

Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 gene editing is an emerging therapeutic modality that shows promise in Huntington’s disease and spinocerebellar ataxia (SCA) mouse models. However, advancing CRISPR-based therapies requires methods to fully define in vivo editing outcomes. Here, we use polymerase-free, targeted long-read nanopore sequencing and evaluate single- and dual-gRNA AAV-CRISPR editing of human ATXN2 in transgenic mouse models of SCA type 2 (SCA2). Unbiased high sequencing coverage showed 10%–25% editing. Along with intended edits there was AAV integration, 1%–2% of which contained the entire AAV genome and were largely unmethylated. More than 150 kb deletions at target loci and rearrangements of the transgenic allele (1%) were also found. In contrast, PCR-based nanopore sequencing showed bias for partial AAV fragments and inverted terminal repeats (ITRs) and failed to detect full-length AAV. Cumulatively this work defines the spectrum of outcomes of CRISPR editing in mouse brain after AAV gene transfer using an unbiased long-read sequencing approach.
Keywords: CRISPR/Cas9 editing, nanopore long-read sequencing, AAV integration, spinocerebellar ataxia, ATXN2
Graphical abstract
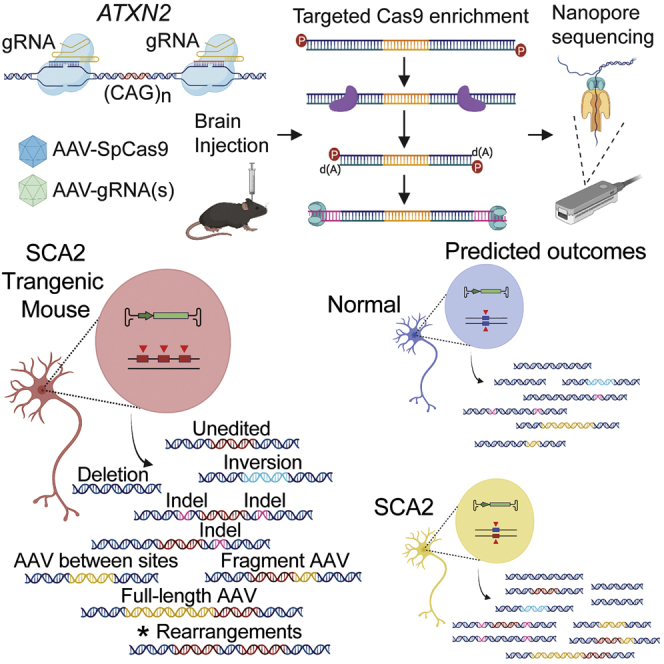
Here we present methods to define the spectrum of CRISPR editing outcomes in mouse brain after AAV delivery using unbiased long-read sequencing. Our method detected intended deletions and unintended large deletions and rearrangements at target loci, and fulllength and fragment AAV integrations, while PCR-based sequencing detected only AAV fragments and ITRs.
Introduction
The success of gene editing therapies relies on research in animal models as well as methods to accurately assess and predict editing outcomes, which are variable depending on gene targets, cell types, and delivery modalities. In efforts to develop adeno-associated virus (AAV)-delivered clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 (CRISPR-associated protein 9) (AAV-CRISPR) gene editing strategies to treat spinocerebellar ataxia type 2 (SCA2), an autosomal-dominant neurodegenerative disease caused by a CAG trinucleotide repeat expansion in exon 1 of ATXN2, and also associated with amyotrophic lateral sclerosis (ALS) risk,1 requires we first understand the nature of editing events in vivo. Indeed, tools that help to define the nature of AAV-based editing in brain could be applied to other repeat expansion disorders where CRISPR editing has shown promise including Huntington’s disease (HD),2,3 fragile X syndrome (FXS),4,5 SCA type 3 (SCA3),6 C9orf72-mediated ALS/frontotemporal dementia (FTD),7,8 and myotonic dystrophy type 1 (DM1).9,10 Notably, however, evaluating editing near long repetitive sequences is error prone and can be biased when using PCR-based methods.
AAV vectors carrying a variety of cargo have been shown to integrate into double-strand breaks (DSBs) in host genomes,11,12 with, frequencies of 1%–3% recently shown in the liver of a humanized mouse model.13 Because CRISPR/Cas9 induces DSBs, it stands to reason that these same vectors can integrate at these sites. Indeed, using PCR-based methods, AAV integration has been found after AAV-CRISPR delivery,14,15,16,17,18,19,20 with AAV integration occurring more frequently than the intended editing event.20 Reports of AAV integrations using PCR methods also show that they predominantly involve the inverted terminal repeat (ITR).
As reliable analysis of DNA repeats is achieved using polymerase-free long-read sequencing,21 and our intended editing is near an expanded repeat region of ATXN2, we reasoned that long-read nanopore Cas9-targeted sequencing (nCATS) could be applied to similarly evaluate native and AAV-CRISPR edited DNA without amplification bias.22 We assessed the utility of our methods in two transgenic mouse models of SCA2 after AAV-CRISPR delivery.23,24 Here, we show that our modified nCATS methodology provides high on-target coverage, a requirement for interrogating AAV-CRISPR editing, and captured editing outcomes missed by standard PCR methods. Our data support applying targeted long-read sequencing methods to define the spectrum of in vivo editing outcomes and advance AAV and CRISPR-based therapies for inherited disorders.
Results
Cas9-targeted enrichment yields high read coverage after in vivo editing
CRISPR/Cas9 guide RNAs (gRNAs) were designed to target human ATXN2 using a single gRNA or dual-gRNA strategy. For the first approach, gRNA5 targets downstream of the CAG repeat to induce indels and premature termination of transcripts (Figures 1A, S1, and Table S1). For the dual-gRNA strategy, gRNA4+5 flanked the CAG repeat tract for targeted deletion (Figures 1A, S1, and Table S1). To validate the approach, Cas9-targeted nanopore sequencing of editing events were assessed in two distinct SCA2 mouse models after delivery of the AAV-CRISPR machinery to brain. One SCA2 model is Pcp2-ATXN2-127Q, where the transgene contains human ATXN2 cDNA with 127 CAG repeats.23 The second is BAC-ATXN2-72Q, which has a BAC transgenic genome harboring full-length human ATXN2 (approximately 169 kb) with 72 CAG repeats.24 As an important step to understand editing outcomes, we used digital droplet PCR (ddPCR) to define transgene copy numbers in both models (Figure S2; there are six BAC-72Q transgene copies and three Pcp2-127Q transgene copies). In vivo editing was performed by the co-delivery of AAV-SpCas9 and AAV-gRNA(s) vectors (Figure 1B) injected at equal doses (Figure 1C). Four weeks later, mice were euthanized and enhanced green fluorescent protein (eGFP)-positive brain tissue micro-dissected (Figure 1D).
Figure 1.

AAV-CRISPR editing in the brain of SCA2 mice and nCATS workflow
(A) Schematic of CRISPR/Cas9 editing strategies with flanking dual-gRNA4+5 to delete the CAG repeat and a single gRNA5 downstream to knockout ATXN2 expression. (B) AAV-SpCas9 vector with the neuronal-specific Mecp2 promoter driving SpCas9 expression, AAV-gRNA5, and AAV-gRNA4+5 vectors with the human U6 promoter(s) driving gRNA(s) expression and the CMV promoter driving eGFP reporter expression. (C) BAC-72Q and Pcp2-127Q mouse models of SCA2 were bilaterally injected in the striatum with an equal 1:1 ratio of AAV-SpCas9 and AAV-gRNA(s) at 2.5E10 vg per AAV vector per hemisphere (1E11 vg total) at 8 weeks old. (D) At 4 weeks post-injection, transduced GFP positive brain tissue was micro-dissected and gDNA was extracted with gravity-flow columns. (E) gDNA was sheared to 20 kb by Covaris g-TUBE centrifugation and small fragments (<4 kb) were removed by Circulomics small read elimination XS kit. (F) Libraries were prepared by dephosphorylating gDNA, Cas9-enrichment gRNAs flanking the ATXN2 transgene region of interest (ROI) targeted SpCas9 cleavage, DNA ends were dA-tailed, and sequencing adapters were ligated. (G) Libraries were sequenced with nanopore Flongle or MinION flow cells (Oxford Nanopore Technologies, ONT).
