

Abstract
Editing efficiency is pivotal for the efficacies of CRISPR-based gene therapies. We found that fusing an HMG-D domain to the N terminus of SpCas9 (named efficiency-enhanced Cas9 [eeCas9]) significantly increased editing efficiency by 1.4-fold on average. The HMG-D domain also enhanced the activities of non-NGG PAM Cas9 variants, high-fidelity Cas9 variants, smaller Cas9 orthologs, Cas9-based epigenetic regulators, and base editors in cell lines. Furthermore, we discovered that eeCas9 exhibits comparable off-targeting effects with Cas9, and its specificity could be increased through ribonucleoprotein delivery or using hairpin single-guide RNAs and high-fidelity Cas9s. The entire eeCas9 could be packaged into an adeno-associated virus vector and exhibited a 1.7- to 2.6-fold increase in editing efficiency targeting the Pcsk9 gene in mice, leading to a greater reduction of serum cholesterol levels. Moreover, the efficiency of eeA3A-BE3 also surpasses that of A3A-BE3 in targeting the promoter region of γ-globin genes or BCL11A enhancer in human hematopoietic stem cells to reactivate γ-globin expression for the treatment of β-hemoglobinopathy. Together, eeCas9 and its derivatives are promising editing tools that exhibit higher activity and therapeutic efficacy for both in vivo and ex vivo therapeutics.
Keywords: Cas9, base editors, genome editing, epigenetic regulation, double-strand DNA binding domain, HMG-D, gene therapy, Pcsk9, β-hemoglobinopathy
Graphical abstract
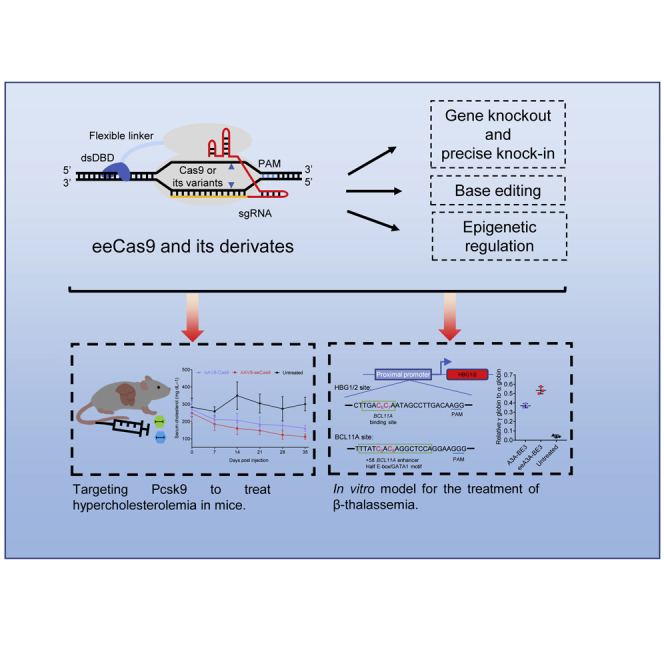
Li and colleagues improved the efficiencies of Cas9 and its derivatives by enhancing their DNA binding affinity. They applied this strategy to an in vivo gene therapy experiment targeting Pcsk9 to treat hypercholesterolemia in mice and an in vitro model for the treatment of β-thalassemia.
Introduction
The CRISPR/Cas system is a revolutionary technology for the achievement of efficient and precise genome editing in multiple cell types, organisms, and human gene therapies.1 SpCas9 (Streptococcus pyogenes Cas9, hereafter referred to as Cas9) is the most efficient and commonly used nuclease that binds a single-guide RNA (sgRNA) and recognizes an NGG sequence as the protospacer adjacent motif (PAM) in the genome.2,3,4 Once Cas9 binds an NGG PAM, the double-strand DNA is unwound to facilitate single-guide RNA (sgRNA) pairing with the target.5,6 When the sgRNA perfectly matches the target, Cas9 undergoes a conformational change, and two nuclease domains (HNH and RuvC) adapt to an active state, catalyzing a blunt-end double-stranded break (DSB).5,6 DSBs are repaired either by error-prone non-homologous end joining to generate small insertions and deletions (indels) or by homology-directed repair (HDR) with the presence of repair templates for precise sequence alterations.7 In addition to generating DSBs, Cas9 and its variants have been adapted to a broad spectrum of applications, such as epigenome regulation,8 base editing,9 and prime editing.10 Although the CRISPR/Cas system is the most efficient technology compared with ZFNs (zinc-finger nucleases) and TALENs (transcription activator-like effector nucleases), its activity varies in different loci. Therefore, highly active Cas9 variants are eagerly demanded not only for basic research but also for numerous applications, especially for gene therapy.
To increase gene-editing efficiency, researchers devoted their efforts to two essential components of the CRISPR/Cas system, the sgRNA and the Cas9 protein. Chemically modified sgRNA tends to be more stable in cells, enabling much higher efficiencies in cleavage and base editing in human cells and animal embryos.11,12 Through electroporation of chemically modified sgRNA and Cas9 protein, over 90% editing efficiency has been achieved in human hematopoietic stem cells (HSCs).13 This strategy has also been successfully applied in clinical investigations to treat β-hemoglobinopathies.14,15 Besides, modifications on Cas9, such as optimization of the nuclear localization signals and codon usage,13,16 have also proved very effective. Another strategy is to enhance Cas9's ability to access complex chromatin regions. Since eukaryotic genomic DNA is assembled into higher-order chromatin structures, chromatin remodeling proteins or peptides, such as yChd1,17 chromatin-modulating peptides,18 or synthetic transcription activation domains,19 were respectively tested on Cas9. Although these strategies are effective their efficacy is not well characterized, especially in living animals and for gene therapy purposes.
Binding and interrogation of the target sequences are two crucial steps for Cas9-mediated DNA cleavage.5,6 Thus, we hypothesized that the activity of Cas9 might be elevated when the association between Cas9 and its DNA substrate is enhanced, as previously reported on cytosine base editor (CBE) activity.20 Here, we developed an efficiency-enhanced Cas9 (eeCas9) by fusing a double-strand DNA binding domain (dsDBD) with Cas9. The dsDBD is compatible with multiple Cas9 variants, other Cas9 orthologs, Cas9-based epigenetic editors, and base editors. Importantly, eeCas9 exhibits comparable off-targeting effects with Cas9 while presenting a higher editing efficiency in both in vivo and ex vivo gene therapy models. Our results demonstrate that enhancing the DNA binding activity of Cas9 could significantly increase the editing efficiency, providing powerful tools for basic research and gene therapies.
Results
Screening of non-sequence-specific DNA binding domains to increase Cas9 editing activity
We previously demonstrated that the increased affinity between CBEs and single-strand DNA substrates dramatically improved base-editing efficiency.20 Inspired by this idea, we hypothesized that the fusion of a non-sequence-specific dsDBD with the Cas9 protein could also enhance the activity of Cas9. There are three major types of dsDBD proteins among which the high-mobility group (HMG) family of chromosomal proteins and archaeal chromosomal proteins are relatively compact.21 Since smaller-sized Cas9 fusion proteins are friendlier for future engineering and application, we screened 11 candidates derived from the compact dsDBDs by fusing each of them to the N terminus of Cas9 via a flexible linker (Figures 1A, S1A, and S1B). The fusion proteins were evaluated via a modified fluorescent reporter system in which an mCherry reporter was out of frame with its start codon (Figure 1B). Once Cas9 cleaves the target region, the resulting indel will correct the reading frame of the mCherry randomly and then restore the fluorescence (Figure 1B). Seventy-two hours after transfection, mCherry+ cells of each group were counted by flow cytometry and normalized by their EGFP+ cell number, which was expressed by sgRNA plasmid as a transfection control (Figure 1B). The results showed that almost all dsDBDs increased the percentage of mCherry+ cells, indicating elevated Cas9 activity (Figure 1C). In addition, we also tested the 11 candidates on endogenous EMX1-2 and VEGF-1 target sites in HEK293T cells. High-throughput sequencing (HTS) results showed that all the 11 candidates increased their indel efficiency when compared with Cas9 (Figure 1D). Overall, we observed that the HMG-D from Drosophila melanogaster resulted in the highest activity increase both on the fluorescent reporter target (a 1.5-fold increase) and two endogenous targets (a 1.8- and 1.6-fold increase, respectively) (Figures 1E and 1F). These results suggested that enhancing the DNA binding affinity of Cas9 by fusing an extra dsDBD significantly increased the Cas9 activity.
