

Abstract
BRD4 is a well‐recognized transcriptional activator, but how it regulates gene transcriptional repression in a cell type‐specific manner has remained elusive. In this study, we report that BRD4 works with Polycomb repressive complex 2 (PRC2) to repress transcriptional expression of the T‐helper 2 (Th2)‐negative regulators Foxp3 and E3‐ubiqutin ligase Fbxw7 during lineage‐specific differentiation of Th2 cells from mouse primary naïve CD4+ T cells. Brd4 binds to the lysine‐acetylated‐EED subunit of the PRC2 complex via its second bromodomain (BD2) to facilitate histone H3 lysine 27 trimethylation (H3K27me3) at target gene loci and thereby transcriptional repression. We found that Foxp3 represses transcription of Th2‐specific transcription factor Gata3, while Fbxw7 promotes its ubiquitination‐directed protein degradation. BRD4‐mediated repression of Foxp3 and Fbxw7 in turn promotes BRD4‐ and Gata3‐mediated transcriptional activation of Th2 cytokines including Il4, Il5, and Il13. Chemical inhibition of the BRD4 BD2 induces transcriptional de‐repression of Foxp3 and Fbxw7, and thus transcriptional downregulation of Il4, Il5, and Il13, resulting in inhibition of Th2 cell lineage differentiation. Our study presents a previously unappreciated mechanism of BRD4's role in orchestrating a Th2‐specific transcriptional program that coordinates gene repression and activation, and safeguards cell lineage differentiation.
Keywords: BRD4, gene repression, gene transcriptional regulation, PRC2, Th2
Subject Categories: Chromatin, Transcription & Genomics; Immunology
BRD4 regulates T‐helper 2 differentiation via the recruitment of PRC2 to promote transcriptional repression of the T‐helper 2‐negative regulators Foxp3 and Fbxw7.
Introduction
T‐helper (Th) cells play an important role in adaptive immunity and are involved not only in host defense against different types of infectious agents, but also in pathogenesis of autoimmunity and inflammatory diseases (Paul & Zhu, 2010; Abraham & Medzhitov, 2011; Iwasaki & Medzhitov, 2015). After stimulation with antigens, different combinations of cytokines guide naïve CD4+ T cells to differentiate into various subsets of T‐helper cells such as Th1, Th2, Th17, and regulatory T (Treg) cells (Mosmann & Coffman, 1989; Rengarajan et al, 2000; Murphy & Reiner, 2002; Park et al, 2005; Zhu & Paul, 2010). Th2 cells are crucial in host immunity to fight against extracellular parasites, however, dysregulation of Th2 cells also induces airway inflammation such as allergic asthma, characterized by an overproduction of interleukin‐4 (IL‐4), IL‐5 and IL‐13 (Mosmann & Coffman, 1989; Cohn et al, 2004). During Th2 cell differentiation, multiple mechanisms coordinately regulate the expression of key cytokines. T cell receptor signaling is indispensable for both Th1 and Th2 cell differentiation (Ouyang et al, 2000; Rengarajan et al, 2000; Paul, 2010). In addition, IL‐4‐induced STAT6 activation stimulates transcriptional expression of GATA3, a crucial transcription factor for Th2 cell differentiation, and induces chromatin remodeling at loci of key Th2 genes to further amplify IL‐4 production (Zhang et al, 1997; Lee et al, 2001; Zhu et al, 2006; Ho et al, 2009; Paul & Zhu, 2010; Tanaka et al, 2011).
The bromodomain (BrD) and extra‐terminal (BET) family proteins, consisting of BRD2, BRD3, BRD4, and testis‐specific BRDT, are major transcription regulators, which are characterized by tandem bromodomains (BD1 and BD2) that function in gene transcription through binding to acetyl‐lysine residues of histones and transcription factors (Dhalluin et al, 1999; Mujtaba et al, 2007). Small molecular compounds targeting the BrDs effectively inhibit the transcriptional activation of genes mediated by BET proteins (Dhalluin et al, 1999; Filippakopoulos et al, 2010; Belkina et al, 2013; Hammitzsch et al, 2015; Ghosh & Lora, 2016; Cheung et al, 2017b). Recent studies found that BD1 and BD2 domains in BET proteins have distinct but complementary functions in the regulation of gene transcription (Shi et al, 2014; Cheung et al, 2017b). In general, BRD4‐BD2 domain interacts with lysine‐acetylated transcription factors (Shi et al, 2014), while BRD4‐BD1 interacts with acetylated H4 to cooperatively facilitate the transcriptional activation of target genes (Filippakopoulos et al, 2010; Schroder et al, 2012; Zhang et al, 2012).
