

Abstract
In drug research, developing a sound understanding of the key mechanistic drivers of pharmacokinetics (PK) for new molecular entities is essential for human PK and dose predictions. Here, characterizing the absorption, distribution, metabolism, and excretion (ADME) processes is crucial for a mechanistic understanding of the drug–target and drug–body interactions. Sufficient knowledge on ADME processes enables reliable interspecies and human PK estimations beyond allometric scaling. The physiologically based PK (PBPK) modeling framework allows the explicit consideration of organ‐specific ADME processes. The sum of all passive and active ADME processes results in the observed plasma PK. Gene expression information can be used as surrogate for protein abundance and activity within PBPK models. The absolute and relative expression of ADME genes can differ between species and strains. This is affecting both, the PK and pharmacodynamics and is therefore posing a challenge for the extrapolation from preclinical findings to humans. We developed an automated workflow that generates whole‐body gene expression databases for humans and other species relevant in drug development, animal health, nutritional sciences, and toxicology. Solely, bulk RNA‐seq data curated and provided by the Swiss Institute of Bioinformatics from healthy, normal, and untreated primary tissue samples were considered as an unbiased reference of normal gene expression. The databases are interoperable with the Open Systems Pharmacology Suite (PK‐Sim and MoBi) and enable seamless access to a central source of curated cross‐species gene expression data. This will increase data transparency, increase reliability and reproducibility of PBPK model simulations, and accelerate mechanistic PBPK model development in the future.
INTRODUCTION AND MOTIVATION
During drug research and development, gaining insights into the mechanistic drivers of the observed pharmacokinetics (PK) is crucial to enable solid human PK extrapolations as well as predicting human efficacious doses. The absorption, distribution, metabolism, and excretion (ADME) processes build the foundation for a mechanistic understanding of the drug–body and drug–target interactions. Sufficient knowledge of the underlaying ADME processes is needed to enable cross‐species and human PK translations that go beyond the scaling of PK parameters with empiric allometric factors. 1 The physiologically based PK (PBPK) modeling framework can be a powerful tool to predict the PK of an arbitrary compound or particle based on the underlying biophysical, biochemical, and physiological processes.
The PBPK framework is commonly used in academic research 2 and the pharmaceutical industry 3 and by regulatory authorities. 4 PBPK modeling integrates the passive and active ADME processes of a compound and later are mediated by enzymatic or transporter processes at their respective side of action in various tissues. 5 Together, the passive and active processes are the determinants of the overall PK profile in plasma and tissues. In the context of PBPK modeling, gene expression information can be used as a surrogate for protein abundance and hence catalytic activity. 6 Notably, the absolute and relative expression of ADME genes and their ortholog counterparts in nonhuman species can substantially differ between species 7 and strains 8 after a toxicant challenge 9 or between healthy and disease states. 10 This can affect both, the observed PK behavior and the pharmacodynamic (PD) effects, leading to substantially different PK‐PD profiles. 11 Accounting for these differences reduces the inherent uncertainty of cross‐species extrapolations and improves overall PK predictions. 1 , 12 , 13
Efforts to resolve ADME processes down to the individual enzyme contribution (e.g., cytochrome P450 [CYP], uridine diphosphate glucuronosyltransferase, adenosine triphosphate–binding cassette) are investigated rather late during candidate profiling, when the metabolic profiles between human and nonhuman species are compared. 14 Also, explicit ADME phenotyping or kinetic investigations are usually conducted only for humans and pose another challenge for cross‐species extrapolations. 11 In addition, the manual collection of required expression information can be a tedious and an error‐prone task, which can take up a substantial fraction of the overall model development. As a consequence, the explicit consideration of transporter and metabolizing enzymes is not commonly considered in PBPK modeling, especially in early drug discovery and for later cross‐species extrapolations. 15 , 16
Furthermore, without a common reference or standard, the prediction quality of a PBPK model considering expression information might be limited by the quality of the used data. Usually, the used sources are individually chosen and collected from different literature sources. If not explicitly provided together with the PBPK model, this poses a risk for reproducibility and can lead to inconsistent model simulations across or even within an organization.
