

Abstract
Limited T cell persistence restrains chimeric antigen receptor (CAR)-T cell therapy in solid tumors. To improve persistence, T cells have been engineered to secrete proinflammatory cytokines, but other possible methods have been understudied. Runx3 has been considered a master regulator of T cell development, cytotoxic T lymphocyte differentiation, and tissue-resident memory T (Trm)-cell formation. A study using a transgenic mouse model revealed that overexpression of Runx3 promoted T cell persistence in solid tumors. Here, we generated CAR-T cells overexpressing Runx3 (Run-CAR-T cells) and found that Run-CAR-T cells had long-lasting antitumor activities and achieved better tumor control than conventional CAR-T cells. We observed that more Run-CAR-T cells circulated in the peripheral blood and accumulated in tumor tissue, indicating that Runx3 coexpression improved CAR-T cell persistence in vivo. Tumor-infiltrating Run-CAR-T cells showed less cell death with enhanced proliferative and effector activities. Consistently, in vitro studies indicated that AICD was also decreased in Run-CAR-T cells via downregulation of tumor necrosis factor (TNF) secretion. Further studies revealed that Runx3 could bind to the TNF promoter and suppress its gene transcription after T cell activation. In conclusion, Runx3-armored CAR-T cells showed increased antitumor activities and could be a new modality for the treatment of solid tumors.
Keywords: Runx3, CAR-T therapy, hepatocellular carcinoma, TNF, AICD
Graphical abstract
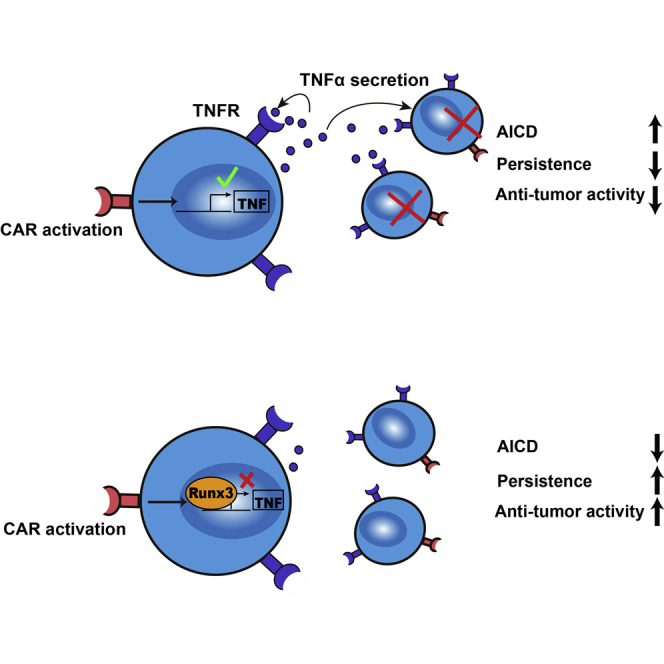
Improving the T cell persistence in tumor is an important issue in cellular immunotherapy. In this study, Z. Li and colleagues reported that enforced expression of Runx3 in CAR-T cells had achieved better tumor control with enhanced T cell persistence by reducing TNF-dependent activation-induced cell death.
Introduction
Chimeric antigen receptor (CAR)-engineered T cell therapy has produced highly encouraging successes in hematological malignancies, but the progress in treating solid tumors is still limited.1,2 One of the major obstacles to improving the efficacy of CAR-T cell therapy in solid tumors is the hostile tumor microenvironment (TME). Tumor cells, together with immunosuppressive cells, produce inhibitory signals and a metabolic state that constrain CAR-T cell function and survival.3,4
To overcome these challenges, CAR-T cells can be further engineered to secrete proinflammatory cytokines, such as IL-7,5,6 IL-12,7,8 or IL-18,9,10 to support T cell proliferation and effector functions in tumors. These cells are termed fourth-generation CAR-T cells or T cells redirected for universal cytokine killing (TRUCKs) by some researchers. Although TRUCKs exhibit increased efficacy in preclinical models, the potential for cytokine-associated activation of other immune cells leading to unexpected toxicity remains a concern for clinical use.11,12 CAR-T cells have also been armed with many other elements to augment T cell persistence and activity, but most of them have focused on a specific signaling pathway or inhibitory factor. The approach of coexpressing transcription factors, which would profoundly affect the cell state by reprogramming gene expression, is still poorly studied.
Runt domain-related transcription factor 3 (Runx3) is a master regulator in T cell development induced by IL-7 and IL-15.13,14 It has been identified to promote CD8+ T cell maturation15,16 and cytotoxic T lymphocyte (CTL) differentiation17,18 and to repress terminal CTL differentiation by transcriptional programming.19 A recent study showed that the Runx3-driven effector differentiation of T cells played a critical role in the control of viral infections and tumors.20 Interestingly, Runx3 is a central driver of tissue-resident memory T (Trm)-cell differentiation, which could reprogram cells and induce gene expression related to Trm-cell differentiation.21 In a B16 melanoma mouse model, Runx3 was required for the tumor residency of tumor-infiltrating lymphocytes (TILs), and overexpression of Runx3 in tumor-specific T cells led to improved tumor control.21 Hence, we hypothesized that exogenously expressing Runx3 could be a way to alter CAR-T cells to achieve long-term persistence and improved effector function against solid tumors.
Our previous clinical study indicated that the area under the copies approximate time curve from time zero to time of last measurable value (AUClast) of GPC3-redirected CAR-T cells in the peripheral blood tended to be higher in responders than in nonresponders, suggesting a close correlation between CAR-T cell persistence and therapeutic efficacy.22 In this study, we developed Runx3-coexpressing CAR-T cells (Run-CAR) and evaluated the antitumor potential of these CAR-T cells in vitro and in vivo.
Results
Run-CAR-T cells displayed increased antitumor activities in vitro and in vivo
We generated a Run-CAR construct by fusing the nucleotide fragments encoding human Runx3 and the GPC3-BBz CAR with the 2A peptide sequence (Figure 1A). The overexpression of Runx3 in Run-CAR-T cells was verified (Figures S1A and S1B). To examine tumor-killing activity, CAR-T cells were cocultured with SK-Hep1 (GPC3-negative), PLC/PRF/5 (GPC3-positive), or Huh-7 (GPC3-positive) cells. The expression of GPC3 in these cell lines was detected in a previous study.23 The Run-CAR-T cells induced modestly higher in vitro tumor lysis than conventional GPC3 CAR-T cells (Con-CAR-T cells) (Figure 1B). No significant difference between the Con- and Run-CAR-T cell products was found in the central memory or tissue-resident memory phenotype or in the expression of inhibitory receptors (Figures S1C–S1E). Additionally, the expression of Fas and FasL was not significantly different in vitro (Figure S1F). Cell expansion in the absence of antigen stimulation was similar for both CAR-T cells (Figure S1G). To investigate the antitumor activities of Run-CAR-T cells in vivo, we established a PLC/PRF/5 xenograft model, in which tumor growth was significantly suppressed during the early phase after Con-CAR-T cell infusion but became uncontrolled in the late stage.23 We speculated that long-term persistence of CAR-T cells should be essential to preventing relapse in this model. As expected, both Con-CAR-T cells and Run-CAR-T cells dramatically inhibited tumor growth in the first 2 weeks after infusion, but only Run-CAR-T cells had sustained antitumor effects and eliminated tumors (Figures 1C–1E). Furthermore, we explored the efficacy of Run-CAR-T cells against a high tumor burden. The results showed that Run-CAR-T cells exhibited more durable antitumor responses and led to a longer mouse survival time than Con-CAR-T cells (Figures 1F–1H). The levels of cytokines in the peripheral blood (PB) were low in the NSG mouse model. To further evaluate Run-CAR-T cell function in the early phase, tumor-bearing mice were administered a high dose of CAR-T cells to increase cytokine levels (Figure 1I). Except for that of IL-2, the levels of functional cytokines, including IL-6, IL-10, and IFNγ, in the PB peaked on day 3 during the first 6 days. The cytokine levels in the Run-CAR-T cell group were comparable to those in the Con-CAR-T cell group, with the exception of the decrease in IL-2 on day 1 (Figure 1J). In addition, the cytokine levels in tumor tissues were lower in the Run-CAR-T cell group (Figure 1K).