In contrast to its well‐known role in gene transcriptional activation, BRD4 has also been implicated in gene transcriptional repression with limited mechanistic understanding. For example, BRD4 was reported for its function for transcriptional repression of oxidative phosphorylation (OXPHOS) genes, leading to bioenergetic deficiency caused by mitochondrial disease complex I mutations (Barrow et al, 2016). BRD4 short isoform (BRD4S) interacts via its BrDs and ET domain with BRG1, a catalytic unit of BAF chromatin remodeling complex for repression of HIV‐1 transcription (Conrad et al, 2017). BRD4 can also bind to histone lysine methyltransferase G9a to repress genes involved in autophagy and lysosome biogenesis (Sakamaki et al, 2017; Sakamaki & Ryan, 2017). More recently, it has also been shown that BRD4 interacts with lysine‐specific demethylase (LSD1) to demethylate H3K4me2 for gene transcriptional repression (Liu et al, 2022). However, it has remained elusive as to how BRD4 represses gene transcription through H3K27me3, the most prevalent form of repressive histone mark, underlying the need to identify and characterize BRD4 repressor complexes. In this study, we showed that BRD4 works with Polycomb repressor complex 2 (PRC2) to repress transcriptional expression of Th2‐negative regulators Foxp3 and Fbxw7 in lineage‐specific differentiation of Th2 cells from mouse primary naïve CD4+ T cells. Our study presents a new mechanistic model of a repressor function of BRD4‐PRC2 complex in regulation of gene transcription in chromatin.
Results
Brd4 promotes Th2 cell differentiation and murine airway inflammation
We first confirmed the role of Brd4 in Th2 cell differentiation using a pan‐BET BrD inhibitor JQ1 that inhibits both BD1 and BD2 of Brd4, and repressed lineage differentiation of Th2 cells from mouse primary naïve CD4+ T cells in a dose‐dependent manner (Fig EV1A). JQ1 inhibition of Th2 cell differentiation is likely a result of its downregulation of transcriptional expression of Il4, Il5 and Il13, and IL‐4 secretion of Th2 cells (Fig EV1B–D). In contrast to Brd4, the protein expression levels of Brd2 and Brd3 are significantly lower in Th2 than other Th cells (Fig EV1E). shRNA lentiviral knockdown of Brd4, but not Brd2 or Brd3 in Th2 cells caused a reduction of IL‐4 expressing Th2 cells (Fig EV1F). This effect is independent of cell survival and viability, as assessed by gating on live/dead cells and PI/Annexin V staining (Fig EV2A and B).
Figure EV1. Brd4 promotes Th2 cell differentiation.
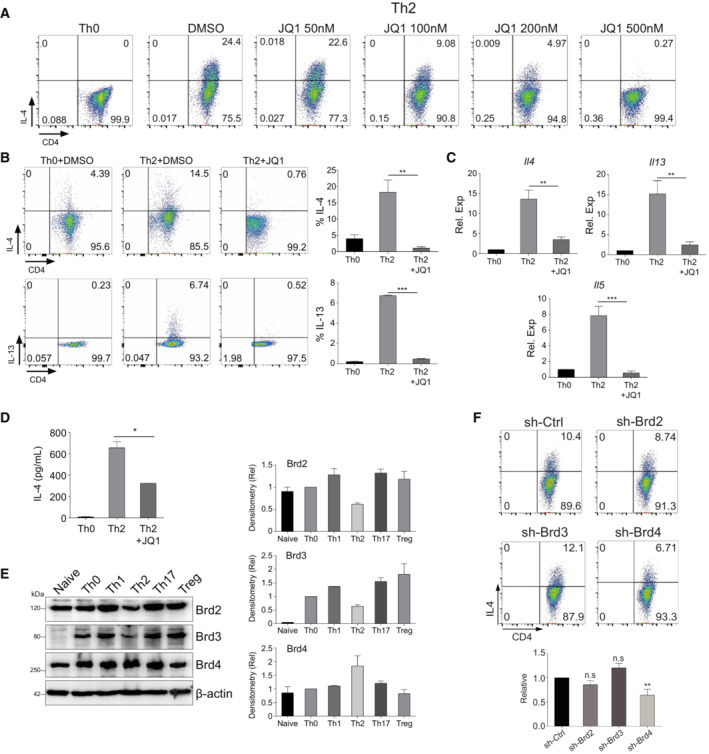
- Flow cytometry analysis of mouse Th2 cells treated with JQ1 as indicated. Mouse Th2 cells were differentiated from mouse primary naïve CD4+ T cells for 6 days before analysis.
- Flow cytometric (left) and statistical analysis (right) of IL‐4 and IL‐13 in Th2 cells derived from mouse primary naïve CD4+ T cells treated with or without JQ1 (500 nM).
- qPCR analysis of Il4, Il5, and Il13 in mouse Th2 cells treated with or without JQ1.
- ELISA analysis of IL‐4 secretion into the supernatant of mouse Th2 cells treated with or without JQ1.
- Western blotting (left) and densitometry analysis (right) of Brd2, Brd3, and Brd4 in mouse naïve CD4+ T cells, Th0, Th1, Th2, Th17, and Treg cells.
- Flow cytometric (upper) and statistical analysis (lower) of IL‐4 in mouse Th2 cells infected with sh‐Ctrl, sh‐Brd2, sh‐Brd3, or sh‐Brd4 lentivirus.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All data represent mean ± SD and average of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
Figure EV2. Brd4 promotes murine airway inflammation.