Figure 1.

Design of eeCas9 through fusion of a non-sequence-specific DNA binding domain
(A) Schematic of Cas9 with N-terminal dsDBD. dsDBD, double-strand DNA binding domain. (B) Schematic diagram of the modified fluorescent reporter system. This reporter system contains an mCherry protein which is out of frame with its start codon (left). FACS analysis of Cas9 induced mCherry expression in EGFP+ HEK293T cells (right). (C) Quantification of the mCherry+ HEK293T cells induced by Cas9 with different N-terminal dsDBDs. (D) Quantification of the gene-editing efficiencies at two endogenous EMX1-2 and VEGF-1 sites by HTS analysis. (E) Ratio of mCherry+ cells between Cas9 and Cas9 with different N-terminal dsDBDs on fluorescent reporter system. (F) Ratio of indels between Cas9 and Cas9 with different N-terminal dsDBDs on two endogenous EMX1-2 (left) and VEGF-1 (right) targets. Data in graphs (C), (D), (E), and (F) represent mean ± SD (n = 3 independent experiments).
Next, we tried to optimize the construct of HMG-D-Cas9 to further increase its efficiency. We replaced the 32-amino-acid N-terminal flexible linker (L4) with other linkers (Figures S2A and S2B), but no other linkers outperformed the L4 linker (Figure S2C), and further increasing the copy number of HMG-D did not elevate or even impair editing efficiency (Figure S2C). We also tested fusing the HMG-D domain to the C terminus of Cas9 with a long linker. The HTS results showed that the two C-terminal HMG-D constructs had similar efficiencies comparable with the N-terminal HMG-D construct via an L4 linker (Figure S2C). These data demonstrated that a single N-terminal HMG-D with a flexible L4 linker is the optimized construct, which was named efficiency-enhanced Cas9 (eeCas9).
To understand how HMG-D increases Cas9 activity, we tested whether the HMG-D domain increased the expression of the fusion protein. Western blot assay suggested that the N-terminal fusion of HMG-D did not increase the protein level of eeCas9 (Figure S3A). Next, we introduced three point mutations,22 which weaken the DNA binding of HMG-D to eeCas9 (called 3m-HMG-D-L4-Cas9) (Figure S3B). As expected, the enhancement effect of HMG-D was abolished in 3m-HMG-D-L4-Cas9, indicating that the DNA binding capability of HMG-D was critical to the elevation of Cas9 activity (Figure S3B). These data suggest that the enhancement effect of HMG-D comes from its DNA binding affinity.
HMG-D enhances the activities of Cas9 and its variants with non-canonical PAM compatibility in multiple cell lines
Cas9 achieves moderate to high efficiencies in a wide range of target sites in multiple cell lines while only eking out low indel rates in some refractory sites.19 To confirm that the effect of HMG-D is not site specific, we tested an additional 39 endogenous targets in HEK293T cells. The HTS results showed that eeCas9 consistently led to higher indel frequencies (Figure 2A), with a mean indel frequency 1.4 times that of the Cas9 group (Figure 2B). Although only mild boosts were detected at sites where Cas9 exerted high indel frequencies (e.g., 10% increase at MECP2-2 site), significant increases of up to 110% (e.g., transforming growth factor β1 site) were observed at some sites refractory to Cas9 cleavage (Figure 2A). This characteristic of being more potent at low-efficiency target sites is advantageous if some clinically related targets are resistant to Cas9. Besides this, we also observed that the number of target sites with frequencies greater than 50% increased from 14 (34%) in the Cas9 group to 30 (73%) in the eeCas9 group (Figure 2C). Next, we compared the efficiency of eeCas9 and Cas9 in other cell lines, including HeLa, Hep-2, and SH-Sy5y, at five endogenous target sites. As shown in Figure S4, eeCas9 activity was increased significantly, up to 1.4-fold in HeLa cells, 1.8-fold in Hep2 cells, and 1.7-fold in SH-Sy5y cells (Figures S4A and S4B), suggesting that the elevation of activity was broadly applicable in variant cell types. To test whether eeCas9 could introduce higher HDR efficiencies than Cas9, we evaluated the introduction of specific DNA fragment deletions or insertions at three endogenous sites using single-stranded oligodeoxynucleotide (ssODN) donors. Sequencing results indicated that eeCas9 achieved a 1.2- to 2.1-fold increase of HDR efficiency (Figures 2D and S5), suggesting that eeCas9 also had advantages to introduce precise editing through HDR.
Figure 2.

Characterization of HMD-G fusion to Cas9, multiple Cas9 variants, and other Cas9 orthologs in HEK293T cells
(A) Indel efficiencies of Cas9 and eeCas9 at 39 endogenous targets in HEK293T cells. (B) Summary of editing efficiencies of Cas9 and eeCas9 for targets at 41 endogenous targets (39 targets in A and two targets in D) in HEK293T cells. (C) Summary of target sites with an indel rate of over 50%. (D) Comparison of HDR-mediated precise editing efficiencies of Cas9 and eeCas9 in HEK293T cells. (E) Summary of editing efficiencies of Cas9 variants with or without HMG-D domain at multiple endogenous targets for non-NGG PAM in HEK293T cells. (F) Indel efficiencies of eCas9 1.1 and ee-eCas9 1.1 at 25 endogenous targets in HEK293T cells. (G) Summary of editing efficiencies of three high-fidelity Cas9 variants with or without HMG-D domain in HEK293T cells. (H) Indel efficiencies of CjCas9 and eeCjCas9 at eight endogenous targets in HEK293T cells. (I) Indel efficiencies of Nme2Cas9 and eeNme2Cas9 at six endogenous targets in HEK293T cells. Data in (A), (D), (F), (H), and (I) represent mean ± SD (n = 3 independent experiments). Because editing efficiencies were highly variable among all tested targets, statistical analysis of the average editing efficiencies including (E), (G), (H) (right). and (I) (right) were performed by using paired two-tailed Student's t test, while the remaining graphs were performed by using unpaired two-tailed Student's t test. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
PAM restriction limits the targeting scope of the CRISPR/Cas system. Several studies have shown that engineered Cas9 variants such as xCas9,23 Cas9-NG,24 SpG,25 and SpRY exhibit broader PAM compatibility.25 However, their activities on non-NGG PAM targets are compromised.26 To increase the activity of the above Cas9 variants, we fused HMG-D to their N terminus. Twenty non-NGG targets for eexCas9, eeCas9-NG, and eeSpG, respectively, and ten non-NGG targets for eeSpRY were evaluated. HTS results showed that HMG-D fusion increased indel frequencies of all these variants by over 1.3-fold (Figures 2E, S6A, and S6B), especially for eeCas9-NG and eeSpG, the average editing frequencies of which were comparable with Cas9 (Figures 2B and 2E).
Similarly, we tested HMG-D fusion on high-fidelity Cas9 variants, which displayed improved accuracy but lowered activity.27,28 We fused HMG-D to the N-terminal eCas9 1.1,29 Cas9-HF1,30 or HypaCas931 to test their activities at multiple endogenous target sites in HEK293T cells. As expected, fusion of HMG-D significantly increased the activities of each high-fidelity variant on all tested target sites (Figures 2F, S7A, and S7B). We observed an average increase of 2.8-, 1.7-, and 1.5-fold for ee-eCas9 1.1, eeCas9-HF1, and eeHypaCas9, respectively (Figure 2G).
Given the remarkable effect of HMG-D, we wondered whether it could be applied to other Cas9 orthologs such as the compact CjCas9 and Nme2Cas9.32,33 In the manner of eeCas9 construction, we generated eeCjCas9 and eeNme2Cas9 nucleases and tested them at the endogenous targets. The next-generation sequencing data demonstrated that fusion of HMG-D increased the activities of both eeCjCas9 and eeNme2Cas9 to approximately 1.3-fold compared with the controls (Figures 2H and 2I). Together, these data suggest that fusion of the HMG-D domain is applicable not only to the Cas9 but also to its variants and orthologs.
