

Abstract
Introduction:
Necrotizing enterocolitis (NEC) is a gastrointestinal disease of premature neonates. We previously validated a NEC enteroid model derived from human infant intestinal tissue. Typical enteroid configuration is basolateral-out (BO) without direct access to the luminal (apical) surface. Apical access is necessary to allow physiologic comparison of pathogen interaction with the intestinal epithelial barrier. We hypothesize that apical-out (AO) enteroids will provide a relevant NEC model to study this relationship.
Methods:
Following institutional review board approval (#11610-11611), neonatal intestinal tissue was collected from surgical specimens. Stem cells were collected, enteroids were generated and grown to maturity in BO conformation then everted to AO. Enteroids were untreated or treated for 24 hours with 100ug/mL lipopolysaccharide (LPS) and hypoxia. Protein and gene expression were analyzed for inflammatory markers, tight junction (TJ) proteins and permeability characteristic of NEC.
Results:
Apical TJ protein zonula occludens-1 (ZO-1) and basolateral protein β-catenin immunofluorescence confirmed AO configuration. Treated AO enteroids had significantly increased mRNA (p=0.001) and protein levels (p<0.0001) of TNF-α compared to controls. Corrected total cell fluorescence of TLR-4 was significantly increased in treated AO enteroids compared to control (p=0.002). Occludin was found to have significantly decreased mRNA in treated AO enteroids (p=0.003). Expression of other TJ proteins claudins-1, −4 and ZO-1 was significantly decreased in treated AO enteroids (p<0.05).
Conclusion:
AO enteroids present an innovative model for NEC with increased inflammation and gut barrier restructuring. This model allows for a biologically relevant investigation of the interaction between pathogen and the intestinal epithelial barrier in NEC.
Keywords: necrotizing enterocolitis, enteroids, apical-out, model
Introduction
Necrotizing enterocolitis (NEC) is a devastating disease that primarily affects the gastrointestinal tract of premature neonates. The pathophysiology of NEC is multifactorial and includes intestinal microbial dysbiosis, a hyperinflammatory response and a dysfunctional intestinal epithelial barrier (1). Cronobacter sakazakii (previously known as Enterobacter sakazakii) is a gram-negative pathogen that has been associated with outbreaks of NEC in infants (2). C. sakazakii is resistant to desiccation, which has contributed to its ability to contaminate dry infant formula (3). It has been linked to several cases of NEC, with the most recent outbreak occurring in May 2022 in the United States, resulting in four illnesses and two deaths (4). C. sakazakii is able to modulate pyroptotic cellular responses leading to increased inflammation seen in NEC, specifically demonstrated with upregulation of TLR4 signaling (5). It has also been demonstrated to increase intestinal epithelial injury through induction of apoptosis (6). Understanding the interaction between pathogens that are relevant to human disease, and their host intestine are useful tools in advancing our understanding of the pathogenesis of NEC.
The hallmarks of NEC include increased inflammatory markers such as tumor necrosis factor (TNF)-α and interleukin (IL)-1β, increased intestinal permeability and a breakdown of the intestinal epithelial barrier associated with a decrease in select tight junction (TJ) protein expression (1). Although there have been many animal models of NEC, the utility of these systems is limited by species-specific differences (7). Immortalized human cell lines are another model for NEC. However, these lack a complex, three-dimensional structure with diverse epithelial cell types allowing for a more physiologic study of this disease process (8).
Enteroids bridge this gap in that they can be human-derived, develop a three-dimensional assembly mimicking in vivo conditions and contain all the major intestinal epithelial cell types (9). Our lab has previously demonstrated that enteroids derived from premature neonates provide an in vitro model for NEC which more closely imitates clinical NEC when compared to two-dimensional human primary cell lines and animal models (10, 11).
Enteroids maintain the characteristics of their host and retain physiologic polarity organizing into a three-dimensional structure with apical and basolateral surfaces (8, 9). Typically, in vitro enteroids are grown in a basolateral-out (BO) conformation, where the apical side faces the enclosed luminal compartment and the basolateral surface is exposed to the surrounding media (12). In vitro induction of NEC in enteroids involves hypoxia and the addition of either bacteria, such as C. sakazakii, or lipopolysaccharide (LPS) into the media surrounding the basolateral surface. Conversely, in vivo, bacteria and their products come in contact with the apical surface of epithelial cells lining the intestine. It is this interaction between the apical surface and these pathogens, or their by-products, that initiates the cascade of events which ultimately leads to NEC (13). The challenge with the traditional BO enteroid NEC model is acquiring experimental access to the luminal compartment.
Co et al. were the first to describe the ability to reverse the polarity of enteroids so that the apical surface comes in contact with the surrounding media (12). They demonstrated that these apical-out (AO) enteroids maintain correct epithelial polarity, contain all the major intestinal cell types, preserve epithelial barrier integrity and provide easy access to the apical (luminal) surface (12).
Ours is the first study to investigate the role of AO enteroids in the study of NEC. Our lab has previously shown the utility of using enteroids as a model for NEC. Thus, we hypothesize that AO enteroids will provide an innovative NEC model allowing easier access to the apical surface providing benefit in studying pathogen-host interactions.
Material and Methods
Human Samples
After obtaining IRB approval (IRB #11610-11611) and parental consent, human infant intestinal tissue was collected from patients undergoing bowel resections at Oklahoma Children’s Hospital (Oklahoma City, OK). Specimens were collected from patients undergoing surgery for NEC or other intestinal surgery such as intestinal atresia repair. Gestational age, intestinal section (i.e., ileum), and type of surgical procedure were recorded for each specimen. Enteroids derived from six patients were used for experimentation. Enteroids were derived from both male and female patients. Corrected gestational age (cGA) of infants at the time of collection ranged from 36 weeks and 1 day to 51 weeks and 5 days. All but one patient had a history of prematurity with estimated gestational age at birth ranging from 25 weeks and 1 day to 34 weeks. The one patient without a history of prematurity was collected at 2 months of age. Among these, two infants (both with a history of prematurity) underwent resection due to NEC. The remaining four infants underwent resection for gastroschisis, ostomy takedown or intestinal atresia. Tissue specimens were divided following collection and subsequently snap frozen in liquid nitrogen or washed in cold Dulbecco’s Phosphate Buffered Saline (DPBS, Sigma Life Science, #D1408, St. Louis, MO) then kept in RPMI media (Gibco, #11875-093, Waltham, MA) for 24 hours at 4°C until processed for enteroid culture.
In Vitro Human Neonatal Enteroid Cultures
Crypts were isolated from intestinal tissue samples and cultured for enteroid generation within 24 hours of collection. The protocol used for crypt isolation and enteroid processing was previously described by our lab (11). Enteroid cultures were suspended in Matrigel (Corning, #CB-40230C, Corning, NY) domes and grown in 50% L-WRN conditioned media prepared in our lab, supplemented with 50ng/mL Epidermal Growth Factor (EGF, Millipore Sigma, #GF144, Burlington, MA), 1mM N-Acetylcysteine (Millipore Sigma, #A9165-5G, Burlington, MA), 500nM A-83-01 (R&D Systems, #2939/10, Minneapolis, MN), 10μM SB202190 (Millipore Sigma, #S7067-5MG, Burlington, MA), 10mM Nicotinamide (Millipore Sigma, #N0636-100G, Burlington, MA), and 10nM [leu] 15-gastrin 1 (Millipore Sigma, #G9145-.1MG, Burlington, MA). Enteroids were passaged every 5–10 days and used at maturity, between passages 4 and 22 for all experiments.
Enteroid NEC Experimentation
Enteroid cultures were grown embedded in basement membrane matrix (Matrigel, Corning, #CB-40230C, Corning, NY) with growth media for 7–20 days. Untreated enteroids from patients of comparable cGA with active NEC at the time of resection were compared to those derived from patients without NEC. For all experiments, there were approximately 300 enteroids grown per well. Three of these wells were pooled into one sample which was then repeated in duplicate or triplicate. All experiments were repeated for a minimum of two separate experiments for validation. At maturation, enteroids underwent transformation to the AO configuration and subsequent NEC experimentation. Individual NEC experiments were run comparing untreated versus 100μg/mL lipopolysaccharide (LPS) and hypoxia treated enteroids derived from the same patient to ensure adequate control. Enteroids derived from five different patients were used for all NEC experiments (both comparing between NEC and non-NEC patients as well as comparing between control and treated from the same patient). Two of the five patients had intestinal resection due to NEC. The remaining three patients were resected for gastroschisis, ostomy takedown and intestinal atresia. All five of these patients had a history of prematurity and their cGA at the time of collection ranged from 36 weeks 1 day to 51 weeks 5 days. Enteroids derived from a term neonate was used to determine hypoxia in enteroids via qRT-PCR for hypoxia inducible factor (HIF)-1α.
