

Summary
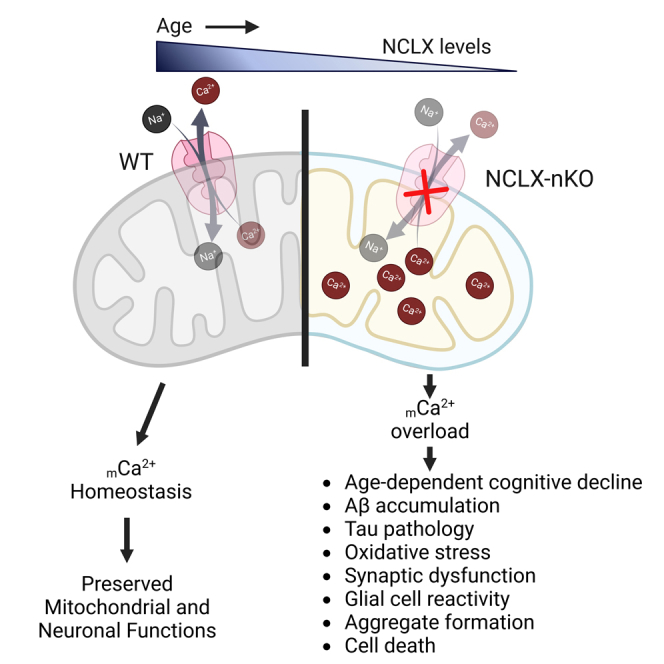
Mitochondrial calcium overload contributes to neurodegenerative disease development and progression. We recently reported that loss of the mitochondrial sodium/calcium exchanger (NCLX), the primary mechanism of mCa2+ efflux, promotes mCa2+ overload, metabolic derangement, redox stress, and cognitive decline in models of Alzheimer’s disease (AD). However, whether disrupted mCa2+ signaling contributes to neuronal pathology and cognitive decline independent of pre-existing amyloid or tau pathology remains unknown. Here, we generated mice with neuronal deletion of the mitochondrial sodium/calcium exchanger (NCLX, Slc8b1 gene), and evaluated age-associated changes in cognitive function and neuropathology. Neuronal loss of NCLX resulted in an age-dependent decline in spatial and cued recall memory, moderate amyloid deposition, mild tau pathology, synaptic remodeling, and indications of cell death. These results demonstrate that loss of NCLX-dependent mCa2+ efflux alone is sufficient to induce an Alzheimer’s disease-like pathology and highlights the promise of therapies targeting mCa2+ exchange.
Subject areas: Behavioral neuroscience, Molecular neuroscience, Cellular neuroscience, Cognitive neuroscience
Graphical abstract
Highlights
-
•
Reduced mitochondrial calcium efflux (mCa2+) promotes age-associated cognitive decline
-
•
Deletion of neuronal NCLX is sufficient to promote Alzheimer’s-like pathology
-
•
Neuronal mitochondrial dysfunction is sufficient to drive Aβ and tau pathology
-
•
Mitochondrial calcium exchange is a target for neurodegeneration, as occurs in AD
Behavioral neuroscience; Molecular neuroscience; Cellular neuroscience; Cognitive neuroscience
Introduction
A decline in mitochondrial function is associated with normal aging and contributes to the development of various progressive neurodegenerative diseases. Decades ago, the mitochondrial theory of aging was proposed because of the significant role of mitochondria-derived reactive oxygen species (ROS) in aging.1 Recent studies suggest that mitochondrial function directly impacts several essential aspects of aging and age-associated neurodegeneration, such as energetics, inflammation, cellular senescence, and redox stress.2 Alzheimer’s disease (AD) is the most common age-associated neurodegenerative disease and is characterized by memory loss, neuronal dysfunction, and the deposition of amyloid-beta (Aβ) and tau neurofibrillary tangles (NFTs) in the brain, specifically in the cortex and hippocampus. AD is classified as either familial or sporadic. Familial AD accounts for less than 5% of cases and is most often triggered by mutations in one of three genes, amyloid precursor protein (APP) and the presenilin encoding genes (PSEN1 and PSEN2). Sporadic AD has no known specific etiology and accounts for 90–95% of all AD diagnoses.3 Discouragingly, following decades of research, this multifactorial disease remains incurable and the cellular and molecular mechanisms of AD pathogenesis remain poorly defined. The majority of AD phase II/III trials have targeted components of the amyloid-β cascade or tau pathway and have been largely unsuccessful.4,5 Furthermore, many studies suggest that cognitive decline does not always correlate with Aβ and NFT deposition.6,7,8,9 Taken together, these findings underscore the importance of defining alternative disease mechanisms driving the progression of AD.
Alterations in intracellular calcium (iCa2+) homeostasis10,11,12,13 and mitochondrial function14,15 are critical to AD pathogenesis and occur before plaque deposition.16,17,18,19iCa2+ signaling plays a crucial role in pre- and postsynaptic neurotransmission and membrane excitability. Impaired iCa2+ signaling has been linked to AD pathogenesis via several different mechanisms including alterations in the inositol 1,4,5-trisphosphate receptor (IP3R),20,21,22 ryanodine receptor (RyR),23,24 store-operated Ca2+ entry,25,26 voltage-operated channels, and SERCA.27 Furthermore, presenilin mutations and Aβ have been shown to increase the activity of RyR28 and IP3R,29 suggesting exaggerated ER Ca2+ release in AD.30iCa2+ enters the mitochondrial matrix via the mitochondrial calcium uniporter channel (mtCU).31,32mCa2+ is a key regulator of various mitochondrial processes and modulates the activity of multiple dehydrogenases (PDH, α-KGDH, and ICDH) in the Krebs cycle and ETC complexes to increase ATP production.33,34mCa2+ and the enzymatic activity of Ca2+-dependent dehydrogenases are reported to be altered in neurodegenerative diseases, including AD,15,18,35,36,37,38 highlighting the essential role of mCa2+ in the regulation of oxidative energy metabolism. In the brain, mCa2+ not only maintains energetics but also buffers iCa2+ and thus can regulate processes such as synaptic neurotransmission, synaptic vesicle exocytosis and cycling, neuronal excitability and axonal trafficking.39,40 However, excessive uptake leads to mCa2+ overload and can cause rupture of the outer mitochondria membrane, opening of mitochondrial permeability transition pore (mPTP), loss of ATP production, excessive reactive oxygen species (ROS) generation, calpain activation and cell death.41
We recently reported that decreased expression of neuronal NCLX (the main mechanism formCa2+ efflux42) causes mCa2+ overload in sporadic AD patients, 3xTg-AD mice, and APPswe cell lines.18 Complete deletion of neuronal NCLX in the 3xTg-AD mouse model accelerated memory deficits and AD progression.18 Furthermore, reducing mCa2+ overload by rescuing neuronal NCLX expression impedes AD-associated pathology and cognitive decline.18 In support of this, multiple studies have reported mCa2+ overload as a feature of neurodegeneration. For example, Stavsky et al.43 reported that a genetic loss of function of NCLX leads to mCa2+ overload and is linked with severe mental retardation. In addition, a recent study showed increased neuronal mCa2+ levels in the APP/PS1-Tg mouse model correlated with plaque deposition and neuronal demise.35 Similarly, an increase in mCa2+ by overexpression of MCU in the cortex of mice resulted in neuronal dysfunction and gliosis.36 In addition, increased ER-mitochondria interaction, which can augment mCa2+ overload, is reported in familial AD,44 sporadic AD,45 and in sel-12 (PSEN ortholog) mutants in C.elegans.46 In addition, loss of acute mCa2+ uptake via genetic ablation of MCU in cortical neurons is protective against ischemia-reperfusion injury without any metabolic abnormalities.47 Altogether, these studies demonstrate that mCa2+ overload may be a central contributor to neuronal dysfunction.
In this study, we examined whether mCa2+ overload alone is sufficient to cause AD-like pathology. We tracked age-associated changes in memory and development of neuronal histopathology in mice with neuron-specific deletion of NCLX (NCLX-nKO) and controls. Loss of neuronal mCa2+ efflux caused a decline in cued recall and spatial working memory with a moderate increase in Aβ levels, tau phosphorylation, oxidative stress, and synaptic dysfunction. Our results demonstrate that loss of neuronal mCa2+ efflux and mitochondrial dysfunction promotes an AD-like phenotype, including age-associated cognitive decline.
Results
Loss of NCLX-dependent neuronal mCa2+ efflux promotes cognitive decline
We have previously reported that loss of neuronal NCLX accelerates AD-pathology in the 3xTg-AD mouse model.18 To examine NCLX expression during aging, we isolated protein from cortex of 2, 9, and 15-month-old control (Camk2a-cre) mice and noted an age-dependent reduction in NCLX expression (Figures S1A andS1B). To test whether impaired neuronal mCa2+ efflux alone is sufficient to elicit neuropathology we generated a neuron-restricted knockout of Slc8b1, the gene encoding the mitochondrial Na+/Ca2+ exchanger, hereafter referred to as NCLX, by crossing NCLXfl/fl mice48 with mice expressing a neuronal-restricted Cre recombinase (Camk2a-Cre) that primarily targets pyramidal neurons of the hippocampus, amygdala, and cerebral cortex49 (Figure 1A). qPCR analysis of NCLX mRNA expression in the frontal cortex of 2-month-old NCLXfl/fl x Camk2a-Cre mice (NCLX-nKO) confirmed loss of NCLX compared to Camk2a-Cre controls (Figure 1B). Immunoblot analysis of NCLX in mitochondrial fractions purified from mouse brain identifies 50- and 65-kDa bands.42 Western blot analysis confirmed loss of both the 50-kDa and 65-kDa forms of NCLX in the frontal cortex of 2-month-old mice without any significant alternation in the expression of components of the mitochondrial calcium uniporter channel (mtCU) (Figures 1C, 1D, and S1C–S1K).
Figure 1.