We adapted the nCATS protocol to isolate high quality genomic DNA (gDNA) from brain by gravity-flow purification (Figure 1D), followed by shearing to approximately 20 kb and eliminating small gDNA fragments by size selection (Figures 1E and S3). The size selection step acted as an additional cleaning step prior to Cas9 enrichment and library preparation. These modifications reduced small reads and improved sequencing output and target coverage.
Cas9-enrichment gRNAs flanking the ATXN2 transgene region of interest (ROI) were tested and optimized using Flongle flow cells (Oxford Nanopore Technology [ONT]) (Figures 1F and 1G). In BAC-72Q mice, we initially followed the nCATS protocol recommending four Cas9-enrichment gRNAs to enrich for target ROI reads at high coverage. However, Cas9-targeted enrichment of the Pcp2-127Q ROI presented unforeseen challenges: the short transgene (5.7 kb) had a limited sequence window for Cas9-enrichment gRNA design and targeting the Pcp2 promoter in the transgene would enrich for the off-target mouse Pcp2 gene (chr8:3625324-3625343) (Figure S4A). With follow-up Cas9-enrichment gRNA tests, the Pcp2-127Q transgene was located at three sites in chromosome 19 (Figure S4B). Two transgene copies were inserted in Gldc intronic (chr19: 30177215) and promoter (chr19:30195946) regions and one copy was inserted in a Mbl2 intronic region (chr19:30237764). Pcp2-127Q insertions were confirmed with higher coverage (417×) and the Mbl2 insertion was PCR validated (Figures S4B and S4C). These aligned chr19 insertion reads provided additional targetable Pcp2-127Q sequence, which included backbone sequence from the plasmid used to generate the transgenic mouse. Using the additional targetable sequence, we designed and tested multiple Cas9-enrichment gRNAs to ascertain the most efficient. Ultimately, we found that two Cas9-enrichment gRNAs could provide high target coverage in Pcp2-127Q mice.
Targeted sequencing was performed with Cas9-enrichment gRNAs (Table S1) on treated SCA2 mouse samples (Figure 1F) and libraries sequenced with high throughput MinION flow cells (ONT) (Figure 1G). To provide a better understanding of potential off-target sequences targeted with the Cas9-enrichment gRNAs, we performed in silico analysis with Cas-OFFinder.25 The number of putative targets ranged from 2-5 with three mismatches and up to 343–702 with five mismatches (Table S2). The tool confirmed the mouse Pcp2 off-target with 0 mismatches. Nonetheless, resulting reads (read length N50 approximately 10 kb; see materials and methods) aligned to the target transgenes with high average sequencing coverage of 2,765× and 3,940× for Pcp2-127Q and BAC-72Q, respectively (Table S3).
Editing outcomes after dual-gRNA delivery to SCA2 mice
The transgenic alleles for the Pcp2-127Q and BAC-72Q models were enriched using two or four Cas9-enrichment gRNAs, respectively, for targeted sequencing (Figures 2A and 2B). With dual-gRNAs, full CAG repeat deletion was, as expected, inefficient, at 1.7% (Figure 2C) and 3.5% (Figure 2D) of relevant reads in the Pcp2-127Q or BAC-72Q mice, respectively. Of the full-length CAG deletions (Figures 2C and 2D), 57.3% and 64.3% (Figure 2E) and 41.5% and 43.2% (Figure 2F) were precise deletions of 779 bp and 614 bp in Pcp2-127Q and BAC-72Q mice, respectively. Thus, while deletions were infrequent, they were of the correct size and were confirmed by end-point PCR, Sanger sequencing, and PCR-targeted amplicon nanopore sequencing (Figure S5).
Figure 2.

Frequency of CAG deletions after dual-gRNA AAV-CRISPR editing
(A and B) Representative IGV alignment plots of nCATS reads aligned to Pcp2-127Q (A) and BAC-72Q (B) transgene ROIs. The IGV plots were cropped to highlight the relevant data, coverage is in green, maximum base coverage is on the left, positive strand reads are red, negative strand reads are blue, and reads with intended CAG deletions are shown by the red arrowheads. AAV gRNA4 and gRNA5 cleavage sites (+3-nt upstream of the PAM) are shown by black arrowheads and Cas9-enrichment gRNA sites are shown by open arrowheads; Pcp2-127Q Cas9-target enrichment with two gRNAs and BAC-72Q Cas9-target enrichment with four gRNAs. (C and D) Frequency of target reads with intended CAG deletions in Pcp2-127Q (C) (n = 2; closed circles; 4,242× and 1,843× coverage) and BAC-72Q (D) (n = 2; open circles; 4,798× and 2,520× coverage). Each point represents a single mouse and bars represent the group mean. (E and F) Distribution of intended CAG deletion sizes. Dashed line represents precise intended CAG deletion sizes of 779 bp and 614 bp in Pcp2-127Q mice (E) and BAC-72Q mice (F), respectively. Blue and red dots represent the deletion sizes in the two mice tested and the frequency of intended CAG deletions that were precise are shown below. (G and H) Frequency of target reads with inversions between the intended cleavage sites in Pcp2-127Q mice (G) (n = 2; 3,850× and 1,890× coverage) and BAC-72Q mice (H) (n = 2; 4,130× and 2,100× coverage). (I and J) Frequency of indels in Pcp2-127Q (n = 3, ctrl-gRNA and gRNA5; n = 4, gRNA4+5) at gRNA4 site (I; gRNA4+5, ∗∗∗∗p < 0.0001) and gRNA5 site (J; gRNA5, ∗∗∗∗p < 0.0001; gRNA4+5, ∗∗∗p = 0.0003) assayed by drop-off ddPCR. (K and L) Frequency of indels in BAC-72Q (n = 3, ctrl-gRNA and gRNA5; n = 5, gRNA4+5) at gRNA4 site (K) (gRNA4+5, ∗∗p = 0.0052) and gRNA5 site (L) (gRNA5, ∗p = 0.0107; gRNA4+5, ∗p = 0.0247) assayed by drop-off ddPCR. Indels were normalized to a transgene reference. Bars represent the group mean and error bars represent the standard deviation. Significance was determined by one-way ANOVA with Dunnett’s test for multiple comparisons for all ddPCR data; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
To determine if the dual-gRNA strategy caused inversions as previously reported,16,20 reads were aligned to a new reference transgene harboring a predicted inversion with CTG repeats. We observed 0.9% (Figure 2G) and 1.5% (Figure 2H) inversion events in SCA2 mice after editing. In non-targeting control (ctrl)-gRNA treated mice, CAG repeats were intact in target reads (Figure S6). Additionally, alignments to the mouse Atxn2 locus and the human ATXN2 gene or cDNA transgenes did not reveal any CRISPR-mediated translocations in all treated mice.
To quantify indels at gRNA target site(s) in dual- and single-gRNA-treated SCA2 mice, we developed a drop-off ddPCR assay to detect small indels within the predicted cleavage window (Figure S7A). In dual-gRNA4+5 treated groups compared to the ctrl-gRNA groups, we observed significant indel efficiencies of 8% (Figure 2I) and 12% (Figure 2K) at gRNA4 site in Pcp2-127Q and BAC-72Q mice, respectively, and 14% (Figure 2J) and 18% (Figure 2L) at gRNA5 site in Pcp2-127Q and BAC-72Q, respectively. Additionally, we compared the single gRNA5 group with the ctrl-gRNA group and observed significant indel efficiencies of 19% and 23% at gRNA5 site in Pcp2-127Q and BAC-72Q, respectively. There was 5%–6% higher gRNA5 efficiency at gRNA5 site with single gRNA5 treatment compared with dual-gRNA4+5 treatment (Figures 2J and 2L); however, this difference was not significant. Overall, these data indicate that DNA repair after editing favored small indels over larger CAG deletions.
Editing outcomes unique to transgenic alleles and CAG repeat stability
Long CAG repeats are susceptible to somatic expansions through DNA damage and repair mechanisms.26,27 Therefore, we monitored repeat length after DSB induction by CRISPR/Cas9 and the subsequent repair using RepeatHMM.28 In all treatment groups, Pcp2-127Q mice showed a single peak at approximately 120 CAG repeats (Figures 3A, 3C, and 3E) and BAC-72Q mice showed four peaks ranging from approximately 50–120 CAG repeats (Figures 3B, 3D, and 3F), indicative of multiple transgene copies. The CAG sizing peaks agreed with PCR-based fragment analysis (Figure S8). In all groups, we observed similar CAG repeat sizes, with averages of 119 in Pcp2-127Q and 86 in BAC-72Q (Figures 3G and 3H). The data indicate that CRISPR-mediated DSBs neither expanded nor contracted repeat expansions in SCA2 mouse models.
