

Abstract
Despite recent advances in molecular therapeutics, lung cancer is still a leading cause of cancer deaths. Currently, limited targeted therapy options and acquired drug resistance present significant barriers in the treatment of patients with lung cancer. New strategies in drug development, including those that take advantage of the intracellular ubiquitin-proteasome system to induce targeted protein degradation, have the potential to advance the field of personalized medicine for patients with lung cancer. Specifically, small molecule proteolysis targeting chimeras (PROTACs), consisting of two ligands connected by a linker that bind to a target protein and an E3 ubiquitin ligase, have been developed against many cancer targets, providing promising opportunities for advanced lung cancer. In this review, we focus on the rationale for PROTAC therapy as a new targeted therapy and the current status of PROTAC development in lung cancer.
Keywords: targeted therapy, argeted protein degradation, proteolysis targeting chimeras, PROTACs, lung cancer
Graphical abstract
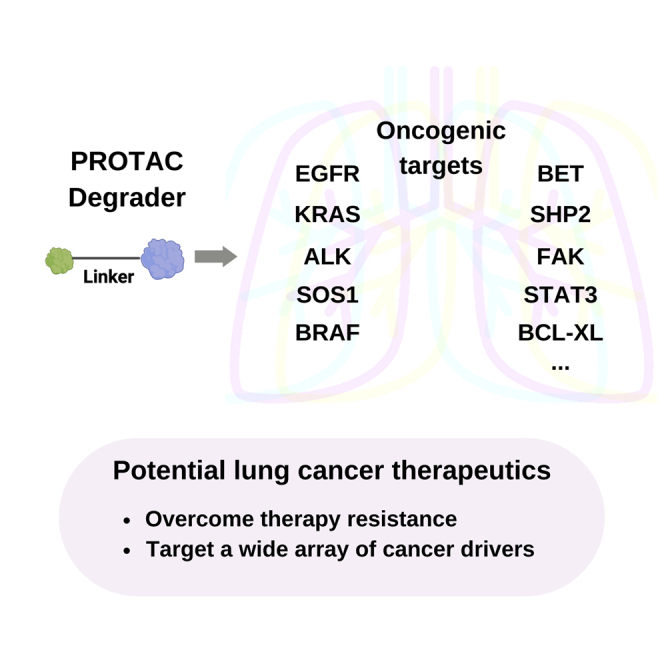
This review focuses on the rationale for PROTAC therapy as a new targeted therapy and the current status of PROTAC development in lung cancer.
Introduction
There has been a reduction in new lung cancer diagnoses and lung cancer deaths due to decreases in smoking, early screening, and advances in treatment.1,2 However, lung cancer remains the leading cause of cancer deaths, contributing to almost 20% of all cancer deaths. Only 10% to 20% of all patients diagnosed with lung cancer have >5-year survival rate, and more people die of lung cancer than of breast cancer, colon cancer, and prostate cancer combined.1
Lung cancer is classified into two major histological types: small cell lung cancer (SCLC), which makes up around 15% of all lung cancers, and non-small cell lung cancer (NSCLC), which makes up the majority at 85%.3 NSCLC includes three main histological subtypes: lung adenocarcinoma (around 40% of NSCLC), squamous cell carcinoma (25%), and large cell carcinoma (10%). The remaining types include adenosquamous carcinoma, sarcomatoid carcinomas, salivary gland type tumors, carcinoid tumors, and other unclassified carcinomas. Despite these subclassification efforts, molecular analyses within each of these histological lung cancer subtypes, such as adeno-, squamous, and small cell carcinoma, have shown a highly heterogeneous genetic landscape as revealed by the Cancer Genome Atlas.4,5,6,7,8 This heterogeneity continues to challenge the interpretation of large therapeutic trials that average clinical outcomes and, accordingly, may miss important treatment opportunities that are limited to specific mutational profiles.
Therefore, it is increasingly recognized that both tumor, node, metastasis (TNM) staging and detailed genetics are important in guiding treatment of patients with lung cancer. Patients undergo biopsy, staging and next-generation sequencing to help guide lung cancer treatment, whether it be surgical resection, radiation, systemic chemotherapy, targeted therapy, and/or immunotherapy.3,9 For example, a subset of patients with NSCLC harboring actionable mutations, such as those in EGFR, ALK, ROS1, BRAF, or MET, receive targeted small molecule inhibitor therapy as the first-line treatment, whereas those without driver mutations may receive combinations of standard cytotoxic chemotherapy and immunotherapy or immunotherapy alone. However, most patients who present with advanced disease ultimately develop tumor progression through clonal selection of drug-resistant tumor cells and new strategies that overcome drug resistance are urgently needed to improve patient outcomes.
The concept of harnessing the endogenous ubiquitin-proteasome system to degrade pathogenic proteins through proteolysis targeting chimeras (PROTACs) was initially introduced and demonstrated in a proof-of-concept study in 2001.10 This technology has since emerged as a paradigm-shifting approach in small molecule drug discovery and has been driving unprecedented innovations in drug development in the past several years.11,12 Excitingly, multiple PROTACs are being evaluated in phase I and phase II clinical trials.12,13,14,15 With these advances, PROTAC technology is increasingly poised to lead to new targeted therapies that benefit patients with lung cancer. In this review, we introduce the PROTAC technology, summarize the progress of PROTAC development in lung cancer, and discuss the promises and challenges in moving PROTACs toward clinical applications in advancing lung cancer treatment.
The PROTAC technology offers exciting opportunities for developing novel targeted cancer therapeutics
PROTACs are hetero-bifunctional molecules that co-opt the intrinsic ubiquitin-proteasome system to specifically destroy proteins implicated in cancers and other diseases.10,12,14,16 PROTACs consist of two ligands, one that binds to a target protein of interest (POI) and the other that binds to an E3 ubiquitin ligase, with a linker connecting the two ligands (Figure 1). By forming a ternary complex with a POI and an E3 ligase, a PROTAC brings the POI to close proximity to the E3 ligase, inducing POI poly-ubiquitination and subsequent proteasome-mediated degradation. The PROTAC is then recycled to target and degrade another POI molecule.
Figure 1.

Schematic diagram of the PROTAC technology. A typical PROTAC molecule consists of two ligands, one binding to a protein of interest (POI) and the other recruiting an E3 ligase, which are held together via a chemical linker.
The PROTAC molecules induce proximity between the POI and an E3 through the formation of ternary complexes, leading to poly-ubiquitin chain formation on the POI and ultimately proteasomal-mediated POI degradation. The figure was created with BioRender.
The modular nature of PROTAC design offers great potential, flexibility, and speed in the development of novel PROTACs to target and degrade essentially any intracellular protein targets. In terms of ligands for POIs for degradation, small molecules that can bind to any surfaces of target proteins can potentially be used for PROTAC development, unlike the conventional small molecule inhibitors (SMIs) that require high affinity to block protein function. Available small molecule compounds such as those Food and Drug Administration (FDA)-approved small molecule targeted inhibitors, those that failed clinical trials, or tool compounds have been used in the design and development of many PROTACs. For example, as discussed below, the available small molecule epidermal growth factor receptor (EGFR) inhibitors and ALK inhibitors were used to develop PROTACs that target and degrade EGFR and ALK, respectively. With many available small molecule libraries, the identification of new ligands that bind to many cancer targets will expand degradable POIs to those undruggable targets for PROTAC development.
Due to a very limited number of available E3 ligands, currently developed PROTACs mainly recruit E3 ligases cereblon (CRBN), von Hippel-Lindau (VHL), mouse double minute-2 (MDM2), and inhibitor of apoptosis proteins (IAPs), despite a collection of more than 600 E3 ligases encoded by the human genome. Most published PROTACs recruit ubiquitously expressed CRBN and VHL E3 ligases, which have the potential to cause on-target toxicity. PROTACs targeting the same POI, however, with either a CRBN or a VHL E3 ligase have demonstrated differing rates of degradation. Identification of tumor-specific/enriched E3 ligases and the development of their specific ligands will have great impact on enhancing tumor-specific/selective therapeutic effects.