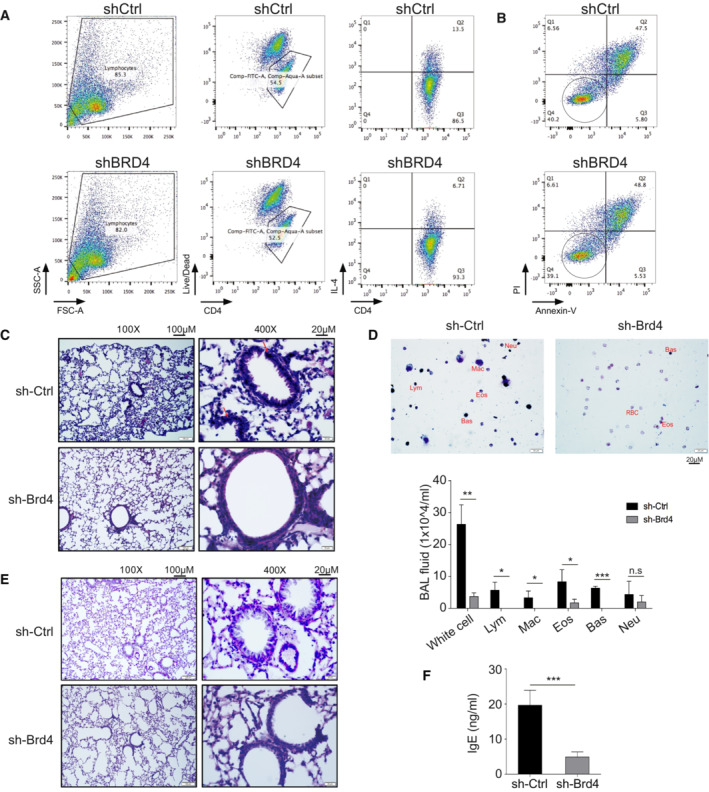
- Representative gating strategies using side‐scatter, live‐dead, followed by assessment of IL‐4‐expressing Th2 cells, treated with control shRNA or shRNA targeting BRD4.
- Representative PI/Annexin V staining of Th2 cells, treated with control shRNA or shRNA targeting BRD4. Live cells are circled.
- Representative lung H&E staining showing infiltration of inflammatory cells indicated by arrows.
- Quik‐Tiff staining of BALF (upper) and quantification of inflammatory cells (lower) in the BALF of mice transferred with mouse Th2 cells and Brd4‐knockdown Th2 cells.
- Representative PAS staining showing proliferation of mucin producing cells in the airway epithelia in Rag1−/− hosts adoptively transferred with mouse Th2 cells and Brd4‐knockdown Th2 cells.
- Quantitation of IgE in BALF with ELISA.
Data information: All data are representative of two experiments with n = 4. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
We verified Brd4 function in Th2 cell development in vivo in an ovalbumin (OVA)‐sensitized and challenged mouse model that recapitulates Th2‐associated airway inflammation in human asthma (Brusselle et al, 1994). In this study, syngeneic Rag1 −/− mice were injected with Th2 cells with or without Brd4 knockdown by shRNA, and challenged with aerosolized OVA daily for 6 days, which induced asthma features (Lambrecht & Hammad, 2015). Histological analysis revealed that the mice transferred with Th2 cells of Brd4 knockdown displayed strong inhibition of inflammatory cell infiltration (eosinophils, H&E staining) in the lung and airway (Fig EV2C and D), hyperplasia of mucus‐producing goblet cell (PAS staining) in the airway (Fig EV2E), and IgE level in the bronchoalveolar lavage fluid (BALF) (Fig EV2F). Collectively, these results established Brd4's functional role in Th2 cell differentiation and OVA‐induced airway inflammation, likely facilitated by Th2 cytokines such as IL‐4, IL‐5, and IL‐13.
Brd4 represses a key gene transcriptional program during Th2 cell differentiation
To gain mechanistic insights into how Brd4 promotes cell type‐specific Th2 response, we performed comprehensive RNA‐seq and ChIP‐seq studies of mouse Th2 cells treated with or without JQ1. The RNA‐seq data revealed that JQ1 caused transcriptional downregulation of 3,459 genes, and surprisingly, upregulation of a significant number of genes (1,533 genes) in Th2 cells (Figs 1A and EV3A). Our ChIP‐seq analysis revealed that Brd4 binds predominantly to distal intergenic and promoter regions of Th2 genome (Figs 1B and EV3B). We further established direct gene targets that are BRD4‐transcriptionally activated (1,197 genes) or repressed (449 genes) (Fig 1C), of which associated BRD4 peaks were displaced globally by JQ1 treatment (Fig 1D).
Figure 1. Brd4 represses a key gene transcriptional program during Th2 cell differentiation.