Loss of NCLX-dependent neuronal mCa2+ efflux promotes cognitive decline
(A) Schematic of NCLX-nKO mice (NCLXfl/fl x Camk2a-Cre) mutant mouse strategy.
(B and C) (B) NCLX mRNA expression in tissue isolated from the frontal cortex of NCLX-nKO and age-matched controls (Camk2a-Cre). mRNA expression corrected to the housekeeping gene Rps13; expressed as fold change versus control, n = 3 for both groups. All data presented as mean ± SEM; ∗∗∗p<0.001; two-tailed, unpaired t-test(C). Western blots for NCLX expression in tissue isolated from the cortex of 2-month-old NCLXfl/fl x Camk2a-Cre mice compared to age-matched control Camk2a-Cre mice. VDAC, voltage-dependent anion channel, served as mitochondrial loading controls.
(D) NCLX protein expression expressed as fold-change versus Camk2a-Cre con. corrected to a mitochondrial loading control VDAC in brain cortex of 2-month-old mice. All data presented as mean ± SEM; ∗∗p<0.01; two-way ANOVA with Sidak’s multiple comparisons test.
(E and F) Y-maze spontaneous alternation test. (E) Percentage of spontaneous alternation. (F) Total number of arm entries.
(G–I) Fear-conditioning test. (G) Freezing responses in the training phase. (H) Contextual recall freezing responses, (I). Cued recall freezing responses. n = individual dots shown for each group in all graphs. All data presented as mean ± SEM. Data for percentage alternations, contextual and cued recall freezing response was analyzed using Prism (GraphPad) two-way ANOVA multiple comparison testing for an age effect with Dunnett’s post-hoc test for comparison to age 6 months and comparison of genotype across all ages using a Bonferroni’s multiple comparisons test, ∗∗p < 0.01. All comparisons were non-significant except those denoted. To ensure equivalent motor activity and behavior in the Y-maze and equivalent training for fear-conditioning behavioral testing data for Y-maze number of entries and freezing during training was analyzed using two-way ANOVA testing for comparison of genotype across all ages using a Bonferroni’s multiple comparisons test. No statistical differences were noted.
Our recently published work showed that rescue of neuronal NCLX expression was sufficient to attenuate cognitive decline in 3xTg-AD mice.18 To determine whether the loss of neuronal mCa2+ efflux alone is sufficient to promote age-associated cognitive decline we tested spatial working memory and cued/contextual recall in NCLX-nKO mice using a Y-maze spontaneous alternations test and fear-conditioning paradigm, respectively, at 6-, 9-, 12-, and 15-month of age. The Y-maze test revealed decreased spontaneous alternations in NCLX-nKO mice compared to controls at 15 months of age (Figure 1E), suggesting a decline in spatial working memory. No significant differences were observed in activity or motor function with the number of arm entries being similar between controls and NCLX-nKO mice (Figure 1F). Evaluation of associative memory by the fear-conditioning test revealed a decline in cued recall in 12- to 15-month-old NCLX-nKO mice compared to controls (Figure 1I). NCLX-nKO mice showed no impairments in contextual recall during training or recall phases (Figures 1G and 1H). We further validated motor function and coordination by challenging mice on a rotarod apparatus. 15-month-old NCLX-nKO mice showed no impairment and performed similarly to controls in the accelerating rotarod test (Figure S1L). These findings indicate a progressive age-associated decline in cognitive function in mice with loss of neuronal mCa2+ efflux in the forebrain.
Loss of neuronal NCLX increases Aβ accumulation
Aβ accumulation in the brain is a central feature of AD pathogenesis. We have previously reported that loss of neuronal mCa2+ efflux accelerates amyloidosis in 3xTg-AD mice.18 Therefore, to determine whether genetic ablation of neuronal NCLX impacts Aβ metabolism or amyloidosis, we performed an Aβ ELISA and immunohistochemistry assays. NCLX-nKO and controls were euthanized at 16 months of age and brain cortex homogenates were assayed for total Aβ1-40 and Aβ1-42 peptide levels in the RIPA-soluble and insoluble fractions. NCLX-nKO cortical samples were found to have significantly increased total Aβ1-40 and Aβ1-42 in soluble fraction as compared to controls (Figures 2A and 2B). These results suggest that the loss of NCLX increases the levels of the amyloidogenic peptide. Next, we examined the protein levels of Aβ precursor protein (APP) and proteases involved in its metabolism. NCLX-nKO cortical samples showed a significant increase in the expression of β-secretase (BACE-1) (Figures 2C and 2D), as compared to controls at 16 months. No differences in the expression of total APP or γ-secretase complex associated proteins (PS1, NCT, and APH1 subunit) were observed between groups (Figures 2C and S2A–S2E).
Figure 2.

Loss of neuronal NCLX increases Aβ accumulation
(A and B) Soluble and insoluble Aβ1–40 and Aβ1–42 levels in the cortex of 16-month-old mice, measured by sandwich ELISA. n = individual dots are shown for each group in all graphs. All data presented as mean ± SEM; ∗∗∗∗p<0.001, ∗∗p<0.01, ∗p<0.05; two-way ANOVA with Sidak’s multiple comparisons test.
(C) Western blots for full-length APP, ADAM-10, BACE1, PS1, Nicastrin, APH, and tubulin (loading control) from cortex of 16-month-old NCLX-nKO and control mice, n = 3 for all groups.
(D) BACE-1 protein expression expressed as fold change versus Camk2a-Cre con. corrected to tubulin loading control from frontal cortex of 2-month-old mice. All data presented as mean ± SEM; ∗p<0.05; two-tailed, unpaired t-test.
Neuronal loss of NCLX increases tau pathology
Hyperphosphorylation of microtubule-associated protein (tau) and the formation of neurofibrillary tangles (NFTs) are prominent characteristics of various neurodegenerative diseases, such as frontotemporal dementia, chronic traumatic encephalopathy, and AD. To determine if loss of neuronal mCa2+ efflux impacts tau pathology we performed Western blots to examine the total expression and phosphorylation of tau at various residues, including S202/T205, T231/S235, and T181 and S396 as recognized by the antibodies AT8, AT180, AT270, and PHF-13, respectively. We observed an increase in phosphorylation at T231/S235 (AT180 immunoreactivity) and at S202/T205 (AT8 immunoreactivity) in cortical lysates from NCLX-nKO mice with no significant change in total tau or phosphorylation of other residues (Figures 3A–3E). These results were corroborated by our immunohistochemistry results showing increased T231/S235 tau phosphorylation (Figures 3F–3H) in the CA1 pyramidal region of the hippocampus. These results suggest genetic ablation of neuronal mCa2+ efflux is sufficient to increase tau pathology with aging.
Figure 3.

Loss of NCLX increases tau-pathology
(A) Representative western blots of soluble total tau (HT7), and phosphorylated tau at residues S202/T205 (AT8), T231/S235 (AT180), T181 (AT270), and S396 (PHF13) in cortex homogenates of 16-month-old mice, n = 3 for all groups.
(B–E) Densitometric analysis of western blots shown in Figure 3A expressed as fold-change versus Camk2a-Cre con. Corrected to a loading control tubulin.
(F) Representative immunohistochemical staining for total tau (HT7) and phospho-tau T231/S235 (AT180) in hippocampus of NCLX-nKO and control mice; scale bar = 50 μM.
(G and H) Quantification of the integrated optical density area of HT7 and AT180 immunoreactivity, n = 4 for all groups. All data presented as mean ± SEM; ∗∗p<0.01, ∗p<0.05; two-tailed, unpaired t-test.
Neuronal loss of mCa2+ efflux leads to redox imbalance, decreased synaptic stability and neuronal loss
Increased lipid peroxidation is indicative of oxidative stress that occurs early in neurodegenerative diseases, such as AD.50,51 To determine the effect of neuronal loss of mCa2+ efflux on redox status, we examined lipid peroxidation by 4-hydroxy-2-nonenal (4-HNE) staining in 16-month-old NCLX-nKO brains and controls. NCLX-nKO mice displayed a ∼35% increase in 4-HNE staining in the cortex, as compared to controls (Figures 4A and 4B).
Figure 4.

Neuronal loss of mCa2+ efflux leads to redox imbalance, decreased synaptic stability and neuronal loss
(A) Representative images of 4-HNE immunohistochemical staining in cortex and hippocampus of 16-month-old NCLX-nKO and control mice.
(B) Percent change in 4-HNE-integrated optical density area corrected to Camk2a-Cre controls. N = 4 for all groups, scale bar = 50 μM.
(C) Western blots for SYP and PSD-95 expression in tissue isolated from the cortex of 16-month-old-mice, n = 3 for all groups.
(D and E) Densitometric analysis of western blots shown in Figure 4C, expressed as fold change versus Camk2a-Cre con. corrected to loading control tubulin.
(F) Representative image of Nissl staining in cortex and hippocampus of 16-month-old mice to detect neuronal density.
(G and H) Quantitative analysis of Nissl positive cells in cortex and hippocampus areas of brain sections expressed as percent change versus Camk2a-Cre controls. n = 4 for all groups, scale bar = 50 μM.
(I) Western blots for GFAP and IBA1 expression in cortex of 16-month-old-mice, n = 3 for all groups.
(J and K) Densitometric analysis of western blots shown in Figure 4I, expressed as fold change versus Camk2a-Cre con. corrected to loading control β-actin. All data presented as mean ± SEM; ∗∗p<0.01, ∗p<0.05; two-tailed, unpaired t-test.
AD is characterized by loss of synaptic function.52,53,54 Loss of NCLX in primary hippocampal neurons is reported to cause massive synaptic impairment and deficits in long-term potentiation.43 Considering the important role of NCLX in regulating neuronal transmission, we next examined how loss of neuronal NCLX affects synaptic integrity and stability. Postsynaptic density protein 95 (PSD-95) is a major synaptic scaffold protein regulating glutamate receptor trafficking and localization and plays an important role in synaptic plasticity, development, and learning.55,56 Synaptophysin (SYP) is a highly abundant presynaptic vesicular membrane glycoprotein implicated in biogenesis and endocytosis of synaptic vesicles, synapse formation, and regulation of synaptic transmission.57,58 Loss of either PSD-95 or SYP is indicative of synaptic instability and is an early marker of neurodegeneration.59,60 Western blot analysis revealed significant decreases in both SYP and PSD-95 expression in the frontal cortex of NCLX-nKO mice (Figures 4C–4E). These findings suggest that loss of neuronal mCa2+ efflux alone promotes synaptic dysfunction.
