

Abstract
This study aimed to identify a recommended phase II dose and evaluate the safety, tolerability, pharmacokinetics/pharmacodynamics, and preliminary clinical activity of JNJ‐63709178, a CD123/CD3 dual‐targeting antibody, in patients with relapsed or refractory acute myeloid leukemia. Intravenous (i.v.) and subcutaneous (s.c.) administration of JNJ‐63709178 were evaluated. The i.v. infusions were administered once every 2 weeks (cohorts 1–5 [n = 17]) or twice weekly (cohorts 6–11 [n = 36]). A twice‐weekly s.c. dosing regimen with step‐up dosing was also studied (s.c. cohorts 1–2 [n = 9]). Treatment‐emergent adverse events (TEAEs) greater than or equal to grade 3 were observed in 11 (65%) patients in cohorts 1–5 and 33 (92%) patients in cohorts 6–11. At the highest i.v. dose (4.8 μg/kg), 5 (71%) patients discontinued treatment due to TEAEs. For s.c. administration (n = 9), eight (89%) patients experienced TEAEs greater than or equal to grade 3 and injection site reactions (≤ grade 3) emerged in all patients. At 4.8 μg/kg (i.v. and s.c.), the mean maximum serum concentrations were 30.3 and 3.59 ng/ml, respectively. Increases in multiple cytokines were observed following i.v. and s.c. administrations, and step‐up dosing strategies did not mitigate cytokine production or improve the safety profile and led to limited duration of treatment. Minimal clinical activity was observed across all cohorts. The i.v. and s.c. dosing of JNJ‐63709178 was associated with suboptimal drug exposure, unfavorable safety profiles, limited clinical activity, and inability to identify a recommended phase II dose.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
The 5‐year overall survival rate for acute myeloid leukemia (AML) is ~30%; thus, there is an unmet need for effective treatment options, particularly in the relapsed setting. The α‐chain of the interleukin‐3 receptor (IL‐3Rα/CD123) is overexpressed in a large proportion (40%–93%) of patients with AML, and CD123‐targeting agents have shown some initial success. JNJ‐63709178 is a humanized IgG4‐PAA bispecific antibody that targets CD123 and CD3 and was developed with the rationale of bringing T cells and leukemic cells into proximity with the intention of promoting the activation of T cells and subsequent leukemic cell lysis.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study aimed to identify a recommended phase II dose and evaluate the safety, tolerability, and pharmacokinetics/pharmacodynamics of JNJ‐63709178 in patients with relapsed or refractory AML.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Despite varying full treatment, step‐up dosing schedules, and routes of administration, an unfavorable safety profile and suboptimal pharmacokinetic profile (short half‐life; average exposures below the target) were observed for JNJ‐63709178; therefore, a recommended phase II dose could not be identified.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Although JNJ‐63709178 treatment was found to have limited exposures and clinical activity with an unfavorable safety profile, these findings are important to shape continued exploration of other similar CD123 targeting agents in AML.
INTRODUCTION
Despite success with frontline treatments for acute myeloid leukemia (AML), the 5‐year survival rate is ~30%. 1 , 2 There is an unmet need for effective treatment options, particularly in the relapsed setting. The α‐chain of the interleukin‐3 receptor (IL‐3Rα/CD123) is overexpressed in a large proportion (40–93%) of patients with AML, 3 , 4 and CD123‐targeting agents have shown initial success. 5 , 6 , 7 , 8 , 9
JNJ‐63709178 is a humanized IgG4‐PAA bispecific antibody that targets CD123 and CD3 developed with the rationale of bringing T cells and leukemic cells into proximity with the intention of promoting the activation of T cells and subsequent leukemic cell lysis. 10 This trial was designed to identify a recommended phase II dose (RP2D), evaluate the safety, tolerability, and pharmacokinetics/pharmacodynamics (PKs/PDs), and assess for preliminary clinical activity of JNJ‐63709178.
