

Abstract
Heart failure has reached epidemic proportions in a progressively ageing population. The molecular mechanisms underlying heart failure remain elusive, but evidence indicates that DNA damage is enhanced in failing hearts. Here, we tested the hypothesis that endogenous DNA repair in cardiomyocytes is critical for maintaining normal cardiac function, so that perturbed repair of spontaneous DNA damage drives early onset of heart failure. To increase the burden of spontaneous DNA damage, we knocked out the DNA repair endonucleases xeroderma pigmentosum complementation group G (XPG) and excision repair cross‐complementation group 1 (ERCC1), either systemically or cardiomyocyte‐restricted, and studied the effects on cardiac function and structure. Loss of DNA repair permitted normal heart development but subsequently caused progressive deterioration of cardiac function, resulting in overt congestive heart failure and premature death within 6 months. Cardiac biopsies revealed increased oxidative stress associated with increased fibrosis and apoptosis. Moreover, gene set enrichment analysis showed enrichment of pathways associated with impaired DNA repair and apoptosis, and identified TP53 as one of the top active upstream transcription regulators. In support of the observed cardiac phenotype in mutant mice, several genetic variants in the ERCC1 and XPG gene in human GWAS data were found to be associated with cardiac remodelling and dysfunction. In conclusion, unrepaired spontaneous DNA damage in differentiated cardiomyocytes drives early onset of cardiac failure. These observations implicate DNA damage as a potential novel therapeutic target and highlight systemic and cardiomyocyte‐restricted DNA repair‐deficient mouse mutants as bona fide models of heart failure.
Keywords: apoptosis, cardiac function, congestive heart failure, DNA damage, DNA repair
Loss of DNA repair permitted normal heart development but subsequently caused progressive deterioration of cardiac function and dilation, resulting in overt congestive heart failure and premature death within 6 months.

1. INTRODUCTION
Heart failure has become a global disease epidemic, particularly in the elderly (Bui et al., 2011; Lloyd‐Jones et al., 2002). Notwithstanding major advances in treatment, the exact molecular mechanisms underlying the pathogenesis of heart failure remain incompletely understood hampering a more mechanism‐based approach to effective treatment and prevention. Experimental and clinical evidence indicates that DNA damage, for example, due to oxidative stress or following chemotherapy (Octavia et al., 2012), is associated with heart failure (Bartunek et al., 2002; Higo et al., 2017; Shukla et al., 2010). In addition, accumulation of unrepaired DNA damage is linked with segmental premature ageing phenotypes in several organ systems (Hoeijmakers, 2009; Lopez‐Otin et al., 2013; Vermeij, Hoeijmakers, & Pothof, 2016). However, the precise effects of unrepaired endogenous DNA damage, and its role in the pathogenesis of heart failure, remain elusive. In the present study, we tested the hypothesis that DNA repair in cardiomyocytes is critical for maintaining normal cardiac function. For this purpose, we generated mice with genetically depleted DNA repair mechanisms in either all cell types or restricted to cardiomyocytes. XPG and ERCC1‐XPF, two major players in DNA repair, are structure‐specific endonucleases responsible for excision of DNA lesions in nucleotide excision repair (NER) and transcription‐coupled repair (TCR). Moreover, XPG is implicated in promoting base excision repair of oxidative DNA damage and double‐strand break (DSB) repair (Marteijn et al., 2014; Trego et al., 2016). ERCC1‐XPF is additionally involved in interstrand cross‐link repair, and sub‐pathways of DSB repair (Gregg et al., 2011). Xpg −/− , Ercc1 −/− and Ercc1 Δ/− mice (and corresponding human syndromes) show many progressive progeroid characteristics (e.g., cachexia, neurodegeneration, osteoporosis and impaired growth) and a severely shortened lifespan (Barnhoorn et al., 2014; Weeda et al., 1997). In addition to these systemic mouse mutants, we generated mice with genetically depleted Xpg and Ercc1 in differentiated cardiomyocytes using an alpha‐myosin heavy chain (αMHC)‐promoter‐driven Cre transgene (mice referred as αMHC‐Xpg c/− and αMHC‐Ercc1 c/− respectively). The cardiomyocyte‐restricted mutants were employed to circumvent the potentially confounding influence on the heart of premature ageing in other organs associated with systemic loss of Xpg and Ercc1. Both systemic and cardiomyocyte‐restricted mutants were investigated to elucidate the cell‐autonomous effect of endogenous DNA damage on cardiac function.
2. RESULTS
2.1. Deficient DNA repair results in time‐dependent deterioration of global LV function and severely shortened lifespan in all mutants
Ercc1 −/− mice showed severe growth retardation and a dramatically reduced lifespan. Consequently, these mice could only be studied at 8 weeks at which time point survival rate was 39% (Figure S1). We therefore also included Ercc1 Δ/− mice, in which the Δ allele only partially inactivates the function of Ercc1 due to a seven amino‐acid carboxy‐terminal truncation; consequently, lifespan, compared with Ercc1 −/− mice, is extended to 4–6 months and Ercc1 Δ/− mice exhibit a milder degenerative phenotype, consistent with a milder mutation (Weeda et al., 1997). To achieve cardiomyocyte‐restricted Xpg and Ercc1 gene inactivation, mice with conditional Xpg and Ercc1 alleles were crossed with a transgenic line with Cre recombinase under control of the αMHC promotor (Figure S2a,b). These mice were heterozygous for Xpg and Ercc1, respectively, in all cell types, except for cardiomyocytes, which were homozygous for loss of Xpg and Ercc1, and are referred to as αMHC‐Xpg c/− and αMHC‐Ercc1 c/− , respectively. Littermates, heterozygous for the floxed allele and the Cre transgene, were used as controls and are indicated as αMHC‐Xpg Ctrl and αMHC‐Ercc1 Ctrl. Left ventricular (LV) dimension and function were indistinguishable between these mice and the control littermates heterozygous for the floxed allele only (Figure S3a), indicating that the presence of Cre recombinase itself did not lead to a heart failure phenotype. Control littermates heterozygous for Xpg or Ercc1 in all cell types (referred to as Xpg fl/− αMHC‐Cre − and Ercc1 fl/− αMHC‐Cre − , Figure S2a,b) did not show cardiac alterations compared with control littermates wildtype in all cell types (referred to as Xpg fl/+ αMHC‐Cre − and Ercc1 fl/+ αMHC‐Cre − , Figures S2a,b and S3b), indicating that Xpg or Ercc1 heterozygosity in all cell types did not affect cardiac function. This is compatible with the autosomal recessive nature of these genetic defects and the general absence of overt phenotypes in obligate carriers of defects in most human nucleotide excision repair disorders, such as Xeroderma Pigmentosum and Cockayne syndrome (Hoeijmakers, 2009; Vermeij, Hoeijmakers, & Pothof, 2016).
Myocardial Xpg and Ercc1 mRNA expression levels in, respectively, αMHC‐Xpg c/− and αMHC‐Ercc1 c/− mice, were significantly attenuated compared with corresponding control mice (Figure S4a).
Xpg −/− and Ercc1 Δ/− mice exhibited reduced growth, followed by body weight loss after 8 weeks of age, and premature death, in striking contrast to apparent normal body growth of αMHC‐Xpg c/− and αMHC‐Ercc1 c/− mice (Figure 1a and Figure S4b,c).
FIGURE 1.
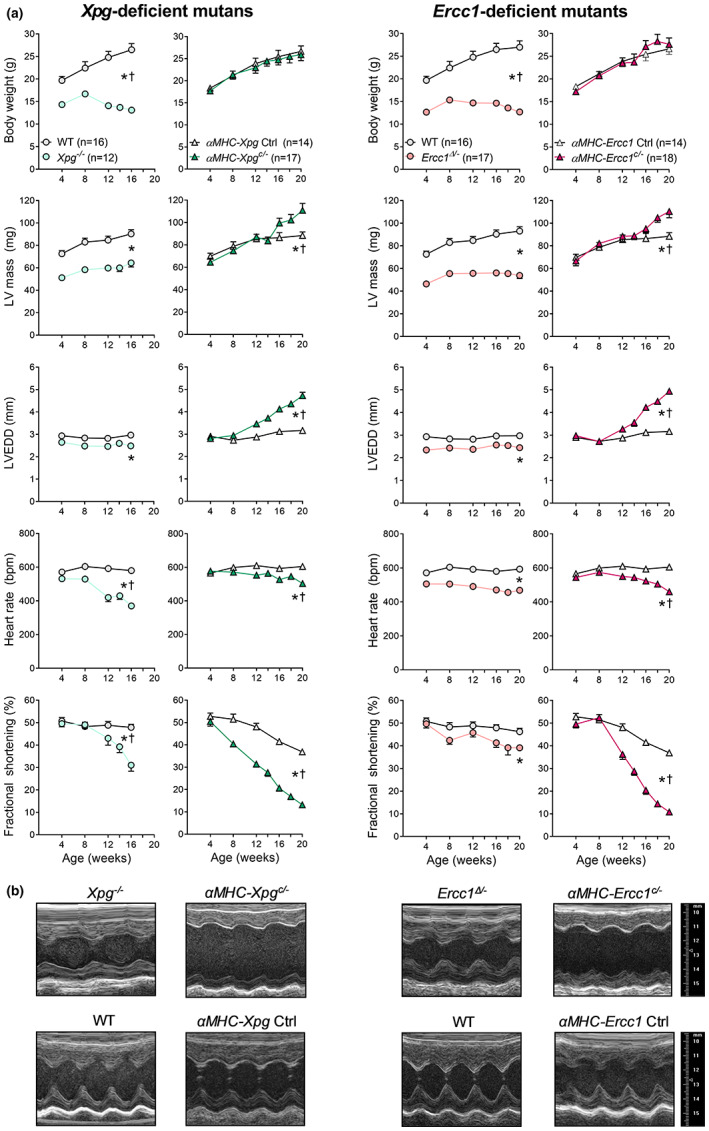
Impaired DNA repair resulted in time‐dependent deterioration of global LV function in all mutants (Study I, lifespan studies). (a) Effect of Xpg and Ercc1 deficiency on body weight, LV mass and geometry and hemodynamic parameters during age. LV mass, left ventricular mass, calculated using the VisualSonics Cardiac Measurements Package; LVEDD, LV end‐diastolic lumen diameter. The number of animals is indicated in the body weight graph. All mutants have their own corresponding control littermates. Data are presented as mean ± SEM. (b) Representative LV short axis M‐mode images of the different DNA repair‐deficient mutants and corresponding control at age 16 weeks. *p < 0.05 vs. corresponding control; †p < 0.05 genotype × age using mixed linear model–repeated measures.
