

Abstract
Establishing the functionality, reproducibility, robustness, and reliability of the microphysiological systems is a critical need. A high throughput microphysiological system for liver studies was recently proposed in which induced pluripotent stem cell-derived hepatocytes (iHeps) and non-parenchymal cells (endothelial cells and THP-1 cells differentiated into macrophage-like cells) were co-cultured in OrganoPlate® 2-lane 96 plates. The goal of this study was to evaluate this platform using additional cell types and conditions and characterize its utility and reproducibility. Primary human hepatocytes or iHeps, with and without non-parenchymal cells, were cultured for up to 17 days. Image-based cell viability, albumin and urea secretion into culture media, CYP3A4 activity and drug metabolism were assessed. The iHeps co-cultured with non-parenchymal cells demonstrated stable cell viability and function up to 17 days; however, variability was appreciable both within and among studies. The iHeps in monoculture did not form clusters and lost viability and function over time. The primary human hepatocytes in monoculture also exhibited low cell viability and hepatic function. Metabolism of various drugs was most efficient when iHeps were co-cultured with non-parenchymal cells. Overall, we found that the OrganoPlate® 2-lane 96 plate, when used with iHeps and non-parenchymal cells, is a functional liver microphysiological model; however, the high-throughput nature of this model is somewhat dampened by the need for replicates to compensate for high variability.
Introduction
Testing for the potential adverse effects of drugs and chemicals on the liver is among the most common types of toxicity investigations (LeCluyse et al., 2012; Soldatow et al., 2013). The liver is a crucial organ for metabolism and performs a wide range of other essential physiological functions; it stores and synthesizes proteins, bile acids, hormones and other biologically active molecules, and plays a role in immune responses to gut-derived pathogens and ammonia detoxification. The liver is also a common target for harmful xenobiotics and pathogens, and a source of injurious metabolites. Both drugs and other chemicals may target the liver through a variety of mechanisms (Rusyn et al., 2021). Due to the liver’s prominent role in both physiology and disease, studies of hepatotoxicity are a very active area of research and discovery; however, existing clinical, animal, or cell-based model systems have limitations (Kullak-Ublick et al., 2017; Monticello et al., 2017; Sistare et al., 2016).
Because of the great need to improve the physiological relevance and reliability of testing drug candidates and chemicals for potential adverse liver effects, a variety of novel cell-based models have been proposed, including microphysiological systems, which include “tissue chips” (Collins et al., 2019; Taylor et al., 2019). Liver tissue chip models typically include media flow and allow for studies of different cell compartments (Underhill and Khetani, 2018). Additional functionalities have also been established to allow for oxygen zonation, as well as inclusion of immune components and other cell types (Taylor et al., 2019). While a number of publications have provided exciting examples for how these models can be used in drug safety evaluation, liver disease modeling, and to decipher species-specific liabilities (Low et al., 2021), the wider adoption of these approaches has been hampered by their relatively low throughput, high complexity and cost. The translatability of these models from scholarly publications to regulatory submissions is also difficult because there is a general lack of consistency in the characterization of these models and examples of how they can deliver value in the context of decision-making (Baudy et al., 2020).
A related challenge in the field of in vitro liver models, including microphysiological systems, has been the lack of human cells with sufficient metabolic function and quantity. Primary human hepatocytes from non-transplant grade post mortem liver tissues are known to be highly variable in their functionality and are a finite resource from a particular donor (Hewitt et al., 2007). Immortalized cells with some residual liver function are also commonly used; these can be cultured in monolayers, sandwich or as spheroids, with or without non-parenchymal cells (Godoy et al., 2013). More recently, human induced pluripotent stem cell (iPSC)-derived hepatocytes have become a cell source that is gaining acceptance for studies that aim to minimize donor variability (Bulutoglu et al., 2020; Sirenko et al., 2014). iPSC-derived hepatocytes have been tested in several recent studies of liver microphysiological systems. In a study of a human microfluidic four-cell “liver acinus microphysiology system” (LAMPS), a comparable performance was achieved when using either primary human hepatocytes or iPSC-derived hepatocytes for both basic function of the cells and their response to drug-related toxicity (Sakolish et al., 2021b). The LAMPS model seeded with iPSC-derived hepatocytes exhibited higher precision for predicting hepatic drug clearance as compared to LAMPS with primary cells, or 2D cultures of either cell type (Sakolish et al., 2021a). In addition, a microfluidic high-throughput liver model, the OrganoPlate® 2-lane 96 plate, was recently developed based on iPSC-derived hepatocytes co-cultured with endothelial cells and macrophages (Bircsak et al., 2021).
This study aimed to determine if the OrganoPlate® 2-lane 96 plate device is a reproducible model for studies of the liver and can be replicated in another laboratory. We also compared hepatocyte function in this model using human primary and iPSC-derived hepatocytes cultured with and without non-parenchymal cells, as well as evaluated the degree of intra-laboratory reproducibility of the liver model based on the OrganoPlate® 2-lane 96 plate. To accomplish these goals, we evaluated basic liver functions, drug metabolism, and drug toxicity phenotypes.
Materials and Methods
Chemicals and materials
Phenacetin (77440, CAS#62-44-2), coumarin (C4261, CAS#91-64-5), diclofenac (D6899, CAS#15307-79-6), terfenadine (T9652, CAS#50679-08-8), phenolphthalein (105945, CAS#77-09-8), acetaminophen (PHR1005, CAS#103-90-2) and 4-hydroxy diclofenac (32412, CAS#64118-84-9), fexofenadine (PHR1685, CAS#153439-40-8), lipopolysaccharide (LPS; L2630), troglitazone (T2573, CAS#97322-87-7), and trovafloxacin (TVX; PZ0015, CAS#147059-75-4) were purchased from Millipore Sigma (Burlington, MA). Midazolam (19391, CAS#59467-70-8), 1’-hydroxymidazolam (10385, CAS#59468-90-5), and phenolphthalein β-D-glucuronide (30424, CAS#15265-26-6) were purchased from Cayman Chemical (Ann Arbor, MI).
The microfluidic tissue chips used in this study were purchased from Mimetas (OrganoPlate® 2-lane 96, Leiden, Netherlands). Each chip of this 96-well platform contains one gel channel and one perfusion channel. This configuration enables the culture of a perfused tubule adjacent to extracellular matrix (ECM) of choice without a membrane (Bircsak et al., 2021). Black-walled, clear-bottom, tissue culture-treated 96-well (3603, Corning, Corning, NY) and 384-well plates (3765, Corning) were used for 2D cell culture experiments.
Cells and reagents
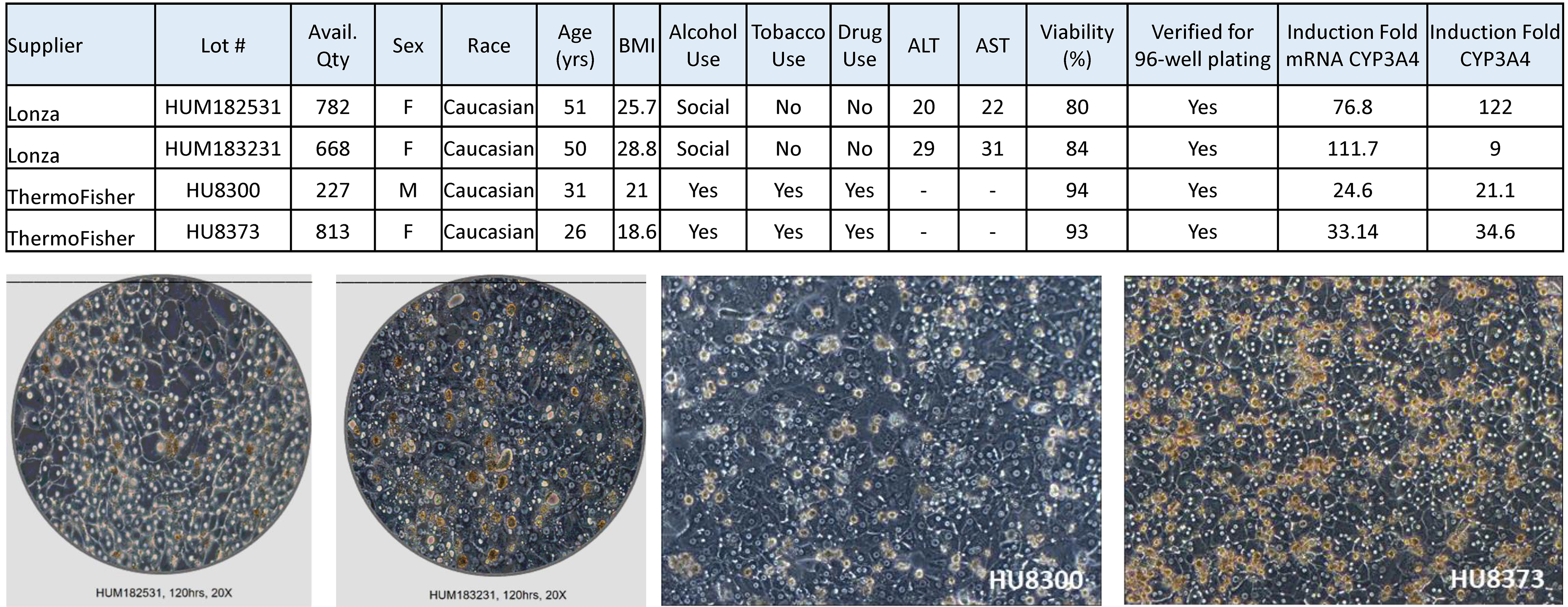
Human induced pluripotent stem cell-derived hepatocytes (iCell Hepatocytes 2.0, abbreviated herein as iHeps) were purchased from FujiFilm-Cellular Dynamics International (C1023, lot#103934, Santa Ana, CA). iHeps plating media consisted of DMEM/F12 (21041025, ThermoFisher, Waltham, MA) supplemented with 2% B-27 supplement (17504044, ThermoFisher), 100 nM dexamethasone (265005, Millipore Sigma), 25 μg/mL gentamicin (15710072, ThermoFisher), and 20 ng/mL oncostatin M (295-OM-010, R&D Systems, Minneapolis, MN); it was used for pre-differentiation of iHeps according to the manufacturer’s protocol. iHeps maintenance media consisted of DMEM/F12, 2% B-27, 100 nM dexamethasone, and 25 μg/mL gentamicin; it was used for cell culture in the OrganoPlate® 2-lane 96 plates and in the 384-well plates. Primary human hepatocytes (female Caucasian donor, 26 years of age, prior history of alcohol, tobacco and drug use) were obtained from ThermoFisher (HMCPIS, lot #HU8373). This lot of cells was selected among several available options (Supplemental Figure 1) based on the following criteria: sufficient inventory (200 or more vials), cells were verified for plating efficiency, high cell viability after thawing (93%), verified for induction of major CYPs (CYP3A4 induction fold 34.6), and superior function in preliminary experiments in 2D cell culture as detailed below (Supplemental Figure 2).