Figure 3.

CAG repeat sizes and large deletions and rearrangements of transgenes after AAV-CRISPR editing
(A–F) Representative plots of CAG repeat size counts in Pcp2-127Q mice (A, C, E) and BAC-72Q mice (B, D, F); ctrl-gRNA (red) (A, B), gRNA5 (orange) (C, D), and gRNA4+5 (blue) (E, F) in nanopore Cas9-targeted reads from RepeatHMM analysis. Box plots represent CAG repeat size distribution and outliers are shown by dots. (G and H) Average CAG repeat size count for Pcp2-127Q mice (G) (n = 2, ctrl-gRNA; n = 2, gRNA5; n = 2, gRNA4+5) and BAC-72Q mice (H) (n = 2, ctrl-gRNA; n = 2, gRNA5; n = 2, gRNA4+5). Each point represents a single mouse and bars represent the group mean. (I) Frequency of target reads containing multiple CAG repeats within single reads in BAC-72Q mice (917× and 378× coverage). Bars represent the group mean. (J) Schematic of tandem transgene copies in BAC-72Q mice showing gRNA4 and gRNA5 target sites (red arrowheads) in each transgene copy. A gRNA5-gRNA4 deletion, where edit sites could be contiguous or noncontiguous, between transgene copies would yield a read containing two CAG repeats (left). Two consecutive gRNA5-gRNA4 deletions between transgene copies would cause a read containing three CAG repeats (right).
Interestingly, outlier reads with repeats longer than the normal distribution were identified in dual-gRNA-treated mice (Figures 3E and 3F). This prompted further examination of human ATXN2 reads containing CAG repeats, which showed that 1% of target reads from BAC-72Q contained multiple CAG repeat sequences within proximity in single reads (Figure 3I). These contain repaired gRNA5 to gRNA4 sites near CAG repeats, separated by intervening sequences, suggesting that the repair of large deletions (approximately ≥169 kb) between tandem transgene copies led to complex rearrangements (Figures 3J, S9, and S10). Figure S11 shows that large deletions can result from the repair of transgene copies between gRNA4 and gRNA4 edit sites, as well as inversions. These examples highlight how unbiased, PCR-free long-read sequencing can capture events commonly missed by standard methods, including editing outcomes unique to transgenic animals with multiple editable copies on a single chromosome.
Editing strategies and gRNA target impact AAV site integration frequency
PCR-based analysis of tissues after AAV delivery has shown evidence of AAV integration. Alignments of reads from Cas9-targeted enriched gDNA showed that AAV integration occurred at high frequency after AAV-SpCas9 and AAV-gRNA(s) delivery to brain. Total AAV integration events were 26% (22.5% indels; Figures 2I and 2J) and 13% (19.4% indels; Figure 2J) of target reads in Pcp2-127Q mice with dual-gRNA and single-gRNA treatments, respectively, and 22% (29.8% indels; Figures 2K and 2L) and 20% (23.5% indels; Figure 2L) in BAC-72Q with dual-gRNA and single gRNA treatments, respectively (Figure 4A). In ctrl-gRNA-treated mice, reads aligned to the AAV vector sequences at low coverage (6–43×) (Figure S12 and Table S4) and did not supplementally align to the on-target gRNA sites, supporting low background contamination from AAV episomal DNA in the sequencing libraries. In the dual-gRNA-treated mice, we observed AAV-SpCas9 and AAV-gRNA(s) combined total AAV integration frequencies of 16.6% (8.4% indels; Figure 2I) and 11.7% (12.0% indels; Figure 2K) at gRNA4 site compared to 9.1% (14.1% indels; Figure 2J) and 10.2% (17.8% indels; Figure 2L) at the gRNA5 site in Pcp2-127Q and BAC-72Q, respectively (Figure 4B). Thus, AAV integration frequencies were higher at the gRNA4 site compared to the gRNA5 site. Conversely, indel efficiency was lower at the gRNA4 site compared with the gRNA5 site in BAC-72Q and Pcp2-127Q (Figures 2I–2L). AAV integrations occurring between the gRNA sites in dual-gRNA-treated mice were 0.4%–2% (Figure 4C), suggesting that CAG deletions followed by insertion of AAV sequence occurred less frequently than indels. Interestingly, AAV-SpCas9 sequences integrated at higher frequencies compared with AAV-gRNA(s) in both dual- and single-gRNA-treated mice (Figures 4B–4D). Because both AAVs were delivered at the same dose, the data indicate that AAV-SpCas9 (4.8 kb) provided more template sequence to integrate than AAV-gRNA(s) (2.3 and 2.7 kb). Overall, editing outcomes observed between gRNA sites were not equivalent among editing strategies.
Figure 4.

Frequency of AAV integrations
(A) Frequency of target reads with AAV integrations (AAV-SpCas9 + AAV-gRNA[s]) after dual-gRNA4+5 (n = 2, dark blue and red closed circles, 4,750× and 2,205× coverage) and single gRNA5 (n = 2, light blue and orange closed circles, 2,900× and 2,550× coverage) treatments in Pcp2-127Q and dual-gRNA4+5 (n = 2, dark blue and red open circles, 5,300× and 2,630× coverage) and single gRNA5 (n = 2, light blue and orange open circles, 4,400× and 3,450×) treatments in BAC-72Q. Each point represents a single mouse. Percent indels from Figures 2I–2L are shown below. (B) Frequency of target reads with AAV-SpCas9 (squares) or AAV-gRNA(s) (triangles) integrations at gRNA4 (n = 2, 5,200× and 2,460× coverage) and gRNA5 (n = 2, 4,300× and 1,950× coverage) sites in dual-gRNA4+5-treated Pcp2-127Q mice (closed shapes) and at gRNA4 (n = 2, 5,500× and 2,710× coverage) and gRNA5 (n = 2, 5,100× and 2,550× coverage) sites in dual-gRNA4+5-treated BAC-72Q mice (open shapes); sum of the AAV-SpCas9 mean and AAV-gRNA(s) mean integration frequency is denoted. Percent indels from Figures 2I–2L are shown below. (C) Frequency of target reads with AAV-SpCas9 and AAV-gRNA(s) integrations between gRNA4 and gRNA5 sites in dual-gRNA4+5-treated Pcp2-127Q mice (n = 2, 4,750× and 2,205× coverage) and BAC-72Q mice (n = 2, 5,300× and 2,625× coverage). (D) Frequency of target reads with AAV-SpCas9 and AAV-gRNA(s) integrations at the gRNA5 site in single gRNA5 treated Pcp2-127Q mice (n = 2, 2,900× and 2,550× coverage) and BAC-72Q mice (n = 2, 4,400× and 3,450× coverage); the sum of the AAV-SpCas9 mean and AAV-gRNA(s) mean integration frequency is denoted. Percent indels from Figures 2I–2L are shown below.
AAV integrations are full-length genomes, ITR-less fragments, and unmethylated
Next, AAV integrants were assessed by nCATS- and PCR-based methods in edited brain tissues. The former identified full-length genomes, while nanopore PCR-targeted sequencing did not (Figures 5A and S13). PCR was biased toward short fragments and ends, primarily containing ITRs, before coverage dropped off across the AAV cargo. We did not observe reads aligned to AAV vector sequences in ctrl-gRNA-treated mice, which confirms that PCR was specific for the target region. To our knowledge, this is the first direct comparison between PCR-based and PCR-free sequencing demonstrating the intrinsic bias associated with PCR-based methods for AAV integration analysis.
Figure 5.