METHODS
Study design
This phase I, first‐in‐human, open‐label, multicenter, dose escalation study with dose expansion was conducted in adult patients with relapsed or refractory AML who were ineligible for or had exhausted standard therapeutic options. The study occurred between June 2, 2016, and March 24, 2021. The trial initiated with intravenous (i.v.) dosing once every 2 weeks without step‐up dosing (i.v. cohorts 1–4) or a single step‐up dose (i.v. cohort 5), followed by twice‐weekly i.v. dosing with less than or equal to five step‐up doses (i.v. cohorts 6–11; Table S1). To improve the safety profile and exposure, a twice‐weekly subcutaneous (s.c.) dosing regimen with step‐up dosing was studied (s.c. cohorts 1–2). Dose escalation was guided by Bayesian Logistic Regression Model with Escalation with Overdose Control principle. 11 The study design is further described in Figure S1.
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Protocols were reviewed and approved by an institutional review board.
Safety analyses
Descriptive statistics were used to summarize the incidence, severity, and type of adverse events which were graded using Common Toxicity Criteria for Adverse Events (CTCAE) 4.03. PK analysis was done using Phoenix WinNonlin (version 8.1; 1998–2018; Certara LP, USA).
PK/PD analysis
Descriptive statistics were used to summarize serum concentrations and PK parameters at each timepoint and to summarize cytokine levels (measured via a meso scale discovery platform), T cell activation, and reduction in CD123+ blasts (measured by flow cytometry).
Efficacy analysis
Investigator‐assessed response was evaluated according to study‐specific response criteria based on European LeukemiaNet 2017. 2 Modifications included stable disease, which was defined as absence of any response without meeting criteria for progression.
RESULTS
Sixty‐two patients with AML across 13 dosing cohorts were treated. Patient and disease characteristics are included in Table S2. Of these cohorts, 11 evaluated i.v. administration (i.v. cohorts 1–11, n = 53) and two evaluated s.c. administration (s.c. cohorts 1–2, n = 9). Two dosing regimens were explored: biweekly (i.v. cohorts 1–5) and twice weekly (i.v. cohorts 6–11 and s.c. cohorts 1–2) with varied step‐up schedules.
Across all cohorts, the most common (≥10% patients) treatment‐emergent adverse events (TEAEs) considered related to study drug were cytokine release syndrome (CRS; 43.5%), alanine aminotransferase increase (19.4%), aspartate aminotransferase increase (14.5%), pyrexia (12.9%), and infusion‐related reactions (11.3%). TEAEs leading to dose interruptions occurred in 53.2%, whereas those leading to dose reductions occurred in 4.8% of patients.
For i.v. cohorts, dose‐limiting toxicities (DLTs) included a grade 5 CRS in cohort 2 (6 μg/kg, without step‐up dosing) and a grade 3 infusion‐related reaction (IRR) in cohort 11 (4.8 μg/kg, with multiple step‐up doses). Overall incidence of CRS was five (29%) patients in cohorts 1–5 and 16 (44%) in cohorts 6–11. The most common symptom associated with CRS was pyrexia (3 [18%] in cohort 1–5; 15 [42%] in cohorts 6–11). In cohort 11, five (71%) patients experienced greater than or equal to one drug‐related TEAE greater than or equal to grade 3 and three (43%) patients discontinued due to TEAEs greater than or equal to grade 3 (Table 1). Across the bi‐weekly i.v. cohorts (6–11), there was a clear progression of the incidence and severity of drug‐related toxicity, including the emergence of a second DLT (Grade 3 IRR) in cohort 11. Regarding clinical activity, the median duration of treatment was 22 days in cohorts 1–5 and 34 days in cohorts 6–11 with one (1.6%) participant achieving stable disease.
TABLE 1.
