

Abstract
Light passes through biological tissue, and so is used for imaging biological processes in situ. Such observation is part of the very essence of science, but mechanistic understanding requires intervention. For more than 50 years a “second function” for light has emerged, namely that of photochemical control. Caged compounds are biologically inert signaling molecules that are activated by light. These optical probes enable external instruction of biological processes by stimulation of an individual element in complex signaling cascades in its native environment. Cause and effect are linked directly in spatial, temporal, and frequency domains in a quantitative manner by their use. I provide a guide to the basic properties required to make effective caged compounds for the biological sciences.
Keywords: caged compounds, photochemistry, biological hypotheses, protecting groups, signal transduction
All scientists know the absorption of light is fundamental for life. Photosynthesis and visual transduction are the prime examples of this truism. Further, within science itself light microscopy has played an irreplaceable role in the profound discoveries of Galileo, Copernicus, Kepler, Hooke, van Leeuwenhoek, etc[1]. More recently, photochemistry has helped to change modern life with the invention of the photographic negative by Talbot in 1830s[2]. The recording of images independently of the human eye, using a photochemical reaction, was a Copernican revolution to match that of many other inventions of the Industrial Revolution. Talbot even went on to perform the first “flash photolysis” experiment in 1851[3]. Nowadays the recording of events using light microscopy remains the dominant optical technique in life and science (smart phones, confocal microscopes, Hubble, etc.). However, beginning the 1960s, scientists started to use light to control functionality at the single atom level. Thus, the potential for light to have a dual role in science, namely, to help to monitor and manipulate events, became apparent[4].
In this Perspective I provide a guide to an important area of photochemical manipulation, namely that of caged compounds[4–6] (Figure 1). This method uses photosensitive protecting groups as the light sensor to create optical probes that have been widely used in the biological sciences at all levels of complexity, from single proteins (Figure 2a) or protein complexes (Figure 2b), to cultured cells (Figures 2c,d), and even whole animals (Figure 2e). This Perspective is designed as a guide to the fundamentals of the field, which I hope will be helpful both to those curious about, new to, or even experienced within this large, multi-faceted discipline.
Figure 1.
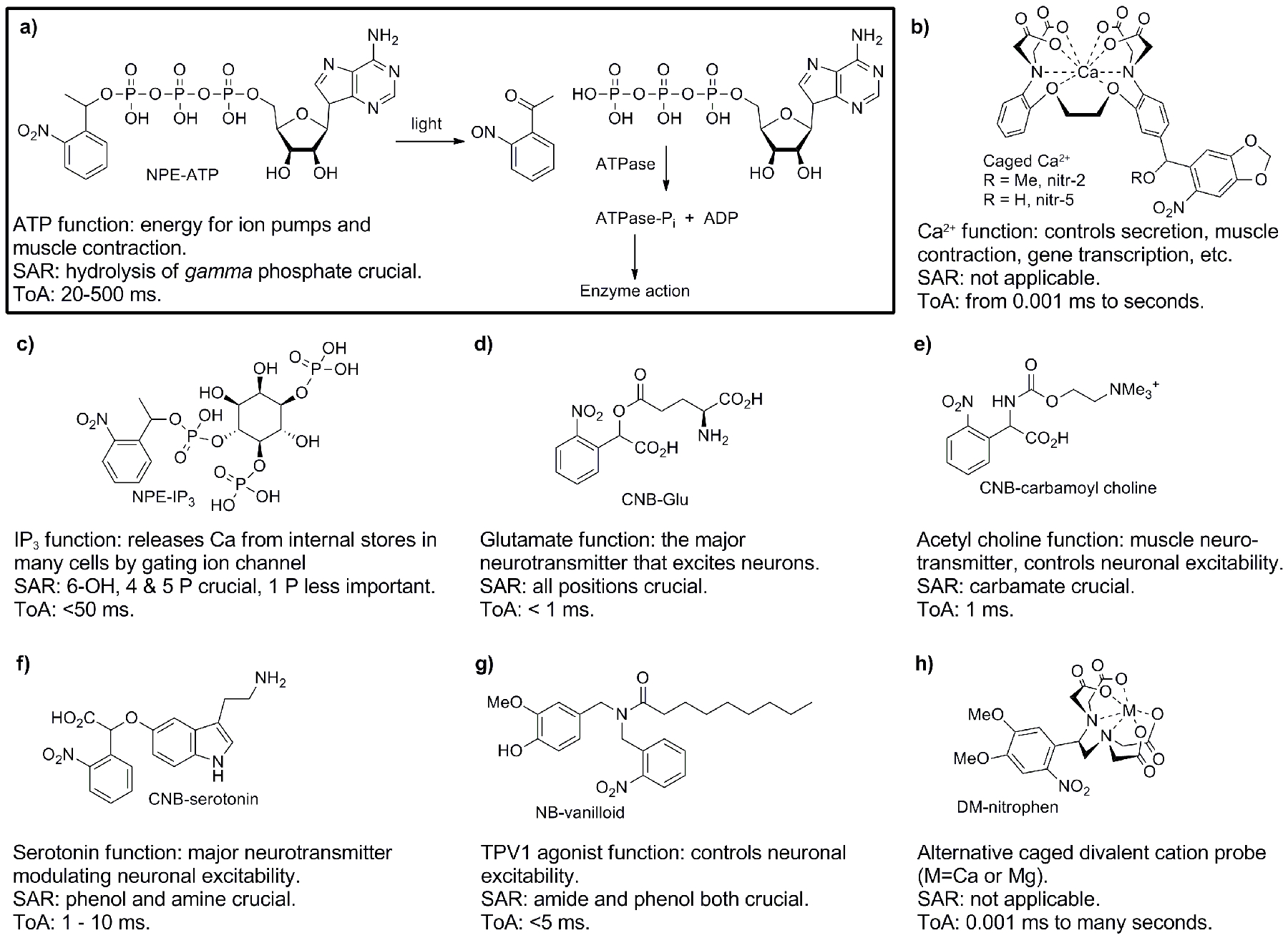
Representative caged compounds used in the biological sciences. From these examples it can be seen that many types of biological signaling molecules have been caged. Further, a wide range of C-X bonds can be cut photochemically. Simple summaries of the structure activity relationship (SAR), and the time of action (ToA) of the biological signaling system are given for each probe. a) Structure and photoreaction of NPE-ATP[11], here one sees most of the core ideas in the uncaging technique; b) Caged Ca2+ probes based on BAPTA[12, 13], cutting C-O bond decreases the electron density on the Ca2+-coordinating N atom, releasing some Ca2+; c) NPE-IP3, cutting the C-phosphate bond liberates the important second messenger[14]; d) CNB-Glu, cutting the C-acid bond releases the most important neurotransmitter, L-glutamate[32]; e) CNB-carbamoyl choline, cutting C-carbamate bond releases an acetyl choline receptor agonist[91]; f) CNB-serotonin, cutting C-phenol bond releases this neuromodulator[92]; g) NB-vanilloid, cutting C-amide releases a stimulant of nociception[93]; h) DM-nitrophen, alternative caged Ca2+, which also functions as caged Mg2+, cutting a C-N bond decreases Ca2+ affinity 600,000 fold[8].
Figure 2.