Cardiomyocyte‐restricted inactivation of Xpg and Ercc1 also reduced lifespan in both male and female mice, albeit less than in Xpg −/− and Ercc1 Δ/− mice, respectively (Figure S4c,d), suggesting that defective DNA repair in the heart is not lifespan limiting in the systemic mutants. As reported previously (Vermeij, Dollé, et al., 2016; Vermeij, Hoeijmakers, & Pothof, 2016), the more severe repair defect in the absence of Ercc1 relative to Xpg correlates with a shorter lifespan compared with Xpg inactivation, consistent with an inverse dose–response relationship between unrepaired DNA damage and lifespan (Figure S4c). Importantly, echocardiography at 4 weeks of age showed normal cardiac function in all mutants, indicating unaffected heart development (Figure 1a). Over time, all DNA repair‐deficient mutants gradually developed bradycardia and contractile dysfunction compared with corresponding control (Figure 1a,b), demonstrating that loss of DNA repair induces a degenerative process. Time‐dependent contractile deterioration was more severe in αMHC‐Xpg c/− and αMHC‐Ercc1 c/− mice than in systemic mutants, demonstrated by progressive increases in LV end‐diastolic lumen diameter and decreases in fractional shortening starting after 8 weeks of age (Figure 1a,b). Systemic DNA repair‐deficient mutants display macro‐ and microvascular vasodilator dysfunction, which could potentially have contributed to the observed LV‐remodeling. Thus, similar to what we previously observed in Ercc1 Δ/ (Durik et al., 2012), 16‐week‐old Xpg −/− mice showed reductions in acetylcholine‐ and sodium nitroprusside‐induced aortic relaxation (Figure S5a). Importantly, however, no vascular dysfunction was observed in αMHC‐Xpg c/− mice (Figure S5b), despite severe cardiac dysfunction. This indicates that conditional disruption of Xpg in cardiomyocytes left vascular function unaffected and conversely that the perturbed vascular function observed in Xpg −/− mice did not contribute to the heart failure phenotype.
2.2. Xpg‐ and Ercc1‐deficiency produces LV‐remodeling and LV dysfunction
Detailed analysis of LV function at age 16 weeks (except for Ercc1 −/− mice that were studied at 8 weeks) revealed increased LV end‐diastolic lumen diameters (LVEDD, normalized to LV weight) in all DNA repair‐deficient mutants (Figure 2a). In addition, systolic LV function (fractional shortening, LVdP/dtmax) and early (LVdP/dtmin, tau) and late (LVEDP) diastolic LV function were markedly impaired (Figure 2b).
FIGURE 2.
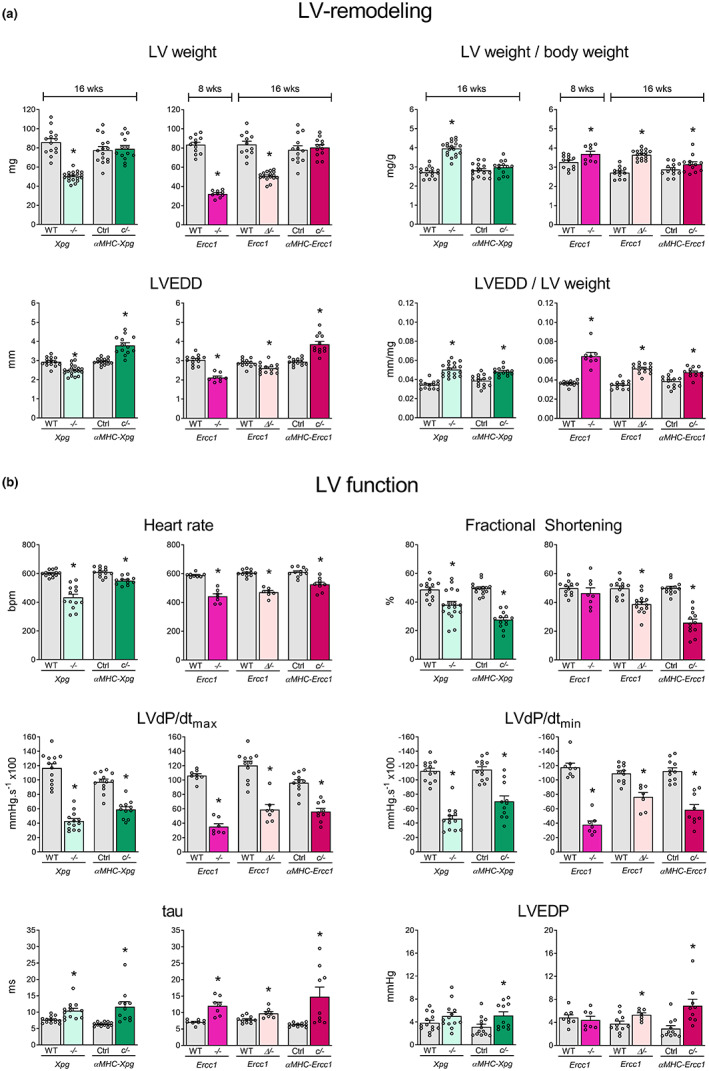
LV‐remodeling and LV dysfunction in DNA repair‐deficient mutants at a specific time point (study II). (a) Effect of Xpg and Ercc1 deficiency on LV mass and geometry and (b), hemodynamic parameters in 16‐week‐old Xpg −/− and αMHC‐Xpg c/− , 8‐week‐old Ercc1 −/− , 16‐week‐old Ercc1 Δ/− and αMHC‐Ercc1 c/− mice and corresponding control (n = 7–16 animals/group). LV weight, left ventricular weight; BW, body weight; LVEDD, LV end‐diastolic lumen diameter; LVdP/dtmax, maximum rate of rise of LV pressure; LVdP/dtmin, maximum rate of fall of LV pressure; tau, relaxation time constant; LVEDP, LV end‐diastolic pressure. Data are presented as mean ± SEM. *p < 0.05 vs. corresponding control by two‐way ANOVA followed by SNK post hoc testing.
2.3. Increased cardiac expression of foetal genes in Xpg mutants
Based on the survival curves (Figure S4c) and the similar degree of cardiac dysfunction in Xpg −/− and αMHC‐Xpg c/− mice at 16 weeks (Figure 1), we elected to further study the Xpg mutants in more detail. LV dilation in 16‐week‐old αMHC‐Xpg c/− mice was accompanied by increased re‐expression of the foetal genes atrial natriuretic peptide (Anp), brain natriuretic peptide (Bnp) and α‐skeletal actin (Acta1), whereas in 16‐week‐old Xpg −/− mice, only Anp levels were increased (Figure S6a). At 8 weeks, expression of these marker genes was not yet elevated (Figure S6b), further supporting the degenerative (rather than developmental) nature of the disease process.
2.4. Altered cardiomyocyte contractile properties in Xpg −/− mice
To explore whether functional properties of cardiomyocytes contributed to perturbed LV function, force measurements were performed in membrane‐permeabilized single LV cardiomyocytes isolated from Xpg mutant mice. Contractile properties of single cardiomyocytes obtained by mechanical isolation and attached to a force transducer and a piezoelectric motor (Figure 3a) were studied at a sarcomere length of 2.2 μm. Unexpectedly, maximum force generating capacity (Fmax) was maintained in cardiomyocytes from both Xpg mutants, indicating that the observed global systolic LV dysfunction was not due to a loss of force development of individual cardiomyocytes. A small elevation in passive tension (F pas) was observed in Xpg −/− mice only (Figure 3b). Similarly, in Xpg −/− mice, increases in myofilament Ca2+ sensitivity were noted (Figure 3c), which were accompanied by a lower protein kinase A (PKA)‐mediated phosphorylation of cardiac troponin I (cTnI), at the PKA sites Ser23/24 (Figure 3e; Figure S7). In addition, increased protein levels of β‐MHC and decreased maximal rate of force redevelopment (max Ktr; changes typically associated with LV hypertrophy and failure (Hamdani et al., 2008)) were observed in Xpg mutants, reaching statistical significance in Xpg −/− mice (Figure 3c,d). These changes explain, at least partly, the diastolic dysfunction in the Xpg mutants.
FIGURE 3.
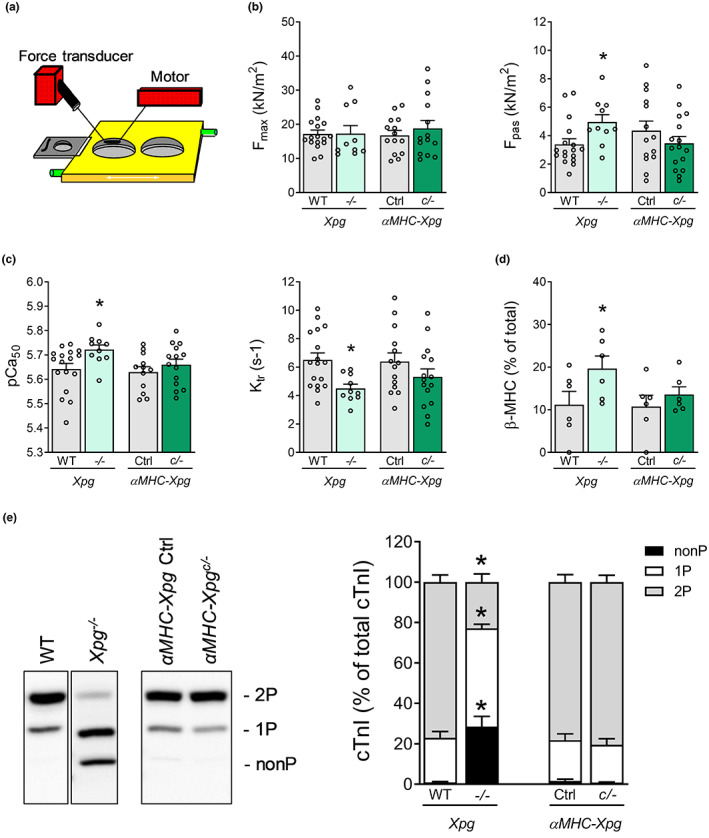
Altered cardiomyocyte contractile properties in 16‐week‐old Xpg −/− mice, but not in 16‐week‐old αMHC‐Xpg c/− mice (Study II). (a) Schematic experimental myocyte set‐up. (b) Isometric force measurements in single permeabilized cardiomyocytes (n = 6 animals/group; 1–4 cardiomyocytes/animal). F max, maximal force; F pas, passive force. (c) Determination of Ca2+ sensitivity of force (pCa50) and maximal rate of force redevelopment (Ktr) (n = 6 animals/group; 1–4 cardiomyocytes/animal). (d) Myosin heavy chain isoform composition was determined by protein levels of myosin heavy chain isoforms (n = 6 animals/group). Beta‐myosin heavy chain (β‐MHC) content is expressed as % of total MHC. (e) Phos‐tag analysis of cardiac troponin I (cTnI) species, expressed as % of total phosphorylated cTnI (n = 6–7 animals/group). Phosphorylated cTnI species, of similar molecular weight, were separated into three forms: Non‐phosphorylated (nonP), mono‐phosphorylated (1P) and bis‐phosphorylated (2P). These forms were visualized with a specific antibody against troponin I, which recognizes non‐phosphorylated and phosphorylated forms. Data are presented as mean ± SEM. *p < 0.05 vs. corresponding control by two‐way ANOVA followed by SNK post hoc testing.
2.5. Enhanced extracellular matrix turnover and increased levels of cell loss in Xpg mutants
Histological analysis revealed a small decrease in cardiomyocyte cross‐sectional area in Xpg −/− , but not in αMHC‐Xpg c/− mice, paralleling changes in heart weight (Figure 4a). Collagen fraction tended to increase (Figure 4a) and extracellular matrix turnover was enhanced (Figure S8a,b), which may have contributed not only to the cardiac remodelling but also to the fractionated QRS pattern observed on surface ECG analysis in vivo (Figure S8c), suggestive of cardiac conduction delay (Rizzo et al., 2012). Moreover, αMHC‐Xpg c/− mice displayed bradycardia and significantly increased PR interval, P duration and QRS interval (Figure S8d). These ECG parameters were already affected at the age of 8 weeks, while these parameters were unaffected at the age of 4 weeks in αMHC‐Xpg c/− mice (Figure S8e). Collagen content was still unchanged in hearts of 8‐week‐old αMHC‐Xpg c/− mice (αMHC‐Xpg c/− 1.54 ± 0.13%, n = 5 vs. corresponding control 1.39 ± 0.24%, n = 5; p = 0.60), indicating that cardiac conduction abnormalities preceded the development of structural alterations and were not the consequence of the latter. While (atrio‐)ventricular conduction was not affected in Xpg −/− mice, significant QT prolongation was observed. This was likely the consequence of the low heart rate in these mice since no differences were observed in QT interval corrected for heart rate (QTc; Figure S8d).
FIGURE 4.
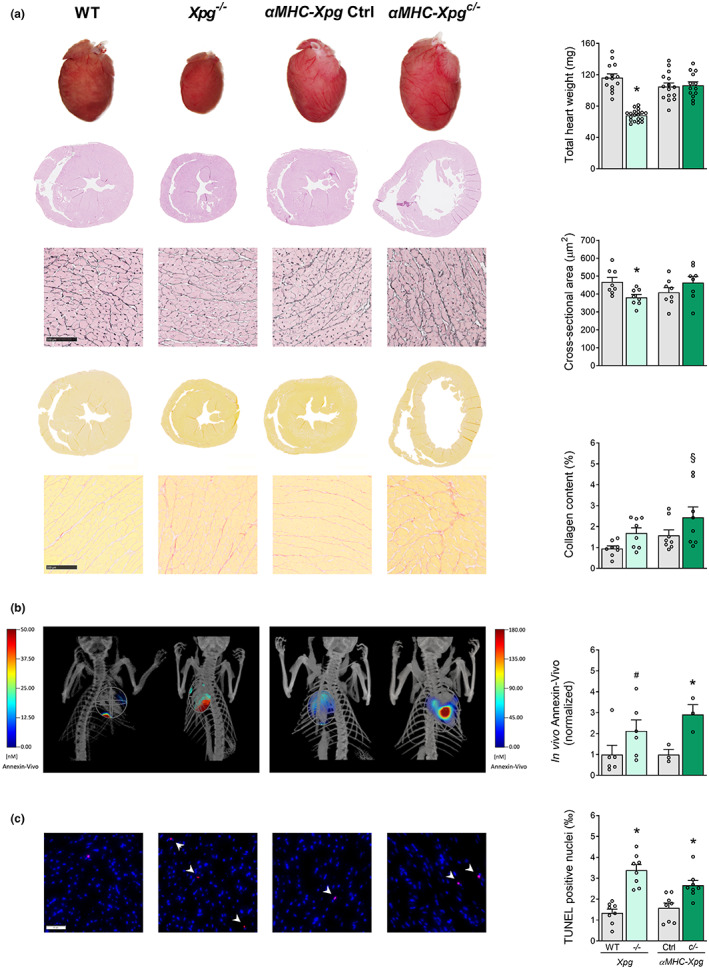
Characterization of LV phenotype in 16‐week‐old Xpg mutants (Study II). (a) Representative images of whole hearts and haematoxylin and eosin‐stained two‐chamber view sections and examination of total heart weight (n = 13–19 animals/group). Representative gomori‐ and picro‐sirius red‐stained LV sections and quantification of, respectively, cardiomyocyte cross‐sectional area and myocardial collagen content (n = 8 animals/group). (b) Representative images and quantification of the in vivo visualization of the membrane‐bound phospholipid phosphatidylserine to detect early apoptosis using FMT combined with μCT (n = 3–6 animals/group). (c) Representative TUNEL‐stained LV sections and quantification of TUNEL‐positive nuclei (indicated by arrows) to detect late apoptotic cells (n = 8 animals/group). Data are presented as mean ± SEM. *p < 0.05, §p = 0.062, #p = 0.087 vs. corresponding control by two‐way ANOVA followed by SNK post hoc testing.
To assess the purported role of oxidative stress in heart failure, basal superoxide generation was measured in homogenates using lucigenin‐enhanced chemiluminescence. Both NOX activity (using NADPH as substrate) and NOX‐dependent superoxide production (using NOX inhibitor VAS2870) were increased, particularly in Xpg −/− mice, resulting in elevations in total superoxide production (Figure S9). Since oxidative stress is a major cause of apoptosis (Kannan & Jain, 2000), molecular imaging was performed to determine the level of apoptosis in the in vivo heart using the near‐infrared fluorescent Annexin‐Vivo™ 750 probe, combined with contrast‐enhanced μCT for anatomical reference. This probe recognizes and attaches to phosphatidylserine residues displayed on the cell surface during early stages of apoptosis. Both Xpg mutants showed increased in vivo levels of apoptosis in the heart (Figure 4b). Finally, Xpg −/− and αMHC‐Xpg c/− mice showed elevated levels of TUNEL‐positive nuclei in LV myocardium (Figure 4c), indicating increased apoptotic cell death. Loss of cells likely contributed to the observed deterioration in cardiac function. Surprisingly, this loss was not accompanied by increased myocardial expression of the cyclin‐dependent kinase inhibitors p21 and p16 in Xpg mutants compared with control. These observations contrast with our present and previous (Barnhoorn et al., 2014; Durik et al., 2012) findings in the liver of Xpg −/− mice, which exhibited increased expression of p21 and p16 (Figure S10a). Moreover, evaluation of phosphorylation of the H2A.X histone (γH2A.X), an indicator of DNA damage (Greenberg, 2008), revealed increased γH2A.X‐positive nuclei in Xpg −/− liver compared with control, but not in Xpg −/− and αMHC‐Xpg c/− hearts (Figure S10b). These observations could be interpreted to suggest that myocardial cells appear highly sensitive to DNA damage, responding with apoptotic cell death.
2.6. Congestive heart failure, enhanced fibrosis and dramatically elevated levels of cell loss in advanced age αMHC‐Xpg c /− mice
The observed LV dysfunction at 16 weeks of age was not accompanied by LV hypertrophy and pulmonary congestion. However, αMHC‐Xpg c/− mice at an advanced age (18–26 weeks) displayed overt congestive LV failure, demonstrated by increases in relative LV weight, left atrial (LA) weight, lung fluid (LF) weight and right ventricular (RV) weight (Figure 5a). Echocardiographic analysis at 22 weeks of age (Study I: Lifespan study) revealed a progressively enlarged LV lumen and impairment of fractional shortening compared with 16‐week‐old αMHC‐Xpg c/− mice, showing further disease progression, and in addition, the expression level of Anp was further elevated (Figure 5b). The LV hypertrophy displayed in advanced age αMHC‐Xpg c/− mice was accompanied by significantly increased cardiomyocyte cross‐sectional area compared with 16‐week‐old control mice (data not shown). In addition, collagen fraction was markedly elevated and TUNEL staining revealed a dramatic loss of cells (Figure 5c; Figure S11a). Importantly, at 8 weeks of age, no differences in the level of TUNEL‐positive cells (αMHC‐Xpg c/− 1.82 ± 0.32‰, n = 6 vs. corresponding control 1.73 ± 0.29‰, n = 5; p = 0.85) and collagen content (as mentioned previously) were observed between αMHC‐Xpg c/− and corresponding control, indicating that apoptosis and fibrosis start at a later age, further underscoring the degenerative nature of the disease process. Cellular stress, such as DNA damage, activates the intrinsic apoptotic pathway (Giam et al., 2008). Indeed, apoptosis in advanced age αMHC‐Xpg c/− mice was accompanied by increased expression of phorbol‐12‐myristate‐13‐acetate‐induced protein 1 (Pmaip1), Bcl‐ associated X protein (Bax), Bcl2‐related ovarian killer protein (Bok) and Bcl2‐modifying factor (Bmf) (Figure S11b).
FIGURE 5.
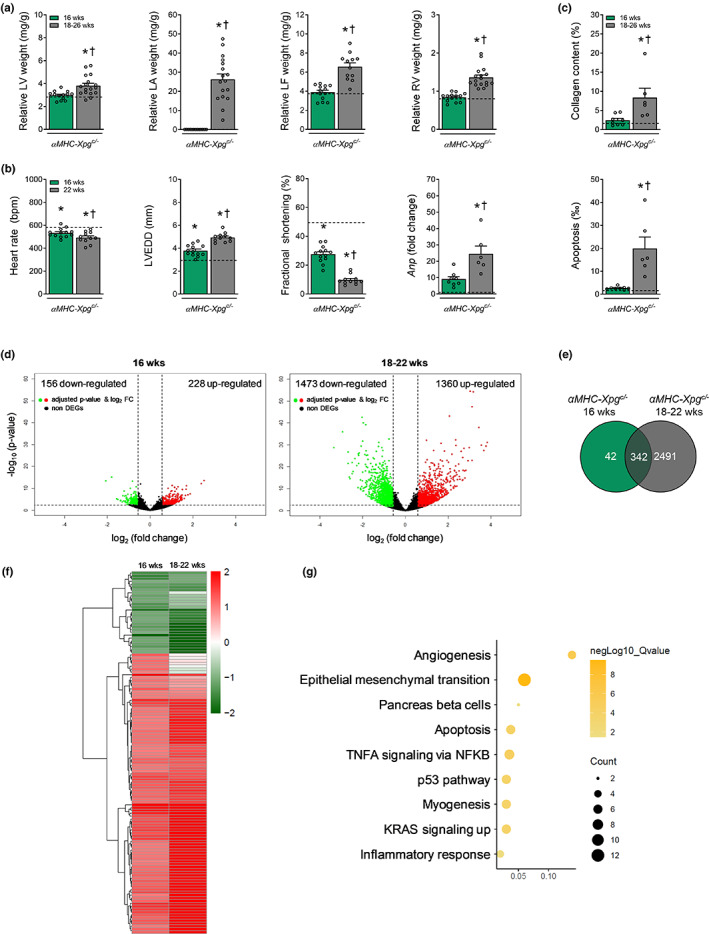
Progressive congestive heart failure and activation of TP53 in advanced age αMHC‐Xpg c/− mice. (a) Cardiomyocyte‐restricted loss of Xpg resulted in increased LV, LA, LF and RV weight during aging (n = 12–17 animals/group). LV weight, left ventricular weight; LA weight, left atrial weight; LF weight, lung fluid weight; RV weight, right ventricular weight. Weights are normalized to body weight. (b) Echocardiographic analysis at age 22 weeks (Study I, lifespan study) revealed progressive enlarged LV lumen diameter and aggravated loss of factional shortening compared with 16‐week‐old αMHC‐Xpg c/− mice and corresponding control (Study II) (n = 11–15 animals/group). In addition, quantitative real‐time PCR analysis revealed an elevated expression level of Anp (n = 6–8 animals/group). Expression is corrected for Hprt expression and normalized to control. LVEDD, LV end‐diastolic lumen diameter, Anp, atrial natriuretic peptide. (c) Quantification of respectively myocardial fibrosis and TUNEL‐positive nuclei to detect apoptotic cells (n = 6–8 animals/group). (d) Volcano plots present the overall gene expression of 16‐week and 18‐ to 22‐week‐old αMHC‐Xpg c/− compared with 16‐week‐old control (n = 3 animals/group). The dashed lines denote the cut‐off values for a differentially expressed gene (DEG; FDR adjusted P‐value <0.05 and absolute fold change >1.5). Significantly up‐regulated genes are highlighted in red and significantly down‐regulated genes in green. The non‐DEGs are represented in black. (e) Venn diagram display the total amount of overlapping DEGs in both αMHC‐Xpg c/− groups compared with 16‐week‐old control. (f) Heat map of hierarchically clustered overlapping DEGs in 16‐week and 18‐ to 22‐week‐old αMHC‐Xpg c/− mice compared with 16‐week‐old control. (g) Hallmark pathways enriched in αMHC‐Xpg c/− mice. Dot size represents the number of genes in each involved pathway. Dot colour show −log10 (Q‐value) in each term enrichment. X‐axis shows the ratio of the genes to all DEGs. (a–c) For the sake of comparison we have repeated 16‐week‐old αMHC‐Xpg c/− mice (green bars) and corresponding control (black dotted line) in this figure, both from Study II. (d–g) 16‐week‐old αMHC‐Xpg c/− mice: Study II; 18‐ to 22‐week‐old αMHC‐Xpg c/− mice: Study I, lifespan study. Data are presented as mean ± SEM. *p < 0.05 vs. corresponding control; †p < 0.05 vs. αMHC‐Xpg c/− 16 weeks by one‐way ANOVA followed by SNK post hoc testing.
2.7. TP53 activation and enrichment of apoptosis, DNA damage and impaired DNA repair‐associated pathways in αMHC‐Xpg c /− mice
To explore the molecular mechanisms related to DNA damage, messenger RNA sequencing was performed in LV tissues of αMHC‐Xpg c/− mice, revealing 384 differentially expressed genes (DEGs; FDR adjusted p‐value <0.05 and absolute fold change >1.5) in 16‐week‐old αMHC‐Xpg c/− compared with corresponding control. Strikingly, in the advanced age (18–22 weeks) αMHC‐Xpg c/− , 2833 DEGs were found, of which 342 overlapped with 16‐week‐old αMHC‐Xpg c/− DEGs (Figure 5d,e). Analysis of DEGs using ingenuity pathway analysis (IPA) identified the tumour suppressor gene TP53 as one of the top active upstream transcription regulators in 16‐week‐old (overlap p‐value 2.34 E‐14; activation z‐score 2.563) and in 18‐ to 22‐week‐old (overlap p‐value 2.06 E‐52; activation z‐score 6.449) αMHC‐Xpg c/− . TP53 is known to trigger cell‐cycle arrest, apoptosis or DNA repair in response to DNA damage and thereby activates specific genes (Lakin & Jackson, 1999). In advanced age αMHC‐Xpg c/− , 438 TP53‐target genes were activated (Table S2). This observation in our cardiomyocyte‐restricted DNA repair‐deficient Xpg mouse mutant is in excellent agreement with the observation of TP53 activation in another tissue‐restricted DNA repair‐deficient mouse mutant (accompanying manuscript by Henpita et al. (2023)). The overlapping DEGs shown in the heat map were enriched for pathways involved in angiogenesis, apoptosis and inflammation (Figure 5f,g).
Gene ontology (GO) terms enrichment analysis considering all DEGs (p‐value <0.05) in αMHC‐Xpg c/− at 16 and at 18–22 weeks of age, resulted in seven networks including mitochondrial pathway, transport, signaling, metabolic process, response to stimulus, development and homeostasis (Figure S12). Detailed analysis revealed apoptosis and DNA damage as significant subnetworks (Figure S13). In addition, gene set enrichment analysis based on the biological pathways supplied by KEGG showed enrichment of pathways which are associated with impaired DNA repair, such as Huntington's (adjusted p‐value 2.75 E‐17), Parkinson's (adjusted p‐value 2.07 E‐24) and Alzheimer's (adjusted p‐value 5.82 E‐25) disease in advanced age αMHC‐Xpg c/− , which are hallmarks of DNA repair disorders (Sepe et al., 2013).
2.8. Association of genetic variants in ERCC1 and XPG with cardiac remodelling and dysfunction
To study the effect of genetic variation in the ERCC1 and XPG gene on cardiac function in humans, a genome‐wide association study (GWAS) of LVEDD and fractional shortening was performed in up to 7653 individuals from the population‐based Rotterdam Study (Ikram et al., 2020). The effect estimates for the common genetic variants located in the genes and those 50kB up‐ and downstream were extracted. A total of 26 genetic variants in ERCC1 were associated with LVEDD (p‐value <0.05), of which two single‐nucleotide polymorphisms (SNPs; 19:45967369 and 19:45976718) showed an opposite effect for LVEDD and fractional shortening (Table S3). Conversely, a total of 13 SNPs in ERCC1 were associated with lower fractional shortening (p‐value <0.05), of which six SNPs (19:45877788, 19:45879195, 19:45884546, 19:45888808, 19:45896834 and 19:45914174) showed an opposite effect for fractional shortening and LVEDD (Table S4). For XPG, a total of 14 SNPs were associated with LVEDD, of which four SNPs (13:103467184, 13:103457497, 13:103527113 and 13:103548761) showed an opposite effect for LVEDD and fractional shortening (Table S3). Conversely, five SNPs were associated with fractional shortening (p‐value <0.05), none of which showed an effect with LVEDD (Table S4). Taken together, these findings reveal genetic variants that may be implicated in the development of heart failure in humans. Of notice, the observation that some of these genetic variants have opposite effects on LVEDD and fractional shortening makes these variants interesting candidates for further study.
3. DISCUSSION
Despite major advances in treatment, the exact molecular mechanisms underlying the pathogenesis of heart failure remain incompletely understood hampering a more mechanism‐based approach to effective treatment and prevention. Experimental and clinical evidence indicates that DNA damage—due to oxidative stress (e.g., ischemia–reperfusion injury) or after chemotherapy (Octavia et al., 2012)—is associated with heart failure (Bartunek et al., 2002; Higo et al., 2017; Shukla et al., 2010). However, the precise effects of unrepaired DNA damage, and its role in the pathogenesis of heart failure, are still poorly understood. In the present study, we tested the hypothesis that DNA repair in cardiomyocytes is critical for maintaining normal cardiac function. For this purpose, we employed mice with systemic and cardiomyocyte‐restricted depletion of Xpg and Ercc1.
The main findings of the present study were that (i) impaired DNA repair resulted in progressive cardiac remodelling and dysfunction, ultimately leading to severe congestive heart failure and premature death. (ii) In both Xpg mutants (i.e., Xpg −/− mice and αMHC‐Xpg c/− mice), systolic LV dysfunction could not be explained by a loss of cardiac myofilament force development. In addition, the lack of loss of peripheral vasodilator function in αMHC‐Xpg c/− mice excludes chronic increase in afterload as a mechanism for cardiac remodelling. (iii) Only in Xpg −/− mice, diastolic LV dysfunction could be explained by increases in F pas and myofilament Ca2+ sensitivity, and a reduction in max Ktr, which could be attributed to decreased protein phosphorylation and a shift in myosin heavy chain isoform composition, respectively. (iv) Myocardial collagen content was progressively increased. (v) Apoptosis was markedly and progressively elevated in both Xpg mutants, which was accompanied by increases in reactive oxygen species, expression of pro‐apoptotic genes, activation of TP53 and enrichment of DNA damage‐related pathways and diseases. (vi) In support of the observed cardiac phenotype in mutant mice, several genetic variants in the ERCC1 and XPG gene in human GWAS data were found to be associated with cardiac remodelling and dysfunction. The implications of these findings will be discussed.
3.1. Importance of sufficient DNA repair for the maintenance of cardiac function
Unrepaired DNA damage can lead to pronounced cellular dysfunction and promote disease development (Lopez‐Otin et al., 2013). The structure‐specific endonucleases XPG and ERCC1‐XPF, two major players in DNA repair, are responsible for the excision of DNA lesions and are involved in several DNA repair mechanisms (Gregg et al., 2011; Marteijn et al., 2014; Trego et al., 2016). Previously, using cardiomyocyte‐restricted Ercc1 knockout mice, genome instability in the heart, measured by variant calling in RNA and genomic DNA, was found to be strongly elevated compared with wildtype mice, and several other prematurely ageing mouse models (De Majo et al., 2021). The finding of enhanced mutations is consistent with the repair deficiency. However, small‐scale mutations are now being recognized not to be a major driver of ageing (Robinson et al., 2021), in line with the growing notion that DNA damage rather than mutations is relevant for ageing‐associated functional decline in post‐mitotic organs and tissues (Schumacher et al., 2021) and consistent with our finding of DNA damage‐driven transcription stress, which we also found to be enhanced as apparent from gene expression data sets of ageing in the heart (Gyenis, Chang, et al., Nat. Genetics in press). In the present study, both systemic and cardiomyocyte‐restricted loss of Xpg and Ercc1 (i.e., Xpg −/− , αMHC‐Xpg c/− , Ercc1 Δ/− and αMHC‐Ercc1 c/− mice) resulted in significant cardiac dysfunction. In conjunction with recent observations no or only minimal cardiac dysfunction in endothelium‐restricted (Bautista‐Nino et al., 2020) and smooth‐muscle‐restricted (Ataei Ataabadi et al., 2021) Ercc1 mutant mice, respectively, the present findings imply a critical role for cardiomyocyte DNA repair in maintaining cardiac function.
Importantly, we observed normal cardiac function in all DNA repair‐deficient mutants at 4 weeks of age, after which all mutants gradually developed contractile dysfunction. These observations indicate that loss of DNA repair induces a degenerative rather than a developmental process in the heart. These results are consistent with observations in mice in which deficient DNA repair is induced by depletion of Xrcc1, an essential protein for single‐strand break repair. In pressure overload‐induced heart failure, accumulation of single‐strand breaks was associated with more activated DNA damage response and deteriorated cardiac dysfunction in Xrcc1 deficient mutants (Higo et al., 2017).
In the present study, cardiac dysfunction in both systemic and cardiomyocyte‐restricted mutants was associated with a markedly reduced lifespan. Interestingly, cardiomyocyte‐restricted loss of Xpg and Ercc1 resulted in a slightly longer lifespan compared with mice with systemic loss of Xpg and Ercc1, suggesting that the heart is not lifespan limiting in the systemic mutants. This is also supported by the relatively milder degree of cardiac dysfunction in the systemic (fractional shortening ~30%) versus the cardiomyocyte‐restricted (~10%) mutants just prior to death. Conversely, the cardiomyocyte‐restricted mutants displayed severe signs of congestive heart failure in the final week, suggesting that mortality in these mice was related to the severe loss of cardiac function. It has been shown that survival rates of DNA repair‐deficient mouse models depend on the extent to which DNA repair mechanisms are affected, with Ercc1 mutants showing an even more severe phenotype than Xpg mutants (Vermeij, Dollé, et al., 2016). Indeed, we observed a slightly worse survival in the cardiomyocyte‐restricted Ercc1 mutants compared with the Xpg mutants. From the present study, we cannot determine the exact cause of death, that is progressive pump failure or fatal ventricular arrhythmias, in the cardiomyocyte‐restricted mutants. However, the observation that the surface ECG analysis did not show any sign of ventricular arrhythmias, together with the severe signs of congestive heart failure, including dyspnoea, pulmonary oedema and ascites, suggests that the cause of death most likely was progressive pump failure.
3.2. Mechanism of cardiac dysfunction
The mechanisms underlying the observed cardiac dysfunction in the Xpg and Ercc1 mutants could be several‐fold, including altered cardiomyocyte contractile properties, enhanced extracellular matrix deposition and increased levels of senescence and myocardial cell loss.
DNA repair‐deficient mutants demonstrated severe systolic LV dysfunction, which could not be explained by a loss of maximum force development of individual cardiomyocytes, as Fmax was maintained in both Xpg mutants. We also observed diastolic LV dysfunction in Xpg mutants, which could be explained, at least in the systemic Xpg mutants, by significant elevations in Fpas, myofilament Ca2+ sensitivity, and a decrease in max Ktr. The increased Ca2+ sensitivity was likely due to the lower PKA‐mediated cTnI phosphorylation, while reduced max Ktr is explained by the shift towards the β‐MHC isoform. The maintained force development, the increased Ca2+ sensitivity and lower cTnI phosphorylation are in excellent agreement with the observed alterations in cardiomyocyte function in patients with end‐stage dilated cardiomyopathy (DCM; Hamdani et al., 2008, 2010).
We also found evidence of extracellular matrix remodelling, as we observed a 50%–80% increase in LV myocardial collagen content at 16 weeks of age, with an additional 240% increase by the end of the cardiomyocyte‐restricted Xpg mutants' lifespan. These observations are similar to the elevated collagen volume fraction in ventricular biopsies of end‐stage DCM patients (Hamdani et al., 2010) and may have contributed to the progressive deterioration of cardiac function.
DNA damage can interfere with the vital process of transcription or induce replication arrest, which can trigger cellular senescence or cell death (Hoeijmakers, 2009). Several studies with different DNA repair‐deficient mouse mutants concerning other organ systems showed elevated levels of senescence markers, including increased expression of p21 and p16, and increased senescence‐associated β galactosidase activity (Barnhoorn et al., 2014; Vermeij, Dollé, et al., 2016). In line with those previous studies, we observed increased expression of p21 and p16 in the liver of 16‐week‐old systemic Xpg mutants. Interestingly, expression of p21 and p16 was not elevated in the Xpg mutant heart at 16 weeks of age, which could be interpreted to suggest that differentiated cardiomyocytes with depleted Xpg appear may be highly sensitive to DNA damage, responding with early apoptotic cell death. Indeed, examination of apoptosis showed significantly elevated levels of apoptotic cell death in both Xpg mutants, already at 16 weeks, which was markedly (fivefold) further increased in the cardiomyocyte‐restricted Xpg mutants at the end of their lifespan. These observations support the concept that the transition from cardiac dysfunction to overt heart failure is due to increased apoptosis (Li et al., 1997; Marin‐Garcia, 2016).
There is evidence that DNA damage can drive apoptosis in cardiomyocytes (Higo et al., 2017; Shukla et al., 2011). The mechanisms underlying DNA damage‐induced apoptosis may involve activation of TP53, a key regulator of apoptosis. Thus, in the accompanying manuscript by Henpita et al. (2023), unrepaired DNA damage in differentiated cardiomyocytes resulted in activation of p53, while genetic depletion of p53 attenuated the level of apoptosis. In line with their observations, the dramatic loss of cardiomyocytes at the end of the cardiomyocyte‐restricted Xpg mutants' lifespan was associated with activation of TP53 and elevated expression of the pro‐apoptotic genes Bax, Pmaip1, Bok and Bmf. Oxidative stress has been implicated in the pathogenesis of heart failure. In the failing human heart, due to dilated cardiomyopathy and acute myocardial infarction, elevated levels of the oxidative DNA damage marker 8‐oxo‐7,8‐dihydro‐2′‐deoxyguanosine and DNA repair enzymes were detected in serum and in cardiomyocytes (Bartunek et al., 2002; Kono et al., 2006). Interestingly, both Xpg mutants showed NOX‐dependent increases in superoxide production, which may have contributed to apoptosis, possibly via activation of TP53, and the progressive deterioration of cardiac pump function in the DNA repair‐deficient mutants.
Gene set enrichment analysis showed that the subnetworks of apoptosis and DNA damage were enriched. The NER pathway plays a critical role in neurodegenerative diseases, such as Alzheimer's, Parkinson's and Huntington's disease. Impaired DNA repair, due to defects in the NER pathway, causes severe neurodevelopmental abnormalities (Sepe et al., 2013). Identification of diseases related to the differentially expressed genes in our dataset revealed that cardiomyocyte‐restricted loss of Xpg, affected genes that are involved in these DNA damage‐related neurodegenerative diseases, consistent with the concept that DNA damage is present in our Xpg‐mutant mice.
3.3. Association of genetic variants in ERCC1 and XPG with cardiac remodelling and dysfunction
To study the effect of genetic variation in the ERCC1 and XPG gene on cardiac remodelling and dysfunction in humans, a genome‐wide association study of LVEDD and fractional shortening was performed in the Rotterdam Study. These results revealed that some of the genetic variants in ERCC1 and XPG genes were associated with an increase in LVEDD and a decrease in fractional shortening (corresponding with the cardiac phenotype in our mutant mice), suggesting that these variants might be implicated in the development of heart failure in humans.
3.4. Conclusions
The present study, in conjunction with the accompanying study by Henpita et al. (2023), demonstrates that unrepaired endogenously generated DNA damage in differentiated cardiomyocytes drives early onset of cardiac failure. Since Xpg and Ercc1 are both involved in NER, TCR and pathways of DSB repair, these genome stability mechanisms protect against cardiac failure. Importantly, even in the presence of these potent repair mechanisms, DNA damage inevitably accumulates over time even in normal cells, as some types of DNA damage are not recognized by global genome repair systems, while others are irreparable (Hoeijmakers, 2009). Moreover, age‐related decline in repair also occurs (Moriwaki & Takahashi, 2008; Vermeij, Dollé, et al., 2016). Hence, time‐dependent accumulation of DNA lesions may well contribute to the aetiology of heart failure, implicating DNA damage as a novel therapeutic target. Moreover, our work highlights systemic and cardiomyocyte‐restricted DNA repair‐deficient mutants as bona fide models of heart failure well suited for identifying novel therapeutic interventions.
4. EXPERIMENTAL PROCEDURES
For detailed experimental procedures, see Extended Methods in Appendix S2.
4.1. Animals
Generation and characterization of Xpg −/− , Ercc1 Δ/− and Ercc1 −/− mice have been described previously (Barnhoorn et al., 2014; Weeda et al., 1997). Briefly, Xpg −/− mice were obtained by crossing C57BL/6J Xpg +/− with FVB/N Xpg +/− animals, Ercc1 Δ/− mice by crossing FVB/N Ercc1 Δ/+ with C57BL/6J Ercc1 +/− and Ercc1 −/− mice by crossing C57BL/6J Ercc1 +/− with FVB/N Ercc1 +/− animals, resulting in experimental animals in C57BL6/FVB F1 hybrid background. In order to study cardiomyocyte‐restricted effects of DNA repair deficiency on cardiac geometry, function and structure, mice with cardiomyocyte‐restricted inactivation of Xpg and Ercc1 genes were generated (Figure S2a,b). The generation of Cre‐inducible Xpg and Ercc1 knockout alleles has been described previously (Barnhoorn et al., 2014; Doig et al., 2006). To achieve cardiomyocyte‐restricted Xpg and Ercc1 gene inactivation, a transgenic line with Cre recombinase under control of the alpha‐myosin heavy chain (αMHC) promotor was used (Agah et al., 1997). αMHC‐Cre +/− female mice were crossed with Xpg +/− male mice or with Ercc1 +/− male mice to generate, respectively, Xpg +/− αMHC‐Cre +/− and Ercc1 +/− αMHC‐Cre +/− mice in a C57BL/6J background. The obtained females were subsequently crossed with Xpg fl/fl and Ercc1 fl/fl male mice in a FVB/N background to yield Xpg fl/− αMHC‐Cre + and Ercc1 fl/− αMHC‐Cre + mice in a C57BL6/FVB F1 hybrid background. Xpg fl/− αMHC‐Cre + and Ercc1 fl/− αMHC‐Cre + mice, respectively, were heterozygous for Xpg and Ercc1 in all cell types, except for cardiomyocytes, which were homozygous for Xpg and Ercc1, after Cre‐mediated excision of the floxed allele. These mice are referred to as αMHC‐Xpg c/− and αMHC‐Ercc1 c/− , respectively, throughout this manuscript. Xpg fl/+ αMHC‐Cre + and Ercc1 fl/+ αMHC‐Cre + (referred to as αMHC‐Xpg Ctrl and αMHC‐Ercc1 Ctrl) were used as controls. These controls were wildtype in all cell types, except for cardiomyocytes, which were heterozygous. All animals described in this study were born at a predicted Mendelian frequency and have the same genetic C57BL6/FVB F1 hybrid background to minimize background‐specific effects. All animals were bred and maintained with ad libitum access to water and AIN93G synthetic pellets (Research Diet Services B.V., Wijk bij Duurstede, The Netherlands; gross energy content 4.9 kcal/g dry mass, digestible energy 3.97 kcal/g). A total of 342 mice of either sex entered the study (randomized). Each experiment included males and females, except the mRNA sequencing of LV tissue, which was performed only in males. No animals were excluded. Experiments were performed in accordance with the Council of Europe Convention (ETS123) and the Directive (2010/63/EU) for the protection of vertebrate animals used for experimental and other scientific purposes, and with approval of the Animal Care Committee of Erasmus MC, University Medical Center Rotterdam.
4.2. Experimental design
Two study protocols were performed. Study I consisted of a lifespan study, based on which we designed Study II, in which animals were studied in great detail and sacrificed after a pre‐determined follow‐up period.
4.2.1. Study I
Lifespan studies were performed to characterize body growth, as well as global cardiac geometry and function in the various DNA repair‐deficient mutants described above. For this purpose, animals were weighed and subsequently underwent transthoracic echocardiography, every 2‐4 weeks, until death (DNA repair‐deficient mutants) or 30 weeks of age (control mice).
4.2.2. Study II
To study cardiac geometry, function and structure in more detail, Xpg −/− , αMHC‐Xpg c/− , Ercc1 Δ/− , αMHC‐Ercc1 c/− mice and corresponding control littermates were sacrificed at the age of 16 weeks. Since αMHC‐Ercc1 c/− mice are deficient for Ercc1 in their cardiomyocytes, we added an additional group of Ercc1 −/− animals to Study II. However, as these animals have a much shorter lifespan (~8–10 weeks), we studied this additional group at 8 weeks of age.
4.2.3. Statistical analysis animal studies
Data from study I (lifespan study) were compared using mixed linear model–repeated measures (SPSS Statistics 21.0). Data from study II were compared using one‐way or two‐way ANOVA, followed by Student–Newman–Keuls (SNK) post hoc testing when appropriate (SigmaPlot 11.0). Comparison of variables between two groups at a single time point was performed by unpaired Student's t‐test (SigmaPlot 11.0). Vascular function curves (Figure S5) were compared with general linear model for repeated measures (GLM‐RM), assuming sphericity (SPSS Statistics 21.0). Since male and female mice demonstrated very similar LV responses to DNA repair deficiency (Figure S14), males and females were pooled. Statistical significance was accepted when p < 0.05 (two‐tailed). All data are presented as mean ± SEM.
4.3. Human genetic studies
4.3.1. Study cohort
In accordance with the observed phenotype in Xpg and in Ercc1 mutants, the association of common single‐nucleotide polymorphisms (SNPs) in both genes with enlarged LV lumen diameter and deteriorated fractional shortening were investigated. To this end, a genome‐wide association study (GWAS) of these two measures was performed in the population‐based Rotterdam Study (RS). The RS is a prospective cohort study, initially comprising 7983 individuals (78% of 10,215 invitees), 55 years of age or over, living in the well‐defined Ommoord district in the city of Rotterdam, The Netherlands (RS‐I, recruitment 1990–1993). The RS aims to examine determinants of disease and health in older subjects focussing on neurogeriatric, cardiovascular, bone and eye diseases (Ikram et al., 2020). The RS has been extended several times. In 2000, RS was extended to include individuals who had become 55 years of age or moved into the study district since the start of the study (RS‐II, including 3011 participants out of 4472 invitees). In 2006, the cohort was further extended to include subjects aged 45 years or over (RS‐III, 3932 subjects out of 6057 invitees). By the end of 2008, RS comprised 14,926 subjects, aged 45 years or over, with an overall response figure of 72.0% (14,926 out of 20,744). Participants of the RS are being revisited at the research centre every 3–4 years. Current analyses included participants from the fourth examination of the original cohort (RS‐I‐4), the second examination of the second cohort (RS‐II‐2) and the baseline examination of the third cohort (RS‐III‐1) for whom both genetic data and echocardiographic assessment were available. The RS has been approved by the Medical Ethics Committee of the Erasmus Medical Center and by the Ministry of Health, Welfare and Sport of the Netherlands, implementing the Wet Bevolkingsonderzoek: ERGO (Population Studies Act: Rotterdam Study). All participants provided written informed consent to participate in the study and to obtain information from their treating physicians.
4.3.2. Statistical analysis
Genome‐wide association was done among 7653 individuals for LVEDD (RS1: N = 2926, RS2: N = 2206, RS3: N = 2521) and a total of 7499 for fractional shortening (RS1: N = 2807, RS2: N = 2181, RS3: N = 2511). Genetic variants were tested for association with markers of cardiac function by linear regression adjusted for age, sex and four principal components (Rvtests). The study was imputed using the HRC reference v1.1. panel. Quality control of the data excluded genetic variants with a minor allele frequency (MAF) of <0.05 and imputation quality of <0.3. Summary‐level results for each of the three RS cohorts were meta‐analysed jointly with METAL (released on March 25, 2011) using a fixed‐effects model with an inverse‐variance weighted approach, correcting for genomic control. We extracted information for the genetic variants that were located in the ERCC1 and XPG (plus 50kB up and downstream of the gene) gene region from the genome‐wide association results to assess their association with LVEDD and fractional shortening. In view of the hypothesis‐driven nature of these analyses, we considered a p‐value <0.05 as statistically significant.