Primary human hepatocyte plating media consisting of William’s E (A1217601, ThermoFisher) supplemented with thawing and plating supplements (3.6% cocktail A, 5% FBS, 10 nM dexamethasone (CM3000, ThermoFisher) was used for spheroid formation and seeding into the OrganoPlate 2-lane plates and the 96-well plates. Primary human hepatocyte maintenance media consisted of William’s E with primary hepatocyte maintenance supplements (4% cocktail B, 1 nM dexamethasone; CM4000, ThermoFisher); it was used for cell culture in the OrganoPlate® 2-lane 96 plates and 96-well plates. THP-1 monocytes and HMEC-1 endothelial cells were obtained from ATCC (Manassas, VA). THP-1 monocytes were cultured in RPMI (30–2001, ATCC) with 10% fetal bovine serum (30–2020, ATCC) and 50 nM 2-mercaptoethanol (M3148, Millipore Sigma). THP-1 monocytes were differentiated (Lee-Montiel et al., 2017) into adherent macrophages via treatment with 250 nM phorbol 12-myristate-13-acetate (356150050, ThermoFisher) for 48 h prior to seeding into OrganoPlate® 2-lane 96 plates or multi-well plates. HMEC-1s were cultured in MCDB 131 medium (10372019, ThermoFisher) with 10 % fetal bovine serum (30–2020, ATCC), 2 mM L-glutamine (30–2214, ATCC), 100 units/mL penicillin-streptomycin (P0781, Millipore Sigma), 1 μg/mL hydrocortisone (H0888, Millipore Sigma), and 10 ng/mL epidermal growth factor recombinant human protein (PHG0314, ThermoFisher).
iHep culture methods in the OrganoPlate® 2-lane 96 plates and 384-well plates
The day when the cells were seeded into OrganoPlate® 2-lane 96 plates and 384-well plates was defined as Day 0. iHeps were cultured in OrganoPlate® 2-lane 96 plates using the protocol described (Bircsak et al., 2021). Briefly, thawed iHeps were seeded at a density 2.5×106 cells/well on a 6-well plate pre-coated with type 1 collagen (657950–005, Greiner Bio-One North America, Monroe, NC) in iHeps plating media. The cells were cultured for 4 h and unattached cells were removed when the media were replaced with fresh iHep plating media. The cells were differentiated for 5 days with daily changes of plating media. The differentiated iHep clusters were collected by centrifugation (200 g, 3 min) and resuspended into 3.33 mg/mL collagen (Cultrex 3-D Culture Matrix Rat Collagen-I, 3447-020-01, R&D Systems; 5 mg/mL type 1 collagen, 1 M HEPES, 37 g/L sodium bicarbonate at a ratio of 4:1:1, respectively) at a density of approximately 8.0×106 cells/mL. The iHep/collagen suspension (2.5 μL/device) was subdivided into two aliquots that were kept on ice. Aliquots, each sufficient for 48 devices on the plate, were used consecutively whereby the suspension was gently injected into the inlet of the gel channels using multi-channel electronic pipettor making sure that the time it takes to complete this task is no longer than 3 minutes. After both aliquots were injected, the whole plate was placed at 37°C, 5% CO2 for 15 min to allow polymerization of the type 1 collagen.
For co-culture conditions, THP-1 monocytes and HMEC-1 endothelial cells were seeded into the perfusion channels of the OrganoPlate® 2-lane 96 plates. A mixture of HMEC-1s at 40×106 cells/mL and differentiated THP-1s at 3×106 cells/mL was prepared in iHep maintenance media. After that, 2.5 μL HMEC-1/THP-1 cell suspension was injected into the inlets of the perfusion channel using multichannel electronic pipettor. The plates were incubated elevated at a 70° angle at 37 °C, 5% CO2 to allow HMEC-1s and THP-1s to attach to the iHeps/collagen in the gel channel above the Phaseguide, a meniscus-pinning barrier that is part of the microfluidic tissue culture plates manufactured by Mimetas (Vulto et al., 2011). After 15 min incubation, 50 μL iHeps maintenance media was added into medium inlets and outlets of the perfusion channel (for both co-cultured and mono-cultured chips) and the plates were incubated elevated at a 70° angle at 37 °C, 5% CO2 for an additional 45 min. The plates were then placed on the perfusion rocker platform (Mimetas) set to cycle every 4 min to a maximum angle of approximately 15° to induce gravity-driven media flow through the perfusion channel. The media was collected and exchanged every 1–2 days by aspirating and replacing media from medium inlets and outlets (50 μL in each).
For the iHep 2D model, differentiated iHeps clusters were collected as previously described and resuspended into 3.33 mg/mL collagen at a density of approximately 2.0×106 cells/mL. The iHep/collagen suspension (10 μL/well) was pipetted into wells on the 384-well plate using multichannel electronic pipettor. The plate was placed at 37°C, 5% CO2 for 15 min to allow polymerization of the type 1 collagen gel. After that, 50 μL iHep maintenance media was added into each well and plates were incubated at 37°C, 5% CO2. The media was exchanged every 1–2 days.
Primary human hepatocyte culture methods in the OrganoPlate® 2-lane 96 plates and 96-well plates
In these studies, primary human hepatocytes were seeded directly into the individual devices of the OrganoPlate® 2-lane 96 plates as follows. Thawed cells were collected by centrifugation (100 g, 10 min) and were resuspended into 3.33 mg/mL collagen at a density of approximately 8.0×106 cells/mL. The cell/collagen suspension (2.5 μL/device) was injected into the inlet of the gel channel using the same procedure as detailed above for iHeps. A mixture of HMEC-1s at 40×106 cells/mL and differentiated THP-1s at 3×106 cells/mL was prepared in the plating media. After that, 2.5 μL HMEC-1/THP-1 cell suspension was injected into the medium inlets of the perfusion channel using multichannel electronic pipettor. After 15 min incubation under a 70° angle at 37 °C, 5% CO2, 50 μL primary human hepatocytes plating media was dispensed into medium inlets and outlets including mono-cultured chips and incubated under a 70° angle at a 37 °C, 5% CO2 for an additional 45 min. The plates were placed on the perfusion rocker platform set to same cycle as iHeps cultures. The media was collected and exchanged every 1–2 days by aspirating and replacing with the maintenance media.
For the primary human hepatocyte sandwich culture model, cells were cultured using the protocol previously described (Keemink et al., 2015). Briefly, thawed cells were collected by centrifugation (100 g, 10 min) and resuspended with the plating media. Cells were plated into 96-well plates at a density of 6.0×104 cells/well and incubated for 6 h at 37°C, 5% CO2. Media was replaced with 100 μL of plating media with 0.35 mg/mL matrigel (CB-40234C, Corning). After incubation overnight, media were exchanged to the maintenance media. Media were collected and exchanged every 1–2 days.
Primary human hepatocyte spheroids culture methods in the OrganoPlate® 2-lane 96 plates
Thawed primary human hepatocytes were incubated in AggreWell™400 6-well plate (34425, Stemcell Technologies, Vancouver, Canada) for 7 days at a cell density of 3.0×105 cells/well (approximately 50 cells/microwell) in the plating media. Cells were allowed to aggregate for 7 days with daily media changes. Spheroidized primary human hepatocytes were harvested using 37 μm reversible strainer (27250, Stemcell Technologies) to separate single cells from cell aggregates, and the latter were collected from the strainer and centrifuged for 10 min at 100 g. Spheroids were resuspended into 3.33 mg/mL collagen at a density of approximately 1.6×105 spheroids/mL to have the match cell density in other culture conditions and were injected into OrganoPlate® 2-lane 96 plates using the same procedure as that described above for iHeps. For co-culture conditions, THP-1 and HMEC-1 cells were seeded into the perfusion channels in the same manner as in primary human hepatocyte cultures detailed above.
Biochemical Assays
Cell culture media collected in the experiments was evaluated for a variety of biomarkers. The ELISA assays for albumin (E88-129, Bethyl Laboratories, Montgomery, TX), urea (EIABUN, ThermoFisher), lactate dehydrogenase (ab102526, abcam, Cambridge, UK), and interleukin-6 (IL-6; D6050, R&D Systems) were conducted using manufacturer’s instructions. The P450 Glo 3A4 with Luciferin-IPA assay (V9001, ProMega, Madison, WI) was conducted using manufacturer’s recommendations with the IPA substrate added to the perfusion channels or wells for 1 h prior to luminescence plate reading (SpectraMax iD3, Molecular Devices, San Jose, CA).
Cell viability assessment
To evaluate cell viability, iHeps or primary human hepatocyte maintenance media containing 2 μg/mL propidium iodide (P4864, Millipore-Sigma) and 10 μg/mL Hoechst 33342 (H3570, Thermo Fisher) was added to the perfusion channels or wells, respectively, and cells were incubated for 3 hrs (30 min for primary human hepatocyte sandwich culture model) before confocal imaging in an ImageXpress Micro Confocal (Molecular Devices) using a 10× objective. MetaXpress High-Content Image Acquisition and Analysis Software (Molecular Devices) was used to acquire a stack of 30 images separated by 5 μm, starting at the bottom and covering most hepatocytes. A stack that was well-focused on many hepatocytes was selected within the range of the phase-guide location. ImageJ (National Institutes of Health, Bethesda, MD) was used to analyze cell viability when comparing culture conditions for either liver parenchymal cell type with or without THP-1 and HMEC-1 cells. Images containing hepatocytes in gel lane were extracted with the phase-guide as the boundary, and the number of viable hepatocytes and other cell types was counted.
Immunocytochemical staining
Cells in the OrganoPlate® 2-lane 96 plates were fixed by adding 4% neutral buffered formalin solution for 10 min. The formalin fixative was removed by rinsing with phosphate-buffered saline (PBS) and the fixed cultures were treated for 10 min with 0.1% Triton X-100 (BP151, ThermoFisher) in PBS to permeabilize cells. Next, cells were washed, and incubated for 60 min in a 2 % bovine serum albumin (A8806, Millipore-Sigma) PBS solution. Mouse anti-human CD68 antibodies (556059, BD Biosciences, San Jose, CA) were added to perfusion channels to label THP-1 monocytes and plates were incubated for 2 days at 4°C. Goat anti-mouse IgG secondary antibody (ab150115, AF647, abcam) was added to visualize CD68 and plates were incubated overnight at 4°C. All cultures were counterstained with Hoechst 33342 and FITC-phalloidin (F432, ThermoFisher) to visualize nuclei and cytoskeleton, respectively. Cells were imaged using the ImageXpress microscope.
Drug treatments in the OrganoPlate® 2-lane 96 plates
For evaluation of drug metabolism, iHeps co-cultured with THP-1/HMEC in the OrganoPlate® 2-lane 96 plates were exposed on day 6 of culture to either a 5-drug cocktail (100 μM phenacetin, 20 μM coumarin, 10 μM diclofenac, 10 μM terfenadine, and 10 μM phenolphthalein) or to 5 μM midazolam in iHep maintenance media. Drugs in the cocktail solution were first dissolved in in dimethyl sulfoxide (final concentration 0.5%). Midazolam was dissolved in methanol (final concentration 0.16%). After drug addition, media was collected after 24 hrs and replaced with fresh iHep maintenance media. The same devices were re-exposed for 24 hrs on days 12 and 16 of culture. Separate devices were used in experiments with troglitazone. Devices were treated on day 7 of culture with either vehicle (0.5 % dimethyl sulfoxide) or troglitazone (180 μM) for 24 hrs, media was collected and fresh media was added with the same condition (vehicle of drug) for 24 hrs. Three treatments were conducted consecutively. In the experiments with trovafloxacin, treatments were conducted on day 8 of culture with either vehicle (0.5 % dimethyl sulfoxide), trovafloxacin (50 or 150 μM), and/or lipopolysaccharide (LPS, 1 μg/mL). Effects of these treatments were evaluated after 48 hrs.
Determination of drug concentrations using liquid chromatography (LC) - tandem mass spectrometry (MS/MS) analyses
Media samples (50 μL) were combined with 100 μL of 0.1 μM internal standard (Caffeine-13C3) solution in chilled acetonitrile. To allow for protein precipitation, samples were vortexed and centrifuged (12,000 g, 10 min, room temperature). The supernatant was dried under vacuum, and reconstituted in 50 μL de-ionized water with 0.1 % (v/v) formic acid (mobile phase A). Samples were then transferred to autosampler vials with 200 μL inserts and stored at −20°C prior to analysis. Samples (8 μL) were automatically injected and chromatographed on a ZORBAX SSHD Eclipse Plus C18 column (3.0×50 mm, 1.8 μm, 959757-302, Agilent, Santa Clara, CA) with a guard column (2.1×5 mm, 1.8 μm, 821725-901, Agilent) using a 1290 Infinity II LC (Agilent). Column temperature and flow rate were set at 40°C and 0.4 mL/min, respectively. Initial chromatographic condition was maintained at 90% mobile phase A (water with 0.1% (v/v) formic acid) and 10% mobile phase B (acetonitrile with 0.1% (v/v) formic acid) for 1 minute, then increased to 80% B by 3 min, then to 95% B by 4 min, and then returned to initial condition at 5 min until 8 min for sufficient equilibration prior to next run. MS/MS analyses were performed using an Agilent 6470 triple quadrupole MS (Agilent) in positive ion mode with an electrospray ionization source. Capillary voltage, sheath gas pressure, and sheath gas temperature were set at 3500 V, 40 psi and 350°C, respectively.