Figure 1.

Run-CAR-T cells displayed increased antitumor activities in vitro and in vivo
(A) Schematic of the Con- and Run-GPC3-CAR constructs. (B) Cytotoxic activities of Con- and Run-CAR-T cells. CAR-T cells were coincubated with target cells for 18 h, and specific lysis was measured by a standard LDH cytotoxicity assay. SK-Hep1 cells are GPC3 negative and were used as control cells. (C) PLC/PRF/5 cells (2.0 × 106) were subcutaneously implanted into mice (NOD-Prkdcscid Il2rgnull). 11 days after tumor implantation, when the tumor volume had reached approximately 150 mm3, the mice were intravenously infused with 2.5 × 106 CAR-T cells (n = 6). (D) Tumor size was monitored every 2–4 days.
(E) Tumor weight was measured at the end of the experiment. (F) PLC/PRF/5 cells (2.0 × 106) were subcutaneously implanted into mice. 14 days after tumor implantation, when the tumor volume had reached approximately 250 mm3, the mice were intravenously infused with 2.0 × 106 CAR-T cells (n = 6). (G and H) Tumor volume and animal survival were monitored every 2–3 days. (I) PLC/PRF/5 cells (2.0 ×106) were subcutaneously implanted into mice. 17 days after tumor implantation, when the tumor volume had reached approximately 350 mm3, the mice were intravenously infused with 1.0 × 107 CAR-T cells (n = 3). (J) Cytokine levels in the peripheral blood were measured at the indicated times. (K) Cytokine levels in tumor tissue were measured on day 6 after T cell infusion. Data are presented as the mean ± SD (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, nonsignificant; one-way ANOVA with Tukey’s post-test for multiple comparisons).
Enhanced persistence of Run-CAR-T cells in vivo
T cell survival in the PB is a general indicator of CAR-T cell persistence during hematological malignancy treatment. Indeed, more CAR-T cells were observed in the PB of mice in the Run-CAR-T cell group than in the Con-CAR-T cell group (Figure 2A) on day 24 after T cell infusion. Additionally, Run-CAR-T cells were more abundant in the bone marrow and spleen (Figures S2A and S2B). To investigate the survival of CAR-T cells in tumors, mice were sacrificed on day 14 after CAR-T cell infusion. As expected, more Run-CAR-T cells accumulated in the tumors, as confirmed by anti-CD3 immunohistochemistry (IHC) and an RNA scope assay targeting the GPC3-scFv (Figure 2B). Memory T cell differentiation has been associated with CAR-T cell persistence. We also examined the memory subsets of T cells in the PB, bone marrow, spleen, and tumor. Overexpression of Runx3 in CAR-T cells resulted in a modest yet statistically significant increase in the subset of effector memory T cells (TEM, CD95+, CD45RA–, CCR7–) and a reduction in the subset of terminal effector T cells (TTE, CD95+, CD45RA+, CCR7–) in all these tissues except the PB (Figure 2C). Consistent with this, CD8+ TILs in the Run-CAR-T cell group expressed higher levels of memory-associated genes, such as BACH2,24,25 BCL6,26 and TCF727,28,29 (Figure 2D). Exhaustion, however, was almost unchanged, as the CAR-T cells in the different groups expressed PD1 and TIM3 at similar levels (Figure S2C). We also tried to evaluate the Trm-cell subset in tumor tissues using the markers CD69 and CD103,30 which are classic markers for Trm cells distributed in the skin. However, no significant difference was found in TILs between the Con-CAR and Run-CAR groups (Figure S2D), probably because these markers might not be definitive for Trm cells in tumor tissues.31 Nevertheless, we found higher proportions of TILs expressing the proliferation marker Ki-67 (Figure 2E) and the effector molecule granzyme B (Figure 2F) in the Run-CAR-T cell group, which indicated that Run-CAR-T cells retained strong effector function in solid tumors. Correspondingly, a larger proportion of Con-CAR-T cells in tumors were stained by annexin V, suggesting a higher level of cell death in Con-CAR TILs (Figures 2G and 2H). These results clearly indicated that the addition of Runx3 greatly improved the persistence of Run-CAR-T cells in solid tumors.
Figure 2.

Run-CAR-T cells showed enhanced persistence in vivo
(A) T cells in the peripheral blood were stained with anti-CD3, anti-CD4, and anti-CD8 antibodies or an anti-9F2-scFv antibody and counted on day 24 after T cell infusion. (B) Tumor tissues were harvested on day 14 after T cell infusion, and CAR-T cell infiltration was determined by CD3 IHC staining and an RNA scope assay. (C–H) The memory phenotypes of T cells in the indicated tissues were determined on day 14 after T cell infusion; CD45RA+CCR7+CD95+ for TSCM cells, CD45RA–CCR7+CD95+ for TCM cells, CD45RA–CCR7–CD95+ for TEM cells, and CD45RA+CCR7-CD95+ for TTE cells. CD8+ TILs were isolated from tumors on day 14 after T cell infusion for (D) RNA extraction and real-time qPCR analysis, (E) Ki-67 and (F) granzyme B staining, and (G and H) cell death analysis. Data are presented as the mean ± SD (n = 3; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; one-way ANOVA with Tukey’s post hoc test for multiple comparisons).
Reduced AICD in Run-CAR-T cells
The differences in gene expression in CD8+ TILs between Con- and Run-CAR-T cells were analyzed by RNA sequencing. Gene set enrichment analysis (GSEA) revealed that the TCR signaling pathway, the Jak-Stat interleukin pathway, glycolysis, and DNA replication were highly active in Run-CAR TILs (Figure 3A), which suggested the strong effector function and high viability of these cells. Although the gene expression in TILs displayed a core residency signature21 compared with that in T cells in the spleen (Figure S3A), the difference between Con- and Run-CAR-T cells was not significant (Figure S3B). Intriguingly, the expression of apoptosis-associated genes was greatly changed (Figure 3B), which supported that Runx3 could regulate apoptosis in CAR-T cells in solid tumors.
Figure 3.

