of rifampin administered at 600, 900, and 1,200 milligrams daily in patients with pulmonary tuberculosis. Antimicrob Agents Chemother. 2017;61:e01054‐e01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Qian Y, Markowitz JS. Prediction of carboxylesterase 1‐mediated in vivo drug interaction between methylphenidate and cannabinoids using static and physiologically based pharmacokinetic models. Drug Metab Dispos. 2022;50:968‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vuong S. Development of Liquid Chromatography‐Tandem Mass Spectrometry Methods of Cannabinoids For Pediatric Patient Samples. Master of Science thesis. University of Saskatchewan; 2020. [Google Scholar]
- 44. Center for Drug Evaluation and Research. Non‐Clinical Review(s) Epidiolex 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000PharmR.pdf. [Accessed March 31, 2022].
- 45. Taylor L, Crockett J, Tayo B, Morrison G. A phase 1, open‐label, parallel‐group, single‐dose trial of the pharmacokinetics and safety of cannabidiol (CBD) in subjects with mild to severe hepatic impairment. J Clin Pharmacol. 2019;59:1110‐1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mazina J, Spiljova A, Vaher M, Kaljurand M, Kulp M. A rapid capillary electrophoresis method with LED‐induced native fluorescence detection for the analysis of cannabinoids in oral fluid. Anal Methods. 2015;7:7741‐7747. [Google Scholar]
- 47. Bansal S, Maharao N, Paine MF, Unadkat JD. Predicting the potential for cannabinoids to precipitate pharmacokinetic drug interactions via reversible inhibition or inactivation of major cytochromes P450. Drug Metab Dispos. 2020;48:1008‐1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ohlsson A, Lindgren JE, Andersson S, Agurell S, Gillespie H, Hollister LE. Single‐dose kinetics of deuterium‐labelled cannabidiol in man after smoking and intravenous administration. Biomed Environ Mass Spectrom. 1986;13:77‐83. [DOI] [PubMed] [Google Scholar]
- 49. Tayo B, Taylor L, Sahebkar F, Morrison G. A phase I, open‐label, parallel‐group, single‐dose trial of the pharmacokinetics, safety, and tolerability of cannabidiol in subjects with mild to severe renal impairment. Clin Pharmacokinet. 2020;59:747‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schoedel KA, Szeto I, Setnik B, et al. Abuse potential assessment of cannabidiol (CBD) in recreational polydrug users: a randomized, double‐blind, controlled trial. Epilepsy Behav. 2018;88:162‐171. [DOI] [PubMed] [Google Scholar]
- 51. Center for Drug Evaluation and Research. Other Review(s): Sponsor's Submission, GWEP1541 . 2018. . Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/210365Orig1s000OtherR.pdf. [Accessed March 31, 2022].
- 52. 7‐Hydroxycannabidiol [Internet]. PubChem. 2022. Available from: https://pubchem.ncbi.nlm.nih.gov/compound/7‐Hydroxycannabidiol. [Accessed May, 21 2022].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: