

Graphical abstract

Keywords: 3-phenyl-1,2,4-oxadiazole derivatives; SARS-CoV-2; Main protease inhibitor
Abstract
The ongoing COVID-19 pandemic has led to massive infections and deaths and caused tremendous grief among the people. Although vaccines have played an important role in fighting COVID-19, the situation that the protective effect of current vaccines significantly decreases against mutated strains reminds us of the pressing need for developing effective antiviral therapeutics. The main protease (Mpro) is a key enzyme for SARS-CoV-2 viral replication and transcription and an attractive target for drug development. In this research, we report a new series of Mpro inhibitors containing 3-phenyl-1,2,4-oxadiazole. Structure-activity relationship (SAR) studies led to the discovery of the most active compound, 16d, which showed an IC50 value of 5.27 ± 0.26 μM. Collectively, we obtained a new small molecular inhibitor targeting SARS-CoV-2 Mpro, which contains a new scaffold. This compound could be taken as a lead compound for subsequent drug discovery against SARS-CoV-2.
An infectious respiratory disease with atypical pneumonia emerged in 2019.1 Its rapid and global spread has been officially declared as a pandemic by the World Health Organization (WHO).2 This global threat has been proven to be caused by a coronavirus named severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), due to its similarities to SARS-CoV both in genomics and clinical symptoms.3 SARS-CoV-2 is a kind of RNA virus. Two open reading frames (ORFs) of the SARS-CoV-2 genome, ORF1a and ORF1b, respectively encodes two partially overlapping polyproteins, pp1a and pp1ab, which is processed into 16 non-structural proteins (NSPs) by two self-encoding cysteine proteases, papain-like protease (PLpro) and main protease Mpro (also referred to as 3CLpro).4, 5 These NSPs, together with four structural proteins (spike, envelope, membrane, and nucleocapsid) which encoded by other ORFs, play key roles in maintaining the normal life cycle of the virus.6 Thus, some of them are considered as potential drug targets for the treatment of COVID-19, including Mpro, PLpro, spike protein, etc.7, 8 Among these targets, Mpro has been considered as an ideal target for the development of antiviral drugs due to its crucial role in the viral life cycle, its distinctiveness from human proteases, and its highly conserved both in its variants and other coronaviruses.9, 10.
Currently, a variety of advanced Mpro inhibitors have been reported, such as 13b 11, 11a 12, MI-09 13, 23R 14, Y180 15 and Nirmatrelvir (PF-07321332)16 (Fig. 1 ). Covalent inhibitors 13b and 11a were investigated for their pharmacokinetic properties in mice, and exhibited low toxicity.11, 12 MI-09 was reported to be the first orally effective drug candidate and significantly reduced viral load in the lungs and lung lesions in a transgenic mouse model of SARS-CoV-2 infection.13 23R is a selective non-covalent inhibitor, whereas, with just moderate activity which needs to be further improved.14 Y180, a potent reversible covalent Mpro inhibitor with a scaffold similar to 23R, further advanced the development of highly specific antiviral therapeutics for COVID-19.15 Nirmatrelvir, developed by Pfizer Inc., has been granted an emergency use authorization (EUA) for the treatment of mild to moderate COVID-19 by the US Food and Drug Administration (FDA).16, 17 Because of its peptide-like structure, Nirmatrelvir has unsatisfied oral bioavailability and short plasma half-life and needs to be used in combination with the ritonavir (an CYP3A4 inhibitor), which limits its applicable population.16, 18 Therefore, it is still necessary to discover non-peptide Mpro inhibitors with new chemical scaffolds. Herein we identified a class of Mpro inhibitors with 3-phenyl-1,2,4-oxadiazole scaffold, providing a new option for the development of small molecule antivirals.
Fig. 1.

Structures and bioactivities of representative Mpro inhibitors.
In order to discover novel Mpro inhibitors, we conducted a screening experiment against our in-house chemical library containing 2000 compounds by fluorescence resonance energy transfer (FRET) assay. Hit-01, N-((3-(4-(trifluoromethyl)phenyl)-1,2,4-oxadiazol-5-yl)methyl)cinnamamide), were found to be able to inhibit the enzymatic activity of Mpro with an IC50 value of 46 μM (Fig. 2 A and 2B). Subsequently, structure optimization and structure–activity relationship studies were performed on this hit compound.
Fig. 2.

(A) The chemical structure of Hit-01; (B) Dose-activity curve of Hit-01 against SARS-CoV-2 Mpro in the FRET assay.
Firstly, we adopted molecular docking to predict the binding mode between Hit-01 and Mpro, which provided guidance for us on further optimization. The three-dimensional (3D) structure was taken from RCSB Protein Data Bank (PDB entry: 7M8P). As shown in Fig. 3 , Hit-01 properly occupied S1 and S3/4 active pocket of Mpro with S2 unoccupied. The nitrogen atom on 2-position of 1,2,4-oxadiazole forms a hydrogen bond with the side-chain of Q192, and the fluorine atom of trifluoromethyl group forms another hydrogen bond with the main-chain amide of Q192. The phenyl group of Hit-01 occupies S1 pocket and forms the π-anion interaction with the side-chain of E166. In S3/4 pocket, two π-alkyl interactions are formed between Hit-01 and the side chain of M165, P168, respectively. According to the information above, a series of analogues were designed to explore the structure–activity relationship and the structural optimization of compound Hit-01, which principally focused on three regions (Fig. 4 ): cinnamyl (region I), 4-trifluoromethyl (region II), and methylene (region III).
Fig. 3.

Binding mode of Hit-01 is predicted by molecular docking. Predicted binding mode of Hit-01 (in orange) with SARS-CoV-2 Mpro (in gray). Key residues are shown as gray sticks. Hydrogen bonds are represented as red dashed lines. π-alkyl interactions are represented as purple solid lines. π-anion interaction is represented as magenta dashed line.
Fig. 4.

Regions that are the focuses of structural optimization.
In the first round optimization, according to the binding mode of Hit-01 and Mpro shown in Fig. 3, we first explored whether the π-anion interaction which is formed by the phenyl group of Hit-01 and E166 is important. So we used different substituents to replace cinnamyl subgroups (region I) and synthesized 4 compounds (7a-d). Moreover, the binding mode shows that there is some extra space in region I, especially around the vinyl linker of Hit-01. Then we synthesized compounds 7e-h to explore the influence of different linkers on bioactivities. Synthetic routes for these compounds are depicted in Scheme 1 . Commercially available reagent 4-(trifluoromethyl)benzonitrile (1) reacted with hydroxylamine hydrochloride to give intermediate 2, which then cyclized with (tert-butoxycarbonyl)glycine (3) to obtain intermediate 4. Deprotection of intermediate 4 delivered intermediate 5. Target compounds (7a-h) were acquired via condensation reaction between compound 5 and acids 6a-h (Supplementary Scheme 1).
Scheme 1.

Synthetic routes of compounds 7a-h . Reagents and conditions: (i) NH2OH·HCl, EtOH, K2CO3, reflux, 6 h; (ii) a: EDCI, HOBT, DIEA, DMF, rt, 6 h; b: toluene, K2CO3, rt, overnight; (iii) HCl in 1,4-dioxane, rt, 1 h; (iv) HATU, DIEA, DCM, rt, 4 h, 70-90% yield.
The chemical structures and bioactivities of compounds 7a-h are shown in Table 1 . Removing the phenyl group caused significantly decreased potency (7a). Then we replaced phenyl group with several heterocycles including pyridyl (7b), furyl (7c) and thienyl (7d), all the resulted compounds displayed reduced bioactivity compared to Hit-01. Thus, we kept the terminal benzene ring of cinnamyl and replaced its vinyl linker. 4 compounds were then synthesized (7e-h). To our delight, compound 7h, containing (2-methylcycloprop-1-en-1-yl)benzene, exhibited obviously increased potency, with an IC50 value of 6.71 μM.
Table 1.
Chemical structures and bioactivities of compounds 7a-h.a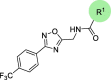
| Compound | R1 |
SARS CoV-2 Mpro IC50 (μM)a |
|---|---|---|
| Hit-01 | 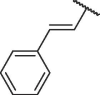 |
46.29 ± 3.48 |
| 7a |  |
249.99 ± 3.44 |
| 7b | 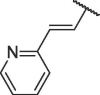 |
102.44 ± 0.22 |
| 7c | 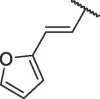 |
60.52 ± 6.13 |
| 7d |  |
54.56 ± 4.41 |
| 7e |  |
62.03 ± 9.34 |
| 7f | 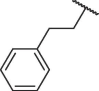 |
90.48 ± 0.51 |
| 7g |  |
46.87 ± 0.43 |
| 7h | 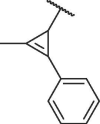 |
6.71 ± 0.24 |
Each value represented the average results from three independent experiments.
In the second round of optimization, we explored whether the change in the position of trifluoromethyl and electronic effects of groups makes a difference. According to the predicted binding mode shown in Fig. 3, the fluorine atom of trifluoromethyl group forms one hydrogen bond with the main-chain amide of Q192. Thus, we fixed region I as (2-methylcycloprop-1-en-1-yl)benzene and discussed the effects of 4-trifluoromethyl (region II) on biological activity. 9 compounds (12a-i) were synthesized in this part. The synthetic routes are showed in Scheme 2 . Commercially available reagents 8a-i reacted with hydroxylamine hydrochloride delivered intermediates 9a-i, which then cyclized with (tert-butoxycarbonyl)glycine (3) to give intermediates 10a-i. Removal of Boc protection generated amines 11a-i. Target compounds 12a-i were finally obtained through condensation of 11a-i and 6h.
Scheme 2.

Synthetic routes of compounds 12a-i. Reagents and conditions: (i) NH2OH·HCl, EtOH, K2CO3, reflux, 6 h; (ii) a: EDCI, HOBT, DIEA, DMF, rt, 6 h; b: toluene, K2CO3, rt, overnight; (iii) HCl in 1,4-dioxane (4 M), rt, 1 h; (iv) 6 h, HATU, DIEA, DCM, rt, 4 h, 70-80% yield.
The activities of compounds 12a-i are shown in Table 2 . Compared with 7h, 12a and 12b with changing position of trifluoromethyl led to decreased activities. Then removing the trifluoromethyl on the phenyl ring resulted in a significantly decrease in potency, as evidenced by 12c. So we used different groups to replace trifluoromethyl at para-position of phenyl ring. Replacing the trifluoromethyl with various electron-donating groups (12d-e), halogen (12f-g), or electron-withdrawing groups (12h-i) also displayed decreased bioactivities. We thus fixed region II as trifluoromethyl in the following optimization step.
Table 2.
Chemical structures and bioactivities of compounds 7j and 12a-ia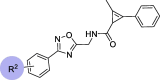
| Compound | R2 |
SARS CoV-2 Mpro IC50 (μM)a |
|---|---|---|
| 7h |  |
6.71 ± 0.24 |
| 12a | 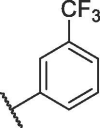 |
14.26 ± 0.16 |
| 12b |  |
66.92 ± 1.48 |
| 12c |  |
79.62 ± 1.68 |
| 12d | 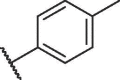 |
35.22 ± 1.05 |
| 12e |  |
39.32 ± 3.34 |
| 12f | 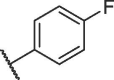 |
66.96 ± 1.70 |
| 12 g |  |
21.24 ± 2.70 |
| 12h | 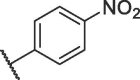 |
19.66 ± 0.23 |
| 12i | 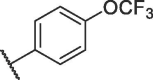 |
19.64 ± 2.99 |
Each value represented the average results from three independent experiments.
In the final optimization, according to the binding mode shown in Fig. 3, S2 pocket is unoccupied, which suggests it might be helpful to increase activity through introducing groups to occupy S2. Then we explored the influence of region III (R3) on the bioactivity and synthesized 9 compounds (16a-i). The synthetic routes are described as Scheme 3 . Intermediates 2 reacted with racemate of amino acids 13a-i to give intermediates 14a-i, and then underwent a deprotection reaction to afford intermediates 15a-i. Condensation of 15a-i with 6h yielded target racemate compounds 16a-i.
Scheme 3.

Synthetic routes of compounds 16a-i . Reagents and conditions: (i) a: EDCI, HOBT, DIEA, DMF, rt, 6 h; b: toluene, K2CO3, rt, overnight; (ii) a: EDCI, HOBT, DIEA, DMF, rt, 6 h; b: toluene, K2CO3, rt, overnight; (iii) HCl in 1,4-dioxane (4 M), rt, 1 h; (iv) 6 h, HATU, DIEA, DCM, rt, 4 h, 70-92% yield.
Table 3 exhibits the chemical structures and bioactivities of compounds 16a-i. Various substituents were introduced on R 3 position to occupy the S2 pocket. As shown by 16a-c, the bioactivity increased with the size of the alkyl substituents. Compound 16c with a cyclohexylmethyl group displayed an IC50 value of 19.37 μM, but was not as active as 7h. We then replaced the cyclohexylmethyl group with benzyl group, and compound 16d exhibited significantly increased Mpro inhibitory activity, which was better than that of 7h. Exchanging benzyl group to larger groups, including 2-naphthylmethyl and 2-indolylmethyl led to reduced bioactivities (16e-f). Introduction of a fluorine atom on orthro, meta or para position of the benzene ring also resulted in decreased activities (16 g-i). Thus, benzyl might be the most suitable subgroup to occupy the S2 pocket. All in all, 16d was the most favorable compound.
Table 3.
Chemical structures and bioactivities of compounds 16a-ia
| Compound | R3 |
SARS CoV-2 Mpro IC50 (μM)a |
|---|---|---|
| 7h |  |
6.71 ± 0.24 |
| 16a |  |
96.93 ± 1.02 |
| 16b |  |
76.79 ± 0.14 |
| 16c |  |
19.37 ± 1.38 |
| 16d | 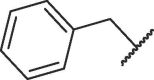 |
5.27 ± 0.26 |
| 16e |  |
6.47 ± 0.13 |
| 16f | 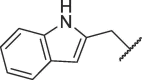 |
14.74 ± 1.20 |
| 16g | 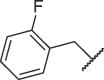 |
7.48 ± 0.26 |
| 16h | 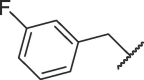 |
14.15 ± 1.44 |
| 16i | 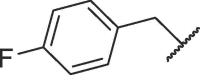 |
16.51 ± 0.33 |
Each value represented the average results from three independent experiments.
To explore the effect of the configuration of 16d on bioactivity, we tried to purify the enantiomers of 16d with two chiral centers. Unfortunately, we failed to separate every enantiomerically pure compound. We obtained two mixture Tmo-1 and Tmo-2 eventually (Supplementary Figs. 1–3). Bioactivities of Tmo-1 and Tmo-2 are 16.74 ± 1.01 μM and 3.64 ± 0.26 μM respectively.
Finally, we used molecular docking to predict the binding mode of compound 16d and SARS CoV-2 Mpro to explain the high potency of 16d. Compared with S configuration, we reasonably chose the R configuration for docking (Supplementary Fig. 4). As shown in Fig. 5 , the compound R-16d properly occupies the active pocket of Mpro and has a similar binding mode to Hit-01. Meanwhile, the benzyl group extends to the hydrophobic S2 pocket to form the π-alkyl interaction with the side-chain of M49, which might be the reason why inhibitory activity of 16d is improved. Overall, the predicted binding mode of compound 16d with Mpro is consistent with our expectation and explains its improved potency against SARS-CoV-2 Mpro.
Fig. 5.