Frequency of full-length AAV genome and ITR integrations
(A) Comparison of representative IGV alignments of nanopore Cas9-targeted and PCR-targeted sequencing reads aligned to AAV-gRNA4+5 and AAV-SpCas9 in dual-gRNA4+5 treated BAC-72Q mice. Full-length AAV genome integrations at on-target gRNA sites detected by Cas9-targeted sequencing are shown by red arrowheads. PCR-targeted sequencing shows decreasing coverage (green) across the AAV cargo between ITR sequences. The IGV plots were cropped to highlight the relevant data and maximum base coverage is shown on the left. (B) Frequency of Cas9-targeted reads with full-length AAV genome integrations from AAV-SpCas9 (squares), AAV-gRNA(s) (triangles), and their combined total (circles) in dual-gRNA4+5-treated (red and blue) Pcp2-127Q mice (n = 2, closed shapes, 4,750× and 2,205× coverage) and BAC-72Q mice (n = 2, open shapes, 5,300× and 2,630× coverage), and single gRNA5 (orange and light blue) treated Pcp2-127Q mice (n = 2, closed shapes, 2,900× and 2,550× coverage) and BAC-72Q mice (n = 2, open shapes, 4,400× and 3,450× coverage). Each point represents a single mouse and bars represent the group mean. (C) Frequency of AAV-SpCas9 and AAV-gRNA(s) integrations that were full-length AAV genomes in dual-gRNA4+5 and single gRNA5-treated Pcp2-127Q and BAC-72Q mice. (D) Representative IGV alignment of coverage across the wild-type AAV2 ITR sequence. The preferred breakpoint (red arrowhead) is shown by reduced coverage and the AAV ITR hairpin schematic (flop orientation) on the right shows the location of the breakpoint between the B′–B and A′–A arms. Black boxes around the ITR sequence regions shown below the coverage. (E–H) Frequency of AAV ITR integration at gRNA4 site (E) (gRNA4+5, ∗∗∗∗p < 0.0001) and gRNA5 site (F) (gRNA5, ∗∗∗p = 0.0003; gRNA4+5, ∗∗∗∗p < 0.0001) in Pcp2-127Q mice (n = 3, ctrl-gRNA and gRNA5; n = 4, gRNA4+5) and gRNA4 site (G) (gRNA4+5, ∗∗p = 0.0035) and gRNA5 site (H) (gRNA5, ∗∗p = 0.0034; gRNA4+5, ∗∗p = 0.0045) in BAC-72Q mice (n = 3, ctrl-gRNA and gRNA5; n = 5, gRNA4+5). AAV-ITR integrations were normalized to the transgene reference. Bars represent the group mean and error bars represent the standard deviation. Significance was determined by one-way ANOVA with Dunnett’s test for multiple comparisons for all data; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001.
With Cas9-targeted nanopore sequencing, full-length AAV genomes were observed in 1%–2% of target reads (Figure 5B). Of the total AAV integrations in target reads (Figure 4A), a higher frequency of full-length AAV-gRNA(s) (10%–29%) was observed compared with full-length AAV-SpCas9 (4%–7%), and was most striking in the single gRNA treated mice (26%–29%) (Figure 5C). Thus, the smaller AAV-gRNA(s) genome integrates as full-length more efficiently than AAV-SpCas9.
The AAV ITR sequence can drive integration.12,19 In brain, we observed partial fragment integrations with and without ITRs (Figure 5A), and overall higher AAV cargo coverage compared with ITR coverage with Cas9-targeted sequencing (Table S4). In contrast, PCR showed higher ITR coverage, in most cases, compared with AAV cargo coverage (Table S5). The coverage between the ITRs was constant across the AAV cargos suggesting no sequence hotspots or specific microhomologies shared with the on-target sites and AAV cargo. We did observe a significant drop in coverage between the palindromic B–B′ short arm and A–A′ long arm of the WT AAV2 ITR hairpin indicating a favored breakpoint (Figures 5A and 5D).29 Interestingly, prior reports using PCR-based, short-read sequencing also identified preferential breakpoints in the palindromic short arms B and C of the ITR hairpin proximal to the payload.19,30 Using the region of high ITR coverage for ddPCR assay design (Figure S7B), we confirmed ITR containing AAV integrations at gRNA4 and gRNA5 sites at frequencies of 4%–5.5% (Figures 5E–5H). Compared with the ctrl-gRNA group, we observed significant ITR integration in Pcp2-127Q at gRNA4 site and gRNA5 site in the dual-gRNA group (Figures 5E and 5F), and gRNA5 site in the single gRNA group (Figure 5F). While the BAC-72Q mice showed more variability, there was significant ITR integration at gRNA4 site and gRNA5 site in the dual-gRNA group (Figures 5G and 5H), and gRNA5 site in the single gRNA group (Figure 5H), when compared with the ctrl-gRNA group. As expected, ITR containing integration (4%–5.5%) was significant. Taken together with total AAV integrations (10%–25%; Figure 4A), the data indicate ITR-less fragment AAV integration occurs in brain after editing. Overall, full-length AAV genome integrations represented a small subset of total integrations and ITR containing sequence was frequently observed at gRNA target sites in the brain.
Next, the methylation status of AAVs was assessed in the brain after editing. DNA methylation is an epigenetic mechanism that regulates gene expression at promoters by repressing transcription. CpG dinucleotides in AAV-packaged DNA are predominately unmethylated (0%–2% methylation)31,32; however, epigenetic silencing can occur through de novo CpG methylation. Moreover, integrated AAV genomes can become hypermethylated, and the methylation status of the target cells can influence AAV integration and transgene expression.33 The post-injection methylation status of AAVs has not been deeply surveyed in vivo and is unknown in the brain. To address this, we used Megalodon, a computational tool with high methylation calling performance in repetitive sequence regions.34,35 In AAV-ctrl-gRNA and AAV-gRNA(s)-treated mice, we observed high CpG 5mC methylation in the promoter region upstream of exon 1 in reads aligned to the BAC-72Q ROI showing tool detection sensitivity (Figure S14A). Similar CpG methylation patterns were visualized at the gRNA target sites and proximal genetic landscape of the transgenes, indicating that CRISPR editing and AAV integration did not cause overt epigenetic changes (Figures S14A and S15A). AAV-SpCas9 and AAV-gRNA(s) fragment and full-length aligned reads were predominately unmethylated (frequency of 5mC methylation 0%–2%) at CpG sites across the AAV genomes (Figures S14B, S14C, S15B, and S15C). Aligned AAV sequences were at lower coverage in AAV-ctrl-gRNA mice compared with AAV-gRNA(s) treated mice (Table S6) and our Cas9-targeted enrichment of the edited loci captured high AAV integration (Figure 4A), suggesting the majority of unmethylated AAV sequence reads represented AAV integrations, including partial fragments and full-length genomes. The data extend the utility of Cas9-targeted nanopore sequencing to evaluate the methylation status of AAV vectors and integrations in vivo.
In summary, editing outcomes were similar in two transgenic mouse models of SCA2 (Figure 6A). Unbiased long-read sequencing allowed for a categorical breakdown of AAV integrations demonstrating preferential integrations of AAV fragments over full-length genomes (Figure 6B). Overall, the preclinical evaluation of editing outcomes in the brain of SCA2 transgenic mice can be used to predict editing in humans with SCA2 (Figure 6C), with the caveat that transgenic mouse models contain multiple editable copies on a single chromosome, leading to large deletions and rearrangements, a unique and unintended outcome not expected in humans.
Figure 6.

Summary of editing outcomes in the brain of SCA2 transgenic mice
(A) Editing outcomes from nCATS (Figures 2, 3, 4, and 5) or from ddPCR (Figures 2I–2L; indels). (B) Frequency of integrations that were fragments or full-length AAVs. (C) Data observed in SCA2 transgenic mice brain offer predictions for expected editing outcomes in normal and SCA2 humans. SCA2 transgenic mice with multiple copies have multiple target sites (red arrowheads) on a single chromosome compared to humans with targets sites on two chromosomes. The asterisk denotes the large deletions and transgene rearrangements unique to editing in a transgenic mouse model.
Discussion
We adapted unbiased long-read nCATS to evaluate AAV-CRISPR editing outcomes near expanded CAG repeats in SCA2 mouse model brains. This allowed evaluation of CRISPR-mediated deletions and detection of partial and full-length AAV integrations. The data show that available gRNA sites and AAV genome size influence full-length AAV genome integrations frequencies. As two Cas9-enrichment gRNAs effectively enriched for the three Pcp2-127Q transgenic alleles, high coverage (>1,000×) of endogenous genes could likely be done with two enrichment gRNAs.
AAV fragment integrations and small indels occurred more frequently than the overall intended CAG deletions with dual-gRNAs. Deletion efficiency has been shown to inversely correlate with deletion size.36 However, unintended large deletions (approximately ≥169 kb) were observed at frequencies similar to the smaller intended CAG deletions. Interestingly, this outcome was only observed with dual-gRNA4+5, and not gRNA5 alone. We speculate that the higher indel efficiencies at the gRNA5 site suggest a temporal and spatial relationship, whereby DNA repair occurred more efficiently at the gRNA5 site compared with the gRNA4 site, preventing large deletions and rearrangements between gRNA5 to gRNA5 sites. Notably, the large deletions were due to the presence of multiple tandem transgene copies in the SCA2 model used, which would be applicable to editing repeated motifs in genes that contain the same sequence on the same chromosome. This highlights the advantage of our unbiased long-read sequencing approach to unravel large complex editing events.