Summary of TEAEs and DLTs in i.v. and s.c. dosing cohorts
| IV cohorts 1–5 (Q2W) | IV cohorts 6–11 (twice weekly) | SC cohorts 1–2 (twice weekly) | Overall | |
|---|---|---|---|---|
| Analysis set: all treated | 17 | 36 | 9 | 62 |
| Any TEAE (%) | 15 (88.2) | 35 (97.2) | 9 (100) | 59 (95.2) |
| Drug‐related a | 9 (52.9) | 21 (58.3) | 9 (100) | 39 (62.9) |
| Any serious TEAE (%) | 10 (58.8) | 23 (63.9) | 7 (77.8) | 40 (64.5) |
| Drug‐related a | 4 (23.5) | 8 (22.2) | 1 (11.1) | 13 (21) |
| Infection | 6 (35.3) | 13 (36.1) | 4 (44.4) | 23 (37.1) |
| Maximum severity of a TEAE (%) | ||||
| Grade 1 | 0 | 1 (2.8) | 0 | 1 (1.6) |
| Grade 2 | 4 (23.5) | 1 (2.8) | 1 (11.1) | 6 (9.7) |
| Grade 3 | 6 (35.3) | 7 (19.4) | 1 (11.1) | 14 (22.6) |
| Grade 4 | 2 (11.8) | 22 (61.1) | 6 (66.7) | 30 (48.4) |
| Grade 5 | 3 (17.6) | 4 (11.1) | 1 (11.1) | 8 (12.9) |
| Treatment discontinuations due to TEAE b (%) | 2 (11.8) | 6 (16.7) | 2 (22.2) | 10 (16.1) |
| Drug related a | 1 (5.9) | 3 (8.3) | 0 | 4 (6.5) |
| Any DLT TEAE (%) | 1 (5.9) | 1 (2.8) | 0 | 2 (3.2) |
| Drug related a | 1 (5.9) | 1 (2.8) | 0 | 2 (3.2) |
| Any CRS (%) | 5 (29.4) | 16 (44.4) | 6 (66.7) | 27 (43.5) |
| Drug related a | 5 (29.4) | 16 (44.4) | 6 (66.7) | 27 (43.5) |
| Serious | 3 (17.6) | 6 (16.7) | 0 | 9 (14.5) |
| Time to CRS onset (days) c , n d | 5 | 16 | 6 | 27 |
| Mean (SD) | 3.8 (6.3) | 23.0 (17.6) | 2.3 (1.4) | 14.9 (16.9) |
| Median | 1.0 | 23.0 | 2.0 | 6.0 |
| Range | (1; 15) | (1; 51) | (1; 5) | (1; 51) |
| Death due to TEAE (%) | 3 (17.6) | 4 (11.1) | 1 (11.1) | 8 (12.9) |
| Drug related a (%) | 2 (11.8) | 0 | 0 | 2 (3.2) |
| Infusion reaction TEAE (%) | 0 | 7 (19.4) | NA | 7 (11.3) |
| Drug related a (%) | 0 | 7 (19.4) | NA | 7 (11.3) |
| Serious (%) | 0 | 2 (5.6) | NA | 2 (3.2) |
| Injection site reaction TEAE (%) | NA | NA | 9 (100.0) | 9 (14.5) |
| Drug related a (%) | NA | NA | 9 (100.0) | 9 (14.5) |
| Serious | NA | NA | 0 | 0 |
Abbreviations: CRS, cytokine release syndrome; DLT, dose limiting toxicity; Q2W, once every 2 weeks; TEAE, treatment emergent adverse event.
An adverse event is categorized as related if assessed by the investigator as possibly, probably, or very likely related to study agent.
Treatment discontinuation due to adverse event based on treatment disposition CRF page.
Time to CRS is the first occurrence of CRS event relative to the date of first dose of study agent.
This n refers to the number of CRS events in each group.
In the s.c. cohorts (n = 9), all patients experienced greater than or equal to one drug‐related TEAE (Table 1). A new safety signal of less than grade 3 injection site reaction (ISR) emerged in nine (100%) patients. The majority of ISRs was less than or equal to grade 2 (67%), however, the clinical severity of the ISRs increased upon dose escalation from s.c. cohort 1 (treatment dose, 2.4 μg/kg) to s.c. cohort 2 (treatment dose, 4.8 μg/kg) and was associated with pyrexia (6 [67%] patients) and reported CRS with worsening injection site pain (6 [67%] patients). The most common clinical descriptions of ISRs included erythema (56%), irritation (33%), general reaction (33%), pruritis (22%), and rash (22%). No clinical activity (including stable disease) was observed in any of the s.c. patients and the overall median duration of treatment was limited to 30 days.