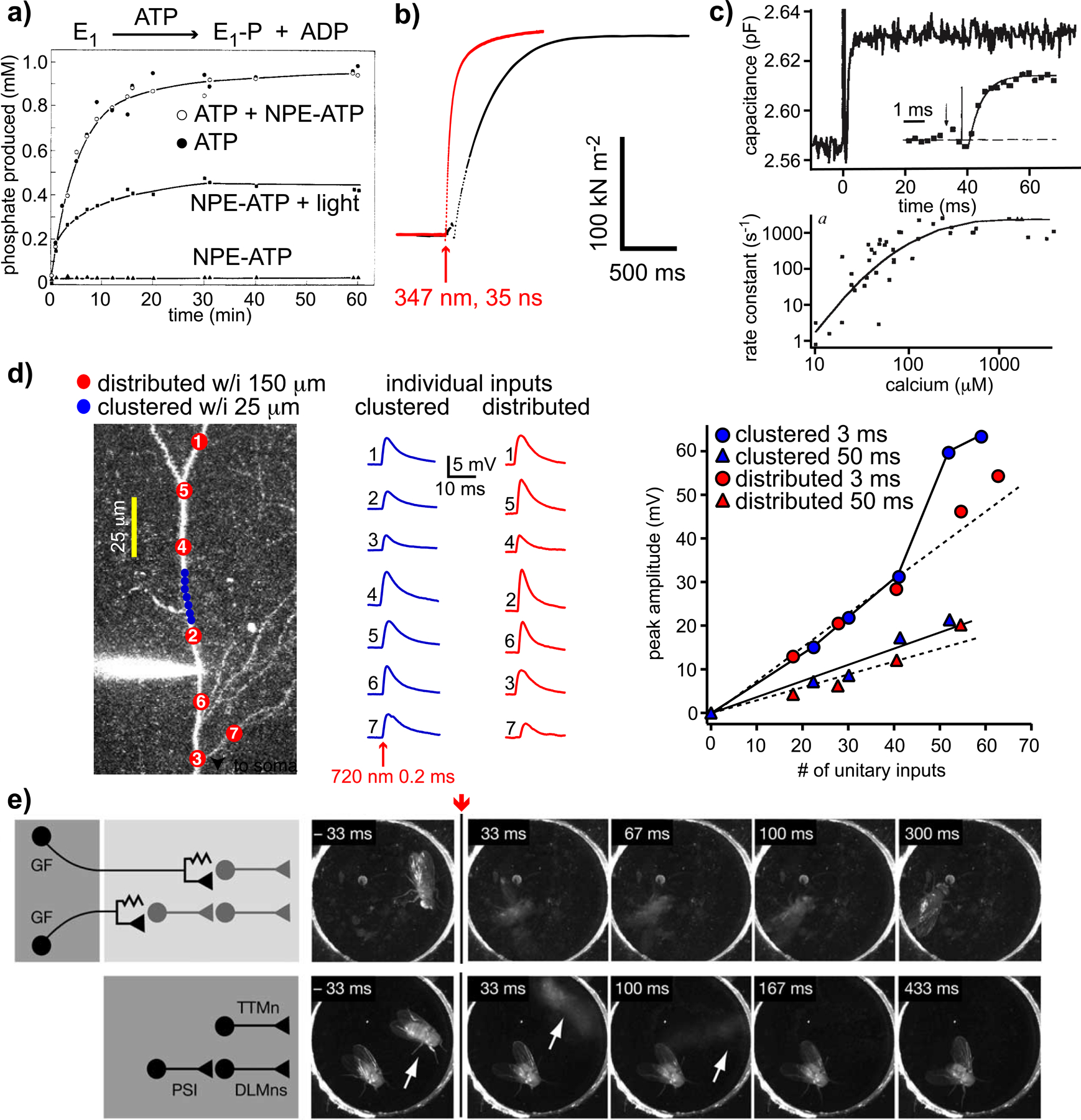
Examples of biology with caged compounds not possible with traditional methods. a) The original caged ATP experiments[11] showed NPE-ATP was not a substrate for the Na+,K+-ATPase (E1), and did not block ATP hydrolysis; b) Ca2+ uncaging from DM-nitrophen stimulates skeletal muscle contraction (red), and overcomes the diffusional delays associated with rapid mixing[8] (black); c) Intracellular uncaging of Ca2+ from DM-nitrophen generates large, step increases within 1 ms, stimulating rapid membrane fusion of glutamate-containing vesicles in retinal bipolar nerve terminals (upper). The quantitative relation between [Ca2+] and rate of membrane fusion is shown in the lower panel; d) Patterned 2-photon uncaging of MNI-glutamate reveals synchronous, multisite uncaging produces non-linear responses when clustered spatially and temporally[9] (blue circles); e) ATP uncaging using UV light in freely moving drosophila induces flight, even in headless animals (lower panel) when “alien” ATP channels are genetically targeted to the appropriate circuits[10]. This work was even the subject of a joke on the Tonight Show with Jay Leno on NBC in 2005[94]. I watched that show when it was broadcast. Part a) copyright 1978 ACS, used by permission; part c) copyright 1994 Nature, used by permission; part d) copyright 2006 Society for Neuroscience, used by permission; part e) copyright 2005 Elsevier Inc., used by permission.
Why use caged compounds?
The raison d’être for these probes is that they offer biologists photochemical tools that enable them to carry out experiments which cannot be done any other way[4] (Figure 2). Thus, it should not surprise chemists that it is the biological question that has prime place in the design process.
The first step in development of any new caged compound must be “Is this photochemical technology necessary for the biological experiment?” Can simpler methods to produce concentration changes of biological substrates or drugs, such as dialysis or iontophoresis[7], be used? Next we should ask: “What biological hypothesis will you test with your new probe?” In the USA, science supported by the NIH is famously “hypothesis driven”. Thus, it is crucial to look not merely at how to make a proof of principle biological test, but envisage what new biological experiments will be carried out with the caged compound. The next step in the process is to ask, “Is there an adequate caged compound already available for the biology?” Analogously, no major pharmaceutical company invests in the development of a new drug, if they cannot improve on what their competitors offer. Similarly, one must justify why one would make a new photosensitive protecting group, if current chromophores work well already. Having given these notes of caution, chemists can make invaluable photochemical probes that enable their biology colleagues to carry out beautiful experiments, many of which would be impossible without caged compounds. A few representative examples of these are illustrated in Figure 2. Here we can see uncaging can be: faster than even the most rapid solution change[8] (Figure 2b); intracellular (Figure 2c); optically patterned with submicron precision in complex biological tissue[9] (Figure 2d); and carried out in freely moving animals, for remote control of genetically-targeted neuronal circuits[10] (Figure 2e). At this point we can start to detect a real challenge for chemists working on caged compounds, namely that physiology is complex, with the many particulars being not part of the training of a “typical organic chemist”. Nevertheless, it is these very details which are crucial for the understanding of the development of caged compounds.
Reverse engineering caged compounds.
In section 43 of his Philosophical Investigations Ludwig Wittgenstein stated: “For a large class of cases—though not for all—in which we employ the word “meaning” it can be defined thus: the meaning of a word is its use in the language.” This is often paraphrased as the aphorism “Meaning is Use”. Following this idea, the many hundreds of publications using caged ATP, caged neurotransmitters and caged second messengers (some examples are shown in Figure 1) suggest that these probes have properties from which we can reverse engineer a set of guiding principles for all caged compounds.
1. Caged compounds must be biologically inert before photolysis.
Basic idea: caged compounds must be neither agonists nor antagonists.
This is by far the most important property of any caged compound. Why? If you want to target and turn on a specific biological process by uncaging, the biological activity encapsulated within the caged compound must be completely latent. All chemists appreciate the idea that pharmaceuticals are built on target binding with high affinity and specificity, caged compounds can be thought of being the “exact opposite” of this, before photolysis. A concrete example helps the non-specialist understand this basic idea, see ortho-nitrophenethyl(NPE)-ATP[11] in Figure 1a. This first caged compound was not a substrate the Na+,K+-ATPase, as the covalent bond prevented ATP hydrolysis (Figure 2a, triangles). Furthermore, NPE-ATP did not antagonize ATP hydrolysis by the enzyme (Figure 2a, circles). Note that Kaplan et at., did report some toxicity of the side product on isolated enzyme, but they solved this issue[11]. They detected no toxicity in cells; a result supported by many thousand of subsequent physiological studies with similar probes. Often, when properties of caged compounds are presented, biological inertness is not mentioned, but for me it is the most important property. Figure 1 shows a range of caged compounds, all of which are biologically inert. The caged Ca2+ probes developed by Roger Tsien[12, 13] (nitr-2 and nitr-5, Figure 1b) chelate free Ca2+ inside cells to non-activating levels. Irradiation decreases their buffering capacity, releasing some bound Ca2+. Inositoltrisphosphate (IP3) binds to a ligand-gated, Ca2+ release channel on the endoplasmic reticulum, thus activating a wide variety of Ca2+-driven processes. NPE-IP3 (Figure 1c) only induced smooth muscle contraction after intracellular uncaging[14]. Neurotransmitters activate ligand-gated ion channels which conduct Na+ and/or Ca2+ to excite neurons, or Cl- to hyperpolarize membrane potential[15]. These are the ionotropic class of receptors. Further, a different set of cell surface receptors, called metabotropic, stimulate G-protein coupled signal cascades[15]. These are activated by the same set of neurotransmitters, leading modulation of the former. Glutamate and acetylcholine (Figures 1d,e) are used on both pathways, whereas serotonin (Figure 1f) is mostly modulatory. Attachment of a caging chromophore to any heteroatom of these neurotransmitters produces an inert probe. The photolabile EDTA (ethylenediaminetetraacetic acid) probe, DM-nitrophen[8] (Figure 1h), chelates Ca2+ and/or Mg2+, so can be used as either a caged Ca2+, or a caged Mg2+, depending on the conditions. Essentially every other important biological signaling molecule has been caged using the ATP strategy[16, 17] (Figure1a).
In terms of physiological inertness, it turned out that two important classes of caged compounds, namely caged ATP and γ-aminobutyric acid (GABA), do antagonize receptors[18, 19]. In the initial report on α-carboxy-2-nitrobenzyl(CNB)-GABA in 1994, Hess stated this probe had zero antagonism at 0.5 mM towards GABA receptors[20]. However, in 2000 the IC50 of CNB-GABA was shown to be about 0.03 mM[19]. Further, in 2001, Ogden reported that 4-methoxy-7-nitroindolinyl(MNI)-Glu was not an antagonist of GABA receptors[21]. Thus, we were very surprised when we discovered MNI-Glu was indeed a strong antagonist of the GABA receptor[22]. It turns out all the widely used caged GABA and Glu probes are antagonists of the GABA receptor at working concentration levels (Figure 3a). While such antagonism is never so strong as to prevent receptor activation, it must limit to some degree the physiologists’ ability to mimic perfectly naturalistic synaptic signaling. Recently, my group has solved the antagonism problem with the “cloaking” method[23, 24] (Figure 3).
Figure 3.