Statistical analyses
General descriptive statistical analyses were conducted using GraphPad Prism 9.0 (San Diego, CA). Statistical significance (p<0.05 was selected as a threshold) was tested with paired or unpaired t-test with Welch’s correction or one-way ANOVA with Dunnett’s multiple-comparisons test as indicated in Figure and Table legends. Power analyses for sample size needed to reach statistical significance with 80% statistical power were estimated using 1) the two-sided paired t-tests, and 2) two-sided two sample t-tests with different variances, for paired and independent samples, respectively. MATLAB, SAS, and PASS software packages were used to estimate the effect size and calculate the required sample size (Machin et al., 2011; Zar, 1984).
Data availability
Detailed experimental protocols, images and raw data for all experiments included in this study can be obtained using hyperlinks included in Supplemental Table 1.
Results
Comparison of liver function by cell source and culture conditions
The originally proposed OrganoPlate® 2-lane 96 plate devices for studies of the liver is based on iPSC-derived hepatocytes that are co-cultured with endothelial cells and macrophages separated into two compartments (Bircsak et al., 2021). Because a number of challenges remain with the metabolic competency of iHeps and their fetal-like characteristics (Corbett and Duncan, 2019), we tested if primary human hepatocytes can be used in this model. A related research question was the need for inclusion of non-parenchymal cells in this model to achieve the most optimal performance. To address these questions, pre-differentiated iHeps, freshly thawed primary human hepatocytes, or primary human hepatocyte spheroids were cultured for up to 17-days in OrganoPlate® 2-lane 96 plates, with and without THP-1 monocytes and HMEC-1 endothelial cells (Figure 1). As previously demonstrated by (Bircsak et al., 2021), under co-culture condition with non-parenchymal cells, iHeps remained as cell aggregates in the gel channel while HMEC-1 endothelial cells formed and maintained vascular-like structures for up to 17 days; however, iHeps cultured in the gel lane without non-parenchymal cells retained fewer cell clusters. When primary human hepatocytes were seeded into the devices immediately after thawing, small cell aggregates formed over time in the gel channel, regardless of whether non-parenchymal cells were present. Interestingly, the non-parenchymal cells did not remain viable and sloughed off over time and none were present by day 17. When primary human hepatocytes were pre-cultured to form spheroids before seeding into the devices, they maintained their morphology for up to 14 days, regardless of the presence of non-parenchymal cells. Similarly, non-parenchymal cells were not retained in the perfusion channel in this culture condition.
Figure 1. Bright field and immunocytochemical images in OrganoPlate® 2-lane 96 plate.
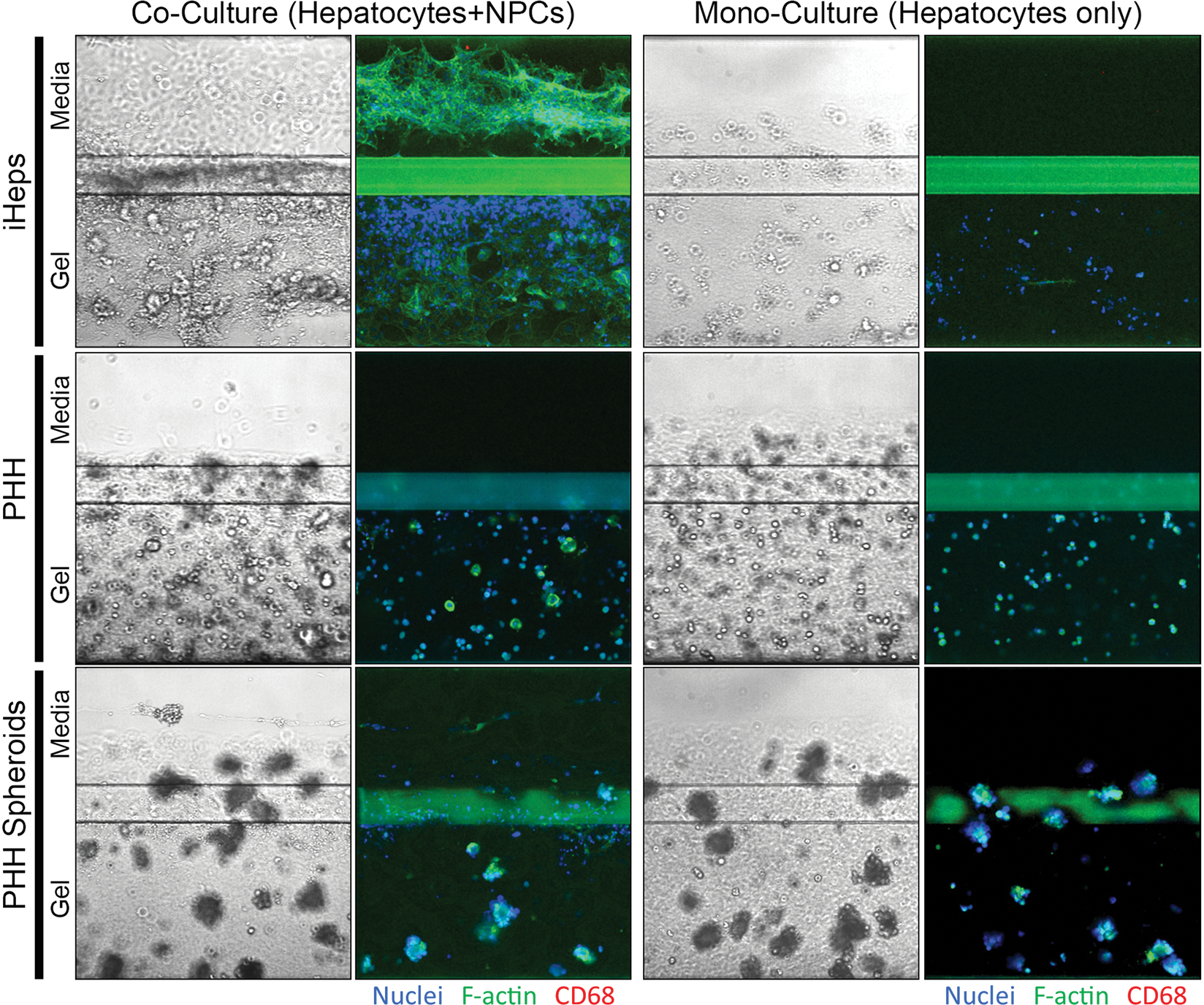
OrganoPlate® 2-lane 96 plates consist of the perfusion and organ lanes separated by a phase guide. Under all conditions, differentiated iHeps, primary human hepatocytes (PHH), or primary human hepatocyte spheroids were cultured in the gel lane with type 1 collagen. Under co-culture conditions, HMEC-1 endothelial cells and THP-1 derived macrophages were cultured in the perfusion lane. Images are shown for iHeps and primary human hepatocytes after 17 days of culture and for primary human hepatocyte spheroids after 14 days of culture. Some hepatocytes may appear to be on top of the Phaseguide or inside the perfusion channel; however, because the gel forms a meniscus they are still trapped in the gel. For immunocytochemistry, nuclei were stained with Hoechst-33342, cytoplasm was stained with F-actin, and THP-1 monocytes were stained with anti-CD68 antibodies.
Cell viability of hepatocytes, while variable between the devices, was the highest for iHeps co-cultured with non-parenchymal cells (Figure 2A). Primary human hepatocytes plated immediately after thawing in presence of non-parenchymal cells were the second-best condition, albeit only about one quarter viable cells remained as compared to iHeps co-cultured with non-parenchymal cells. Under 2D culture conditions, viability of iHeps and primary human hepatocytes did not differ (Figure 2B). When iHeps or primary human hepatocytes were cultured in the devices in the presence of non-parenchymal cells, lactate dehydrogenase leakage was significantly higher in the initial 1–4 days of culture as compared to days 7 through 13 (Figure 2C). This marker began to increase for iHeps with non-parenchymal cells on day 17. In 2D cultures of either parenchyma cell type, lactate dehydrogenase release was low and marginally higher only on day 1 after seeding (Figure 2D).
Figure 2. Cell viability, LDH, albumin, urea, and CYP3A4 activity in OrganoPlate® 2-lane 96 plate (left panels A, C, E, G, and I) and traditional 2D cultures (right panels B, D, F, H, and J).
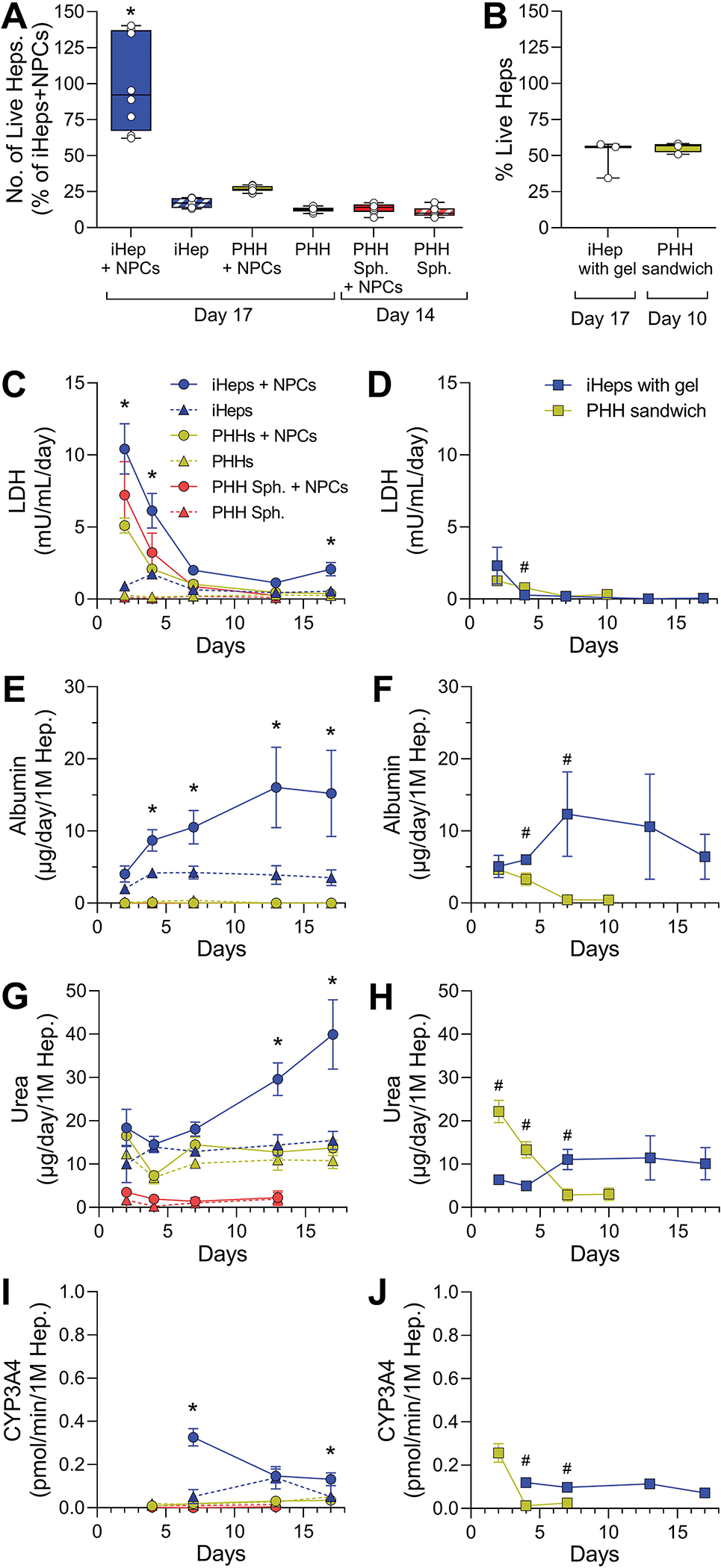
(A and B) Imaging-based cell viability at the end of cell culture. Data are graphed as box (interquartile range) and whiskers (min-max range) with individual data points shown. (C and D) LDH leakage, (E and F) albumin secretion, (G and H) urea secretion, and (I and J) CYP3A4 activity data are plotted as a function of time. Data are plotted as mean ± standard deviation (n = 3–4). For OrganoPlate® 2-lane 96 plate data, asterisks (*) denote statistical differences at p < 0.05 (one-way ANOVA with Dunnett’s multiple comparisons test). For 2D models, asterisks (#) denote statistical differences at p < 0.05 (unpaired t-test with Welch’s correction). Abbreviations: iHeps, human induced pluripotent stem cell-derived hepatocytes; PHH, primary human hepatocytes; NPCs, non-parenchymal cells; Sph., spheroids.