Next, we examined if NCLX-nKO mice experience neuronal loss, we performed Nissl staining using cresyl violet acetate. The neuronal density was measured in 16-month-old NCLX-nKO and controls brain section. NCLX-nKO mice exhibited significantly reduced Nissl-positive staining in the cortex and hippocampus relative to controls. (Figures 4F–4H). Increased glial cell reactivity is usually associated with the death of neurons and the activation of astrocytes and microglia is a critical pro-inflammatory mechanisms in AD59,60 that can be evaluated by the expression of biomarkers. We analyzed protein levels of the glial fibrillary acidic protein (GFAP), a marker of astrocyte reactivity,61 and Ionized calcium-binding adaptor molecule 1 (Iba1), a marker for microglia activation. The expression of Iba1 was increased in brain homogenates isolated from NCLX-nKO mice compared to controls, without significant changes in GFAP expression (Figures 4I–4K), suggesting that loss of neuronal NCLX may elicit glial activation.
Loss of NCLX increases mCa2+ overload, aggregate formation, and cell death
To evaluate how the loss of NCLX contributes to AD progression and mitochondrial dysfunction, we generated stable NCLX knockout (Slc8b1 deletion; NCLX−/−) neuroblastoma (N2a) cell lines using CRISPR/SpCas9. SpCas9 was targeted to exon 8 of Slc8b1 by using a double guide RNA strategy (Figure 5A). After hygromycin selection, clonal NCLX−/− cell lines were examined for loss of mRNA by qPCR. NCLX−/− cell lines showed a significant loss of NCLX expression compared to wild-type (WT) N2a controls (Figure 5B). Next, matrix Ca2+ levels in NCLX−/− and WT cells were measured in a permeabilized cell system using the ratiometric reporter Fura2 (Figures 5C and 5D). Equal numbers of cells from both groups were treated with digitonin and thapsigargin and the protonophore, FCCP was added after reaching a steady state recording, to collapse ΔΨ and initiate the release of all matrix-free Ca2+ (Figure 5C). Consistent with previous study,43 quantification of mCa2+ content showed that loss of NCLX causes a significant increase in mCa2+ content compared to WT (Figure 5D). Next, we employed a mCa2+ retention capacity assay (mitoCRC) using the ratiometric reporters FuraFF (Ca2+) and added repetitive boluses of 10-μM Ca2+ until the collapse of ΔΨ. NCLX−/− cells showed decreased mitoCRC by ∼65% compared to WT cells, suggesting increased susceptibility to Ca2+-dependent permeability transition pore opening (Figures 5E and 5F). Overall, these data suggest that loss of NCLX causes mCa2+ overload and decreases CRC.
Figure 5.

Loss of NCLX increases mCa2+ overload, aggregate formation, and cell death
(A) Schematic for generation of NCLX knockout cell line (NCLX−/−) using CRISPR/SpCas9.
(B) NCLX mRNA expression in NCLX−/− and controls (WT) N2a cells, corrected to the housekeeping gene, Rps13; expressed as fold change versus control, n = 3 for both groups. All data presented as mean ± SEM; ∗∗∗∗p<0.001; two-tailed, unpaired t-test.
(C) Representative traces for basal mCa2+ content.
(D and E) (D) Quantification of mCa2+ content, n = 3 for both groups. All data presented as mean ± SEM; ∗∗p<0.001; two-tailed, unpaired t-test(E). Representative recordings of mCa2+ retention capacity.
(F and G) (F)Percent change in mCa2+ retention capacity versus N2a control cells, n = 3 for both groups. All data presented as mean ± SEM; ∗p<0.05; two-tailed, unpaired t-test(G). Representative images of intracellular protein aggregates in NCLX−/− and WT cells stained with Proteostat aggresome detection reagent (red) and Hoechst 33,342 nuclear stain (blue), scale bars = 20-μm.
(H) Total aggregates per cell, n = 96 for NCLX−/− and n = 58 for WT N2a control. All data presented as mean ± SEM; ∗∗∗∗p<0.001; two-tailed, unpaired t-test.
(I and J) NCLX−/− and WT cells were assessed for plasma membrane rupture, Sytox Green, after treatment with (I). Ionomycin (10–40 μM), (J). thapsigargin (10–50 μM), n = 10 for both groups. All data presented as mean ± SEM; ∗∗∗p<0.001, ∗∗p<0.01, ∗p<0.05; two-way ANOVA with Sidak’s multiple comparisons test.
Protein misfolding and aggregation is a hallmark feature of neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, dementia with Lewy bodies, and Amyotrophic Lateral Sclerosis, among others. To test if loss of NCLX impacts protein aggregation, we performed a ProteoStat Aggresome detection assay. NCLX−/− cells exhibited a significantly increased abundance (∼50% increase) of intracellular ‘amyloid-like’ aggregates compared to WT (Figures 5G and 5H). These results suggest that loss of NCLX alone can drive protein aggregation.
mCa2+ overload is strongly associated with cell death. To further evaluate how loss of NCLX modulates cell death, cells from both groups were treated with varying concentrations of necrotic agonists ionomycin (general intracellular calcium stress,10–40 μM) and thapsigargin (ER Ca2+ stress, 10–50 μM) and examined plasma membrane rupture, a hallmark of cell death using Sytox Green. NCLX−/− cells demonstrated significantly increased plasma membrane rupture compared to WT at most doses (Figures 5I and 5J). Altogether, these results suggest that the inhibition of mCa2+ efflux causes mCa2+ overload, amyloid-like protein aggregation, and potentiates cell death.
Discussion
In this study, we utilized a genetic loss-of-function model to show that impaired mitochondrial calcium efflux is sufficient to promote an AD-like phenotype. We have previously reported that diminished mCa2+ efflux contributes to mCa2+ overload and accelerates pathogenesis of AD, including cognitive decline, in Aβ and tau genetic models. In addition, we previously reported that rescue of neuronal NCLX expression was sufficient to attenuate cognitive deficits and neuropathology in 3xTg-AD mouse model.18 It is important to note that 3xTg-AD mutant mice harbor three gene mutations associated with familial AD: Presenilin 1 (Psen1, M146V homozygous knock-in), amyloid beta precursor protein (APPswe, KM670/671NL transgene) and microtubule-associated protein tau (MAPT, P301L transgene) and that these mice demonstrate progressive mCa2+ overload with aging. Thus, cognitive decline is more rapid and profound in the 3xTg-AD mice after neuronal deletion of NCLX compared to NCLX-nKO as it is exacerbating pre-existing mCa2+ overload, a pathologic feature of disease. Intriguingly, here we found that loss of neuronal NCLX expression causes AD-like dysfunction, including memory loss, Aβ plaque burden, tau hyperphosphorylation, oxidative stress, and loss of synaptic integrity in aged mice (12–16 months old) in the absence of any AD or neurodegenerative genetic predisposition (i.e., no expression of familial AD mutant genes). Our study demonstrates that loss of neuronal NCLX and resulting mCa2+ overload causes neurodegenerative pathology and age associated cognitive decline.
Our hypothesis, that mCa2+ overload is a driver of AD-pathology, is supported by several studies. Recently, NCLX-knockout primary hippocampal neurons showed mCa2+ overload, depolarization of mitochondrial membrane potential and reduced calcium clearance during neuronal activity.43 Even more striking the same group showed that an NCLX loss-of-function mutation is linked with severe mental retardation and that loss of NCLX causes synaptic impairment and long-term plasticity deficits.43 These data strongly support our findings that NCLX-nKO mice experience an age-dependent decline in spatial memory and cued recall, moderate amyloid deposition, mild tau hyperphosphorylation, and synaptic remodeling. mCa2+ overload is a trigger for mPTP opening that leads to cell death.62,63,64,65 Inhibition of mPTP has shown beneficial effects against neuronal cell death resulting from glutamate excitotoxicity,66 traumatic brain injury,67 ischemia-reperfusion injury,68 Parkinson’s disease,69 and AD.62,70 The loss of either MCUB (a negative regulator of the mtCU)71 or MICU1 (gatekeeper of the mtCU at low cytosolic Ca2+ levels)72,73 promotes mCa2+ overload. Important to the current study, reduced expression of mtCU-associated proteins, including MICU1 and MCUB, is reported in the frontal cortex of sporadic AD patients and 3xTg-AD mice.18 In addition, loss of MICU1 (homozygous deletion of exon 1) results in sporadic neurological and muscular defects in humans, which is characterized by the mCa2+ overload, diminished metabolism, early muscle weakness, myofiber damage, impaired cognition, and an extrapyramidal movement disorder.74,75,76 Furthermore, MICU1 variants are implicated in congenital brain malformation, characterized by white matter abnormalities, cerebellar dysplasia, and acute encephalopathy.77 Recently, a parallel line of research has found myopathy with extrapyramidal symptoms in patients carrying nonsense mutations in the MICU1 gene (c.385C>T; p.(R129∗)),78 (c.52C>T; p.(Arg18∗) and c.553C>T; p.(Arg185∗)).79 Consistent with human studies, deletion of Micu1 in mice causes marked ataxia and muscular defects.80,81 Recently, our group reported that reducing mCa2+ influx by neuronal-specific deletion of MCU in the 3xTg-AD mouse model reduces AD progression.82 In summary, a growing body of evidence implicates mCa2+ exchange dysregulation in a plethora of neurodegenerative diseases.