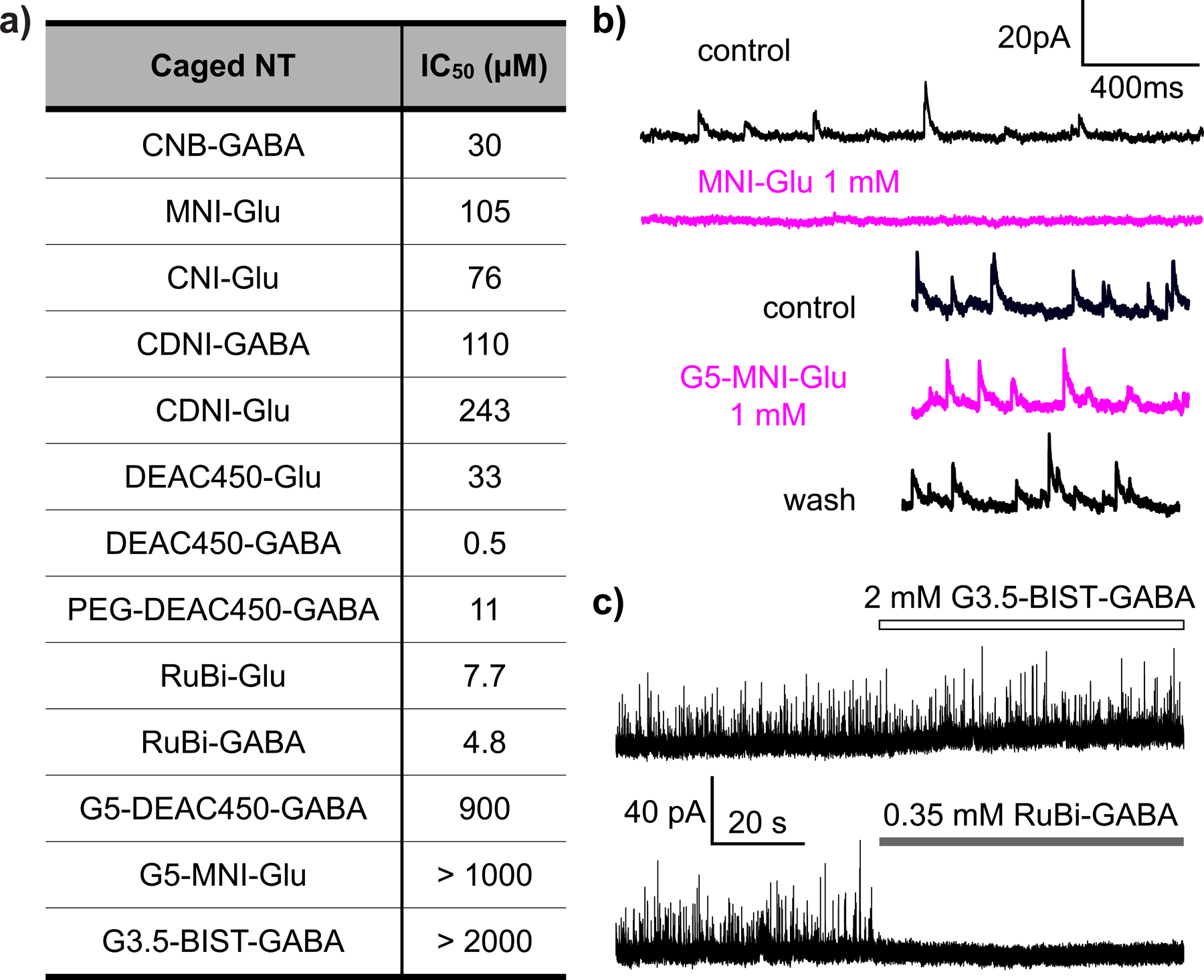
Most caged Glu and GABA probes are antagonistic to GABA receptors. Caged Glu probes do not block their target Glu receptors, but almost all these probes have off target antagonism towards GABA receptors. The Table in a) summarizes the IC50 values for several such probes, as well as caged GABA probes; b) Bath application of MNI-Glu to a neuron shows graphically the antagonism problem by complete blockade of recordings of miniature inhibitory post-synaptic currents (mIPSCs). In contrast, “cloaked” caged Glu (G5-MNI-Glu[95]) had no antagonism at the same concentration; c) “on target” antagonism can been for RuBi-caged GABA, which was not detected with a “cloaked” caged GABA[24]. Part c) copyright 2019 Wiley, used by permission.
After the initial success of NPE-ATP in time-resolved muscle studies[25, 26], it was discovered that many caged nucleotides bind to their receptors[18, 27]. One of the most elegant publications using a caged compound actually took advantage of this disadvantage, by crystallizing NPE-GTP bound to ras p21. Flash-photolysis and time-resolved X-ray crystallography allowed Goody and co-workers to follow the atomic movement of the protein during GTP hydrolysis[28]. To my knowledge no fully inert caged ATP probes have been developed. Importantly, kinetic modeling can be used to deconvolve this problem for many ATPases[25, 26].
Summary: if a good structure activity relationship (SAR) is known for the target molecule to be caged, then production of a biologically inert probe should be straightforward.
Breaking the rule: some time-resolved X-ray studies use receptor antagonism[28, 29].
2. Rates of uncaging must be fast.
Basic idea: uncaging must be at least 10x faster than the biology.
Clearly, if we wish to use photochemistry to “switch on” a biological signal cascade, the speed of the photochemical event must be significantly faster than the process under study. However, for most important biological signaling molecules (glutamate, GABA, IP3, etc), an appropriate optical indicator does not exist. Thus, mechanistic photochemical studies can play a useful role in probe development. Trentham and co-workers set the standard for the field with a seminal paper on caged ATP, published in 1980[30]. It was known already that the primary photochemical process of ortho-nitrobenzyl photochemistry was benzylic proton abstraction, as irradiation of toluene produces a transient, aci-nitro intermediate, which decayed back to toluene[31]. Trentham used a fluorescent sensor to detect that ATP appeared with a rate similar to the aci-nitro decay. In 1994, following the work of Trentham, Hess showed[32] that the aci-nitro intermediate (Figure 4a) for CNB-Glu decayed with time constant of 23 μs (Figure 4b), much faster than the time to activate ionotropic Glu receptors (Figure 4c). However, the kinetic data shown in Figure 4b,c do not prove aci-nitro decay is the rate limiting step for uncaging, such evidence was provided by subsequent time-resolved-IR study[33]. Note Hess also showed CNB-Glu was not an antagonist or agonist, and was stable at physiological pH. This seminal 1994 paper remains a model for the field[32].
Figure 4.
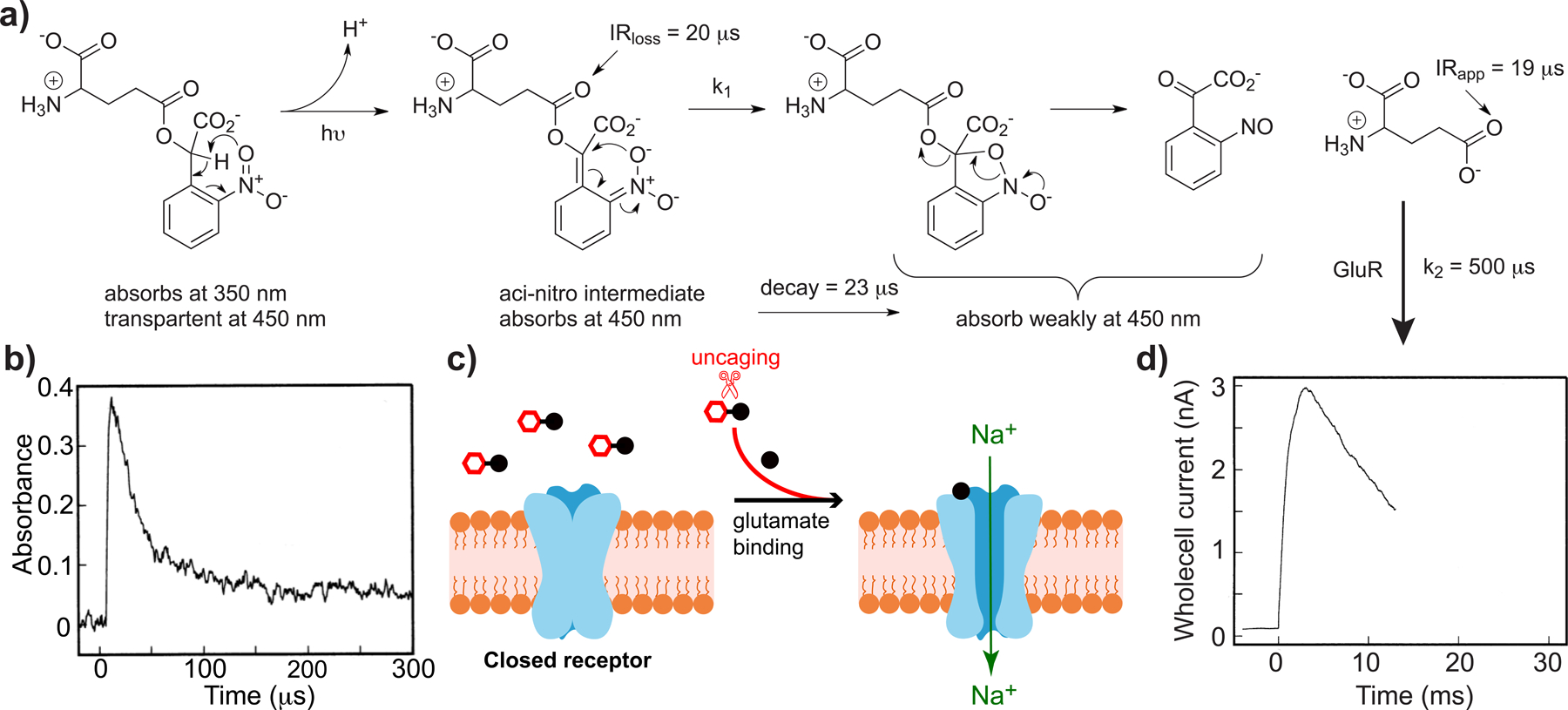
Uncaging of CNB-Glu is faster than naturalistic receptor activation[32].
a) Photochemical reaction mechanism of CNB-Glu uncaging, the primary photochemical step is benzylic H abstraction, producing the aci-nitro intermediate, which decays by C-O bond scission. This rate (k1) that matches C=O bond stretch loss in the IR, and C=O bond appearance of glutamate, implying k1 < k2; b) The aci-nitro intermediate absorption decay monitored at 450 nm; c) Cartoon of ion channel opening; d) Glutamate receptors currents show rapid rise times after CNB-Glu laser photolysis, these mimic physiological rates, proving CNB-Glu can be used for fast physiology, as k1 > k3. Parts b) and d) copyright 1994 National Academy of Science, used by permission.
