

Abstract
Background
Myocardial fibrosis (MF) is an advanced pathological manifestation of many cardiovascular diseases, which can induce heart failure and malignant arrhythmias. However, the current treatment of MF lacks specific drugs. Ginsenoside Re has anti-MF effect in rat, but its mechanism is still not clear. Therefore, we investigated the anti-MF effect of ginsenoside Re by constructing mouse acute myocardial infarction (AMI) model and AngⅡ induced cardiac fibroblasts (CFs) model.
Methods
The anti-MF effect of miR-489 was investigated by transfection of miR-489 mimic and inhibitor in CFs. Effect of ginsenoside Re on MF and its related mechanisms were investigated by ultrasonographic, ELISA, histopathologic staining, transwell test, immunofluorescence, Western blot and qPCR in the mouse model of AMI and the AngⅡ-induced CFs model.
Results
MiR-489 decreased the expression of α-SMA, collagenⅠ, collagen Ⅲ and myd88, and inhibited the phosphorylation of NF-κB p65 in normal CFs and CFs treated with AngⅡ. Ginsenoside Re could improve cardiac function, inhibit collagen deposition and CFs migration, promote the transcription of miR-489, and reduce the expression of myd88 and the phosphorylation of NF-κB p65.
Conclusion
MiR-489 can effectively inhibit the pathological process of MF, and the mechanism is at least partly related to the regulation of myd88/NF-κB pathway. Ginsenoside Re can ameliorate AMI and AngⅡ induced MF, and the mechanism is at least partially related to the regulation of miR-489/myd88/NF-κB signaling pathway. Therefore, miR-489 may be a potential target of anti-MF and ginsenoside Re may be an effective drug for the treatment of MF.
Keywords: Acute myocardial infarction, Angiotensin Ⅱ, Ginsenoside re, miR-489, Myocardial fibrosis
Graphical abstract
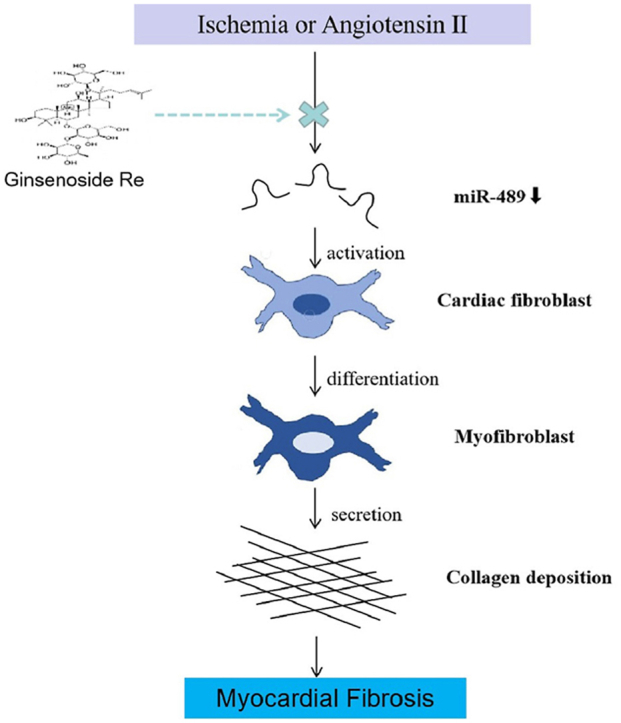
1. Introduction
Myocardial fibrosis (MF) is an advanced pathological manifestation of various cardiovascular diseases, including acute myocardial infarction (AMI), hypertension and pulmonary hypertension [1]. The main pathological changes are the imbalance of synthesis, metabolism and degradation of collagen in the extracellular matrix (ECM). In the pathological process, cardiac fibroblasts (CFs) are abnormally activated, collagen fibers are excessive abnormal accumulation in the ECM, and collagen composition is changed [2]. Type Ⅰ and Ⅲ collagen are the main components of ECM, accounting for more than 90% of the total amount of myocardial interstitial collagen. The continuous deposition of ECM severely affects the normal systolic and diastolic function of the heart, which ultimately leads to refractory heart failure, malignant arrhythmia, even sudden death [3]. MF has become a pathological manifestation and a pathogenic factor of various cardiovascular diseases, and is also an important reason for the irreversible and continuous development of ventricular remodeling [4].
CFs are the main effector cells that secrete the ECM of the myocardium [5]. The adult mammalian myocardium contains abundant CFs [6]. Under normal circumstances, these cells maintain a state of equilibrium and are responsible for homeostasis of the ECM. However, when stimulated by pathological conditions (like ischemia, inflammation), CFs are abnormally activated, proliferate and transdifferentiate into myocardial fibroblasts expressing α-SMA [[7], [8], [9]]. The transformation from CFs to myocardial fibroblasts further secrets large amounts of collagen-related proteins. As a consequence, a large amount of deposition and reduction of collagen degradation lead to the imbalance and disorder of collagen structure, which ultimately leads to MF [10,11].
After MI, the repair of fibrotic scar in the damaged area of heart has irreplaceable significance for maintaining the integrity of heart structure, maintaining the heart pump function and effectively delivering blood to various organs [12]. However, after the fibrotic scar repair in the infarction zone, the process of fibrosis in the heart tissue does not stop but continues, affecting the infarct edge and even the non-infarct area [13]. Secondary interstitial fibrosis of myocardium in the non-infarction zone will aggravate myocardial ischemia and hypoxia, increase ventricular stiffness and decrease ventricular compliance, resulting in systolic dysfunction and greatly increasing the incidence of heart failure [14]. Therefore, inhibiting the hyperfibrotic process after MI, especially that in the non-infarction zone, is an effective method to relieve left ventricular remodeling and improve cardiac function.
MF can be induced by many factors, such as angiotensin Ⅱ (AngⅡ), transforming growth factors and inflammatory factors [2]. These factors influence the occurrence and development of MF through various pathways [15]. However, studies on these factors have failed to find effective drugs for the treatment of MF due to different reasons. Therefore, it is necessary for the treatment of MF to find key factors in the pathogenesis of MF.
MicroRNAs (miRNAs) are a class of endogenous, non-coding single-stranded RNAs with 18–22 bases. Although miRNAs can not encode proteins due to the lack of open reading frame, they can degrade mRNAs at transcriptional level by binding to the 3′untranslated region (UTR) sequences of target genes [16]. Functionally, miRNAs regulate multiple biological processes, including cell proliferation, differentiation, apoptosis and development [17,18]. They are widespread in eukaryotes and regulate large amounts of mRNAs. It has been reported that miRNAs are able to regulate one-third of human genes [15]. Thousands of miRNAs are abnormally expressed in human heart tissue with heart failure [19]. Thus, it is believed that miRNAs may serve as key regulators in cardiac fibrotic diseases.