The complex rearrangements noted herein raise important points for interpreting editing in transgenic animal models, relevant in part because clinical development pipelines often require testing in transgenic animal models that have multiple editable copies. For example, AAV integrations could occur after one CRISPR-mediated DSB in a single transgene copy or after multiple DSBs in multiple transgene copies. In our study, we find that, while the BAC-72Q and Pcp2-127Q models have varying transgene copy numbers (Figure S1), the editing and AAV integration results were similar. Whether the editing outcomes would be similar to an endogenous gene with two trans copies is unknown. The rearrangements would certainly be different, but the frequencies of AAV fragment or full genome insertions may be similar. Future efficacy studies will address the limitations of editing in transgenic animals and if the unintended editing outcomes compromise any potential therapeutic benefit. Nonetheless, cataloging these events can help to inform toxicities arising from long-term in vivo testing.
Our study corroborates previous findings of partial fragment and ITR-less AAV integrations,13,20,37 and extends this work to show full-length integrations are possible. AAV integrates as ssDNA through homologous recombination (HR); in vitro studies showed that AAV does not undergo HR as double-strand DNA (dsDNA).38 One hypothesis to explain partial fragment and ITR-less integrations is that degraded linear AAV genomes are undergoing HR. It remains unclear whether AAV integrations occur as dsDNA after concatemeric episome formation in vivo. DNA damage, such as nicking of the AAV episome, could make it a template for integration through recruitment of DNA repair proteins. Controlling SpCas9 expression after AAV episome formation may offer insights into these unknown mechanisms and potentially decrease CRISPR-mediated AAV integration.39 As long-read nanopore sequencing accuracy and depth continue to improve, and more data are generated among various laboratories, we may better understand how DNA repair and AAV integration occurs after the induction of DSBs in the transduced neurons.
The emergence of new sequencing approaches to analyze large editing outcomes (>100 bp) overcome the limitations of previous targeted PCR and next-generation sequencing approaches. For instance, uni-directional targeted sequencing (UDiTaS) evaluated on- and off-target indels and genome rearrangements and translocations in cultured cells.40 Individual DNA molecule sequencing (IDMseq), a PCR-based, long-read nanopore sequencing approach, revealed large deletions and complex rearrangements in human embryonic stem cells.41 However, these approaches introduce biases from tagmentation or PCR. Finally, an unbiased long-read sequencing approach profiled the integration of large CRISPR-guided transposition products (≤5,066 bp) genome-wide in Escherichia coli.42 While these approaches are complementary to our unbiased targeted nCATS approach, each has blind spots that the other can identify. Our approach aimed to define on-target editing, thereby missing off-target editing and AAV integration genome wide. Compared with short-read sequencing with high accuracy and depth, small indels and rare editing outcomes, such as translocations, are additional blind spots with our approach. The continued development and adoption of new sequencing approaches, or combinations of sequencing approaches, will further strengthen our understanding of intended and unintended, large editing events.
It will be interesting in future work to determine the biodistribution of the editing outcomes we observed. Given current technologies, assessing specific events in situ will be challenging given the complexity and diversity of editing outcomes and AAV integration species, including vector deletions and rearrangements.
Quantifying genome-wide AAV integrations is difficult; current tools are designed for short-read amplicon sequencing. Thus, we manually counted on-target AAV integrations from alignments. While tedious, this approach could also be used in follow-up studies with Cas9-targeted enrichment of the AAV genomes and mouse genomic sequences to capture off-target AAV integration. Additionally, deep amplicon sequencing could be used for genome-wide profiling to inform focused evaluation with Cas9-targeted long-read sequencing.
CpG depletion of AAV vectors decreases TLR9-mediated and CD8+ T cell immune responses.43,44 However, the methylation status of AAVs after integration has not been well-studied in vivo. If integrated AAVs remain unmethylated, as noted here, the possibility of those sequences being expressed remains. If unmethylated CpGs are present in AAV vector genomes, they can also activate immune responses. Conversely, the occurrence or consequence of de novo DNA methylation after AAV integration are also unknown in vivo. Targeted insertion without nucleases rely on AAV integration via HR.45 These events may cause methylation of nearby promoters disrupting gene expression. For these reasons, more data are needed to understand DNA methylation of AAV integrations, expression from those integrants, and the impact on host genomes in vivo after the delivery of active nucleases to brain.
In summary, we present new insights for the fields of gene editing and gene therapy and, importantly, show the relevance of applying unbiased long-read sequencing to analyze editing outcomes as the field advances into the clinic. We expect polymerase-based and polymerase-free methods can work together to evaluate and monitor gene editing outcomes of CRISPR-based therapies for SCA2 and other diseases.
Materials and methods
AAV preparation
For in vivo mouse studies, four different recombinant AAV (rAAV) vectors were generated by the Research Vector Core at the Raymond G. Perelman Center for Cellular and Molecular Therapeutics at The Children’s Hospital of Philadelphia. PX551 AAV shuttle plasmid expressed SpCas9 under the control of the neuronal-specific Mecp2 promoter and upstream of an SV40pA (PX551 was a gift from Feng Zhang; Addgene plasmid # 60957; http://n2t.net/addgene:60957; RRID:Addgene_60957).46 gRNA expression cassettes were moved into the G0619 AAV shuttle plasmid with eGFP gene under the control of the CMV promoter and upstream of an SV40pA signal (G0619 was from the University of Iowa Viral Vector Core). The non-targeting ctrl-gRNA sequence used was from the GeCKO v2 CRISPR screening library.47 All rAAV plasmid shuttles have AAV2 ITR sequences. rAAV vectors were produced by the standard calcium phosphate transfection method in HEK293 cells with the AdHelper plasmid, AAV1 Rep2/Cap1 packaging plasmid, and AAV shuttle plasmids with double CsCl purification.48 Vector titers were determined by ddPCR and were 1E13 vg/mL. Vector purity was tested by silver stain.
In vivo administration of AAV-CRISPR in mice
Mouse studies and protocols were approved by The Children’s Hospital of Philadelphia Institutional Animal Care and Use Committee. SCA2 mice, Bl6/Tg(Pcp2-ATXN2∗127Q) and FVB/Tg(ATXN2∗72Q), were obtained from Stefan Pulst at the University of Utah. Mice were housed in a temperature-controlled environment on a 12 h light/dark cycle. Food and water were provided ad libitum. Mice were injected at 8 weeks of age with an equal ratio (1:1) of rAAV2/1-Mecp2-SpCas9 vector and either rAAV2/1-hU6-gRNA5-CMV-eGFP vector or rAAV2/1-hU6-gRNA4-hU6-gRNA5-CMV-eGFP vector or rAAV2/1-hU6-ctrlgRNA-CMV-eGFP vector. For rAAV injections, mice were anesthetized with isoflurane and 10 μL rAAV mixture was injected bilaterally into the striatum at 0.22 mL/min (coordinates: +0.86 mm rostral to bregma, −1.8 mm lateral to medial, and −2.5 mm ventral from brain surface). After 4 weeks, mice were anesthetized with a ketamine and xylazine mixture and perfused with 15 mL of ice-cold 1× PBS. Brains were removed, placed on ice-cold petri dishes, and GFP-positive tissues were micro-dissected under a fluorescent stereomicroscope. All tissue was flash frozen in liquid nitrogen and stored at −80°C.
gDNA preparation
Frozen tissue samples were ground with a pestle on ice in lysis buffer and gDNA was extracted using a DNA Genomic-tip kit (Qiagen, catalog no. 13343). gDNA was sheared to 20 kb using a g-TUBE (Covaris, catalog no. 520079). Sheared gDNA was size selected using Circulomics Short Read Eliminator XS kit (PacBio, catalog no. SS-100-121-01), concentrations were quantified using the Qubit fluorometer (Thermo Fisher Scientific), and gDNA was visualized on agarose gel with ethidium bromide.