An important inference can be made from the data shown in Figure 4, namely we can, under certain circumstances, use biology with known rates (such as glutamate-induced currents) as surrogate for an independent optical measure of release rates for compounds that have no optical indicator (e.g. glutamate, Figure 5a). Thus, since the rate of current rise after 2-photon photolysis of MNI-Glu with a 50 μs laser flash was found to match that recorded for synaptic secretion (both were 0.5 ms, Figure 5b,c), we concluded that nitro-indolinyl uncaging was not too slow for synaptic studies using this probe[34]. Note also that our data were the first examples to show that 2-photon uncaging can be used to mimic natural physiological inputs. Taken together, the chemistry and biology of CNB-Glu and MNI-Glu still set a standard for the development of caged compounds, for both linear and non-linear excitation, even though the studies are more than 20 years old.
Figure 5.
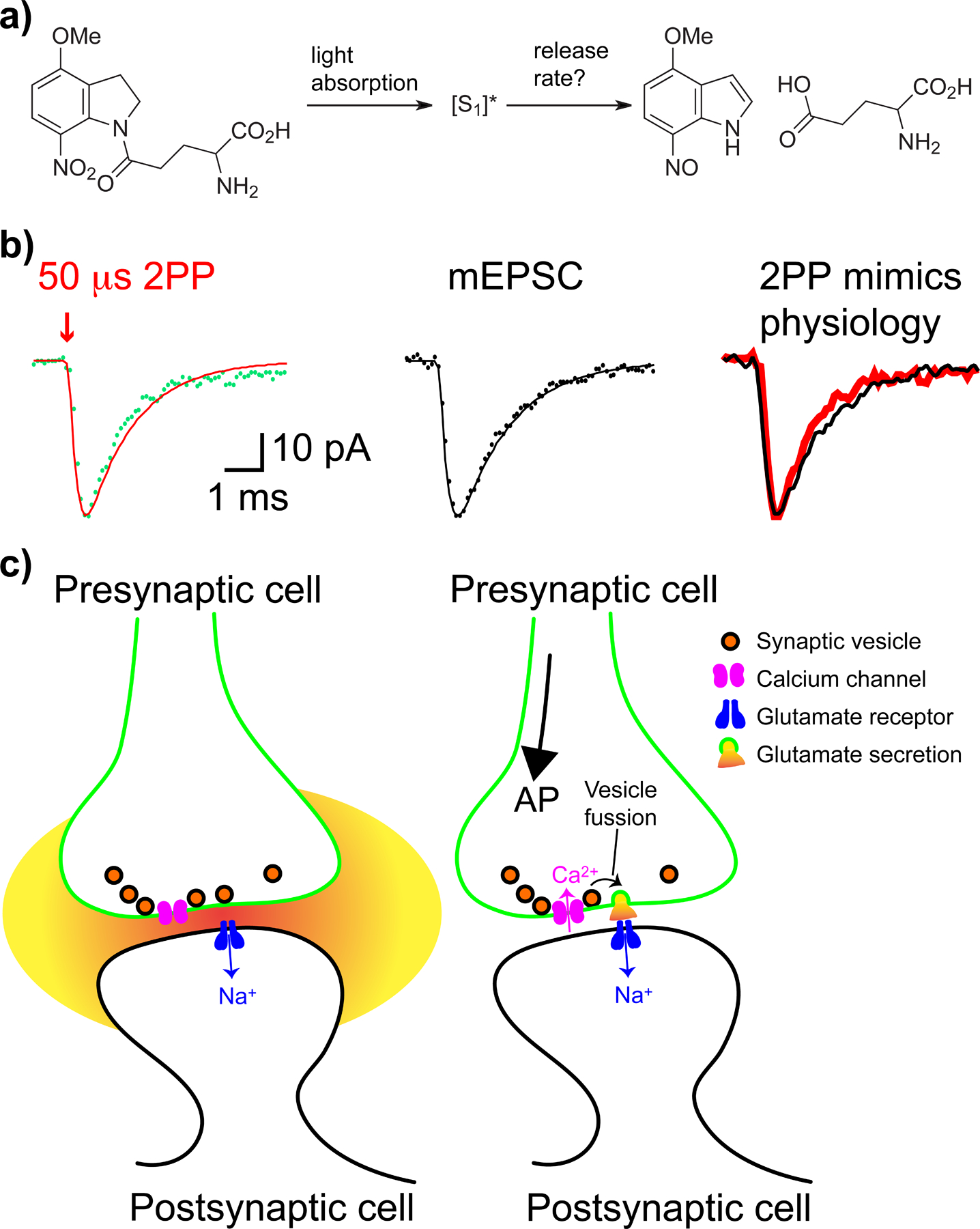
Fast biology can be used to test the rate of uncaging.
a) Glutamate release from MNI-Glu, unlike CNB-Glu, using time-resolved absorption analysis is not well characterized; b) However, 2-photon photolysis (2PP) of MNI-Glu with short pulses (50 μs) of 720 nm light produces rapid responses at spine heads (red), which mimic naturalistic responses (mEPSC, black), the time to peak was 0.5 ms[34]. Such data demonstrate fast biology can be used to estimate release rates; c) Cartoons of focal 2-photon uncaging at glutamate receptors on a spine head (left), and Ca2+-driven single vesicle fusion, causing the mEPSC currents shown above (right). mEPSC - miniature post-synaptic excitatory current. Part b) adapted from ref. 34, copyright 2001 GCRE-D.
So does time-resolved physiology require caged compounds that are much faster than CNB-Glu and MNI-Glu? Wittingstein’s postulate would suggest that if this were the case, we would see older probes being replaced in biological experiments by newer ones, if the old were not adequate. For genetically-encoded Ca2+ imaging this has happened. Sensors such as GCaMP2 and GCaMP3 are no longer being used, as they are too insensitive and too slow for faithful detection of [Ca2+] during single action potentials[35]. In other words, knowledge of the biology is crucial for chemists to help focus scientific efforts in a fruitful way. All the ortho-nitrobenzyl caged compounds shown in Figure 1 can be used to interrogate their target receptors, showing that cutting most C-X bonds using this photochemistry has adequate kinetics for the biological sciences. Other photosensitive protecting groups can be much faster (e.g. p-hydroxyphenacyl(pHP)-caged acids[27], ruthenium-bipyridyl(RuBi)-caged amines[36] and coumarin-caged acids[37]), but this extra speed confers no advantage in most biological experiments[27, 38–40].
This discussion of release rates has presupposed full and open access of the probe to the receptor. Put simply, probes such as CNB-Glu can be applied to the outside of a cell, and reach equilibrium, before irradiation, in a very facile manner[32]. Such open access also allows rapid solution changes to be accomplished very effectively, so for processes taking many second, minutes, let alone hours[41], caged compounds do not confer a clear advantage. This is not the case for receptors within the cell interior, as the cytosol is closed off by the plasma membrane. The fact that light passes though cells has not been discussed explicitly here so far, but this property has enabled many hundreds of physiological studies with caged compounds loaded into cells[42] (e.g. Figure 2c[43]). The importance of both inertness and fast release is utilized in these studies. Cell loading takes several minutes, so quiescence is crucial to permit full equilibration of the caged compound. Fast uncaging enables synchronization of the photo-stimulated cell receptors by rapid release[43] (see Figure 2c). Also, some biological preparations are either too sensitive to mechanical perturbation or diffusional delays are too long for rapid activation by traditional means[8, 32] (see Figs. 2b, 4c). Here again, in such circumstances, uncaging can help biological studies.
Summary: an adequate (AKA “fast”) rate of uncaging is as important as the previous property (inertness). We know enough about the photochemical kinetics of the major photosensitive protecting groups such that the rate of uncaging should never be a hindrance to biology.
Breaking the rule: red, single-photon uncaging in vivo (see section 9 below) does not need to be fast.
3. Aqueous solubility.
Basic idea: caged compounds must be soluble in aqueous buffer, with no organic co-solvent.
Life takes place in water, not water with 1% DMSO. So caged compounds must be fully soluble in (100%) physiology buffer. This issue is not new(s) for the pharmaceutical industry. For example: “Solubility, the phenomenon of dissolution of solute in solvent to give a homogenous system, is one of the important parameters to achieve desired concentration of drug in systemic circulation for desired (anticipated) pharmacological response. Low aqueous solubility is the major problem encountered with formulation development of new chemical entities (NCEs) as well as for the generic development. More than 40% NCEs developed in pharmaceutical industry are practically insoluble in water. Solubility is a major challenge for formulation science.[44]” This challenge is equally true for caged compounds, yet there is almost no discussion of this property in other reviews. The ideal for the biologist is to make a 100x solution of a probe in water, freeze multiple aliquots indefinitely, and use them as required, without any concern. Of course the required level of solubility will vary depending on receptor affinity for the effector. ATP and glutamate have a very low affinity for receptors (ca. 0.1 – 1.0 mM), whereas other ligands tend to have higher affinities. But, the photolytic conversion in time-resolved biological experiments is seldom > 30–50%, and often only 5–10%. Consequently, one must plan to apply any caged compound at least a 10-fold higher concentration than the receptor affinity. This is significant constraint, if the biomolecule solubility is dramatically reduced by addition of the photosensitive protecting group. Further, in some experiment one might seek to perform repetitive stimulations meaning one would not want to deplete fully the probe concentration in one or two light flashes.
Summary: aqueous solubility of caged compounds is crucial.