However, numerous fundamental questions remain unanswered. For example, why are neurons in aged mice susceptible to mCa2+ overload and how is the loss of NCLX or remodeling of the mtCU coincide with the development of AD? What are the molecular mechanisms underlying the proteomic remodeling of mCa2 machinery? Furthermore, it remains enigmatic whether mCa2+ dysregulation is specific to AD or central to other neurodegenerative diseases. We hypothesize that increased mCa2+ content is a mechanism for ‘stressed’ neurons to meet their energetic demands, enhancing mitochondrial metabolism by augmenting dehydrogenase activity (PDH and αKGD), Krebs cycle flux, and ATP production.33,83 This early adaptive response ultimately becomes maladaptive through sustained stress, leading to mCa2+ overload and mitochondrial dysfunction. Previous research suggests that the calcium buffering capacity of mitochondria declines with aging, making mitochondria more prone to mCa2+ overload.84 These observations further support our hypothesis of mCa2+ mishandling during aging that ultimately can contribute to the development of neurodegenerative diseases.
It’s clear that mCa2+ overload is a primary upstream mechanism in AD-like pathology; yet it remains unknown how loss of NCLX results in Aβ and tau aggregation. There are likely many key contributors such as oxidative stress, metabolic dysfunction, and cell death that get activated in response to mCa2+ dysregulation, causing AD progression. It’s also possible that there is a feedback loop between mCa2+ dysregulation and AD-pathology. For example, it is reported that increased mCa2+ uptake and glutamate excitotoxicity lead to oxidative stress in a number of studies.72,85,86 This paradigm further corroborates our finding of increased lipid peroxidation following loss of neuronal NCLX. Intracellular Ca2+ signaling is reported to modulate BACE1 expression and turnover. Furthermore, reports suggest that oxidative stress increases BACE1 expression, Aβ accumulation, tau hyperphosphorylation, and synaptic dysfunction.87,88 Given the noted relationship between mCa2+ overload and ROS, its plausible that a maladaptive pathway may contribute to Aβ processing. Also, oxidative stress has been shown to impair respiration by reducing glucose uptake and limiting substrate availability.89 In addition, iCa2+ signaling is also believed to enhance reactive nitrogen species (RNS),90 which can impair mitochondrial metabolism through inhibitory S-nitrosylation of Krebs cycle enzymes such as aconitase, isocitrate dehydrogenase, alpha-ketoglutarate dehydrogenase91,92 and electron transport chain (ETC) complexes.93,94 The resulting dysfunctional mitochondrial metabolism can further disrupt axonal transport of mitochondria and other APP-associated proteins, including BACE-1.95,96 Of interest, impaired axonal transport is an early contributor of AD95,96 that has diverse effects including compromised mitochondrial quality control mechanisms, leading to protein aggregation, neuronal dysfunction, and cell death.97 We hypothesize that impaired mitochondrial quality control and protein aggregation can potentiate mitochondrial dysfunction such as oxidative stress and cell death, creating a vicious cycle underlying the progression of AD. In addition, the mechanism of synaptic dysfunction and gliosis in a mCa2+ overloaded state might be associated with Aβ deposition. It is known that Aβ impacts glial activation that can subsequently drive synapse dysfunction and neuronal death, and that vice versa glial cell activation can modulate Aβ production through several mechanisms. These mechanisms need to be explored in the future and might include changes in neuronal ionic homeostasis and an alternation of various transporters, exchangers (present on the plasma membrane, and ER), and Ca2+-handling proteins that are responsible for various neuronal functions.98,99 In conclusion, mCa2+ dysregulation is sufficient to perturb neuronal mitochondrial function and drive age-associated neurodegeneration and cognitive decline. This study, coupled with the reports mentioned above, advocates for correcting mCa2+ dysregulation by restoring NCLX-dependent mCa2+ efflux capacity as a promising therapeutic strategy in treating neurodegenerative diseases. The phosphorylation of NCLX by PKA, which potentiates NCLX mediated mCa2+ efflux, is effective in preventing dopamine-dependent demise of PINK1 deficient neurons,100 a cellular Parkinson’s model. In combination with past work demonstrating that genetic rescue of NCLX slows cognitive decline in mouse models of AD, these findings further support that modulation of mCa2+ handling, specifically via activation of NCLX, could be an effective strategy to treat neurodegenerative diseases.
Limitations of the study
This report suggests that impaired mCa2+ efflux is sufficient to drive Alzheimer’s disease-like, age-associated dementia. One limitation is that the current results did not identify the downstream molecular mechanisms by which loss of mCa2+ efflux results in neuropathology. Another caveat of this study is the utilization of mouse models that fail to recapitulate all the features of cognitive decline in humans. In general the field is hampered by a lack of animal models that faithfully capture non-familial or sporadic AD. Such models are needed to test if augmenting mCa2+ efflux is indeed a viable therapeutic strategy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Experimental models: Organisms/strains | ||
| NCLXfl/fl mice | Luongo et al.,48 | N/A |
| B6.Cg-Tg(Camk2a-cre)T291-Stl/J | The Jackson Laboratory, USA | Stock # 005359 |
| NCLXfl/fl x Camk2a-Cre | This Study | N/A |
| Antibodies | ||
| MCU (1:1000) | Sigma | Cat # HPA016480 |
| MCUb (1:1000) | Abgent | Cat # AP12355b |
| MICU1 (1:500) | Tomar et al.,101 | Custom generation by Yenzyme |
| MICU2 (1:500) | Abcam | Cat # ab101465 |
| MICU3 (1:500) | Sigma Aldrich | Cat # HPA024771 |
| EMRE (1:1000) | Santa Cruz Biotechnology Inc. | Cat # sc-86337 |
| NCLX (1:500) | Santa Cruz Biotechnology Inc. | Cat # sc-161921 |
| VDAC (1:2500) | Abcam | Cat # ab15895 |
| OxPhos Cocktail | Abcam | Cat # MS604 |
| Total APP (22C11, 1:1500) | Chemicon International | Cat # MAB348 |
| BACE1 (1:500) | Sigma | Cat # MAB5308 |
| ADAM10 (1:500) | Chemicon International | Cat # AB19026 |
| PS1 (1:500) | Sigma | Cat #S182 |
| Nicastrin (1:200) | Cell Signaling | Cat # 5665 |
| APH-1 (1:200) | Millipore | Cat # AB9214 |
| HT7 (1:200) | Thermo Fisher Scientific | Cat # MN1000 |
| AT180 (1:200) | Thermo Fisher Scientific | Cat # MN1040 |
| AT8 (1:200) | Thermo Fisher Scientific | Cat # MN1020 |
| AT270 (1:200) | Thermo Fisher Scientific | Cat # MN1050 |
| PHF13 (1:200) | Cell Signaling | Cat # 9632 |
| 4-HNE | Abcam | Cat # ab48506 |
| SYP (1:500) | Santa Cruz Biotechnology | Cat # sc-55507 |
| IBA-1 (1: 250) | Proteintech | Cat # 10904-1-AP |
| PSD-95 (1:250) | Invitrogen | Cat # MA1-045 |
| GFAP | Proteintech | Cat # 60190-1-Ig |
| Beta-Tubulin (1: 1000) | Abcam | Cat # ab6046 |
| Anti-Mouse (1:10000) | Licor | Cat # 925–68070 |
| Anti-Goat (1:10000) | Licor | Cat # 926-32214 |
| Anti-Rabbit (1:10,000) | Licor | Cat # 926–32211 |
| Oligonucleotides | ||
| Rps13-F | GCACCTTGAGAGGAACAGAA | N/A |
| Rps13-R | GAGCACCCGCTTAGTCTTATAG | N/A |
| Nclx-F | GCCATCTCCACTAACCTCAAA | N/A |
| Nclx-R | GGGTCTGAGAAAGCCACTAAA | N/A |
| Recombinant DNA | ||
| gRNA #1 | GGGCCTCTACGTGTTCTACGGTTTTAGAG CTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCG AGTCGGTGC |
N/A |
| gRNA #2 | CTCACCTGGTGTCTCCGATAGTTTTAGAG CTAGAAATAGCAAGTTAAAATAAGGCTAG TCCGTTATCAACTTGAAAAAGTGGCACCG AGTCGGTGC |
N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Paraformaldehyde | Sigma | Cat #P6148 |
| Vector Elite ABC HRP Kit | Vector Laboratory | Cat # PK-6100 |
| DAB Substrate Kit | BD Biosciences | Cat # 550880 |
| RIPA buffer | EMD-Millipore | Cat # 20-188 |
| SIGMAFAST™ Protease Inhibitor Cocktail | Sigma-Aldrich | Cat #S8830 |
| Phosphatase inhibitor | Sigma-Aldrich | Cat # 04906837001 |
| Formic acid | Sigma | Cat # 33015 |
| Human Aβ (1-40) ELISA Kit | Wako Chemicals USA | Cat # 298-64601 |
| Human Aβ (1-42) ELISA Kit | Wako Chemicals USA | Cat # 298-62401 |
| Protein Assay Dye Reagent | Bio-Rad | Cat # 22660 |
| PVDF Immobilon-FL membrane | EMD Millipore | Cat # IPFL00010 |
| Blocking buffer | Rockland | Cat # MB-070 |
| Dihydroethidium (DHE) | Thermo Fisher Scientific | Cat #D11347 |
| KCl | Sigma-Aldrich | Cat #P9333 |
| Cresyl Violet | Sigma-Aldrich | Cat# C5042 |
| Glacial acetic acid | Sigma-Aldrich | Cat# A6283 |
| KH2PO4 | Sigma-Aldrich | Cat #P5655 |
| MgCl2 | Sigma-Aldrich | Cat # 449172 |
| NaCl | Sigma-Aldrich | Cat # 746398 |
| DAPI | Thermo Fisher Scientific | Cat #P36981 |
| Software and algorithms | ||
| Fiji ImageJ software | Fiji ImageJ | https://imagej.net/software/fiji/ |
| Zen 2010 | Carl Zeiss | https://www.zeiss.com/microscopy/en/products/software/zeiss-zen.html |
| GraphPad Prism 9 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ Pro plus software | Meyer Instruments | https://www.meyerinst.com/mediacybernetics/image-pro-plus/ |
| PACKWIN software | Harvard Apparatus | https://www.harvardapparatus.com/packwin-software-panlab.html |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the corresponding author, John W. Elrod (elrod@temple.edu).