Apical-Out Enteroid NEC Model
Enteroid cultures were grown in the BO configuration and embedded in basement membrane matrix (Matrigel, Corning, #CB-40230C, Corning, NY) with growth media for 7–20 days. Subsequently, the media was removed and 5mM Ethylenediaminetetraacetic acid (EDTA, Millipore Sigma, #EDS-500G, Burlington, MA) in PBS was added then placed on a rotator for 1 hour at 4°C to solubilize. After centrifugation at 200g for 3 minutes at 4°C, the supernatant was discarded and the enteroid pellet was resuspended in growth media in ultra-low attachment 24-well tissue culture plates (Corning Costar #3473, Glendale, AZ). The enteroids were then incubated at 37°C with 5% carbon dioxide for 72 hours.
NEC was induced in the treatment group via exposure to 24 hours of 100μg/mL LPS (Millipore Sigma, #L260-25MG, Burlington, MA) and 6–24 hours of hypoxic conditions (1% oxygen, 5% carbon dioxide, 94% nitrogen). Controls were maintained at 37°C under normoxic conditions (room air) with 5% carbon dioxide.
Bacterial Culture and GFP Insertion into Cronobacter sakazakii
C. sakazakii strains BAA-894 and 29544 (obtained from ATCC) were cultured on solid luria broth (LB) agar. Both C. sakazakii strains were prepared for electroporation following the protocol in the MicroPulser Electroporation Apparatus user manual (BioRad, Hercules, CA). Prepared C. sakazakii bacteria were transformed with the pRSET/EmGFP (Invitrogen, Waltham, MA) plasmid using the MicroPulser Electroporation Apparatus. C. sakazakii colonies were selected for transformants by growing on LB + 100μg/mL ampicillin plates and GFP expression was confirmed by observation under an Olympus BX43 fluorescent microscope. A 5mL culture of LB + ampicillin was inoculated with an overnight culture of transformed C. sakazakii and grown to early- to mid-log phase. The OD600 was used to determine bacterial culture concentration. Mature AO enteroids were treated with 3×105 CFUs/mL of bacteria and incubated for 3 hours. Enteroids were fixed, stained for F actin (Alexa Fluor™ 647 Phalloidin, Invitrogen A22287, 1:400 dilution, Waltham, MA) and evaluated as a whole mount using an Olympus BX43 to qualitatively assess association of bacteria to the apical surface.
Cronobacter sakazakii Treatment and Adherence
To test for bacterial adherence to the apical surface of enteroids, polycarbonate transwell inserts (Corning, #3413, Glendale, AZ) were incubated in basement membrane matrix (Matrigel, Corning, #CB-40230C, Corning, NY) diluted 1:20 in DMEM/F12 media (Life Technologies Corporation, #11320–033, Grand Island, NY) for two hours prior to seeding. Mature NEC and non-NEC derived enteroids were seeded to the transwells in duplicate. After reaching confluence at 8 days, intestinal monolayers were treated with 1×107 CFU/mL of C. sakazakii. Treated cultures were incubated for 3 hours then washed four times with media and twice with DPBS to remove free floating bacterial cells. Transwell membranes were carefully removed from the plate inserts and washed in 0.4% Triton X (Invitrogen, #HFH10, Waltham, MA) for 8 minutes to lyse enteroids. The membrane surface was scraped with a cell scraper to suspend all cells in preparation for plating. Cell suspensions were serially diluted, plated to LB plates, and incubated overnight. Colony counts were recorded the following day. The experiment was repeated with the C. sakazakii strain BAA-894.
Escherichia coli Treatment and Adherence
Mature BO enteroids from NEC and non-NEC derived premature neonatal intestinal samples were grown to maturity in basement membrane matrix domes (Matrigel, Corning, #CB-40230C, Corning, NY). Three days prior to bacterial treatment, half of the wells were treated with Corning Cell Recovery Solution (CRS, Corning, #354253, Glendale, AZ) and incubated at 4°C to dissolve the domes. Enteroids were then transferred to microcentrifuge tubes and centrifuged at 300g for 3 minutes. Supernatant was removed and enteroids were resuspended in 50% LWRN media. Suspensions were transferred to ultra-low attachment plates (Corning, #3473) and placed in a CO2 incubator for 3 days to allow the enteroids to flip from a BO to AO configuration. For determination of multiplicity of infection (MOI), a well from each AO and BO group was harvested for cell counting. Domes for BO groups were dissolved in CRS (Corning, #354253, Glendale, AZ). BO and AO enteroids were then dissociated to single cells using a 27-gauge needle and treatment with Trypsin-EDTA (0.25%). Live cells were counted using 0.4% Trypan Blue stain (Invitrogen, #T10282) and a Countess3 automated cell counter (Invitrogen). Remaining BO wells were dissolved in CRS then both BO and AO enteroids were pelleted by centrifugation at 300g for 3 minutes. Supernatant was removed and enteroids were resuspended in 500uL of media containing 250 μg/mL of ampicillin and E. coli (ATCC 35150GFP) at a MOI = 100. Suspensions were transferred to ultra-low attachment 24-well plates (Corning, #3473) and incubated at 37°C for 3 hours. One well from each group was left untreated to serve as a negative control. Additionally, wells containing E. coli only were incubated to serve as wash controls for each group at the respective bacterial concentration. Following treatment, the contents of each well were transferred to microcentrifuge tubes. Enteroids were allowed to settle by gravity then the supernatant was discarded. Enteroids were resuspended in 500μL antibiotic free media and allowed to settle by gravity. After removing the supernatant, the enteroids were again washed in antibiotic free media five additional times to remove non-adhered bacteria. Following the final wash, one tube of enteroids from each group was set aside for immunofluorescent microscopy via whole mount. For the remaining samples, supernatant was removed and enteroids were resuspended in 500μL of 0.4% Triton X and incubated at room temperature for eight minutes. Enteroids were further dissociated by drawing through a 27-gauge needle eight times. Suspensions were then serially diluted, plated to LB plates, and incubated overnight at 37°C. Colony counts were taken the following day. Enteroids saved for whole mount were fixed and stained for F actin (Alexa FluorTM 647 Phalloidin, Invitrogen A22287, 1:400 dilution, Waltham, MA) and 1 μg/mL DAPI (Millipore Sigma, 50-874-10001).
Immunofluorescence Microscopy
AO and BO enteroids were harvested for both whole mount and paraffin embedding. For whole mount, AO enteroids were centrifuged at 300g for 3 minutes and media was removed. BO enteroids had media removed and then were placed in 500μL of Corning Cell Recovery Solution (Corning, #354253, Glendale, AZ) at 4°C for 30–45 minutes to dissolve the basement membrane dome. Both AO and BO enteroids were resuspended in basement membrane matrix and spread in a thin layer over a microscope slide. This was allowed to solidify at room temperature for 15 minutes. Slides were then fixed and stained with the primary antibody to the apical protein zonula occludens (ZO)-1 (Cell Signaling, #D6L1E, rabbit, 1:200 dilution, Danvers, MA), basolateral protein β-catenin (Cell Signaling, #14-2567-82, mouse, 1:100 dilution, Danvers, MA) or TLR-4 (Santa Cruz, #sc-293072, mouse, 1:50 dilution, Dallas, TX) overnight at 4°C. They were then treated with a secondary antibody (Invitrogen #A11032, Alexa Fluor 594 goat anti-mouse and/or Invitrogen #A11034 goat anti-rabbit both 1:1000, Waltham, MA) for 1 hour. Slides were imaged using an Olympus BX43 microscope (Olympus, Tokyo, Japan). Paraffin embedding and imaging of the slides was performed per our previously described protocol (11).
Quantification of immunofluorescence was calculated via corrected total cell fluorescence (CTCF) using ImageJ software (14).