We established a computational workflow (Figure 1) that enables the reproducible generation of whole‐body gene expression databases that are interoperable with the Open Systems Pharmacology (OSP) Suite components PK‐Sim and MoBi. 17 , 18 We used a single data source (https://bgee.org) that contains publicly available and manually curated RNA‐seq data from healthy, normal, untreated primary tissue samples of multiple species. 19 A seamless access to a central source of curated cross‐species ADME and target gene expression to increase transparency of data usage, ensure reproducibility of PBPK model simulations, and accelerate overall PBPK model development. The workflow is currently implemented for 17 species including humans and is relevant for drug development, animal health, nutritional sciences, and toxicology. To ensure maintenance, open access, and further development, the provided workflow is also part of the OSP community (www.open‐systems‐pharmacology.org). 20
FIGURE 1.
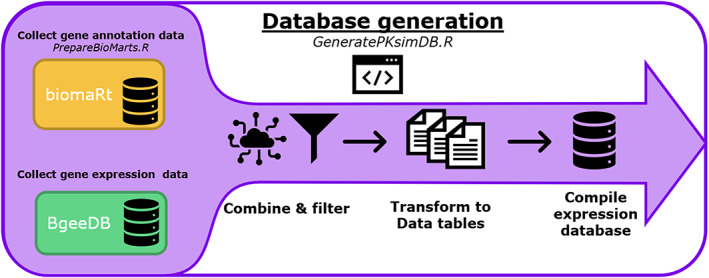
Workflow to generate Open Systems Pharmacology Suite interoperable gene expression databases. First, raw gene expression data from BgeeDB is merged with gene annotation information from biomaRt. After filtering and reformatting, the annotated expression data are split into data tables (Figure 2) and compiled into a local SQL database.
Structure
A gene expression database underlaying the OSP Suite (PK‐Sim and MoBi) consists of 19 interconnected tables holding the gene expression values as well as meta‐information of the genes and experimental sources (Figure 2). Three tables hold information on (i) global expression statistics, (ii) mapping between PK‐Sim organ‐container names and names of the experimentally sampled tissues, and (iii) interconnection information of the different tables (TAB_GLOBAL_STATISTICS, Appendices S5 and S6). The explicit gene expression values are stored in the TAB_EXPRESSION_DATA_VALUES tables and alternative gene and protein name annotations are stored in the TAB_GENES, TAB_GENE_NAMES, TAB_GENE_NAME_TYPES, and TAB_GENE_VARIANTS tables. Information about the data sources is stored in the TAB_EXPRESSION_DATA_RECORDS table.
FIGURE 2.
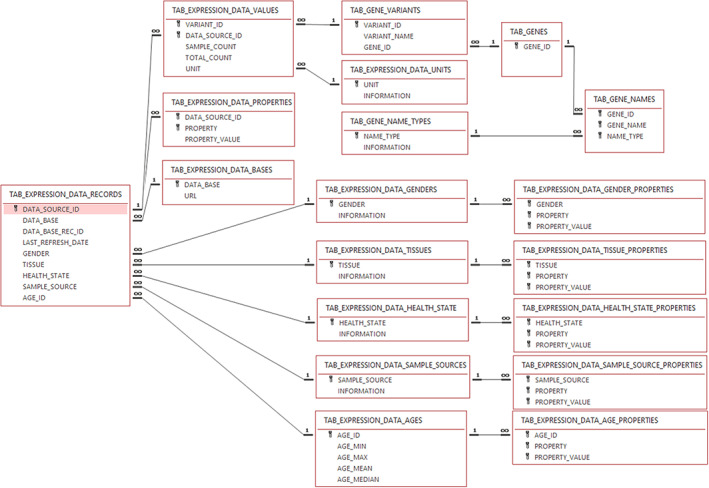
Layout of the Open Systems Pharmacology Suite interoperable gene expression databases. Rectangles represent distinct data tables with the first row being the table name. Key symbols indicate the unique key variables of the respective table. Lines indicate how individual variables are interconnected over multiple tables. Symbols indicate if an entry is mapped to multiple (∞) or a single entry (1).