The ligands for a POI and E3 ligase are held together through a chemical linker. Research has demonstrated that the type, the length, the rigidity, and the positions of linkage on the POIs and E3 ligands could affect PROTAC functions in terms of the induced ternary complex formation, the efficacy and selectivity of protein degradation, pharmacokinetics (PK), and bioavailability.16 Therefore, the optimization of the chemical linker is an important step for PROTAC development.
PROTACs offer several advantages in comparison with traditional SMIs.12,14,16,17 First, PROTACs degrade protein targets, instead of inhibiting the target activity. This likely achieves better inhibition in a scenario where oncogenic activity of a target can have activity independent of the enzymatic activity. For example, PROTACs can abrogate the scaffolding function of the target proteins to potentially override resistance and improve efficacy of the warheads. Second, PROTACs can be developed to degrade “difficult to drug” proteins that do not possess well-defined pockets, such as transcription factors (e.g., STAT3, MYC) and RAS (e.g., KRASG12C). It can also target and degrade multicomponent protein complexes, which are conventionally considered as “undruggable” because inhibition of one subunit does not necessarily remove the complex’s function. Third, PROTACs have the potential to overcome resistance by degrading upregulated POIs (induced by SMIs), or POIs that arise from mutations in the targets. Fourth, and perhaps most importantly, PROTACs have unique event-driven pharmacology and can mediate multiple rounds of degradation, unlike occupancy-driven pharmacology for SMIs. The unique mechanism of action allows one single PROTAC molecule to degrade multiple POIs, thus enabling the use of low drug concentrations to reach a desired therapeutic effect. Last, the modular nature of PROTACs enables relative ease and flexibility in the design and optimization.
However, there are several challenges associated with the PROTAC drugs. PROTAC molecules are generally large, which leads to poor pharmaceutical properties such as bioavailability. It has been reported that some PROTACs have excellent activity in cellular models, yet have no activity in mouse models in vivo due to issues with PK and tissue penetration. In addition, protein degradation potentially confers severe on-target toxicity as compared with the inhibition of protein function in certain settings. It is also difficult to achieve specific degradation of altered proteins in tumors, as a result, potential on- and off-target toxicity could arise, which should be carefully evaluated.
Progress in the PROTAC development for lung cancer treatment
PROTACs are under rapid development as novel therapeutics of lung cancer and novel strategies to overcome drug resistance in the past few years. Multiple PROTAC molecules have been developed for validated therapeutic targets in NSCLC such as EGFR, KRAS, ALK, BRAF, and BCL-XL, with promising compounds showing antitumor efficacy in cell models and preclinical tumor models, which are summarized below (Table 1). Further efforts are required to optimize these PROTAC molecules and to conduct systematic preclinical evaluation before clinical studies.
Table 1.
A list of PROTACs that have been developed to target key cancer drivers and tested at different preclinical phases for lung cancer
| Gene target (POI) | PROTAC name | Ligand for POI | E3 ligand | Specificity | Lung cancer type | Study type | In vivo lung cancer model | Ref. |
|---|---|---|---|---|---|---|---|---|
| EGFR | Compound 3 | Gefitinib | VHL ligand | Del19; L858R | LUAD | In vitro | NT | Burslem et al.18 |
| Compound 4 | Afatinib | VHL ligand | L858R/T790M | LUAD | In vitro | NT | Burslem et al.18 | |
| MS39 | Gefitinib | VHL ligand | Del19; L858R | LUAD | In vitro | NT | Cheng et al.19 | |
| MS154 | Gefitinib | CRBN ligand | Del19; L858R | LUAD | In vitro | NT | Cheng et al.19 | |
| Compound 14o | XFT-262 | VHL ligand | L858R/T790M | LUAD | In vitro | NT | Zhang et al.20 | |
| DDC-01-163 | JBJ-07-149 | CRBN ligand | L858R/T790M; L858R/T790M/C797S; L858R/T790M/L718Q |
LUAD | In vitro | NT | Jang et al.21 | |
| SIAIS125;126 | Canertinib | CRBN ligand | Del19; L858R/T790M | LUAD | In vitro | NT | Qu et al.22 | |
| Compound P3 | Compound F | VHL ligand | Del19; L858R | LUAD | In vitro | NT | Zhao et al.23 | |
| CP17 | Osimertinib | VHL ligand | Del19; L858R/T790M | LUAD | In vitro | NT | Zhao et al.24 | |
| Compound 6h | Brigatinib analog | VHL ligand | Del19/T790M/C797S | LUAD | In vitro | NT | Zhang. et al.25 | |
| Compound 1q | CO-1686 | CRBN ligand | Del19; L858R | LUAD | In vitro | NT | Li et al.26 | |
| HJM-561 | Brigatinib | CRBN ligand | Del19/T790M/C797S; L858R/T790M/C797S | LUAD |
In vitro; In vivo |
PDX-LUAD (Del19/T790M/C797S) | Du et al.27 | |
| Compound 13 | Dacomitinib | VHL ligand | Del19 | LUAD |
In vitro; In vivo |
HCC827 xenograft | Shi et al.28 | |
| 13a;13b | Osimertinib | CRBN ligand | L858R/T790M | LUAD |
In vitro; In vivo |
H1975 xenograft | Zhang et al.29 | |
| CFT8919 | EGFR allosteric inhibitor (not disclosed) | CRBN ligand | L858R; L858R/C797S; L858R/T790M; L858R/T790M/C797S | LUAD |
In vitro; In vivo |
H1975 xenograft | C4 Therapeutics30 | |
| KRAS | XY-4-88 | ARS-1620 | CRBN ligand | GFP-KRAS | LUAD | In vitro | NT | Zeng et al.31 |
| LC-2 | MRTX849 | VHL ligand | KRASG12C | LUAD | In vitro | NT | Bond et al.32 | |
| YF135 | MRTX849 | VHL ligand | KRASG12C | LUAD | In vitro | NT | Yang et al.33 | |
| SOS1 | Compound 9d | BI 1701963 | VHL ligand | SOS1 | LUAD |
In vitro; In vivo |
NCI-H358 xenograft | Zhou et al.34 |
| ALK | Compound 9 | TAE684 | CRBN ligand | ALK or ALK fusions | LUAD | In vitro | NT | Powell et al.35 |
| Compound 11 | Ceritinib | CRBN ligand | ALK or ALK fusions | LUAD | In vitro | NT | Powell et al.35 | |
| Compound 5;6 | Ceritinib | CRBN ligand | EML4-ALK; NPM-ALK | LUAD | In vitro | NT | Zhang et al.36 | |
| Opto-dALK | Ceritinib | CRBN ligand (opto-pomalidomide) | EML4-ALK; NPM-ALK | LUAD | In vitro | NT | Liu et al.37 | |
| IAIS091; SIAIS001 | Alectinib | CRBN ligand | NPM-ALK | NT | In vitro | NT | Ren et al.38 | |
| SIAIS117 | Brigatinib | VHL ligand | EML4-ALK | LUAD, SCLC | In vitro | NT | Sun et al.39 | |
| TD-004 | Ceritinib | VHL ligand | EML4-ALK | LUAD |
In vitro; In vivo |
H3122 xenograft | Kang et al.40 | |
| Compound 17 | Alectinib | CRBN ligand | EML4-ALK | LUAD |
In vitro; In vivo |
NT | Xie et al.41 | |
| Compound B3 | LDK378 | CRBN ligand | ALK fusion | LUAD |
In vitro; In vivo |
H3122 xenograft | Yan et al.42 | |
| BRAF | P4B | BI 882370 | CRBN ligand | BRAF-V600E | LUAD |
In vitro; In vivo |
NT | Posternak et al.43 |
| SJF-0628 | Vemurafenib | VHL ligand | 3 classes of BRAF mutations | LUAD |
In vitro; In vivo |
NT | Alabi et al.44 | |
| Compound 12 | Vemurafenib | CRBN ligand | BRAF-V600E | LUAD | In vitro | NT | Han et al.45 | |
| Compound 23 | BI 882370 | CRBN ligand | BRAF-V600E | LUAD | In vitro | NT | Han et al.45 | |
| BET | ARV-825 | OTX015 | CRBN ligand | BRD4 | LUAD |
In vitro; In vivo |
NT | Vartak et al46; Lu et al.47 |