RNA Isolation, Reverse Transcription and qRT-PCR
Total RNA was isolated using TRIzol™ (Life Technologies, Carlsbad, CA) and quantified using a NanoDrop Lite spectrophotometer (Thermo Scientific, Waltham, MA). RNA was reverse transcribed to 0.5–2μg of cDNA using the high capacity cDNA reverse transcription kit (Applied biosystems, #4368814, Waltham MA). Real time PCR results were obtained using the CFX Opus 96 system and iQ SYBR Green Supermix (BioRad, Hercules, CA) with 4ng of cDNA template and a 0.5μM final primer concentration. Expression levels were quantitated using the 2−ΔΔCT method and normalized to GAPDH. The following primers were used for amplification of target cDNA: HIF1-α, 5’ TTCACCTGAGCCTAATAGTCC 3’ and 5’ CAAGTCTAAATCTGTGTCCTG 3’; TNF-α, 5’ CTCGAACCCCGAGTGACAAG 3’ and 5’ TGAGGTACAGGCCCTCTGAT 3’; IL-8, 5’ GGCCGTGGCTCTCTTGGCAG 3’ and 5’ TGTGTTGGCGCAGTGTGGTCC 3’; occludin, 5’ TCAGGGAATATCCACCTATCACTTCAG 3’ and 5’ CATCAGCAGCAGCCATGTACTCTTCAC 3’; claudin-1, 5’ TGAGGATGGCTGTCATTGGG 3’ and 5’ AAAGTAGGGCACCTCCCAGA 3’; claudin-3, 5’ GCCACCAAGGTCGTCTACTC 3’ and 5’ CCTGCGTCTGTCCCTTAGAC 3’; claudin-4, 5’ GGCCGGCCTTATGGTGATAG 3’ and 5’ AGTAAGGCTTGTCTGTGCGG 3’; ZO-1, 5’ AGGGGCAGTGGTGGTTTTCTGTTCTTT 3’ and 5’ GCAGAGGTCAAAGTTCAAGGCTCAAG 3’; GAPDH, 5’ ACCACAGTCCATGCCATCAC 3’ and 5’ TCCACCACCCTGTTGCTGTA 3’. Data shown are fold changes of target gene compared to GAPDH.
Protein Isolation, Western Blot and Enzyme Linked Immunosorbent Assay (ELISA)
Collected enteroid samples were homogenized via sonification on ice with RIPA buffer (Cell Signaling #9806S, Danvers, MA) supplemented with phosphatase inhibitor and protease inhibitor cocktail (Sigma-Aldrich, #P5726 and #P8340, St. Louis, MO). The homogenate was centrifuged, supernatant quantified via Pierce™ BCA protein assay kit (ThermoScientific, #23225, Waltham, MA) and 8–10μg of total protein was loaded into each well of a 10–12% SDS-page gel. After electrophoresis and transfer, the nitrocellulose membrane was blocked with 5% dry milk in phosphate buffered saline with tween-20 (PBST) buffer. Primary antibodies were added to the membranes and allowed to incubate at 4°C overnight. Following washing with PBST, secondary antibodies were added and allowed to incubate at room temperature for 1 hour. Primary antibodies used were occludin (Invitrogen, #33–1500, mouse, 0.5μg/mL dilution, Waltham, MA and Invitrogen, #71–1500, rabbit, 2μg/mL dilution, Waltham, MA), claudin-1 (abcam, #AB15098, rabbit, 1:1000 dilution, Cambridge, UK), TLR-4 (Santa Cruz, #sc-293072, mouse, 1:1000 dilution, Dallas, TX), GAPDH (Cell Signaling, #2118L, rabbit, 1:1000 dilution, Danvers, MA) and β-actin (Cell Signaling, #8H10D10, mouse, 1:1000 dilution, Danvers, MA). Secondary antibodies used were horse anti-mouse IgG HRP-linked antibody (Cell Signaling, #7076S, 1:1000– 1:10,000, Danvers, MA) and goat anti-rabbit (Invitrogen #31460, 1:10,000 dilution, Danvers, MA). Membrane development, band detection and imaging were conducted as previously reported (15).
Enzyme linked immunosorbent assay (ELISA) was performed on enteroid media for TNF-α per the standard kit protocol (Invitrogen, #KCH3011, Waltham, MA).
Intestinal Permeability Assessment
Collected mature AO enteroids were centrifuged at 300g for 3 minutes. The media was removed and enteroids were washed twice with 500μL of warm DPBS. Following wash steps, 500μL of warm growth media with 50μL of 2.5mg/mL Lucifer Yellow (Cat # L-453, ThermoFisher) was added to each well and incubated for 2 hours. The enteroids were then centrifuged at 300g for 3 minutes with gentle removal of the media with Lucifer Yellow without dissociating the enteroids. The enteroids were then washed 4 times with 500μL of warm DPBS followed by centrifugation at 300g for 3 minutes each time. Each well was resuspended in 1000μL of DPBS and the enteroids were lysed by vigorous pipetting to release the intraluminal Lucifer Yellow. 150μL of each sample was used to measure Lucifer Yellow fluorescence intensity in triplicate on a plate reader with excitation at 428nm and emission at 536nm (SpectraMax iD3, Molecular Devices, San Jose, CA).
Statistical Analysis
Graph Pad Prism 9.2.0 was used for statistical analysis and graphing the results. Continuous data was tested for a normal distribution using the Shapiro-Wilk normality test. Student’s t-test and analysis of variance (ANOVA) were used as appropriate for data with a normal distribution. For non-normally distributed data, a Wilcoxon rank sum test was performed. Statistical significance was accepted if p<0.05.
Results
Enteroid Characteristics from NEC and non-NEC Samples
Untreated, BO enteroids derived from NEC and non-NEC samples were evaluated prior to experimentation for inflammatory markers and TJ proteins to document baseline characteristics. Untreated enteroids derived from infants with NEC demonstrated significantly decreased gene expression of the TJ proteins occludin and claudin-1 when compared to non-NEC derived enteroids (p<0.05, n=3 in each group, Figure 1A). This was confirmed via western blot (p<0.05, n=3 in the control group, n =2 in the NEC group, Figure 1B, 1C and 1D). The inflammatory markers TNF-α and IL-8 were not significantly increased (p=0.42 for both, n=3 in each treatment group, Figure 1E). Protein analysis via ELISA confirmed these results (p=0.6, n=3 in each group, Figure 1F). The proliferation marker, ki-67, was significantly decreased in NEC derived enteroids compared to non-NEC derived enteroids (p=0.04, n=3 in each group, Figure 1E). Due to these innate differences, enteroids derived from the same patient were used as an untreated control compared to LPS and hypoxia treated enteroids for all NEC experiments. This allowed for an internal control to ensure that only those characteristics that changed in response to treatment were captured.
Figure 1.
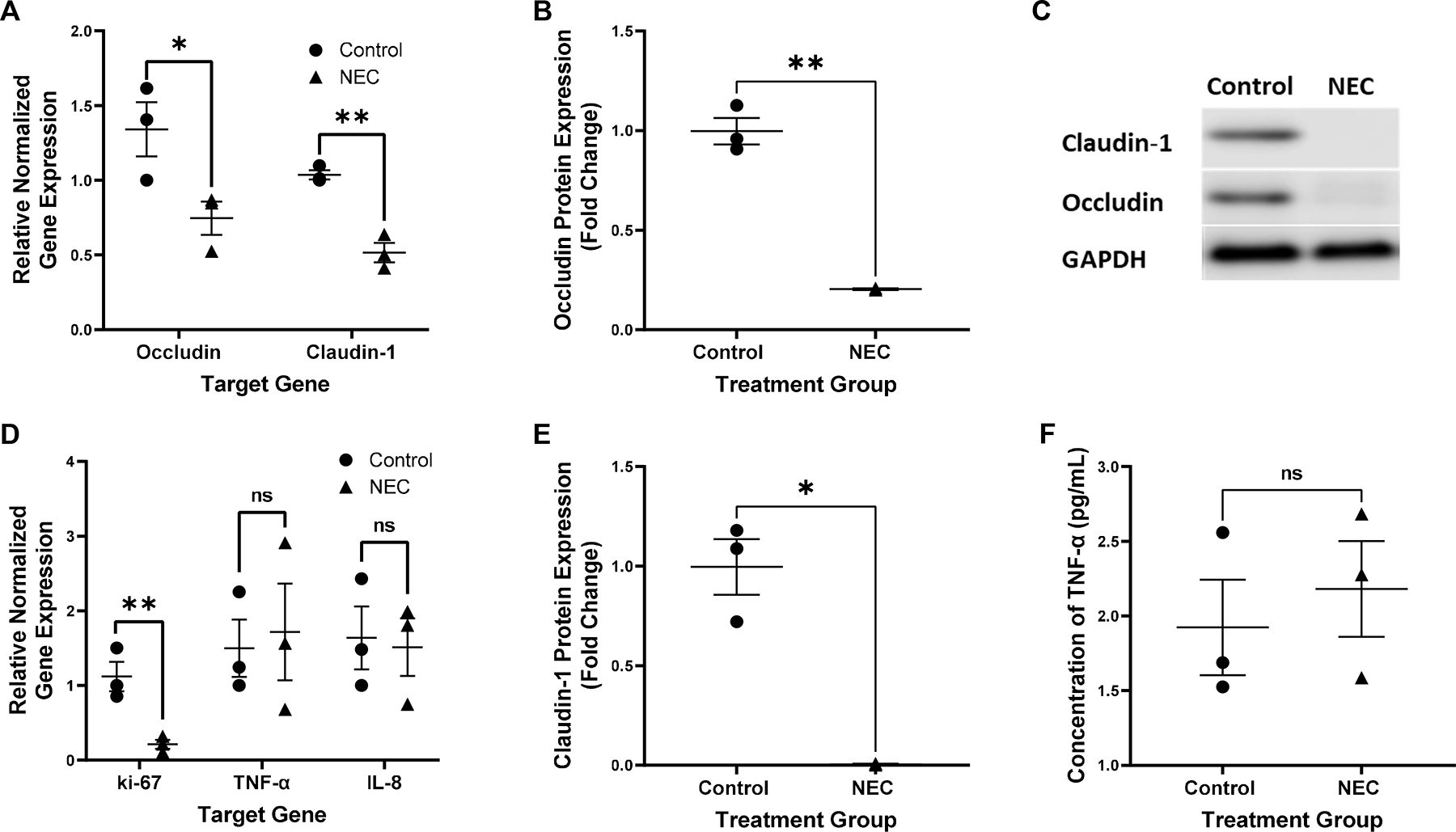
Relative gene expression of occludin and claudin-1 normalized to GAPDH in control vs untreated NEC derived enteroids, *p=0.04 for occludin, **p=0.006 for claudin-1, n=3 in each treatment group (A). Expression of the protein occludin relative to control and normalized to GAPDH in control vs NEC derived enteroids, **p=0.003, n=3 in the control group, n=2 in the NEC group (B). Representative western blot of occludin, claudin-1 and GAPDH in control vs NEC derived enteroids, n=3 in the control group, n=2 in the NEC group (C). Expression of the protein claudin-1 relative to control and normalized to GAPDH in control vs NEC derived enteroids, *p=0.01, n=3 in the control group, n=2 in the NEC group (D). Relative gene expression of ki-67, TNF-α and IL-8 normalized to GAPDH in control vs untreated NEC derived enteroids, *p=0.04 for ki-67, p=0.42 for TNF-α and IL-8, n=3 for each treatment group (E). Concentration of TNF-α in the media of untreated control vs NEC derived enteroids, p=0.60, n=3 in each group (F).