PK results with i.v. administration showed that the serum concentration profiles and parameter values (maximum plasma concentration [C max] and area under the curve) had an approximately dose‐proportional increase within the dose range (0.15–4.8 μg/kg), with a mean C max of 30.3 ng/ml observed at the highest full treatment dose in i.v. cohort 11 (4.8 μg/kg; Figure 1; Table S3).The terminal elimination half‐life was estimated in four of five patients after 4.8 μg/kg and averaged 39.6 (range: 23.8–46.5) h. With s.c. administration, a mean C max of 3.59 ng/ml was observed at the highest full treatment dose in s.c. cohort 2 (4.8 μg/kg). Based on ex vivo models, the exposure required to achieve even the low end of a potential therapeutic range was not clinically attainable due to the emerging safety profile. 10
FIGURE 1.
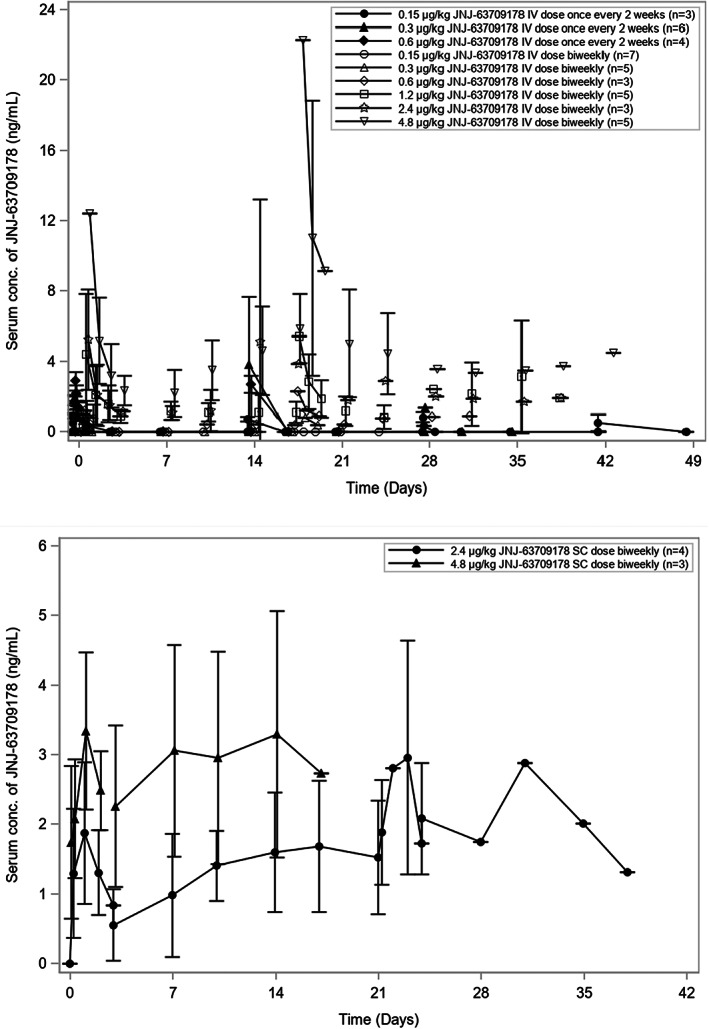
Mean serum concentration‐time curves of JNJ‐63709178. (a) Mean serum concentration following i.v. administration. The i.v. doses of JNJ‐63709178 were administered once every 2 weeks for cohort 3 (0.15 μg/kg), cohort 4 (0.30 μg/kg), and cohort 5 (0.6 μg/kg after priming dose of 0.3 μg/kg). Doses were administered twice weekly for cohort 6 (0.15 μg/kg after 4 priming doses of 0.15 μg/kg), cohort 7 (0.3 μg/kg after 4 priming doses of 0.15 μg/kg), cohort 8 (0.6 μg/kg after 4 priming doses of 0.15/0.30 μg/kg), cohort 9 (1.2 μg/kg after priming doses of 0.15, 0.3 and 0.6 μg/kg), cohort 10 (2.4 μg/kg after priming doses of 0.15, 0.3, 0.6, 1.2 μg/kg), and cohort 11 (4.8 μg/kg after priming doses of 0.15, 0.3, 0.6, 1.2 μg/kg and, if applicable, 2.4 μg/kg). Serum concentrations of JNJ‐63709178 were measured by an electrochemiluminescence‐based immunoassay. (b) Mean serum concentration following s.c. administration. The s.c. doses of JNJ‐63709178 were administered twice weekly for s.c. cohort 1 (2.4 μg/kg after priming doses of 0.3, 0.6, 1.2 μg/kg) and s.c. cohort 2 (4.8 μg/kg after priming doses of 0.6, 1.2, 2.4 μg/kg). Serum concentrations of JNJ‐63709178 were measured by an electrochemiluminescence‐based immunoassay. Mean concentration values are connected with lines from dose groups with at least three patients with samples taken at the specified times post dose. When mean concentrations fell below the limit of quantitation the dots could not be connected. Error bars = SD.