In recent years, a large number of miRNAs have been confirmed to have anti-fibrotic functions, pro-fibrotic functions, or serve as fibrotic biomarkers. For example, miR-208b improves post-infarction MF via directly targeting GATA binding protein 4 [20]. MiR-199a promotes the proliferation and migration of CFs through regulating the secretion of frizzled-related protein 5 [21].
Recently, miR-489 has gradually attracted wide attention, which plays an important role as tumor suppressor in various kinds of tumors, such as hypopharyngeal squamous cell carcinoma [22], gastric cancer [23], bladder cancer [24], glioma [25]. MiR-489 is also involved in the pathological process of fibrosis. The expression of miR-489 levels were decreased in a mouse model of silica-induced pulmonary fibrosis. MiR-489 overexpression supperssed pulmonary fibrosis both in vivo and vitro by directly regulating myeloid differentiation primary response gene 88 (myd88) which is critical mediator in the inflammation [26]. Myd88 is related to MI induced by ischemia/reperfusion, and knockdown of myd88 is able to reduce myocardial infarct sizes [27,28]. Nuclear factor-kappa B (NF-κB) is a vital downstream effector of myd88 [29]. The myd88-dependent signaling pathway activates NF-κB, thereby inducing the production of inflammatory cytokines (like IL-6, IL-1β, and TNF-α) [30]. Inflammation plays a crucial role in the overall process of MF [7,31]. Moreover, miR-489 is widely expressed in myocytes and fibroblasts. AngⅡ-induced miR-489 downregulation in cardiomyocytes. The overexpression of miR-489 reduces hypertrophic responses induced by AngⅡ in vivo and vitro. Importantly, MF was alleviated in miR-489 transgenic mice and myd88-knockout mice induced by AngⅡ [32]. These data indicate that miR-489 is an anti-fibrotic factor. Considering the anti-myocardial hypertrophy and anti-MF effects of miR-489, it may become an important target for the treatment of ventricular remodeling. However, it is not yet clear whether miR-489 has the same effect in the pathological process of MF induced by AMI, and whether its effect is related to target genes myd88.
Ginseng has been used as a medicinal herb for thousands of years against various diseases. There is increasing evidence that ginseng has beneficial effects on cardiac disease [33]. Among the complex constituents of ginseng, ginsenosides are the major pharmacological active components. Ginsenoside Re (G-Re) is distributed in ginseng leaf, berry and root, accounting for about 30% of total ginsenosides [34,35]. G-Re has many pharmacological activities, such as reducing oxidative stress and inflammation [36], inhibiting vascular smooth muscle cells proliferation [37], attenuating neuroinflammation [38]. Importantly, G-Re possesses multifaceted beneficial pharmacological effects on cardiovascular system, such as anti-ischemic effect, anti-arrhythmic effect and angiogenic regeneration [39]. Recently, it has been reported that G-Re has anti-fibrotic effect. G-Re can improve cardiac function and MF induced by isoproterenol and AMI, and the mechanism may be related to regulating TGF-β1/Smad3 pathway in rats [40,41]. Whether the anti-fibrotic effect of G-Re is related to the miR-489/myd88/NF-κB pathway remains to be further studied.
In this study, we transfected miR-489 mimic and inhibitor into CFs to observe the anti-MF effect of miR-489 and its regulatory effect on downstream myd88. In addition, we constructed AMI mouse model and AngⅡ-induced CFs model to observe the anti-MF effect of G-Re and its regulatory effect on miR-489 related signaling pathway.
2. Materials and methods
2.1. Establishment and treatment of AMI model in mice
All the animal experiments were approved by the ethics committee of the Fujian University of Traditional Chinese Medicine (IACUC Issue No: FJTCM IACUC 2019044). The C57BL/6 mice (8 weeks, weighing 20–25g) were purchased from the Shanghai Slake Laboratory Animal Co., LTD. Experimental Animal Center of Fujian University of Traditional Chinese Medicine provided with a standard diet and free access to water. Mice were maintained in a temperature-controlled (20–22 °C) environment under a 12 h light-dark cycle. The experimental mice were randomly divided into four groups (n = 10/group), as follows: Sham operation group (Sham), MI group (MI), MI + G-Re low-dose(19.5 mg/kg/d) group (L), and MI + G-Re high-dose (39 mg/kg/d) group (H). In this study, a AMI mouse model was constructed by ligating the anterior descending branch of the left coronary artery according to the previous study [42]. In Sham group, mice were only threaded without ligation, and other operations were the same as those in MI group. At the next day of AMI surgery, mice were intragastric administrated with 19.5 mg/kg/day or 39 mg/kg/day of G-Re (Solarbio, China) for 4 weeks. Meanwhile, mice in Sham group and MI group were orally administrated with the same volume of normal saline.
2.2. Isolation of cardiac fibroblasts
Cardiac fibroblasts (CFs) were isolated as previously reported [3]. Briefly, C57BL/6 mice (3–7 days) were anesthetized and sacrificed. After disinfection with 75% ethanol, mice hearts were harvested and washed with normal saline. Heart tissues were cut with an ophthalmic scissors. After 4 mL of 0.125% Collagenase typeⅡ (Gibco, USA) and Trypsin-EDTA (Gibco, USA) mixture were added, tissues are digested on a water bath shaker at 37 °C for 20 min each time until tissues are completely digested. After centrifugation, the supernatant was discarded. Collected CFs were cultured in Dulbecco's modified eagle Medium (DMEM) (Gibco, USA) containing 10% fetal bovine serum (FBS) (Gibco, USA) and 1% penicillin streptomycin solution (Hyclone, USA) in a 5% CO2 incubator at 37 °C. Non-adherent cells were removed by aspiration and discarded after 90 min. The third-generation CFs were used for experiments. The MF phenotype was induced by AngⅡ. CFs were co-treated with AngⅡ (10−6 M) [43] and different concentrations of G-Re (50 μmol/L, 100 μmol/L, 200 μmol/L) for 24 h.
2.3. Transfection of mimic and inhibitor
The miR-489–3p mimic, mimic negative control (mimic-NC), miR-489–3p inhibitor and inhibitor negative control (inhibitor-NC) were purchased from Guangzhou Ruibo Biotechnology Co. LTD (RiboBio, China). Cells were transfected with 50 nM mimic, 150 nM inhibitor and corresponding negative control, respectively. The transfection was performed using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instruction.