Content
Gene expression data is provided by Bgee (https://bgee.org/), a public database hosted by the Swiss Institute of Bioinformatics that holds curated gene expression information from publicly available sources originating from primary tissue samples of healthy untreated individuals. 19 Thus, no in vitro cell lines, disease states, gene knock‐out strains, or samples collected after a treatment or challenge of chemicals or drugs are included. Bgee was set up to enable the comparison of gene expression patterns across and within multiple species and holds data generated by multiple methods such as bulk RNA‐seq, single‐cell RNA‐seq, Affymetrix, in situ hybridization, and expressed sequence tags (ESTs). Furthermore, Bgee provides information on gene orthology and homology between organs and species to enable the comparison of tissue‐specific expression patterns. We solely used bulk RNA‐seq data and neglected information originating from other methods such as microarray, in situ hybridization, or EST data. The raw data were accessed with the R‐Bioconductor package BgeeDB 21 and filtered for high‐quality entries with significant expression (“high quality” and “present” annotations). The resulting entries were annotated with the R‐Bioconductor package biomaRt 22 to enable the search across multiple namespaces and to link human genes to their animal orthologues.
The used version of the Bgee repository (Version 15.0) enables access to gene expression information of 52 species. The presented workflow makes use of gene expression information for a subset of 17 species relevant in drug research and development, toxicology testing, animal health, and nutritional sciences. An overview of the total experimental libraries and unique sampled tissues for each species is given in Table 1. Notably, the number of unique sampled tissues can be higher than the number of organs in a species because the sampling information mentions different tissue subsections such as "left lung" and "right lung".
TABLE 1.
Species‐specific content of the Open Systems Pharmacology Suite interoperable gene expression databases
| Species | Binomial name | Total library count | Unique sampled tissues a |
|---|---|---|---|
| Human | Homo sapiens | 5975 | 75 |
| Cattle | Bos taurus | 1985 | 78 |
| Turkey | Meleagris gallopavo | 590 | 17 |
| Mouse | Mus musculus | 566 | 26 |
| Pig | Sus scrofa | 527 | 26 |
| Sheep | Ovis aries | 432 | 54 |
| Guinea pig | Cavia porcellus | 284 | 11 |
| Monkey macaque | Macaca mulatta | 264 | 9 |
| Horse | Equus caballus | 248 | 6 |
| Dog | Canis lupus familiaris | 162 | 30 |
| Zebrafish | Danio rerio | 161 | 12 |
| Rat | Rattus norvegicus | 116 | 13 |
| Rabbit | Oryctolagus cuniculus | 104 | 10 |
| Chicken | Gallus gallus | 84 | 9 |
| Goat | Capra hircus | 64 | 17 |
| Crab‐eating macaque | Macaca fascicularis | 37 | 14 |
| Cat | Felis catus | 34 | 7 |
Note: Total library counts represent the number of experimental gene expression measurements used to construct the data base. Unique sampled tissues indicate the number of sample body sides.
Includes different tissue subsections, resulting in high numbers of unique sampled tissues.
The bulk RNA‐seq gene expression values are reported as transcripts per kilobase million (TPM), fragments per kilobase million (FPKM), and read counts. Apart from read counts, the other measures normalize for sequencing depth and gene length, 23 and information about their technical differences can be found elsewhere. 24 , 25 To enable a comparison with other data sets, expression values can be translated into reads per kilobase million (RPKM). Here, the total read counts of an experiment are calculated and scaled to read counts per million, and next the read count of each gene is divided by the total read count of the respective experiment and divided by the gene length in kilobase (Equation 1).
| (1) |
Before expression data can be used in a PBPK model in PK‐Sim, normalization is needed to enable the integration of expression data from multiple sources (experiments) as well as to calculate the relative expression reflecting the different expression activity across multiple tissues. The normalization procedure and calculation of the relative expression values is defined in the code of the PK‐Sim software itself, and only the unit‐specific geometric mean (Equation 3) of an average gene (gAVG) estimated over all studies is calculated in the presented workflow and stored in the respective expression database table (TAB_GLOBAL_STATISTICS).
Sample counts (SC) represent the gene‐specific quantified expression value of a specific expression unit (e.g., TPM, FPKM, RPKM, read counts). Total counts (TC) represent the sum of all SC in one experiment. The relative individual ratio (RIR) of a gene is calculated for each experiment as the ratio between the SCs and TCs (Equation 2).
| (2) |
Thus, the RIR stands for the relative contribution of a single gene to the total measured signal in an experimental data set (similar to the TPM unit). In a database, the gAVG expression of a gene is estimated over all studies n with the same expression unit and stored in the TAB_GLOBAL_STATISTICS table (Equation 3).
| (3) |
The final normalized expression value () for a gene is calculated with Equation (4) as the ratio of the RIR and gAVG (Equation 4).
| (4) |
For a gene , the organ compartment with the largest normalized expression value is arbitrarily set as 1. The remaining expression values of other organ compartments are scaled relative to this maximal value (<1).