AUTHOR CONTRIBUTIONS
M.B. provided experimental design, generated cardiomyocyte‐specific mutants, performed the majority of the experiments, analysed data and wrote the manuscript. M.L.H. provided technical assistance in animal experiments and assistance in figure design. J.C., J.P., L.M., L.J.W. and M.S. provided experimental design and performed gene expression analysis. B.S.T. and Y.R. performed and analysed in vivo μCT‐FMT and ex vivo FMT imaging. M.G.J.K. provided technical assistance. Y.O. performed and analysed lucigenin experiments. E.D.D. and J.V. designed myofilament function experiments. L.A.B. performed and analysed quantitative RT‐PCR experiments. R.M.C.B. and S.B. generated systemic mutant mice. P.K.B. and A.J.M.R. quantified vascular function. I.K.P. performed histology and immunohistochemistry and analysed images. R.W. and C.A.R. analysed ECG data. B.S.T. and J.E. designed the in vivo μCT‐FMT and ex vivo FMT imaging experiments. M.M.B. and B.A. performed genome‐wide association studies in the Rotterdam Study. M.L.H., J.C., M.M.B., E.D.D., B.A., M.G., C.K., A.H.J.D., I.P., C.A.R., A.J.M.R., M.K., J.E. and J.V. critically revised manuscript. J.H.J.H. and D.J.D. conceived the study, designed the study and wrote the manuscript. D.J.D. coordinated the study.
FUNDING INFORMATION
This work was supported by the Dutch CardioVascular Alliance: An initiative with financial support of the Dutch Heart Foundation [Grants 2017B018‐ARENA‐PRIME (to L.J.W., J.V. and D.J.D.), 2018B030‐PREDICT2 (to C.A.R.) and 2021B008‐RECONNEXT (to D.J.D.)], Lijf en leven [Grant DIVERS (to J.E.)], National Institute of Health (NIH)/National Institute of Ageing (NIA) [Grant PO1 AG017242 (to J.H.J.H. and J.P.)], European Research Council Advanced Grants [Grants DamAge (to J.H.J.H.) and Dam2Age (to J.H.J.H.)], Dutch Cancer Society [Grant ONCODE (to J.H.J.H.)], Memorabel and Chembridge (ZonMW; to J.H.J.H. and J.P), BBoL (NWO‐ENW; to J.H.J.H.) and the Deutsche Forschungsgemeinschaft [Grant SFB 829 (to J.H.J.H.).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
Supporting information
Appendix S1.
Appendix S2.
ACKNOWLEDGMENTS
Alpha‐MHC‐Cre mice were kindly provided by Johannes Backs (University of Heidelberg, Heidelberg, Germany); floxed Ercc1 mice were kindly provided by David W. Melton (MRC Institute of Genetics and Molecular Medicine, University of Edinburgh, MRC Human Genetics Unit, Western General Hospital, Crewe Road, Edinburgh EH4 2XU, UK). Molecular imaging was enabled by the use of imaging equipment provided by the Applied Molecular Imaging Erasmus MC facility. We gratefully acknowledge Hans Bosch for the use of the Vevo770 High‐Resolution Imaging System (FUJIFILM VisualSonics Inc.), the Core Facility Genomics of the Medical Faculty Münster for performing RNA sequencing and Robert Beurskens, Wies Lommen, Ruud Zaremba, Jennifer Sarikaya and Rene de Vries for their expert technical support.
de Boer, M. , te Lintel Hekkert, M. , Chang, J. , van Thiel, B. S. , Martens, L. , Bos, M. M. , de Kleijnen, M. G. J. , Ridwan, Y. , Octavia, Y. , van Deel, E. D. , Blonden, L. A. , Brandt, R. M. C. , Barnhoorn, S. , Bautista‐Niño, P. K. , Krabbendam‐Peters, I. , Wolswinkel, R. , Arshi, B. , Ghanbari, M. , Kupatt, C. … Duncker, D. J. (2023). DNA repair in cardiomyocytes is critical for maintaining cardiac function in mice. Aging Cell, 22, e13768. 10.1111/acel.13768
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on reasonable request to the corresponding author.
REFERENCES
- Agah, R. , Frenkel, P. A. , French, B. A. , Michael, L. H. , Overbeek, P. A. , & Schneider, M. D. (1997). Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac‐restricted, site‐specific rearrangement in adult ventricular muscle in vivo. The Journal of Clinical Investigation, 100(1), 169–179. 10.1172/JCI119509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataei Ataabadi, E. , Golshiri, K. , van der Linden, J. , de Boer, M. , Duncker, D. J. , Jüttner, A. , de Vries, R. , van Veghel, R. , van der Pluijm, I. , Dutheil, S. , Chalgeri, S. , Zhang, L. , Lin, A. , Davis, R. E. , Snyder, G. L. , Danser, A. H. J. , & Roks, A. J. M. (2021). Vascular ageing features caused by selective DNA damage in smooth muscle cell. Oxidative Medicine and Cellular Longevity, 2021, 2308317. 10.1155/2021/2308317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnhoorn, S. , Uittenboogaard, L. M. , Jaarsma, D. , Vermeij, W. P. , Tresini, M. , Weymaere, M. , Menoni, H. , Brandt, R. M. , de Waard, M. C. , Botter, S. M. , Sarker, A. H. , Jaspers, N. G. , van der Horst, G. , Cooper, P. K. , Hoeijmakers, J. H. , & van der Pluijm, I. (2014). Cell‐autonomous progeroid changes in conditional mouse models for repair endonuclease XPG deficiency. PLoS Genetics, 10(10), e1004686. 10.1371/journal.pgen.1004686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartunek, J. , Vanderheyden, M. , Knaapen, M. W. , Tack, W. , Kockx, M. M. , & Goethals, M. (2002). Deoxyribonucleic acid damage/repair proteins are elevated in the failing human myocardium due to idiopathic dilated cardiomyopathy. Journal of the American College of Cardiology, 40(6), 1097–1103; discussion 1104‐1095. 10.1016/s0735-1097(02)02122-8 [DOI] [PubMed] [Google Scholar]
- Bautista‐Nino, P. K. , Portilla‐Fernandez, E. , Rubio‐Beltran, E. , van der Linden, J. J. , de Vries, R. , van Veghel, R. , de Boer, M. , Durik, M. , Ridwan, Y. , Brandt, R. , Essers, J. , Menzies, R. I. , Thomas, R. , de Bruin, A. , Duncker, D. J. , van HMM, B. , Ghanbari, M. , JHJ, H. , Sedlacek, R. , … Roks, A. J. M. (2020). Local endothelial DNA repair deficiency causes aging‐resembling endothelial‐specific dysfunction. Clinical Science, 134(7), 727–746. 10.1042/CS20190124 [DOI] [PubMed] [Google Scholar]
- Bui, A. L. , Horwich, T. B. , & Fonarow, G. C. (2011). Epidemiology and risk profile of heart failure. Nature Reviews. Cardiology, 8(1), 30–41. 10.1038/nrcardio.2010.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Majo, F. , Martens, L. , Hegenbarth, J. C. , Ruhle, F. , Hamczyk, M. R. , Nevado, R. M. , Andrés, V. , Hilbold, E. , Bär, C. , Thum, T. , de Boer, M. , Duncker, D. J. , Schroen, B. , Armand, A. S. , Stoll, M. , & De Windt, L. J. (2021). Genomic instability in the naturally and prematurely aged myocardium. Proceedings of the National Academy of Sciences of the United States of America, 118(36), e2022974118. 10.1073/pnas.2022974118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig, J. , Anderson, C. , Lawrence, N. J. , Selfridge, J. , Brownstein, D. G. , & Melton, D. W. (2006). Mice with skin‐specific DNA repair gene (Ercc1) inactivation are hypersensitive to ultraviolet irradiation‐induced skin cancer and show more rapid actinic progression. Oncogene, 25(47), 6229–6238. 10.1038/sj.onc.1209642 [DOI] [PubMed] [Google Scholar]
- Durik, M. , Kavousi, M. , van der Pluijm, I. , Isaacs, A. , Cheng, C. , Verdonk, K. , Loot, A. E. , Oeseburg, H. , Bhaggoe, U. M. , Leijten, F. , van Veghel, R. , de Vries, R. , Rudez, G. , Brandt, R. , Ridwan, Y. R. , van Deel, E. , de Boer, M. , Tempel, D. , Fleming, I. , … Roks, A. J. (2012). Nucleotide excision DNA repair is associated with age‐related vascular dysfunction. Circulation, 126(4), 468–478. 10.1161/CIRCULATIONAHA.112.104380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giam, M. , Huang, D. C. , & Bouillet, P. (2008). BH3‐only proteins and their roles in programmed cell death. Oncogene, 27(Suppl 1), S128–S136. 10.1038/onc.2009.50 [DOI] [PubMed] [Google Scholar]
- Greenberg, R. A. (2008). Recognition of DNA double strand breaks by the BRCA1 tumor suppressor network. Chromosoma, 117(4), 305–317. 10.1007/s00412-008-0154-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, S. Q. , Robinson, A. R. , & Niedernhofer, L. J. (2011). Physiological consequences of defects in ERCC1‐XPF DNA repair endonuclease. DNA Repair (Amst), 10(7), 781–791. 10.1016/j.dnarep.2011.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani, N. , Borbély, A. , Veenstra, S. P. , Kooij, V. , Vrydag, W. , Zaremba, R. , dos Remedios, C. , Niessen, H. W. , Michel, M. C. , Paulus, W. J. , Stienen, G. J. , & van der Velden, J. (2010). More severe cellular phenotype in human idiopathic dilated cardiomyopathy compared to ischemic heart disease. Journal of Muscle Research and Cell Motility, 31(4), 289–301. 10.1007/s10974-010-9231-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdani, N. , Kooij, V. , van Dijk, S. , Merkus, D. , Paulus, W. J. , Remedios, C. D. , Duncker, D. J. , Stienen, G. J. M. , & van der Velden, J. (2008). Sarcomeric dysfunction in heart failure. Cardiovascular Research, 77(4), 649–658. 10.1093/cvr/cvm079 [DOI] [PubMed] [Google Scholar]