2.4. Echocardiography
At the 2nd and 4th week postoperatively, mice were anesthetized with 3% isoflurane and fixed on an ultrasound test table. The echocardiography examination was performed with Vevo 2100 High-Resolution Imaging System (Visual Sonics, Toronto, ON, Canada) for the mice. Two-dimensional guided M-mode tracings were recorded in both parasternal long and short axis views at the level of papillary muscles. Left ventricular ejection fraction (LVEF) and left ventricular fractional shortening (LVFS) were calculated with the established standard equation. All the measurements were made from more than three beats and averaged [32].
2.5. Assessment of heart weight index (HWI) and heart weight to tibial length ratio (HW/TL)
Mice hearts were removed rapidly and heart tissues were washed with precooled phosphate-buffered saline (PBS). Hearts were weighed after separating the aorta and adipose tissue. The body weight and left tibial length of mice were also recorded. The HWI was calculated by dividing the heart weight by body weight and HW/TL was calculated by dividing the heart weight by tibial length.
2.6. Enzyme-linked immunosorbent assay (ELISA)
Blood samples were collected from the orbital venous plexus after anesthesia. The expression levels of creatine kinase-MB (CK-MB), AngⅡ, brain natriuretic peptide (BNP), alanine aminotransferase (ALT) and serum creatinine (S–Cr) were measured. All serological detections were performed using related commercial kits purchased from the Jiangsu Enzyme Industry Co., Ltd. (MEIMIAN, China).
2.7. Histological analysis
Histological analysis was performed as previously reported [42]. Briefly, heart tissues were fixed in 4% paraformaldehyde, embedded in paraffin and sectioned into 5 μm slices. Hematoxylin-eosin (HE) staining and standard Masson trichrome staining (Solarbio, China) were performed according to manufacturer's instructions.
2.8. Transwell assay
600 μL of DMEM with AngⅡ (10−6 M) and various concentrations of G-Re were added in each well of the 24-well plate, and transwell chambers were placed into each well. Cells in different groups were prepared for cell suspension in serum-free DMEM, and then about 105 cells were added in each chamber. After incubation at 37 °C incubator for 24 h, the chamber was taken out and cells were fixed for 30 min. Crystal violet staining assay was performed to evaluate the cellular migration ability under microscope. Cells in 5 randomly selected fields were counted.
2.9. Immunofluorescence staining
CFs were inoculated in laser confocal petri dishes, washed with PBS after intervention for a corresponding time, and fixed with 4% paraformaldehyde at room temperature. The corresponding primary antibodys were added and 4 °C incubated overnight. Rabbit anti-vimentin (1:100; SAB), rabbit anti-p–NF–κB p65 (1:200; Immunoway), rabbit anti-Collagen1A1 (1:100; SAB), mouse anti-Collagen Ⅲ (1:200; SAB), rabbit anti-α-SMA (1:100; CST). Incubate corresponding secondary antibodies at room temperature and avoid light for 1 h. Random images were taken using a laser confocal scanning microscope. Image J software analyzes the images.
2.10. Western blot analysis
The hearts of mice and CFs in each group were harvested and homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime, Shanghai, China). The supernatant was retained for standby application. The protein concentration in the specimen was measured using the bicinchoninic acid protein assay kit (Thermo Scientific, USA). Proteins were transferred to polyvinylidene fluoride (PVDF) membranes (GE Healthcare Life Sciences, Germany). After the membranes were sealed with 5% nonfat milk, corresponding primary antibodies were incubated overnight at 4 °C. Rabbit anti-myd88 (1:1000; Abbkine), rabbit anti–NF–κB p65(1:1000; SAB), rabbit anti-p–NF–κB p65 (1:1000; Immunoway), anti-collagenⅠ (1:1000; Proteintech), rabbit anti-collagen Ⅲ (1:1000; Proteintech), rabbit anti-α-SMA (1:1000; CST), rabbit anti-GAPDH (1:5000; Abbkine). Membranes were immersed in secondary antibody solution at room temperature for 2 h. Subsequently, bands were visualized using enhanced chemiluminescence detection system (Bio-Rad Laboratories, USA). The relative protein expression levels were normalized as the ratio of the grayscale value of each protein to that of GAPDH or the ratio of the phosphorylated protein to total protein with Image Lab software version 5.0.
2.11. Real-time quantitative RT-PCR
Total RNA from tissues or cells were extracted by TRIzol reagent (TaKaRa), transcribed into complementary deoxyribose nucleic acid (cDNA) following the reverse transcription kit instruction (TaKaRa). The target genes were amplified by qRT-PCR according to the instructions of kits (TaKaRa), reversely. The relative expression level of target genes were analyzed by the 2−ΔΔCT method. GAPDH and U6 were used as internal references in the quantitative analysis of myd88, ANF, BNP, β-MHC and miR-489 expression, respectively. Primer sequences used were as follows: miR-489–3p, 5′-CGCCCGAATGACACCACATATATG-3’; myd88, F: 5′-GCATGGTGGTGGTTGTTTCT-3′, R: 5′-TCTGTTGGACACCTGGAGAC-3’; ANP, F: 5′-CCCTCCGATAGATCTGCCCT-3’; R: 5′-GTCCACTCTGGGCTCCAATC-3’; BNP, F: 5′-GGTGCTGTCCCAGATGATTCT-3’; R: 5′-AATTGCTCTGGAGACTGGCT-3’; β-MHC, F: 5′-CGCATGGACCTAGAGCGAG-3’; R: 5′-GCTTGCTCATCCTCAATCCT-3’; U6, F: 5′-GCTTCGGCAGCACATATACTAA-3’; R: 5′-AACGCTTCACGAATTTGCGT-3’; GAPDH, F: 5′-TGCCCCCATGTTTGTGATG-3’; R: 5′-TGTGGTCATGAGCCCTTCC-3’.
2.12. Statistical analysis
SPSS 22.0 and GraphPad prism 5.0 were used for data analysis. Results are expressed as mean ± SD. Statistical comparison among different groups was performed by one-way ANOVA. Two groups were evaluated by Student t-test. P < 0.05 was considered statistically significant.