Breaking the rule: how soluble do caged lipids[45, 46] need to be in buffer?
4. Aqueous stability.
Basic idea: water at physiological pH must not liberate the caged species.
This property is related to the previous, and chemists should keep in mind the biological ideal described there. For certain types of bonds we know as chemists that they are hydrolytically stable at pH 7.2–7.4. For example, for nitrobenzyl-caged C-N bonds with amines and C-O bonds with ethers and phenols, such C-X cannot be hydrolyzed. For nitrobenzyl-caged amides and carbamates this also is true at neutral pH. Synthetic organic chemists will know that the stability of benzylic esters is more complex. Fortunately, simple NPE-caged phosphate esters (Figure 1a,c) are very stable a pH 7.2. In comparison, cyclic phosphate esters are less stable, especially in the case of the electron rich nitroveratyl (NV) versions. However, more electron deficient versions of these probes show excellent stability[47]. Carboxylate esters are less stable than phosphate esters, but, again, the electron deficient CNB-Glu was reported to have excellent stability[32]. The fact that this probe can be used for long-term mapping experiments proves it has adequate physiological stability. In the case of MNI-caged acids, we reported that MNI-D-Asp was completely stable at pH 7.4 for 3 days at 4oC[48]. I have found that solutions of MNI-Glu and MNI-Glu.TFA in water are stable for many years when frozen. In contrast, the more electron deficient 4-methoxy-5,7-dinitroindolinyl(MDNI)-Glu.TFA is quite unstable at neutral pH. The RuBi chromophore offers a useful alternative to caging transmitters, in that it forms bonds with primary amines. These Ru-N bonds are extremely stable at physiological pH. In terms of enzymatic stability, caged second messengers such as, for example, IP3 can be loaded into intact cells without any significant degradation before photolysis. This can be inferred from the fact there is no change in resting intracellular Ca2+ concentrations before irradiation[49–51].
Summary: most nitrobenzyl-caged compounds are either inherently stable, or show usable aqueous stability at physiological pH to present little practical concern for biologists. Further, most chemists who develop new caged compounds are diligent, and present careful characterization of probe stability in the primary literature.
Breaking the rule: do not.
5. High quantum yield.
Basic idea: more bang for your buck is good.
The quantum yield (QY) of a photochemical probe is the % of excited-state molecules that give the desired product. Following the idea “meaning is use”, which caged compound in wide use in biology has the lowest QY? This value could provide a starting point for reverse engineering QYs. I believe probes such as MNI-Glu might qualify in this regard. The QY of photolysis is now thought to be 0.065[52], suggesting that for fast acting, low affinity biological signaling molecules, an uncaging QY of much less than this might be problematic for biological studies. However, looking at the QY values for the most widely used caged compounds in physiological studies, we discover all are high performing. For example, NPE-ATP has a QY = 0.5[11]; NPE-IP3, 0.5[49]; CNB-Glu[32], 0.15; NP-EGTA, 0.23[53]; DM-nitrophen, 0.18[8]; NV-IP3, 0.09; RuBi-GABA, 0.20[54]; etc. So reverse engineering the QY property suggests probes with low efficient of release (QY < 0.02) will not be useful in practice.
Summary: no doubt a high quantum yield makes the biology easier.
Breaking the rule: applications of caged probes to very slow processes, such as in vivo drug release with red light, do not need a high QY.
6. Non-toxic photon dosage.
Basic idea: too much light kills cells.
Photolysis on live cells can be affected in several different ways, with quite different photon flux densities. The simplest uses full field illumination through the microscope objective with incoherent light from a flash lamp (Figure 2c) or LED, which produces uniform excitation of a sample, with the size depending on the magnification of the microscope objective[55]. The second method uses laser beams pointed to specific region in the field of view with galvanometer scanning systems with a diffraction limited spot[55]. Holographic beam shaping offers the ability to control coherent light in 3D shapes which can match cell segments and so offer an unique alternative to other methods[56]. To my knowledge there has been no systematic comparison of these methods to define the threshold for phototoxic damage of cells. Furthermore, my experience tells me that such thresholds depend greatly on the cell type and preparation. So I am sorry to say generalizations are really difficult to provide, and, realistically, must be determined for each biological field, excitation method and probe. Of course, irradiating cells with too much light can cause heating of samples, and care must be taken to monitor such side effects.
Advances in technology only become clear when a side-by-side comparison of old and new approaches is made. An example of this can be seen in our development of new caged compounds for 2-photon uncaging[57]. We showed that using MNI-Glu and 4-carboxymethoxy-5,7-dinitroindolinyl(CDNI)-Glu, synaptic rundown could be detected for the former using much higher powers, whereas no rundown was detected with the latter at lower powers[57]. This improvement was because CDNI has a quantum yield 5x higher than MNI. When such direct comparisons are not made, it will unclear exactly what advantage any proposed new technology will deliver for the biologist. Note that photon dosage can be adjusted (lower power and longer irradiation) with MNI-Glu to produce no run down in response (Figure 6b). Indeed, single spine, high frequency 2-photon uncaging of MNI-Glu has been used to test many important hypotheses by several groups[58], proving this approach is not phototoxic.
Figure 6.
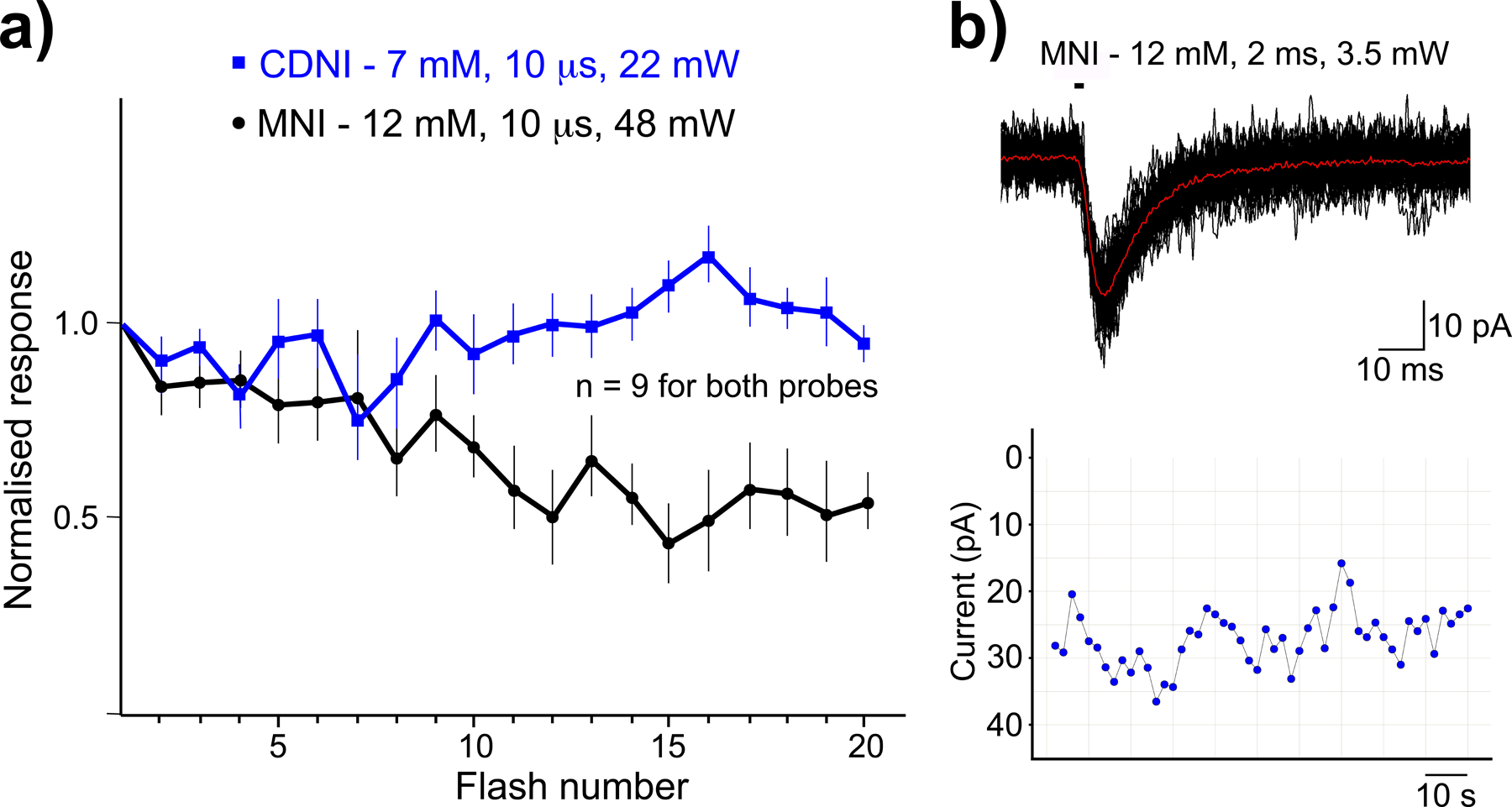
Phototoxicity thresholds for 2-photon uncaging on neurons.
Light of any wavelength can be phototoxic to live cells. We can establish appropriate levels for uncaging power using the highly sensitive electrophysiology technique. 720 nm-light from a mode-locked, Ti:sapphire laser focused at single spine heads on neurons in brain slices were used for these tests. a) Comparative photolysis MNI-Glu and CDNI-Glu[57]. Power levels were set so responses from both probes were the same. MNI required higher energy and run down became apparent (black). Significantly lower energies for CNDI resulted in no run down (blue); b) Uncaging MNI-Glu at a single spine 60 times with lower powers but longer periods showed no run down, proving such powers are not phototoxic. Part a) adapted from ref. 57, copyright 2007 GCRE-D.