Materials availability
All reagents and mice generated in this study are available from the lead contact, J.W.E. (elrod@temple.edu).
Experimental model and subjectdetails
Neuronal-specific NCLX knockout (NCLXfl/fl x Camk2a-Cre) mouse
NCLX floxed mice were generated by our lab by acquiring targeted ES cells made by recombinant insertion of a construct containing loxP sites flanking exons 5 - 7 of the NCLX, Slc8b1 gene (ch12: 113298759- 113359493).48 NCLXfl/fl mice were crossed with Camk2a-Cre mice (expression of Cre recombinase directed by calcium/calmodulin-dependent protein kinase II alpha promoter, primarily in the prefrontal cortex and CA1 layer of the hippocampus)49 to generate neuron-specific NCLX knockouts (NCLX-nKO; NCLXfl/fl x Camk2a-Cre). Animal studies were approved by Temple University’s IACUC and followed AAALAC guidelines. We used both male and female mice of different ages (2, 6, 9, 15, and 16m, depending on the assay) for this study.
Method details
Immunohistochemistry
Mouse brains from NCLX-nKO (NCLXfl/fl x Camk2a-Cre) and control (Camk2a-Cre) mice were prepared for immunohistochemistry.18 In brief, brains were fixed in 4% paraformaldehyde overnight, embedded in paraffin, and sectioned. 6-μm thick coronal sections were then deparaffinized, hydrated, and blocked in 2% fetal bovine serum. The sections were incubated with primary antibody overnight at 4°C. The primary antibodies and dilutions were as follows: HT7 dilution 1:150 (Thermo Fisher Scientific, Catalog # MN1000), phospho-tau (pThr231) monoclonal AT180 dilution 1:50 (Thermo Fisher Scientific, Catalog # MN1040) and anti-4 hydroxynonenal antibody (4-HNE) dilution 1:20 (Abcam, Catalog # ab48506). The sections were then incubated with secondary antibodies and developed using the Vector Elite ABC (Avidin-Biotin Complex) system (Vector Laboratories Inc., Burlingame, CA).
Biochemical and western blot analysis
Brains harvested from NCLX-nKO (NCLXfl/fl x Camk2a-Cre) and control mice (Camk2a-Cre) were homogenized in 1x RIPA lysis buffer with SIGMAFAST™ Protease Inhibitor Cocktail and phosphatase inhibitor. Lysates were centrifuged and the supernatant (soluble fraction) collected. The pellet was further lysed in 70% formic acid, sonicated, and centrifuged to collect the insoluble fraction. The resulting supernatant (insoluble fraction) was neutralized with 6 N sodium hydroxide. The lysates were centrifuged for 45 min at 4°C, and the supernatant was used as the soluble fraction. To get the insoluble fraction, the pellet was further lysed in 70% formic acid, sonicated, and centrifuged for 45 min at 4°C. The resulting supernatant (representing the insoluble fraction) was neutralized with 6 N sodium hydroxide. The soluble and insoluble fractions were assayed for Aβ1–40 and Aβ1–42 levels using sandwich ELISA (Wako Chemicals USA, Inc.) according to manufacturer instructions. Data were presented as a percentage change of Aβ1–40 and Aβ1–42 relative to controls.
For Western blot analysis, the protein concentration in the soluble fraction was determined by a Bio-Rad Protein Assay Dye Reagent. Equal amounts of protein (25-30 μg) were resolved using SDS-polyacrylamide gel electrophoresis (SDS-PAGE). After transferring protein from the gel to the PVDF Immobilon-FL membrane, the membrane was incubated in blocking buffer for 1 hrat room temperature, followed by incubation with primary antibody at 4°C overnight and then with specific Licor IR secondary antibody for 1 hr at room temperature. Licor Odyssey system was used to scan all blots and all full-length western blots are available in Figure S3.
Memory tests
Mice from NCLX-nKO (NCLXfl/fl x Camk2a-Cre) and controls (Camk2a-Cre) at 6-, 9-, 12-, and 15-months of age were assessed in the following cognition function tests.
Y-maze of spontaneous alternation
The Y-maze was used to assess spontaneous alternation, a measure of spatial working memory in mice, by allowing the test animals to explore all three arms of the Y-shaped maze (San Diego Instruments, 32 cm (long) 610 cm (wide) with 26-cm walls) for 5 min. The mice were tested for the total number of arms entries and for the three-consecutive sequence of entries (i.e., 1, 2, 3, or 2, 3, 1, or 3,1,2) to calculate percentage alternations using the formula “total alternation number/(total number of entries-2) × 100”.18
Contextual and cued fear conditioning
This test took place in a fear-conditioning apparatus (StartFear, Panlab Harvard Apparatus, 25 cm height × 30 cm width × 25 cm depth) on two consecutive days as described.18 During the training phase (Day 1), animals were placed in the chamber, and baseline freezing was recorded during a 6-min time interval with three cycles of 30s of sound and 10 s of electric shock (1.5mA). During the memory test phase (Day 2), mice were assessed for two trials, contextual and cued memory. For the contextual test, mice spent 5 min in the same chamber used during training, but without tones or electric shocks, and freezing behavior was recorded. For the cued test, two hours following the contextual test mice were placed in the same chamber with a modified environment such as different walls, smells, lighting, flooring, sound and freezing behavior was recorded for 6 min. Differences in freezing time between groups were analyzed via PACKWIN (Panlab, Harvard Apparatus, USA).
Rotarod test
To assess motor coordination, mice from controls and NCLX-nKO groups were tested using the rotarod test. During the training phase (Day 1-3), mice were placed on the rotarod apparatus initially at speed 0 rpm (30 s) and then at a constant speed 4 rpm (60 s), up to 6 trials per day with a 30 min rest period between each trial. During the test phase (Day 4), the rotarod started at speed 0 and accelerated to 90 rpm over a period of 90 s. The latency to falling off the rod was recorded on each day.
Assessment of lipid peroxidation
To measure redox stress, we performed 4-hydroxy-2-nonenal (4-HNE) staining. Mouse brains were prepared for immunohistochemistry as described above. The deparaffinized brain tissue sections of 16-month-old NCLX-nKO and controls were subjected to endogenous peroxidase quenched with 5% H2O2 in methanol for 30 minutes, followed by washing thrice in TBS-X (TBS and 0.3% Triton X-100) buffer for 5 minutes. Sections were treated with blocking buffer (2 % fetal bovine serum in TBS-X for overnight at 4°C) and incubated with primary 4-HNE antibody (dilution 1:20) overnight at 4°C in a humidified chamber. The next day, after washing with TBS-X thrice for 5 minutes, tissues were incubated with a biotinylated anti-mouse IgG for 1 hour in a humidified chamber and subsequently developed using an avidin-biotin-peroxidase complex, and the peroxidase activity was visualized using a stable DAB (diaminobenzidine) solution. Immunoreactivity was visualized using a light microscope and images were quantified using the Image-Pro Plus software.
Nissl staining
Nissl staining to examine neuronal density was performed in NCLX-nKO and control brain sections using 0.5% Cresyl violet acetate solution that stains Nissl substance in the cytoplasm of neurons.102 Imaging was performed using a whole slide imager Olympus VS-110 at the 40x resolution. Neuronal density was calculated in a given area of cells/mm2 using Fiji Image J software and presented as percentage change vs. Camk2a-Cre controls.
Cell culture and generation of NCLX knockout cells
Mouse neuroblastoma Neuro-2a cells (N2a) were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (peak serum) and 1% penicillin/streptomycin (gibco) at 37 °C in the presence of 5% CO2. For the generation of NCLX knockout (NCLX-/-) cells, N2a cells were transfected with a mammalian expression plasmid (VectorBuilder) encoding 2 gRNA’s under the direction of the U6 promoter, human codon-optimized cas9 (also known as SpCas9) under the direction of the CBh promoter (CMV early enhancer fused to modified chicken β-actin promoter), and hygromycin resistance via electroporation. 48 hours following electroporation, hygromycin was added to culture media (200 μg/ml). Clonal populations were grown up from the bulk transfected population and screened via qPCR.
qPCR mRNA analysis
RNA was extracted using the Qiagen RNeasy kit. cDNA was generated from 1ug of RNA using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was conducted according to manufacturer instructions (PowerUp SYBR Green Master Mix, Applied Biosystems). All reactions were run in triplicate and RPS13 was used as a housekeeping gene. For validation of NCLX knock-out cell lines, Exon-spanning qPCR primers were designed to amplify the junction between exons 8 and 9. 2ˆΔΔCt was calculated to determine NCLX mRNA expression relative to WT control.
Quantification of mCa2+ content and retention capacity
For evaluation of mCa2+ retention capacity and content, cells were cultured and transferred to an intracellular-like medium (120mM KCl, 10mM NaCl, 1mM KH2PO4, 20mM HEPES-Tris, pH 7.2) containing digitonin (80-μg/ml) to permeabilize the plasma membrane, thapsigargin (3 μM) to inhibit SERCA so that the movement of Ca2+ was only influenced by mitochondria, protease inhibitors (Sigma EGTA-Free cocktail), and succinate (5 mM). All solutions used were cleared of trace Ca2+ with Chelex 100 (Sigma). For evaluation of mCa2+ content: 2 million digitonin-permeabilized neuronal cells were loaded with the ratiometric calcium reporter Fura-2 (Invitrogen, 1μM) to monitor extramitochondrial Ca2+ and Ru360 (10 μM) and CGP-37157 (10 μM) to inhibit mCa2+ uptake and efflux, respectively. At 250 s. Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) was added to rapidly collapse mitochondrial membrane potential, causing Ca2+ to evacuate the matrix. Fluorescent signals were monitored in a spectrofluorometer (Delta RAM, Photon Technology Int.) at 380/510 excitation/emmision For evaluation of mCa2+ retention capacity cells were transferred to an intracellular-like medium containing thapsigargin and digitonin and loaded with the ratiometric Ca2+ reporter FuraFF (Cayman Chemical Company, 1 μM) to monitor extramitochondrial Ca2+ and JC-1 (Enzo Life Sciences, 4.8 μM) to monitor mitochondrial membrane potential. At 400s a repetitive series of 10μM Ca2+ boluses were added at the indicated time points until spontaneous Ca release, at which point 1 μM FCCP was added.