HMG-D enhances the activities of Cas9-based transcriptional activators and base editors
Cas9 is a versatile protein for different gene-editing purposes, such as transcriptional regulation, base editing, and prime editing. To investigate whether HMG-D fusion could be adapted to Cas9-based transcriptional activators, we fused a synthetic transcriptional activator, VP64-p65-Rta (VPR),34 to the nuclease-dead eeCas9 (eedCas9) to generate eedCas9-VPR (Figure S8A). We set up a transcriptional induction assay at five endogenous promoter sites to compare the activity of eedCas9-VPR and dCas9-VPR. As shown by quantitative PCR (qPCR), eedCas9-VPR exhibited stronger induction ability than dCas9-VPR at all target sites (ranging from 1.7- to 2.9-fold higher; Figure S8B).
Next, we investigated whether HMG-D could be adapted to base editing. Considering that the deaminases were always fused to the N terminus of Cas9 nickase (Cas9n), we fused the HMG-D domain to the C terminus of BE4max or ABEmax via the L5 linker (Figures S2A and S2C),16 resulting in eeBE4max and eeABEmax (Figures S9A and S9B). We compared eeBE4max and eeABEmax with their respective counterparts at multiple endogenous target sites. HTS analysis demonstrated that the fusion of HMG-D to the C terminus of BE4max or ABEmax had an overall enhancement effect for each substrate base in its corresponding editing windows (Figures 3A and 3B). The average fold increase in editing efficiency was 1.4-fold for eeBE4max and 1.6-fold for eeABEmax (Figures 3C and 3D). Likewise, we generated a series of CBE and ABE variants with non-NGG PAM compatibility. We observed a similar enhancement effect by HMG-D (Figures S9C–S9F) and a 1.2- to 1.5-fold increase on all tested targets (Figures 3C and 3D). Interestingly, the fusion of an HMG-D to BEmax or ABEmax did not change its editing characteristics, including product purity, indel frequencies, and editing windows (Figures 3A, 3B, 3E, 3F, S10A. and S10B). Collectively, these results demonstrated that the HMG-D dsDBD could be broadly adapted to various Cas9-based genome-editing tools with enhanced efficiency.
Figure 3.

Characterization of HMD-G fusion to base editors in HEK293T cells
(A) Comparison of the C-to-T base-editing efficiencies of BE4max and eeBE4max at 24 endogenous targets in HEK293T cells. (B) Comparison of the A-to-G base-editing efficiencies of ABEmax and eeABEmax at 15 endogenous targets in HEK293T cells. (C) Summary of the cumulative C-to-T base-editing efficiencies of multiple BE4max variants and multiple eeBE4max variants at endogenous targets for “NGG” PAM and “non-NGG” PAM in HEK293T cells. (D) Summary of the cumulative A-to-G base-editing efficiencies of multiple ABEmax variants and multiple eeABEmax variants at endogenous targets for “NGG” PAM and “non-NGG” PAM in HEK293T cells. (E) Frequency of indels by BE4max and eeBE4max. Each point represents the average indel frequency at a target site. (F) Frequency of indels by ABEmax and eeABEmax. Each point represents the average indel frequency at a target site. Data in (A) and (B) represent means ± SD (n = 3 independent experiments). The statistical analysis of (C), (D), (E), and (F) was performed by using paired two-tailed Student’s t test. ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Off-target analysis of eeCas9 and a series of base editors in human cells
As eeCas9 had higher activity because of the enhanced affinity between Cas9 and DNA, this could also increase the possibility that Cas9 would bind to the non-target sequence. Therefore, it is critical to investigate whether HMG-D could also enhance the off-target cleavage of Cas9. We first designed a gRNA-target mismatch tolerance experiment at the VEGF-1 target site. We constructed two sets of sgRNAs with all possible single-base mismatches or two-consecutive-base mismatches in the protospacer (Figure 4A). As previously demonstrated,3 Cas9 maintained moderate activity if single-base mismatches were in the PAM distal region (Figure 4A, sgRNAs m18-m20), while the activity gradually declined as the mismatch was moved toward PAM. The enhancement effect by HMG-D was observed in the single-base mismatched sgRNA group, especially in those with a mismatch in the PAM distal region (sgRNAs m13-m20, increase of 1.4- to 2.3-fold) (Figure 4A). However, we did not observe noticeable enhancement effects when consecutive mismatches were in the PAM-proximal region (Figure 4A, sgRNAs m23-m30), indicating that the enhancement on off-target could be greatly avoided by careful sgRNA design that excludes PAM distal mismatches.
Figure 4.

Evaluation of the off-target effects of eeCas9 in HEK293T cells
(A) gRNA-target mismatch tolerance experiment to evaluate the specificity of Cas9 and eeCas9. VEGF-targeting sgRNAs with single mismatch or two consecutive mismatches in the protospacer region were generated to evaluate gene-editing efficiencies by HTS analysis. The PAM sequences are shown in blue. The mismatched nucleotides are shown in red. n.c., not calculated because of no significance between eeCas9 and Cas9. (B) Comparison of on- and off-target efficiencies of Cas9 or eeCas9 with sgRNA plasmid delivery, Cas9 or eeCas9 with hp-sgRNA plasmid delivery, and Cas9 or eeCas9 RNP delivery at EMX1-6 site and FANCF-2 site. (C) Specificity metric of eeCas9 and Cas9 with different delivery strategies in (B). (D) On- and off-target efficiencies by high-fidelity Cas9 with or without HMG-D domain were measured by HTS analysis. (E) Specificity metric of high-fidelity Cas9 with or without HMG-D domain for indicated target sites. (F) Number of PEM-seq detected off-target sites for eeCas9 and Cas9. The values above the columns represent the number of detected off-target sites. In (C) and (E), the specificity metric is defined as on-target editing rate divided by the sum of all off-target editing rates. Data in (A) and (D) represent means ± SD (n = 3 independent experiments), and statistical analysis was performed by using unpaired two-tailed Student’s t test in (A). ∗∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Next, we analyzed 40 previously reported off-target candidates calculated from two endogenous sites in HEK293T cells to compare the off-target activity between eeCas9 and Cas9 (Figures S11A and S11B).35 Consistent with the sgRNA mismatch assay, we observed only slightly higher off-target activity (0.3%–2.3% higher indels on OT4, OT11, and OT15 of EMX1-6 site, and OT8 of FANCF-2 site) at candidate sites with single or double mismatches in the PAM distal region (Figures 4B, S11A, and S11B). However, the enhanced off-target activities were barely detectable if three or more mismatches were present in the protospacer, confirming that sgRNA-target pairing is critical for the cleavage process. Hence, we tried to counteract the off-target enhancement via hairpin (hp)-sgRNA,36 which impedes the formation of the R loop due to RNA-DNA mispairing. Indeed, replacing the FANCF-sg2 site with hp-FANC2-sg2 significantly reduced the off-target indel efficiencies while still maintaining a higher specificity ratio (on-target versus. off-target ratio) (Figures 4B and 4C). In addition to modifying the structure of the sgRNA, we also tried the strategy of electroporating the purified eeCas9 protein because the purified protein would have fewer chances of cutting the off-target sites due to a shorter intracellular half-life.37 As expected, when compared with plasmid delivery, ribonucleoprotein (RNP) electroporation of eeCas9/sgRNA reduced its off-target efficiencies (Figure 4B), resulting in much higher specificity ratios at EMX1-6 and FANCF-2 sites, respectively (Figure 4C).
To test whether the HMG-D fusion could create a better balance between on-target and off-target efficiencies for those high-fidelity Cas9s, we tested ee-eCas9 1.1, eeCas9-HF1, eeHypaCas9, and the controls on five endogenous targets. As expected, HMG-D fusion exhibited more dramatic on-target activity than the activity on the off-target sites (Figure 4D), leading to higher specificity ratios for the HMG-D fused high-fidelity Cas9 variants (Figure 4E).
To further evaluate the specificity of eeCas9 at the whole-genome-wide level, we performed primer-extension-mediated sequencing (PEM-seq),38,39 an HTS method based on detecting the translocation junctions between on-target and off-target sites. Our results showed that eeCas9 did not significantly increase the number of off-target sites for the five tested targets compared with Cas9 (Figures 4F and S12; see materials and methods).