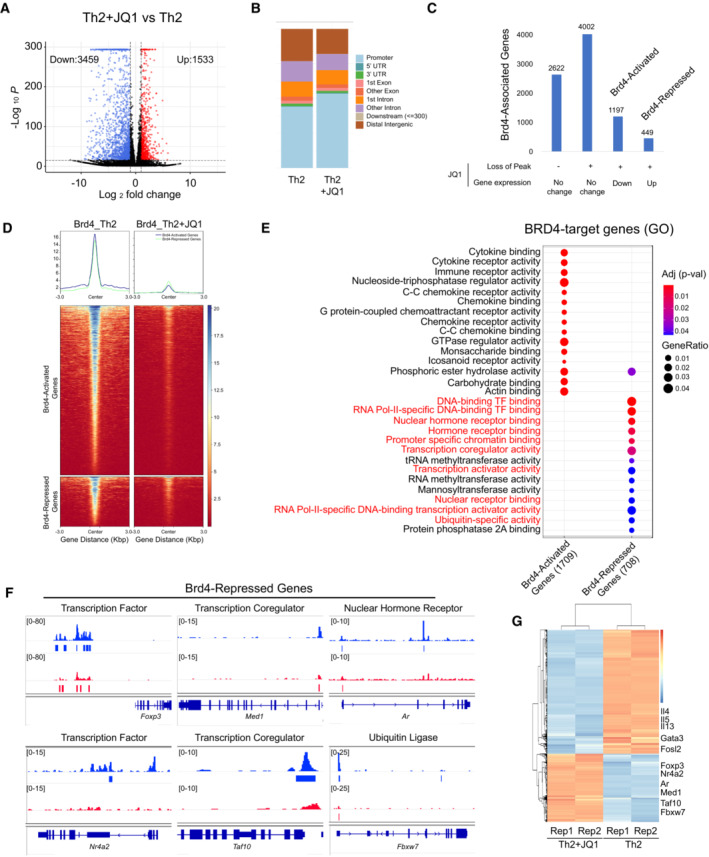
- Volcano plot of RNA‐seq data showing differential expression of mRNA transcripts in mouse Th2 cells treated with or without JQ1 (250 nM). Data are representative of two biological replicates.
- Global ChIP‐seq analysis of Brd4 occupancy at the genomic regions of mouse Th2 cells treated with or without JQ1 (250 nM). Data are representative of three biological replicates.
- Identification of direct targets of BRD4‐activated and ‐repressed genes in Th2 cells.
- Brd4 ChIP‐seq intensity associated with BRD4‐activated and ‐repressed genes in Th2 cells, treated with or without JQ1.
- Gene ontology (GO) analysis of direct targets of BRD4‐activated and ‐repressed genes in Th2 cells.
- ChIP‐seq tracks Brd4 on select genes (Foxp3, Nr4a2, Med1, Taf10, Ar, and Fbxw7) in Th2 cells, treated with or without JQ1.
- Heatmap of RNA‐seq analysis showing up‐ and down‐regulation of select genes in Th2 cells, treated with or without JQ1.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified.
Figure EV3. RNA‐seq and ChIP‐seq analysis of Brd4 in Th2 cells.
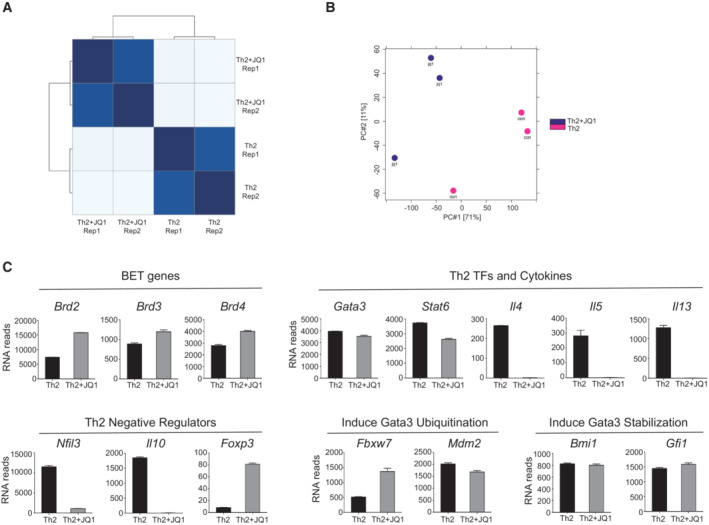
- Heatmap of sample‐to‐sample distance of RNA‐seq analysis in mouse Th2 cells treated with or without JQ1.
- Principal component analysis of ChIP‐seq peaks in mouse Th2 cells treated with or without JQ1.
- RNA‐seq data analysis showing expression of key genes in mouse Th2 cells treated with or without JQ1 (250 nM).
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. Data are representative of two to three biological replicates.
Importantly, BRD4 represses a key gene program that consists of DNA‐binding transcription factors (TFs), nuclear hormone receptors, transcriptional coregulators, and ubiquitin‐specific regulators (Fig 1E), suggesting important functional role of BRD4‐repressed genes during Th2 cell differentiation. We showed that JQ1 treatment diminished Brd4 binding to the cis‐regulatory regions of the key repressed‐genes including Foxp3, Nr4a2, Med1, Taf10, Ar, and Fbxw7, leading to their upregulation of mRNA expression (Figs 1F and G, and EV3C). We confirmed that while IL‐4 protein expression was abolished in Th2 cells treated by JQ1, Foxp3 mRNA and protein levels were dramatically increased (Fig EV4A–C). ChIP‐seq of Brd4 further revealed strong Brd4 binding on enhancer sites of Foxp3, which was reduced by JQ1 treatment (Fig 1F), as further confirmed by ChIP‐qPCR (Fig EV4D). Similarly, Brd4 also represses Fbxw7 gene expression, as JQ1 treatment displaced Brd4 binding on the promoter of Fbxw7 locus and induced Fbxw7 mRNA in Th2 cells (Fig 1F and G). As shown in a dual‐luciferase assay, Brd4 binding on Fbxw7 promoter exerted an inhibitory effect on gene activation, which was reversed by an acetyl‐lysine binding‐deficient Brd4 mutant N140A/N433A (Ren et al, 2018) (Fig EV4E). We further performed kinetic studies of the de‐repression of BRD4‐repressed genes identified from our ChIP‐seq and RNA‐seq studies, including Nr4a2, Foxp3, and Fbxw7. Th2 cells differentiated for 3 days were treated with BRD4 inhibitor JQ1 and degrader MZ1, and the genes were de‐repressed 6–24 h post‐treatments (Fig EV4F and G). Collectively, these results suggested that Brd4 acts directly in transcriptional repression of Foxp3 and Fbxw7 in Th2 cells.