| dBET | JQ-1 | CRBN ligand | BRD2, BRD3, BRD4 | LUAD; SCC; SCLC |
In vitro; In vivo |
NT | Zong et al.; Zhou et al.; Winter et al.48,49,50 | |
| ZBC260 | JQ-1 | CRBN ligand | BRD2, BRD3, BRD4 | LUAD; SCC; SCLC |
In vitro; In vivo |
H1975 xenograft; lung PDX | Zong et al.; Zhou. et al.; Winter et al.48,49,50 | |
| CFT-2718 | JQ-1 analog | CRBN ligand | BRD4 | SCLC |
In vitro; In vivo |
SCLC PDX | Zong et al.48, Sun et al.51 | |
| QCA570 | QCA276 | CRBN ligand | BRD2, BRD3, BRD4 | LUAD; LSCC |
In vitro; In vivo |
HCC827/AR xenograft | Qin et al.52; Liu et al.53 | |
| SHP2 | SHP2-D26 | Compound 5 | VHL ligand | SHP2 | LUAD; LSCC |
In vitro; In vivo |
PC9 and PC9/AR xenografts | Wang et al.54; Deng et al.55 |
| FAK | D-PROTAC | Defactinib | VHL ligand | FAK | LUAD |
In vitro; In vivo |
A427 xenograft | Liu. et al.56 |
| PROTAC B5 | Compound 2 | CRBN ligand | FAK | LUAD | In vitro | NT | Sun et al.57 | |
| PROTAC-3 | Defactinib | VHL ligand | FAK | NT | In vitro | NT | Cromm et al.58 | |
| STAT3 | SD-36 | SI-109 | CRBN ligand | STAT3 | NT | In vitro; in vivo | NT | Bai et al.59 |
| BCL-XL | DT2216 | ABT263 | VHL ligand | BCL-XL | LUAD; SCLC |
In vitro; In vivo |
H358 and H146 xenografts | Khan et al.60,61 |
| BCL-XL/2 | 753b | ABT263 | VHL ligand | BCL-XL/2 | SCLC |
In vitro; In vivo |
H146 xenograft (unpublished) | Lv et al.62 |
CRBN, cereblon; LSCC, lung squamous cell carcinoma; LUAD, lung adenocarcinoma; NT, not tested; PC9/AR and HCC827/AR, osimertinib-resistant LUAD cell lines; PDX, patient-derived xenograft; SCC, small cell lung cancer; VHL, von Hippel-Lindau.
EGFR-targeting PROTACs
The EGFR signaling pathway is critical in regulating cell growth, differentiation, and survival.9,63 EGFR is mutated in approximately 15% of patients with lung adenocarcinoma with more frequent mutation rates in lifelong nonsmokers, women, and patients of East Asian descent. EGFR tyrosine kinase inhibitors (EGFR-TKIs) are standard care for patients with NSCLC who harbor activating mutations in EGFR, based on the paradigm-shifting discovery that tumor responses are tightly linked with tumor EGFR mutational status.64,65,66,67 The EGFR activating mutations include exon 19 in-frame deletions (del19) in approximately 45% mutant EGFR lung cancer cases, point mutations in exon 21 such as L858R in another 40% to 45% cases, and assorted additional “atypical” mutations in 5% to 10% mostly localized within exon 18. To date, three generations of FDA-approved EGFR-TKIs have been developed with fourth-generation agents on the horizon.68,69,70,71,72,73,74,75,76,77,78 The first-generation EGFR-TKIs, gefitinib and erlotinib, which bind to the ATP site, showed good therapeutic effect on patients with EGFR 19del and EGFR L858R mutations. However, disease invariably progresses within 13 months, due to acquired resistance from new mutations such as T790M, a gatekeeper mutation that increases the affinity of ATP to the protein. The second-generation EGFR-TKIs, afatinib and dacomitinib, are irreversible inhibitors forming covalent bonds with C797, and were shown to be superior to gefitinib in terms of primary end-point and overall survival. However, they failed to overcome T790M resistance. The third-generation osimertinib and almonertinib demonstrated improved survival outcomes compared with first-generation EGFR-TKIs. However, almost all patients who initially respond to osimertinib again developed acquired resistance due to secondary mutations in EGFR, structural mutations, and amplification of MET. Around 40% to 50% of patients developed new EGFR mutations such as in C797, G796, and L718 residues, causing tumor relapse due to drug resistance. Overall, resistance limits the efficacy of the EGFR-TKIs in essentially all advanced-stage lung cancer patients, highlighting the need for new therapeutic strategies targeting acquired resistance mechanisms such as selection for new EGFR C797S and T790M mutations.
The PROTAC technology represents an effective strategy to overcome EGFR resistant mutations through targeted degradation of EGFR mutants. The first EGFR PROTACs that degraded EGFR mutant proteins were reported from the Crews group in 2018.18 A mutant EGFR-selective gefitinib-based, VHL-recruiting PROTAC molecule, Compound 3, was shown to effectively degrade EGFR mutant proteins in HCC827 cells carrying 19del and H3255 cells harboring L858R mutation at low nanomolar concentrations, while sparing wild-type EGFR. Another afatinib-based PROTAC molecule, Compound 4, degraded gefitinib-resistant L858R/T790M mutated EGFR in the H1975 cell line. This study demonstrated the feasibility of using PROTACs to specifically degrade membrane-bound mutated EGFR to overcome drug-resistant EGFR mutations.
Inspired by this exciting study, multiple groups have developed several novel EGFR PROTACs, with some showing in vivo antitumor efficacy in preclinical models.19,20,21,22,23,24,25,26,27,28,29,30,79 For example, the Zhang group recently reported an orally bioavailable EGFR PROTAC, HJM-561, as a promising therapeutic strategy for overcoming EGFR triple mutation-mediated drug resistance in NSCLC, because it specifically degraded EGFR mutant proteins and induced potent antitumor activity in EGFR Del19/T790M/C797S-driven Ba/F3 cell line-derived xenograft (CDX) and patient-derived xenograft (PDX) models that were resistant to osimertinib treatment.27 In another study,28 the Zhu group generated novel potent covalent inhibitor dacomitinib-based EGFR degraders and identified a promising compound, 13, which potently induced degradation of EGFR(Del19) with low nanomolar DC(50) value in HCC827 cells, but not other EGFR mutant, wild-type EGFR proteins or HER2 and HER4 receptors from the EGFR family. This compound exhibited excellent antitumor efficacy with no observable toxic effects. The Li group also reported two highly potent and selective CRBN-recruiting EGFR(L858R/T790M) degraders, 13a and 13b, which effectively and selectively blocked the growth of the NCI-H1975 xenograft tumors with good PK properties in vivo.29 Moreover, CFT8919, developed by C4 Therapeutics, is an orally bioavailable, allosteric, selective degrader of the EGFR L858R mutation, which accounts for more than a third of mutant EGFR lung cancer diagnoses. CFT8919 was reported to induce tumor regression in preclinical tumor models resistant to first-, second-, and third-generation EGFR-TKIs and has the potential to target CNS metastasis in the preclinical model.30 Collectively, these promising EGFR PROTACs are excellent candidates that can be further developed and evaluated in clinical studies as novel therapeutics to overcome EGFR-TKI-induced resistance.
KRAS-targeting PROTACs
The GTPase KRAS is a signal transducer in the RAS/MAPK pathway with a crucial role in regulating multiple cellular processes such as cell growth and survival.80 KRAS mutations have been observed in around 30% of lung adenocarcinomas, which cluster in several hotspots such as residues 12, 13, 61, and 141.6 Specifically, KRASG12C mutation makes up around half of KRAS mutations found in patients with lung adenocarcinomas.