Recent studies have shown that base editors could induce sgRNA-dependent and -independent off-target events.40 We first evaluated sgRNA-dependent off-target events of eeBE4max and eeABEmax on 73 previously reported off-target sites41,42 and 28 web-predicted (https://www.benchling.com/) off-target sites. The results indicated that off-target enhancements were not detectable at sites where BE4max or ABEmax achieved low off-target editing rates (<0.5%) (Figures S13A–S13D and S14A–S14D). Similar to the eeCas9, we also reduced the off-target enhancements of eeBE4max by delivering the RNP complex (Figure 5A), resulting in much higher specificity ratios (Figure 5B). Most strikingly, although the ABEmax seems to increase the off-target editing on multiple off-target sites (Figures S13A and S13B), the specificity of eeABEmax was comparable with or even slightly improved by ABEmax (Figure 5C). Next, we analyzed sgRNA-independent DNA off-target events of eeBE4max and eeABEmax using an orthogonal R-loop assay.43 After HTS analysis of six R-loop sites generated by catalytically inactivated dSaCas9 and corresponding sgRNAs, we observed that neither eeBE4max nor eeABEmax induced significantly higher sgRNA-independent off-target efficiency than their corresponding controls (Figures 5D and 5E). Collectively, these results fully demonstrated that eeCas9 and its derivatives maintained comparable specificity at sites where the off-target editing rates were low or almost undetectable when compared with their respective wild-type editors.
Figure 5.

Evaluation of the off-target effects of eeBE4max and eeABEmax in HEK293T cells
(A) Comparison of on- and off-target efficiencies of BE4max or eeBE4max with sgRNA plasmid delivery, and BE4max or eeBE4max RNP delivery at EMX1-6, FANCF-2, FANCF-3, and HBB 02 sites. (B) Specificity metric of eeBE4max and BE4max with different delivery strategies in (A). (C) Specificity metric of eeABEmax and ABEmax for indicated target sites. (D) Evaluation of sgRNA-independent DNA off-target efficiencies of BE4max and eeBE4max via using an orthogonal R-loop assay. (E) Evaluation of sgRNA-independent DNA off-target efficiencies of ABEmax and eeABEmax using an orthogonal R-loop assay. In (B) and (C), the specificity metric is defined as on-target editing rate divided by the sum of all off-target editing rates. Data in (A), (D), and (E) represent means ± SD (n = 3 independent experiments).
eeCas9 increased editing efficiency on the Pcsk9 gene in the mouse model
Higher editing efficiencies often lead to better therapeutic efficacies in gene therapies, although it is challenging to improve genomic editing efficiencies in vivo. To test the efficiency of eeCas9 in vivo, we aimed to target the proprotein convertase subtilisin/kexin type 9 (Pcsk9) gene in mice, since Pcsk9 is a prime therapeutic target for the prevention of cardiovascular disease.44 We designed two Pcsk9-targeting sgRNAs (sg1 and sg2) and tested, respectively, their efficiencies with eeCas9 or Cas9 in NIH3T3 cells (Figure 6A). eeCas9 achieved higher indel efficiencies than Cas9 at both target sites (mean 1.7-fold higher at Pcsk9-1 site and 1.9-fold higher at Pcsk9-2 site; Figure 6B).
Figure 6.

AAV delivery of eeCas9 for in vivo genome editing
(A) Schematic depicting the experimental procedure to edit Pcsk9 gene in wild-type mice. Two sgRNAs (sg1 and sg2) were selected to target exon 3 and exon 5 of Pcsk9 gene, respectively. (B) sgRNA validation in NIH3T3cells via HTS analysis (n = 3 independent experiments). (C) Schematic diagram of dual AAV vector system for liver-directed gene editing. MiniCMV, minimal cytomegalovirus; short PolyA, short polyadenylation signal; ITR, inverted terminal repeat. (D) Time course of total serum cholesterol in animals treated with AAV8-Cas9 or AAV8-eeCas9. n = 6 for sg1, n = 3 for sg2. (E) Indels at Pcsk9-1(n = 6) and Pcsk9-2 (n = 3) targeting sites of Pcsk9 in mice. (F) Analysis of Pcsk9 protein expression in mouse liver by western blot. Psck9 level was determined by densitometry of western blot bands normalized by that of actin in each sample. (G) WGS analysis of potential off-target of two representative mice (1# and 2#) for Pcsk9-1 target. SNV, single-nucleotide variant; Indels, insertions and deletions. (H) PEM-seq analysis of the number of off-target sites of two representative mice (1# and 2#) for Pcsk9-1 target. Data represent means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Next, to target Pcsk9 in vivo, we cloned eeCas9 or Cas9 into an adeno-associated virus 8 (AAV8) construct. Minimal-cytomegalovirus-promoter (miniCMV) and a short polyadenylation signal (short polyA) were used to minimize the cargo size to 4.9 kb/4.5 kb for the eeCas9/Cas9 construct (Figure 6C and Table S6). Targeting sgRNAs (sg1 or sg2) were cloned into a separate AAV8 construct containing an EGFP gene for labeling of the infected hepatocytes (Figure 6C). On day 0, AAV8 particles of Cas9 or eeCas9 were injected with AAV8-sg1 or AAV8-sg2 (5 × 1011 vg/mouse for each vector) into 6-week-old mice via tail vein injection. Fourteen days later, a partial hepatectomy was performed to assess the infection rate and the expression level of eeCas9/Cas9. We observed a comparable amount of GFP-expressing cells between the groups of eeCas9 and Cas9 (Figure S15A), which indicated that the infection efficiencies were similar between the two groups. This was also supported by similar levels of eeCas9 and Cas9 mRNA expression (Figure S15B). We monitored the total serum cholesterol level at different time points from day 0 to day 35. Lower serum cholesterol levels were observed on day 7 of the experiment in all treated groups. More dramatic decreases were observed over time. However, treatment with eeCas9 led to a more significant reduction in cholesterol level at each time point, regardless of whether combined with sg1 or sg2 (Figure 6D). On day 35, mice treated with eeCas9 exhibited a 55% (with sg1) and a 65% (with sg2) reduction in serum cholesterol levels. However, the reductions in the Cas9-treated groups were only 40% and 30% at this time point, suggesting that eeCas9 had a higher editing efficiency than Cas9 (Figures 6D, S16A, and S16B). To further analyze the in vivo editing efficiency, we amplified the targeted region from the liver genomic DNA of each group followed by HTS. Efficiencies of eeCas9 groups were about 1.7- and 2.6-fold higher than that of Cas9 groups at Pcsk9-1 and Pcsk9-2 sites, respectively (mean 38.4% at the Pcsk9-1 site and 31.2% at the Pcsk9-2 site for eeCas9 versus 22.7% at the Pcsk9-1 site and 11.9% at the Pcsk9-2 site for Cas9) (Figures 6E, S16A, and S16B). As expected, the Pcsk9 protein levels in eeCas9-treated mice were dramatically reduced compared with Cas9-treated mice or control mice, as detected by western blot (Figure 6F). Finally, we evaluated the in vivo off-target rates of eeCas9 and Cas9. We first checked the top 20 web-predicted (https://www.benchling.com/) off-target sites for sg1 or sg2 by HTS and detected no apparent (>0.1% efficiency) off-target event among those sites (Figures S17A and S17B). Then we performed whole-genome sequencing (WGS) and PEM-seq analysis using genomic DNA from the liver tissue of each group to reveal the potential off-target events more sensitively. Interestingly, WGS and PEM-seq did not detect prominent off-target events in groups treated with either eeCas9 or Cas9 (Figures 6G, 6H, S17C, and S17D).