PD findings show that s.c. and i.v. administrations led to increases in multiple cytokines including IFN‐γ among others (IL‐6, IL‐10, and TNF‐α). A greater induction of cytokines (Figure S3A,B) was observed in patients with AEs including IRR, CRS, and elevated liver enzymes compared with patients with no events. The s.c. administration resulted in a decreased induction of cytokines and a delay in the timing of peak levels compared to i.v. administration (IV, end‐of‐infusion‐6 h postdose vs. s.c., 24‐72 h postdose), as peak levels were observed with last step‐up or full treatment dose levels following both i.v. and s.c. administrations. Whereas T‐cell activation in the periphery showed a correlation with CRS, a concomitant reduction in blasts was not sustained in bone marrow or periphery (Figures S2, S3E,F). The non‐sustainable reduction in blasts was likely multifactorial including the relatively short half‐life and limited exposures due to toxicity. Expression of CD123 on blasts were similar in patients with CRS versus no event (Figure S4).
CONCLUSIONS
JNJ‐63709178 was evaluated in 13 dosing cohorts (62 patients). The i.v. escalation was associated with a progressively unfavorable safety profile (including CRS and IRR), limited clinical activity, and suboptimal drug exposures which prompted a pivot to s.c. dosing. The s.c. administration was investigated at two doses (2.4 and 4.8 μg/kg); however, emergence of ISR prevented attainment of a therapeutic window, and the median duration on treatment was not improved. Although T cell activation following treatment suggested target engagement, this did not correlate with meaningful median duration of treatment or subsequent clinical activity as indicated by sustained expression of CD123 on peripheral blood blasts. Despite varying full treatment, step‐up dosing schedules, and routes of administration, an unfavorable safety profile and suboptimal PK profile (short half‐life; average exposures below the target half‐maximal effective concentration) were observed; therefore, an RP2D could not be identified.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript, designed and performed the research, and analyzed the data.
FUNDING INFORMATION
This work was supported by Janssen Research and Development, LLC.
CONFLICT OF INTEREST
N.D., L.F., J.G., C.G., X.L., T.T., and X.X. are employees of Janssen Research and Development and may hold company stock in the company. M.B. is an employee of Genentech and may hold stock in the company. M.R.G. has received consulting fees from Abbvie, Agios, Amgen, Astellas, Blueprint Medicines, Bristol Myers Squibb, Cardinal Health, CTI Biopharma, Daiichi Sankyo, Gamida Cell, Gilead, Incyte, Invitae, Karius, Ono Pharmaceutical, Pfizer, Pharmacosmos, Premier, Sierra Oncology, Stemline, and Trovagene; research support from Incyte, Genentech/Roche, and Janssen Research and Development; and owns stock in Medtronic. J.A.P.S. has participated in advisory boards and/or educational activities and/or grants supported by Janssen Research and Development, Jazz, BMS, Takeda, Novartis, Gilead, Alexion. O.S. has received honoraria from Jazz, BMS‐Celgene, Novartis, Pfizer, Abbvie. J.M.A.D. has received research support from Incyte, Pfizer, Astellas and Celgene. P.D. received research support from Janssen Research and Development, and his spouse is employed at Johnson & Johnson. W.D. has received grant funding from Abbvie, Aileron Therapeutics, Astex Pharmaceuticals, AstraZeneca, Bellicum Pharmaceuticals, Bristol‐Myers Squibb, Celgene, CTI Biopharma, Forma Therapeutics, Forty