PCR assays
For ATXN2 CAG deletions, PCR amplification through the CAG repeats of ATXN2 transgenes was performed using brain gDNA template with betaine and Biolase DNA polymerase (Bioline, catalog no. BIO-21066). For BAC-72Q mice, the PCR cycle conditions were: 94°C 5 min (94°C 30 s, 60°C 30 s, 72°C 1.5 min) ×34, 72°C 10 min, and 4°C hold. For Pcp2-127Q mice, the PCR cycle conditions were: 94°C 5 min (94°C 30 s, 60°C 30 s, 72°C 2.75 min) ×34, 72°C 10 min, and 4°C hold. PCR products were separated and visualized on an agarose gel with ethidium bromide stain. The unedited PCR products were 2,317 bp for BAC-72Q and 2,622 bp for Pcp2-127Q mice. The edited PCR products were 1,703 bp for BAC-72Q mice and 1,843 bp for Pcp2-127Q mice. For Pcp2-127Q transgene insertion at mouse Mbl2, a forward primer targeted Mbl2 and a reverse primer targeted the 5′ end of the Pcp2-127Q transgene and a forward primer targeted the 3′ end of the Pcp2-127Q transgene and a reverse primer targeted Mbl2. PCR was performed using PrimeSTAR GXL DNA polymerase (Takara Bio, catalog no. R050A) with PCR conditions: 98°C 10 s, 60°C 15 s, 68°C 130 s [5′ PCR] or 90 s [3′ PCR] ×29, 4°C hold. The PCR products were 1,835 bp for 5′ Pcp2-127Q and 1,506 bp for 3′ Pcp2-127Q. The PCR products were gel purified and Sanger sequenced. The custom PCR assay sequences are in Table S7. For fragment analysis, PCR amplification through CAG repeats of ATXN2 transgenes was performed using Pcp2-127Q and BAC-72Q mouse tail gDNA template extracted with MyTaq Extract-PCR kit (Bioline, catalog no. BIO-21127). PCR was performed with 5′-FAM-labeled SCA2-A forward and SCA2-B reverse primers24 with PCR cycle conditions: 95°C 3 min (95°C 30 s, 58°C 30 s, 72°C 1 min) ×34 and 4°C hold. Capillary electrophoresis with MapMarker 1,000 bp standard was done by the Napcore facility at the Research Institute of the Children’s Hospital of Philadelphia. Fragment analysis was analyzed with Applied Biosciences GeneMapper Software 5 (Thermo Fisher Scientific).
ddPCR assays
ddPCRs were performed on the QX200 (Bio-Rad) according to the manufacturer’s instructions for probe-based assays with 35 ng gDNA. All ddPCR assays were multiplexed for target and reference (Figure S7). Using QX Manager Software (Bio-Rad, v1.2), drop off-assay was used for analyzing indel efficiency and direct quantification was used for analyzing AAV-ITR frequency and transgene copy number. For measuring transgene copy number in SCA2 mice, mouse Tfrc reference assay (Thermo Fisher Scientific, catalog no. 4458366) was used. The remaining custom assays are in Table S7.
Cas9-targeted enrichment library preparation for nanopore sequencing
Cas9-enrichment gRNAs were designed using CHOPCHOP (chopchop.cbu.uib.no) with more than 1 kb of sequence flanking both sides of the target SCA2 transgene ROI.49 In BAC-72Q mice, the nCATS protocol was initially followed with the recommended four Cas9-enrichment gRNAs for optimal target coverage. In the Pcp2-127Q mice, 17 Cas9-enrichment gRNAs were tested, and ultimately, 2 gRNAs provided high coverage. Upstream Cas9-enrichment gRNAs targeted the (+) strand and downstream Cas9-enrichment gRNAs targeted the (−) strand. gRNAs were assembled using an equimolar Alt-R CRISPR-Cas9 crRNA (IDT, custom order) pool and tracrRNA (IDT, catalog no. 1072532) by denaturation at 95°C and cooled at room temperature for 20 min. Cas9 RNPs were formed using assembled gRNAs and Alt-R S.p. HiFi Cas9 Nuclease V3 (IDT, catalog no. 1081060) in 1× CutSmart Buffer (NEB catalog no. B7204) for 30 min at room temperature and stored at 4°C until use. Sheared and size selected input gDNA, 5 μg, was dephosphorylated using Quick CIP (NEB, catalog no. M0525) in 1× CutSmart Buffer by incubation at 37°C for 10 min, 80°C for 2 min, and then held at 20°C. Dephosphorylated gDNA was cleaved with Cas9 RNPs and dA-tailed using dATP and Taq polymerase (NEB, catalog no. M0273) by incubation at 37°C for 15 min, then 72°C for 5 min, and held at 4°C. Adapter ligation was performed with Ligation sequencing kit (ONT, SQK-LSK110) using NEBNext Quick T4 DNA ligase (NEB, catalog no. E6056) by incubating for 10 min at room temperature. Samples were cleaned up using 0.3× volume AMPure XP beads (Beckman Coulter, catalog no. A63881), washed twice on a magnetic rack with short fragment buffer (ONT, SQK-LSK110) and DNA libraries were eluted in 8 μL (Flongle) or 13 μL (MinION) elution buffer (ONT, SQK-LSK110) at 37°C for 15 min. The above method was modified from the detailed Cas9-mediated PCR-free enrichment protocol (version: ENR_9084_v109_revL_04Dec2018) available through ONT.
PCR-targeted amplicon library preparation for nanopore sequencing
Using the ATXN2 CAG deletion PCR method, PCR products were pooled from two 50 μL reactions and cleaned up with 1.8× volume AMPure XP beads (Beckman Coulter, catalog no. A63881), washed twice with 70% ethanol on a magnetic rack and eluted with nuclease-free H20. PCR products were phosphorylated using T4 Polynucleotide Kinase (NEB, catalog no. M0201S) in 1× T4 ligase buffer, cleaned up with 1.8× volume AMPure XP bead, washed twice with 70% ethanol on a magnetic rack and eluted with nuclease-free H20. Adapter ligation was performed with Ligation sequencing kit (ONT, SQK-LSK110) using NEBNext Quick T4 DNA ligase. Samples were cleaned up using 0.4× volume AMPure XP beads washed twice on a magnetic rack with short fragment buffer (ONT, SQK-LSK110) and DNA libraries were eluted in 7 μL elution buffer (ONT, SQK-LSK110) at room temperature for 10 min.
Nanopore sequencing
The sequencing library was prepared with 7 μL (Flongle) or 12 μL (MinION) DNA library, 15 μL (Flongle), or 37.5 μL (MinION) sequencing buffer II (ONT, SQK-LSK110), and 10 μL (Flongle) or 25.5 μL (MinION) loading beads II (ONT, SQK-LSK110). Flow cell priming mix was prepared with 3 μL (Flongle) or 30 μL (MinION) of flush tether (ONT, SQK-LSK110) and 117 μL (Flongle) or a tube (MinION) of flush buffer (ONT, SQK-LSK110). Libraries were loaded onto Flongle flow cells with R9.4.1 nanopores (ONT, catalog no. FLO-FLG001) for optimizing Cas9-enrichment gRNAs and PCR-targeted amplicon sequencing. Libraries were loaded onto MinION flow cells with R9.4.1 nanopores (ONT, catalog no. FLO-MIN106D) for sequencing treated SCA2 mice with optimized gRNAs. One flow cell was used per animal and run on a Mk1B or Mk1C using MinKNOW software for 24 h (Flongle) or 72 h (MinION).