eeA3A-BE3 induced higher γ-globin gene expression in human hematopoietic stem cells
Reactivation of γ-globin is an alternative strategy to treat β-hemoglobinopathies. Studies have shown several therapeutic target sites, including the promoter region of γ-globin genes (HBG1/2) and the BCL11A DHS +58 functional core sequences, to reactivate γ-globin by base editors.45,46 Base-editing efficiency is critical for an excellent therapeutic effect, but our previous study has shown that the editing efficiency of A3A-BE3 is limited at the site on HBG promoter in HSCs.45 Therefore, we tried to compare the editing efficiency and potential efficacy of eeA3A-BE3 with those of the classical A3A-BE3 via base editing of the HBG1/2 promoter or the BCL11A DHS +58 functional core (Figures 7A and 7B). We electroporated the purified eeA3A-BE3 or A3A-BE3 protein with the chemically modified sgRNA targeting the HBG1/2 promoter or BCL11A enhancer into human HSCs. Base-editing efficiencies were evaluated 72 h after electroporation via HTS. The results demonstrated that eeA3A-BE3 achieved higher C>T conversion rates within the editing windows at both the HBG-2 and the BCL11A-1620 sites (Figure 7C). The average fold increases were 1.2 at HBG-2 site (mean 41.3% at C6, 42.3% at C7 for A3A-BE3 versus 49.3% at C6, 50.9% at C7 for eeA3A-BE3) and 1.2–1.3 at the BCL11A-1620 site (mean 59.1% on C6, 44.4% on C8 for A3A-BE3 versus 69.5% on C6, 59.6% on C8 for eeA3A-BE3) (Figure 7C), which indicated that the eeA3A-BE3-treated cells would have a higher γ-globin mRNA level. Indeed, when these cells were subjected to erythropoiesis to evaluate the γ-globin expression by qPCR, cells treated with eeA3A-BE3 exhibited a 1.2-fold (with HBG-2 site) and 1.4-fold (with BCL11A-1620 site) higher γ-globin mRNA level compared with cells treated with A3A-BE3 (Figure 7D). Furthermore, similar to previous results, we did not observe significant variations in the product purity, indel frequencies, and editing windows in the eeA3A-BE3-treated group (Figures 7E and 7F).
Figure 7.

Efficient base editing with eeA3A-BE3 in CD34+ HSCs
(A) Schematic of the base-editing strategy in the promoter of HBG1/2 and in the enhancer of BCL11A. The core sequence of BCL11A repressor binding site is boxed in red. The editing C base (C6 and C7 for HBG-2, or C6 and C8 for BCL11A-1620) in windows are boxed in black. The PAM sequence is highlighted in blue. (B) Schematic diagram of the construct of A3A-BE3 and eeA3A-BE3 proteins. (C) Analysis of C-to-T editing efficiency of eeA3A-BE3 and A3A-BE3 by HTS. (D) Analysis of γ-globin mRNA expression relative to α-globin mRNA by qPCR. (E) Analysis of C-to-T, or -A, or -G base conversion efficiency for A3A-BE3 and eeA3A-BE3 at HBG-2 site and BCL11A-1620 site by HTS. (F) Analysis the indel efficiency induced by A3A-BE3 and eeA3A-BE3 at HBG-2 site and BCL11A-1620 site. Data represent means ± SD (n = 3 independent experiments). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001; ns, not significant.
Discussion
Editing efficiency, targeting scope, and fidelity are the major issues that affect the broad applications of Cas9-based genome-editing technology. Previous studies have demonstrated substantial improvement in targeting scope by identifying novel Cas9 variants with distinct PAMs or engineering well-characterized Cas9 to relax its PAM stringency.47 Although these novel Cas9 variants had been engineered, their activity is generally reduced compared with wild-type Cas9.26,27 In this study, through the fusion of a non-sequence-specific HMG-D dsDBD to Cas9 protein, a series of eeCas9 tools with significantly enhanced activity has been engineered. This strategy is compatible with multiple Cas9 variants and other Cas9 orthologs and can be adapted for base editing and epigenetic regulation. More importantly, eeCas9 exhibited substantial elevation of editing activity for gene therapy in both an in vivo model targeting Pcsk9 to treat hypercholesterolemia and an in vitro model for the treatment of β-thalassemia.
Molecular evolution is the most commonly used and successful strategy to improve the performance of genome-editing tools.48 However, mutagenic library-based screening is difficult in eukaryotic cells, especially in mammalian cells. As Cas9 is super-active in prokaryotic cells, it is difficult to filter out mutations with limited activity enhancement. Thus, fusing dsDBD to Cas9 is an important strategy for engineering genome-editing tools. Our previous report has demonstrated that insertion of RAD51 ssDBD between Cas9n and cytosine deaminase resulted in hyperactive CBEs (hyCBEs), which exhibited dramatically increased activity and targeting scope.20 RAD51 ssDBD has two functions in hyCBEs, as a linker making deaminase more flexible and as a DNA binding domain to increase affinity for the DNA substrate. In eeCas9, this suggests that HMG-D dsDBD only functions in increasing the DNA binding activity because of weakening DNA binding affinity through mutation of HMG-D protein eliminating its enhancement effects. It would inspire us to enhance Cas9 activity through structure-based rational design of its DNA binding domains to elevate the DNA affinity of Cas9 proteins. In a study by Bolukbasi et al.,49 a programmable DNA binding domain (pDBD, a sequence-specific zinc-finger protein or transcriptional activator-like effector) was fused to Cas9 and enhanced its editing efficiency on multiple sites with non-NGG PAM, thus expanding its targeting range. Compared with pDBD, HMG-D in eeCas9 is a non-sequence-specific dsDBD independent of the targeted locus. In our experiment, HMG-D exhibited compatibility with multiple Cas9 variants and orthologs.
DNA binding affinity is a double-edged sword to Cas9 tools. Since HMG-D is a non-sequence-specific dsDBD, it may function on both on-target and off-target sites. Slightly increased off-target editing (4 of the 40 off-target sites only increase 0.3%–2.3% indels) was observed on some previously reported targets in eeCas9-treated cells compared with Cas9. The specificity of eeCas9 was higher than Cas9 when combined with other strategies to reduce the off-target effect, such as RNP delivery, usage of hp-sgRNA, or high-fidelity Cas9 variants (Figures 4B–4E). Moreover, no more off-target sites were detected through genome-wide PEM-seq and WGS analysis (Figures 4F, 6G, and 6H). In this study, we further analyzed the sgRNA-dependent and -independent off-target events of HMG-D fused base editors, which were not significantly increased (Figures 5A–5E). This is a very exciting observation, since the off-target effect of eeCas9 and its derivatives, including base editors and epigenetic editors, is able to be avoided through careful design and analysis of sgRNA.
In summary, we developed a generic approach to improve Cas9-mediated genome-editing efficiencies, such as gene knockout, precise HDR, base editing, and epigenetic regulation. Importantly, eeCas9 can be packaged into AAV vectors and exhibits very high activity in vivo and ex vivo. As lipid nanoparticles-mediated mRNA delivery has achieved amazing success for the first time in transthyretin amyloidosis patients,50 the increased size of eeCas9 or eeCas9-based base editors would not be a concern. eeCas9-based editing tools exhibit higher activity and comparable specificity and would be important for both basic research and gene therapy, especially for those targets inefficiently edited by other Cas9 variants.
Materials and methods
Plasmid construction
Plasmids, sgRNAs, and primer sequences used in this study are listed in the Tables S1 and S2 and S3–S5. The dsDBD (Table S1), oligonucleotides (Table S2), ssODN (Table S3), and primers (Tables S2, S4 and S5) were synthesized by Azenta (Suzhou, China). Px330 (#42230), BE4max (#112093), and ABEmax (#112095) were purchased from Addgene. The DNA sequence of CjCas9 and Nme2Cas9 were also synthesized by Azenta. Cas9 variants with non-NGG PAM compatibility (xCas9, Cas9-NG, SpG, and SpRY) were generated by introducing corresponding mutations via PCR. Cas9-mediated gene activation tool (dCas9-VPR) was generated by fusing a synthetic VPR activation domain to catalytically dead Cas9 (dCas9). The eeCas9, its variants, eeCjCas9, eeNme2Cas9, and eedCas9-VPR were generated by fusing HMG-D to the N terminus of Cas9. The eeBE4max, eeABEmax, and its variants were constructed by fusing HMG-D to the C terminus of the Cas9n domain of a base editor. AAV expression plasmids were generated by cloning the corresponding sequence into the AAV2 backbone plasmid with two intact inverted terminal repeats (Table S6). Protein expression plasmids were generated by cloning bacterium codon-optimized sequence into pET-28a. All sgRNA oligonucleotides were annealed and ligated into BbsI-linearized sgRNA expression plasmid (PB: U6-sgRNA-EF1a-GFP). PCR was performed using TransStart Pfu DNA polymerase (TransGene, China), and the standard molecular cloning experiments were performed using T4 ligase (Beyotime, China) or a ClonExpress MultiS One Step Cloning Kit (Vazyme, China).