Synthetic function of liver parenchymal cells under different culture conditions was estimated using production of albumin and urea. In OrganoPlate® 2-lane 96 plate devices, the highest albumin production was observed for iHeps in the presence of non-parenchymal cells (Figure 2E). Over time, median levels reached up to 20 μg/day/million iHeps, which is similar to the report by Bircsak et al (2021) who demonstrated levels between 4 and 20 μg/day/million iHeps. Other studies with liver tissue chips seeded with primary human hepatocytes reported similar values, albeit 2–3-fold variability among donors (Kang et al., 2020; Sakolish et al., 2021b; Vernetti et al., 2016). These values are below albumin production levels in human liver that is estimated at 37–105 μg/day/million hepatocytes (Baudy et al., 2020). Among other conditions tested herein in OrganoPlate® 2-lane 96 plate devices, only iHeps cultured without non-parenchymal cells sustained some (~5 μg/day/million) albumin secretion, comparable to iHeps cultured in 2D condition (Figure 2F). The data for urea production in the OrganoPlate® 2-lane 96 plate devices was similar; iHeps cultured with non-parenchymal cells showed the highest and upward-trending production reaching ~40 μg/day/million iHeps. These values were about 2-fold higher than those reported by Bircsak et al (2021) in this device, and comparable to the data in LAMPS model with primary human hepatocytes (Sakolish et al., 2021b; Vernetti et al., 2016). Human liver urea production is 2–5 times higher (Baudy et al., 2020). iHeps cultured alone or freshly-thawed primary human hepatocytes with or without non-parenchymal cells exhibited a steady-state urea production of about 15 μg/day/million cells, and spheroidized primary human hepatocytes produced negligible amounts (Figure 2G). In 2D cultures, typical patterns of urea production were observed with primary human hepatocytes rapidly losing their synthetic functions and iHeps maintaining urea production at a low level (Figure 2H).
Activity of CYP3A4, one of the most important P450 isoforms for drug metabolism, was used as a representative marker for the metabolic competency of the cells in different culture conditions. CYP3A4-mediated drug metabolism is highly variable (Dorne et al., 2003) and wide variability in CYP3A4 activity (from 1 to 600 pmol/million/min) was reported in freshly isolated human hepatocytes (Ramaiahgari et al., 2017; Utkarsh et al., 2016). In prolonged culture, primary human hepatocytes show much lower activities of CYP3A4 (from less than 1 to 10 pmol/million/min) (Lu et al., 2015; Ramaiahgari et al., 2017), unless they are maintained as spheroids (Bell et al., 2018). Human iPSC-derived hepatocytes show enzyme activity in the range of primary human hepatocytes (Lu et al., 2015). We found that iHeps cultured with non-parenchymal cells showed the highest CYP3A4 activity among other conditions when cultured in OrganoPlate® 2-lane 96 plate devices (Figures 2I–J); however, even the highest activity at day 5 of culture was at the low end of that in primary human hepatocytes from different donors (Lu et al., 2015; Ramaiahgari et al., 2017).
Evaluation of reproducibility in the experiments with co-cultured iHeps and non-parenchymal cells
Because OrganoPlate® 2-lane 96 plate devices seeded with primary human hepatocytes, with or without non-parenchymal cells, demonstrated sub-optimal functionality (e.g., CYP3A4 activity, albumin and urea production), we only used iHeps in all subsequent studies. To evaluate the reproducibility of independent experiments, a total of six independent studies were performed in OrganoPlate® 2-lane 96 plate devices seeded with iHeps and non-parenchymal cells. These studies comprised of a total of 425 individual devices that included vehicle controls (0.5% DMSO or 0.16% methanol); thus, these data were used to evaluate the reproducibility and robustness of the basal cell function in this platform. First, we evaluated the cell seeding success rate in our studies, as suspensions were manually loaded using an electronic pipettor. Successful loading was determined by observing under a microscope that each chip was correctly filled. In the first step of assembly, where 2.5 μL iHep/collagen solution is injected into the gel channel, our overall success rate was 98.6% (419/425 chips, Supplemental Table 2). Following collagen polymerization in the gel channel, HMEC-1 and differentiated THP-1 cells were added to the perfusion channel with 96.7% (411/425 chips) success. These results were comparable to those achieved with automated loading (Bircsak et al., 2021).
Next, we evaluated the overall trends in the basal function of iHeps over time when all data were combined and the coefficients of variability within each experiment (Figure 3 and Supplemental Tables 3–4). The overall time-related trends in the basal functionality of iHeps were similar across independent experiments – we observed gradual decline in cell viability, decrease in albumin secretion, and increase in LDH production after 2 weeks in culture, while secretion of urea and activity of CYP3A4 remained high. However, the intra-plate variability was high in most experiments, generally at 20% or greater, mostly consistent with the published expectations for such liver microphysiological systems to be at 30% or less (Baudy et al., 2020). We also performed power analysis to determine the number of replicates that would be needed to observe significant differences in the basal phenotypes over time (Table 1). When using a standard assumption of requiring 80% power and a 5% significance level, we found that more than 7 replicates are needed in vehicle control group to discern the time-related differences in basal phenotypes. Based on these analyses, it is evident that repeated collection of the media from the same devices (paired analyses) results in greater reproducibility; however, overall time trends are difficult to ascertain beyond pair-wise comparisons between early and later time points.
Figure 3. Time trends and variability in repeat control experiments for cell viability, LDH, albumin, urea, and CYP3A4 activity parameters in OrganoPlate® 2-lane 96 plates seeded with iHeps and non-parenchymal cells.
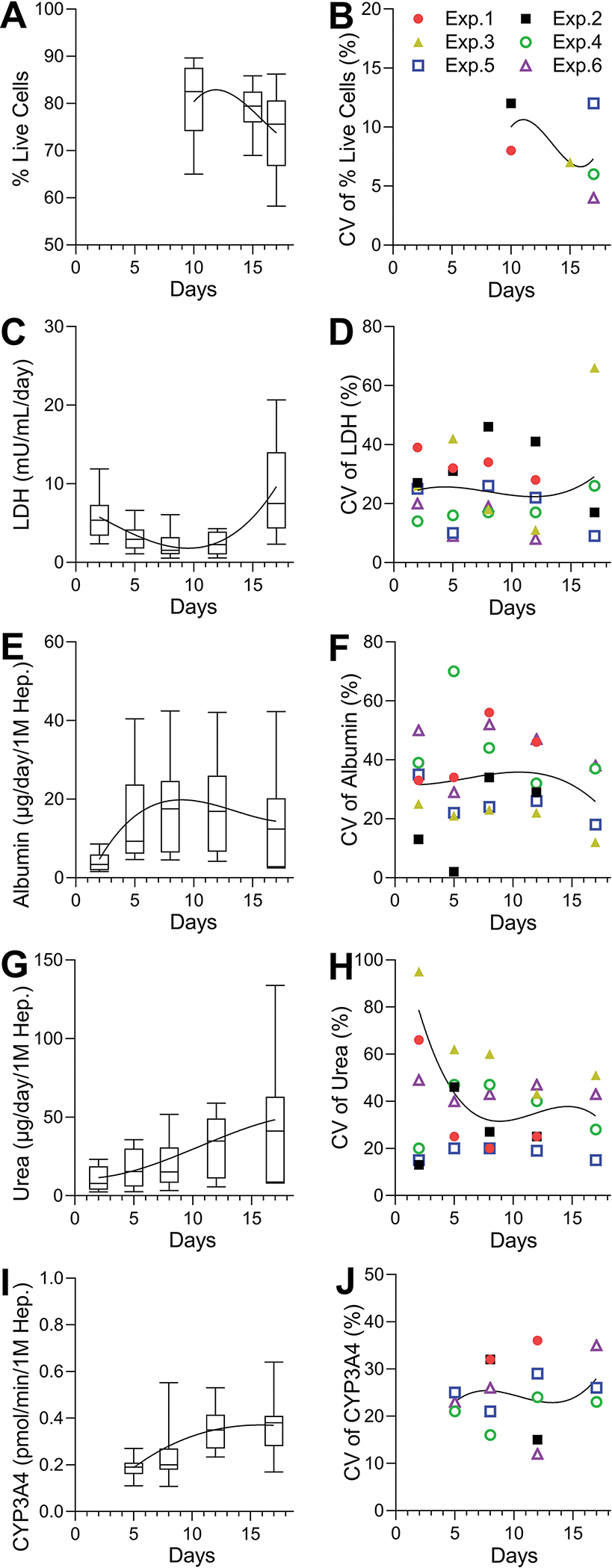
(A, C, E, G, I) Variability for each phenotype as indicated in the y-axis. The values were graphed as box and whiskers (10–90 percentile) by combining data from all experiments (n = 16–31). (B, D, F, H, J) Coefficients of variation and their trends for each phenotype. Data were plotted for each experiment (n = 3–8). Data were combined into 5 representative time points: days 1–3, 4–6, 7–9, 10–13, and 14–17.
Table 1.
Sample size estimates for observing time-related differences in basic hepatic function in experiments with OrganoPlate® 2-lane 96 plates seeded with iHeps and non-parenchymal (vehicle-treated groups). Shown are the number of replicate devices needed for detecting significant (p<0.05 with 80% power) differences between timepoints or in a trend test.
| Endpoint | Un-paired samples | Paired samples |
|---|---|---|
|
| ||
| Lactate dehydrogenase | ||
| Days 1–3 vs Days 14–17 | 66 (33/group) | 28 |
| Days 7–9 vs Days 14–17 | 20 (10/group) | 9 |
| Time trend (regression) | >100 | >100 |
|
| ||
| Albumin | ||
| Days 1–3 vs Days 4–6 | 26 (13/group) | 13 |
| Days 1–3 vs Days 7–9 | 20 (10/group) | 10 |
| Time trend (regression) | >100 | >100 |
|
| ||
| Urea | ||
| Days 1–3 vs Days 4–6 | 54 (27/group) | 20 |
| Days 1–3 vs Days 14–17 | 28 (14/group) | 14 |
| Time trend (regression) | 33 | >100 |
|
| ||
| CYP3A4 activity | ||
| Days 4–6 vs Days 7–9 | 76 (38/group) | 35 |
| Days 4–6 vs Days 14–17 | 16 (8/group) | 7 |
| Time trend (regression) | 65 | >100 |
Evaluation of metabolic capacity in experiments with co-cultured iHeps and non-parenchymal cells
Metabolic function of iHeps was evaluated using two approaches, with a 5-drug cocktail (phenacetin, 100 μM (CYP1A2); coumarin, 20 μM (CYP2A6); diclofenac, 10 μM (CYP2C9); terfenadine, 10 μM (CYP3A4); phenolphthalein, 10 μM (β-glucuronidase)), or 5 μM midazolam (CYP3A4). Drugs were added for 24 hrs on days 6, 12, and 16 of culture. We evaluated both disappearance of the parent compounds as well as the formation of primary metabolites (Figure 4). Parent compound concentrations were normalized to chips containing the gel without cells (blank chips) to account for non-specific binding and retention in the gel. Of all compounds tested, terfenadine showed the greatest degree of metabolism (Figure 4A), with significant reduction of the parent in at least one of the time points was observed for all compounds. However, the reduction in midazolam concentration was not significant. Primary metabolites of phenacetin, diclofenac, terfenadine, phenolphthalein and midazolam were detected (Figure 4B), most exhibiting a significant increase in metabolite formation with time. Next, we used these data to estimate the number of replicates that would be necessary to detect the loss of the parent molecule in this model at each time point (Table 2). We found that the most optimal time point for studies of metabolism is day 12 in culture; 6 to 11 replicates would be needed to detect significant change (p<0.05) with 80% power.
Figure 4. Drug metabolism in OrganoPlate® 2-lane 96 plate seeded with iHeps and non-parenchymal cells over time.