AICD in Run-CAR-T cells was reduced in vitro and in vivo
(A) CD8+ TILs were isolated on day 14 after T cell infusion for RNA-seq analysis and GSEA. (B) Heatmap of apoptosis-related genes differentially expressed between Con- and Run-CAR-T TILs. (C and D) CAR-T cells were serially stimulated with HCC cells for two rounds. The cell death level was measured by annexin V staining after the second round of coculture. (E and F) CAR-T cells were stimulated with a precoated GPC3 peptide (GCCT, 5 μg/mL) for 24 or 48 h, and the ROS level was measured using a DCFH-DA probe. (G–I) CAR-T cells were stimulated with a precoated GPC3 peptide (GCCT, 5 μg/mL) for 48 h, and the cleavage of caspases 3 and 8 was determined by western blotting. The relative protein levels of active caspase 3/8 compared with that of GAPDH were analyzed by grayscale quantification. Data are presented as the mean ± SD (n = 3; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; one-way ANOVA with Tukey’s post hoc test for multiple comparisons).
Then, we investigated whether Runx3 could regulate CAR-T cell death in vitro. Stimulation with target cells induced cell death in both CAR-T cell lines. Although the levels of cell death were comparable between the two CAR-T cell populations after the first-round stimulation (Figure S4A), more cell death occurred in Con-CAR-T cells than in Run-CAR-T cells after the second round of stimulation (Figures 3C and 3D). Consistent with the results for TILs, there was no significant difference in the expression of the checkpoint molecules PD1 and TIM3 (Figure S4B). In addition, the intracellular level of reactive oxygen species (ROS), which have been considered an activation-induced cell death (AICD) regulator in T cells,32,33 was significantly decreased, although no more than 10%, in Run-CAR-T cells upon antigen stimulation (Figures 3E and 3F). Upon tumor rechallenge, Run-CAR-T cells exhibited higher tumor-killing activity and cell expansion than Con-CAR-T cells in vitro (Figures S4E and S4F). Whereas the expression levels of the B cell lymphoma-2 (Bcl-2) family of proteins were similar (Figures S4C and S4D), the activation-induced cleavage of caspase 8 and caspase 3 was decreased in Run-CAR-T cells (Figures 3G and 3I). These data suggested that enforced expression of Runx3 played an inhibitory role in AICD and favored the long-term antitumor activities of CAR-T cells.
Runx3 regulated AICD via TNF signaling
As a pivotal regulator of T cell development and differentiation, Runx3 has been reported to regulate the expression of multiple genes through transcriptional reprogramming.18,19,21,34 However, few specific pathways orchestrated by Runx3 have been elucidated. Based on the analysis of cytokines in tumors and the TIL RNA sequencing data, we noticed that the expression of TNF, a cytokine associated with caspase 8 cleavage and AICD in T cells,35,36 was downregulated in Run-CAR-T cells (Figure 1, Figure 3K and 3B). Consistently, Run-CAR-T cells also produced lower levels of TNF upon tumor stimulation in vitro (Figures 4A and S4G). It has been reported that monocytes can be activated by T cells and secrete TNF.37 To investigate whether TNF levels were affected by monocytes, we further incubated CAR-T cells with tumor cells and monocytes from the same donor (Figure 4B). Less TNF was detected in the culture of the Run-CAR-T cell group (Figure 4C), suggesting that enforced expression of Runx3 could constrain TNF production in the presence of autologous monocytes. Profiling of the transcription factor-binding site with the JASPAR database revealed two potential Runx3 binding elements in the TNF promoter (Figure 4D). A ChIP-qPCR assay revealed that Runx3 could bind to the TNF promoter and that this binding was increased upon antigen stimulation (Figure 4E). Consistently, the mRNA level of TNF following stimulation was lower in Run-CAR-T cells (Figure 4F), suggesting that Runx3 regulated TNF expression at the transcriptional level. To determine whether Runx3 directly regulates the transcriptional activity of the TNF promoter, we generated a TNF promoter-luciferase reporter system in Jurkat cells. The reporter cells were transduced with the CAR or Run-CAR (Figures 4G and 4H) and stimulated with tumor cells or a precoated antigen. As expected, TNF promoter activity was significantly decreased in Run-CAR Jurkat cells (Figure 4I). These data demonstrated that Runx3 could repress the transcriptional activity of the TNF promoter and thus inhibit TNF expression.
Figure 4.

Runx3 regulated TNF expression at the transcriptional level
(A) CAR-T cells were stimulated with HCC cells at a ratio of 1:1 for 24 h, and cytokine levels in the supernatant were measured using a cytometric bead array. (B) Schematic of the experimental design in which CAR-T cells were cocultured with HCC cells and autologous monocytes. (C) Detection of cytokines in the supernatant after T cells and monocytes were cocultured with HCC cells at a ratio of 1:1:1 for 48 h using a cytometric bead array. (D) Two predicted Runx3 binding elements (RBEs, by the JASPAR database) were located in the promoter region of the human TNF gene. (E) CAR-T cells were stimulated with a precoated GPC3 peptide (GCCT, 5 μg/mL) for 24 h, and the binding between Runx3 and the TNF promoter was determined by ChIP-qPCR. Isotype IgG was used as a mock control. (F) CAR-T cells were stimulated with a precoated GPC3 peptide (GCCT, 5 μg/mL) for 24 or 48 h. The relative mRNA expression of TNF was measured by real-time qPCR. (G and H) Generation of Con-CAR- and Run-CAR-expressing Jurkat cells. The expression of the CAR and Runx3 was determined by flow cytometry and western blot analysis. (I) CAR-expressing Jurkat cells electroporated with TNF-promoter-luciferase and Renilla-luciferase vectors were stimulated with precoated GCCT (5 μg/mL) or HCC cells for 24 h. Luciferase activity was measured by a dual-luciferase reporter assay system. Data are presented as the mean ± SD (n = 3; ∗∗∗p < 0.001; one-way ANOVA with Tukey’s posttest for multiple comparisons).
To evaluate the effect of TNF on AICD in CAR-T cells, we used an anti-TNF monoclonal antibody (adalimumab) to block TNF binding with its receptors in the tumor rechallenge assay. TNF blockade partially inhibited AICD in Con-CAR-T cells to a level similar to that observed for Run-CAR-T cells after target cell stimulation, indicating the role of TNF in AICD induction (Figures 5A and 5B). To avoid any side effects of the anti-TNF antibody, we generated TNF-deficient CAR-T cells via CRISPR-Cas9-mediated knockout (KO). The number of TNF-positive cells was reduced from approximately 60% to 7% upon phorbol 12-myristate 13-acetate (PMA) and ionomycin stimulation following TNF KO (Figure 5C). As a control, IFNγ expression was not significantly influenced by knocking out TNF (Figure 5C). To further confirm the KO efficiency, we sequenced the edited TNF gene. The KO efficiencies of the three guide RNAs were 80%, 35%, and 13%, calculated by ICE analysis with the SYNTHEGO website (Figure S5A). TNF production was completely impaired in KO CAR-T cells, whereas IL-2 and INFγ were not significantly affected (Figures 5D and S5B). As expected, knocking out TNF reduced AICD in Con-CAR-T cells but had almost no effects on Run-CAR-T cells (Figure 5E). Con-CAR-T cells with TNF knocked out retained higher tumor-killing activity and cell expansion upon stimulation (Figures S5C and S5D). These results suggested that TNF signaling played an important role in the repression of CAR-T cell AICD by Runx3.
Figure 5.