Cell culture and hematopoietic stem cell differentiation
The HEK293T, HeLa, Hep2, SH-Sy5y, and NIH3T3 cell lines were purchased from American Type Culture Collection and cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (Thermo Fisher Scientific, USA). CD34+ HSCs were enriched from mobilized peripheral blood and cultured in X-VIVO 15 serum-free medium (Lonza, Switzerland) supplemented with 100 ng/mL human stem cell factor (SCF) (Peprotech, USA), 100 ng/mL human thrombopoietin (Peprotech), and 100 ng/mL human Flt3-ligand (Peprotech). For the erythroid differentiation assay, 48 h after electroporation CD34+ cells were subjected to three-phase differentiation. During phase I (0–7 days), cells were cultured in Iscove’s modified Dulbecco’s medium (Cellgro, USA) supplemented with 330 μg/mL holo-human transferrin (Sigma, USA), 10 μg/mL recombinant human insulin (Sigma), 2 IU/mL heparin (Sigma), 5% human solvent detergent pooled plasma AB (Beau Tompkins), 3 IU/mL erythropoietin (Amgen, USA), 1% L-glutamine (Life Technologies, USA), 1% penicillin/streptomycin (Thermo Fisher Scientific), 1 μM hydrocortisone (Sigma), 100 ng/mL human SCF (R&D Systems, USA), and 5 ng/mL human interleukin-3 (IL-3) (R&D Systems). During phase II (7–11 days) culturing, hydrocortisone and IL-3 were withdrawn from the medium. During phase III (11–18 days), SCF was withdrawn. Eighteen days later, cells were harvested for subsequent analysis.
All cells used in this study were cultured in an incubator maintained at 37°C and 5% CO2.
Cell transfection and genomic DNA extraction
For the fluorescent reporter assay, HEK293T cells were seeded onto a 24-well plate and transfected with the mCherry reporter plasmid (200 ng per well), nuclease plasmids (1,200 ng per well), and sgRNA plasmid (600 ng per well) at approximately 80% confluence. After 72 h, cells were analyzed by fluorescence-activated cell sorting (FACS) (BD, USA), and the percentage of mCherry+ cells among the GFP+ cells was calculated. For gene-editing assays, HEK293T cells, HeLa cells, Hep2 cells, and SH-Sy5y cells were seeded onto 24-well plates and transfected with nuclease plasmids (1,200 ng per well), sgRNA plasmids (600ng per well), with or without ssODN (50μM) at approximately 80% confluence. For VPR-mediated transcriptional activation assays, HEK293T cells were seeded into 24-well plates and transfected with Cas9-mediated activator plasmids (1,200 ng per well) and sgRNA plasmids (600 ng per well) at approximately 80% confluence. For base-editing assays, HEK293T cells were seeded onto 24-well plates and transfected with nuclease plasmids (1,200 ng per well) and sgRNA plasmids (600 ng per well), with or without uracil glycosylase inhibitor-expressed plasmid (200 ng per well) at approximately 80% confluence. In all transfection assays, polyethyleneimine (Polysciences, USA) was used according to the manufacturer's protocol. Seventy-two hours post transfection the cells were collected, and genomic DNA was extracted using the Genomic Extraction Kit (Tiangen Biotech, China) according to the manufacturer's instructions.
Animal experiments
C57BL/6 mouse strains were housed in standard cages and supplied with sufficient food and water. All animal experiments were performed following East China Normal University Center for Animal Research approved protocol. AAV8 vectors were packaged in HEK293T cells and purified by iodixanol density gradient centrifugation. AAV titer was determined using qPCR. For virus injection, 6-week-old C57BL/6 wild-type mice were injected with AAV8-Cas9 (or AAV8-eeCas9) (5 × 1011 genome copies) and AAV8-sgRNA (5 × 1011 genome copies) via tail vein. To detect the levels of total cholesterol in serum, mice were fasted overnight for 12 h before blood testing. Serum was separated by centrifugation and stored at −20°C for subsequent analysis. At the endpoint of the experiment, mice were euthanized by carbon dioxide inhalation and the liver tissue collected.
Serum analysis and immunofluorescence assay
Total cholesterol levels in serum were detected using the Total Cholesterol Assay Kit (NJJCBIO, China) following the manufacturer's protocol. To detect GFP expression levels of liver tissue using immunofluorescence assay, liver tissue was collected, fixed with 4% paraformaldehyde (Sangon, China) for 2 h, embedded with optimal cutting temperature compound (OCT) (Leica, Germany), and cryosectioned at 4 μm. After removing the OCT by PBS, tissue sections were stained with DAPI (Sigma, USA) and analyzed by fluorescence microscopy (Olympus, Japan) following the standard protocols.
Western blot
Cells or smashed liver tissue were lysed with RIPA solution (supplemented with proteinase inhibitors) for 30 min on ice, vortexed every 10 min, and centrifuged at 10,000 rpm at 4°C for 10 min to remove the sediment. The protein solution was quantified using the BCA kit (Thermo Fisher Scientific). For Cas9 detection, 40 μg/well of total protein loaded on 8% SDS-PAGE gel, and Cas9 protein was determined by anti-FLAG (Sigma, #F1804). For Pcsk9 detection, 20 μg/well of total protein was loaded on a 12% SDS-PAGE gel, and Pcsk9 protein was determined by anti-Pcsk9 (R&D Systems, #AF3985). β-Actin protein was taken as a loading control and determined by anti-β-actin (Cell Signaling Technology, USA, #3700).
Protein purification and RNP electroporation
For Cas9 or base-editor protein purification, corresponding plasmids were transformed into E. coli strain BL21 (DE3) (TransGen, China), and a single colony was cultured in 2× TY medium at 37°C until OD 0.8–1.0. The cultures were then cultured at 18°C, induced with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (Sangon, China), and shaken at 170 rpm for 18 h. Protein purification procedures were performed at 4°C. Cells were collected by centrifugation and lysed using a homogenizer in buffer A (20 mM Tris [pH 8.0], 500 mM NaCl, 10% glycerol, 1 mM tris(2-carboxyethyl)phosphine [TCEP]). Cell lysate was centrifuged at 12,000 rpm for 1 h to remove cell debris. The supernatant was slowly loaded onto an Ni-NTA column (GE Life Sciences, USA) and eluted via a linear gradient of 0%–100% buffer B (20 mM Tris [pH 8.0], 500 mM NaCl, 10% glycerol, 250 mM imidazole, 1 mM TCEP) using AKTA Pure FPLC (GE Life Sciences, USA). Eluted protein fractions were pooled and subjected to ion-exchange purification on a 5-mL SP HiTrap column (GE Life Sciences) with a linear gradient of 100 mM to 1 M KCl, then further purified by gel filtration (GE Life Sciences) via buffer C (20 mM Tris [pH 8.0], 500 mM KCl, 10% glycerol, 1 mM TCEP) and stored in liquid nitrogen. Protein solution was filtered via a 0.22-μm membrane (Millipore, USA) before use.
The Lonza 4D Nucleofector Kit was used for nucleofection according to the manufacturer’s recommendations. In brief, Cas9 (20 μg) or base editor (20 μg for HEK293T cells and 90 μg for CD34+ cells) protein with guide RNA (300 pmol for HEK293T cells and 800 pmol for CD34+ cells) (Genscript, China) were prepared as RNP complex and incubated at room temperature for 10 min. The 20-μL electroporation buffer was then prepared according to the V4XP-3032 kit (Lonza), and cells (HEK293T or CD34+ cells) were resuspended with the 20-μL electroporation buffer containing the RNP complex. Electroporation mix was transferred in 16-well cuvettes for electroporation with program EO-100. After electroporation, cells were washed once using 1× PBS and finally cultured in an incubator at 37°C.
On- and off-target editing efficiency determination
All on-target sites and off-target sites used in this study are listed in Tables S2 and S4. On- and off-target sites were amplified from 50–100 ng of genomic DNA from corresponding cell lines or tissues. PCR was performed with TransStart Fast Pfu DNA polymerase (TransGene Biotech, China) using site-specific primers containing an adapter sequence. All primers used in this study are listed in Tables S2 and S4. The above PCR products were subjected to a second round of PCR using primers containing unique barcode sequences. The final PCR products were mixed and purified to generate an HTS amplification library and sequenced on an Illumina HiSeq platform. The indels (30-bp window surrounding the cut site), HDR, and base conversions (including C-to-T, A-to-G, or others) in HTS files were analyzed using Cas-Analyzer and BE-Analyzer (www.rgenome.net), respectively. The accumulative base-editing efficiency was determined by calculating the frequencies of modified target sites with at least one edit within the editing window (positions 3–9 for CBE and positions 4–8 for adenine base editor).