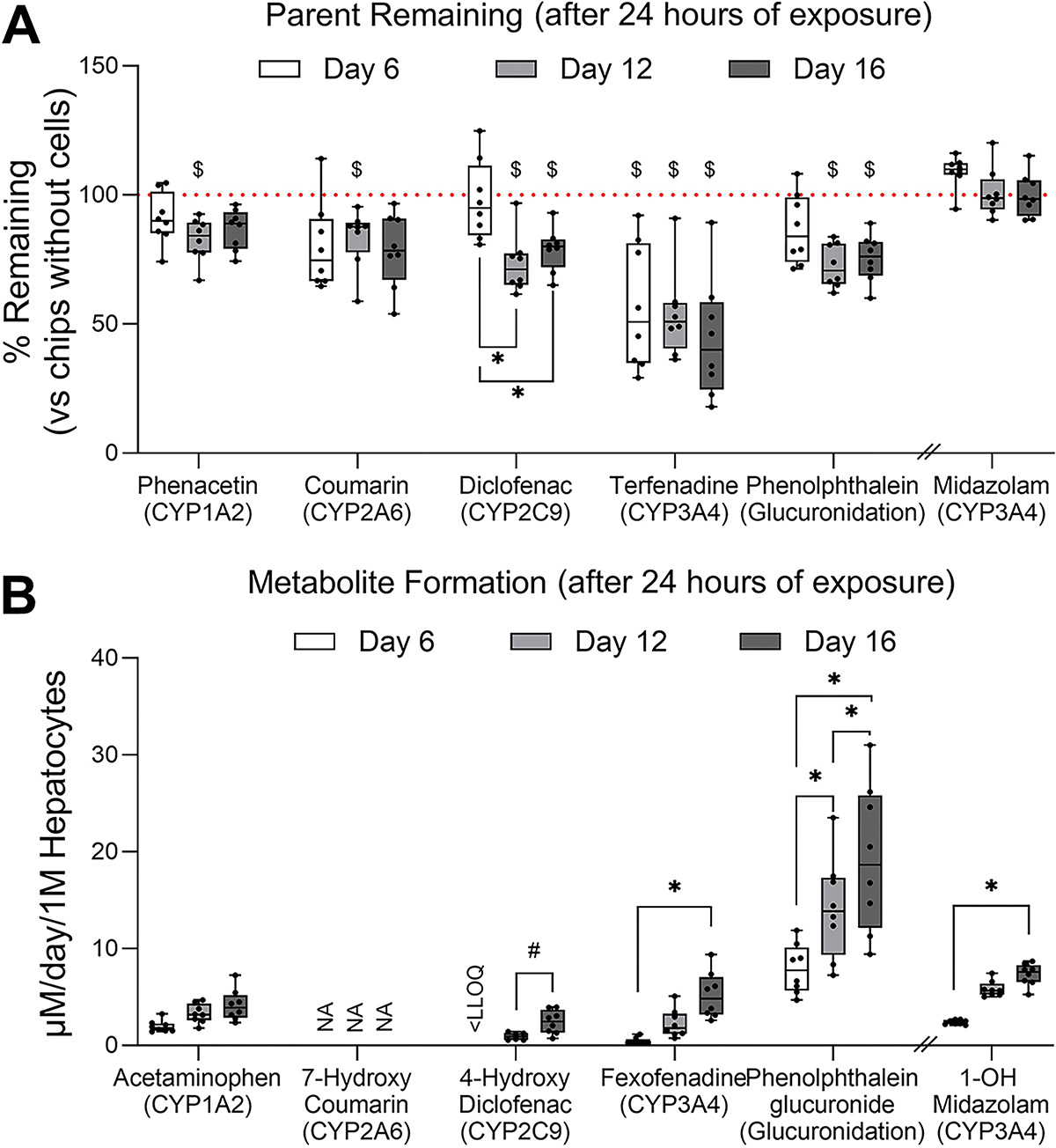
(A) A 5-chemical cocktail (phenacetin, 100 μM; coumarin, 20 μM; diclofenac, 10 μM; terfenadine, 10 μM; phenolphthalein, 10 μM) or 5 μM midazolam were added to OrganoPlate® 2-lane 96 plates for 24 h on days 6, 12, and 16 of culture (n = 8). The drug probe media were also injected to chips without cells (only gel) for 24 h at the same timepoints and these values were used as reference (horizontal dotted lines). Data are graphed as box and whiskers (min-max). Asterisks indicate statistical significance as follows: (*) is for p < 0.05 by one-way ANOVA with Dunnett’s multiple comparisons test within each compound; ($) is for p < 0.05 by unpaired t-test with Welch’s correction compared to blank devices at each sampling point. (B) Metabolite formation from each parent compound. Data are graphed as box and whiskers (min-max). Asterisks indicate statistical significance as follows: (*) is for p < 0.05 by one-way ANOVA with Dunnett’s multiple comparisons test within each compound; (#) is for p < 0.05 by paired t-test at each sampling point. NA, not available; LLOQ, lower limit of quantification.
Table 2.
Sample size estimates for observing time-related differences in drug metabolism (disappearance of the substrate) in experiments with OrganoPlate® 2-lane 96 plate seeded with iHeps and non-parenchymal cells. Shown are the number of replicate devices needed for detecting significant (p<0.05 with 80% power) differences at each timepoint.
| Drug (Primary metabolic pathway) | Day 6 | Day 12 | Day 16 |
|---|---|---|---|
|
| |||
| Phenacetin (CYP1A2) | 19 | 8 | 16 |
| Coumarin (CYP2A6) | 14 | 11 | 13 |
| Diclofenac (CYP2C9) | >100 | 8 | 10 |
| Terfenadine (CYP3A4) | 9 | 7 | 8 |
| Phenolphthalein (Glucuronidation) | 18 | 6 | 10 |
| Midazolam (CYP3A4) | 12 | >100 | >100 |
Hepatotoxicity studies in experiments with co-cultured iHeps and non-parenchymal cells
Most in vitro models of the liver are used for studies of drug and chemical toxicity. Indeed, the model studied herein was proposed for high-throughput compound toxicity screening (Bircsak et al., 2021). To evaluate the utility of this model to detect hepatotoxicity, iHeps co-cultured with non-parenchymal cells for 7 days were first treated with 180 μM troglitazone (about 30× of human Cmax) every 24 hrs for 3 consecutive days. Troglitazone treatment significantly reduced cell viability as compared to vehicle controls after 3 days of treatment (Figure 5A). When other biomarkers were evaluated from daily media collections, we observed clear time-dependent trends in the loss of cell functionality (Figures 5B–D); the loss of albumin production was the most robust endpoint with significant effects observed after 48 and 72 hrs of treatment (Figure 5C). Overall, the observed effects of troglitazone were in accordance with those reported by Bircsak et al (2021). Under conditions of overt toxicity as in the case with troglitazone, we found that 4–9 replicates would be sufficient to detect adverse effects on cell viability, LDH and albumin production, especially after 48 or 72 hrs of treatment (Table 3). We found that urea production was highly variable across this experiment and would be less preferable biomarker of toxicity in this model.
Figure 5. Changes of cell viability, LDH, albumin, and urea after 180 μM troglitazone exposure in OrganoPlate® 2-lane 96 plate seeded with iHeps and non-parenchymal cells.
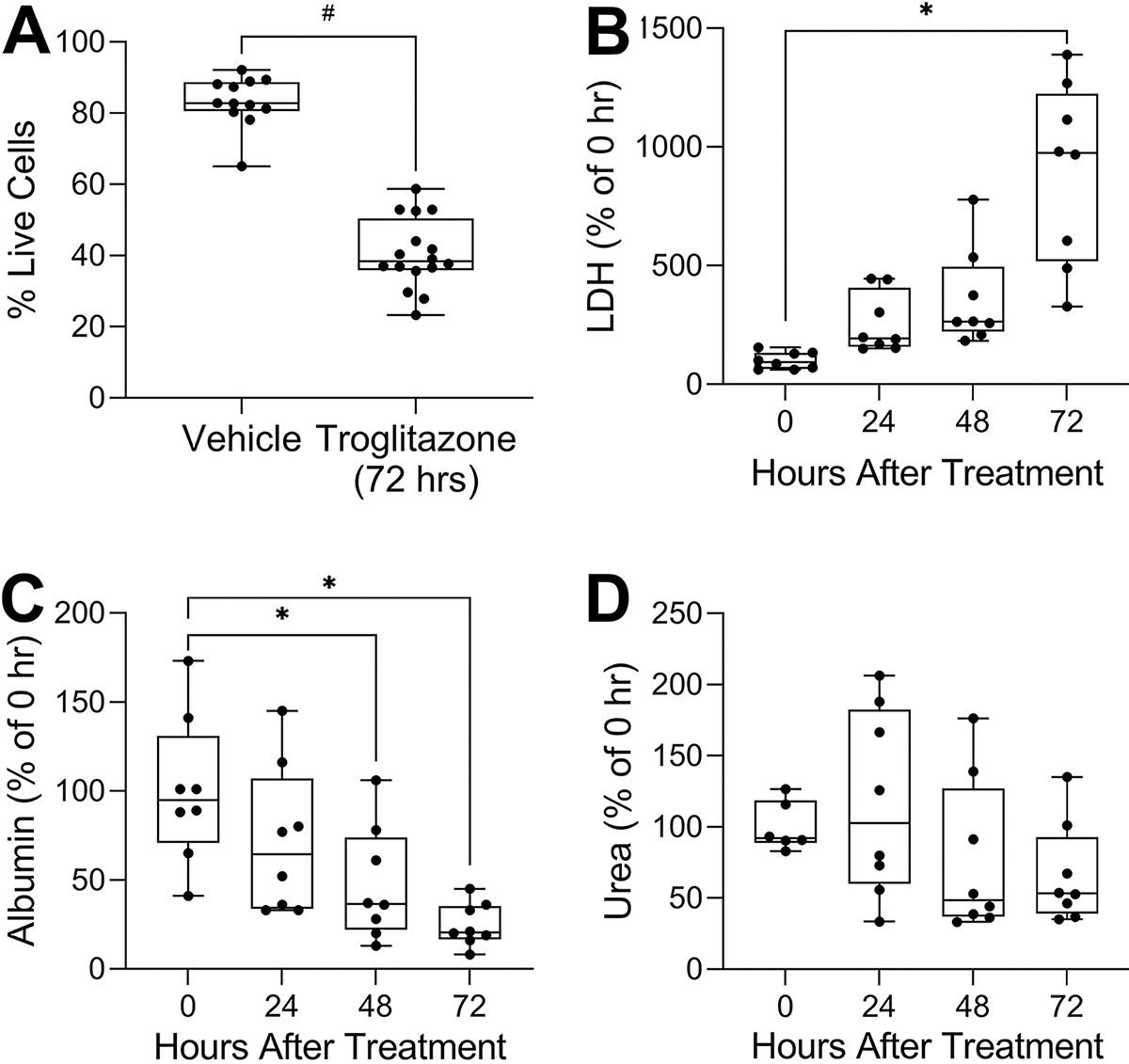
(A) Image-based cell viability, (B) LDH leakage, (C) albumin secretion, (D) urea secretion. Except for cell viability, endpoints were normalized to the mean of the data at 0 h. Data are plotted as box and whiskers (min-max, n = 8–16). Asterisks indicate statistical significance as follows: (*) is for p < 0.05 by one-way ANOVA with Dunnett’s multiple comparisons test; (#) is for p < 0.05 by unpaired t-test with Welch’s correction.
Table 3.
Sample size estimates for observing time-related differences in troglitazone (180 μM) toxicity in experiments with OrganoPlate® 2-lane 96 plate seeded with iHeps and non-parenchymal cells. Shown are the number of paired devices needed for detecting significant (p<0.05 with 80% power) differences at each timepoint.
| Endpoint | 24 h | 48 h | 72 h |
|---|---|---|---|
|
| |||
| Live cells (%) | 6 (3/group) | ||
| Lactate dehydrogenase | 8 | 6 | 4 |
| Albumin | 30 | 9 | 5 |
| Urea | 41 | >100 | 7 |
Because this model contains both iHeps and non-parenchymal cells, we determined whether immune-mediated adverse effects of drugs or other molecules could be observed. The original study by Bircsak et al (2021) reported that treatment with lipopolysaccharide resulted in production of TNFα; in this study we evaluated production of interleukin (IL)-6 in addition to other measures of cell functionality (Figure 6). Treatment with lipopolysaccharide (1 μg/mL) alone led to significant increases in albumin and urea secretion and production of IL-6, as well as a reduction in CYP3A4 activity. Next, we used a combined treatment of trovafloxacin and lipopolysaccharide, a classic model of immune-mediated liver toxicity (Beggs et al., 2014; Shaw et al., 2007; Waring et al., 2006). We tested trovafloxacin at two concentrations (50 and 150 μM, about 10× and 30× human Cmax, respectively), with and without addition of lipopolysaccharide. While expected effects were observed such as a decrease in cell viability, production of urea, and loss of CYP3A4 function, commensurate with known suppression of hepatic metabolic enzymes by cytokines (Dickmann et al., 2011); high variability between individual devices (n=4 to 8 for each phenotype) resulted in few of these effects reaching statistical significance. In addition, effects of lipopolysaccharide on trovafloxacin-mediated toxicity were not evident.