KRAS is difficult to target due to the lack of deep binding pockets, high affinity to GTP and the abundant level of cellular GTP.80,81 The recent breakthrough involves the development of potent covalent KRASG12C inhibitors, leading to the first FDA-approved agent, sotorasib to treat patients with KRASG12C-mutated NSCLC in 2021.82 Adagrasib (MRTX849) is another oral, highly selective, covalent small molecule inhibitor of KRASG12C and has demonstrated clinical efficacy against KRASG12C-mutated NSCLC with an acceptable safety profile in phase 1 clinical trial.83 However, similar to other targeted therapies, tumor cells acquire resistance to KRASG12C inhibition through multiple mechanisms, including selection of new KRAS mutations such as G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C, and amplification of the KRASG12C allele as well as other KRAS-independent mechanisms such as MET amplification and activating mutations in NRAS, BRAF, MAP2K1, and RET.84
KRAS PROTACs have been developed to overcome KRAS12C resistance and to target other KRAS mutants. The first KRAS PROTAC targeting KRASG12C was reported by the Gray group in 2020.31 The PROTACs used ARS-1620 that binds to KRASG12C, and thalidomide derivatives, thus targeting KRAS G12C mutant to CRBN E3-ligase for degradation. However, the lead degrader XY-4-88 failed to degrade endogenous KRASG12C, despite it being able to degrade GFP-KRAS fusion proteins. Subsequently, the Crews group designed VHL-based PROTACs using MRTX849 as the covalent KRASG12C warhead.32 Their lead molecule LC-2 induced KRAS degradation and impaired the downstream MAPK signaling, as demonstrated by reduced p-ERK level in several human KRASG12C-mutated lung cancer cell lines. However, this PROTAC did not induce better antiproliferative effects as compared with MRTX849, which is likely due to its covalent irreversible nature that compromises the catalytic mechanism of action typically associated with PROTACs. To reinstate the substoichiometric activity of PROTACs, the Lu group developed a reversible-covalent PROTAC that efficiently targeted endogenous KRASG12C degradation in KRASG12C-mutated H358 and H23 lung cancer cells.33 To date, no KRAS PROTACs have been evaluated in vivo. Another strategy to indirectly treat KRAS-driven cancer is through targeted degradation of Son of Sevenless Homologue 1 (SOS1), the primary guanine nucleotide exchange factor for regulating the GDP-GTP cycle of KRAS. The Xu group developed the first-in-class agonist-based SOS1 PROTACs by connecting a VHL ligand to a SOS1 agonist.34 They identified a compound that induced SOS1 degradation, displayed superior antiproliferative activity to the agonist itself in various KRAS-driven cancer cells, and showed antitumor potency against human lung cancer H358 xenograft models. Therefore, targeted SOS1 degradation represents a promising therapeutic strategy for overcoming KRAS-driven cancers.
ALK-targeting PROTACs
ALK fusion oncogenes are detected in about 3% to 5% of NSCLC, occurring in non or former smokers, and those of a younger age.85,86 EML4-ALK fusion is the most important ALK fusion and a validated oncogenic driver. ALK TKIs are first-line treatment in patients with ALK-rearranged lung cancer. To date, three generations of ALK inhibitors have been approved by the FDA.67 Second-generation ALK TKIs, such as alectinib and brigatinib, are preferred over first-generation ALK TKI crizotinib. Alectinib also showed improved outcomes in the treatment of brain metastases as compared with crizotinib. NSCLC develops resistance to ALK TKIs through acquiring secondary mutations within the ALK tyrosine kinase domain, amplification of the ALK fusion gene or activation of bypass signaling pathways.67 Lorlatinib, a third-generation ALK TKI, is used for those patients who progress on previous generations. It has activity against most known ALK inhibitor resistance mutations such as G1202R. However, patients can also develop resistance to lorlatinib.67
Currently, the PROTAC technology is being used to develop ALK degraders.35,36,37,38,39,41,42 In 2018, the Gray group reported the first ALK-targeting PROTAC molecules by conjugating known pyrimidine-based ALK inhibitors, TAE684 or LDK378, and the cereblon ligand pomalidomide.35 They showed that one PROTAC, Compound 9, degraded ALK in EML4-ALK fusion-positive H3122 lung cancer cells at nanomolar concentrations and had antiproliferative activity in vitro; yet the compound non-specifically degraded other kinases as well. In the same year, the Hwang group reported a VHL-recruiting, ceritinib-based ALK PROTAC molecule, TD-004, which induced ALK degradation and blocked the proliferation of fusion-positive lung cancer cell line H3122 and lymphoma cell line SU-DHL-1.66 Importantly, this PROTAC inhibited the growth of H3122 xenograft tumors with no significant effect on mouse body weight. In 2020, the Wei group reported a light-activated ALK-targeting PROTAC molecule.37 When exposed to UV light irradiation, the photosensitive “cage” group on pomalidomide was removed from the PROTAC, allowing it to bind and degrade ALK proteins. In 2021, three new PROTACs with oral bioavailability, specific ALK degradation, and/or in vivo antitumor activities in a mouse model were reported from the Jiang, Li, and Xu groups.38,41,42 These PROTAC compounds are promising therapeutics that could delay the emergence of resistance mutations to improve patient outcomes.
Other PROTACs relevant to lung cancer
Many PROTACs currently under preclinical or clinical development for other protein targets or other cancer types have the potential to be used for lung cancer. BRAF mutations account for about 4% of NSCLC and the currently developed BRAF-targeting PROTACs43,44,45 could be evaluated in BRAF-mutated NSCLCs. The bromodomain and extraterminal domain (BET) proteins BRD2, BRD3, and BRD4 are epigenetic readers and critically regulate gene expression. High levels of BRD4 were associated with NSCLC progression, which is consistent with the finding that BRD4 PROTACs blocked 3D lung cancer cell growth in vitro.46 Multiple BRD4 or pan-BRD PROTACs have been developed that showed promising antitumor efficacy in the xenograft tumors from EGFRi-resistant cell lines and lung cancer PDXs.46,47,48,49,50,51,52,53 The protein tyrosine phosphatase SHP2 is frequently activated in lung cancer to maintain a fully activating oncogenic signaling and thus serves as an attractive therapeutic target. A potent SHP2 PROTAC, D26, was developed54 and shown to have therapeutic efficacy alone, or in combination with the third-generation EGFR inhibitor, osimertinib, in blocking osimertinib-resistant lung cancer cells.55 Focal adhesion kinase (FAK) is overexpressed in many types of tumors, including lung cancer. FAK-targeting PROTAC was reported to have antiproliferative effects in lung cancer cells in vitro or antitumor activity in KRAS mutant NSCLC A427 xenograft model.56,57,58 Up-regulation of total or phosphorylated STAT3 was associated with poor prognosis in patients with NSCLCs and thus STAT3 is an attractive therapeutic target.87 The Wang group developed a potent and selective STAT3 PROTAC, which can be evaluated in lung cancer.59 BCL-XL is a key antiapoptotic protein in the Bcl-2 family and its up-regulation is widely observed in human lung cancer cells. The Zhou and Zheng group developed BCL-XL PROTAC degrader DT2216,85 which is currently in phase 1 clinical trials. They recently reported that DT2216 synergized with the FDA-approved KRASG12C inhibitor sotorasib to block tumor growth in preclinical models of KRASG12C-mutated cancers.61 This study thus identified a potential effective combination therapy for patients with KRASG12C-mutated NSCLC, although the safety of this combination requires further evaluation. Since many SCLC cells depend on BCL-XL and BCL-2, DT2216 in combination with ABT-199 (a BCL-2 selective SMI) was tested, which showed antitumor effect in an SCLC H146 xenograft model.60 Furthermore, they recently developed a BCL-XL/BCL-2 dual degrader,62 which had antitumor efficacy as a single agent in SCLC xenograft tumors (unpublished). Collectively, PROTAC development has opened up new therapeutic opportunities in addition to alternative approaches to conventional chemical inhibition.