Runx3 regulated AICD via TNF signaling
(A and B) CAR-T cells were stimulated with HCC cells with or without an anti-TNF antibody (5 μg/mL) for two rounds, and the cell death level was measured by annexin V staining. (C) TNF knockout in CAR-T cells was validated. CAR-T cells were stimulated with PMA and ionomycin in the presence of brefeldin A. TNF expression was measured using flow cytometry, and IFNγ expression was measured as a control. (D) CAR-T cells were stimulated with HCC cells at a ratio of 1:1 for 24 h. The TNF level in the medium was measured with a cytometric bead array. (E) WT and TNF-KO CAR-T cells were stimulated with HCC cells for two rounds. The cell death level was measured by annexin V staining after the second round of coculture. Data are presented as the mean ± SD (n = 3; ∗∗∗p < 0.001; NS, nonsignificant; one-way ANOVA with Tukey’s posttest for multiple comparisons).
Improved antitumor effects of Run-CAR-T cells in an immunocompetent animal model and patient-derived tumor xenograft model
To better evaluate the therapeutic efficacy of Run-CAR-T cells, further studies were carried out in immunocompetent and patient-derived xenograft (PDX) mouse models. A mouse-derived CAR-T cell therapeutic model based on Hepa1-6 allografts in immunocompetent mice was established as described in a previous study.38 Murine Run-CAR-T cells were generated by retroviral transduction (Figures S6A and S6B). Murine Run-CAR-T cells showed significantly enhanced potency against these tumors, and survival was obviously prolonged in the mRun-CAR-T cell group compared with the mCon-CAR-T cell treatment group (Figures 6A–6C), even with a higher tumor burden (Figures 6D–6F), suggesting that the enhancement in the antitumor activities of Run-CAR-T cells was not compromised even in the immunocompetent tumor microenvironment. Moreover, Run-CAR-T cells performed better than Con-CAR-T cells in the PDX model (Figures 6G–6I), with increased CAR-T cell accumulation in tumor tissues (Figures 6J and 6K). In addition, no body weight loss or obvious damage to or T cell infiltration in the liver, lungs, or spleen of mice was observed after treatment with Run-CAR-T cells (Figures S6C–S6E), supporting that Run-CAR-T cells did not induce off-tumor toxicities in these models.
Figure 6.

Antitumor effects of Run-CAR-T cells in an immunocompetent animal model and a patient-derived tumor xenograft (PDX) model
(A) A total of 1.0 × 107 Hepa1-6-GPC3 cells were subcutaneously implanted into C57BL/6 mice. 5 days after tumor implantation, when the tumor volume had reached approximately 300 mm3, the mice were intravenously infused with 1.5 × 106 murine CAR-T cells (n = 6). (B) Tumor size was monitored every 2–4 days. (C) Tumor weight was measured at the end of the experiment. (D) A total of 1.0 × 107 Hepa1-6-GPC3 cells were subcutaneously implanted into C57BL/6 mice. 8 days after tumor implantation, when the tumor volume had reached approximately 400 mm3, the mice were intravenously infused with 1.0×106 murine CAR-T cells (n = 6). (E and F) Tumor volume and animal survival were monitored every 2–4 days. (G) Patient-derived tumor tissues were subcutaneously implanted into mice (NOD-Prkdcscid Il2rgnull). 26 days after tumor implantation, when the tumor volume had reached approximately 160 mm3, the mice were intravenously infused with 2.0 × 106 CAR-T cells (n = 6). (J and K) T cell infiltration in tumor tissues was determined by CD3 IHC. Data are presented as the mean ± SD (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, nonsignificant; one-way ANOVA with Tukey’s post-test for multiple comparisons).
Discussion
CAR-T cell therapy against solid tumors is restrained by limited persistence. Evidence supports that Trm cells can be maintained for a long time in tissues once they become resident. Previous studies have demonstrated that donor Trm cells can persist in transplanted organs for years,39,40,41 suggesting that Trm cells are highly persistent and tissue specific. Furthermore, adoptive transfer of mouse T cells directed to undergo Trm-cell differentiation by Runx3 overexpression was shown to lead to better tumor control with long-term T cell persistence in tumors.21 These results suggested that overexpressing Runx3 in CAR-T cells would enhance therapeutic efficacy by promoting Trm-cell differentiation.
Unexpectedly, in our study, no obvious difference in Trm-cell differentiation was observed between Con-CAR-TILs and Run-CAR-TILs using either classic biomarkers or a gene expression signature. It has been suggested that phenotypic markers are inadequate to define Trm cells among other memory T cells in the TME42 and that the transcriptional profile, despite providing some insight into Trm cells in tumors, still needs to supplemented with additional phenotypic or functional studies to define the different Trm-cell subsets in tumors.43 Nevertheless, in this study, Run-CAR-T cells showed potent antitumor activity with increased persistence, proliferation, and effector molecule expression, displaying a Trm-like function in solid tumors. Therefore, we concluded that overexpression of Runx3 in CAR-T cells might enhance Trm-cell formation, but the subsets were difficult to identify using the methods included in our study.
T cell apoptosis has been considered a new immune checkpoint in the context of antitumor immunity in solid tumors.44,45 Some researchers have focused on ameliorating apoptosis in next-generation CAR-T cells in solid tumors. CAR-T cells expressing dominant-negative Fas can resist FasL-induced cell death and achieve long-term persistence in tumors.46 The efficacy of this method depends on the FasL level in tumors, and this approach needs to be tested in clinical trials. Interestingly, subtle changes in the CAR construct, such as tuning the extracellular spacer length, can modulate cell susceptibility to AICD.47 However, this method provides this improvement at the cost of reduced antitumor activities of CAR-T cells, as indicated by in vitro assays. Our study clearly demonstrated that Run-CAR-T cells were resistant to AICD in solid tumors and maintained potent antitumor activity in vitro and in vivo, providing a new way to reduce T cell AICD in tumors by reprogramming gene expression associated with apoptosis.
Our study suggested that resistance to AICD was associated with longer persistence in solid tumors. Although the mechanism was not fully elucidated, we discovered that TNF signaling played an important role in Runx3-mediated AICD resistance. Our data demonstrated that Runx3 directly repressed TNF transcription and thus inhibited TNF-induced cell death signaling. TNF is one of the functional cytokines of activated T cells. It has multifaceted effects that regulate both survival and apoptosis according to the cell context.48,49 TNF has both protumor and antitumor effects in cancers, and whether TNF benefits tumor therapy depends on the type of tumor and state of cancer immunity.50 In regard to effects on T cells, TNF signaling has been reported to be crucial in inducing AICD, and TNFR2−/− T cells displayed prolonged survival at the tumor site and provided enhanced protection against tumor growth.51,52 Moreover, T cell-derived TNF has been recognized as one of the causes of monocyte-mediated cytokine release syndrome,37 which is one of the major safety concerns for CAR-T cell therapy. Our study indicated that TNF secretion is not crucial for the therapeutic efficacy of CAR-T cells in hepatocellular carcinomas (HCC) but could increase AICD in CAR-T cells.
Nevertheless, our study still had limitations. We did not perform an in vivo study using an orthotopic HCC animal model. Tumor cells transplanted into the liver may produce a different microenvironment than that of tumors growing subcutaneously. It is unknown whether the persistence of Run-CAR-T cells would also be longer in situ. In addition, the effect of Run-CAR-T cells on antitumor activities and cytokine release should be further investigated in the clinic since our animal models could not fully mimic the interaction between CAR-T cells and immunocytes in patients.