Figure EV4. Brd4 Inhibits Foxp3 transcription and enhances Gata3 stability by repressing Fbxw7 transcription to facilitate Th2 cell differentiation.
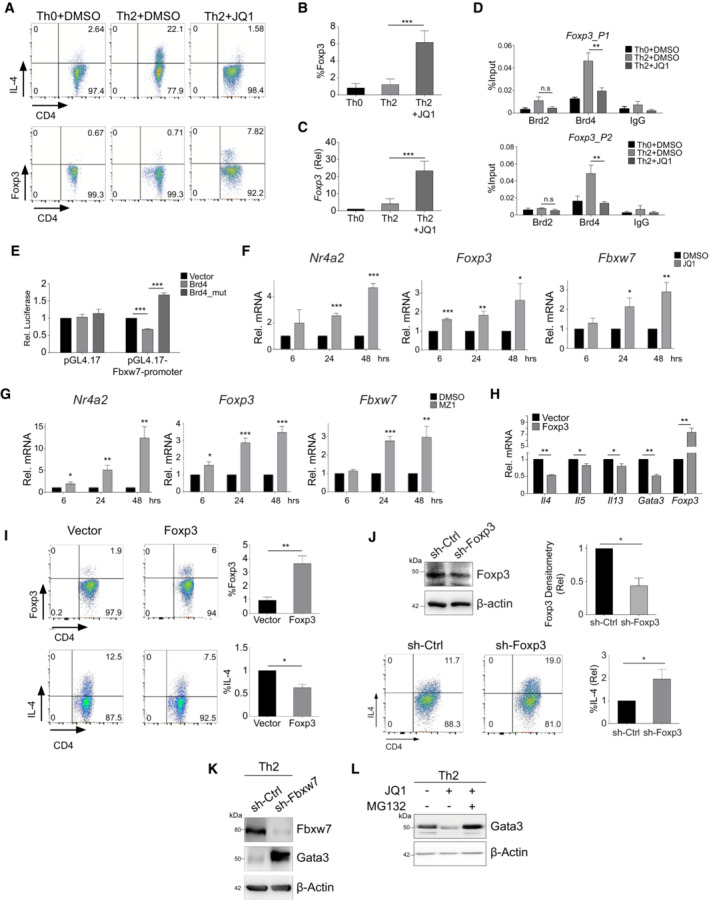
- Flow cytometric analysis of IL‐4 and Foxp3 protein in mouse Th2 cells treated with or without JQ1 (500 nM).
- Statistical analysis of Foxp3 protein in mouse Th2 cells treated with or without JQ1 (500 nM).
- mRNA expression of Foxp3 in mouse Th2 cells treated with or without JQ1 (500 nM).
- ChIP‐qPCR analysis of the binding of Brd2 and Brd4 on the gene loci of Foxp3 in mouse Th2 cells treated with or without JQ1 (250 nM).
- Dual‐luciferase reporter assay of HEK293T cells transfected with Fbxw7‐promoter, Brd4 and Brd4‐mutant (N140A/N433A).
- Th2 cells were differentiated for 3 days and then treated with DMSO and JQ1 (250 nM) for 6, 24, and 48 h, followed by qPCR analysis of Nr4a2, Foxp3, and Fbwx7.
- Th2 cells were differentiated for 3 days and then treated with DMSO and MZ1 (1 μM) for 6, 24, and 48 h, followed by qPCR analysis of Nr4a2, Foxp3, and Fbwx7.
- qPCR analysis of Il4, Il5, Il13, Gata3, and Foxp3 in mouse Th2 cells overexpressed with empty vector or Foxp3.
- Flow cytometric (left) and statistical analysis (right) of Foxp3 and IL‐4 in Th2 cells overexpressed with empty vector or Foxp3.
- Western blotting (upper left) and densitometry statistical analysis (upper right) of Foxp3 in Th2 cells infected with sh‐Ctrl and sh‐Foxp3 lentivirus. Flow cytometric (lower left) and statistical analysis (lower right) of IL‐4 in Th2 cells infected with sh‐Ctrl and sh‐Foxp3 lentivirus.
- Western blotting of Fbxw7 and Gata3 in mouse Th2 cells infected with sh‐ctrl and sh‐Fbxw7.