Light in the UV-A range (i.e. 340–390 nm) is often said to be highly phototoxic by chemists who develop probes that respond to visible light. However, it is apparent from the 100s of studies using UV lasers and flash lamps[42, 59], such problems are not normally observed by biologists who use such light sources (e.g. Figure 2c). During UV uncaging experiments we could easily challenge the same neuron over 100 times with sufficient UV light to cause action potentials (10 ms, 5 mW) with caged Glu, but we could never kill cells with such power trains[60]. Since UV light can be used for uncaging in drosophila (Figure 2f[10, 61]), it crucial for probe developers to establish which experiments really cannot be done with such probes by comparison with newer, visible light sensitive probes.
Summary: power levels required for biological experiments with caged compounds are normally significantly lower than the phototoxic threshold. This is true for UV and 2P-sensitive probes.
Breaking the rule: do not.
7. Two-photon cross-section.
Basic idea: stronger light absorption makes biology easier.
Linear excitation by UV or visible light induces uncaging wherever the probe absorbs light. Two-photon excitation produces the S1 state by the simultaneous absorption of two photons[62]. The simplest aromatic chromophores (e.g. ortho-nitrobenzyl) have small 2-photon cross-sections (2PCSs), however two studies published in 1998 revealed how chemists can improve the 2PCS of chromophores significantly[63, 64]. This work catalyzed a large effort by many groups to develop fluorophores and other organic compounds with improved non-linear absorption properties[65, 66]. Translating the ideas from 1998 into photosensitive protecting groups with improved 2PCSs has been a challenge (but see[24, 67]). Fortunately a modest increase in the electron density of nitroaromatic chromophores yields reasonably effective photosensitive protecting groups for some 2P applications[68] (Figure 5).
In practice, 2-photon uncaging requires a very high photon flux density, produced from specialized Ti:sapphire lasers, which produce axially confined excitation of 2–3 microns. Since glutamate acts in nanodomains naturalistically, this can be mimicked with sub-micron precision using non-linear excitation.[34] Many studies of glutamate function which are only possible using 2-photon uncaging, especially using multisite photolysis (Figure 2d), have enabled biologists to test many important hypotheses with respect to glutamate function[69]. Other biomolecules do not work in this way. For example dopamine and serotonin work mainly by what is called “volume transmission”, meaning secretion causes a diffuse release, in a “cloud like” manner. Thus, developing probes for 2P release of these and similar biomolecules has little sense for biology, i.e. Meaning is Use.
Furthermore, the importance of fast photochemical release can be seen when we compare the MNI-Glu current data with that from 2-photon photolysis of bromohydroxycoumarin(Bhc)-Glu. The latter has a 2PCS about 16x larger than the former (0.95 vs 0.06 GM, and 15x larger QY), but glutamate release takes about 10 ms from Bhc-Glu, so the evoked postsynaptic currents showed a rise-time of 100 ms, and required very high powers[70]. These data illustrate that fast uncaging is crucial for 2-photon photolysis because of the size of a diffraction-limited laser beam, molecules photolyzed diffuse out of the focal volume with a half-time of about 0.3 ms[71]. Thus, to take full advantage of the 3D resolution of the method, uncaging rates of 20–50,000 s−1 are ideal.
Summary: 2-photon uncaging is now a mature technique in the physiological sciences. Improvements in the properties of photosensitive protecting groups should deliver interesting biology. Many apparent photochemical improvements (e.g. ref[72]) have not been reflected in terms of enabling quantitatively new biology[73].
Breaking the rule: probes with what appear[74] to be an inadequate 2-photon cross-sections (ca. 0.06 GM) have proved surprisingly useful for biology studies[34, 69].
8. Compatibility with other chromophores.
Basic idea: clean separation of different optical channels facilitates multi-color photochemical biology.
In many biological experiments, especially those at the cellular level, fluorescence microscopy is used for visualization of a cell to allow targeted uncaging (e.g. Figure 2d[9]). Ideally, the chromophores for uncaging and imaging work in separate optical channels, so the wavelengths used for manipulation and monitoring do not produce optical cross-talk. Biologists typically use green and red fluorophores for visualization[9], this leaves the UV-violet channel for “clean” uncaging. Thus, if chemists want biologists to use blue or longer wavelengths of light for uncaging, they must realize this starts to limit monitoring to the red channel. Many of the photosensitive protecting groups developed by chemists for uncaging with long wavelength are based on fluorophores (e.g. coumarins, BODIPYs, etc.). These probes introduce an additional complication for biologists, one which is not often discussed by chemists, namely that emission from the “caging chromophore” can interfere significantly with emission from green and red dyes. Notable exceptions in this regard are the RuBi-caged compounds[36]. These react with blue light very well, but unlike coumarins and BODIPYs, are essentially non-fluorescent in the green channel, allowing their use with standard green and red dyes. It should be noted that there are examples of using protecting group fluorescence to advantage in terms of measuring intracellular uncaging[75], or tracking caged lipids for membrane or organelle localization[45, 46]. The latter is an elegant twist of an apparent disadvantage into a strong positive advantage for this type of probe.
Most nitro-aromatic chromophores that are used as photosensitive protecting groups are also non-fluorescent, so have been widely used for uncaging experiments by physiologists[42, 69]. Thus, probes like MNI-Glu have been used many times with green and red fluorophores with dual laser, 2-photon microscopes with uncaging at 720 nm, and two-color imaging at 820 nm[69]. RuBi-GABA fits nicely into such experiments as a source of GABA by uncaging with blue light, enabling two-color uncaging (720 nm and 470 nm) and two-color, 2-photon imaging[76, 77] at 820 nm. It is useful to note that most fluorescent and incandescent lights used in rooms do not produce much violet or near-UV light, making the handling of probes like NPE-ATP or MNI-Glu really quite facile in a standard room environment. In contrast, caged compounds using chromophores that absorb blue to red wavelengths have to be handled with caution.
The pioneering biological experiments with caged compounds used near-UV light for which microscopes were not required. However, the vast majority of physiological studies with caged compounds do utilize such optical devices. Microscopes glass is incompatible with wavelengths of < 330 nm, precluding the use of UV lasers. Furthermore, with the development of 2-photon microscopy the optical path has been tuned to favor the 400–1300 nm range. Thus, modern microscope objectives are not parfocal below this range. Also, standard tube and scan lenses transmit the near-UV very inefficiently, making the use of near-UV lasers problematic. Thus, uncaging with near-UV normally involves full-field illumination of samples, an optical path that by-passes such complexities.
Summary: photosensitive protecting groups that are only photolyzed with near-UV or violet light offer the widest compatibility with fluorophores normally used by biologists for imaging live cells.
Breaking the rule: spectral overlap can be circumvented if imaging is much more sensitive than uncaging (e.g. Figure 2c[43]).
Intermediate summary: reverse engineering the most widely used caged compounds in cell physiology[8, 11, 14, 32, 34, 50, 53] reveals such probes are biologically inert, uncage in the 0.02 to 20 ms time domain with a good quantum yield (>0.06). Further, high solubility in physiological buffer (>100 mM with no organic co-solvent) and stability against spontaneous hydrolysis are also standard features. Surprisingly[78] modest 2-photon absorption cross sections[34] of electron-rich nitroaromatic chromophores enable high-resolution photophysiology, in conjunction with widely used blue, green and red fluorophores[76].
9. Red light uncaging.
The importance of methods such as “photodynamic therapy” (PDT) for treatment of some human diseases[79] may have induced chemists to become interested in using light for what is being called “photopharmacology”[80]. Since light in the 650–900 nm range penetrates biological tissue best, red light uncaging of drugs is a topic of current research by several groups. Google scholar reveals only about 150 hits for “photopharmacology” between 1960–2010, with relatively few citations for these studies. (Note there is a similar situation for the term “caged compounds” in the 1960–1980 period.) But there are about 1,000 hits for “photopharamacology” in the 2011–2020 period (many highly cited). While hits in google scholar might be similar to “likes” on social media, it gives us a feeling for the rise of the term “photopharmacology” in science. To date, I am not aware of any clinical trial using drug uncaging (or photoswitching), so the jury is still out on this therapeutic use of photochemistry.
Schnermann and coworkers developed the first system for uncaging with red light that worked for targeted drug delivery in vivo. Proof of principle appeared in 2014, with tests in cultured cells[81]. Importantly, these experiments were followed up in 2015 with studies in vivo using a rodent cancer model[82]. These papers set the standard for work in this field. Using red light for uncaging in vivo is far more challenging in terms of probe properties than the “photophysiology” discussed here so far. Some properties are common to both areas, namely aqueous stability, solubility, non-toxic photon dosage, and biological inertness before photolysis. But drug release in vivo does not need to be in the millisecond time domain. Further, a high quantum yield is not required, and it may even be a disadvantage. The additional challenges for “photopharmacology” with red light start with light delivery to freely moving animals. Additionally, the pharmacokinetic (PE) profile of any drug (i.e. its absorption, distribution, metabolism, and excretion in mammals) is a fundamental and complex property which does not apply to probes like caged glutamate. Unfortunately, the PE profile is normally not examined in most “photopharmacology” studies[83], even though it is common to show cartoons of human bodies in these reports[84].
Summary: uncaging drugs in mammals with red light is even more challenging than uncaging physiological signaling molecules with UV light. Proof of principle in rodent models of human diseases show promise for this approach.
Breaking the rule: when clinical trials show success, the rules will become clear[83].
The following sections are not actual “properties” of caged compounds. But they are topics which I think are important when considering which photosensitive protecting group to use when making a new caged compound. Or how to think about the development of new protecting groups for this field.