- Henpita, C. , Vyas, R. , Healy, C. L. , Kieu, T. L. , Gurkar, A. U. , Yousefshahi, M. J. , … Niedernhofer, L. J. (2023). Loss of DNA repair mechanisms in cardiac myocytes induces dilated cardiomyopathy. Aging Cell, e13782. 10.1111/acel.13782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higo, T. , Naito, A. T. , Sumida, T. , Shibamoto, M. , Okada, K. , Nomura, S. , Nakagawa, A. , Yamaguchi, T. , Sakai, T. , Hashimoto, A. , Kuramoto, Y. , Ito, M. , Hikoso, S. , Akazawa, H. , Lee, J. K. , Shiojima, I. , McKinnon, P. , Sakata, Y. , & Komuro, I. (2017). DNA single‐strand break‐induced DNA damage response causes heart failure. Nature Communications, 8, 15104. 10.1038/ncomms15104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers, J. H. (2009). DNA damage, aging, and cancer. The New England Journal of Medicine, 361(15), 1475–1485. 10.1056/NEJMra0804615 [DOI] [PubMed] [Google Scholar]
- Ikram, M. A. , Brusselle, G. , Ghanbari, M. , Goedegebure, A. , Ikram, M. K. , Kavousi, M. , Kieboom, B. C. T. , Klaver, C. C. W. , de Knegt, R. J. , Luik, A. I. , Nijsten, T. E. C. , Peeters, R. P. , van Rooij, F. , Stricker, B. H. , Uitterlinden, A. G. , Vernooij, M. W. , & Voortman, T. (2020). Objectives, design and main findings until 2020 from the Rotterdam study. European Journal of Epidemiology, 35(5), 483–517. 10.1007/s10654-020-00640-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan, K. , & Jain, S. K. (2000). Oxidative stress and apoptosis. Pathophysiology, 7(3), 153–163. 10.1016/s0928-4680(00)00053-5 [DOI] [PubMed] [Google Scholar]
- Kono, Y. , Nakamura, K. , Kimura, H. , Nishii, N. , Watanabe, A. , Banba, K. , Miura, A. , Nagase, S. , Sakuragi, S. , Kusano, K. F. , Matsubara, H. , & Ohe, T. (2006). Elevated levels of oxidative DNA damage in serum and myocardium of patients with heart failure. Circulation Journal, 70(8), 1001–1005. 10.1253/circj.70.1001 [DOI] [PubMed] [Google Scholar]
- Lakin, N. D. , & Jackson, S. P. (1999). Regulation of p53 in response to DNA damage. Oncogene, 18(53), 7644–7655. 10.1038/sj.onc.1203015 [DOI] [PubMed] [Google Scholar]
- Li, Z. , Bing, O. H. , Long, X. , Robinson, K. G. , & Lakatta, E. G. (1997). Increased cardiomyocyte apoptosis during the transition to heart failure in the spontaneously hypertensive rat. The American Journal of Physiology, 272(5 Pt 2), H2313–H2319. 10.1152/ajpheart.1997.272.5.H2313 [DOI] [PubMed] [Google Scholar]
- Lloyd‐Jones, D. M. , Larson, M. G. , Leip, E. P. , Beiser, A. , D'Agostino, R. B. , Kannel, W. B. , Murabito, J. M. , Vasan, R. S. , Benjamin, E. J. , Levy, D. , & Framingham Heart Study . (2002). Lifetime risk for developing congestive heart failure: The Framingham Heart study. Circulation, 106(24), 3068–3072. 10.1161/01.cir.0000039105.49749.6f [DOI] [PubMed] [Google Scholar]
- Lopez‐Otin, C. , Blasco, M. A. , Partridge, L. , Serrano, M. , & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217. 10.1016/j.cell.2013.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Garcia, J. (2016). Cell death in the pathogenesis and progression of heart failure. Heart Failure Reviews, 21(2), 117–121. 10.1007/s10741-016-9538-7 [DOI] [PubMed] [Google Scholar]
- Marteijn, J. A. , Lans, H. , Vermeulen, W. , & Hoeijmakers, J. H. (2014). Understanding nucleotide excision repair and its roles in cancer and ageing. Nature Reviews. Molecular Cell Biology, 15(7), 465–481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Moriwaki, S. , & Takahashi, Y. (2008). Photoaging and DNA repair. Journal of Dermatological Science, 50(3), 169–176. 10.1016/j.jdermsci.2007.08.011 [DOI] [PubMed] [Google Scholar]
- Octavia, Y. , Tocchetti, C. G. , Gabrielson, K. L. , Janssens, S. , Crijns, H. J. , & Moens, A. L. (2012). Doxorubicin‐induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. Journal of Molecular and Cellular Cardiology, 52(6), 1213–1225. 10.1016/j.yjmcc.2012.03.006 [DOI] [PubMed] [Google Scholar]
- Rizzo, S. , Lodder, E. M. , Verkerk, A. O. , Wolswinkel, R. , Beekman, L. , Pilichou, K. , Basso, C. , Remme, C. A. , Thiene, G. , & Bezzina, C. R. (2012). Intercalated disc abnormalities, reduced Na(+) current density, and conduction slowing in desmoglein‐2 mutant mice prior to cardiomyopathic changes. Cardiovascular Research, 95(4), 409–418. 10.1093/cvr/cvs219 [DOI] [PubMed] [Google Scholar]
- Robinson, P. S. , Coorens, T. H. H. , Palles, C. , Mitchell, E. , Abascal, F. , Olafsson, S. , Lee, B. C. H. , Lawson, A. R. J. , Lee‐Six, H. , Moore, L. , Sanders, M. A. , Hewinson, J. , Martin, L. , Pinna, C. M. A. , Galavotti, S. , Rahbari, R. , Campbell, P. J. , Martincorena, I. , Tomlinson, I. , & Stratton, M. R. (2021). Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nature Genetics, 53(10), 1434–1442. 10.1038/s41588-021-00930-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher, B. , Pothof, J. , Vijg, J. , & Hoeijmakers, J. H. J. (2021). The central role of DNA damage in the ageing process. Nature, 592(7856), 695–703. 10.1038/s41586-021-03307-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepe, S. , Payan‐Gomez, C. , Milanese, C. , Hoeijmakers, J. H. , & Mastroberardino, P. G. (2013). Nucleotide excision repair in chronic neurodegenerative diseases. DNA Repair (Amst), 12(8), 568–577. 10.1016/j.dnarep.2013.04.009 [DOI] [PubMed] [Google Scholar]
- Shukla, P. , Singh, K. , Yanagawa, B. , Teoh, H. , & Verma, S. (2010). DNA damage repair and cardiovascular diseases. The Canadian Journal of Cardiology, 26, 13A–16A. 10.1016/s0828-282x(10)71055-2 [DOI] [PubMed] [Google Scholar]
- Shukla, P. C. , Singh, K. K. , Quan, A. , al‐Omran, M. , Teoh, H. , Lovren, F. , Cao, L. , Rovira, I. I. , Pan, Y. , Brezden‐Masley, C. , Yanagawa, B. , Gupta, A. , Deng, C. X. , Coles, J. G. , Leong‐Poi, H. , Stanford, W. L. , Parker, T. G. , Schneider, M. D. , Finkel, T. , & Verma, S. (2011). BRCA1 is an essential regulator of heart function and survival following myocardial infarction. Nature Communications, 2, 593. 10.1038/ncomms1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trego, K. S. , Groesser, T. , Davalos, A. R. , Parplys, A. C. , Zhao, W. , Nelson, M. R. , Hlaing, A. , Shih, B. , Rydberg, B. , Pluth, J. M. , Tsai, M. S. , Hoeijmakers, J. H. J. , Sung, P. , Wiese, C. , Campisi, J. , & Cooper, P. K. (2016). Non‐catalytic roles for XPG with BRCA1 and BRCA2 in homologous recombination and genome stability. Molecular Cell, 61(4), 535–546. 10.1016/j.molcel.2015.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij, W. P. , Dollé, M. E. , Reiling, E. , Jaarsma, D. , Payan‐Gomez, C. , Bombardieri, C. R. , Wu, H. , Roks, A. J. , Botter, S. M. , van der Eerden, B. , Youssef, S. A. , Kuiper, R. V. , Nagarajah, B. , van Oostrom, C. , Brandt, R. M. , Barnhoorn, S. , Imholz, S. , Pennings, J. L. , de Bruin, A. , … Hoeijmakers, J. H. (2016). Restricted diet delays accelerated ageing and genomic stress in DNA‐repair‐deficient mice. Nature, 537(7620), 427–431. 10.1038/nature19329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij, W. P. , Hoeijmakers, J. H. , & Pothof, J. (2016). Genome integrity in aging: Human syndromes, mouse models, and therapeutic options. Annual Review of Pharmacology and Toxicology, 56, 427–445. 10.1146/annurev-pharmtox-010814-124316 [DOI] [PubMed] [Google Scholar]
- Weeda, G. , Donker, I. , de Wit, J. , Morreau, H. , Janssens, R. , Vissers, C. J. , Nigg, A. , van Steeg, H. , Bootsma, D. , & Hoeijmakers, J. H. (1997). Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Current Biology, 7(6), 427–439. 10.1016/s0960-9822(06)00190-4 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Appendix S2.
Data Availability Statement
The data that support the findings of this study are available on reasonable request to the corresponding author.