- Western blotting of Gata3 in mouse naïve CD4+ T cells cultured in Th2 polarization were treated with JQ1 (500 nM) at Day 0, and then treated with DMSO or MG132 (20 μM) for 6 h before harvest.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All Western blotting data are representative of three independent experiments. All data represent mean ± SD and average of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
Foxp3 inhibits Gata3 gene transcription while Fbxw7 promotes Gata3 protein degradation
ATAC‐seq analysis of chromatin accessibility of Th2 cells (Puleston et al, 2021) revealed that Brd4 binds to open cis‐regulatory regions of Foxp3, Fbwx7, Gata3, and Il4‐13 (Fig 2A). Notably, the Brd4‐binding sites along the Foxp3 and Fbxw7 loci are readily accessible not only in CD4+ naïve T cells but also in differentiated Th2 and other Th1, Th17, and Treg subtypes, whereas the Brd4‐binding regions in the Il4‐13 loci only selectively become accessible in Th2 cells after differentiation from naïve T cells, but not in other Th subtypes (Fig 2A). This result suggests that Brd4 control of Foxp3 and Fbxw7 transcription precedes its regulation of transcriptional expression of Th2‐cytokines Il4‐Il13 during lineage‐specific differentiation of Th2 cells from naïve T cells.
Figure 2. Foxp3 inhibits Gata3 gene transcription while Fbxw7 promotes Gata3 protein degradation.
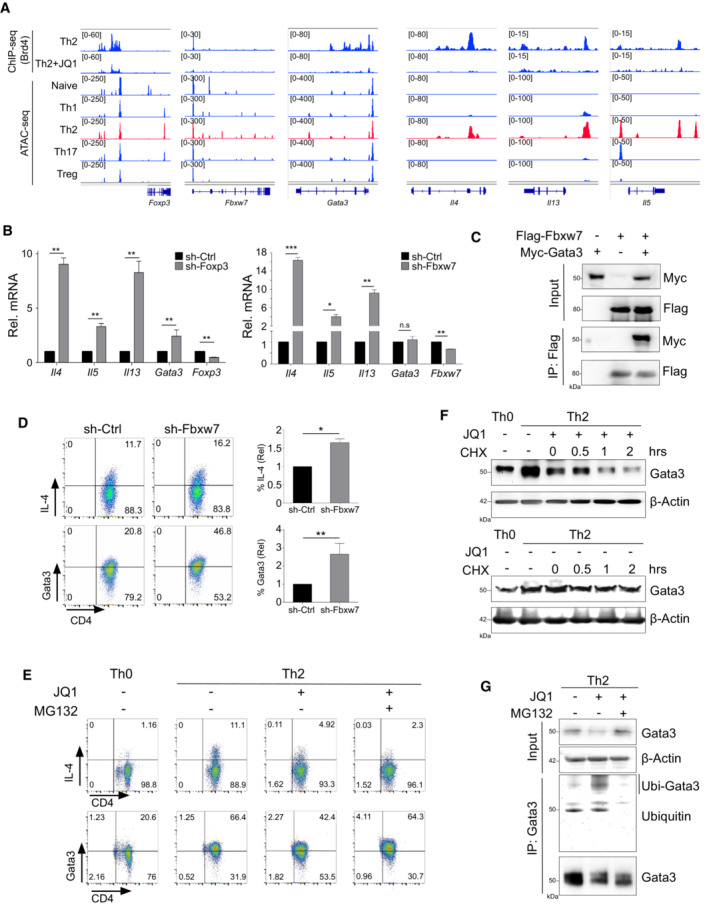
- ATAC‐seq tracks of accessible cis‐regulatory regions of Foxp3, Fbxw7, Gata3, and Il4‐13 in mouse naïve, Th1, Th2, Th17, and Treg cells.
- qPCR analysis of Il4, Il5, Il13, Gata3, Foxp3, and Fbxw7 in mouse Th2 cells infected with sh‐Ctrl, sh‐Foxp3, and sh‐Fbxw7 lentivirus.
- Immunoprecipitation of Flag‐tagged Fbxw7 with anti‐Flag affinity gel, followed by Western blotting of Myc to detect Myc‐Gata3 in HEK293T cells transfected with Myc‐Gata3 and/or Flag‐Fbxw7 plasmids.
- Flow cytometric (left) and statistical analysis (right) of IL‐4 and Gata3 in mouse Th2 cells infected with sh‐ctrl and sh‐Fbxw7.
- Flow cytometric analysis of IL‐4 and Gata3 in mouse naïve CD4+ T cells cultured in Th2 polarization were treated with JQ1 (500 nM) at Day 0, and then treated with DMSO or MG132 (20 μM) for 6 h before harvest.
- Western blotting of Gata3 in mouse Th2 cells treated with or without JQ1 (500 nM), and then treated with cycloheximide (CHX; 25 μg/ml) for 0 h, 0.5 h, 1 h, and 2 h before harvest to stop protein synthesis.