10. Functional group generality.
In 1966 Barltrop tested the ortho-nitrobenzyl photosensitive protecting group with carboxylic acid and acid-like functionalities[85]. Subsequently, many studies extended this work, going on to show all hetero-atom functionality can be deprotected[86]. Figure 1 illustrates that many useful caged compounds exploited this generality. This feature is unmatched by any other category of photosensitive protecting groups. In particular, my own work developing caged Ca2+ probes takes advantage of the unique ability the ortho-nitrobenzyl to cut C-N bonds in tertiary amines[8, 53, 87] and C-O bonds in ethers[87]. Gene chip manufacturing uses the intramolecular photo-redox chemistry of the ortho-nitrobenzyl chromophore, as it can take place in air[88]. Deprotection of acids by the ortho-nitrobenzyl group is matched by several other photosensitive protecting groups such as coumarin, desyl, pHP, etc. However, these photosensitive protecting groups use photosolvolysis[89], so do not work with amines and alcohols, without the addition of the oxycarbonyl linker. RuBi photosensitive protecting groups form bonds with N atoms via their lone pair donating electrons to Ru d orbitals[36]. This approach is different from cutting C-N bonds with the ortho-nitrobenzyl chromophore, but is useful for many types of biomolecules, but could not be used for caged Ca2+.
Summary: if you seek to develop new photosensitive protecting groups, you must ask if you cannot match the generality of the ortho-nitrobenzyl chromophore, what gap in the current arsenal you are seeking to fill?
Breaking the rule: the success of specialist protecting groups shows generality is not required to be useful.
11. Synthetic flexibility.
The most impactful caged compounds used by biologists needed to be tolerant of a wide range of chemicals and conditions (e.g. TFA, TBAF, NaOH, borane-based reductants, strong acids, BuOOH, O3, cross-couplings, etc.) during their multi-step synthesis, making Barltrop’s original chromophore[85] a key player for much photophysiology[68]. Why do we not see TFA or organic bases used in synthetic routes with, for example, many BODIPY groups? Why are many reducing agents missing from applications of coumarin and phenacetyl groups? Organic chemists know synthetic flexibility as a key property of a protecting group, but this factor is never mentioned in major reviews of photosensitive protecting groups. However, I feel it does merit inclusion when planning the development of new protecting groups for caged compounds
Summary: The ability to withstand a wide range of standard synthetic conditions is an important feature of chromophores used as photosensitive protecting groups.
Breaking the rule: not a deal breaker, but surely this will restrict utility?
Summary: the Photochemical Catechism.
The biological sciences are continuously transformed and invigorated by adopting techniques from physics and chemistry. Since WWII sequencing of proteins and DNA, NMR, mass spectrometry, lasers, electron microscopy, chromatography, PCR, etc. all received Nobel Prizes for Physics or Chemistry, and these technological revolutions have had stunning impact in the biological sciences.
The inventor of optogenetics, Gero Miesenböck, wrote a primer for that field in 2009 entitled “The Optogenetic Catechism”[90]. Just like genetically encoded sensors and actuators, photochemical probes can form a similar interrogative partnership that question and answer cell function (the “catechism”). Caged compounds, unlike DNA delivered actuators, are unlimited in which molecule they can control photochemically. Thus, both natural and non-natural products can be subjected to the uncaging method. Their scope is only limited by our imagination and ability as chemists to deliver optical probes for the biological sciences.
Acknowledgements.
I want to thank the editor, Dr Eric Castro, for many constructive criticisms during the preparation of this article. My laboratory has been continuously supported a RO1 award from the NIH, GM53395, since 1995. Recently, I obtained an “Outstanding Investigator Award” from the NIH (R35, NS111600). I want to acknowledge the visionary support of the HFSP for projects that the NIH will not fund because they are considered too risky. I am privileged to have my third HFSP currently (RGP0035/2020).
Biography.
I obtained my Ph.D. in chemistry at the University of Reading in 1982, under the supervision of Prof. Derek Bryce-Smith (he discovered aromatic photochemistry in 1957). The external examiner for my Ph.D. was John Coyle, a mentee of Prof. John Barltrop, the inventor of photosensitive protecting groups. After this, I did postdoctoral studies at the University of Pennsylvania with Prof. Jack Kaplan, the inventor of caged compounds. From 1996 I was a Professor of Physiology (Drexel University, Philadelphia), until 2010, when I became a Professor of Neuroscience (Mount Sinai, NYC).
References.
- [1].Kuhn TS, The Structure of Scientific Revolutions, 3rd ed., The University of Chicago Press, Chicago, USA, 1996. [Google Scholar]
- [2].Talbot W, Phil Trans Royal Soc 1839, 4, 120–121. [Google Scholar]
- [3].Talbot W, Phil Trans Royal Soc 1851, 6, 82. [Google Scholar]
- [4].Miesenböck G, Kevrekidis IG, Annu Rev Neurosci 2005, 28, 533–563. [DOI] [PubMed] [Google Scholar]
- [5].Paoletti P, Ellis-Davies GCR, Mourot A, Nat Rev Neurosci 2019, 20, 514–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kaplan JH, Somlyo AP, Trends Neurosci 1989, 12, 54–59. [DOI] [PubMed] [Google Scholar]
- [7].Liu G, Nat Neurosci 2004, 7, 373–379. [DOI] [PubMed] [Google Scholar]
- [8].Kaplan JH, Ellis-Davies GCR, Proc Natl Acad Sci U S A 1988, 85, 6571–6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gasparini S, Magee JC, J Neurosci 2006, 26, 2088–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lima SQ, Miesenböck G, Cell 2005, 121, 141–152. [DOI] [PubMed] [Google Scholar]
- [11].Kaplan JH, Forbush B, Hoffman JF, Biochemistry 1978, 17, 1929–1935. [DOI] [PubMed] [Google Scholar]
- [12].Tsien RY, Zucker RS, Biophys J 1986, 50, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Adams SR, Kao JPY, Grynkiewicz G, Minta A, Tsien RY, J Am Chem Soc 1988, 110, 3212–3220. [Google Scholar]
- [14].Walker JW, Somlyo AV, Goldman YE, Somlyo AP, Trentham DR, Nature 1987, 327, 249–252. [DOI] [PubMed] [Google Scholar]
- [15].Boron W, Boulpaep E, Medical Physiology, 2nd ed., Elsivier, New York, 2004. [Google Scholar]
- [16].Ellis-Davies GCR, Nat Methods 2007, 4, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mayer G, Heckel A, Angew Chem Int Ed 2006, 45, 4900–4921. [DOI] [PubMed] [Google Scholar]
- [18].Thirlwell H, Sleep JA, Ferenczi MA, J Muscle Res Cell Motil 1995, 16, 131–137. [DOI] [PubMed] [Google Scholar]
- [19].Molnár P, Nadler JV, Eur J Pharmacol 2000, 391, 255–262. [DOI] [PubMed] [Google Scholar]
- [20].Gee KR, Wieboldt R, Hess GP, J Am Chem Soc 1994, 116, 8366–8367. [Google Scholar]
- [21].Canepari M, Nelson L, Papageorgiou G, Corrie J, Ogden D, J Neurosci Meth 2001, 112, 29–42. [DOI] [PubMed] [Google Scholar]
- [22].Matsuzaki M, Hayama T, Kasai H, Ellis-Davies GCR, Nat Chem Biol 2010, 6, 255–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Richers MT, Amatrudo JM, Olson JP, Ellis-Davies GCR, Angew Chem Int Ed 2017, 56, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Richers MT, Passlick S, Agarwal H, Ellis-Davies GCR, Angew Chem Int Ed 2019, 58, 12086–12090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Goldman YE, Hibberd MG, McCray JA, Trentham DR, Nature 1982, 300, 701–705. [DOI] [PubMed] [Google Scholar]
- [26].Hibberd MG, Dantzig JA, Trentham DR, Goldman YE, Science 1985, 228, 1317–1319. [DOI] [PubMed] [Google Scholar]
- [27].Geibel S, Barth A, Amslinger S, Jung AH, Burzik C, Clarke RJ, Givens RS, Fendler K, Biophys J 2000, 79, 1346–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Schlichting I, Almo S, Rapp G, Wilson K, Petratos K, Lentfer A, Wittinghofer A, Kabsch W, Pai EF, Petsko GA, Goody RS, Nature 1990, 345, 309–315. [DOI] [PubMed] [Google Scholar]
- [29].Martin-Garcia JM, Crystals 2021, 11, 521. [Google Scholar]
- [30].McCray JA, Herbette L, Kihara T, Trentham DR, Proc Natl Acad Sci U S A 1980, 77, 7237–7241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Morrison H, Migdalof BH, J. Org. Chem 1965, 30, 3996. [Google Scholar]