Conclusion and perspective: PROTAC therapy as a promising targeted strategy for lung cancer treatment
PROTACs have the potential to be developed as new lung cancer therapeutics. Currently, two PROTACs, ARV-471, which targets the estrogen receptor (ER), and ARV-110, which targets the androgen receptor (AR), have been advanced into phase II clinical trials.12,13,14,15 Multiple other PROTACs are currently in phase I clinical trials, including NX-2127 (a Bruton Tyrosine Kinase, BTK degrader), KT474 (an IRAK4 degrader), and DT2216 (a BCL-XL degrader). Significant progress has been made in the development of PROTAC molecules for validated therapeutic targets in NSCLC such as EGFR, KRAS, and ALK fusions, leading to promising compounds with antitumor efficacy in preclinical models. These important advances demonstrate the feasibility and exciting potential of PROTACs as new therapeutics to improve lung cancer outcomes.
Lung cancer represents a highly heterogeneous collection of tumors, each carrying different combinations of genetic alterations for which there are only limited SMIs in the current clinical toolbox. In addition, even when highly specific SMIs are available, the invariable selection of tumor cells with acquired resistance is an ongoing challenge for patients and their clinicians. Unlike SMIs, PROTACs have the capacity to degrade oncogenic proteins, including resistant mutants, with very low intracellular concentration requirements due to the ability of single molecules to undergo multiple rounds of degradation. Thus, PROTACS have the potential to overcome current treatment barriers in lung cancer and fulfill the promise of personalized therapy.
However, to expand the therapeutic landscape to more patients, it is crucial that ligands for the full oncogenic landscape of POI targets are available for development. PROTAC research has been focused, to date, on those cancer targets with available small molecule compounds, such as previous FDA-approved molecules and those that have entered clinical trials. It will be important to expand and evaluate small molecules that can be used to bind and degrade conventionally “undruggable” oncogenic proteins. In addition, Schneider et al created a library with a systematic assessment of >1,000 potential drug targets to guide researchers in further PROTAC development.88
It is also important to design PROTACs to recruit an E3 ligase that is minimally expressed in normal tissues to reduce toxicity. There are currently more than 600 E3 ligases, and research is warranted to identify tumor-specific or tissue-specific E3 ligases and their ligands that could be used for PROTACs. Event-driven PROTAC molecules do not necessarily need high-affinity ligands for activity, thus, low micromolar E3 recruiting elements identified from screens could be linked to already existing high-affinity ligands to develop next-generation PROTACs. Further efforts should also be made to control PROTAC activity in degrading proteins on demand. Novel approaches have been explored in activating PROTACs with light (opto-PROTAC)37 or in response to tumor hypoxic environment.89
PROTACs as a class have poor physicochemical properties that pose challenges in drug development. For example, PROTACs may have poor penetration to the disease tissue. Thus, for the treatment of lung cancer, it is critical to evaluate the tissue distribution of the PROTACs to the lung. In addition, PROTACs are known to have pharmacodynamic (PD) effects beyond their PK, which would require the use of proper PD biomarkers to determine dosing regimen. Since the PROTAC technology is new, more work is also needed to understand how to integrate these investigational agents with other standard anti-cancer therapeutics to optimize clinical benefit. New biomarker assays designed to monitor PROTAC activity both in vitro and in vivo using lung cancer xenograft models and immunocompetent model models are needed in preclinical phases. More importantly, careful evaluations for PK and PD, toxicity profiles, and efficacy in human clinical studies are needed in order to move this new class of drugs to patient treatment.
For the past two decades, lung cancer has been at the forefront of molecular oncology therapeutics. New preclinical and early clinical development testing of an expanding portfolio of PROTAC candidates will continue the promise of personalized and targeted therapy to improve the outcome of patients with advanced lung cancer.
Acknowledgments
L.W. was supported by R01CA234351 and R01DE023641. We thank Ms. Leah Buletti at the UF Health Cancer Center for the help in generating the cartoon of the graphic abstract.
Author contributions
L.W. and F.J.K. designed the manuscript. J.W.L. drafted the manuscript and prepared the figure. L.W., F.J.K., G.Z., and J.W.L. edited the manuscript. All authors approved the final version of the manuscript.
Declaration of interests
G.Z. is co-founder of and has equity in Dialectic Therapeutics, which develops BCL-XL/2 PROTACs as cancer therapeutics.
Contributor Information
Frederic J. Kaye, Email: fkaye@ufl.edu.
Lizi Wu, Email: lzwu@ufl.edu.
References
- 1.Sung H., Ferlay J., Siegel R.L., Laversanne M., Soerjomataram I., Jemal A., Bray F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2021;71:209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA. Cancer J. Clin. 2022;72:7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 3.Nicholson A.G., Tsao M.S., Beasley M.B., Borczuk A.C., Brambilla E., Cooper W.A., Dacic S., Jain D., Kerr K.M., Lantuejoul S., et al. The 2021 WHO classification of lung tumors: impact of advances since 2015. J. Thorac. Oncol. 2022;17:362–387. doi: 10.1016/j.jtho.2021.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research Network Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–525. doi: 10.1038/nature11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.George J., Lim J.S., Jang S.J., Cun Y., Ozretić L., Kong G., Leenders F., Lu X., Fernández-Cuesta L., Bosco G., et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53. doi: 10.1038/nature14664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cancer Genome Atlas Research Network Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell J.D., Alexandrov A., Kim J., Wala J., Berger A.H., Pedamallu C.S., Shukla S.A., Guo G., Brooks A.N., Murray B.A., et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016;48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carrot-Zhang J., Yao X., Devarakonda S., Deshpande A., Damrauer J.S., Silva T.C., Wong C.K., Choi H.Y., Felau I., Robertson A.G., Castro M.A.A., et al. Whole-genome characterization of lung adenocarcinomas lacking the RTK/RAS/RAF pathway. Cell Rep. 2021;34:108707. doi: 10.1016/j.celrep.2021.108707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553:446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 10.Sakamoto K.M., Kim K.B., Kumagai A., Mercurio F., Crews C.M., Deshaies R.J. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc. Natl. Acad. Sci. USA. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Békés M., Langley D.R., Crews C.M. PROTAC targeted protein degraders: the past is prologue. Nat. Rev. Drug Discov. 2022;21:181–200. doi: 10.1038/s41573-021-00371-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu Z., Crews C.M. Recent developments in PROTAC-mediated protein degradation: from bench to clinic. Chembiochem. 2022;23:e202100270. doi: 10.1002/cbic.202100270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schapira M., Calabrese M.F., Bullock A.N., Crews C.M. Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug Discov. 2019;18:949–963. doi: 10.1038/s41573-019-0047-y. [DOI] [PubMed] [Google Scholar]