Predicted binding mode of R-16d (in green) with SARS-CoV-2 Mpro (in gray). Key residues are shown as gray sticks. Hydrogen bonds are represented as red dashed lines. π-alkyl interactions are represented as purple solid lines. π-anion interaction is represented as magenta dashed line.
In summary, structural optimization of compound Hit-01 led to the discovery of a number of 3-phenyl-1,2,4-oxadiazole derivatives that showed inhibitory activity against Mpro. Compound 16d was the most potent one, which displayed an IC50 value of 5.27 μM. Collectively, this study provides a promising lead compound for drug discovery targeting SARS CoV-2 Mpro, and further optimization of this series of inhibitors is ongoing.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NO. 82130104, 82273787 and 22107081).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmcl.2023.129238.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Wang C., Horby P.W., Hayden F.G., et al. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.WHO. Coronavirus (COVID-19) Dashboard. https://covid19.who.int/ (2022).
- 3.Wu F., Zhao S., Yu B., et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y., Xu Z., Wang P., et al. New-onset autoimmune phenomena post-COVID-19 vaccination. Immunology. 2022;165:386–401. doi: 10.1111/imm.13443. [DOI] [PubMed] [Google Scholar]
- 5.Lee J., Kenward C., Worrall L.J., et al. X-ray crystallographic characterization of the SARS-CoV-2 main protease polyprotein cleavage sites essential for viral processing and maturation. Nat Commun. 2022;13:5196. doi: 10.1038/s41467-022-32854-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brant A.C., Tian W., Majerciak V., et al. SARS-CoV-2: from its discovery to genome structure, transcription, and replication. Cell Biosci. 2021;11:136. doi: 10.1186/s13578-021-00643-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raj R. Analysis of non-structural proteins, NSPs of SARS-CoV-2 as targets for computational drug designing. Biochem Biophys Rep. 2020;25 doi: 10.1016/j.bbrep.2020.100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trougakos I.P., Stamatelopoulos K., Terpos E., et al. Insights to SARS-CoV-2 life cycle, pathophysiology, and rationalized treatments that target COVID-19 clinical complications. J Biomed Sci. 2021;28:9. doi: 10.1186/s12929-020-00703-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin Z., Du X., Xu Y., et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 10.Ullrich S., Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg Med Chem Lett. 2020;30 doi: 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L., Lin D., Sun X., et al. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368 doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dai W., Zhang B., Jiang X.M., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qiao J.X., Li Y.S., Zeng R., et al. SARS-CoV-2 Mpro inhibitors with antiviral activity in a transgenic mouse model. Science. 2021;371 doi: 10.1126/science.abf1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kitamura N., Sacco M.D., Ma C., et al. Expedited Approach toward the Rational Design of Noncovalent SARS-CoV-2 Main Protease Inhibitors. J Med Chem. 2022;65:2848–2865. doi: 10.1021/acs.jmedchem.1c00509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quan B.X., Shuai H.P., Xia A.J., et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat Microbiol. 2022;7:716–725. doi: 10.1038/s41564-022-01119-7. [DOI] [PubMed] [Google Scholar]
- 16.Owen D.R., Allerton C.M.N., Anderson A.S., et al. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science. 2021;374:1586–1593. doi: 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- 17.Arbel R., Wolff S.Y., Hoshen M., et al. Nirmatrelvir Use and Severe Covid-19 Outcomes during the Omicron Surge. N Engl J Med. 2022;387:790–798. doi: 10.1056/NEJMoa2204919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eng H., Dantonio A.L., Kadar E.P., et al. Disposition of Nirmatrelvir, an Orally Bioavailable Inhibitor of SARS-CoV-2 3C-Like Protease, across Animals and Humans. Drug Metab Dispos. 2022;50:576–590. doi: 10.1124/dmd.121.000801. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.