Seven, Genentech/Roche, H3 Biomedicine, Incyte, Janssen Research and Development, Karyopharm Therapeutics, Kite Pharma, MedImmune, Pfizer, PTC Therapeutics, Daiichi Sankyo, Seattle Genetics, Takeda, TCR2 Therapeutics, Celularity, Ryvu Therapeutics; provided consultancy to Abbvie, Amgen, PTC Therapeutics, Seattle Genetics. J.Y. has received research funding from AROG, Seagen and H3 Biomedicine. N.P. has received consulting fees from Pacylex Pharmaceuticals, ImmunoGen, Bristol Myers Squibb, Blueprint Medicines, Clearview Healthcare Partners, Astellas Pharmaceuticals, Protagonist Therapeutics, Triptych Health Partners, and CTI Biopharma; research support from Affymetrix and SagerStrong Foundation; honoraria from Incyte, Novartis, LFB Biotechnologies, Stemline Therapeutics, Celegene, AbbVie, MustangBio, Roche Diagnostics, Blueprint Medicines, DAVA Oncology, Springer Science and Business Media, Apptitude Health, NeoPharm Israel, and CareDx; and serves on the ASH Communications Committee and ASCO Leukemia Advisory Panel.
Supporting information
Table S1
ACKNOWLEDGMENTS
The authors would like to extend their gratitude to all the study participants and their caregivers. The authors thank the investigators and their study staff. Medical writing and editorial support were provided by Colleen Elliott, PhD (CME Science Writers, LLC).
Boyiadzis M, Desai P, Daskalakis N, et al. First‐in‐human study of JNJ‐63709178, a CD123/CD3 targeting antibody, in relapsed/refractory acute myeloid leukemia. Clin Transl Sci. 2023;16:429‐435. doi: 10.1111/cts.13467
REFERENCES
- 1. NIH, National Cancer Institure . Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia (AML); 2021. https://seer.cancer.gov/statfacts/html/amyl.html
- 2. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Munoz L, Nomdedeu JF, Lopez O, et al. Interleukin‐3 receptor alpha chain (CD123) is widely expressed in hematologic malignancies. Haematologica. 2001;86(12):1261‐1269. [PubMed] [Google Scholar]
- 4. Testa U, Riccioni R, Militi S, et al. Elevated expression of IL‐3Ralpha in acute myelogenous leukemia is associated with enhanced blast proliferation, increased cellularity, and poor prognosis. Blood. 2002;100(8):2980‐2988. [DOI] [PubMed] [Google Scholar]
- 5. Espinoza‐Gutarra MR, Green SD, Zeidner JF, Konig H. CD123‐targeted therapy in acute myeloid leukemia. Expert Rev Hematol. 2021;14(6):561‐576. [DOI] [PubMed] [Google Scholar]
- 6. Pemmaraju N, Konopleva M. Approval of tagraxofusp‐erzs for blastic plasmacytoid dendritic cell neoplasm. Blood Adv. 2020;4(16):4020‐4027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Slade MJ, Uy GL. CD123 bi‐specific antibodies in development in AML: what do we know so far? Best Pract Res Clin Haematol. 2020;33(4):101219. [DOI] [PubMed] [Google Scholar]
- 8. Uy GL, Aldoss I, Foster MC, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. 2021;137(6):751‐762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Venugopal S, Daver N, Ravandi F. An update on the clinical evaluation of antibody‐based therapeutics in acute myeloid leukemia. Curr Hematol Malig Rep. 2021;16(1):89‐96. [DOI] [PubMed] [Google Scholar]
- 10. Nair‐Gupta P, Rudnick SI, Luistro L, et al. Blockade of VLA4 sensitizes leukemic and myeloma tumor cells to CD3 redirection in the bone marrow microenvironment. Blood Cancer J. 2020;10(6):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Neuenschwander B, Branson M, Gsponer T. Critical aspects of the Bayesian approach to phase I cancer trials. Stat Med. 2008;27(13):2420‐2439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1