Induction of Apical-Out Enteroid Conformation
Immunofluorescence of AO enteroids demonstrated eversion of the luminal surface after 72 hours in suspension compared to BO enteroids maintained in basement membrane matrix (Figure 2). This was confirmed in both whole mount and paraffin sections where the apical protein, ZO-1 (green), is visualized on the outer surface of the AO enteroid. The basolateral protein, β-catenin (red), is seen on the inner surface of the AO enteroid. This conformation is reversed in enteroids maintained in basement membrane matrix.
Figure 2.
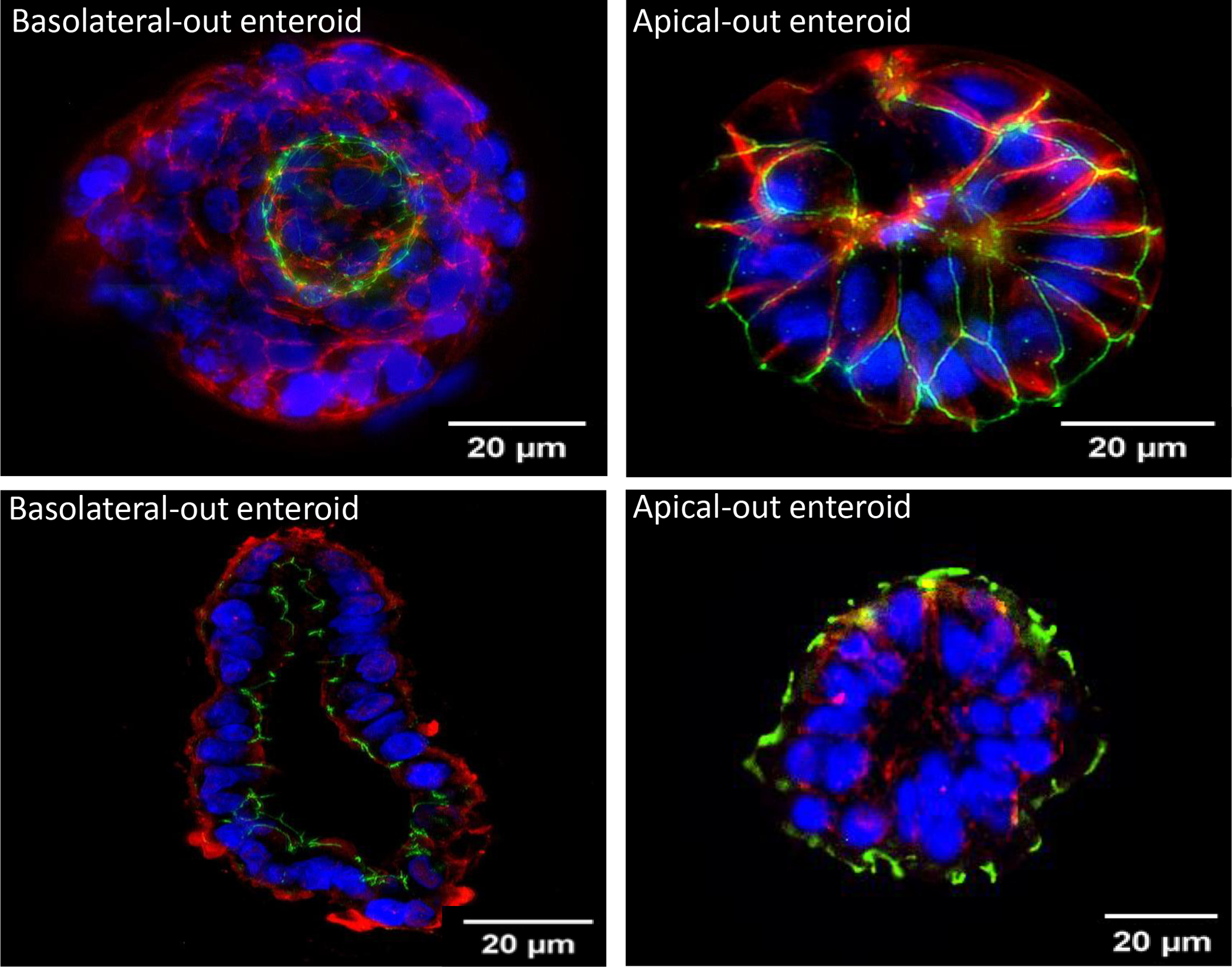
Immunofluorescence of whole mount basolateral-out (BO) vs apical-out (AO) enteroids imaged at 20μm (Top). Immunofluorescence of paraffin embedded BO vs AO enteroids imaged at 20μm (Bottom). Green: Zonula occludens-1, Red: β-catenin, Blue: DAPI.
Hypoxia Confirmed in Treated Apical-Out Enteroids
AO enteroids cultured for 24 hours in hypoxic conditions (1% oxygen, 5% carbon dioxide, 94% nitrogen) showed significantly decreased gene expression of HIF-1α compared to AO enteroids maintained in normoxic conditions (p=0.04, n=3 in each treatment group, Figure 3A). HIF-1α has frequently been used as a marker for hypoxia although the pathways are incompletely understood. It is constitutively transcribed and translated under normoxic conditions where it undergoes hydroxylation to allow it to bind to the ubiquitin ligase complex ultimately resulting in proteasomal degradation leading to low levels of HIF-1α (Figure 3B) (16). Hypoxic conditions inhibit hydroxylation of HIF-1α which stabilizes it, allowing build-up of the protein in the cytosol. Under sustained hypoxia, HIF-1α binds to HIF-1β which then moves into the nucleus and acts as negative feedback to suppress transcription of HIF-1α (16, 17). This results in decreased messenger RNA (mRNA) expression alongside increased protein levels of HIF-1α.
Figure 3.