As previously described by Meyer et al., 6 relative expression values in combination with a scaling factor (SF)—an arbitrary reference concentration—can be used as a surrogate for the protein concentration of a gene in an organ compartment (Equation 5). Notably, the default reference concentration is currently defined as 1 μmoL/L in PK‐Sim and can be changed by the user.
| (5) |
The total catalytic activity in an organ is defined as the product of an enzymatic catalytic rate and the organ‐specific protein concentration (Equation 6).
| (6) |
In addition to gene expression values, Bgee provides UBERON ontologies for anatomical entities (i.e., organs and tissues) and life cycle stages (ontogeny). 26 The mapping of gene expression data from external sources (like Bgee) to the respective PK‐Sim organ containers is enabled by a mapping table (Appendix S6). Notably, besides complete organ samples (homogenates), subtissue parts, such as “left lung,” “skin of left leg,” or “lateral live lobe,” can be mapped to their respective PK‐Sim organ container. The current version of PK‐Sim (Version 11) allows the user, besides other properties, to filter for numeric ages (non‐numeric entries are arbitrarily set to 0). Here, negative values indicate the time before, and positive values indicate the time after birth in years, respectively. The life cycle stages from Bgee, however, also contain non‐numeric values such as “adult” or “juvenile state.” Where possible, numeric ages were extracted from Bgee for all species, scaled to years, and used as mean ages (Appendix S3). For non‐numeric stages, the age is arbitrary set to zero. Due to the implemented numeric age restriction in the software, explicit filtering for non‐numeric age stages is currently not possible. However, the non‐numeric stages can be accessed by filtering for the age zero or by using the default setting where all age properties are considered.
CONSTRUCTION
The presented workflow generates gene expression databases interoperable with the OSP Suite (PK‐Sim and MoBi) and is outlined in Figure 1. The underlaying data originates from publicly available sources, accessible through the BgeeDB R package. 21 Currently, gene expression databases for 17 species can be automatically generated with the presented workflow (Table 1). The required R code executing the workflow can be found in the Appendix S3. In addition, the latest maintained version of the code and derived databases are available via the OSP user community GitHub page: https://github.com/Open‐Systems‐Pharmacology/Gene‐Expression‐Databases/releases.
For many users, only ADME‐relevant genes are of interest when developing PBPK models. As suggested by Zhao et al., 25 we compiled a subset of genes empirically known to contribute to ADME processes from public sources. 27 , 28 , 29 , 30 The resulting set consists of 80 search terms based on the first three to four letters of the official HUGO Gene Nomenclature Committee gene symbols 31 to consider all subvariants of a gene family (Figure 3, Appendix S1).
FIGURE 3.
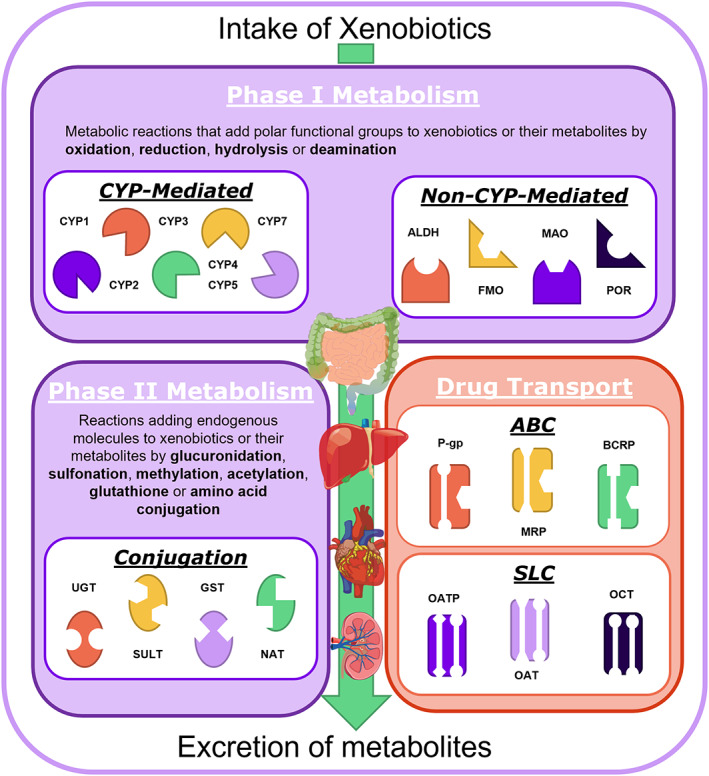