- Co‐IP to immunoprecipitated Gata3, followed by Western blotting of ubiquitin in mouse naïve CD4+ T cells cultured in Th2 polarization treated with JQ1 (500 nM) at Day 0, and then treated with DMSO or MG132 (20 μM) for 6 h before harvest.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All Western blotting data are representative of three independent experiments. All data represent mean ± SD and average of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
To determine the functional role of Foxp3 and Fbxw7 in Th2 cell differentiation, we ectopically expressed Foxp3 in mouse Th2 cells, which showed significantly decreased expression of Th2 key genes Il4, Il5, Il13, and Gata3 (Fig EV4H). This was confirmed by downregulation of IL‐4 expression in Th2 cells (Fig EV4I). Furthermore, loss of Foxp3 by shRNA knockdown exerted an opposite effect (Figs 2B and EV4J), confirming that Foxp3 inhibits gene transcription of these Th2‐key cytokines and Gata3.
Next, we showed that Fbxw7 knockdown by shRNA increased transcriptional expression of Il4, Il5, and Il13, but had no effect on transcriptional level of Gata3 (Fig 2B), a Th2 lineage‐specific transcription factor that controls the expression of key Th2 genes such as Il4, Il5, and Il13 (Zheng & Flavell, 1997). Importantly, we found that Fbxw7 binds with (Fig 2C) and ubiquitinates Gata3 (Song et al, 2018; Suehiro et al, 2020), and Fbxw7 knockdown induced protein level of Gata3 and IL‐4 in Th2 cells (Figs 2D and EV4K), suggesting the role of Fbxw7 in protein degradation of Gata3 in Th2 cells. Consistently, JQ1 caused a reduction of the protein expression level of Gata3 in Th2 cells (Figs 2E and EV4L), which is correlated with the induction of Fbxw7 by JQ1 (Figs 1G and EV3C). This is in contrast to nearly no changes in expression of other select genes known for protein ubiquitination or stabilization such as Mdm2 (Yamashita et al, 2005), Bmi1 (Hosokawa et al, 2006) and Gfi1 (Shinnakasu et al, 2008) (Fig EV3C). JQ1‐induced protein reduction of Gata3, but not IL‐4, was blocked by a proteasome inhibitor MG132 (Fig 2E), suggesting Gata3 degradation via ubiquitination. The latter was supported by the observation that JQ1‐induced Gata3 ubiquitination caused a faster degradation of Gata3 in Th2 cells when new protein synthesis was inhibited by cycloheximide (CHX) (Fig 2F). Our Co‐IP data further demonstrated that JQ1‐induced Gata3 ubiquitination was markedly decreased with MG132 treatment (Fig 2G). Taken together, these results showed that Brd4 represses the transcription of Fbxw7 through direct binding to the promoter of Fbxw7, leading to increased Gata3 stability in Th2 cell differentiation. Notably, Brd4's transcriptional repression of Th2‐negative regulators Foxp3 and Fbxw7 ensures the transcriptional expression of Th2 key genes through the regulation of Gata3 mRNA expression and protein stability.
Brd4 recruits PRC2 subunits to repress Th2‐negative regulators through H3K27me3
To determine how Th2 transcription factors work with Brd4, we examined the binding of Gata3 on gene loci of Il4, Il5, Il13, Foxp3, and Fbxw7 using the available ChIP‐seq data (Fang et al, 2018). Interestingly, Gata3 binds closely to or colocalizes with Brd4 binding sites on Foxp3 and Fbxw7, in addition to Il4, Il5, and Il13 gene loci (Fig 3A). Previous studies showed that Gata3 and YY1 interact with each other in Th2 cells to regulate transcriptional expression of Th2 key cytokines (Il4, Il5, and Il13) (Guo et al, 2001, 2008; Hwang et al, 2013). Using a ChIP‐reChIP assay, we confirmed that Gata3/YY1/Brd4 form a complex on the gene loci of Il4, Foxp3 and Fbxw7 (Fig EV5A). Knockdown of Gata3 or YY1 reduced Brd4 binding to chromatin (Fig 3B), suggesting that these transcription factors recruit Brd4 to chromatin in Th2 cells.
Figure 3. Brd4 recruits PRC2 subunits to repress Th2 negative regulators through H3K27me3.
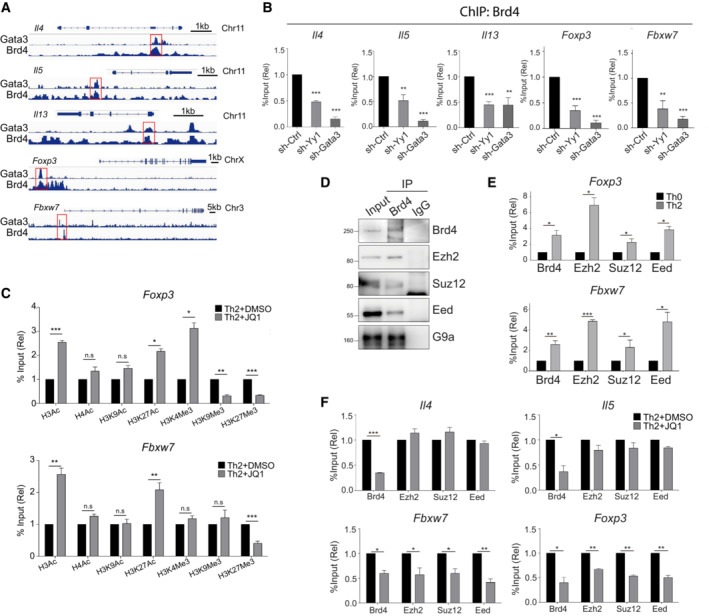
- ChIP‐seq tracks of Gata3 and Brd4 co‐occupancy on the gene loci of Il4, Il5, Il13, Foxp3, and Fbxw7 in mouse Th2 cells.