In conclusion, the present study provided evidence that overexpressing Runx3 in CAR-T cells could enhance therapeutic efficacy with long-term persistence in solid tumors. In addition, we identified a TNF-associated mechanism by which Runx3 regulated T cell AICD and thus promoted T cell survival in tumors. Run-CAR-T cells may serve as a promising therapeutic agent for future clinical use in solid tumor treatment.
Materials and methods
Mice
5- to 6-week-old female immunodeficient mice (NOD-Prkdcscid Il2rgnull) and C57BL/6 mice were housed under specific pathogen-free conditions. The animal study was carried out in accordance with the recommendations of the Animal Research: Reporting in vivo Experiments (ARRIVE) criteria. The protocol was approved by the Shanghai Cancer Institute Experimental Animal Care Commission.
Cell lines
SK-Hep1, PLC/PRF/5, and HEK-293T cells were obtained from American Type Culture Collection. Huh-7 and Jurkat cells were obtained from Cell Bank of the Shanghai Institute of Cell Biology, Chinese Academy of Sciences. SK-Hep1, PLC/PRF/5, Huh-7, and HEK-293T cells were cultured in DMEM medium supplemented with 10% FBS (Gibco). Jurkat cells were cultured in RPMI-1640 medium supplemented with 10% FBS. All cells were maintained in 5% CO2.
Plasmids
To generate lentiviral vector expressing Run-CAR, the nucleotide fragments coding for human Runx3 (GenBank: NM_004350.3) and 9F2-BBz CAR were amplified by PCR, respectively. A self-cleaving 2A (F2A) sequence was introduced into the PCR product. The two fragments were linked together by the overlapped F2A sequence and integrated into pRRLSIN lentiviral vector using ClonExpress cloning kit (Vazyme Biotech). The vector for murine Run-CAR was constructed in a similar method according to our previous study.5 The sequences coding murine Runx3 (GenBank: NM_001369050.1) and murine GPC3-CAR were fused by F2A sequence. For luciferase reporter construct, human TNF promoter sequence (−1,000 ∼ +100 nt) was cloned and inserted into pGL3-Enhancer vector.
CAR-T cell generation
Peripheral blood mononuclear cells from healthy donors were purified by density gradient centrifugation with Ficoll-Paque (GE Healthcare). T cells were activated with Dynabeads (Invitrogen) at a T cell:beads ratio of 1:2 for 48 h and transduced with lentivirus (multiplicity of infection = 5) by 1,200 g centrifugation for 40 min in a retronectin-precoated 24-well plate. The Dynabeads were removed using Dynamag spin-magnet (Invitrogen) 7 days after stimulation. Unless otherwise mentioned, CAR-T cells were maintained in AIM-V medium (Gibco) supplemented with 2% AB-type human serum and 500 U/mL recombinant human IL-2 (Huaxin Biotech, Shanghai). The generation of murine CAR-T cells has been described in a previous study.5
Tumor rechallenge assay
CAR-T cells were incubated with target cells at E to T ratio of 1:1. The number of total T cells was normalized by adding UTD cells. Tumor cells were supplemented 2 days after the first-round stimulation.
Flow cytometry
Cells were harvested and washed with ice-cold phosphate buffered saline (PBS) followed by incubation with antibodies for 30 min on ice. After being rinsed with PBS twice, cells were subjected to analysis by flow cytometry. CAR expression was measured using biotin-labeled GCCT peptide (CARsgen Therapeutics, Shanghai, China) that contained the epitope recognized by 9F2 scFv and detected by PE-streptavidin (Affymetrix eBioscience). Other antibodies used for flow cytometry were purchased from BD Biosciences and Affymetrix eBioscience (Table S1). For apoptosis analysis, the membrane phospholipid phosphatidylserine was detected by Annexin-V-FITC (BD Biosciences) according to the instructions. For ROS detection, cells were incubated with 10 μM DCFH-DA (Sigma-Aldrich) at 37°C for 30 min, and then we read the signal at the FITC channel.
Western blot analysis
2 × 106 CAR-T cells were harvested by 400 g centrifugation for 5 min. After being washed with PBS, the cells were lysed in 100 μL pre-cold RIPA buffer (Beyotime Biotech, Shanghai) with protease inhibitor cocktail (Sangon Biotech, Shanghai), 5 mM NaF, and 1 mM Na3VO4. After 12,000 g centrifugation for 10 min at 4°C, supernatant was transferred into new tubes. Protein concentration was determined by BCA Protein Assay Kit (Pierce). The protein was denatured in Laemmli buffer by 5-min heating at 100°C, and then we ran SDS-PAGE. After blotting onto PVDF membrane, protein of interest was detected by monoclonal antibodies listed in Table S1.
Quantitative real-time PCR (qPCR)
For antigen stimulation, CAR-T cells were plated in a GCCT-precoated dish for 24 h. Cells were harvested, and total RNA was extracted by TRIzol reagent (Invitrogen). The total RNA was further reverse-transcribed to cDNA by HiScript II Q RT SuperMix (Vazyme Biotech). The mRNA expression was analyzed by qPCR using AceQ SYBR Green Master Mix (Vazyme Biotech) with primers listed in Table S2. The relative quantification of targeted genes was calculated with the ΔΔCt method.
Cytotoxicity assays in vitro
The cytotoxicity assay has been described in the previous study.23 In brief, CAR-T cells were cocultured with target tumor cells at the indicated ratio for 18 h, and the lactate dehydrogenase activity in supernatant was detected by the CytoTox 96 Non-Radioactive Cytotoxicity Assay (Promega). The cytotoxicity for the second-round stimulation was measured by xCELLigence Real-Time Cell Analyzer according to the manufacturer’s instructions.
Cytometric bead array (CBA) assays
Cytokine release was measured by CBA kit (BD Biosciences). For tumor samples, 50 mg tissue was homogenized in 100 μL tissue protein extraction reagent (Thermo Scientific) containing protease inhibitor cocktail (Sangon Biotech, Shanghai). After centrifugation with 12,000 g at 4°C for 10 min, the supernatant was collected for CBA assay. The assay was performed according to the instruction of the manufacturer. In brief, samples were incubated with capture-bead mixture at room temperature for 1 h. Then PE-labeled detect-antibody mixture was added and incubated for another 2 h. After washing, beads were measured for PE fluorescence by flow cytometry. The cytokine level was calculated with the standard curve.
CRISPR-Cas9-mediated knockout
The guide RNAs were synthesized using GeneArt Precision gRNA Synthesis kit (Invitrogen) according to the manufacturer’s instruction. A mixture of three guide RNA (GCTGAGGAACAAGCACCGCC, CTGATTAGAGAGAGGTCCCT, and ATCTCTCAGCTCCACGCCAT) targeting TNF37 and a nonspecific control guide RNA were incubated with Cas9 nuclease (NEB) for 10 min at room temperature. Then the RNA-Cas9 ribonucleoprotein complexes were electroporated into CAR-T cells using Maxcyte GTx. The electroporated cells were cultured for at least 72 h before assays.