3. Results
3.1. miR-489 regulates myd88/NF-κB pathway in CFs
We transfected miR-489 mimic into CFs to overexpress miR-489. The qPCR detection showed that the expression of miR-489 in CFs was significantly increased after transfection with miR-489 mimic (P < 0.01), while there was no statistical difference between normal and mimic-NC control group (Fig. 1A), indicating that miR-489 mimic was successfully transfected. Overexpression of miR-489 could significantly inhibit the fibrotic reaction of CFs, as evidenced by decreased the expression of α-SMA (Fig. 1B), collagenⅠ (Fig. 1C) and collagen Ⅲ (Fig. 1D) in normal CFs. The same results were found in CFs treated by AngⅡ (Figs. S1A–C). We transfected miR-489 inhibitor into CFs to inhibit miR-489. Inhibition of miR-489 could significantly promote the fibrosis reaction of CFs, as evidenced by increased the expression of α-SMA (Fig. 1E), collagenⅠ (Fig. 1F) and collagen Ⅲ (Fig. 1G) in normal CFs. The same results were found in CFs treated by AngⅡ(Figs. S1D–F). These data suggest that miR-489 exerts an anti-fibrosis function in CFs.
Fig. 1.

MiR-489 modulates the fibrotic response of CFs. (A) The expression of miR-489 was analyzed by qPCR in CFs transfected miR-489 mimic. (B–D) The expression of α-SMA, Collagen Ⅰ and Collagen Ⅲ were analyzed in CFs transfected with miR-489 mimic, ∗P < 0.01 vs Normal group; §P < 0.05 vs Normal group; #P<0.01 vs mimic group, &P<0.05 vs mimic group. (E–G) The expression of α-SMA, Collagen Ⅰ and Collagen Ⅲ were analyzed in CFs transfected with miR-489 inhibitor, ∗P < 0.01 vs Normal group; #P<0.01 vs inhibitor group. All data are presented as the mean ± SD (n = 3).
Myd88 has been confirmed to be a direct downstream target of miR-489, which can promote cardiac hypertrophy and pulmonary fibrosis by activating NF-κB [26,32]. Therefore, we tested whether miR-489 regulates myd88/NF-κB pathway in CFs. Overexpression of miR-489 could down-regulate the expression of myd88 (Fig. 2A) and inhibit the phosphorylation of NF-κB p65 (Fig. 2B) in normal CFs. Moreover, we also observed the same effect in CFs induced by AngⅡ (Figs. S2A and 2B). Inhibition of miR-489 promoted the expression of myd88 (Fig. 2C) and the phosphorylation of NF-κB p65 (Fig. 2D) in normal CFs. The same results were found in CFs treated by AngⅡ (Figs. S2C and 2D). These data suggest that the anti-MF effect of miR-489 may be achieved by regulating the myd88/NF-κB pathway.
Fig. 2.

MiR-489 regulated myd88/NF-κB pathway in CFs. (A, B) The expression of myd88, NF-κB p65 and p-p65 were analyzed in CFs transfected with miR-489 mimic, ∗P < 0.01 vs Normal group; #P<0.01 vs mimic group. (C, D) The expression of myd88, NF-κB p65 and p-p65 were analyzed in CFs transfected with miR-489 inhibitor, ∗P < 0.01 vs Normal group; §P < 0.05 vs Normal group; #P<0.01 vs inhibitor group; &P<0.05 vs inhibitor group. All data are presented as the mean ± SD (n = 3).
3.2. G-Re improves cardiac function in post-AMI mice
Compared with mice in Sham group, AMI lead to significant decrease in LVEF and LVFS levels in both 2 and 4 weeks (P < 0.01) (Figs. S3A and S3B). Compared with mice in model group, administration of G-Re could markedly improve LVEF and LVFS (P < 0.01) (Figs. S3A and S3B), and there was no statistical difference between the two doses (P > 0.05) (Figs. S3A and S3B). Serum BNP is an important clinical marker to evaluate the degree of heart failure. We found that serum BNP level of the model group was significantly higher than that of the normal group (P < 0.01) (Fig. S3C), and the BNP level was obviously reduced after the intervention of G-Re (P < 0.01) (Fig. S3C), suggesting that G-Re can improve heart failure induced by AMI.
3.3. G-Re inhibits compensatory cardiac hypertrophy in post-MI mice
With the mice AMI model established and drug intervention, the myocardial hypertrophy reactions were evaluated through HWI, HW/TL, and the mRNAs expression of ANP, BNP and β-MHC in myocardium. As shown in Fig. 3A, the cardiac volume of mice was significantly enlarged in the MI group. There was no statistically significant difference in body weight among four groups (P > 0.05) (Fig. 3B). Compared with the Sham group, the HWI and HW/TL, as well as the mRNAs expression of ANP, BNP, β-MHC obviously increased in MI group (P < 0.01). However, both low and high doses of G-Re inhibited these effects (P < 0.05), and there was no statistical difference between the two doses (P > 0.05) (Fig. 3C-G). These data indicate that AMI induces compensatory myocardial hypertrophy, and G-Re can effectively inhibit this effect.
Fig. 3.

Effects of G-Re on compensatory cardiac hypertrophy in AMI mice. (A) Alterations of cardiac phenotypes in AMI mice after 4 weeks. (B) Weight of mice 4 weeks after AMI (n = 10). (C) Heart weight to body weight ratios (HWI) (n = 10). (D) Heart weight to tibia length ratios (HW/TL) (n = 10). (E,F,G) Expression levels of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), and β-myosin heavy chain (β-MHC) in myocardial tissue by qRT-PCR (n = 3). ∗P<0.01 versus Sham group; #P<0.01 versus Model group; &P<0.05 versus Model group.
3.4. G-Re reduces myocardial damage without obvious hepatorenal toxicity
The levels of serum markers involved in myocardial injury (CK-MB) and hepatorenal function (ALT and S–Cr) were measured. Compared with the Sham group, the expression of CK-MB was significantly increased in the model group (P < 0.01) (Figs. S4A and S4B), suggesting there was still myocardial damage after 28 days of AMI. G-Re treatment significantly decreased CK-MB expression compared with the MI group (P < 0.01), and there was no statistical difference between the two doses (P > 0.05) (Fig. S4A). The expression level of ALT was not statistically different among four groups (P > 0.05) (Fig. S4B), indicating that G-Re had non-hepatotoxic at the dose of 39 mg/kg/d. Compared with the Sham group, the expression of S–Cr was significantly increased in the model group (P < 0.05) (Fig. S4C), implying AMI can cause renal function injury. Compared with the model group, the expression of S–Cr had no statistically different in low- and high-dose G-Re groups (P > 0.05) (Fig. S4C), suggesting G-Re had no significant effect on renal function at the dose of 39 mg/kg/d.