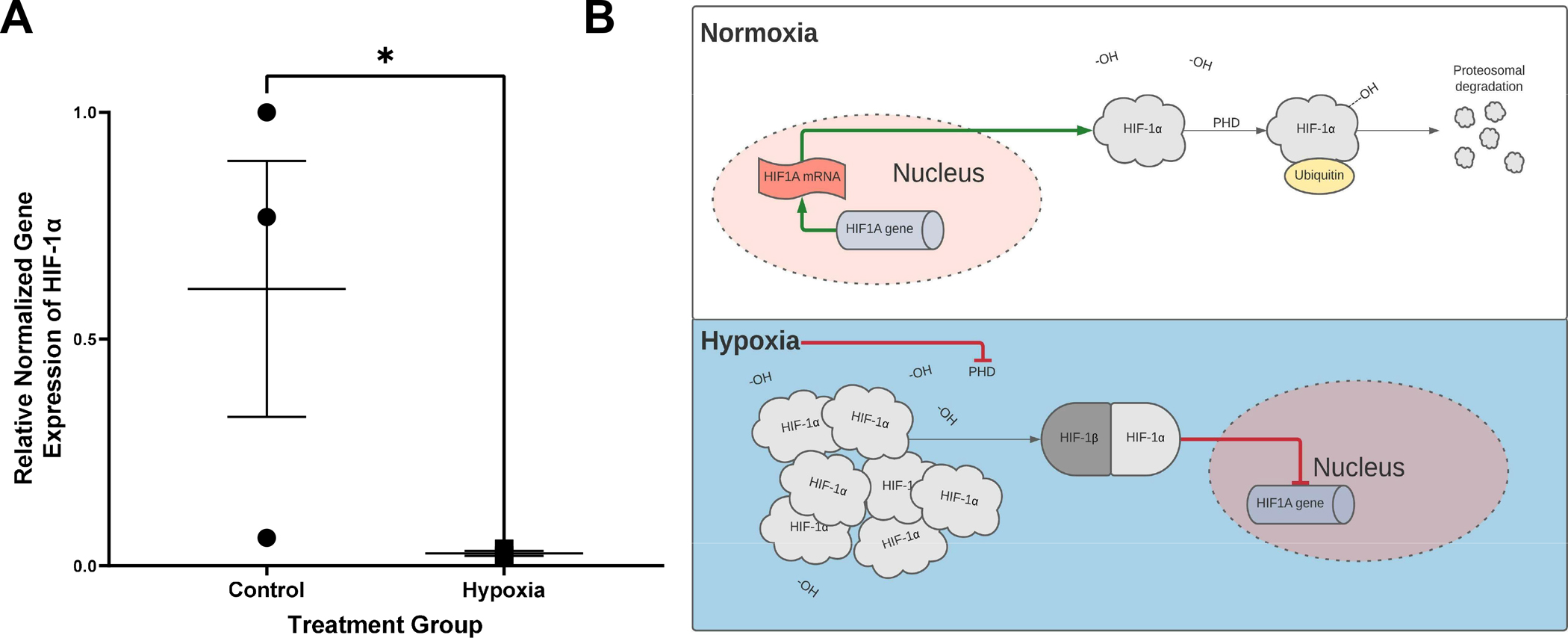
Relative gene expression of hypoxia inducible factor 1α (HIF-1α) normalized to GAPDH, *p=0.04, n=3 in each treatment group (A). Proposed mechanism for HIF-1α regulation leading to decreased gene expression in hypoxic conditions (B).
Treated Apical-Out Enteroids Show Increased Inflammatory Markers Similar to NEC
Necrotizing enterocolitis is known to cause a hyperinflammatory response resulting in increased levels of inflammatory markers such as TNF-α and IL-1β. Treated AO enteroids demonstrated similar findings of increased protein levels of TNF-α when compared to controls (p=0.0003, n=3 in each treatment group, Figure 4A). This was confirmed via significantly increased mRNA expression of TNF-α in treated versus untreated AO enteroids (p=0.0009, n= 3 in each treatment group, Figure 4B). Gene expression of the inflammatory marker, IL-1β was also significantly increased in treated AO enteroids compared to controls (p=0.006, n=3 in each treatment group, Figure 4C). Increased TLR-4 is another well described marker of necrotizing enterocolitis (18). Treated AO enteroids had significantly increased levels of TLR-4 immunofluorescence compared to untreated controls (p=0.002, n=5 in the control group, n=3 in the treated group, Figure 5).
Figure 4.
Protein concentration of TNF-α in control vs treated apical-out (AO) enteroids via ELISA, ***p=0.0003, n=3 in each treatment group (A). Relative gene expression of TNF-α in control vs treated AO enteroids, ***p=0.0009, n=3 in each treatment group (B). Relative gene expression of IL-1β in control vs treated AO enteroids, **p=0.006, n=3 in each treatment group (C).
Figure 5.
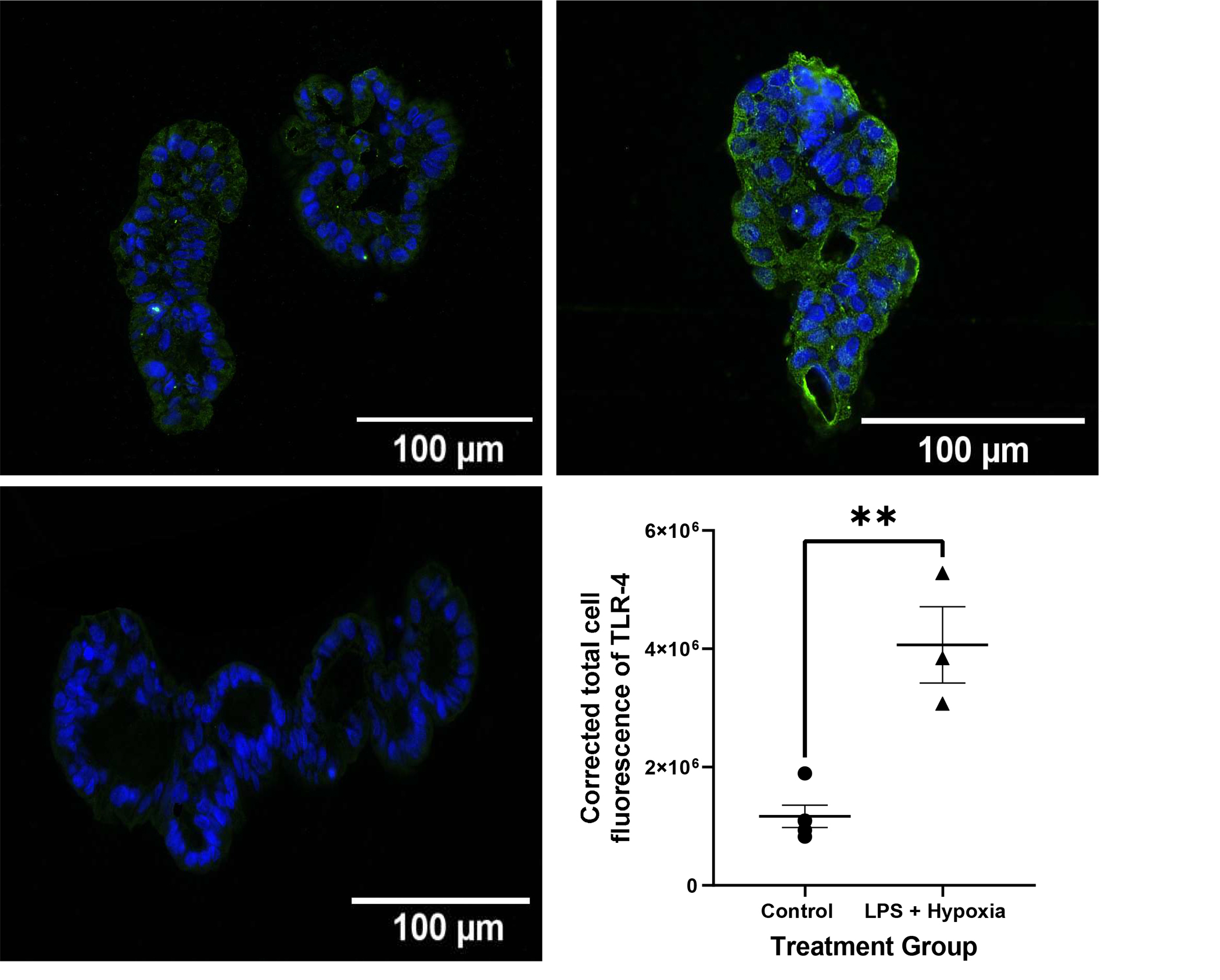
Representative immunofluorescence of paraffin embedded apical-out (AO) control vs LPS and hypoxia treated enteroids imaged at 20X (Top). Corrected total cell fluorescence (CTCF) of TLR-4 in control vs LPS + hypoxia treated AO enteroids, **p=0.002, n=5 in the control group, n=3 in the treated group (Bottom Right). AO enteroid treated with secondary antibody only as a negative control (Bottom Left). Green: TLR-4, Blue: DAPI.
Treated Apical-Out Enteroids Model Decreased TJ Proteins Similar to NEC
Our lab has previously demonstrated that a key feature in the pathophysiology of necrotizing enterocolitis is the breakdown of the intestinal epithelial barrier seen as a decrease in TJ proteins such as occludin, claudin-1, −4 and ZO-1 (19). Treated AO enteroids showed significantly decreased mRNA expression of occludin assessed by qRT-PCR (P=0.008, n=3 in each treatment group, Figure 6A) and was confirmed by significantly decreased protein levels of occludin compared to controls as demonstrated by western blot (p=0.02, n=3 in each treatment group, Figure 6B and 6C). Significantly decreased levels of TJ proteins claudins-1, −4 and ZO-1 were also demonstrated by qRT-PCR (p<0.05, n=3 in each treatment group, Figure 6D).
Figure 6.