Overview of the mechanistic absorption, distribution, metabolism, and excretion processes. Drug metabolism can be categorized into Phase I and Phase II. Here, the Phase I reactions introduce polar functional groups by oxidation, reduction, and hydrolysis, resulting in activated metabolites that can be better excreted or undergo Phase II metabolism with subsequent conjugation to hydrophilic endogenous moieties. Drug transporters facilitate the enhanced uptake and/or export, supporting the drug elimination by metabolism, and play a complemental role to the Phase I and II metabolism. ABC, adenosine triphosphate–binding cassette; ALDH, Aldehyd‐Dehydrogenasen; BCRP, breast cancer resistance protein; CYP, cytochrome P450; FMO, flavin‐containing monooxygenase; GST, glutathion‐s‐transferase; MAO, monoamine oxidases; MRP, multidrug resistance–associated protein; NAT, N‐acetyltransferase; OAT, Organo‐Anion‐Transporter; OATP = Organic anion transporting polypeptides; OCT, Ornithine transcarbamoylase; P‐gp, P‐glycoprotein; SULT, Sulfotransferase; SLC, solute carrier; UGT, uridine diphosphate glucuronosyltransferase.
Before a species‐specific database can be generated, a preparatory step is needed. First, species‐specific gene annotation information is accessed, rearranged, and stored in a local SQL database via the biomaRt 22 R package for further processing (Appendix S2). Next, the main function (Appendix S3) executes the database generation workflow. Here, the species‐specific gene expression data are accessed from the Bgee server and stored in a local SQL database. Next, the expression data are filtered for genes annotated as “high quality” (only for Bgee Versions >13.0 and <15.0) and “present.” The refined expression data are linked to the previously prepared annotation information including gene identifiers (ENSEMBL gene ids, 22 entrez gene ids, 32 Homo sapiens homolog ensemble gene ids 22 ), symbols (uniport gene symbol, 33 synonym), and names (gene description, common gene name, wikigene name, Homo sapiens homolog gene name 22 ) as well as chromosomal start and end positions and Homo sapiens sequence homology in percent. This diverse gene annotation allows a later fuzzy search by a user across different namespaces and for ambiguous search terms. Finally, the annotated expression data are split into separate data tables matching the PK‐Sim database structure and compiled into a local SQL database (Appendix S4).
The main function (Appendix S3) can be customized by the user considering the following specifications: SPECIE, RELEASE, ADME_ONLY, TPM_ONLY, INCLUDE_RPKM and COMPUTE_IN_RAM. The SPECIE variable defines the desired specie (one of human, monkey, minipig, dog, mouse, rat, rabbit, zebrafish, cattle, horse, cat, guineapig, chicken, goat, sheep, turkey, monkey_CrabEatingMacaque). The RELEASE option specifies the Bgee release version (e.g., “13_0,” “14_2,”, “15_0”). If the ADME_ONLY option is set TRUE, only genes categorized as ADME relevant will be used to set up the database (defined in Appendix S1). Setting the TPM_ONLY to TRUE will result in an expression database containing only values in the TPM expression unit (recommended default). The INCLUDE_RPKM option starts the calculation of RPKM values based on read counts (Equation 3). The COMPUTE_IN_RAM option enables data processing in local memory. Here the expression data are first loaded into the local machine memory, which usually requires excessive amounts of ram. For large data sets, we recommend using the default choices, where all calculations are executed on the local hard drive.
Access
All scripts and R code to generate the PK‐Sim compatible gene expression databases are provided in the supplementary material and are available via the official OSP GitHub page: https://github.com/Open‐Systems‐Pharmacology/Gene‐Expression‐Databases/releases.