3.5. G-Re alleviates MF in post-MI mice and CFs induced by AngⅡ
AMI caused markedly pathological changes in mice, including disordered arrangement of the myocardium and abundant deposition of collagen. As demonstrated in H&E staining (Fig. S5), myocardial tissues were arranged orderly with clear texture structure, and the interstitial space was uniform in the Sham group. In the model group, myocardial tissues were ruptured and curled with disordered texture, and many inflammatory cells had infiltrated near the muscle fibers. Compared with the model group, the myocardial tissues structure of G-Re low- and high-dose groups were better, and the infiltration of inflammatory cells were less.
Masson staining was used to evaluate collagen deposition in myocardium (Fig. 4A). There was a large amount of collagen deposition in the myocardium of the model group, while no obvious collagen fibers deposition in the Sham group. After G-Re intervention, the degree of collagen fibers was significantly reduced. Image J software was used for semi-quantitative analysis of collagen volume fraction (CVF). Compared with the Sham group, the CVF increased significantly in MI group (P < 0.01), while the CVF of G-Re (19.5 or 39 mg/kg/d) groups were significantly lower than the model group (P < 0.01) (Fig. 4B).
Fig. 4.

G-Re alleviates myocardial fibrosis in post-MI mice and CFs induced by AngⅡ. (A) Representative images of with Masson trichrome staining, bar, 100 μm. (B) CVF calculated in each group. Data are presented as the mean ± SD (n = 3). (C, D) The expression collagen Ⅰ and collagen Ⅲ were analyzed by Western blot in AMI mice hearts. (E, F) The expression collagen Ⅰ and collagen Ⅲ were analyzed by Western blot in CFs treated by AngⅡ. Data are presented as the mean ± SD (n = 3). ∗P<0.01 versus Sham group; #P<0.01 versus Model group; &P<0.05 versus Model group.
The imbalance extracellular matrix (ECM) is the predominant cause of MF. Abnormal deposition of collagen Ⅰ and collagen Ⅲ, the main components of ECM, can lead to increased cardiac stiffness and decreased ventricular compliance. Western blot analysis showed that the expression of myocardial collagen Ⅰ and collagen Ⅲ were significantly increased in the model group compared with the Sham group (P < 0.01). After treatment with different concentrations of G-Re, collagen Ⅰ and collagen Ⅲ expression were significantly reduced, and there was no dose dependence(P < 0.01) (Fig. 4C and D). The same results were confirmed in AngⅡ-treated CFs by Western blot (Fig. 4E and F) and immunofluorescence (Fig. S 6A-6D) detection. The results of histopathological analysis, Western blot and immunofluorescence suggest that G-Re can attenuate MF induced by AMI and AngⅡ.
3.6. G-Re inhibits the activation of CFs induced by AMI and AngⅡ
The migration of CFs is the main biological behavior leading to MF. In transwell experiment, we found that AngⅡ could significantly promote the migration ability of CFs, and G-Re could inhibit the effect (Fig. 5A). Under pathological conditions, the phenotype of CFs changes lead to increased collagen proteins synthesis and secretion. α-SMA is a typical marker for the transformation of CFs into myofibroblasts. Compared with the Sham group, the expression of α-SMA was significantly increased in myocardial tissue of mice with AMI (P < 0.01), while G-Re reversed the trend (P < 0.01). There was no statistical difference between different doses of G-Re (Fig. 5B). This result was also confirmed in AngⅡ-induced CFs, detected by Western blot (Fig. 5C) and immunofluorescence (Figs. S7A and S7B).
Fig. 5.

G-Re inhibits the migration and activation of CFs. (A) Representative images of the migration ability of CFs in transwell assay, bar, 500 μm (n = 3). (B, C) The expression α-SMA was analyzed by Western blot in AMI mice hearts and CFs treated by AngⅡ. Data are presented as the mean ± SD (n = 3). ∗P<0.01 versus Sham group; #P<0.05 versus Model group.
3.7. G-Re regulates the expression of miR-489
AngⅡ-induced MF was significantly reduced in miR-489 transgenic mice [32], indicating that miR-489 was involved in the pathological process of MF induced by AngⅡ. Our study found that the expression of miR-489 was significantly decreased in CFs stimulated by AngⅡ (P < 0.01) (Fig. 6A), while G-Re could partially counteract the effect of AngⅡ on miR-489 (P < 0.01) (Fig. 6A). At the same time, similar result has been verified in AMI mice model (Fig. 6B). These data suggest that miR-489 is involved in the pathological process of MF after AMI, and G-Re can regulate the expression of miR-489.
Fig. 6.

G-Re regulates the miR-489/myd88/NF-κB pathway. (A, B) The expression miR-489 was measured in CFs treated by AngⅡ and the myocardium of AMI mice. (C, D) The expression of myd88 was analyzed in the myocardium of AMI mice and CFs treated by AngⅡ. (E, F) The expression of NF-κB p65 and p-p65 were analyzed in the myocardium of AMI mice and CFs treated by AngⅡ, ∗P < 0.01 vs Normal group, #P<0.01 vs Model group. All data are presented as the mean ± SD (n = 3).
3.8. G-Re regulates the pathway of myd88/NF-κB
It was proved that myd88 is directly downstream target of miR-489 which can inhibit pulmonary fibrosis and cardiac hypertrophy by suppressing the expression of myd88 [26,32]. Therefore, we hypothesized that myd88 participates in the pathological process of MF after AMI. In support of this hypothesis, myd88 has been found to be upregulated in heart tissue of post-MI mice (Fig. 6C), while G-Re could attenuate this effect (Fig. 6C). The same results were also confirmed in CFs treated with AngⅡ (Fig. 6D).
NF-κB is a important downstream effector of myd88 [29]. As we all know, inflammation runs through the pathological process of MF. Therefore, myd88 may play a pro-fibrotic effect by activating NF-κB. Further studies have shown that the expression of p–NF–κB p65 was obviously increased in AMI mice (Fig. 6E), while G-Re could inhibit the phosphorylation of NF-κB p65 (Fig. 6E). The same results were also confirmed in CFs treated by AngⅡ (Fig. 6F). These data indicates that myd88/NF-κB p65 pathway involved in the pathological process of MF after AMI, and G-Re can regulate their expression.
4. Discussion
MF is a common response to a variety of physiological and pathological stimuli, which can lead to refractory heart failure, malignant arrhythmia and MI. However, the pathogenesis of MF is not yet fully understood, and there is a lack of effective therapeutic drugs. Therefore, it is necessary to search for effective therapeutic targets and drugs of MF. Our work confirmed that miR-489 is an effective anti-fibrosis molecule, and its anti-MF effect is achieved at least in part by inhibiting the myd88/NF-κB pathway. What's more, we found that G-Re has a strong anti-MF effect, and its anti-fibrotic effect may be realized through the regulation of miR-489/myd88/NF-κB pathway. Our results not only provide new insights into the pathogenesis of MF, but also find a potential drug against MF.