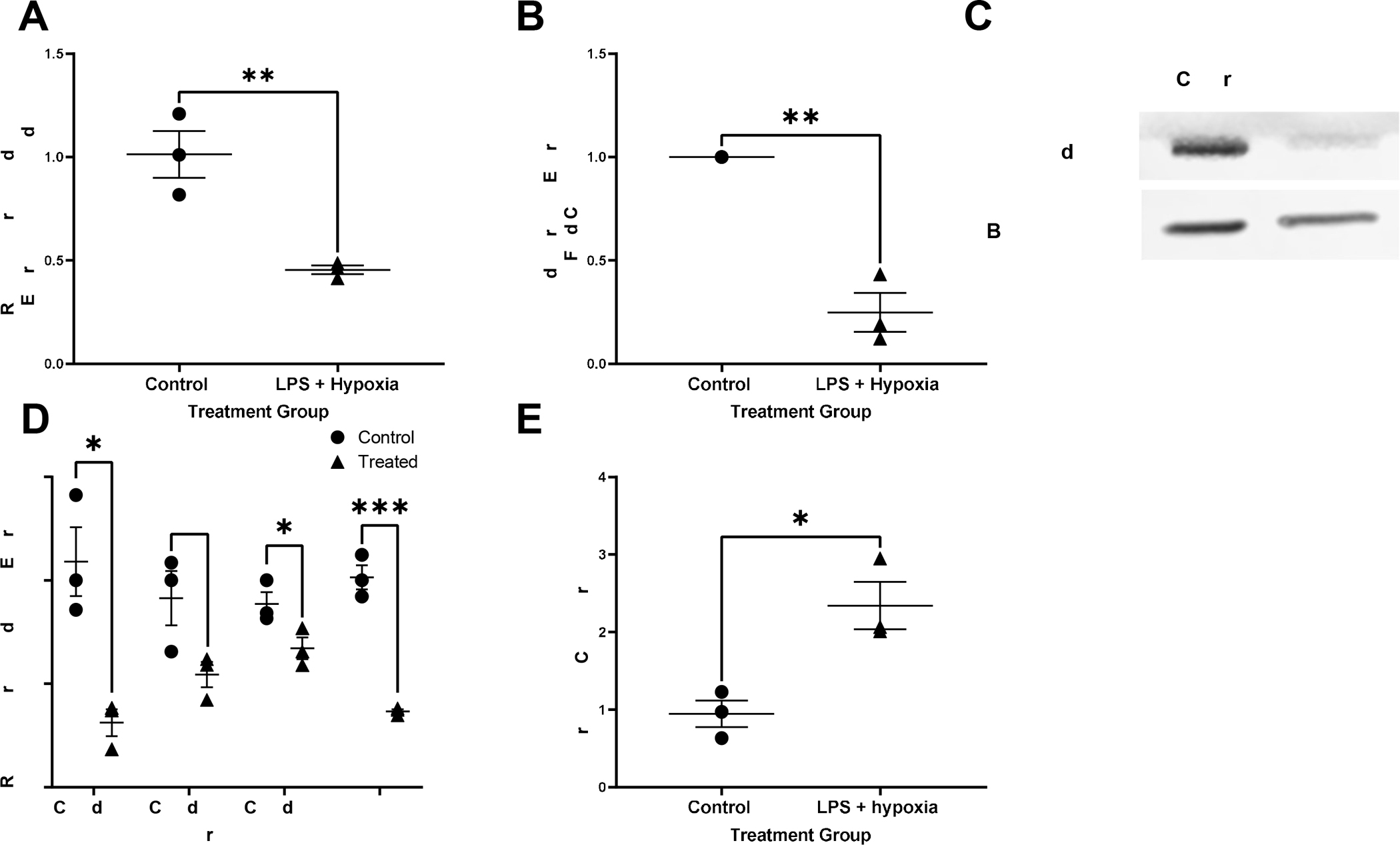
Relative gene expression of occludin normalized to GAPDH in control vs treated apical-out (AO) enteroids, **p=0.008, n=3 in each treatment group (A). Expression of the protein occludin relative to untreated control and normalized to beta-actin in control vs treated AO enteroids, *p=0.001, n=3 in each treatment group (B). Representative western blot of occludin and beta-actin in AO control versus treated enteroids (C). Relative gene expression of tight junction proteins claudins-1,3 and 4 as well as zonula occludens (ZO)-1 normalized to GAPDH in control vs treated AO enteroids, *p=0.01 for claudin-1, p=0.06 for claudin-3, *p=0.05 for claudin 4, ***p=0.0004 for ZO-1, n=3 in each treatment group (D). Increased permeability seen in treated AO enteroids compared to control demonstrated via Lucifer yellow assay, *p=0.02, n=3 in each treatment group (E).
Treated Apical-Out Enteroids Demonstrate Increased Permeability Similar to NEC
Another feature of NEC is increased intestinal permeability (20). Lucifer Yellow permeability assay is used to test for paracellular permeability. This assay demonstrated that treated AO enteroids had significantly increased permeability compared to untreated controls (p=0.02, n=3 in each treatment group, Figure 6E).
C. sakazakii Adheres to the Apical and E. coli Adheres to the Basolateral Surface of Enteroids
C. sakazakii bacteria were observed in close proximity to the surface of AO enteroids, suggesting the bacteria had the potential to adhere to the apical surface thus allowing for study of host-pathogen interactions (Figure 7A). To test for this, bacteria were introduced to the apical surface of the enteroids then washed and plated for adherence. C. sakazakii demonstrated 0.4% to 4.2% adherence to the apical surface of untreated NEC and non-NEC derived enteroids. While greater adherence was observed to the apical surfaces of NEC derived enteroids compared to non-NEC controls, this did not reach significance (p=0.20, Figure 7B). This was repeated with the C. sakazakii strain BAA-894 which demonstrated similar results (0.09% to 0.4% adherence, p=0.3, n=2 in each treatment group). This was further repeated in BO enteroids with E.coli which demonstrated increased adherence in control vs NEC derived enteroids (111% vs 186% adherence in NEC vs control groups, p=0.03, n=3, Figure 8A and 8B). This is in contrast to AO enteroids treated with E.coli which showed a non-significant increase in adherence in NEC derived vs control enteroids (14% vs 6%, p>0.05, n=3, the NEC group had a non-normal distribution with a median=3.1, Q1=2.5, Q3= 36.6, Figure 8C and 8D).
Figure 7.
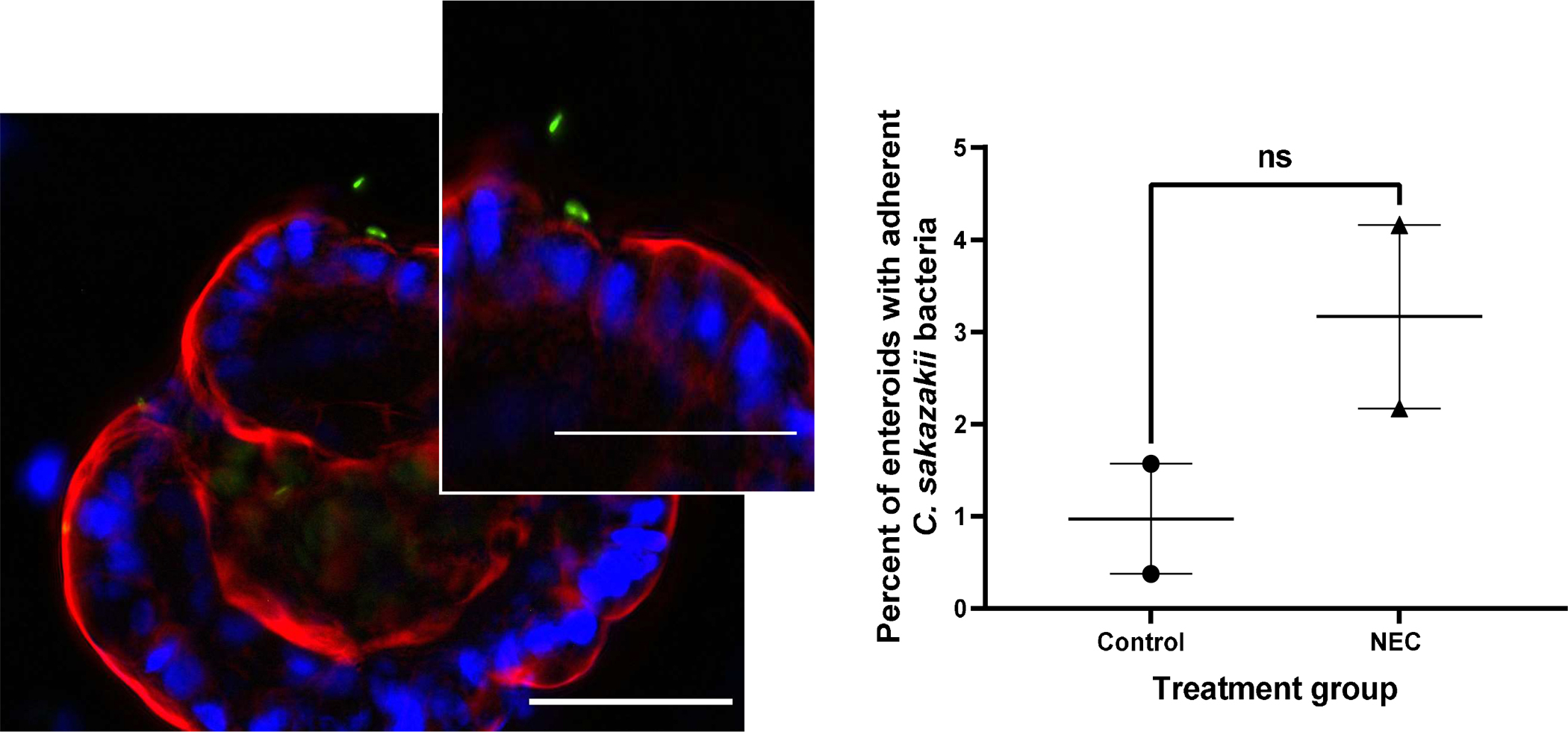
Immunofluorescence of pRSET Cronobacter Sakazakii (CS) expressing GFP adhering to an apical-out enteroid, whole mount images taken at 40X, 50μM scale bar, white arrow points to CS adhering to enteroid (A). Percent of enteroids with adherent C. sakazakii bacteria to the apical surface in NEC derived vs non-NEC derived enteroids, n=2 in each group, p=0.2 (B).