- 14.Mullard A. Targeted protein degraders crowd into the clinic. Nat. Rev. Drug Discov. 2021;20:247–250. doi: 10.1038/d41573-021-00052-4. [DOI] [PubMed] [Google Scholar]
- 15.Nieto-Jiménez C., Morafraile E.C., Alonso-Moreno C., Ocaña A. Clinical considerations for the design of PROTACs in cancer. Mol. Cancer. 2022;21:67. doi: 10.1186/s12943-022-01535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khan S., He Y., Zhang X., Yuan Y., Pu S., Kong Q., Zheng G., Zhou D. PROteolysis TArgeting Chimeras (PROTACs) as emerging anticancer therapeutics. Oncogene. 2020;39:4909–4924. doi: 10.1038/s41388-020-1336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burke M.R., Smith A.R., Zheng G. Overcoming cancer drug resistance utilizing PROTAC technology. Front. Cell Dev. Biol. 2022;10:872729. doi: 10.3389/fcell.2022.872729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burslem G.M., Smith B.E., Lai A.C., Jaime-Figueroa S., McQuaid D.C., Bondeson D.P., Toure M., Dong H., Qian Y., Wang J., et al. The advantages of targeted protein degradation over inhibition: an RTK case study. Cell Chem. Biol. 2018;25:67–77.e3. doi: 10.1016/j.chembiol.2017.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheng M., Yu X., Lu K., Xie L., Wang L., Meng F., Han X., Chen X., Liu J., Xiong Y., Jin J. Discovery of potent and selective epidermal growth factor receptor (EGFR) bifunctional small-molecule degraders. J. Med. Chem. 2020;63:1216–1232. doi: 10.1021/acs.jmedchem.9b01566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Xu F., Tong L., Zhang T., Xie H., Lu X., Ren X., Ding K. Design and synthesis of selective degraders of EGFR(L858R/T790M) mutant. Eur. J. Med. Chem. 2020;192:112199. doi: 10.1016/j.ejmech.2020.112199. [DOI] [PubMed] [Google Scholar]
- 21.Jang J., To C., De Clercq D.J.H., Park E., Ponthier C.M., Shin B.H., Mushajiang M., Nowak R.P., Fischer E.S., Eck M.J., et al. Mutant-selective allosteric EGFR degraders are effective against a broad range of drug-resistant mutations. Angew. Chem. Int. Ed. Engl. 2020;59:14481–14489. doi: 10.1002/anie.202003500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qu X., Liu H., Song X., Sun N., Zhong H., Qiu X., Yang X., Jiang B. Effective degradation of EGFR(L858R+T790M) mutant proteins by CRBN-based PROTACs through both proteosome and autophagy/lysosome degradation systems. Eur. J. Med. Chem. 2021;218:113328. doi: 10.1016/j.ejmech.2021.113328. [DOI] [PubMed] [Google Scholar]
- 23.Zhao H.Y., Yang X.Y., Lei H., Xi X.X., Lu S.M., Zhang J.J., Xin M., Zhang S.Q. Discovery of potent small molecule PROTACs targeting mutant EGFR. Eur. J. Med. Chem. 2020;208:112781. doi: 10.1016/j.ejmech.2020.112781. [DOI] [PubMed] [Google Scholar]
- 24.Zhao H.Y., Wang H.P., Mao Y.Z., Zhang H., Xin M., Xi X.X., Lei H., Mao S., Li D.H., Zhang S.Q. Discovery of potent PROTACs targeting EGFR mutants through the optimization of covalent EGFR ligands. J. Med. Chem. 2022;65:4709–4726. doi: 10.1021/acs.jmedchem.1c01827. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H., Xie R., Ai-Furas H., Li Y., Wu Q., Li J., Xu F., Xu T. Design, synthesis, and biological evaluation of novel EGFR PROTACs targeting del19/T790M/C797S mutation. ACS Med. Chem. Lett. 2022;13:278–283. doi: 10.1021/acsmedchemlett.1c00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q., Guo Q., Wang S., Wan S., Li Z., Zhang J., Wu X. Design and synthesis of proteolysis targeting chimeras (PROTACs) as an EGFR degrader based on CO-1686. Eur. J. Med. Chem. 2022;238:114455. doi: 10.1016/j.ejmech.2022.114455. [DOI] [PubMed] [Google Scholar]
- 27.Du Y., Chen Y., Wang Y., Chen J., Lu X., Zhang L., Li Y., Wang Z., Ye G., Zhang G. HJM-561, a potent, selective, and orally bioavailable EGFR PROTAC that overcomes osimertinib-resistant EGFR triple mutations. Mol. Cancer Ther. 2022;21:1060–1066. doi: 10.1158/1535-7163.Mct-21-0835. [DOI] [PubMed] [Google Scholar]
- 28.Shi S., Du Y., Huang L., Cui J., Niu J., Xu Y., Zhu Q. Discovery of novel potent covalent inhibitor-based EGFR degrader with excellent in vivo efficacy. Bioorg. Chem. 2022;120:105605. doi: 10.1016/j.bioorg.2022.105605. [DOI] [PubMed] [Google Scholar]
- 29.Zhang W., Li P., Sun S., Jia C., Yang N., Zhuang X., Zheng Z., Li S. Discovery of highly potent and selective CRBN-recruiting EGFR(L858R/T790M) degraders in vivo. Eur. J. Med. Chem. 2022;238:114509. doi: 10.1016/j.ejmech.2022.114509. [DOI] [PubMed] [Google Scholar]
- 30.2021. https://ir.c4therapeutics.com/news-releases/news-release-details/c4-therapeutics-presents-pre-clinical-data-cft8919-selective
- 31.Zeng M., Xiong Y., Safaee N., Nowak R.P., Donovan K.A., Yuan C.J., Nabet B., Gero T.W., Feru F., Li L., et al. Exploring targeted degradation strategy for oncogenic KRAS(G12C) Cell Chem. Biol. 2020;27:19–31.e6. doi: 10.1016/j.chembiol.2019.12.006. [DOI] [PubMed] [Google Scholar]
- 32.Bond M.J., Chu L., Nalawansha D.A., Li K., Crews C.M. Targeted degradation of oncogenic KRAS(G12C) by VHL-recruiting PROTACs. ACS Cent. Sci. 2020;6:1367–1375. doi: 10.1021/acscentsci.0c00411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang F., Wen Y., Wang C., Zhou Y., Zhou Y., Zhang Z.M., Liu T., Lu X. Efficient targeted oncogenic KRAS(G12C) degradation via first reversible-covalent PROTAC. Eur. J. Med. Chem. 2022;230:114088. doi: 10.1016/j.ejmech.2021.114088. [DOI] [PubMed] [Google Scholar]
- 34.Zhou C., Fan Z., Zhou Z., Li Y., Cui R., Liu C., Zhou G., Diao X., Jiang H., Zheng M., et al. Discovery of the first-in-class Agonist-based SOS1 PROTACs effective in human cancer cells harboring various KRAS mutations. J. Med. Chem. 2022;65:3923–3942. doi: 10.1021/acs.jmedchem.1c01774. [DOI] [PubMed] [Google Scholar]
- 35.Powell C.E., Gao Y., Tan L., Donovan K.A., Nowak R.P., Loehr A., Bahcall M., Fischer E.S., Jänne P.A., George R.E., Gray N.S. Chemically induced degradation of anaplastic lymphoma kinase (ALK) J. Med. Chem. 2018;61:4249–4255. doi: 10.1021/acs.jmedchem.7b01655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C., Han X.R., Yang X., Jiang B., Liu J., Xiong Y., Jin J. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK) Eur. J. Med. Chem. 2018;151:304–314. doi: 10.1016/j.ejmech.2018.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu J., Chen H., Ma L., He Z., Wang D., Liu Y., Lin Q., Zhang T., Gray N., Kaniskan H.Ü., et al. Light-induced control of protein destruction by opto-PROTAC. Sci. Adv. 2020;6:eaay5154. doi: 10.1126/sciadv.aay5154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren C., Sun N., Kong Y., Qu X., Liu H., Zhong H., Song X., Yang X., Jiang B. Structure-based discovery of SIAIS001 as an oral bioavailability ALK degrader constructed from Alectinib. Eur. J. Med. Chem. 2021;217:113335. doi: 10.1016/j.ejmech.2021.113335. [DOI] [PubMed] [Google Scholar]