Nanopore sequencing analysis
Raw FAST5 sequencing files were base called with Guppy (v5.0.7) high accuracy (HAC) or super high accuracy (SUP) models using a minimum read quality threshold of Q score of 7 to generate passed FASTQ reads (NCBI BioProject accession number PRJNA916868), processed through Porechop (v.0.2.4) (https://github.com/rrwick/Porechop) to remove leading adapter sequences and aligned using MiniMap2 (v2.17-r941) (https://github.com/lh3/minimap2) to create BAM files.50 Alignments were made to reference genome that included mouse (GRCmm38) chromosomes, Pcp2-127Q, or BAC-72Q transgene reference sequences and the AAV-SpCas9 and AAV-gRNA(s) sequences. The BAC-72Q transgene reference sequence was generated from the 150-kb human ATXN2 gene with 16 kb upstream and 3 kb downstream sequences using the UCSC Genome Browser GRCh38/hg38 coordinates: hg38_knownGene_ENST00000643669.2 range=chr12:111,449,214-111618315. From the known human ATXN2 exonic sequence, we assembled the Pcp2-127Q cDNA (exons only) transgene reference sequence using PCR and Sanger sequencing. Total reads and aligned reads were determined with samtools (v.1.10-2) using BAM files (Tables S3–S6). Target reads were defined as those that aligned within the innermost Cas9-enrichment gRNA site coordinates of the ROI. The mean coverage of the ROI and the AAV genomes was determined with samtools using BAM files. Sequencing stats were determined with NanoStat (v1.5.0) using BAM files.51 Read N50 represents one-half of the data are within reads with alignable lengths greater than this value. Guppy super high accuracy was used to base call FAST5 files for CAG repeat size counts detected from BAM files using RepeatHMM (v2.0.3) (https://github.com/WGLab/RepeatHMM) and plotted with R (v4.1.2).28 5mC-modified bases were called from raw FAST5 files using Megalodon (v2.4.2) (https://github.com/nanoporetech/megalodon). Read alignments were viewed with Integrative Genomics Viewer (IGV) (v2.12.0) (https://software.broadinstitute.org/software/igv/) for manually calling CAG deletions, inversions, and AAV integrations.52 CAG deletions were measured by manually counting the total reads that spanned the gRNA4+5 sites and the reads that contained the expected CAG deletions. Inversions were measured by aligning to a new ATXN2 transgene reference genome containing the expected inversion (CTG repeat) between the gRNA4+5 sites, counting the aligned inversion reads, and calculating the percentages from the coverage at the gRNA4+5 sites. AAV integrations were measured by counting AAV reads (primary alignment) with supplemental alignment to the gRNA target sites, and calculating the percentages from the coverage at the gRNA sites. Reads containing multiple CAG repeats within single reads were found by searching for all reads containing CAG repeats with flanking sequences upstream (179-nt upstream of gRNA4 target site) and downstream (186-nt downstream of gRNA5 target site) of the gRNA target sites. IGV was used to visualize 5mC methylation color shading in bisulfite mode from Megalodon alignment mappings.
Statistical analyses
Differences between control gRNA and treatment gRNA groups were compared using one-way ANOVA with Dunnett’s test for multiple comparisons for ddPCR assays. Differences between groups were considered to be significant at a p value of less than 0.05. All results are shown as the mean ± standard deviation. Statistical analyses were performed with GraphPad Prism v9.
Data availability
Sequencing data are available at the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA916868. The following public datasets used: Ensembl, Mus musculus genome (GRCm38.p6), http://ftp.ensembl.org/pub/release-102/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz for Figures 2, 3, 4, 5, S2–S4, and S6–S8. All vectors presented in this work are available on request with approval from the CHOP Office of Technology Transfer.
Acknowledgments
The authors thank Ellie Carrell, Alex Mas Monteys, and Paul Ranum for helpful discussions and careful review of the manuscript, and Stefan Pulst for kindly sharing the SCA2 mouse models. Figure illustrations were created with BioRender.com.
This work was funded by the National Ataxia Foundation Pioneer Translational Research Award 625451 and the Children’s Hospital of Philadelphia Research Institute.
Author contributions
B.P.S., C.M.Y., and B.L.D. designed the study. B.P.S. and A.I performed the experiments. B.P.S. analyzed and evaluated the data. B.P.S. and B.L.D. wrote the paper. B.P.S., C.M.Y., and B.L.D. evaluated the data and edited the paper.
Declaration of interests
B.L.D. is a founder of Spark Therapeutics, Spirovant Sciences and Latus Biosciences. She serves an advisory role and/or receives sponsored research support for her laboratory from Roche, NBIR, Homology Medicines, Resilience, Spirovant Sciences, Patch Biosciences, Saliogen therapeutics, Panorama Medicines, and Voyager Therapeutics. B.P.S., C.M.Y., and A.I. have no competing interests. B.P.S and B.L.D. are co-inventors on U.S. Patent Application No. 17/594,651 entitled: CRISPR/Cas9 Gene Editing of ATXN2 for the Treatment of Spinocerebellar Ataxia Type 2.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.01.004.
Supplemental information
References
- 1.Elden A.C., Kim H.J., Hart M.P., Chen-Plotkin A.S., Johnson B.S., Fang X., Armakola M., Geser F., Greene R., Lu M.M., et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075. doi: 10.1038/nature09320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monteys A.M., Ebanks S.A., Keiser M.S., Davidson B.L. CRISPR/Cas9 editing of the mutant Huntingtin allele in vitro and in vivo. Mol. Ther. 2017;25:12–23. doi: 10.1016/j.ymthe.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ekman F.K., Ojala D.S., Adil M.M., Lopez P.A., Schaffer D.V., Gaj T. CRISPR-Cas9-Mediated genome editing increases lifespan and improves motor deficits in a Huntington's disease mouse model. Mol. Ther. Nucleic Acids. 2019;17:829–839. doi: 10.1016/j.omtn.2019.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park C.Y., Halevy T., Lee D.R., Sung J.J., Lee J.S., Yanuka O., Benvenisty N., Kim D.W. Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 2015;13:234–241. doi: 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 5.Xie N., Gong H., Suhl J.A., Chopra P., Wang T., Warren S.T. Reactivation of FMR1 by CRISPR/Cas9-Mediated deletion of the expanded CGG-repeat of the fragile X chromosome. PLoS One. 2016;11:e0165499. doi: 10.1371/journal.pone.0165499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L., Wang S., Peng L., Zhao H., Li S., Han X., Habimana J.d.D., Chen Z., Wang C., Peng Y., et al. CRISPR/Cas9 mediated gene correction ameliorates abnormal phenotypes in spinocerebellar ataxia type 3 patient-derived induced pluripotent stem cells. Transl. Psychiatry. 2021;11:479. doi: 10.1038/s41398-021-01605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnan G., Zhang Y., Gu Y., Kankel M.W., Gao F.B., Almeida S. CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol. 2020;140:81–84. doi: 10.1007/s00401-020-02154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piao X., Meng D., Zhang X., Song Q., Lv H., Jia Y. Dual-gRNA approach with limited off-target effect corrects C9ORF72 repeat expansion in vivo. Sci. Rep. 2022;12:5672. doi: 10.1038/s41598-022-07746-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Hao L., Wang H., Santostefano K., Thapa A., Cleary J., Li H., Guo X., Terada N., Ashizawa T., et al. Therapeutic genome editing for myotonic dystrophy type 1 using CRISPR/Cas9. Mol. Ther. 2018;26:2617–2630. doi: 10.1016/j.ymthe.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lo Scrudato M., Poulard K., Sourd C., Tomé S., Klein A.F., Corre G., Huguet A., Furling D., Gourdon G., Buj-Bello A. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol. Ther. 2019;27:1372–1388. doi: 10.1016/j.ymthe.2019.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller D.G., Petek L.M., Russell D.W. Human gene targeting by adeno-associated virus vectors is enhanced by DNA double-strand breaks. Mol. Cell. Biol. 2003;23:3550–3557. doi: 10.1128/MCB.23.10.3550-3557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- 13.Dalwadi D.A., Calabria A., Tiyaboonchai A., Posey J., Naugler W.E., Montini E., Grompe M. AAV integration in human hepatocytes. Mol. Ther. 2021;29:2898–2909. doi: 10.1016/j.ymthe.2021.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarrett K.E., Lee C.M., Yeh Y.H., Hsu R.H., Gupta R., Zhang M., Rodriguez P.J., Lee C.S., Gillard B.K., Bissig K.D., et al. Somatic genome editing with CRISPR/Cas9 generates and corrects a metabolic disease. Sci. Rep. 2017;7:44624. doi: 10.1038/srep44624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoon Y., Wang D., Tai P.W.L., Riley J., Gao G., Rivera-Pérez J.A. Streamlined ex vivo and in vivo genome editing in mouse embryos using recombinant adeno-associated viruses. Nat. Commun. 2018;9:412. doi: 10.1038/s41467-017-02706-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeder M.L., Stefanidakis M., Wilson C.J., Baral R., Barrera L.A., Bounoutas G.S., Bumcrot D., Chao H., Ciulla D.M., DaSilva J.A., et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019;25:229–233. doi: 10.1038/s41591-018-0327-9. [DOI] [PubMed] [Google Scholar]