To enable data transfer to PK‐Sim, a generated gene expression database must be manually linked to the respective species physiology within the software. The “Application Tab” in PK‐Sim enables the user to specify the path to a species‐specific expression database. A step‐by‐step manual of how to link the gene expression databases to PK‐Sim can be found in the OSP online documentation: https://docs.open‐systems‐pharmacology.org/working‐with‐pk‐sim/pk‐sim‐documentation/pk‐sim‐options#template‐database‐path. A tutorial on how to incorporate explicit ADME processes and best‐practice recommendation of how to develop and validate mechanistic PBPK models can found in the literature 5 , 6 , 14 and in the OSP online documentation: https://docs.open‐systems‐pharmacology.org/working‐with‐pk‐sim/pk‐sim‐documentation/pk‐sim‐expression‐profile#background‐active‐processes‐in‐pk‐sim.
DISCUSSION
The presented computational workflow enables the reproducible generation of whole‐body gene expression databases interoperable with the OSP Suite PK‐Sim. In total, 17 species relevant for drug development, animal health, nutritional sciences, and toxicology including humans are available (Table 1). Previously, only a single gene expression database for humans was available for PK‐Sim, and thus cross‐species extrapolations of explicit ADME processes was rather challenging. Furthermore, the previous database integrated data from different biological backgrounds, such as cancerous tissue and in vitro cell lines that do not represent a healthy in vivo state. A substantial fraction of the data originated from just a few literature sources, such as for the reverse‐transcription polymerase chain reaction data, 6 or from different experimental methods, such as microarray or EST data, 6 that cannot be used together for an integrated analysis.
The presented whole‐body gene expression databases are based on bulk RNA‐seq data solely derived from healthy, normal, and untreated primary tissue samples. Thus, they provide a reference for healthy normal gene expression as a basis for ADME, target, and off‐target genes. 19
However, the presented databases also come with limitations. Users should be aware that RNA expression is only a surrogate for protein abundance and activity. Gene expression might be influenced by a circadian rhythm, 34 posttranscriptional, 35 and posttranslational modifications 36 that can affect protein amounts and activity. For example, a similar gene expression in distinct organs or tissues might result in different protein abundance due to different capacities of the translation cycle. 37 The used data sets are based on bulk RNA‐seq data from tissue homogenates. Here, the gene expression is averaged over the used tissue mass. Thus, different expression values of specific cell types or subcompartments of the sample cannot be resolved. Furthermore, the underlaying data source does not account for individual expression differences (within population distribution) or alternative gene variants (splicing) that could have profound effects on PK and PD properties. 38 , 39 Considering more detailed information could further support population PK and PK‐PD extrapolations.
Currently, the PK‐Sim interoperable gene expression databases are limited to the integration of relative expression levels. These are translated into actual protein concentrations by multiplication with an arbitrary reference concentration (1 μmoL/L) that is not part of the expression databases but, rather, hard coded in the OSP software. Here, replacing the concept of relative expression values with experimentally measured enzyme tissue concentrations could be of benefit for further development, removing uncertainties about posttranscriptional effects. However, comparing proteome quantifications across different experiments without a shared quantification standard is a challenge in itself and would need substantial experimental investments. 40 A step forward could be the scaling of relative expression values with well‐characterized organ‐specific protein quantifications of ADME enzymes that are available for many species. 7 , 41 , 42 , 43 Here, the combination of relative expression values with measured protein concentrations could be used to replace the arbitrary scaling factor (reference concentration) that is currently used.
OSP users need to be aware of the default software settings when using gene expression information. By default, expression data from the underlaying database for a specific enzyme (e.g., CYP3A4) is averaged over all available entries regardless of age, gender, or health state to derive relative expression values. The user is obliged to review the used expression data, such that only appropriate information suited for the specific modeling task at hand is used.
We provide the established workflow for species‐specific database generation to the OSP GitHub community (https://github.com/Open‐Systems‐Pharmacology) to ensure transparency, open access, and future maintenance. By having seamless access to a central source of curated interspecies gene expression data, we seek to contribute to the development of a community‐wide standard. In practice, we aim to increase data transparency, support a reliable integration of explicit ADME processes in PBPK models, and accelerate overall PBPK model development for interspecies scaling, drug–drug interaction predictions, pediatrics extrapolation, toxicokinetic studies, and PK‐PD considerations in the future.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST
H.C. is an employee at Sanofi‐Aventis Deutschland GmbH and may hold shares and/or stock options of the company. H.C. was an employee at Boehringer Ingelheim Pharma GmbH & Co. KG and finished the manuscript as employee of Sanofi‐Aventis Deutschland GmbH. All other authors declared no competing interests for this work.