- 39.Sun N., Ren C., Kong Y., Zhong H., Chen J., Li Y., Zhang J., Zhou Y., Qiu X., Lin H., et al. Development of a Brigatinib degrader (SIAIS117) as a potential treatment for ALK positive cancer resistance. Eur. J. Med. Chem. 2020;193:112190. doi: 10.1016/j.ejmech.2020.112190. [DOI] [PubMed] [Google Scholar]
- 40.Kang C.H., Lee D.H., Lee C.O., Du Ha J., Park C.H., Hwang J.Y. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC) Biochem. Biophys. Res. Commun. 2018;505:542–547. doi: 10.1016/j.bbrc.2018.09.169. [DOI] [PubMed] [Google Scholar]
- 41.Xie S., Sun Y., Liu Y., Li X., Li X., Zhong W., Zhan F., Zhu J., Yao H., Yang D.H., et al. Development of alectinib-based PROTACs as novel potent degraders of anaplastic lymphoma kinase (ALK) J. Med. Chem. 2021;64:9120–9140. doi: 10.1021/acs.jmedchem.1c00270. [DOI] [PubMed] [Google Scholar]
- 42.Yan G., Zhong X., Yue L., Pu C., Shan H., Lan S., Zhou M., Hou X., Yang J., Li R. Discovery of a PROTAC targeting ALK with in vivo activity. Eur. J. Med. Chem. 2021;212:113150. doi: 10.1016/j.ejmech.2020.113150. [DOI] [PubMed] [Google Scholar]
- 43.Posternak G., Tang X., Maisonneuve P., Jin T., Lavoie H., Daou S., Orlicky S., Goullet de Rugy T., Caldwell L., Chan K., et al. Functional characterization of a PROTAC directed against BRAF mutant V600E. Nat. Chem. Biol. 2020;16:1170–1178. doi: 10.1038/s41589-020-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alabi S., Jaime-Figueroa S., Yao Z., Gao Y., Hines J., Samarasinghe K.T.G., Vogt L., Rosen N., Crews C.M. Mutant-selective degradation by BRAF-targeting PROTACs. Nat. Commun. 2021;12:920. doi: 10.1038/s41467-021-21159-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han X.R., Chen L., Wei Y., Yu W., Chen Y., Zhang C., Jiao B., Shi T., Sun L., Zhang C., et al. Discovery of selective small molecule degraders of BRAF-V600E. J. Med. Chem. 2020;63:4069–4080. doi: 10.1021/acs.jmedchem.9b02083. [DOI] [PubMed] [Google Scholar]
- 46.Vartak R., Saraswat A., Yang Y., Chen Z.S., Patel K. Susceptibility of lung carcinoma cells to nanostructured lipid carrier of ARV-825, a BRD4 degrading proteolysis targeting chimera. Pharm. Res. 2022;39:2745–2759. doi: 10.1007/s11095-022-03184-3. [DOI] [PubMed] [Google Scholar]
- 47.Lu J., Qian Y., Altieri M., Dong H., Wang J., Raina K., Hines J., Winkler J.D., Crew A.P., Coleman K., Crews C.M. Hijacking the E3 ubiquitin ligase cereblon to efficiently target BRD4. Chem. Biol. 2015;22:755–763. doi: 10.1016/j.chembiol.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zong D., Gu J., Cavalcante G.C., Yao W., Zhang G., Wang S., Owonikoko T.K., He X., Sun S.Y. BRD4 levels determine the response of human lung cancer cells to BET degraders that potently induce apoptosis through suppression of mcl-1. Cancer Res. 2020;80:2380–2393. doi: 10.1158/0008-5472.Can-19-3674. [DOI] [PubMed] [Google Scholar]
- 49.Zhou B., Hu J., Xu F., Chen Z., Bai L., Fernandez-Salas E., Lin M., Liu L., Yang C.Y., Zhao Y., et al. Discovery of a small-molecule degrader of bromodomain and extra-terminal (BET) proteins with picomolar cellular potencies and capable of achieving tumor regression. J. Med. Chem. 2018;61:462–481. doi: 10.1021/acs.jmedchem.6b01816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winter G.E., Buckley D.L., Paulk J., Roberts J.M., Souza A., Dhe-Paganon S., Bradner J.E. DRUG DEVELOPMENT. Phthalimide conjugation as a strategy for in vivo target protein degradation. Science. 2015;348:1376–1381. doi: 10.1126/science.aab1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun D., Nikonova A.S., Zhang P., Deneka A.Y., Fitzgerald M.E., Michael R.E., Lee L., Lilly A.C., Fisher S.L., Phillips A.J., et al. Evaluation of the small-molecule BRD4 degrader CFT-2718 in small-cell lung cancer and pancreatic cancer models. Mol. Cancer Ther. 2021;20:1367–1377. doi: 10.1158/1535-7163.Mct-20-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Qin C., Hu Y., Zhou B., Fernandez-Salas E., Yang C.Y., Liu L., McEachern D., Przybranowski S., Wang M., Stuckey J., et al. Discovery of QCA570 as an exceptionally potent and efficacious proteolysis targeting chimera (PROTAC) degrader of the bromodomain and extra-terminal (BET) proteins capable of inducing complete and durable tumor regression. J. Med. Chem. 2018;61:6685–6704. doi: 10.1021/acs.jmedchem.8b00506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu C., Qian L., Vallega K.A., Ma G., Zong D., Chen L., Wang S., Ramalingam S.R., Qin Z., Sun S.Y. The novel BET degrader, QCA570, is highly active against the growth of human NSCLC cells and synergizes with osimertinib in suppressing osimertinib-resistant EGFR-mutant NSCLC cells. Am. J. Cancer Res. 2022;12:779–792. [PMC free article] [PubMed] [Google Scholar]
- 54.Wang M., Lu J., Wang M., Yang C.Y., Wang S. Discovery of SHP2-D26 as a first, potent, and effective PROTAC degrader of SHP2 protein. J. Med. Chem. 2020;63:7510–7528. doi: 10.1021/acs.jmedchem.0c00471. [DOI] [PubMed] [Google Scholar]
- 55.Deng Y., Ma G., Vallega K.A., Wang D., Wang M., Wang C., Wang S., Ramalingam S.S., Sun S.Y. Therapeutic efficacy of the novel SHP2 degrader SHP2-D26, alone or in combination, against lung cancer is associated with modulation of p70S6K/S6, Bim and Mcl-1. Cancer Gene Ther. 2022;29:1558–1569. doi: 10.1038/s41417-022-00472-3. [DOI] [PubMed] [Google Scholar]
- 56.Liu J., Xue L., Xu X., Luo J., Zhang S. FAK-targeting PROTAC demonstrates enhanced antitumor activity against KRAS mutant non-small cell lung cancer. Exp. Cell Res. 2021;408:112868. doi: 10.1016/j.yexcr.2021.112868. [DOI] [PubMed] [Google Scholar]
- 57.Sun Y., Wang R., Sun Y., Wang L., Xue Y., Wang J., Wu T., Yin W., Qin Q., Sun Y., et al. Identification of novel and potent PROTACs targeting FAK for non-small cell lung cancer: design, synthesis, and biological study. Eur. J. Med. Chem. 2022;237:114373. doi: 10.1016/j.ejmech.2022.114373. [DOI] [PubMed] [Google Scholar]
- 58.Cromm P.M., Samarasinghe K.T.G., Hines J., Crews C.M. Addressing kinase-independent functions of fak via PROTAC-mediated degradation. J. Am. Chem. Soc. 2018;140:17019–17026. doi: 10.1021/jacs.8b08008. [DOI] [PubMed] [Google Scholar]