Chromatin immunoprecipitation (ChIP)
1 × 107 CAR-T cells were fixed by 0.75% formaldehyde to cross-link the protein with DNA. After washing, cells were suspended in ChIP buffer and sonicated to break the chromatin. Then 1/10 of the chromatin was kept as input and the rest was 1:10 diluted with RIPA buffer and divided into two parts for immunoprecipitation by isotype IgG or Runx3-specific antibody. Protein A/G beads were blocked by BSA and denatured Salmon sperm DNA and then incubated with the chromatin at 4°C overnight. Next, the beads were washed with low-salt wash buffer, high-salt wash buffer, and LiCl wash buffer. The cross-link was reversed in Elution buffer at 30°C. Following incubation with RNase A and Proteinase K, DNA was purified by DNA mini-prepare kit (Axygen). The quantification of TNF promoter in the chromatin was performed using qPCR.
Luciferase reporter assay
Jurkat cells transduced with CAR and Runx3 were sorted by flow cytometry. Then cells were transfected with luciferase-expressing plasmid by electroporation using Nucleofector (LONZA). Luciferase activity was determined by dual-luciferase reporter assay system (Promega) according to the manufacturer’s instructions. In brief, cells were stimulated by target cells or GCCT for 24 h and harvested by 400 g centrifugation. After washing by ice-cold PBS, cells were lysed in passive lysis buffer (PLB) for 15 min. The lysate was transferred into a 96-well plate and incubated with LAR II and Stop & Glo Reagent, respectively. The luminescence produced by firefly or Renilla luciferase was measured by luminometer. The efficiency of transfection was normalized by Renilla luciferase activity.
Animal models
For the cell line-derived xenograft model, mice (NOD-Prkdcscid Il2rgnull) were subcutaneously inoculated with 2 × 106 PLC/PRF/5 human HCC cells in the right flank. For the Hepa1-6 allograft model, C57BL/6j mice were subcutaneously inoculated with 1 × 107 Hepa1-6-GPC3 cells in the right flank. For the studies in the PDX model, the BALB/c nude mice bearing HCC PDX were kindly provided by Crown Bioscience. The PDX tissues (∼15 mm3) were subcutaneously implanted in the right flank of mice using a Trocar needle. Mice were randomly divided into sub-groups and intravenously injected with indicated dose of CAR-T cells or untransduced T cells. Tumor dimensions and body weight were measured every 3–4 days. The tumor volumes were calculated by the formula: V = (length × width2)/2.
Tumor harvest and dissociation
To analyze tumor-infiltrating cells, tumor tissues were digested using tumor dissociation kit (Miltenyi Biotec) according to the manufacturer’s instructions. Briefly, tumor tissues were minced into ∼2 mm3 pieces and digested with the mixed enzyme solution for 30 min. Cells were filtered by a 70-μm cell strainer and collected by 400 g centrifugation. The red blood cells in the single cell suspension were lysed by ACK lysing buffer (Gibco).
RNA sequencing analysis
TILs were isolated from tumor suspension using EasySep CD8 cell enrichment kit (StemCell) according to the manufacturer’s instructions. Before the separation, the dead cells in suspension were removed by Maglive Dead Cell Removal Kit (Miltenyi Biotec). To make enough cells for RNA-seq analysis, each sample was a mixture of CD8 T cells from two to three tumors within the group. The isolated cells were lysed in TRIzol reagent (Invitrogen) and stored at −80°C till further use. The total RNA extraction, library preparation, sequencing, and GSEA were performed by CapitalBio Technology (Beijing, China). After data cleaning, approximately 45 million clean reads were generated per sample. Reads were mapped using HISAT2, and aligned reads were counted with StringTie.
Immunohistochemistry
Tumor tissues were collected and fixed by formaldehyde solution. After being embedded in paraffin, tissues were sliced at 5-μm thickness. The sections were deparaffined and rehydrated by xylol and ethanol solution. 3% hydrogen peroxide was added to sections to deactivate endogenous peroxidase. For antigen retrieval, slides were merged into pre-heated citrate buffer (pH 6.0) and kept at 95°C for 10 min. After cooling down to room temperature, the slides were moved into PBS for brief wash. PBS with 3% BSA was added to the slides for 20 min at room temperature to block nonspecific binding of antibody. Primary antibody was added to slides at 4°C overnight. Slides were washed with PBS containing 0.5% Tween 20 for 5 min once and with PBS twice. Then the antigen was visualized by peroxidase-DAB EnVision Detection System (Dako). Slides were counterstained with hematoxylin for seconds and dehydrated with ethanol solution for mounting.
RNA scope analysis
A probe targeting RNA sequence encoding 9F2 scFv was designed and synthesized by Advanced Cell Diagnostics. (ACD). The assays were performed with RNA Scope 2.5 HD Assay-Red Kit (ACD) according to the manufacturer’s instructions. In brief, slides were deparaffined by xylol. Hydrogen peroxide was added to sections to deactivate endogenous peroxidase. Then slides were merged into pre-heated 1× Target Retrieval Reagent and kept at 95°C for 15 min. After rinsing, the slides were digested with Protease Plus at 40°C for 30 min. Next, slides were incubated with hybridize probe and Amplifier 1–6 sequentially. The signal was detected by adding a mixture of RED-A and RED-B. After counterstaining, slides were dried in a 60°C oven for 15 min to mount.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 5.0 and SPSS 17.0. Unpaired Student’s t test was used to compare two groups. One-way ANOVA with Tukey post-test was used to determine the statistical significance for three-group comparisons. All experimental data are presented graphically or by mean ± standard deviation (SD).
Acknowledgments
We thank De Xu for the assistance in performing animal experiments. This research was supported by funding from the Program of Shanghai Technology Research Leader (22XD1430700), and the National Natural Science Foundation of China (No. 82073358 and 81871918).
Author contributions
Z.L. and H.J. designed the studies; Y.W. and Y.S. performed the experiments in vitro; Y.W., H.Z., G.D., H.L., J.S., M.Z., and H.Q.X.L. performed in vivo animal studies; B.S. performed the plasmid constructions; Y.W., H.Z., Q.L., and H.L. collected and analyzed the data; Z.L., H.J., and Y.W. wrote the manuscript.
Declaration of interests
Z.L. has ownership interests in Run-CAR-T.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.12.009.
Contributor Information
Hua Jiang, Email: jianghuapy@163.com.
Zonghai Li, Email: zonghaili@shsmu.edu.cn.
Supplemental information
Data and materials availability
The human sequence data generated in this study are not publicly available due to privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.