Figure 6. Changes of cell viability, LDH, albumin, urea, CYP3A4, and IL-6 after 48 h troglitazone exposure with and without LPS in OrganoPlate® 2-lane 96 plate seeded with iHeps and non-parenchymal cells.
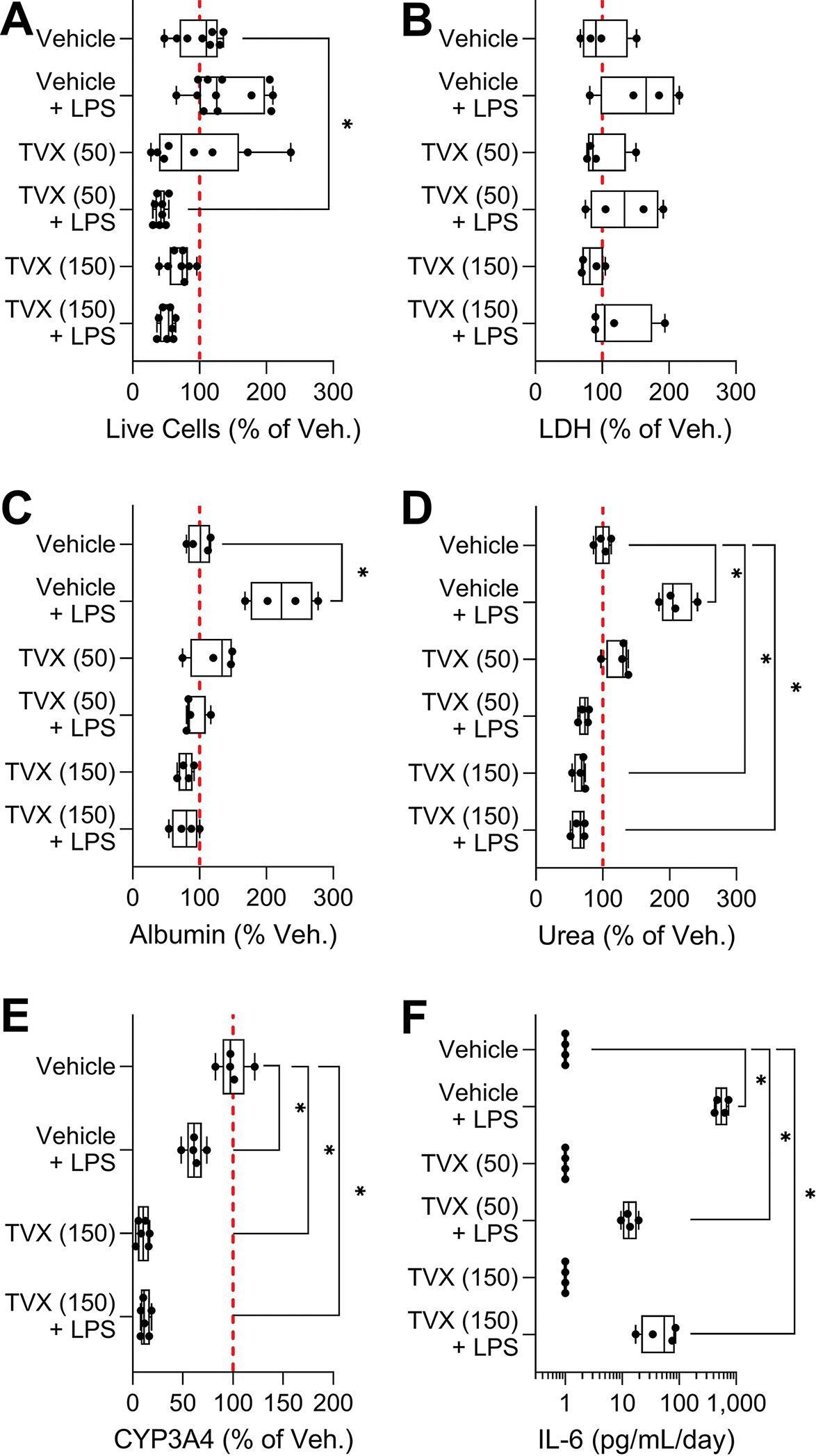
(A) Image-based cell viability, (B) LDH leakage, (C) albumin secretion, (D) urea secretion, (E) CYP3A4 activity, and (F) IL-6 release. Except for IL-6, endpoints were normalized to the mean of the vehicle controls. Data are plotted as box and whiskers (min-max, n = 4–12). The red dotted line shows 100% (mean of the vehicle samples). Asterisks (*) indicate statistical significance at p < 0.05 by one-way ANOVA with Dunnett’s multiple comparisons test as compared to vehicle controls.
Discussion
It is widely appreciated that microphysiological systems offer a number of advantages over traditional in vitro models of the liver; however, their utility to exhibit basic liver function, differentiate toxic from non-toxic structural analogues, and determine toxicity for new compounds needs to be established (Baudy et al., 2020). In addition, for these models to become more routinely used in drug and other chemical safety testing, their throughput, functionality and robustness should be characterized. Because of the complexity of many liver-focused microphysiological systems and their recent introduction to the fields of toxicology and safety pharmacology (Ewart and Roth, 2021), there are few published examples of their technology transfer, robustness, and the most suitable contexts of use. Most publications of novel liver microphysiological systems present initial case studies by their developers. However, only a handful of studies are available that attempted to perform independent testing of these devices under more realistic end-user experimental scenarios (Rubiano et al., 2021; Sakolish et al., 2021b).
One challenge for testing reproducibility and robustness is the model’s throughput; most liver microphysiological systems offer low double-digit throughput (in terms of the number of conditions that can be interrogated simultaneously) in an experiment because of the need for external device-driven microfluidics (Jang et al., 2019; Sasserath et al., 2020; Tsamandouras et al., 2017; Vernetti et al., 2016). By contrast, OrganoPlate® 2-lane 96 plate devices offer a potentially greater throughput and lower cost, in terms of both materials and personnel time; this model was used to test over 150 compounds for their potential hepatotoxicity (Bircsak et al., 2021). The potential utility of the latter model is thus quite promising because its throughput may be approaching that of traditional in vitro models of hepatotoxicity that utilize 96- or 384-well plates (LeCluyse et al., 2012; Soldatow et al., 2013). While the throughput is not the only determining factor with respect to the model’s usability, the potential utility of liver microphysiological systems for safety testing is likely to be as common or greater than that for the mechanistic studies or species comparisons.
Because of the potentially wide application and the higher-throughput nature of OrganoPlate® 2-lane 96 plate model, we aimed to compare hepatic functionality of different cell types and culture conditions and evaluated the degree of reproducibility of this model in independent experiments. The overall rationale for this study was based on a well-acknowledged challenge with moving promising yet complex microphysiological systems from the developer labs to the end-users (Ewart et al., 2017; Livingston et al., 2016). Our study was designed to follow as closely as possible the published protocols (Bircsak et al., 2021), but to also evaluate the model under additional conditions that would be of interest to the prospective end-users, namely the cell types and sources, replication, device handling, and additional treatments. The latter considerations were based on the input from a group of prospective end-users who comprise the TEX-VAL Consortium, a public-private collaboration with representation from the pharmaceutical and consumer goods companies, a trade association, and several US governmental agencies; established for evaluation and testing of diverse microphysiological systems (Rusyn et al., 2022). Testing of reproducibility of microphysiological systems is needed to ensure the acceptance and use of these alternative non-animal testing approaches for basic research and drug or chemical safety assessments. As microphysiological systems are transferred from the developers to prospective end-users, they need detailed information on their utility and limitations, and examples of applications in decision-making. In addition, studies similar to one detailed herein are needed for building confidence through standardization and qualification of microphysiological systems (Ewart and Roth, 2021).
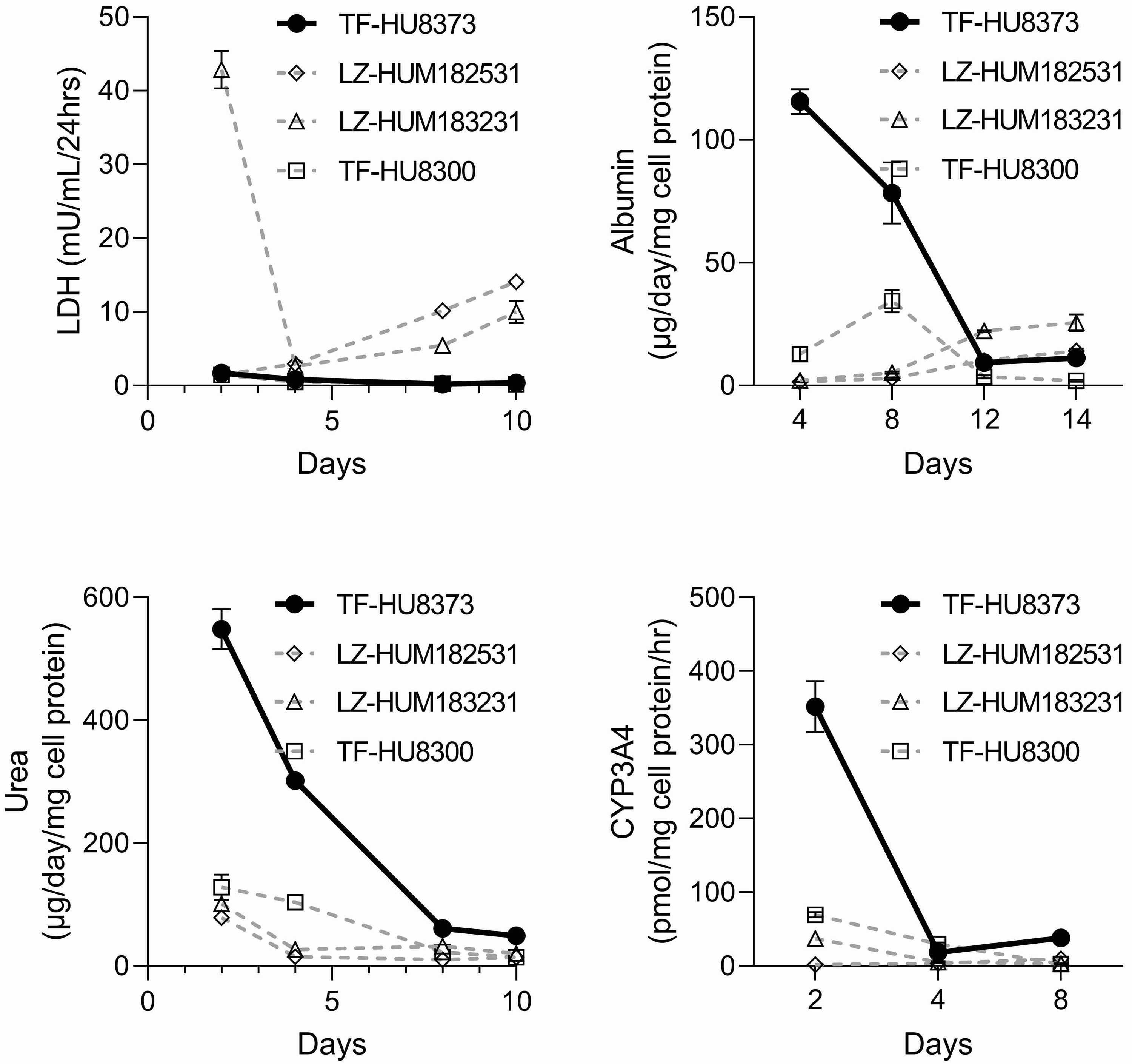
Based on our experience with OrganoPlate® 2-lane 96 plate model, we conclude that it is a robust experimental platform that is fairly straightforward to establish in a new laboratory based on the published protocols from the developers (Bircsak et al., 2021). This model is most physiological in terms of the basic liver function when seeded with both iHeps and non-parenchymal cells, as published (Bircsak et al., 2021). Our data, in accord with the original study, show that most optimal time periods for studies of metabolism and hepatotoxicity are between days 7 and 13; longer culture conditions may be less favored as the function and viability of the hepatocytes begins to fall and few non-parenchymal cells remain in the media perfusion channel. Primary human hepatocytes appear to be a challenging cell type for this model based on the data we provided herein; even though we have selected the most functional lot of primary human hepatocytes for these studies, it is possible that donor variability may have contributed to sub-optimal performance of these cells in the OrganoPlate® 2-lane 96 plate model. While the use of iPSC-derived human hepatocytes is an advantage in terms of future comparisons between studies, most publications use commercially-available iHeps (Sirenko et al., 2014). Therefore the lack of iPSC-derived hepatocytes from different donors, similar to multi-donor iPSC-derived cardiomyocytes (Burnett et al., 2019), is a considerable limitation because of the heterogeneity in basic liver function and metabolic capacity of hepatocytes across a population that is critical for understanding the inter-individual variability in liver toxicity (Ulrich, 2007). In addition, while a number of publications reasoned that iPSC-derived hepatocytes may be comparable in their function to primary human hepatocytes in culture (Lu et al., 2015; Sirenko et al., 2014), major questions remain about their fetal-like characteristics in terms of the metabolic enzyme expression and overall utility in drug development (Yamaguchi et al., 2019).