Membrane rupture assay
The propensity for neuronal death was evaluated using membrane rupture upon exposure to apoptotic agonists. Membrane rupture was quantified using sytox green (life technologies), a cell impermeant dye that enters the cell upon membrane rupture and intercalates DNA causing > 500-fold increase in fluorescent emission. Equal numbers of cells were plated in each well of a 96-well plate. After 24 hours, culture media was replaced with calcium ionophore ionomycin or thapsigargin at the indicated concentrations. After 24 hours, cells were loaded with 10 μM Sytox green for 30 minat 37 °C and measured the fluorescence at 504/523 ex/em using a Tecan Infinite M1000 Pro plate reader. Data are normalized to vehicle control to avoid any differences in cell numbers between the groups.
Detection of protein aggregation
Detection of protein aggregation was done using Proteostat protein aggregation kit (Enzo Life Sciences) according to manufacturer instructions. Proteostat recognizes the β-sheet structure of aggregated and misfolded proteins. In brief, cells were grown on 35 mm glass bottom cell culture dishes, fixed with 4% paraformaldehyde, permeabilized using a buffer containing 0.5% Triton X-100, 3mM EDTA, pH 8.0, and stained using proteostat aggresome detection dye and Hoechst 33342 nuclear counterstain for 30 min at RT. Stained cells were imaged on a ZEISS LSM 900 microscope using a 40x objective with appropriate laser settings for aggresome and nuclear signals. Proteostat staining intensity per cell was quantified using CellProfiler.
Quantification and statistical analysis
For statistical analysis, Graph Pad Prism 9.0 software was used. All experiments were performed thrice, and results were presented as mean ± SEM. Where appropriate, column analyses were performed using an unpaired, two-tailed t-test or two-way ANOVA multiple comparisons testing for an age effect with Dunnett’s post-hoc test for comparison to age 6 months and comparison of genotype across all ages using Bonferroni’s multiple comparisons tests. Results were significant if p values < 0.05 (95% confidence interval).
Acknowledgments
We thank Trevor Tierney and Alycia Hildebrand for technical assistance in the Elrod Lab. The graphical abstract was created with BioRender.com. Support for this work was provided by grants from the NIH: R01NS121379, R01HL136954, R01HL142271, 3R01HL123966-05S1, P01HL147841, P01HL134608; AHA: 20EIA35320226, and Pennsylvania Department of Health CURE Award (420792) to J.W.E.; NIH R00AG065445, P30 AG072947 to P.J., and NIH R00DK120876 to D.T.
Author contributions
J.W.E. conceived the project; J.W.E. and P.J. contributed to study design, data analysis, and writing the paper; P.J., H.C., A.K., D.T., and D.K. performed experiments, data collection, and interpretation, J.W.E., P.J., H.C., and D.T. edited the manuscript and provided expertise with data interpretation.
Declaration of interests
The authors declare no competing financial interests related to this work. J.W.E. is a paid consultant for Mitobridge, Inc., An Astellas Company.
Published: February 28, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.106296.
Supplemental information
Data and code availability
-
•
The data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Harman D. The biologic clock: the mitochondria? J. Am. Geriatr. Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 2.Sun N., Youle R.J., Finkel T. The mitochondrial basis of aging. Mol. Cell. 2016;61:654–666. doi: 10.1016/j.molcel.2016.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack C.R., Jr., Bennett D.A., Blennow K., Carrillo M.C., Dunn B., Haeberlein S.B., Holtzman D.M., Jagust W., Jessen F., Karlawish J., et al. NIA-AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mehta D., Jackson R., Paul G., Shi J., Sabbagh M. Why do trials for Alzheimer's disease drugs keep failing? A discontinued drug perspective for 2010-2015. Expert Opin. Investig. Drugs. 2017;26:735–739. doi: 10.1080/13543784.2017.1323868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Panza F., Lozupone M., Watling M., Imbimbo B.P. Do BACE inhibitor failures in Alzheimer patients challenge the amyloid hypothesis of the disease? Expert Rev. Neurother. 2019;19:599–602. doi: 10.1080/14737175.2019.1621751. [DOI] [PubMed] [Google Scholar]
- 6.Giannakopoulos P., Herrmann F.R., Bussière T., Bouras C., Kövari E., Perl D.P., Morrison J.H., Gold G., Hof P.R. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 7.Guillozet A.L., Weintraub S., Mash D.C., Mesulam M.M. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch. Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 8.Mostafavi S., Gaiteri C., Sullivan S.E., White C.C., Tasaki S., Xu J., Taga M., Klein H.U., Patrick E., Komashko V., et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer's disease. Nat. Neurosci. 2018;21:811–819. doi: 10.1038/s41593-018-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neff R.A., Wang M., Vatansever S., Guo L., Ming C., Wang Q., Wang E., Horgusluoglu-Moloch E., Song W.M., Li A., et al. Molecular subtyping of Alzheimer's disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv. 2021;7:eabb5398. doi: 10.1126/sciadv.abb5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alzheimer's Association Calcium Hypothesis Workgroup Calcium Hypothesis of Alzheimer's disease and brain aging: a framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement. 2017;13:178–182.e17. doi: 10.1016/j.jalz.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Bezprozvanny I., Mattson M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Briggs C.A., Chakroborty S., Stutzmann G.E. Emerging pathways driving early synaptic pathology in Alzheimer's disease. Biochem. Biophys. Res. Commun. 2017;483:988–997. doi: 10.1016/j.bbrc.2016.09.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khachaturian Z.S. Calcium, membranes, aging, and Alzheimer's disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989;568:1–4. doi: 10.1111/j.1749-6632.1989.tb12485.x. [DOI] [PubMed] [Google Scholar]
- 14.Swerdlow R.H. The mitochondrial hypothesis: dysfunction, bioenergetic defects, and the metabolic link to Alzheimer's disease. Int. Rev. Neurobiol. 2020;154:207–233. doi: 10.1016/bs.irn.2020.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jadiya P., Garbincius J.F., Elrod J.W. Reappraisal of metabolic dysfunction in neurodegeneration: focus on mitochondrial function and calcium signaling. Acta Neuropathol. Commun. 2021;9:124. doi: 10.1186/s40478-021-01224-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teo E., Ravi S., Barardo D., Kim H.S., Fong S., Cazenave-Gassiot A., Tan T.Y., Ching J., Kovalik J.P., Wenk M.R., et al. Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. Elife. 2019;8:e50069. doi: 10.7554/eLife.50069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chou J.L., Shenoy D.V., Thomas N., Choudhary P.K., Laferla F.M., Goodman S.R., Breen G.A.M. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer's disease. J. Proteomics. 2011;74:466–479. doi: 10.1016/j.jprot.2010.12.012. [DOI] [PubMed] [Google Scholar]
- 18.Jadiya P., Kolmetzky D.W., Tomar D., Di Meco A., Lombardi A.A., Lambert J.P., Luongo T.S., Ludtmann M.H., Praticò D., Elrod J.W. Impaired mitochondrial calcium efflux contributes to disease progression in models of Alzheimer's disease. Nat. Commun. 2019;10:3885. doi: 10.1038/s41467-019-11813-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Völgyi K., Badics K., Sialana F.J., Gulyássy P., Udvari E.B., Kis V., Drahos L., Lubec G., Kékesi K.A., Juhász G. Early presymptomatic changes in the proteome of mitochondria-associated membrane in the APP/PS1 mouse model of Alzheimer's disease. Mol. Neurobiol. 2018;55:7839–7857. doi: 10.1007/s12035-018-0955-6. [DOI] [PubMed] [Google Scholar]
- 20.Cai C., Lin P., Cheung K.H., Li N., Levchook C., Pan Z., Ferrante C., Boulianne G.L., Foskett J.K., Danielpour D., Ma J. The presenilin-2 loop peptide perturbs intracellular Ca2+ homeostasis and accelerates apoptosis. J. Biol. Chem. 2006;281:16649–16655. doi: 10.1074/jbc.M512026200. [DOI] [PubMed] [Google Scholar]
- 21.Cheung K.H., Mei L., Mak D.O.D., Hayashi I., Iwatsubo T., Kang D.E., Foskett J.K. Gain-of-function enhancement of IP3 receptor modal gating by familial Alzheimer's disease-linked presenilin mutants in human cells and mouse neurons. Sci. Signal. 2010;3:ra22. doi: 10.1126/scisignal.2000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheung K.H., Shineman D., Müller M., Cárdenas C., Mei L., Yang J., Tomita T., Iwatsubo T., Lee V.M.Y., Foskett J.K. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rybalchenko V., Hwang S.Y., Rybalchenko N., Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int. J. Biochem. Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- 24.Stutzmann G.E., Smith I., Caccamo A., Oddo S., Laferla F.M., Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J. Neurosci. 2006;26:5180–5189. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leissring M.A., Akbari Y., Fanger C.M., Cahalan M.D., Mattson M.P., LaFerla F.M. Capacitative calcium entry deficits and elevated luminal calcium content in mutant presenilin-1 knockin mice. J. Cell Biol. 2000;149:793–798. doi: 10.1083/jcb.149.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo A.S., Cheng I., Chung S., Grenfell T.Z., Lee H., Pack-Chung E., Handler M., Shen J., Xia W., Tesco G., et al. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- 27.Green K.N., Demuro A., Akbari Y., Hitt B.D., Smith I.F., Parker I., LaFerla F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell Biol. 2008;181:1107–1116. doi: 10.1083/jcb.200706171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan S.L., Mayne M., Holden C.P., Geiger J.D., Mattson M.P. Presenilin-1 mutations increase levels of ryanodine receptors and calcium release in PC12 cells and cortical neurons. J. Biol. Chem. 2000;275:18195–18200. doi: 10.1074/jbc.M000040200. [DOI] [PubMed] [Google Scholar]