Total mRNA extraction and qPCR quantification of gene expression
Total mRNA was extracted from cell or liver sample using RNAiso Plus (TaKaRa, Japan). The purified mRNA was reverse transcribed using the PrimeScript RT Reagent Kit (TaKaRa) followed by qPCR using SYBR Green Master Mix (Yeasen, China) on the QuantStudio 3 real-time PCR system (Applied Biosystems, USA). Targeted gene expression was normalized to β-actin, and the γ-globin expression was normalized to α-globin. All qPCR primers are listed in Table S5.
Whole-genome off-target analyzed by PEM-seq and WGS
For the PEM-seq assay, first a total of 20 μg of genomic DNA was extracted from cells or mouse liver samples. The genomic DNA was then fragmented to 300–2,000 bp by sonication followed by primer extension with Bst polymerase 3.0 (NEB, USA). Biotinylated PCR product was enriched by Dynabeads MyOne Streptavidin C1 (Thermo Fisher Scientific). Next, the enriched PCR product on C1 beads was washed with 1× B&W buffer (5 mM Tris-HCl [pH 7.4], 1 M NaCl, 1 mM EDTA) and 10 mM Tris-HCl (pH 7.4) in turn, followed by ligating with the bridge adapter containing a random barcode sequence by T4 DNA ligase (Thermo Fisher Scientific) overnight. After ligation, C1 beads were washed twice each with 1× B&W buffer and 10 mM Tris-HCl (pH 7.4). A library was obtained by a nested PCR with I5-nested primer and I7-index primer and then tagged with Illumina adapter sequences. Finally, the library DNA was sequenced on an Illumina HiSeq platform with 2 × 150 bp reads. For off-target hotspot identification, we excluded translocation junctions located within ±250 kb around the on-target cleavage site, identified translocation enriched regions by using MACS2 callpeak with the parameter --extsize 50 -q 0.05 --llocal 10000000, then filtered and removed sites with no on-target site-similar sequence (<8 base substitutions) or fewer than three translocation junctions as previously reported.38,39 Moreover, a bona fide off-target site should be detected in at least two biological replicates.
For the WGS assay, a total of 1.5 μg of genomic DNA was extracted from the mouse liver samples. The sequencing libraries were generated by sonication (∼350 bp), end-repair, and adapter ligation. These libraries were sequenced by the Illumina Nova6000 platform at an average coverage of 30× to 40×. The WGS files were processed through the Genome Analysis Toolkit to generate the total single-nucleotide variants (SNVs) and indels. SNVs and indels with less than three reads or containing homopolymers or near-low-complexity region were filtered. The candidate off-target sites were determined by two characteristics, which were “any sequence not more than six base substitutions of 20-nt target site followed by an NGG or NAG PAM” and “any sequence matching the last 10 nt of the target site followed by an NGG or NAG PAM.”
Statistics
All statistics used in this study were performed on at least n = 3 biologically independent experiments and calculated using an unpaired or paired two-tailed Student's t test using GraphPad Prism v.7. Detailed information on samples and experimental replicates can be found in the figure legends. p values less than 0.05 were considered significant, denoted as ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Data availability
HTS data used in this study have been deposited in the NCBI Sequence Read Archive database (accession codes: PRJNA768303, PRJNA768631, PRJNA769533, PRJNA769747, PRJNA769512, PRJNA769753, and PRJNA882573). WGS data have been deposited in the NCBI Sequence Read Archive database (accession code: PRJNA769256). The PEM-seq data have been deposited in the NCBI Sequence Read Archive database (accession codes: PRJNA771464 and PRJNA771465).
Acknowledgments
We acknowledge the support of the East China Normal University (ECNU) Public Platform for Innovation (011). We also thank Ying Zhang for providing technical support for flow cytometry and the support of the Flow Cytometry Core Facility of School of Life Sciences of ECNU. This work was partially supported by grants from National Key R&D Program of China (2019YFA0110802, 2022YFC3400200, 2019YFA0802800, and 2019YFA0110801), the National Natural Science Foundation of China (no. 32025023 and no. 31971366), the Shanghai Municipal Commission for Science and Technology (21CJ1402200 and 20140900200), and grants from NK2022010207, the Innovation program of Shanghai Municipal Education Commission (2019-01-07-00-05-E00054), the East China Normal University Medicine and Health Joint Fund (2022JKXYD02001), the Fundamental Research Funds for the Central Universities, and support from East China Normal University Outstanding Doctoral Students Academic Innovation Ability Improvement Project (YBNLTS2021-026).
Author contributions
D.L., L.W., and J.H. supervised the research. S.Y. and M.Z. performed most experiments with the help of X.S., X.C., L.Y., Y.H., and J.Y. Y.L. performed PEM-seq and analyzed the data. Y.G., X.Z., H.H., J.Z., M.-M.X., and M.L. provided technical support. D.L., S.Y., and L.W. wrote the manuscript with the help of J.H. and Y.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.11.014.
Contributor Information
Jiazhi Hu, Email: hujz@pku.edu.cn.
Liren Wang, Email: lirenster@163.com.
Dali Li, Email: dlli@bio.ecnu.edu.cn.
Supplemental information
References
- 1.Komor A.C., Badran A.H., Liu D.R. CRISPR-based Technologies for the manipulation of eukaryotic genomes. Cell. 2017;169:559. doi: 10.1016/j.cell.2017.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jinek M., Jiang F., Taylor D.W., Sternberg S.H., Kaya E., Ma E., Anders C., Hauer M., Zhou K., Lin S., et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343 doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ceccaldi R., Rondinelli B., D'Andrea A.D. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26:52–64. doi: 10.1016/j.tcb.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pulecio J., Verma N., Mejía-Ramírez E., Huangfu D., Raya A. CRISPR/Cas9-Based engineering of the epigenome. Cell stem cell. 2017;21:431–447. doi: 10.1016/j.stem.2017.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rees H.A., Liu D.R. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018;19:770–788. doi: 10.1038/s41576-018-0059-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B., et al. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang L., Zhang X., Wang L., Yin S., Zhu B., Xie L., Duan Q., Hu H., Zheng R., Wei Y., et al. Increasing targeting scope of adenosine base editors in mouse and rat embryos through fusion of TadA deaminase with Cas9 variants. Protein Cell. 2018;9:814–819. doi: 10.1007/s13238-018-0568-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu Y., Zeng J., Roscoe B.P., Liu P., Yao Q., Lazzarotto C.R., Clement K., Cole M.A., Luk K., Baricordi C., et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med. 2019;25:776–783. doi: 10.1038/s41591-019-0401-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frangoul H., Altshuler D., Cappellini M.D., Chen Y.S., Domm J., Eustace B.K., Foell J., de la Fuente J., Grupp S., Handgretinger R., et al. CRISPR-Cas9 gene editing for sickle cell disease and beta-thalassemia. N. Engl. J. Med. 2021;384:252–260. doi: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 15.Fu B., Liao J., Chen S., Li W., Wang Q., Hu J., Yang F., Hsiao S., Jiang Y., Wang L., et al. CRISPR-Cas9-mediated gene editing of the BCL11A enhancer for pediatric beta(0)/beta(0) transfusion-dependent beta-thalassemia. Nat. Med. 2022;28:1573–1580. doi: 10.1038/s41591-022-01906-z. [DOI] [PubMed] [Google Scholar]
- 16.Koblan L.W., Doman J.L., Wilson C., Levy J.M., Tay T., Newby G.A., Maianti J.P., Raguram A., Liu D.R. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 2018;36:843–846. doi: 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Horlbeck M.A., Witkowsky L.B., Guglielmi B., Replogle J.M., Gilbert L.A., Villalta J.E., Torigoe S.E., Tjian R., Weissman J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife. 2016;5:e12677. doi: 10.7554/eLife.12677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ding X., Seebeck T., Feng Y., Jiang Y., Davis G.D., Chen F. Improving CRISPR-cas9 genome editing efficiency by fusion with chromatin-modulating peptides. CRISPR J. 2019;2:51–63. doi: 10.1089/crispr.2018.0036. [DOI] [PubMed] [Google Scholar]