MiRNAs are a class of non-coding short-stranded RNAs that are widely expressed in eukaryotic cells. It has been shown that miRNAs can degrade mRNAs at transcriptional level by binding to the 3′UTR sequences of target genes. Two recent reports have shown that miR-489 is involved in the pathological process of fibrosis. The expression of miR-489 was decreased in the mouse model of silica-induced pulmonary fibrosis. MiR-489 overexpression supperssed pulmonary fibrosis by targeting myd88 [26]. Moreover, cardiac hypertrophy and fibrosis were reduced in miR-489 transgenic mice treated with AngⅡ [32]. However, it remains to be proved whether the anti-MF effect of miR-489 is a direct effect or an indirect effect of inhibiting myocardial hypertrophy. In addition, the anti-MF mechanism of miR-489 is still not clear. Our work identified that miR-489 is significantly reduced in post-MI mouse model and CFs treated with AngⅡ, while miR-489 overexpression can inhibit the fibrotic reaction of CFs at both basal level and AngⅡ treatment. Inhibition of miR-489 exerts opposite results. Therefore, miR-489 is a potential anti-MF molecule.
Previous studies have proved that myd88 is the direct downstream target of miR-489. miR-489 inhibits the development of pulmonary fibrosis and myocardial hypertrophy by binding to the 3’ UTR sequences of myd88 [26,32]. Is the anti-fibrotic effect of miR-489 also related to myd88 in MF? Our results showed that myd88 was significantly increased in the AMI mouse model. The same result was also obtained in AngⅡ-treated CFs. In addition, miR-489 overexprssion could significantly decrease the expression of myd88, while miR-489 inhibition produced the opposite result in CFs.
It has demonstrated that NF-κB is a major downstream effector of myd88, and myd88 can activate NF-κB pathway [29]. The activation of NF-κB has been documented to stimulate MF. For example, NF-κB can bind to a specific sequence of TGF-β1 DNA triggering the expression of TGF-β1 [44,45], a powerful fibrogenic cytokine [46,47]. Stevioside exerts a protective effect on mouse MF induced by isoproterenol through inhibiting NF-κB/TGF-β1 signaling pathway [48]. Our results also show that accompanying the increase of myd88, the NF-κB system was activated in AMI mouse model and AngⅡ-treated CFs. Thus, it can be speculated that NF-κB may be a downstream target of myd88 in MF.
G- Re is one of the major active components in ginseng, which has a wide range of biological activities. Recent studies have shown that G-Re can improve rat MF induced by AMI and ISO [40,41]. Our study also confirmed that G-Re can alleviate myocardial injury and improve cardiac function in mice with AMI. Moreover, G-Re suppressed AMI-induced compensatory cardiac hypertrophy, as evidenced by a decreased HWI, HW/TL, and the mRNA expression of ANP, BNP as well as β-MHC, and reversed MF induced by AMI and AngⅡ, as indicated by a decreased CVF and related fibrosis indexes such as collagen Ⅰ, collagen Ⅲ and α-SMA in vivo and vitro. Moreover, we found that G-Re has good safety and no dose-dependent effect.
Re can prevent NF-κB activation and subsequent myocardial inflammatory responses in endotoxemic mice [49]. Another study also showed that G-Re can inhibit IKK-β phosphorylation and NF-κB activation, as well as the expression of proinflammatory cytokines in lipopolysaccharide-stimulated peritoneal macrophages [50]. Therefore, G-Re can inhibit the activation of NF-κB. miR-489 can play an anti-MF effect by regulating the myd88/NF-κB signaling pathway, so is the anti-MF effect of G-Re related to regulating the expression of miR-489. We found that G-Re inhibited down-regulation of miR-489 and activation of myd88/NF-κB in heart of AMI mice and AngⅡ-induced CFs. It is suggested that the anti-MF effect of G-Re is related to the regulation of miR-489/myd88/NF-κB pathway.
This study also has some shortcomings. First of all, the anti-MF effect of miR-489 needs to be further confirmed by in vivo experiments. Secondly, whether myd88/NF-κB pathway is the only pathway of miR-489's anti-MF effect remains to be confirmed. In addition, previous studies have shown that Smad3 is also a direct downstream target of miR-489 [26]. Whether miR-489's anti-myocardial fibrosis effect is related to it remains to be further proved.
5. Conclusions
In summary, our present work identified miR-489 to be an anti-MF miRNA. MiR-489 may inhibit the pathological process of MF by regulating the myd88/NF-κB pathway. Moreover, we demonstrated that G-Re acts as a potential drug against MF through promoting miR-489 expression. The regulation of miR-489 in MF and the therapeutic effect of G-Re may provide a potential treatment method for MF.
Declaration of competing interest
The authors declare that they have no financial conflicts of interest.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant number 81874410); The authors thank the Institute of Integrated Traditional Chinese and Western Medicine, Fujian University of Traditional Chinese Medicine for providing technical help.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jgr.2021.11.009.
Contributor Information
Jun Peng, Email: pjunlab@hotmail.com.