References
- 1.Newick K., O'Brien S., Moon E., Albelda S.M. CAR T cell therapy for solid tumors. Annu. Rev. Med. 2017;68:139–152. doi: 10.1146/annurev-med-062315-120245. [DOI] [PubMed] [Google Scholar]
- 2.Hong M., Clubb J.D., Chen Y.Y. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell. 2020;38:473–488. doi: 10.1016/j.ccell.2020.07.005. [DOI] [PubMed] [Google Scholar]
- 3.Lindo L., Wilkinson L.H., Hay K.A. Befriending the hostile tumor microenvironment in CAR T-cell therapy. Front. Immunol. 2020;11:618387. doi: 10.3389/fimmu.2020.618387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson K.G., Stromnes I.M., Greenberg P.D. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell. 2017;31:311–325. doi: 10.1016/j.ccell.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo H., Su J., Sun R., Sun Y., Wang Y., Dong Y., Shi B., Jiang H., Li Z. Coexpression of IL7 and CCL21 increases efficacy of CAR-T cells in solid tumors without requiring preconditioned lymphodepletion. Clin. Cancer Res. 2020;26:5494–5505. doi: 10.1158/1078-0432.CCR-20-0777. [DOI] [PubMed] [Google Scholar]
- 6.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 7.Chmielewski M., Kopecky C., Hombach A.A., Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 8.Liu Y., Di S., Shi B., Zhang H., Wang Y., Wu X., Luo H., Wang H., Li Z., Jiang H. Armored inducible expression of IL-12 enhances antitumor activity of glypican-3-targeted chimeric antigen receptor-engineered T cells in hepatocellular carcinoma. J. Immunol. 2019;203:198–207. doi: 10.4049/jimmunol.1800033. [DOI] [PubMed] [Google Scholar]
- 9.Hu B., Ren J., Luo Y., Keith B., Young R.M., Scholler J., Zhao Y., June C.H. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18.(2017) Cell Rep. 2017;20:3025–3033. doi: 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chmielewski M., Abken H. CAR T cells releasing IL-18 convert to T-bet(high) FoxO1(low) effectors that exhibit augmented activity against advanced solid tumors. Cell Rep. 2017;21:3205–3219. doi: 10.1016/j.celrep.2017.11.063. [DOI] [PubMed] [Google Scholar]
- 11.Glassman C.R., Mathiharan Y.K., Jude K.M., Su L., Panova O., Lupardus P.J., Spangler J.B., Ely L.K., Thomas C., Skiniotis G., Garcia K.C. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell. 2021;184:983–999.e24. doi: 10.1016/j.cell.2021.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L., Morgan R.A., Beane J.D., Zheng Z., Dudley M.E., Kassim S.H., Nahvi A.V., Ngo L.T., Sherry R.M., Phan G.Q., et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 2015;21:2278–2288. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Etzensperger R., Kadakia T., Tai X., Alag A., Guinter T.I., Egawa T., Erman B., Singer A. Identification of lineage-specifying cytokines that signal all CD8(+)-cytotoxic-lineage-fate 'decisions' in the thymus. Nat. Immunol. 2017;18:1218–1227. doi: 10.1038/ni.3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Park J.H., Adoro S., Guinter T., Erman B., Alag A.S., Catalfamo M., Kimura M.Y., Cui Y., Lucas P.J., Gress R.E., et al. Signaling by intrathymic cytokines, not T cell antigen receptors, specifies CD8 lineage choice and promotes the differentiation of cytotoxic-lineage T cells. Nat. Immunol. 2010;11:257–264. doi: 10.1038/ni.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Woolf E., Xiao C., Fainaru O., Lotem J., Rosen D., Negreanu V., Bernstein Y., Goldenberg D., Brenner O., Berke G., et al. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc. Natl. Acad. Sci. USA. 2003;100:7731–7736. doi: 10.1073/pnas.1232420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sato T., Ohno S.i., Hayashi T., Sato C., Kohu K., Satake M., Habu S. Dual functions of Runx proteins for reactivating CD8 and silencing CD4 at the commitment process into CD8 thymocytes. Immunity. 2005;22:317–328. doi: 10.1016/j.immuni.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 17.Shan Q., Zeng Z., Xing S., Li F., Hartwig S.M., Gullicksrud J.A., Kurup S.P., Van Braeckel-Budimir N., Su Y., Martin M.D., et al. The transcription factor Runx3 guards cytotoxic CD8(+) effector T cells against deviation towards follicular helper T cell lineage. Nat. Immunol. 2017;18:931–939. doi: 10.1038/ni.3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz-Guilloty F., Pipkin M.E., Djuretic I.M., Levanon D., Lotem J., Lichtenheld M.G., Groner Y., Rao A. Runx3 and T-box proteins cooperate to establish the transcriptional program of effector CTLs. J. Exp. Med. 2009;206:51–59. doi: 10.1084/jem.20081242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang D., Diao H., Getzler A.J., Rogal W., Frederick M.A., Milner J., Yu B., Crotty S., Goldrath A.W., Pipkin M.E. The transcription factor Runx3 establishes chromatin accessibility of cis-regulatory landscapes that drive memory cytotoxic T lymphocyte formation. Immunity. 2018;48:659–674.e6. doi: 10.1016/j.immuni.2018.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Z., Arai E., Khan O., Zhang Z., Ngiow S.F., He Y., Huang H., Manne S., Cao Z., Baxter A.E., et al. In vivo CD8(+) T cell CRISPR screening reveals control by Fli1 in infection and cancer. Cell. 2021;184:1262–1280.e22. doi: 10.1016/j.cell.2021.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Milner J.J., Toma C., Yu B., Zhang K., Omilusik K., Phan A.T., Wang D., Getzler A.J., Nguyen T., Crotty S., et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. 2017;552:253–257. doi: 10.1038/nature24993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi D., Shi Y., Kaseb A.O., Qi X., Zhang Y., Chi J., Lu Q., Gao H., Jiang H., Wang H., et al. Chimeric antigen receptor-glypican-3 T-cell therapy for advanced hepatocellular carcinoma: results of phase I trials. Clin. Cancer Res. 2020;26:3979–3989. doi: 10.1158/1078-0432.CCR-19-3259. [DOI] [PubMed] [Google Scholar]
- 23.Gao H., Li K., Tu H., Pan X., Jiang H., Shi B., Kong J., Wang H., Yang S., Gu J., Li Z. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014;20:6418–6428. doi: 10.1158/1078-0432.CCR-14-1170. [DOI] [PubMed] [Google Scholar]
- 24.Roychoudhuri R., Clever D., Li P., Wakabayashi Y., Quinn K.M., Klebanoff C.A., Ji Y., Sukumar M., Eil R.L., Yu Z., et al. BACH2 regulates CD8(+) T cell differentiation by controlling access of AP-1 factors to enhancers. Nat. Immunol. 2016;17:851–860. doi: 10.1038/ni.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsukumo S.i., Unno M., Muto A., Takeuchi A., Kometani K., Kurosaki T., Igarashi K., Saito T. Bach2 maintains T cells in a naive state by suppressing effector memory-related genes.(2013) Proc. Natl. Acad. Sci. USA. 2013;110:10735–10740. doi: 10.1073/pnas.1306691110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ichii H., Sakamoto A., Kuroda Y., Tokuhisa T. Bcl6 acts as an amplifier for the generation and proliferative capacity of central memory CD8+ T cells. J. Immunol. 2004;173:883–891. doi: 10.4049/jimmunol.173.2.883. [DOI] [PubMed] [Google Scholar]