Figure 8.
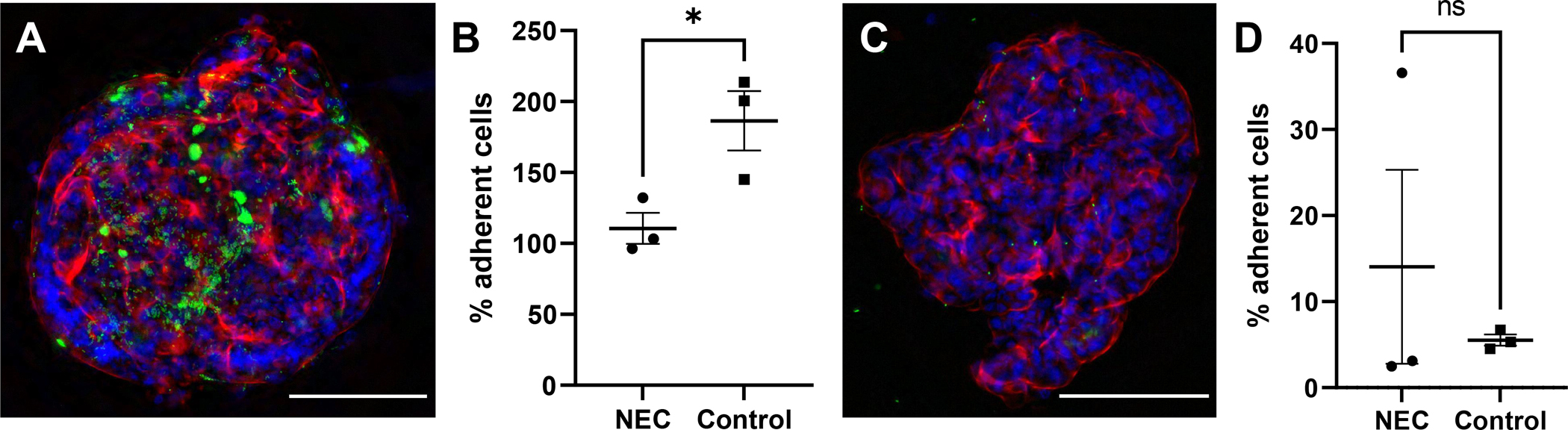
Immunofluorescence of Escherichia coli (ATCC 35150 GFP) expressing GFP adhering to a basolateral-out enteroid, whole mount image taken at 20X, 100μM scale bar (A). Percent of treated E. coli adhering to basolateral-out NEC derived vs non-NEC derived enteroids, n=3 in each group, p=0.0329 (B). Whole mount image of E.coli adhering to apical-out enteroids, taken at 20X, 100μM scale bar (C). Percent of treated E. coli adhering to apical-out NEC derived vs non-NEC derived enteroids, n=3, p>0.05 (D).
Discussion
The pathophysiology of NEC is multifactorial and includes a hyperinflammatory response, increased intestinal permeability and intestinal microbial dysbiosis (1). Among these, increased TLR-4 has been implicated as a part of the hyperinflammatory response (18). These receptors are predominantly on the basolateral surface which requires host-pathogen interactions on the apical surface to alter intestinal epithelial cell permeability thereby allowing pathogen interaction with TLR-4 on the basolateral surface (21). Adding a pathogen to the media of the traditional enteroid model allows direct interaction to TLR-4 and thus induction of NEC. However, this does not allow for study of the interaction between the pathogen and the apical surface of the host enteroid which is seen in vivo. This would require access to the apical surface first in order to allow for natural pathogen-host interaction.
The importance of our ability to study biologically relevant pathogens has become increasingly pressing in the setting of a recent outbreak of C. sakazakii NEC in infants (4, 5, 22). Accessing the luminal compartment in order to study these interactions has been a limitation of the traditional BO enteroid model which places NEC researchers at a disadvantage (23). This study demonstrates that AO enteroids can be used as a model for NEC with easy access of the apical surface to the media. We have further shown through adherence studies and immunofluorescence that C. sakazakii adheres to the apical surface of the enteroid thus allowing for the study of pathogen-host interaction. When repeated with E. coli and using BO enteroids, adherence is greater in the control group suggesting that adherence patterns may be bacteria specific. In addition, AO enteroids may provide greater adherence in NEC enteroids compared to BO enteroids but further studies are needed to determine these interactions. There have been several previously proposed methods to access the apical surface. These include microinjection, fragmentation and enteroid monolayer cultures (24). Microinjection maintains the structural integrity of the enteroid by directly injecting into the lumen in order to contact the apical surface (25). Despite its efficacy, microinjection is a time-consuming process that requires access to expensive equipment such as a micromanipulator and microinjector (26). It would be further challenging to inject enough enteroids for measurable effects (25). This limits its usefulness for applications that require a large volume of cell material although retains its usefulness for smaller applications in labs that have access to the necessary equipment.
Fragmentation involves dissociation of the enteroid into separate cells and infecting them or introducing a pathogen into the media before they re-form their 3D structure (25). This method necessitates the destruction of cellular TJs and loss of structural integrity (24). It does not require special equipment and is relatively simple to perform. However, this process does not allow for the study of host-pathogen interactions since it allows access to both the apical and basolateral surfaces simultaneously. In addition, it loses the architecture of the enteroid as well as the intestinal epithelial barrier at the time of infection limiting the ability to study permeability.
Enteroids can also be grown as 2D monolayers with an apical and basolateral surface. This model allows the enteroid to retain its structural integrity and maintain the benefits of having all of the differentiated intestinal epithelial cell types while allowing access to the apical surface. This provides a useful model for the study of TJs and intestinal permeability. Limitations of this model include loss of 3D architecture and an inability to be expanded through resuspension and passaging (27).
The AO NEC model addresses many of these concerns. It utilizes relatively inexpensive and readily available reagents and equipment. Reversing the polarity is simple, effective and results in high volumes of AO enteroids. In studying NEC specifically, it allows for the simultaneous study of host-pathogen interactions and permeability as seen in vivo, with introduction of pathogens to the apical surface of the intestinal epithelial barrier. This paper demonstrates that AO enteroids are an excellent model for NEC and allow for a more physiologic understanding of permeability and the host-pathogen interactions that are involved in the pathophysiology of NEC.
However, as a relatively new model, the AO model also has limitations. Once the AO conformation is induced, enteroids are destined for terminal experimentation due to their inability to subculture. Longer culturing times are also required to reverse the polarity as compared to BO enteroids. In addition, although bacteria has been shown to attach to the apical surface, there was no difference in adherence in our study which may be due to decreased sample size or bacteria specific characteristics.
Conclusions
AO enteroids present an innovative model for necrotizing enterocolitis, allowing easier access to the apical surface of the epithelium. Enteroids in an AO conformation show increased expression of inflammatory markers and decreased TJ proteins similar to clinical NEC. By exhibiting an AO conformation, this model allows for a biologically relevant investigation of the interaction between pathogen and the intestinal epithelial barrier.