- 59.Bai L., Zhou H., Xu R., Zhao Y., Chinnaswamy K., McEachern D., Chen J., Yang C.Y., Liu Z., Wang M., et al. A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell. 2019;36:498–511.e17. doi: 10.1016/j.ccell.2019.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khan S., Zhang X., Lv D., Zhang Q., He Y., Zhang P., Liu X., Thummuri D., Yuan Y., Wiegand J.S., et al. A selective BCL-X(L) PROTAC degrader achieves safe and potent antitumor activity. Nat. Med. 2019;25:1938–1947. doi: 10.1038/s41591-019-0668-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Khan S., Wiegand J., Zhang P., Hu W., Thummuri D., Budamagunta V., Hua N., Jin L., Allegra C.J., Kopetz S.E., et al. BCL-X(L) PROTAC degrader DT2216 synergizes with sotorasib in preclinical models of KRAS(G12C)-mutated cancers. J. Hematol. Oncol. 2022;15:23. doi: 10.1186/s13045-022-01241-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lv D., Pal P., Liu X., Jia Y., Thummuri D., Zhang P., Hu W., Pei J., Zhang Q., Zhou S., et al. Development of a BCL-xL and BCL-2 dual degrader with improved anti-leukemic activity. Nat. Commun. 2021;12:6896. doi: 10.1038/s41467-021-27210-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Uribe M.L., Marrocco I., Yarden Y. EGFR in cancer: signaling mechanisms, drugs, and acquired resistance. Cancers (Basel) 2021;13:2748. doi: 10.3390/cancers13112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynch T.J., Bell D.W., Sordella R., Gurubhagavatula S., Okimoto R.A., Brannigan B.W., Harris P.L., Haserlat S.M., Supko J.G., Haluska F.G., et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 65.Paez J.G., Jänne P.A., Lee J.C., Tracy S., Greulich H., Gabriel S., Herman P., Kaye F.J., Lindeman N., Boggon T.J., et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 66.Lavacchi D., Mazzoni F., Giaccone G. Clinical evaluation of dacomitinib for the treatment of metastatic non-small cell lung cancer (NSCLC): current perspectives. Drug Des. Devel. Ther. 2019;13:3187–3198. doi: 10.2147/dddt.S194231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cooper A.J., Sequist L.V., Lin J.J. Third-generation EGFR and ALK inhibitors: mechanisms of resistance and management. Nat. Rev. Clin. Oncol. 2022;19:499–514. doi: 10.1038/s41571-022-00639-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Choudhury N.J., Makhnin A., Tobi Y.Y., Daly R.M., Preeshagul I.R., Iqbal A.N., Ahn L.S., Hayes S.A., Heller G., Kris M.G., et al. Pilot study of dacomitinib for patients with metastatic EGFR-mutant lung cancers with disease progression after initial treatment with osimertinib. JCO Precis. Oncol. 2021;5:695–700. doi: 10.1200/po.21.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mitsudomi T., Morita S., Yatabe Y., Negoro S., Okamoto I., Tsurutani J., Seto T., Satouchi M., Tada H., Hirashima T., et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/s1470-2045(09)70364-x. [DOI] [PubMed] [Google Scholar]
- 70.Mok T.S., Wu Y.L., Thongprasert S., Yang C.H., Chu D.T., Saijo N., Sunpaweravong P., Han B., Margono B., Ichinose Y., et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 71.Ramalingam S.S., Vansteenkiste J., Planchard D., Cho B.C., Gray J.E., Ohe Y., Zhou C., Reungwetwattana T., Cheng Y., Chewaskulyong B., et al. FLAURA Investigators Overall survival with osimertinib in untreated, EGFR-mutated advanced NSCLC. N. Engl. J. Med. 2020;382:41–50. doi: 10.1056/NEJMoa1913662. [DOI] [PubMed] [Google Scholar]
- 72.Rosell R., Carcereny E., Gervais R., Vergnenegre A., Massuti B., Felip E., Palmero R., Garcia-Gomez R., Pallares C., Sanchez J.M., et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/s1470-2045(11)70393-x. [DOI] [PubMed] [Google Scholar]
- 73.Sequist L.V., Yang J.C.H., Yamamoto N., O'Byrne K., Hirsh V., Mok T., Geater S.L., Orlov S., Tsai C.M., Boyer M., et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013;31:3327–3334. doi: 10.1200/jco.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 74.Soria J.C., Ohe Y., Vansteenkiste J., Reungwetwattana T., Chewaskulyong B., Lee K.H., Dechaphunkul A., Imamura F., Nogami N., Kurata T., et al. FLAURA Investigators Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N. Engl. J. Med. 2018;378:113–125. doi: 10.1056/NEJMoa1713137. [DOI] [PubMed] [Google Scholar]
- 75.Wood E.R., Shewchuk L.M., Ellis B., Brignola P., Brashear R.L., Caferro T.R., Dickerson S.H., Dickson H.D., Donaldson K.H., Gaul M., et al. 6-Ethynylthieno[3, 2-d]- and 6-ethynylthieno[2, 3-d]pyrimidin-4-anilines as tunable covalent modifiers of ErbB kinases. Proc. Natl. Acad. Sci. USA. 2008;105:2773–2778. doi: 10.1073/pnas.2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu Y.L., Cheng Y., Zhou X., Lee K.H., Nakagawa K., Niho S., Tsuji F., Linke R., Rosell R., Corral J., et al. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol. 2017;18:1454–1466. doi: 10.1016/s1470-2045(17)30608-3. [DOI] [PubMed] [Google Scholar]
- 77.Wu Y.L., Zhou C., Hu C.P., Feng J., Lu S., Huang Y., Li W., Hou M., Shi J.H., Lee K.Y., et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/s1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 78.Yang J.C.H., Wu Y.L., Schuler M., Sebastian M., Popat S., Yamamoto N., Zhou C., Hu C.P., O'Byrne K., Feng J., et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–151. doi: 10.1016/s1470-2045(14)71173-8. [DOI] [PubMed] [Google Scholar]
- 79.Ren C., Sun N., Liu H., Kong Y., Sun R., Qiu X., Chen J., Li Y., Zhang J., Zhou Y., et al. Discovery of a brigatinib degrader SIAIS164018 with destroying metastasis-related oncoproteins and a reshuffling kinome profile. J. Med. Chem. 2021;64:9152–9165. doi: 10.1021/acs.jmedchem.1c00373. [DOI] [PubMed] [Google Scholar]
- 80.Simanshu D.K., Nissley D.V., McCormick F. RAS proteins and their regulators in human disease. Cell. 2017;170:17–33. doi: 10.1016/j.cell.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Papke B., Der C.J. Drugging RAS: know the enemy. Science. 2017;355:1158–1163. doi: 10.1126/science.aam7622. [DOI] [PubMed] [Google Scholar]
- 82.Hong D.S., Fakih M.G., Strickler J.H., Desai J., Durm G.A., Shapiro G.I., Falchook G.S., Price T.J., Sacher A., Denlinger C.S., et al. KRAS(G12C) inhibition with sotorasib in advanced solid tumors. N. Engl. J. Med. 2020;383:1207–1217. doi: 10.1056/NEJMoa1917239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jänne P.A., Riely G.J., Gadgeel S.M., Heist R.S., Ou S.H.I., Pacheco J.M., Johnson M.L., Sabari J.K., Leventakos K., Yau E., et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. N. Engl. J. Med. 2022;387:120–131. doi: 10.1056/NEJMoa2204619. [DOI] [PubMed] [Google Scholar]
- 84.Awad M.M., Liu S., Rybkin I.I., Arbour K.C., Dilly J., Zhu V.W., Johnson M.L., Heist R.S., Patil T., Riely G.J., et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N. Engl. J. Med. 2021;384:2382–2393. doi: 10.1056/NEJMoa2105281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sasaki T., Rodig S.J., Chirieac L.R., Jänne P.A. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur. J. Cancer. 2010;46:1773–1780. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soda M., Choi Y.L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S.i., Watanabe H., Kurashina K., Hatanaka H., et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 87.Harada D., Takigawa N., Kiura K. The role of STAT3 in non-small cell lung cancer. Cancers (Basel) 2014;6:708–722. doi: 10.3390/cancers6020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schneider M., Radoux C.J., Hercules A., Ochoa D., Dunham I., Zalmas L.P., Hessler G., Ruf S., Shanmugasundaram V., Hann M.M., et al. The PROTACtable genome. Nat. Rev. Drug Discov. 2021;20:789–797. doi: 10.1038/s41573-021-00245-x. [DOI] [PubMed] [Google Scholar]
- 89.Cheng W., Li S., Wen X., Han S., Wang S., Wei H., Song Z., Wang Y., Tian X., Zhang X. Development of hypoxia-activated PROTAC exerting a more potent effect in tumor hypoxia than in normoxia. Chem. Commun. 2021;57:12852–12855. doi: 10.1039/d1cc05715d. [DOI] [PubMed] [Google Scholar]