- 27.Wu T., Ji Y., Moseman E.A., Xu H.C., Manglani M., Kirby M., Anderson S.M., Handon R., Kenyon E., Elkahloun A., et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol. 2016;1:eaai8593. doi: 10.1126/sciimmunol.aai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhou X., Yu S., Zhao D.M., Harty J.T., Badovinac V.P., Xue H.H. Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity. 2010;33:229–240. doi: 10.1016/j.immuni.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeannet G., Boudousquié C., Gardiol N., Kang J., Huelsken J., Held W. Essential role of the Wnt pathway effector Tcf-1 for the establishment of functional CD8 T cell memory. Proc. Natl. Acad. Sci. USA. 2010;107:9777–9782. doi: 10.1073/pnas.0914127107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebhardt T., Wakim L.M., Eidsmo L., Reading P.C., Heath W.R., Carbone F.R. Memory T cells in nonlymphoid tissue that provide enhanced local immunity during infection with herpes simplex virus. Nat. Immunol. 2009;10:524–530. doi: 10.1038/ni.1718. [DOI] [PubMed] [Google Scholar]
- 31.Park S.L., Gebhardt T., Mackay L.K. Tissue-resident memory T cells in cancer immunosurveillance.(2019) Trends Immunol. 2019;40:735–747. doi: 10.1016/j.it.2019.06.002. [DOI] [PubMed] [Google Scholar]
- 32.Devadas S., Zaritskaya L., Rhee S.G., Oberley L., Williams M.S. Discrete generation of superoxide and hydrogen peroxide by T cell receptor stimulation: selective regulation of mitogen-activated protein kinase activation and fas ligand expression. J. Exp. Med. 2002;195:59–70. doi: 10.1084/jem.20010659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Norell H., Martins da Palma T., Lesher A., Kaur N., Mehrotra M., Naga O.S., Spivey N., Olafimihan S., Chakraborty N.G., Voelkel-Johnson C., et al. Inhibition of superoxide generation upon T-cell receptor engagement rescues Mart-1(27-35)-reactive T cells from activation-induced cell death. Cancer Res. 2009;69:6282–6289. doi: 10.1158/0008-5472.CAN-09-1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Istaces N., Splittgerber M., Lima Silva V., Nguyen M., Thomas S., Le A., Achouri Y., Calonne E., Defrance M., Fuks F., et al. EOMES interacts with RUNX3 and BRG1 to promote innate memory cell formation through epigenetic reprogramming. Nat. Commun. 2019;10:3306. doi: 10.1038/s41467-019-11233-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng L., Fisher G., Miller R.E., Peschon J., Lynch D.H., Lenardo M.J. Induction of apoptosis in mature T cells by tumour necrosis factor.(1995) Nature. 1995;377:348–351. doi: 10.1038/377348a0. [DOI] [PubMed] [Google Scholar]
- 36.Sytwu H.K., Liblau R.S., McDevitt H.O. The roles of Fas/APO-1 (CD95) and TNF in antigen-induced programmed cell death in T cell receptor transgenic mice. Immunity. 1996;5:17–30. doi: 10.1016/s1074-7613(00)80306-4. [DOI] [PubMed] [Google Scholar]
- 37.Li J., Piskol R., Ybarra R., Chen Y.J.J., Li J., Slaga D., Hristopoulos M., Clark R., Modrusan Z., Totpal K., et al. CD3 bispecific antibody-induced cytokine release is dispensable for cytotoxic T cell activity. Sci. Transl. Med. 2019;11:eaax8861. doi: 10.1126/scitranslmed.aax8861. [DOI] [PubMed] [Google Scholar]
- 38.Wu X., Luo H., Shi B., Di S., Sun R., Su J., Liu Y., Li H., Jiang H., Li Z. Combined antitumor effects of sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol. Ther. 2019;27:1483–1494. doi: 10.1016/j.ymthe.2019.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pallett L.J., Burton A.R., Amin O.E., Rodriguez-Tajes S., Patel A.A., Zakeri N., Jeffery-Smith A., Swadling L., Schmidt N.M., Baiges A., et al. Longevity and replenishment of human liver-resident memory T cells and mononuclear phagocytes. J. Exp. Med. 2020;217:e20200050. doi: 10.1084/jem.20200050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bartolomé-Casado R., Landsverk O.J.B., Chauhan S.K., Richter L., Phung D., Greiff V., Risnes L.F., Yao Y., Neumann R.S., Yaqub S., et al. Resident memory CD8 T cells persist for years in human small intestine. J. Exp. Med. 2019;216:2412–2426. doi: 10.1084/jem.20190414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Snyder M.E., Finlayson M.O., Connors T.J., Dogra P., Senda T., Bush E., Carpenter D., Marboe C., Benvenuto L., Shah L., et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation.(2019) Sci. Immunol. 2019;4:eaav5581. doi: 10.1126/sciimmunol.aav5581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasson S.C., Gordon C.L., Christo S.N., Klenerman P., Mackay L.K. Local heroes or villains: tissue-resident memory T cells in human health and disease. Cell. Mol. Immunol. 2020;17:113–122. doi: 10.1038/s41423-019-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okla K., Farber D.L., Zou W. Tissue-resident memory T cells in tumor immunity and immunotherapy. J. Exp. Med. 2021;218:e20201605. doi: 10.1084/jem.20201605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J., Petit P.F., Van den Eynde B.J. Apoptosis of tumor-infiltrating T lymphocytes: a new immune checkpoint mechanism. Cancer Immunol. Immunother. 2019;68:835–847. doi: 10.1007/s00262-018-2269-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Horton B.L., Williams J.B., Cabanov A., Spranger S., Gajewski T.F. Intratumoral CD8(+) T-cell apoptosis is a major component of T-cell dysfunction and impedes antitumor immunity. Cancer Immunol. Res. 2018;6:14–24. doi: 10.1158/2326-6066.CIR-17-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yamamoto T.N., Lee P.H., Vodnala S.K., Gurusamy D., Kishton R.J., Yu Z., Eidizadeh A., Eil R., Fioravanti J., Gattinoni L., et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest. 2019;129:1551–1565. doi: 10.1172/JCI121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Künkele A., Johnson A.J., Rolczynski L.S., Chang C.A., Hoglund V., Kelly-Spratt K.S., Jensen M.C. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell fas-FasL-dependent AICD. Cancer Immunol. Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 48.Brenner D., Blaser H., Mak T.W. Regulation of tumour necrosis factor signalling: live or let die.(2015) Nat. Rev. Immunol. 2015;15:362–374. doi: 10.1038/nri3834. [DOI] [PubMed] [Google Scholar]
- 49.Mehta A.K., Gracias D.T., Croft M. TNF activity and T cells. Cytokine. 2018;101:14–18. doi: 10.1016/j.cyto.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen A.Y., Wolchok J.D., Bass A.R. TNF in the era of immune checkpoint inhibitors: friend or foe?(2021) Nat. Rev. Rheumatol. 2021;17:213–223. doi: 10.1038/s41584-021-00584-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim E.Y., Teh S.J., Yang J., Chow M.T., Teh H.S. TNFR2-deficient memory CD8 T cells provide superior protection against tumor cell growth. J. Immunol. 2009;183:6051–6057. doi: 10.4049/jimmunol.0803482. [DOI] [PubMed] [Google Scholar]
- 52.Otano I., Alvarez M., Minute L., Ochoa M.C., Migueliz I., Molina C., Azpilikueta A., de Andrea C.E., Etxeberria I., Sanmamed M.F., et al. Human CD8 T cells are susceptible to TNF-mediated activation-induced cell death. Theranostics. 2020;10:4481–4489. doi: 10.7150/thno.41646. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human sequence data generated in this study are not publicly available due to privacy requirements but are available upon reasonable request from the corresponding author. Other data generated in this study are available within the article and its supplementary data files.