- 19.Liu G., Yin K., Zhang Q., Gao C., Qiu J.L. Modulating chromatin accessibility by transactivation and targeting proximal dsgRNAs enhances Cas9 editing efficiency in vivo. Genome Biol. 2019;20:145. doi: 10.1186/s13059-019-1762-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang X., Chen L., Zhu B., Wang L., Chen C., Hong M., Huang Y., Li H., Han H., Cai B., et al. Increasing the efficiency and targeting range of cytidine base editors through fusion of a single-stranded DNA-binding protein domain. Nat. Cell Biol. 2020;22:740–750. doi: 10.1038/s41556-020-0518-8. [DOI] [PubMed] [Google Scholar]
- 21.Murphy F.V., 4th, Churchill M.E. Nonsequence-specific DNA recognition: a structural perspective. Structure. 2000;8:R83–R89. doi: 10.1016/s0969-2126(00)00126-x. [DOI] [PubMed] [Google Scholar]
- 22.Murphy F.V., 4th, Sweet R.M., Churchill M.E. The structure of a chromosomal high mobility group protein-DNA complex reveals sequence-neutral mechanisms important for non-sequence-specific DNA recognition. EMBO J. 1999;18:6610–6618. doi: 10.1093/emboj/18.23.6610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu J.H., Miller S.M., Geurts M.H., Tang W., Chen L., Sun N., Zeina C.M., Gao X., Rees H.A., Lin Z., Liu D.R. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature. 2018;556:57–63. doi: 10.1038/nature26155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishimasu H., Shi X., Ishiguro S., Gao L., Hirano S., Okazaki S., Noda T., Abudayyeh O.O., Gootenberg J.S., Mori H., et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science. 2018;361:1259–1262. doi: 10.1126/science.aas9129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walton R.T., Christie K.A., Whittaker M.N., Kleinstiver B.P. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science. 2020;368:290–296. doi: 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim H.K., Lee S., Kim Y., Park J., Min S., Choi J.W., Huang T.P., Yoon S., Liu D.R., Kim H.H. High-throughput analysis of the activities of xCas9, SpCas9-NG and SpCas9 at matched and mismatched target sequences in human cells. Nat. Biomed. Eng. 2020;4:111–124. doi: 10.1038/s41551-019-0505-1. [DOI] [PubMed] [Google Scholar]
- 27.Kim S., Bae T., Hwang J., Kim J.S. Rescue of high-specificity Cas9 variants using sgRNAs with matched 5' nucleotides. Genome Biol. 2017;18:218. doi: 10.1186/s13059-017-1355-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmid-Burgk J.L., Gao L., Li D., Gardner Z., Strecker J., Lash B., Zhang F. Highly parallel profiling of Cas9 variant specificity. Mol. Cell. 2020;78:794–800.e8. doi: 10.1016/j.molcel.2020.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Slaymaker I.M., Gao L., Zetsche B., Scott D.A., Yan W.X., Zhang F. Rationally engineered Cas9 nucleases with improved specificity. Science. 2016;351:84–88. doi: 10.1126/science.aad5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim E., Koo T., Park S.W., Kim D., Kim K., Cho H.Y., Song D.W., Lee K.J., Jung M.H., Kim S., et al. In Vivo genome editing with a small Cas9 orthologue derived from Campylobacter jejuni. Nat. Commun. 2017;8 doi: 10.1038/ncomms14500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edraki A., Mir A., Ibraheim R., Gainetdinov I., Yoon Y., Song C.Q., Cao Y., Gallant J., Xue W., Rivera-Pérez J.A., Sontheimer E.J. A compact, high-accuracy Cas9 with a dinucleotide PAM for in vivo genome editing. Mol. Cell. 2019;73:714–726.e4. doi: 10.1016/j.molcel.2018.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chavez A., Scheiman J., Vora S., Pruitt B.W., Tuttle M., P R Iyer E., Lin S., Kiani S., Guzman C.D., Wiegand D.J., et al. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kocak D.D., Josephs E.A., Bhandarkar V., Adkar S.S., Kwon J.B., Gersbach C.A. Increasing the specificity of CRISPR systems with engineered RNA secondary structures. Nat. Biotechnol. 2019;37:657–666. doi: 10.1038/s41587-019-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuris J.A., Thompson D.B., Shu Y., Guilinger J.P., Bessen J.L., Hu J.H., Maeder M.L., Joung J.K., Chen Z.Y., Liu D.R. Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yin J., Liu M., Liu Y., Wu J., Gan T., Zhang W., Li Y., Zhou Y., Hu J. Optimizing genome editing strategy by primer-extension-mediated sequencing. Cell Discov. 2019;5:18. doi: 10.1038/s41421-019-0088-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Y., Yin J., Gan T., Liu M., Xin C., Zhang W., Hu J. PEM-seq comprehensively quantifies DNA repair outcomes during gene-editing and DSB repair. STAR Protoc. 2022;3 doi: 10.1016/j.xpro.2021.101088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeong Y.K., Song B., Bae S. Current status and challenges of DNA base editing tools. Mol. Ther. 2020;28:1938–1952. doi: 10.1016/j.ymthe.2020.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim D., Lim K., Kim S.T., Yoon S.H., Kim K., Ryu S.M., Kim J.S. Genome-wide target specificities of CRISPR RNA-guided programmable deaminases. Nat. Biotechnol. 2017;35:475–480. doi: 10.1038/nbt.3852. [DOI] [PubMed] [Google Scholar]
- 42.Liang P., Xie X., Zhi S., Sun H., Zhang X., Chen Y., Chen Y., Xiong Y., Ma W., Liu D., et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat. Commun. 2019;10:67. doi: 10.1038/s41467-018-07988-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doman J.L., Raguram A., Newby G.A., Liu D.R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020;38:620–628. doi: 10.1038/s41587-020-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitzgerald K., Frank-Kamenetsky M., Shulga-Morskaya S., Liebow A., Bettencourt B.R., Sutherland J.E., Hutabarat R.M., Clausen V.A., Karsten V., Cehelsky J., et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–68. doi: 10.1016/S0140-6736(13)61914-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L., Li L., Ma Y., Hu H., Li Q., Yang Y., Liu W., Yin S., Li W., Fu B., et al. Reactivation of gamma-globin expression through Cas9 or base editor to treat beta-hemoglobinopathies. Cell Res. 2020;30:276–278. doi: 10.1038/s41422-019-0267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng J., Wu Y., Ren C., Bonanno J., Shen A.H., Shea D., Gehrke J.M., Clement K., Luk K., Yao Q., et al. Therapeutic base editing of human hematopoietic stem cells. Nat. Med. 2020;26:535–541. doi: 10.1038/s41591-020-0790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anzalone A.V., Koblan L.W., Liu D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020;38:824–844. doi: 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- 48.Cheng C., Zhou M., Su Q., Steigmeyer A., Niu J. J. Genome editor-directed in vivo library diversification. Cell Chem. Biol. 2021;28:1109–1118. doi: 10.1016/j.chembiol.2021.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bolukbasi M.F., Gupta A., Oikemus S., Derr A.G., Garber M., Brodsky M.H., Zhu L.J., Wolfe S.A. DNA-binding-domain fusions enhance the targeting range and precision of Cas9. Nat. Methods. 2015;12:1150–1156. doi: 10.1038/nmeth.3624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gillmore J.D., Gane E., Taubel J., Kao J., Fontana M., Maitland M.L., Seitzer J., O'Connell D., Walsh K.R., Wood K., et al. CRISPR-Cas9 in vivo gene editing for transthyretin amyloidosis. N. Engl. J. Med. 2021;385:493–502. doi: 10.1056/NEJMoa2107454. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
HTS data used in this study have been deposited in the NCBI Sequence Read Archive database (accession codes: PRJNA768303, PRJNA768631, PRJNA769533, PRJNA769747, PRJNA769512, PRJNA769753, and PRJNA882573). WGS data have been deposited in the NCBI Sequence Read Archive database (accession code: PRJNA769256). The PEM-seq data have been deposited in the NCBI Sequence Read Archive database (accession codes: PRJNA771464 and PRJNA771465).