- 29.Stutzmann G.E., Caccamo A., LaFerla F.M., Parker I. Dysregulated IP3 signaling in cortical neurons of knock-in mice expressing an Alzheimer's-linked mutation in presenilin1 results in exaggerated Ca2+ signals and altered membrane excitability. J. Neurosci. 2004;24:508–513. doi: 10.1523/JNEUROSCI.4386-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tu H., Nelson O., Bezprozvanny A., Wang Z., Lee S.F., Hao Y.H., Serneels L., De Strooper B., Yu G., Bezprozvanny I. Presenilins form ER Ca2+ leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baughman J.M., Perocchi F., Girgis H.S., Plovanich M., Belcher-Timme C.A., Sancak Y., Bao X.R., Strittmatter L., Goldberger O., Bogorad R.L., et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Stefani D., Raffaello A., Teardo E., Szabò I., Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Denton R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- 34.Duchen M.R. Mitochondria and calcium: from cell signalling to cell death. J. Physiol. 2000;529:57–68. doi: 10.1111/j.1469-7793.2000.00057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calvo-Rodriguez M., Hou S.S., Snyder A.C., Kharitonova E.K., Russ A.N., Das S., Fan Z., Muzikansky A., Garcia-Alloza M., Serrano-Pozo A., et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat. Commun. 2020;11:2146. doi: 10.1038/s41467-020-16074-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Granatiero V., Pacifici M., Raffaello A., De Stefani D., Rizzuto R. Overexpression of mitochondrial calcium uniporter causes neuronal death. Oxid. Med. Cell. Longev. 2019;2019:1681254. doi: 10.1155/2019/1681254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qiu J., Tan Y.W., Hagenston A.M., Martel M.A., Kneisel N., Skehel P.A., Wyllie D.J.A., Bading H., Hardingham G.E. Mitochondrial calcium uniporter Mcu controls excitotoxicity and is transcriptionally repressed by neuroprotective nuclear calcium signals. Nat. Commun. 2013;4:2034. doi: 10.1038/ncomms3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao J., Irwin R.W., Zhao L., Nilsen J., Hamilton R.T., Brinton R.D. Mitochondrial bioenergetic deficit precedes Alzheimer's pathology in female mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chang K.T., Niescier R.F., Min K.T. Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc. Natl. Acad. Sci. USA. 2011;108:15456–15461. doi: 10.1073/pnas.1106862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryan K.C., Ashkavand Z., Norman K.R. The role of mitochondrial calcium homeostasis in Alzheimer's and related diseases. Int. J. Mol. Sci. 2020;21:9153. doi: 10.3390/ijms21239153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Britti E., Delaspre F., Tamarit J., Ros J. Mitochondrial calcium signalling and neurodegenerative diseases. Neuronal Signal. 2018;2:NS20180061. doi: 10.1042/NS20180061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palty R., Silverman W.F., Hershfinkel M., Caporale T., Sensi S.L., Parnis J., Nolte C., Fishman D., Shoshan-Barmatz V., Herrmann S., et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stavsky A., Stoler O., Kostic M., Katoshevsky T., Assali E.A., Savic I., Amitai Y., Prokisch H., Leiz S., Daumer-Haas C., et al. Aberrant activity of mitochondrial NCLX is linked to impaired synaptic transmission and is associated with mental retardation. Commun. Biol. 2021;4:666. doi: 10.1038/s42003-021-02114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hedskog L., Pinho C.M., Filadi R., Rönnbäck A., Hertwig L., Wiehager B., Larssen P., Gellhaar S., Sandebring A., Westerlund M., et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer's disease and related models. Proc. Natl. Acad. Sci. USA. 2013;110:7916–7921. doi: 10.1073/pnas.1300677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Area-Gomez E., Del Carmen Lara Castillo M., Tambini M.D., Guardia-Laguarta C., de Groof A.J.C., Madra M., Ikenouchi J., Umeda M., Bird T.D., Sturley S.L., Schon E.A. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31:4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sarasija S., Laboy J.T., Ashkavand Z., Bonner J., Tang Y., Norman K.R. Presenilin mutations deregulate mitochondrial Ca(2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife. 2018;7:e33052. doi: 10.7554/eLife.33052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nichols M., Pavlov E.V., Robertson G.S. Tamoxifen-induced knockdown of the mitochondrial calcium uniporter in Thy1-expressing neurons protects mice from hypoxic/ischemic brain injury. Cell Death Dis. 2018;9:606. doi: 10.1038/s41419-018-0607-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luongo T.S., Lambert J.P., Gross P., Nwokedi M., Lombardi A.A., Shanmughapriya S., Carpenter A.C., Kolmetzky D., Gao E., van Berlo J.H., et al. The mitochondrial Na(+)/Ca(2+) exchanger is essential for Ca(2+) homeostasis and viability. Nature. 2017;545:93–97. doi: 10.1038/nature22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tsien J.Z., Chen D.F., Gerber D., Tom C., Mercer E.H., Anderson D.J., Mayford M., Kandel E.R., Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- 50.Bradley-Whitman M.A., Lovell M.A. Biomarkers of lipid peroxidation in Alzheimer disease (AD): an update. Arch. Toxicol. 2015;89:1035–1044. doi: 10.1007/s00204-015-1517-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butterfield D.A., Bader Lange M.L., Sultana R. Involvements of the lipid peroxidation product, HNE, in the pathogenesis and progression of Alzheimer's disease. Biochim. Biophys. Acta. 2010;1801:924–929. doi: 10.1016/j.bbalip.2010.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Almeida C.G., Tampellini D., Takahashi R.H., Greengard P., Lin M.T., Snyder E.M., Gouras G.K. Beta-amyloid accumulation in APP mutant neurons reduces PSD-95 and GluR1 in synapses. Neurobiol. Dis. 2005;20:187–198. doi: 10.1016/j.nbd.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 53.Benarroch E.E. Glutamatergic synaptic plasticity and dysfunction in Alzheimer disease: emerging mechanisms. Neurology. 2018;91:125–132. doi: 10.1212/WNL.0000000000005807. [DOI] [PubMed] [Google Scholar]
- 54.Jackson J., Jambrina E., Li J., Marston H., Menzies F., Phillips K., Gilmour G. Targeting the synapse in Alzheimer's disease. Front. Neurosci. 2019;13:735. doi: 10.3389/fnins.2019.00735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Migaud M., Charlesworth P., Dempster M., Webster L.C., Watabe A.M., Makhinson M., He Y., Ramsay M.F., Morris R.G., Morrison J.H., et al. Enhanced long-term potentiation and impaired learning in mice with mutant postsynaptic density-95 protein. Nature. 1998;396:433–439. doi: 10.1038/24790. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P., Lisman J.E. Activity-dependent regulation of synaptic strength by PSD-95 in CA1 neurons. J. Neurophysiol. 2012;107:1058–1066. doi: 10.1152/jn.00526.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cameron P.L., Südhof T.C., Jahn R., De Camilli P. Colocalization of synaptophysin with transferrin receptors: implications for synaptic vesicle biogenesis. J. Cell Biol. 1991;115:151–164. doi: 10.1083/jcb.115.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kwon S.E., Chapman E.R. Synaptophysin regulates the kinetics of synaptic vesicle endocytosis in central neurons. Neuron. 2011;70:847–854. doi: 10.1016/j.neuron.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Habib N., McCabe C., Medina S., Varshavsky M., Kitsberg D., Dvir-Szternfeld R., Green G., Dionne D., Nguyen L., Marshall J.L., et al. Disease-associated astrocytes in Alzheimer's disease and aging. Nat. Neurosci. 2020;23:701–706. doi: 10.1038/s41593-020-0624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Park J.S., Kam T.I., Lee S., Park H., Oh Y., Kwon S.H., Song J.J., Kim D., Kim H., Jhaldiyal A., et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer's disease. Acta Neuropathol. Commun. 2021;9:78. doi: 10.1186/s40478-021-01180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Caruso D., Barron A.M., Brown M.A., Abbiati F., Carrero P., Pike C.J., Garcia-Segura L.M., Melcangi R.C. Age-related changes in neuroactive steroid levels in 3xTg-AD mice. Neurobiol. Aging. 2013;34:1080–1089. doi: 10.1016/j.neurobiolaging.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Du H., Guo L., Fang F., Chen D., Sosunov A.A., McKhann G.M., Yan Y., Wang C., Zhang H., Molkentin J.D., et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat. Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Szalai G., Krishnamurthy R., Hajnóczky G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999;18:6349–6361. doi: 10.1093/emboj/18.22.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakagawa T., Zhu H., Morishima N., Li E., Xu J., Yankner B.A., Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 65.Luongo T.S., Lambert J.P., Yuan A., Zhang X., Gross P., Song J., Shanmughapriya S., Gao E., Jain M., Houser S.R., et al. The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 2015;12:23–34. doi: 10.1016/j.celrep.2015.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schinder A.F., Olson E.C., Spitzer N.C., Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J. Neurosci. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okonkwo D.O., Büki A., Siman R., Povlishock J.T. Cyclosporin A limits calcium-induced axonal damage following traumatic brain injury. Neuroreport. 1999;10:353–358. doi: 10.1097/00001756-199902050-00026. [DOI] [PubMed] [Google Scholar]