Chenglong Wang, Email: wcl796@sina.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Tschöpe C., Díez J. Myocardial fibrosis as a matter of cell differentiation: opportunities for new antifibrotic strategies. Eur Heart J. 2019;40:979–981. doi: 10.1093/eurheartj/ehy307. [DOI] [PubMed] [Google Scholar]
- 2.Thum T. Noncoding RNAs and myocardial fibrosis. Nat Rev Cardiol. 2014;11:655–663. doi: 10.1038/nrcardio.2014.125. [DOI] [PubMed] [Google Scholar]
- 3.Pan S.C., Cui H.H., Qiu C.G. HOTAIR promotes myocardial fibrosis through regulating URI1 expression via Wnt pathway. Eur Rev Med Pharmacol Sci. 2018;22:6983–6990. doi: 10.26355/eurrev_201810_16169. [DOI] [PubMed] [Google Scholar]
- 4.Tallquist M.D., Molkentin J.D. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. 2017;14:484–491. doi: 10.1038/nrcardio.2017.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Frangogiannis N.G. Can myocardial fibrosis Be reversed? J Am Coll Cardiol. 2019;73:2283–2285. doi: 10.1016/j.jacc.2018.10.094. [DOI] [PubMed] [Google Scholar]
- 6.Rodriguez P., Sassi Y., Troncone L., Benard L., Ishikawa K., Gordon R.E., Lamas S., Laborda J., Hajjar R.J., Lebeche D. Deletion of delta-like 1 homologue accelerates fibroblast-myofibroblast differentiation and induces myocardial fibrosis. Eur Heart J. 2019;40:967–978. doi: 10.1093/eurheartj/ehy188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prabhu S.D., Frangogiannis N.G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. 2016;119:91–112. doi: 10.1161/CIRCRESAHA.116.303577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Travers J.G., Kamal F.A., Robbins J., Yutzey K.E., Blaxall B.C. Cardiac fibrosis: the fibroblast awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van den Borne S.W., Diez J., Blankesteijn W.M., Verjans J., Hofstra L., Narula J. Myocardial remodeling after infarction: the role of myofibroblasts. Nat Rev Cardiol. 2010;7:30–37. doi: 10.1038/nrcardio.2009.199. [DOI] [PubMed] [Google Scholar]
- 10.Daniels A., van Bilsen M., Goldschmeding R., van der Vusse G.J., van Nieuwenhoven F.A. Connective tissue growth factor and cardiac fibrosis. Acta Physiol. 2009;195:321–338. doi: 10.1111/j.1748-1716.2008.01936.x. [DOI] [PubMed] [Google Scholar]
- 11.Martos R., Baugh J., Ledwidge M., O'Loughlin C., Conlon C., Patle A., Donnelly S.C., McDonald K. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–895. doi: 10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 12.Yong K.W., Li Y., Huang G., Lu T.J., Safwani W.K., Pingguan-Murphy B., Xu F. Mechanoregulation of cardiac myofibroblast differentiation: implications for cardiac fibrosis and therapy. Am J Physiol Heart Circ Physiol. 2015;309:H532–H542. doi: 10.1152/ajpheart.00299.2015. [DOI] [PubMed] [Google Scholar]
- 13.Dean R.G., Balding L.C., Candido R., Burns W.C., Cao Z., Twigg S.M., Burrell L.M. Connective tissue growth factor and cardiac fibrosis after myocardial infarction. J Histochem Cytochem. 2005;53:1245–1256. doi: 10.1369/jhc.4A6560.2005. [DOI] [PubMed] [Google Scholar]
- 14.Schirone L., Forte M., Palmerio S., Yee D., Nocella C., Angelini F., Pagano F., Schiavon S., Bordin A., Carrizzo A., et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev. 2017;2017:3920195. doi: 10.1155/2017/3920195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu W.Y., Sun H.H., Sun P.F. MicroRNA-378 attenuates myocardial fibrosis by inhibiting MAPK/ERK pathway. Eur Rev Med Pharmacol Sci. 2019;23:4398–4405. doi: 10.26355/eurrev_201905_17947. [DOI] [PubMed] [Google Scholar]
- 16.Mallory A.C., Vaucheret H. MicroRNAs: something important between the genes. Curr Opin Plant Biol. 2004;7:120–125. doi: 10.1016/j.pbi.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Liu X., Luo G., Bai X., Wang X.J. Bioinformatic analysis of microRNA biogenesis and function related proteins in eleven animal genomes. J Genet Genomics. 2009;36:591–601. doi: 10.1016/S1673-8527(08)60151-4. [DOI] [PubMed] [Google Scholar]
- 19.Yang K.C., Yamada K.A., Patel A.Y., Topkara V.K., George I., Cheema F.H., Ewald G.A., Mann D.L., Nerbonne J.M. Deep RNA sequencing reveals dynamic regulation of myocardial noncoding RNAs in failing human heart and remodeling with mechanical circulatory support. Circulation. 2014;129:1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou C., Cui Q., Su G., Guo X., Liu X., Zhang J. MicroRNA-208b alleviates post-infarction myocardial fibrosis in a rat model by inhibiting GATA4. Med Sci Mon Int Med J Exp Clin Res. 2016;22:1808–1816. doi: 10.12659/MSM.896428. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Chen M.H., Liu J.C., Liu Y., Hu Y.C., Cai X.F., Yin D.C. MicroRNA-199a regulates myocardial fibrosis in rats by targeting SFRP5. Eur Rev Med Pharmacol Sci. 2019;23:3976–3983. doi: 10.26355/eurrev_201905_17827. [DOI] [PubMed] [Google Scholar]
- 22.Kikkawa N., Hanazawa T., Fujimura L., Nohata N., Suzuki H., Chazono H., Sakurai D., Horiguchi S., Okamoto Y., Seki N. miR-489 is a tumour-suppressive miRNA target PTPN11 in hypopharyngeal squamous cell carcinoma (HSCC) Br J Cancer. 2010;103:877–884. doi: 10.1038/sj.bjc.6605811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang B., Ji S., Ma F., Ma Q., Lu X., Chen X. miR-489 acts as a tumor suppressor in human gastric cancer by targeting PROX1. Am J Cancer Res. 2016;6:2021–2030. [PMC free article] [PubMed] [Google Scholar]
- 24.Li J., Qu W., Jiang Y., Sun Y., Cheng Y., Zou T., Du S. miR-489 suppresses proliferation and invasion of human bladder cancer cells. Oncol Res. 2016;24:391–398. doi: 10.3727/096504016X14666990347518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Y., Ma X., Wang Y., Li G. miR-489 inhibits proliferation, cell cycle progression and induces apoptosis of glioma cells via targeting SPIN1-mediated PI3K/AKT pathway. Biomed Pharmacother. 2017;93:435–443. doi: 10.1016/j.biopha.2017.06.058. [DOI] [PubMed] [Google Scholar]