Our data show that OrganoPlate® 2-lane 96 plate model seeded with both iHeps and non-parenchymal cells demonstrates higher basic liver function as compared to same cells cultured in conventional monolayer constructs. We also obtained comparable data on metabolic capacity in this model and replicated the effects of troglitazone and lipopolysaccharide. Additionally, we found that manual seeding of the devices can be successfully performed, albeit with sufficient training and following the precautions detailed here in the Methods. This finding opens the opportunity for wider adoption of this technology by laboratories that may not have access to automated liquid handling that can also be used for seeding of the devices. Overall, these data are encouraging as they demonstrate a success in technology transfer and the ability to maintain long-term (about 2.5 weeks) viability and functionality of the cells in this microphysiological system. However, there are several limitations and additional considerations as listed below that were identified by our study that may serve as additional information for future prospective users of this technology.
Our data is informative for future experimental design considerations in this platform, especially in terms of the number of replicates that may be needed for studies of different “context of use.” We found that 8 or more replicates may be needed to ensure that the experiments are sufficiently powered to detect adverse effects in this model. The number of replicates should be even greater in studies of basal liver function. This finding is potentially counter-balances the prospects of high-throughput studies in this model, albeit comparable data from other liver microphysiological systems of the liver are lacking. The original study reported robust z’-factors, a measure of experimental reproducibility; however, those values were derived from intra-plate reproducibility and no indication of inter-plate reproducibility was provided. Our data show that indeed it is replicate studies on different plates, despite the use of the same lot of iHeps and other factors that could be reasonably standardized (e.g., protocol, operator, media, etc.), that likely contribute to the high variability observed. Indirect evidence of high variability between experiments was also presented by Bircsak et al (2021), specifically, basal albumin production varied about 10-fold between experiments. Experimental variability is a concern not only for this study but for other in vitro studies, or studies in animals and humans; still, clarity about the statistical power is needed to increase confidence in the model and therefore the data provided here should be useful for designing informative studies in the future.
Related to the need for increasing the number of replicates in this model is the consideration of its utility for screening of large libraries of compounds. In this regard, the inclusion of a screening study in the publication by Bircsak et al (2021) is a major positive development in the field of liver microphysiological systems. However, limited replication (n=3) was used for concentration-response study of 21 compounds, and no replication was used for the study of 159 compounds (all tested at 50 μM). The outcomes of these experiments, as reported by Bircsak et al (2021), in comparison to other published in vitro liver model data and clinical classification of the compounds with respect to their potential hepatotoxicity may be characterized as limited success. For example, the accuracy metrics for the data from OrganoPlate® 2-lane 96 plate model (Bircsak et al., 2021) were less favorable than the outcomes from studies in spheroids of primary human hepatocytes (Supplemental Figure 3A), albeit the number of chemicals in these studies varied. Also, the performance of the OrganoPlate® 2-lane 96 plate model (Bircsak et al., 2021), as a predictor of human drug-induced liver injury, improved (Supplemental Figure 3B) when a dose-response study design was used with some replicates (n=3) for each condition. But the generalizability of this finding is yet to be tested because a smaller set of compounds was tested in these experiments. When these results are interpreted in the context of our study, we found that the OrganoPlate® 2-lane 96 plate model could be used to detect compounds that are expected to exhibit key characteristics of hepatotoxicity that originate from hepatocytes themselves (Rusyn et al., 2021). For example, in the case of troglitazone, which is known to be bioactivated to electrophilic moieties, causing cell death through oxidative stress, mitochondria dysfunction and cholestasis (Smith, 2003), we were able to confirm previous findings (Bircsak et al., 2021). However, other microphysiological or simpler in vitro liver models could also be used for this purpose (Rubiano et al., 2021; Sakolish et al., 2021b; Ware et al., 2017; Yokoyama et al., 2018).
For studying hepatotoxicity that involves cytokine-mediated events, another key characteristic for hepatotoxic compounds including drugs (Roberts et al., 2007), the OrganoPlate® 2-lane 96 plate model may not be the most optimal choice. Even though we did observe increase in IL-6 and a decrease in CYP3A4 activity in lipopolysaccharide-treated devices, indicative of the ability of nonparenchymal cells to be activated, we did not observe potentiation of trovafloxacin-mediated toxicity by lipopolysaccharide. Other multi-cellular liver microphysiological systems were able to observe the synergistic effects (Rubiano et al., 2021; Sakolish et al., 2021b). The discordance in these observation between models is likely due to the differences in topology of hepatocytes and non-parenchymal cells between the OrganoPlate® 2-lane 96 plate model and other multi-cellular liver microphysiological systems, the former encapsulates hepatocytes in the gel creating a physical separation between hepatocytes and non-parenchymal cells. This may lead to the challenges in establishing microphysiological conditions in the liver acinus that determine paracrine actions of the non-parenchymal cells on hepatocytes and vice versa (Kmiec, 2001). It is also possible that this was due to the immature state of iHeps, including the lack of adult hepatocyte-like expression of TGF-β which is manipulated during cell de-differentiation and re-differentiation into hepatocytes (Ang et al., 2018).
In conclusion, we note that while the overall transferability of the OrganoPlate® 2-lane 96 plate microphysiological system was a success, the reproducibility with the original study (Bircsak et al., 2021) was greatly dependent on the number of replicates. The role of the differences in the cell source, a typical obstacle to replication of complex cell-based models, was minimal in this study because we used iPSC-derived hepatocytes; still, we showed that the use of other cells such as primary human hepatocytes would have resulted in the lack of reproducibility. Our results will be informative to end-users of this highly promising high-throughput model; however, we caution that careful consideration of replicates and inclusion of dose-response study designs may be needed to increase confidence in interpreting the findings. The ability of this model to detect immune-mediated toxicity, especially under prolonged culture conditions, may be limited in the current implementation of this microphysiological system.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Highlights.
Microphysiological systems are thought to be more human-relevant models for toxicology
Liver microphysiological systems are complex devices that typically have low throughput
This study tested a higher-throughput liver microphysiological system for its robustness
OrganoPlate® 2-lane 96 plates are most functional with iHeps and non-parenchymal cells
This model needs high number of replicates to deliver robust results limiting throughput
Acknowledgements
This work was performed through the TEX-VAL Consortium collaboration funded by equitable monetary contributions from member organizations (American Chemistry Council, Bristol-Myers Squibb, Merck Healthcare KGaA, National Institute of Environmental Health Sciences, Sanofi, Unilever, and United States Environmental Protection Agency). This work was also supported, in part, by a grant from the National Institutes of Health U24 TR002633. The authors greatly appreciate useful discussions and technical support from TEX-VAL Consortium members. We are also grateful to the University of Pittsburgh MPS-Db team for assistance with depositing the data and making them publicly available.
Footnotes
Disclaimer: This work does not represent policy or product endorsement by TEX-VAL Consortium member organizations (American Chemistry Council, Bristol-Myers Squibb, Merck KGaA, National Institute of Environmental Health Sciences, Sanofi, Unilever, and United States Environmental Protection Agency).
References
- Ang LT, Tan AKY, Autio MI, Goh SH, Choo SH, Lee KL, Tan J, Pan B, Lee JJH, Lum JJ, Lim CYY, Yeo IKX, Wong CJY, Liu M, Oh JLL, Chia CPL, Loh CH, Chen A, Chen Q, Weissman IL, Loh KM, Lim B, 2018. A Roadmap for Human Liver Differentiation from Pluripotent Stem Cells. Cell Rep 22, 2190–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudy AR, Otieno MA, Hewitt P, Gan J, Roth A, Keller D, Sura R, Van Vleet TR, Proctor WR, 2020. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip 20, 215–225. [DOI] [PubMed] [Google Scholar]
- Beggs KM, Fullerton AM, Miyakawa K, Ganey PE, Roth RA, 2014. Molecular mechanisms of hepatocellular apoptosis induced by trovafloxacin-tumor necrosis factor-alpha interaction. Toxicol Sci 137, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CC, Dankers ACA, Lauschke VM, Sison-Young R, Jenkins R, Rowe C, Goldring CE, Park K, Regan SL, Walker T, Schofield C, Baze A, Foster AJ, Williams DP, van de Ven AWM, Jacobs F, Houdt JV, Lahteenmaki T, Snoeys J, Juhila S, Richert L, Ingelman-Sundberg M, 2018. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol Sci 162, 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bircsak KM, DeBiasio R, Miedel M, Alsebahi A, Reddinger R, Saleh A, Shun T, Vernetti LA, Gough A, 2021. A 3D microfluidic liver model for high throughput compound toxicity screening in the OrganoPlate(R). Toxicology 450, 152667. [DOI] [PubMed] [Google Scholar]
- Bulutoglu B, Rey-Bedon C, Mert S, Tian L, Jang YY, Yarmush ML, Usta OB, 2020. A comparison of hepato-cellular in vitro platforms to study CYP3A4 induction. PLoS One 15, e0229106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnett SD, Blanchette AD, Grimm FA, House JS, Reif DM, Wright FA, Chiu WA, Rusyn I, 2019. Population-based toxicity screening in human induced pluripotent stem cell-derived cardiomyocytes. Toxicol Appl Pharmacol 381, 114711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SD, Yuen G, Tu T, Budzinska MA, Spring K, Bryant K, Shackel NA, 2019. In Vitro Models of the Liver: Disease Modeling, Drug Discovery and Clinical Applications, In: Tirnitz-Parker JEE. (Ed.), Hepatocellular Carcinoma, Brisbane (AU). [PubMed] [Google Scholar]
- Corbett JL, Duncan SA, 2019. iPSC-Derived Hepatocytes as a Platform for Disease Modeling and Drug Discovery. Front Med (Lausanne) 6, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickmann LJ, Patel SK, Rock DA, Wienkers LC, Slatter JG, 2011. Effects of interleukin-6 (IL-6) and an anti-IL-6 monoclonal antibody on drug-metabolizing enzymes in human hepatocyte culture. Drug Metab Dispos 39, 1415–1422. [DOI] [PubMed] [Google Scholar]