- 17.McCullough K.T., Boye S.L., Fajardo D., Calabro K., Peterson J.J., Strang C.E., Chakraborty D., Gloskowski S., Haskett S., Samuelsson S., et al. Somatic gene editing of GUCY2D by AAV-CRISPR/Cas9 alters retinal structure and function in mouse and macaque. Hum. Gene Ther. 2019;30:571–589. doi: 10.1089/hum.2018.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.György B., Nist-Lund C., Pan B., Asai Y., Karavitaki K.D., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Solanes P., Spataro S., et al. Allele-specific gene editing prevents deafness in a model of dominant progressive hearing loss. Nat. Med. 2019;25:1123–1130. doi: 10.1038/s41591-019-0500-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E., et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H., et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Höijer I., Tsai Y.C., Clark T.A., Kotturi P., Dahl N., Stattin E.L., Bondeson M.L., Feuk L., Gyllensten U., Ameur A. Detailed analysis of HTT repeat elements in human blood using targeted amplification-free long-read sequencing. Hum. Mutat. 2018;39:1262–1272. doi: 10.1002/humu.23580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilpatrick T., Lee I., Graham J.E., Raimondeau E., Bowen R., Heron A., Downs B., Sukumar S., Sedlazeck F.J., Timp W. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat. Biotechnol. 2020;38:433–438. doi: 10.1038/s41587-020-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hansen S.T., Meera P., Otis T.S., Pulst S.M. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum. Mol. Genet. 2013;22:271–283. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dansithong W., Paul S., Figueroa K.P., Rinehart M.D., Wiest S., Pflieger L.T., Scoles D.R., Pulst S.M. Ataxin-2 regulates RGS8 translation in a new BAC-SCA2 transgenic mouse model. PLoS Genet. 2015;11:e1005182. doi: 10.1371/journal.pgen.1005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Massey T.H., Jones L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018;11:031930. doi: 10.1242/dmm.031930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones L., Wheeler V.C., Pearson C.E. Special issue: DNA repair and somatic repeat expansion in Huntington's disease. J. Huntingtons Dis. 2021;10:3–5. doi: 10.3233/JHD-219001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Q., Zhang P., Wang D., Gu W., Wang K. Interrogating the “unsequenceable” genomic trinucleotide repeat disorders by long-read sequencing. Genome Med. 2017;9:65. doi: 10.1186/s13073-017-0456-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilmott P., Lisowski L., Alexander I.E., Logan G.J. A user's guide to the inverted terminal repeats of adeno-associated virus. Hum. Gene Ther. Methods. 2019;30:206–213. doi: 10.1089/hgtb.2019.276. [DOI] [PubMed] [Google Scholar]
- 30.Breton C., Clark P.M., Wang L., Greig J.A., Wilson J.M. ITR-Seq, a next-generation sequencing assay, identifies genome-wide DNA editing sites in vivo following adeno-associated viral vector-mediated genome editing. BMC Genomics. 2020;21:239. doi: 10.1186/s12864-020-6655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toth R., Meszaros I., Huser D., Forro B., Marton S., Olasz F., Banyai K., Heilbronn R., Zadori Z. Methylation status of the adeno-associated virus type 2 (AAV2) Viruses. 2019;11:38. doi: 10.3390/v11010038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rumachik N.G., Malaker S.A., Poweleit N., Maynard L.H., Adams C.M., Leib R.D., Cirolia G., Thomas D., Stamnes S., Holt K., et al. Methods matter: standard production platforms for recombinant AAV produce chemically and functionally distinct vectors. Mol. Ther. Methods Clin. Dev. 2020;18:98–118. doi: 10.1016/j.omtm.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chanda D., Hensel J.A., Higgs J.T., Grover R., Kaza N., Ponnazhagan S. Effects of cellular methylation on transgene expression and site-specific integration of adeno-associated virus. Genes (Basel) 2017;8:232. doi: 10.3390/genes8090232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuen Z.W.S., Srivastava A., Daniel R., McNevin D., Jack C., Eyras E. Systematic benchmarking of tools for CpG methylation detection from nanopore sequencing. Nat. Commun. 2021;12:3438. doi: 10.1038/s41467-021-23778-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Y., Rosikiewicz W., Pan Z., Jillette N., Wang P., Taghbalout A., Foox J., Mason C., Carroll M., Cheng A., et al. DNA methylation-calling tools for Oxford Nanopore sequencing: a survey and human epigenome-wide evaluation. Genome Biol. 2021;22:295. doi: 10.1186/s13059-021-02510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canver M.C., Bauer D.E., Dass A., Yien Y.Y., Chung J., Masuda T., Maeda T., Paw B.H., Orkin S.H. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J. Biol. Chem. 2014;289:21312–21324. doi: 10.1074/jbc.M114.564625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen G.N., Everett J.K., Kafle S., Roche A.M., Raymond H.E., Leiby J., Wood C., Assenmacher C.A., Merricks E.P., Long C.T., et al. A long-term study of AAV gene therapy in dogs with hemophilia A identifies clonal expansions of transduced liver cells. Nat. Biotechnol. 2021;39:47–55. doi: 10.1038/s41587-020-0741-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasileva A., Linden R.M., Jessberger R. Homologous recombination is required for AAV-mediated gene targeting. Nucleic Acids Res. 2006;34:3345–3360. doi: 10.1093/nar/gkl455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Monteys A.M., Hundley A.A., Ranum P.T., Tecedor L., Muehlmatt A., Lim E., Lukashev D., Sivasankaran R., Davidson B.L. Regulated control of gene therapies by drug-induced splicing. Nature. 2021;596:291–295. doi: 10.1038/s41586-021-03770-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giannoukos G., Ciulla D.M., Marco E., Abdulkerim H.S., Barrera L.A., Bothmer A., Dhanapal V., Gloskowski S.W., Jayaram H., Maeder M.L., et al. UDiTaS, a genome editing detection method for indels and genome rearrangements. BMC Genomics. 2018;19:212. doi: 10.1186/s12864-018-4561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bi C., Wang L., Yuan B., Zhou X., Li Y., Wang S., Pang Y., Gao X., Huang Y., Li M. Long-read individual-molecule sequencing reveals CRISPR-induced genetic heterogeneity in human ESCs. Genome Biol. 2020;21:213. doi: 10.1186/s13059-020-02143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vo P.L.H., Acree C., Smith M.L., Sternberg S.H. Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mob. DNA. 2021;12:13. doi: 10.1186/s13100-021-00242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faust S.M., Bell P., Cutler B.J., Ashley S.N., Zhu Y., Rabinowitz J.E., Wilson J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Invest. 2013;123:2994–3001. doi: 10.1172/JCI68205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bertolini T.B., Shirley J.L., Zolotukhin I., Li X., Kaisho T., Xiao W., Kumar S.R.P., Herzog R.W. Effect of CpG depletion of vector genome on CD8(+) T cell responses in AAV gene therapy. Front. Immunol. 2021;12:672449. doi: 10.3389/fimmu.2021.672449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barzel A., Paulk N.K., Shi Y., Huang Y., Chu K., Zhang F., Valdmanis P.N., Spector L.P., Porteus M.H., Gaensler K.M., et al. Promoterless gene targeting without nucleases ameliorates haemophilia B in mice. Nature. 2015;517:360–364. doi: 10.1038/nature13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swiech L., Heidenreich M., Banerjee A., Habib N., Li Y., Trombetta J., Sur M., Zhang F. In vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat. Biotechnol. 2015;33:102–106. doi: 10.1038/nbt.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ayuso E., Mingozzi F., Montane J., Leon X., Anguela X.M., Haurigot V., Edmonson S.A., Africa L., Zhou S., High K.A., et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010;17:503–510. doi: 10.1038/gt.2009.157. [DOI] [PubMed] [Google Scholar]
- 49.Labun K., Montague T.G., Krause M., Torres Cleuren Y.N., Tjeldnes H., Valen E. CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing. Nucleic Acids Res. 2019;47:W171–W174. doi: 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Coster W., D'Hert S., Schultz D.T., Cruts M., Van Broeckhoven C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics. 2018;34:2666–2669. doi: 10.1093/bioinformatics/bty149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data are available at the NCBI Sequence Read Archive (SRA) under the BioProject accession number PRJNA916868. The following public datasets used: Ensembl, Mus musculus genome (GRCm38.p6), http://ftp.ensembl.org/pub/release-102/fasta/mus_musculus/dna/Mus_musculus.GRCm38.dna.primary_assembly.fa.gz for Figures 2, 3, 4, 5, S2–S4, and S6–S8. All vectors presented in this work are available on request with approval from the CHOP Office of Technology Transfer.