- 68.Uchino H., Elmér E., Uchino K., Lindvall O., Siesjö B.K. Cyclosporin A dramatically ameliorates CA1 hippocampal damage following transient forebrain ischaemia in the rat. Acta Physiol. Scand. 1995;155:469–471. doi: 10.1111/j.1748-1716.1995.tb09999.x. [DOI] [PubMed] [Google Scholar]
- 69.Matsuura K., Kabuto H., Makino H., Ogawa N. Cyclosporin A attenuates degeneration of dopaminergic neurons induced by 6-hydroxydopamine in the mouse brain. Brain Res. 1996;733:101–104. doi: 10.1016/0006-8993(96)00686-5. [DOI] [PubMed] [Google Scholar]
- 70.Caspersen C., Wang N., Yao J., Sosunov A., Chen X., Lustbader J.W., Xu H.W., Stern D., McKhann G., Yan S.D. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer's disease. FASEB J. 2005;19:2040–2041. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 71.Raffaello A., De Stefani D., Sabbadin D., Teardo E., Merli G., Picard A., Checchetto V., Moro S., Szabò I., Rizzuto R. The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J. 2013;32:2362–2376. doi: 10.1038/emboj.2013.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mallilankaraman K., Doonan P., Cárdenas C., Chandramoorthy H.C., Müller M., Miller R., Hoffman N.E., Gandhirajan R.K., Molgó J., Birnbaum M.J., et al. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patron M., Checchetto V., Raffaello A., Teardo E., Vecellio Reane D., Mantoan M., Granatiero V., Szabò I., De Stefani D., Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell. 2014;53:726–737. doi: 10.1016/j.molcel.2014.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Debattisti V., Horn A., Singh R., Seifert E.L., Hogarth M.W., Mazala D.A., Huang K.T., Horvath R., Jaiswal J.K., Hajnóczky G. Dysregulation of mitochondrial Ca(2+) uptake and sarcolemma repair underlie muscle weakness and wasting in patients and mice lacking MICU1. Cell Rep. 2019;29:1274–1286.e6. doi: 10.1016/j.celrep.2019.09.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lewis-Smith D., Kamer K.J., Griffin H., Childs A.M., Pysden K., Titov D., Duff J., Pyle A., Taylor R.W., Yu-Wai-Man P., et al. Homozygous deletion in MICU1 presenting with fatigue and lethargy in childhood. Neurol. Genet. 2016;2:e59. doi: 10.1212/NXG.0000000000000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Logan C.V., Szabadkai G., Sharpe J.A., Parry D.A., Torelli S., Childs A.M., Kriek M., Phadke R., Johnson C.A., Roberts N.Y., et al. Loss-of-function mutations in MICU1 cause a brain and muscle disorder linked to primary alterations in mitochondrial calcium signaling. Nat. Genet. 2014;46:188–193. doi: 10.1038/ng.2851. [DOI] [PubMed] [Google Scholar]
- 77.Wilton K.M., Morales-Rosado J.A., Selcen D., Muthusamy K., Ewing S., Agre K., Nickels K., Klee E.W., Ho M.L., Morava E. Developmental brain abnormalities and acute encephalopathy in a patient with myopathy with extrapyramidal signs secondary to pathogenic variants in MICU1. JIMD Rep. 2020;53:22–28. doi: 10.1002/jmd2.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bitarafan F., Khodaeian M., Amjadi Sardehaei E., Darvishi F.Z., Almadani N., Nilipour Y., Garshasbi M. Identification of a novel MICU1 nonsense variant causes myopathy with extrapyramidal signs in an Iranian consanguineous family. Mol. Cell. Pediatr. 2021;8:6. doi: 10.1186/s40348-021-00116-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kohlschmidt N., Elbracht M., Czech A., Häusler M., Phan V., Töpf A., Huang K.T., Bartok A., Eggermann K., Zippel S., et al. Molecular pathophysiology of human MICU1 deficiency. Neuropathol. Appl. Neurobiol. 2021;47:840–855. doi: 10.1111/nan.12694. [DOI] [PubMed] [Google Scholar]
- 80.Antony A.N., Paillard M., Moffat C., Juskeviciute E., Correnti J., Bolon B., Rubin E., Csordás G., Seifert E.L., Hoek J.B., Hajnóczky G. MICU1 regulation of mitochondrial Ca(2+) uptake dictates survival and tissue regeneration. Nat. Commun. 2016;7:10955. doi: 10.1038/ncomms10955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu J.C., Liu J., Holmström K.M., Menazza S., Parks R.J., Fergusson M.M., Yu Z.X., Springer D.A., Halsey C., Liu C., et al. MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep. 2016;16:1561–1573. doi: 10.1016/j.celrep.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jadiya P., Kolmetzky D.W., Tomar D., Thomas M., Khaledi S., Garbincius J.F., Hildebrand A.N., Elrod J.W. SSRN; 2021. Genetic Ablation of Neuronal Mitochondrial Calcium Uptake Halts Alzheimer's Disease Progression. [DOI] [Google Scholar]
- 83.Nichols B.J., Denton R.M. Towards the molecular basis for the regulation of mitochondrial dehydrogenases by calcium ions. Mol. Cell. Biochem. 1995;149–150:203–212. doi: 10.1007/BF01076578. [DOI] [PubMed] [Google Scholar]
- 84.Panel M., Ghaleh B., Morin D. Mitochondria and aging: a role for the mitochondrial transition pore? Aging Cell. 2018;17:e12793. doi: 10.1111/acel.12793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duan Y., Gross R.A., Sheu S.S. Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J. Physiol. 2007;585:741–758. doi: 10.1113/jphysiol.2007.145409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Starkov A.A., Chinopoulos C., Fiskum G. Mitochondrial calcium and oxidative stress as mediators of ischemic brain injury. Cell Calcium. 2004;36:257–264. doi: 10.1016/j.ceca.2004.02.012. [DOI] [PubMed] [Google Scholar]
- 87.Tong Y., Zhou W., Fung V., Christensen M.A., Qing H., Sun X., Song W. Oxidative stress potentiates BACE1 gene expression and Abeta generation. J. Neural. Transm. 2005;112:455–469. doi: 10.1007/s00702-004-0255-3. [DOI] [PubMed] [Google Scholar]
- 88.Tönnies E., Trushina E. Oxidative stress, synaptic dysfunction, and Alzheimer's disease. J. Alzheimers Dis. 2017;57:1105–1121. doi: 10.3233/JAD-161088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gandhi S., Wood-Kaczmar A., Yao Z., Plun-Favreau H., Deas E., Klupsch K., Downward J., Latchman D.S., Tabrizi S.J., Wood N.W., et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nakamura T., Lipton S.A. 'SNO'-Storms compromise protein activity and mitochondrial metabolism in neurodegenerative disorders. Trends Endocrinol. Metab. 2017;28:879–892. doi: 10.1016/j.tem.2017.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chouchani E.T., Hurd T.R., Nadtochiy S.M., Brookes P.S., Fearnley I.M., Lilley K.S., Smith R.A.J., Murphy M.P. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010;430:49–59. doi: 10.1042/BJ20100633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gupta K.J., Shah J.K., Brotman Y., Jahnke K., Willmitzer L., Kaiser W.M., Bauwe H., Igamberdiev A.U. Inhibition of aconitase by nitric oxide leads to induction of the alternative oxidase and to a shift of metabolism towards biosynthesis of amino acids. J. Exp. Bot. 2012;63:1773–1784. doi: 10.1093/jxb/ers053. [DOI] [PubMed] [Google Scholar]
- 93.Sun J., Morgan M., Shen R.F., Steenbergen C., Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ. Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 94.Zhang J., Jin B., Li L., Block E.R., Patel J.M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am. J. Physiol. Cell Physiol. 2005;288:C840–C849. doi: 10.1152/ajpcell.00325.2004. [DOI] [PubMed] [Google Scholar]
- 95.Stokin G.B., Lillo C., Falzone T.L., Brusch R.G., Rockenstein E., Mount S.L., Raman R., Davies P., Masliah E., Williams D.S., Goldstein L.S.B. Axonopathy and transport deficits early in the pathogenesis of Alzheimer's disease. Science. 2005;307:1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 96.Calkins M.J., Manczak M., Mao P., Shirendeb U., Reddy P.H. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum. Mol. Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cai Q., Tammineni P. Alterations in mitochondrial quality control in Alzheimer's disease. Front. Cell. Neurosci. 2016;10:24. doi: 10.3389/fncel.2016.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Boscia F., Pannaccione A., Ciccone R., Casamassa A., Franco C., Piccialli I., de Rosa V., Vinciguerra A., Di Renzo G., Annunziato L. The expression and activity of KV3.4 channel subunits are precociously upregulated in astrocytes exposed to Abeta oligomers and in astrocytes of Alzheimer's disease Tg2576 mice. Neurobiol. Aging. 2017;54:187–198. doi: 10.1016/j.neurobiolaging.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 99.Pannaccione A., Piccialli I., Secondo A., Ciccone R., Molinaro P., Boscia F., Annunziato L. The Na(+)/Ca(2+)exchanger in Alzheimer's disease. Cell Calcium. 2020;87:102190. doi: 10.1016/j.ceca.2020.102190. [DOI] [PubMed] [Google Scholar]
- 100.Kostic M., Ludtmann M.H.R., Bading H., Hershfinkel M., Steer E., Chu C.T., Abramov A.Y., Sekler I. PKA phosphorylation of NCLX reverses mitochondrial calcium overload and depolarization, promoting survival of PINK1-deficient dopaminergic neurons. Cell Rep. 2015;13:376–386. doi: 10.1016/j.celrep.2015.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tomar D., Dong Z., Shanmughapriya S., Koch D.A., Thomas T., Hoffman N.E., Timbalia S.A., Goldman S.J., Breves S.L., Corbally D.P., et al. MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep. 2016;15:1673–1685. doi: 10.1016/j.celrep.2016.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang L., Li J., Lin A. Assessment of neurodegeneration and neuronal loss in aged 5XFAD mice. STAR Protoc. 2021;2:100915. doi: 10.1016/j.xpro.2021.100915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.