- [32].Wieboldt R, Gee KR, Niu L, Ramesh D, Carpenter BK, Hess GP, Proc Natl Acad Sci U S A 1994, 91, 8752–8756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cheng Q, Steinmetz M, Jayaraman V, J Am Chem Soc 2002, 124, 7676–7677. [DOI] [PubMed] [Google Scholar]
- [34].Matsuzaki M, Ellis-Davies GCR, Nemoto T, Miyashita Y, Iino M, Kasai H, Nat Neurosci 2001, 4, 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chen Ti-W, Wardill TJ, Sun Yi, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS, Nature 2013, 499, 295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zayat L, Calero C, Alborés P, Baraldo L, Etchenique R, J Am Chem Soc 2003, 125, 882–883. [DOI] [PubMed] [Google Scholar]
- [37].Eckardt T, Hagen V, Schade B, Schmidt R, Schweitzer C, Bendig J, J Org Chem 2002, 67, 703–710. [DOI] [PubMed] [Google Scholar]
- [38].Geissler D, Kresse W, Wiesner B, Bendig J, Kettenmann H, Hagen V, Chembiochem 2003, 4, 162–170. [DOI] [PubMed] [Google Scholar]
- [39].Nache V, Kusch J, Hagen V, Benndorf K, Biophys J 2006, 90, 3146–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Gilbert D, Funk K, Dekowski B, Lechler R, Keller S, Mohrlen F, Frings S, Hagen V, Chembiochem 2007, 8, 89–97. [DOI] [PubMed] [Google Scholar]
- [41].Kolarski Dusan, Sugiyama Akiko, Breton Ghislain, Rakers Christin, Ono Daisuke, Schulte Albert, Tama Florence, Itami Kenichiro, Szymanski Wiktor, Hirota Tsuyoshi, Feringa BL, J Am Chem Soc 2019, 141, 15784–15791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ellis-Davies GCR, Chem Rev 2008, 108, 1603–1613. [DOI] [PubMed] [Google Scholar]
- [43].Heidelberger R, Heinemann C, Neher E, Matthews G, Nature 1994, 371, 513–515. [DOI] [PubMed] [Google Scholar]
- [44].Savjani Ketan T., Gajjar Anuradha K., Savjani JK, ISRN Pharmaceutics 2012, ID:195727. [DOI] [PMC free article] [PubMed]
- [45].Farley S, Laguerre A, Schultz C, Curr Opin Chem Biol 2021, 65, 42–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hoglinger D, Nadler A, Schultz C, Biochem. Biophys. Acta 2014, 1841, 1085–1096. [DOI] [PubMed] [Google Scholar]
- [47].Wang L, Corrie J, Wootton J, J Org Chem 2002, 67, 3474–3478. [DOI] [PubMed] [Google Scholar]
- [48].Huang YH, Sinha SR, Fedoryak OD, Ellis-Davies GCR, Bergles DE, Biochemistry 2005, 44, 3316–3326. [DOI] [PubMed] [Google Scholar]
- [49].Walker J, Feeney J, Trentham D, Biochemistry 1989, 28, 3272–3280. [DOI] [PubMed] [Google Scholar]
- [50].Liu W, Llopis J, Whitney M, Zlokarnik G, Tsien RY, Nature 1998, 392, 936–941. [DOI] [PubMed] [Google Scholar]
- [51].Crowe SE, Kantevari S, Ellis-Davies GCR, ACS Chem Neurosci 2010, 1, 575–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Corrie J, Kaplan JH, Forbush B, Ogden DC, Trentham DR, Photochem Photobiol Sci 2016, 15, 604–608. [DOI] [PubMed] [Google Scholar]
- [53].Ellis-Davies GCR, Kaplan JH, Proc Natl Acad Sci U S A 1994, 91, 187–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Rial Verde EM, Zayat L, Etchenique R, Yuste R, Front. Neural Circuits 2008, 2, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Passlick S, Ellis-Davies GCR, J Neurosci Meth 2017, 293, 321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Lutz C, Otis TS, DeSars V, Charpak S, DiGregorio DA, Emiliani V, Nat Methods 2008, 5, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ellis-Davies GCR, Matsuzaki M, Paukert M, Kasai H, Bergles DE, J Neurosci 2007, 27, 6601–6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Patterson M, Yasuda R, Br J Pharmacol 2011, 163, 1626–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Eder M, Zieglgansberger W, Dodt HU, Rev Neurosci 2004, 15, 167–183. [DOI] [PubMed] [Google Scholar]
- [60].Passlick S, Kramer PF, Richers MT, Williams JT, Ellis-Davies GCR, PLoS One 2017, 12, e0187732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Claridge-Chang A, Roorda RD, Vrontou E, Sjulson L, Li H, Hirsh J, Miesenböck G, Cell 2009, 139, 405–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Denk W, Strickler JH, Webb WW, Science 1990, 248, 73–76. [DOI] [PubMed] [Google Scholar]
- [63].Albota M, Beljonne D, Bredas J, Ehrlich J, Fu J, Heikal A, Hess S, Kogej T, Levin M, Marder S, McCord-Maughon D, Perry J, Rockel H, Rumi M, Subramaniam C, Webb W, Wu X, Xu C, Science 1998, 281, 1653–1656. [DOI] [PubMed] [Google Scholar]
- [64].Reinhardt B, Brott L, Clarson S, Dillard A, Bhatt J, Kannan R, Yuan L, He G, Prasad P, Chem Mater 1998, 10, 1863–1874. [Google Scholar]
- [65].Rumi M, Barlow S, Wang J, Perry JW, Marder SR, Adv Polym Sci 2008, 213, 1–95. [Google Scholar]
- [66].He GS, Tan LS, Zheng Q, Prasad PN, Chem. Rev 2008, 108, 1245–1330. [DOI] [PubMed] [Google Scholar]
- [67].Agarwal HK, Janicek R, Chi SH, Perry JW, Niggli E, Ellis-Davies GCR, J Am Chem Soc 2016, 138, 3687–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ellis-Davies GCR, Acc Chem Res 2020, 53, 1593–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Ellis-Davies GCR, Front Synaptic Neurosci 2019, 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Furuta T, Wang SS, Dantzker JL, Dore TM, Bybee WJ, Callaway EM, Denk W, Tsien RY, Proc Natl Acad Sci U S A 1999, 96, 1193–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Brown EB, Shear JB, Adams SR, Tsien RY, Webb WW, Biophys J 1999, 76, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Specht A, Bolze F, Donato L, Herbivo C, Charon S, Warther D, Gug S, Nicoud J-F, Goeldner M, Photochem Photobiol Sci 2012, 11, 578–586. [DOI] [PubMed] [Google Scholar]
- [73].Kantevari S, Passlick S, Kwon HB, Richers M, Sabatini BL, Ellis-Davies GCR, ChemBioChem 2016, 17, 953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kiskin NI, Ogden D, Eur Biophys J 2002, 30, 571–587. [DOI] [PubMed] [Google Scholar]
- [75].Hagen V, Dekowski B, Nache V, Schmidt R, Geissler D, Lorenz D, Eichhorst J, Keller S, Kaneko H, Benndorf K, Wiesner B, Angew Chem Int Edit 2005, 44, 7887–7891. [DOI] [PubMed] [Google Scholar]
- [76].Chiu CQ, Lur G, Morse TM, Carnevale NT, Ellis-Davies GCR, Higley MJ, Science 2013, 340, 759–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Hayama T, Noguchi J, Watanabe S, Takahashi N, Hayashi-Takagi A, Ellis-Davies GCR, Matsuzaki M, Kasai H, Nat Neurosci 2013, 16, 1409–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kiskin NI, Chillingworth R, McCray JA, Piston D, Ogden D, Eur Biophys J 2002, 30, 588–604. [DOI] [PubMed] [Google Scholar]
- [79].Brown S, Brown E, Walker I, The Lancet Oncology 2004, 5, 497–508. [DOI] [PubMed] [Google Scholar]
- [80].Broichhagen J, Frank JA, Trauner D, Acc Chem Res 2015, 48, 1947–1960. [DOI] [PubMed] [Google Scholar]
- [81].Gorka AP, Nani RR, Zhu J, Mackem S, Schnermann MJ, J Am Chem Soc 2014, 136, 14153–14159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Nani RR, Gorka AP, Nagaya T, Kobayashi H, Schnermann MJ, Angew Chem Int Ed 2015, 54, 13635–13638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Vickerman BM, Zywot EM, Tarrant TK, Lawrence DS, Nat Rev Chem 2022, 5, 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Velema WA, Szymanski W, Feringa BL, J Am Chem Soc 2012, 136, 2178–2191. [DOI] [PubMed] [Google Scholar]
- [85].Barltrop JA, Plant PJ, Schofield P, Chem. Commun 1966, 822–823.
- [86].Binkley RW, Flechtner TW, in Synthetic Organic Photochemistry (Ed.: Horspool WM), Plenum, New York and London, 1984, pp. 375. [Google Scholar]
- [87].Ellis-Davies GCR, Tetrahedron Letters 1998, 39, 953–956. [Google Scholar]
- [88].McGall GH, Barone AD, Diggelmann M, Fodor SPA, Gentalen E, Ngo N, J. Am. Chem. Soc. 1997, 119, 5081–5090. [Google Scholar]
- [89].Schmidt R, Geissler D, Hagen V, Bendig J, J Phys Chem A 2007. 111, 5768–5774. [DOI] [PubMed]
- [90].Miesenböck G, Science 2009, 326, 395–399. [DOI] [PubMed] [Google Scholar]
- [91].Milburn T, Matsubara N, Billington AP, Udgaonkar JB, Walker JW, Carpenter BK, Webb WW, Marque J, Denk W, Mccray JA, Hess GP, Biochemistry 1989, 28, 49–55. [DOI] [PubMed] [Google Scholar]
- [92].Breitinger HG, Wieboldt R, Ramesh D, Carpenter BK, Hess GP, Biochemistry 2000, 39, 5500–5508. [DOI] [PubMed] [Google Scholar]
- [93].Zhao J, Gover TD, Muralidharan S, Auston DA, Weinreich D, Kao JPY, Biochemistry 2006, 45, 4915–4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].in Yale@Medicine, Vol. 1, Yale University, New Haven, 2005, p. 1. [Google Scholar]
- [95].Durand-de Cuttoli R, Chauhan PS, Petriz Reyes A, Faure P, Mourot A, Ellis-Davies GCR, Proc Natl Acad Sci USA 2020, 117, 6831–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]