Supporting information
Appendix S1.
Appendix S2.
Appendix S3.
Appendix S4.
Appendix S5.
Appendix S6.
ACKNOWLEDGMENTS
The authors thank Dr. Jens Markus Borghardt for his critical review of the manuscript and helpful discussions of the concept as well as Dr. Achim Sauer for his consent to this work. Furthermore, the authors thank Dr. Donato Teutonico, Pavel Balazki, Dr. Sebastian Frechen, and Dr. Stephan Schaller for their in‐depth discussions as well as the Open Systems Pharmacology GitHub community for their user feedback.
Cordes H, Rapp H. Gene expression databases for physiologically based pharmacokinetic modeling of humans and animal species. CPT Pharmacometrics Syst Pharmacol. 2023;12:311‐319. doi: 10.1002/psp4.12904
REFERENCES
- 1. Kuepfer L. Prospects and limitations of physiologically‐based pharmacokinetic modelling for cross‐species extrapolation. SVU Int J Vet Sci. 2019;2:45‐51. [Google Scholar]
- 2. Jamei M. Recent advances in development and application of physiologically‐based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rowland M, Peck C, Tucker G. Physiologically‐based pharmacokinetics in drug development and regulatory science. Annu Rev Pharmacol. 2011;51:45‐73. [DOI] [PubMed] [Google Scholar]
- 4. Shebley M, Sandhu P, Emami Riedmaier A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104:88‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kuepfer L, Niederalt C, Wendl T, et al. Applied concepts in PBPK modeling: how to build a PBPK/PD model. CPT Pharmacometrics Syst Pharmacol. 2016;5:516‐531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Meyer M, Schneckener S, Ludewig B, Kuepfer L, Lippert J. Using expression data for quantification of active processes in physiologically based pharmacokinetic modeling. Drug Metab Dispos. 2012;40:892‐901. [DOI] [PubMed] [Google Scholar]
- 7. Basit A, Fan PW, Khojasteh SC, et al. Comparison of tissue abundance of non‐cytochrome P450 drug metabolizing enzymes by quantitative proteomics between humans and laboratory animal species. Drug Metab Dispos. 2021;50:197‐203. [DOI] [PubMed] [Google Scholar]
- 8. Barr JT, Tran TB, Rock BM, Wahlstrom JM, Dahal UP. Strain‐dependent variability of early discovery small molecule pharmacokinetics in mice: does strain matter? Drug Metab Dispos. 2020;48:613‐621. [DOI] [PubMed] [Google Scholar]
- 9. Schenk A, Ghallab A, Hofmann U, et al. Physiologically‐based modelling in mice suggests an aggravated loss of clearance capacity after toxic liver damage. Sci Rep. 2017;7:6224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lake AD, Novak P, Fisher CD, et al. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:1954‐1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. (CHMP), C. for M. P. for H. U . Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation . 2018. . https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐reporting‐physiologically‐based‐pharmacokinetic‐pbpk‐modelling‐simulation_en.pdf
- 12. Thiel C, Hofmann U, Ghallab A, Gebhardt R, Hengstler JG, Kuepfer L. Towards knowledge‐driven cross‐species extrapolation. Drug Discov Today Dis Model. 2016;22:21‐26. [Google Scholar]
- 13. Thiel C, Schneckener S, Krauss M, et al. A systematic evaluation of the use of physiologically based pharmacokinetic modeling for cross‐species extrapolation. J Pharm Sci. 2015;104:191‐206. [DOI] [PubMed] [Google Scholar]
- 14. Schneckener S, Preuss TG, Kuepfer L, Witt J. A workflow to build PBTK models for novel species. Arch Toxicol. 2020;94:3847‐3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Abouir K, Samer CF, Gloor Y, Desmeules JA, Daali Y. Reviewing data integrated for PBPK model development to predict metabolic drug‐drug interactions: shifting perspectives and emerging trends. Front Pharmacol. 2021;12:708299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sager JE, Yu J, Ragueneau‐Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos. 2015;43:1823‐1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Willmann S, et al. PK‐Sim®: a physiologically based pharmacokinetic ‘whole‐body’ model. Bios. 2003;1:121‐124. [Google Scholar]