- 26.Wu Q., Han L., Yan W., Ji X., Han R., Yang J., Yuan J., Ni C. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci Rep. 2016;6:30921. doi: 10.1038/srep30921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feng Y., Zou L., Si R., Nagasaka Y., Chao W. Bone marrow MyD88 signaling modulates neutrophil function and ischemic myocardial injury. Am J Physiol Cell Physiol. 2010;299:C760–C769. doi: 10.1152/ajpcell.00155.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y., Si R., Feng Y., Chen H.H., Zou L., Wang E., Zhang M., Warren H.S., Sosnovik D.E., Chao W. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and Toll-like receptor 4. J Biol Chem. 2011;286:31308–31319. doi: 10.1074/jbc.M111.246124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li T., Wang Y., Liu C., Hu Y., Wu M., Li J., Guo L., Chen L., Chen Q., Ha T., et al. MyD88-dependent nuclear factor-kappaB activation is involved in fibrinogen-induced hypertrophic response of cardiomyocytes. J Hypertens. 2009;27:1084–1093. doi: 10.1097/HJH.0b013e3283293c93. [DOI] [PubMed] [Google Scholar]
- 30.Wendlandt E.B., Graff J.W., Gioannini T.L., McCaffrey A.P., Wilson M.E. The role of microRNAs miR-200b and miR-200c in TLR4 signaling and NF-kappaB activation. Innate Immun. 2012;18:846–855. doi: 10.1177/1753425912443903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68–69:106–121. doi: 10.1016/j.matbio.2017.11.010. [DOI] [PubMed] [Google Scholar]
- 32.Wang K., Liu F., Zhou L.Y., Long B., Yuan S.M., Wang Y., Liu C.Y., Sun T., Zhang X.J., Li P.F. The long noncoding RNA CHRF regulates cardiac hypertrophy by targeting miR-489. Circ Res. 2014;114:1377–1388. doi: 10.1161/CIRCRESAHA.114.302476. [DOI] [PubMed] [Google Scholar]
- 33.Zheng S.D., Wu H.J., Wu D.L. Roles and mechanisms of ginseng in protecting heart. Chin J Integr Med. 2012;18:548–555. doi: 10.1007/s11655-012-1148-1. [DOI] [PubMed] [Google Scholar]
- 34.Xie J.T., Wang C.Z., Wang A.B., Wu J., Basila D., Yuan C.S. Antihyperglycemic effects of total ginsenosides from leaves and stem of Panax ginseng. Acta Pharmacol Sin. 2005;26:1104–1110. doi: 10.1111/j.1745-7254.2005.00156.x. [DOI] [PubMed] [Google Scholar]
- 35.Joo K.M., Lee J.H., Jeon H.Y., Park C.W., Hong D.K., Jeong H.J., Lee S.J., Lee S.Y., Lim K.M. Pharmacokinetic study of ginsenoside Re with pure ginsenoside Re and ginseng berry extracts in mouse using ultra performance liquid chromatography/mass spectrometric method. J Pharmaceut Biomed Anal. 2010;51:278–283. doi: 10.1016/j.jpba.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Lee G.H., Lee W.J., Hur J., Kim E., Lee H.G., Seo H.G. Ginsenoside Re mitigates 6-hydroxydopamine-induced oxidative stress through upregulation of GPX4. Molecules. 2020;25 doi: 10.3390/molecules25010188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao Y., Zhu P., Xu S.F., Li Y.Q., Deng J., Yang D.L. Ginsenoside Re inhibits PDGF-BB-induced VSMC proliferation via the eNOS/NO/cGMP pathway. Biomed Pharmacother. 2019;115:108934. doi: 10.1016/j.biopha.2019.108934. [DOI] [PubMed] [Google Scholar]
- 38.Cai M., Yang E.J. Ginsenoside Re attenuates neuroinflammation in a symptomatic ALS animal model. Am J Chin Med. 2016;44:401–413. doi: 10.1142/S0192415X16500233. [DOI] [PubMed] [Google Scholar]
- 39.Peng L., Sun S., Xie L.H., Wicks S.M., Xie J.T. Ginsenoside Re: pharmacological effects on cardiovascular system. Cardiovasc Ther. 2012;30:e183–e188. doi: 10.1111/j.1755-5922.2011.00271.x. [DOI] [PubMed] [Google Scholar]
- 40.Wang Q.W., Yu X.F., Xu H.L., Zhao X.Z., Sui D.Y. Ginsenoside Re improves isoproterenol-induced myocardial fibrosis and heart failure in rats. Evid Based Complement Alternat Med. 2019;2019:3714508. doi: 10.1155/2019/3714508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Y., Sun J., Liu J., Wang P., Wang C. Ginsenoside Re preserves cardiac function and ameliorates left ventricular remodeling in a rat model of myocardial infarction. J Cardiovasc Pharmacol. 2020;75:91–97. doi: 10.1097/FJC.0000000000000752. [DOI] [PubMed] [Google Scholar]
- 42.Huang D.D., Huang H.F., Yang Q., Chen X.Q. Liraglutide improves myocardial fibrosis after myocardial infarction through inhibition of CTGF by activating cAMP in mice. Eur Rev Med Pharmacol Sci. 2018;22:4648–4656. doi: 10.26355/eurrev_201807_15524. [DOI] [PubMed] [Google Scholar]
- 43.V H., Titus A.S., Cowling R.T., Kailasam S. Collagen receptor cross-talk determines α-smooth muscle actin-dependent collagen gene expression in angiotensin II-stimulated cardiac fibroblasts. J Biol Chem. 2019;294:19723–19739. doi: 10.1074/jbc.RA119.009744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bowie A., O'Neill L.A. Oxidative stress and nuclear factor-kappaB activation: a reassessment of the evidence in the light of recent discoveries. Biochem Pharmacol. 2000;59:13–23. doi: 10.1016/s0006-2952(99)00296-8. [DOI] [PubMed] [Google Scholar]
- 45.Jain M., Rivera S., Monclus E.A., Synenki L., Zirk A., Eisenbart J., Feghali-Bostwick C., Mutlu G.M., Budinger G.R., Chandel N.S. Mitochondrial reactive oxygen species regulate transforming growth factor-beta signaling. J Biol Chem. 2013;288:770–777. doi: 10.1074/jbc.M112.431973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lijnen P.J., Petrov V.V., Fagard R.H. Induction of cardiac fibrosis by transforming growth factor-beta(1) Mol Genet Metabol. 2000;71:418–435. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 47.Meng X.M., Nikolic-Paterson D.J., Lan H.Y. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325–338. doi: 10.1038/nrneph.2016.48. [DOI] [PubMed] [Google Scholar]
- 48.Wang J., Shen W., Zhang J.Y., Jia C.H., Xie M.L. Stevioside attenuates isoproterenol-induced mouse myocardial fibrosis through inhibition of the myocardial NF-κB/TGF-β1/Smad signaling pathway. Food Funct. 2019;10:1179–1190. doi: 10.1039/c8fo01663a. [DOI] [PubMed] [Google Scholar]
- 49.Chen R.C., Wang J., Yang L., Sun G.B., Sun X.B. Protective effects of ginsenoside Re on lipopolysaccharide-induced cardiac dysfunction in mice. Food Funct. 2016;7:2278–2287. doi: 10.1039/c5fo01357g. [DOI] [PubMed] [Google Scholar]
- 50.Lee I.A., Hyam S.R., Jang S.E., Han M.J., Kim D.H. Ginsenoside Re ameliorates inflammation by inhibiting the binding of lipopolysaccharide to TLR4 on macrophages. J Agric Food Chem. 2012;60:9595–9602. doi: 10.1021/jf301372g. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.