Highlights:
Apical-out enteroids subjected to LPS and hypoxia show NEC characteristics
Chemically everted enteroids (apical-out) allow ready access to their luminal surface
Apical-out enteroids allow study of pathogen-intestinal epithelium interaction
Acknowledgments
We would like to thank the Department of Surgery and the Department of Pediatric Surgery at Oklahoma University for their ongoing support of this project. We are also indebted to our medical colleagues at OU who partner with us in the lab and hospital to continue discovering innovative ways to decrease the burden of disease in children. Finally, we would like to thank the Oklahoma Center for Adult Stem Cell Research and the National Institute of Health for their generous support of our research.
Disclosures/funding:
This work was supported by the National Institute of Health [NIH Grant R03 DK117216-01A1] and the Oklahoma Center for Adult Stem Cell Research. The authors report no proprietary or commercial interest in any product mentioned or concept discussed in this article.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bellodas Sanchez J, Kadrofske M. Necrotizing enterocolitis. Neurogastroenterol Motil. 2019;31(3):e13569. Epub 2019/02/23. doi: 10.1111/nmo.13569. [DOI] [PubMed] [Google Scholar]
- 2.Hunter CJ, Bean JF. Cronobacter: an emerging opportunistic pathogen associated with neonatal meningitis, sepsis and necrotizing enterocolitis. Journal of Perinatology. 2013;33(8):581–5. doi: 10.1038/jp.2013.26. [DOI] [PubMed] [Google Scholar]
- 3.Du XJ, Wang XY, Dong X, Li P, Wang S. Characterization of the Desiccation Tolerance of Cronobacter sakazakii Strains. Front Microbiol. 2018;9:2867. Epub 2018/12/14. doi: 10.3389/fmicb.2018.02867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.CDC - National Center for Emerging and Zoonotic Infectious Diseases (NCEZID) DoF, Waterborne, and Environmental Diseases (DFWED). Cronobacter and powdered infant formula investigation 2022. [updated May 24, 2022]. Available from: https://www.cdc.gov/cronobacter/outbreaks/infant-formula.html.
- 5.Chen Z, Zhang Y, Lin R, Meng X, Zhao W, Shen W, Fan H. Cronobacter sakazakii induces necrotizing enterocolitis by regulating NLRP3 inflammasome expression via TLR4. Journal of Medical Microbiology. 2020;69(5):748–58. doi: 10.1099/jmm.0.001181. [DOI] [PubMed] [Google Scholar]
- 6.Hunter CJ, Singamsetty VK, Chokshi NK, Boyle P, Camerini V, Grishin AV, Upperman JS, Ford HR, Prasadarao NV. Enterobacter sakazakii enhances epithelial cell injury by inducing apoptosis in a rat model of necrotizing enterocolitis. J Infect Dis. 2008;198(4):586–93. Epub 2008/07/01. doi: 10.1086/590186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kovler ML, Sodhi CP, Hackam DJ. Precision-based modeling approaches for necrotizing enterocolitis. Dis Model Mech. 2020;13(6). Epub 2020/08/09. doi: 10.1242/dmm.044388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.In JG, Foulke-Abel J, Estes MK, Zachos NC, Kovbasnjuk O, Donowitz M. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol. 2016;13(11):633–42. Epub 2016/10/26. doi: 10.1038/nrgastro.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ranganathan S, Smith EM, Foulke-Abel JD, Barry EM. Research in a time of enteroids and organoids: how the human gut model has transformed the study of enteric bacterial pathogens. Gut Microbes. 2020;12(1):1795492. Epub 2020/08/17. doi: 10.1080/19490976.2020.1795389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ares GJ, Buonpane C, Yuan C, Wood D, Hunter CJ. A Novel Human Epithelial Enteroid Model of Necrotizing Enterocolitis. J Vis Exp. 2019(146). Epub 2019/04/30. doi: 10.3791/59194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buonpane C, Ares G, Yuan C, Schlegel C, Liebe H, Hunter CJ. Experimental Modeling of Necrotizing Enterocolitis in Human Infant Intestinal Enteroids. J Invest Surg. 2022;35(1):111–8. Epub 2020/10/27. doi: 10.1080/08941939.2020.1829755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Co JY, Margalef-Català M, Li X, Mah AT, Kuo CJ, Monack DM, Amieva MR. Controlling Epithelial Polarity: A Human Enteroid Model for Host-Pathogen Interactions. Cell Rep. 2019;26(9):2509–20.e4. Epub 2019/02/28. doi: 10.1016/j.celrep.2019.01.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel RM, Denning PW. Intestinal microbiota and its relationship with necrotizing enterocolitis. Pediatr Res. 2015;78(3):232–8. Epub 2015/05/21. doi: 10.1038/pr.2015.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, Eliceiri KW. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics. 2017;18(1):529. Epub 2017/12/01. doi: 10.1186/s12859-017-1934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blackwood BP, Wood DR, Yuan C, Nicolas J, De Plaen IG, Farrow KN, Chou P, Turner JR, Hunter CJ. A Role for cAMP and Protein Kinase A in Experimental Necrotizing Enterocolitis. Am J Pathol. 2017;187(2):401–17. Epub 2016/12/13. doi: 10.1016/j.ajpath.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavadas MA, Mesnieres M, Crifo B, Manresa MC, Selfridge AC, Scholz CC, Cummins EP, Cheong A, Taylor CT. REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci Rep. 2015;5:17851. Epub 2015/12/10. doi: 10.1038/srep17851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez CA, Kerr B, Jin C, Cistulli PA, Cook KM. Obstructive Sleep Apnea Activates HIF-1 in a Hypoxia Dose-Dependent Manner in HCT116 Colorectal Carcinoma Cells. Int J Mol Sci. 2019;20(2). Epub 2019/01/24. doi: 10.3390/ijms20020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackam DJ, Sodhi CP. Toll-Like Receptor-Mediated Intestinal Inflammatory Imbalance in the Pathogenesis of Necrotizing Enterocolitis. Cell Mol Gastroenterol Hepatol. 2018;6(2):229–38.e1. Epub 2018/08/15. doi: 10.1016/j.jcmgh.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ares G, Buonpane C, Sincavage J, Yuan C, Wood DR, Hunter CJ. Caveolin 1 is Associated with Upregulated Claudin 2 in Necrotizing Enterocolitis. Sci Rep. 2019;9(1):4982. Epub 2019/03/23. doi: 10.1038/s41598-019-41442-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zani A, Ghionzoli M, Lauriti G, Cananzi M, Smith VV, Pierro A, De Coppi P, Eaton S. Does intestinal permeability lead to organ failure in experimental necrotizing enterocolitis? Pediatr Surg Int. 2010;26(1):85–9. Epub 2009/10/27. doi: 10.1007/s00383-009-2507-7. [DOI] [PubMed] [Google Scholar]
- 21.Yu S, Gao N. Compartmentalizing intestinal epithelial cell toll-like receptors for immune surveillance. Cell Mol Life Sci. 2015;72(17):3343–53. Epub 2015/05/24. doi: 10.1007/s00018-015-1931-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Acker J, de Smet F, Muyldermans G, Bougatef A, Naessens A, Lauwers S. Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol. 2001;39(1):293–7. Epub 2001/01/04. doi: 10.1128/jcm.39.1.293-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Azar J, Bahmad HF, Daher D, Moubarak MM, Hadadeh O, Monzer A, Al Bitar S, Jamal M, Al-Sayegh M, Abou-Kheir W. The Use of Stem Cell-Derived Organoids in Disease Modeling: An Update. Int J Mol Sci. 2021;22(14). Epub 2021/07/25. doi: 10.3390/ijms22147667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stroulios G, Stahl M, Elstone F, Chang W, Louis S, Eaves A, Simmini S, Conder RK. Culture Methods to Study Apical-Specific Interactions using Intestinal Organoid Models. J Vis Exp. 2021(169). Epub 2021/04/13. doi: 10.3791/62330. [DOI] [PubMed] [Google Scholar]
- 25.Dutta D, Heo I, Clevers H. Disease Modeling in Stem Cell-Derived 3D Organoid Systems. Trends Mol Med. 2017;23(5):393–410. Epub 2017/03/28. doi: 10.1016/j.molmed.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 26.Bartfeld S, Bayram T, van de Wetering M, Huch M, Begthel H, Kujala P, Vries R, Peters PJ, Clevers H. In vitro expansion of human gastric epithelial stem cells and their responses to bacterial infection. Gastroenterology. 2015;148(1):126–36.e6. Epub 2014/10/14. doi: 10.1053/j.gastro.2014.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thorne CA, Chen IW, Sanman LE, Cobb MH, Wu LF, Altschuler SJ. Enteroid Monolayers Reveal an Autonomous WNT and BMP Circuit Controlling Intestinal Epithelial Growth and Organization. Dev Cell. 2018;44(5):624–33.e4. Epub 2018/03/06. doi: 10.1016/j.devcel.2018.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]