- Dorne JL, Walton K, Renwick AG, 2003. Human variability in CYP3A4 metabolism and CYP3A4-related uncertainty factors for risk assessment. Food Chem Toxicol 41, 201–224. [DOI] [PubMed] [Google Scholar]
- Ewart L, Fabre K, Chakilam A, Dragan Y, Duignan DB, Eswaraka J, Gan J, Guzzie-Peck P, Otieno M, Jeong CG, Keller DA, de Morais SM, Phillips JA, Proctor W, Sura R, Van Vleet T, Watson D, Will Y, Tagle D, Berridge B, 2017. Navigating tissue chips from development to dissemination: A pharmaceutical industry perspective. Exp Biol Med (Maywood) 242, 1579–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewart L, Roth A, 2021. Opportunities and challenges with microphysiological systems: a pharma end-user perspective. Nat Rev Drug Discov 20, 327–328. [DOI] [PubMed] [Google Scholar]
- Godoy P, Hewitt NJ, Albrecht U, Andersen ME, Ansari N, Bhattacharya S, Bode JG, Bolleyn J, Borner C, Bottger J, Braeuning A, Budinsky RA, Burkhardt B, Cameron NR, Camussi G, Cho CS, Choi YJ, Craig Rowlands J, Dahmen U, Damm G, Dirsch O, Donato MT, Dong J, Dooley S, Drasdo D, Eakins R, Ferreira KS, Fonsato V, Fraczek J, Gebhardt R, Gibson A, Glanemann M, Goldring CE, Gomez-Lechon MJ, Groothuis GM, Gustavsson L, Guyot C, Hallifax D, Hammad S, Hayward A, Haussinger D, Hellerbrand C, Hewitt P, Hoehme S, Holzhutter HG, Houston JB, Hrach J, Ito K, Jaeschke H, Keitel V, Kelm JM, Kevin Park B, Kordes C, Kullak-Ublick GA, LeCluyse EL, Lu P, Luebke-Wheeler J, Lutz A, Maltman DJ, Matz-Soja M, McMullen P, Merfort I, Messner S, Meyer C, Mwinyi J, Naisbitt DJ, Nussler AK, Olinga P, Pampaloni F, Pi J, Pluta L, Przyborski SA, Ramachandran A, Rogiers V, Rowe C, Schelcher C, Schmich K, Schwarz M, Singh B, Stelzer EH, Stieger B, Stober R, Sugiyama Y, Tetta C, Thasler WE, Vanhaecke T, Vinken M, Weiss TS, Widera A, Woods CG, Xu JJ, Yarborough KM, Hengstler JG, 2013. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch Toxicol 87, 1315–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt NJ, Lechon MJ, Houston JB, Hallifax D, Brown HS, Maurel P, Kenna JG, Gustavsson L, Lohmann C, Skonberg C, Guillouzo A, Tuschl G, Li AP, LeCluyse E, Groothuis GM, Hengstler JG, 2007. Primary hepatocytes: current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metab Rev 39, 159–234. [DOI] [PubMed] [Google Scholar]
- Jang KJ, Otieno MA, Ronxhi J, Lim HK, Ewart L, Kodella KR, Petropolis DB, Kulkarni G, Rubins JE, Conegliano D, Nawroth J, Simic D, Lam W, Singer M, Barale E, Singh B, Sonee M, Streeter AJ, Manthey C, Jones B, Srivastava A, Andersson LC, Williams D, Park H, Barrile R, Sliz J, Herland A, Haney S, Karalis K, Ingber DE, Hamilton GA, 2019. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci Transl Med 11, eaax5516. [DOI] [PubMed] [Google Scholar]
- Kang W, Podtelezhnikov AA, Tanis KQ, Pacchione S, Su M, Bleicher KB, Wang Z, Laws GM, Griffiths TG, Kuhls MC, Chen Q, Knemeyer I, Marsh DJ, Mitra K, Lebron J, Sistare FD, 2020. Development and Application of a Transcriptomic Signature of Bioactivation in an Advanced In Vitro Liver Model to Reduce Drug-induced Liver Injury Risk Early in the Pharmaceutical Pipeline. Toxicol Sci 177, 121–139. [DOI] [PubMed] [Google Scholar]
- Keemink J, Oorts M, Annaert P, 2015. Primary Hepatocytes in Sandwich Culture. Methods Mol Biol 1250, 175–188. [DOI] [PubMed] [Google Scholar]
- Kmiec Z, 2001. Cooperation of liver cells in health and disease. Adv Anat Embryol Cell Biol 161, III-XIII, 1–151. [DOI] [PubMed] [Google Scholar]
- Kullak-Ublick GA, Andrade RJ, Merz M, End P, Benesic A, Gerbes AL, Aithal GP, 2017. Drug-induced liver injury: recent advances in diagnosis and risk assessment. Gut 66, 1154–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeCluyse EL, Witek RP, Andersen ME, Powers MJ, 2012. Organotypic liver culture models: meeting current challenges in toxicity testing. Crit Rev Toxicol 42, 501–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Montiel FT, George SM, Gough AH, Sharma AD, Wu J, DeBiasio R, Vernetti LA, Taylor DL, 2017. Control of oxygen tension recapitulates zone-specific functions in human liver microphysiology systems. Exp Biol Med (Maywood) 242, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston CA, Fabre KM, Tagle DA, 2016. Facilitating the commercialization and use of organ platforms generated by the microphysiological systems (Tissue Chip) program through public-private partnerships. Comput Struct Biotechnol J 14, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA, 2021. Organs-on-chips: into the next decade. Nat Rev Drug Discov 20, 345–361. [DOI] [PubMed] [Google Scholar]
- Lu J, Einhorn S, Venkatarangan L, Miller M, Mann DA, Watkins PB, LeCluyse E, 2015. Morphological and functional characterization and assessment of iPSC-derived hepatocytes for in vitro toxicity testing. Toxicological Sciences 147, 39–54. [DOI] [PubMed] [Google Scholar]
- Machin D, Campbell M, Tan S, Tan S-H, 2011. Sample Size Tables for Clinical Studies, Third Edition. Wiley-Blackwell. [Google Scholar]
- Monticello TM, Jones TW, Dambach DM, Potter DM, Bolt MW, Liu M, Keller DA, Hart TK, Kadambi VJ, 2017. Current nonclinical testing paradigm enables safe entry to First-In-Human clinical trials: The IQ consortium nonclinical to clinical translational database. Toxicol Appl Pharmacol 334, 100–109. [DOI] [PubMed] [Google Scholar]
- Ramaiahgari SC, Waidyanatha S, Dixon D, DeVito MJ, Paules RS, Ferguson SS, 2017. Three-Dimensional (3D) HepaRG Spheroid Model With Physiologically Relevant Xenobiotic Metabolism Competence and Hepatocyte Functionality for Liver Toxicity Screening. Toxicol Sci 159, 124–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE, 2007. Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci 96, 2–15. [DOI] [PubMed] [Google Scholar]
- Rubiano A, Indapurkar A, Yokosawa R, Miedzik A, Rosenzweig B, Arefin A, Moulin CM, Dame K, Hartman N, Volpe DA, Matta MK, Hughes DJ, Strauss DG, Kostrzewski T, Ribeiro AJS, 2021. Characterizing the reproducibility in using a liver microphysiological system for assaying drug toxicity, metabolism, and accumulation. Clin Transl Sci 14, 1049–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Arzuaga X, Cattley RC, Corton JC, Ferguson SS, Godoy P, Guyton KZ, Kaplowitz N, Khetani SR, Roberts RA, Roth RA, Smith MT, 2021. Key Characteristics of Human Hepatotoxicants as a Basis for Identification and Characterization of the Causes of Liver Toxicity. Hepatology 74, 3486–3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyn I, Sakolish C, Kato Y, Stephan C, Vergara L, Hewitt P, Bhaskaran V, Davis M, Hardwick R, Ferguson SS, Stanko JP, Bajaj P, Adkins K, Sipes NS, Hunter S, Baltazar MT, Carmichael PL, Sadh K, Becker RA, 2022. Microphysiological Systems Evaluation: Experience of TEX-VAL Tissue Chip Testing Consortium. Toxicol Sci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakolish C, Luo YS, Valdiviezo A, Vernetti LA, Rusyn I, Chiu WA, 2021a. Prediction of hepatic drug clearance with a human microfluidic four-cell liver acinus microphysiology system. Toxicology 463, 152954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakolish C, Reese CE, Luo YS, Valdiviezo A, Schurdak ME, Gough A, Taylor DL, Chiu WA, Vernetti LA, Rusyn I, 2021b. Analysis of reproducibility and robustness of a human microfluidic four-cell liver acinus microphysiology system (LAMPS). Toxicology 448, 152651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasserath T, Rumsey JW, McAleer CW, Bridges LR, Long CJ, Elbrecht D, Schuler F, Roth A, Bertinetti-LaPatki C, Shuler ML, Hickman JJ, 2020. Differential Monocyte Actuation in a Three-Organ Functional Innate Immune System-on-a-Chip. Adv Sci (Weinh) 7, 2000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Hopfensperger MJ, Ganey PE, Roth RA, 2007. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol Sci 100, 259–266. [DOI] [PubMed] [Google Scholar]
- Sirenko O, Hesley J, Rusyn I, Cromwell EF, 2014. High-content assays for hepatotoxicity using induced pluripotent stem cell-derived cells. Assay Drug Dev Technol 12, 43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistare FD, Mattes WB, LeCluyse EL, 2016. The Promise of New Technologies to Reduce, Refine, or Replace Animal Use while Reducing Risks of Drug Induced Liver Injury in Pharmaceutical Development. ILAR J 57, 186–211. [DOI] [PubMed] [Google Scholar]
- Smith MT, 2003. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol 16, 679–687. [DOI] [PubMed] [Google Scholar]
- Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I, 2013. In vitro models for liver toxicity testing. Toxicol Res (Camb) 2, 23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DL, Gough A, Schurdak ME, Vernetti L, Chennubhotla CS, Lefever D, Pei F, Faeder JR, Lezon TR, Stern AM, Bahar I, 2019. Harnessing Human Microphysiology Systems as Key Experimental Models for Quantitative Systems Pharmacology. Handb Exp Pharmacol 260, 327–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsamandouras N, Kostrzewski T, Stokes CL, Griffith LG, Hughes DJ, Cirit M, 2017. Quantitative Assessment of Population Variability in Hepatic Drug Metabolism Using a Perfused Three-Dimensional Human Liver Microphysiological System. J Pharmacol Exp Ther 360, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich RG, 2007. Idiosyncratic toxicity: a convergence of risk factors. Annu Rev Med 58, 17–34. [DOI] [PubMed] [Google Scholar]
- Underhill GH, Khetani SR, 2018. Bioengineered Liver Models for Drug Testing and Cell Differentiation Studies. Cell Mol Gastroenterol Hepatol 5, 426–439 e421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utkarsh D, Loretz C, Li AP, 2016. In vitro evaluation of hepatotoxic drugs in human hepatocytes from multiple donors: Identification of P450 activity as a potential risk factor for drug-induced liver injuries. Chem Biol Interact 255, 12–22. [DOI] [PubMed] [Google Scholar]
- Vernetti LA, Senutovitch N, Boltz R, DeBiasio R, Shun TY, Gough A, Taylor DL, 2016. A human liver microphysiology platform for investigating physiology, drug safety, and disease models. Exp Biol Med (Maywood) 241, 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vulto P, Podszun S, Meyer P, Hermann C, Manz A, Urban GA, 2011. Phaseguides: a paradigm shift in microfluidic priming and emptying. Lab Chip 11, 1596–1602. [DOI] [PubMed] [Google Scholar]
- Ware BR, McVay M, Sunada WY, Khetani SR, 2017. Exploring Chronic Drug Effects on Microengineered Human Liver Cultures Using Global Gene Expression Profiling. Toxicol Sci 157, 387–398. [DOI] [PubMed] [Google Scholar]
- Waring JF, Liguori MJ, Luyendyk JP, Maddox JF, Ganey PE, Stachlewitz RF, North C, Blomme EA, Roth RA, 2006. Microarray analysis of lipopolysaccharide potentiation of trovafloxacin-induced liver injury in rats suggests a role for proinflammatory chemokines and neutrophils. J Pharmacol Exp Ther 316, 1080–1087. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Matsuzaki J, Katsuda T, Saito Y, Saito H, Ochiya T, 2019. Generation of functional human hepatocytes in vitro: current status and future prospects. Inflamm Regen 39, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama Y, Sasaki Y, Terasaki N, Kawataki T, Takekawa K, Iwase Y, Shimizu T, Sanoh S, Ohta S, 2018. Comparison of Drug Metabolism and Its Related Hepatotoxic Effects in HepaRG, Cryopreserved Human Hepatocytes, and HepG2 Cell Cultures. Biol Pharm Bull 41, 722–732. [DOI] [PubMed] [Google Scholar]
- Zar JH, 1984. Biostatistical analysis, 2nd ed. ed. Prentice-Hall, Englewood Cliffs, N.J. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Detailed experimental protocols, images and raw data for all experiments included in this study can be obtained using hyperlinks included in Supplemental Table 1.