- 18. Eissing T, Kuepfer L, Becker C, et al. A computational systems biology software platform for multiscale modeling and simulation: integrating whole‐body physiology, disease biology, and molecular reaction networks. Front Physiol. 2011;2:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bastian FB, et al. The Bgee suite: integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Res. 2020;49:D831‐D847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lippert J, Burghaus R, Edginton A, et al. Open systems pharmacology community—an open access, open source, Open Science approach to modeling and simulation in pharmaceutical sciences. CPT Pharmacometrics Syst Pharmacol. 2019;8:878‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komljenovic A, Roux J, Wollbrett J, Robinson‐Rechavi M, Bastian FB. BgeeDB, an R package for retrieval of curated expression datasets and for gene list expression localization enrichment tests. F1000Res. 2016;5:2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc. 2009;4:1184‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y, Li MC, Konaté MM, et al. TPM, FPKM, or normalized counts? A comparative study of quantification measures for the analysis of RNA‐seq data from the NCI patient‐derived models repository. J Transl Med. 2021;19:269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pimentel, H. Review of RNA‐seq expression units. 2014. Accessed January 11, 2022. https://haroldpimentel.wordpress.com/2014/05/08/what‐the‐fpkm‐a‐review‐rna‐seq‐expression‐units/
- 25. Zhao S, Ye Z, Stanton R. Misuse of RPKM or TPM normalization when comparing across samples and sequencing protocols. RNA. 2020;26:903‐909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mungall CJ, Torniai C, Gkoutos GV, Lewis SE, Haendel MA. Uberon, an integrative multi‐species anatomy ontology. Genome Biol. 2012;13:R5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Whirl‐Carrillo M, Huddart R, Gong L, et al. An evidence‐based framework for evaluating pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2021;110:563‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Elbourne LDH, Tetu SG, Hassan KA, Paulsen IT. TransportDB 2.0: a database for exploring membrane transporters in sequenced genomes from all domains of life. Nucleic Acids Res. 2017;45:D320‐D324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Saier MH, et al. The Transporter Classification Database (TCDB): 2021 update. Nucleic Acids Res. 2020;49:D461‐D467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074‐D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tweedie S, et al. Genenames.Org: the HGNC and VGNC resources in 2021. Nucleic Acids Res. 2020;49:D939‐D946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maglott D, Ostell J, Pruitt KD, Tatusova T. Entrez gene: gene‐centered information at NCBI. Nucleic Acids Res. 2007;35:D26‐D31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Consortium, T. U ., et al. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2020;49:D480‐D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Castelo‐Szekely V, Arpat AB, Janich P, Gatfield D. Translational contributions to tissue specificity in rhythmic and constitutive gene expression. Genome Biol. 2017;18:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao BS, Roundtree IA, He C. Post‐transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18:31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Santos AL, Lindner AB. Protein posttranslational modifications: roles in aging and age‐related disease. Oxid Med Cell Longev. 2017;2017:5716409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gerashchenko MV, Peterfi Z, Yim SH, Gladyshev VN. Translation elongation rate varies among organs and decreases with age. Nucleic Acids Res. 2020;49:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilson JF, Weale ME, Smith AC, et al. Population genetic structure of variable drug response. Nat Genet. 2001;29:265‐269. [DOI] [PubMed] [Google Scholar]
- 39. Ma MK, Woo MH, McLeod HL. Genetic basis of drug metabolism. Am J Health Syst Pharm. 2002;59:2061‐2069. [DOI] [PubMed] [Google Scholar]
- 40. Schork K, Podwojski K, Turewicz M, Stephan C, Eisenacher M. Quantitative methods in proteomics. Methods Mol Biol. 2021;2228:1‐20. [DOI] [PubMed] [Google Scholar]
- 41. Buyssens L, de Clerck L, Schelstraete W, et al. Hepatic cytochrome P450 abundance and activity in the developing and adult Göttingen minipig: pivotal data for PBPK modeling. Front Pharmacol. 2021;12:665644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hammer H, Schmidt F, Marx‐Stoelting P, Pötz O, Braeuning A. Cross‐species analysis of hepatic cytochrome P450 and transport protein expression. Arch Toxicol. 2021;95:117‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang L, Prasad B, Salphati L, et al. Interspecies variability in expression of hepatobiliary transporters across human, dog, monkey, and rat as determined by quantitative proteomics. Drug Metab Dispos. 2015;43:367‐374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Appendix S2.
Appendix S3.
Appendix S4.
Appendix S5.
Appendix S6.