- ChIP‐qPCR analysis of the binding of Brd4 on the gene loci of Il4, Il5, Il13, Foxp3, and Fbxw7 in mouse Th2 cells, retrovirally transduced with vectors expressing shRNA targeting YY1 and Gata3.
- ChIP‐qPCR analysis of histone modifications on the gene loci of Foxp3 and Fbxw7 in mouse Th2 cells treated with or without JQ1 (250 nM).
- Immunoprecipitation of Brd4 in mouse Th2 cells, followed by Western blotting of Ezh2, Suz12, EED, and G9a.
- ChIP‐qPCR analysis of Brd4, Ezh2, Suz12, and EED binding on the gene loci of Foxp3 and Fbxw7 in mouse Th2 cells versus Th0 cells cultured for 3 days.
- ChIP‐qPCR analysis of Brd4, Ezh2, Suz12, and EED binding on the gene loci of Il4, Il5, Foxp3, and Fbxw7 in mouse Th2 cells treat with or without JQ1 (250 nM).
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All Western blotting data are representative of three independent experiments. All ChIP‐qPCR data represent mean ± SD and are representative of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
Figure EV5. Brd4‐BD2 binds to acetylated‐EED to repress Foxp3 and Fbxw7 in Th2 cells.
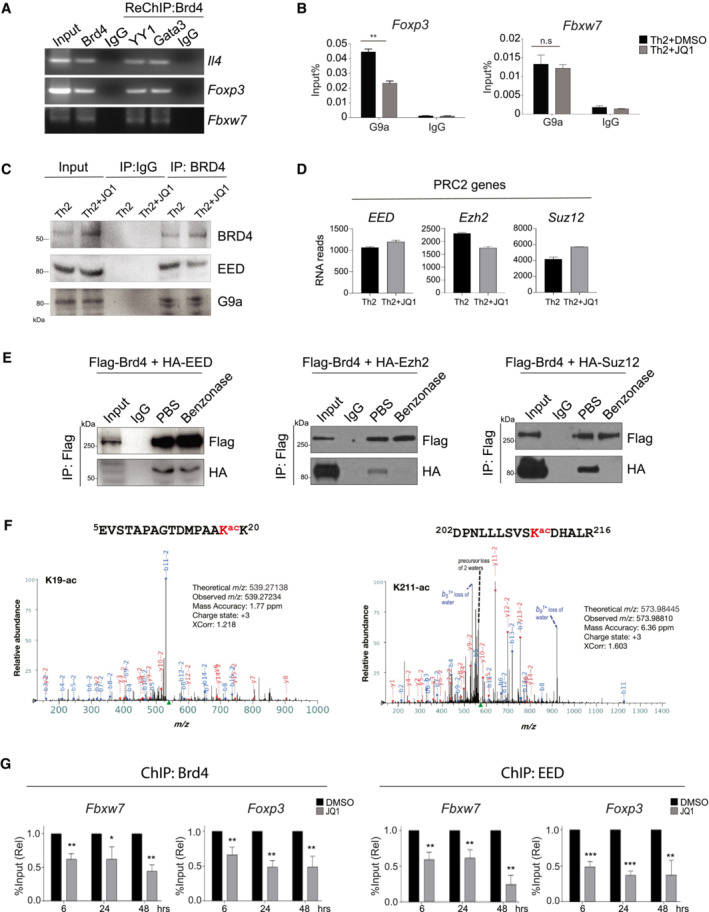
- ChIP‐PCR of the binding of Brd4 and Re‐ChIP‐PCR of YY1 and Gata3 on the gene loci of Il4, Foxp3, and Fbxw7 in mouse Th2 cells.
- ChIP‐qPCR analysis of the binding of G9a on the gene loci of Foxp3 and Fbxw7 in mouse Th2 cells treat with or without JQ1 (250 nM).
- Th2 cells were differentiated for 6 days, treated with JQ1 (250 nM) on Day 0. Lysates are immunoprecipitated with BRD4 followed by Western blotting analysis of BRD4, EED, and G9a.
- RNA‐seq data analysis showing expression of PRC2 key genes in mouse Th2 cells treated with or without JQ1 (250 nM). Data are representative of two biological replicates.
- HEK293T cells are transfected with Flag‐Brd4 and HA‐EED (left), HA‐Ezh2 (middle), or HA‐Suz12 (right) plasmids. Lysates are immunoprecipitated with Flag‐tagged Brd4, treated in vitro with PBS and Benzonase, followed by Western blotting of HA to detect HA‐EED, HA‐Ezh2, and HA‐Suz12.
- HEK293T cells are transfected with Flag‐Brd4 and HA‐EED plasmids. Lysates are immunoprecipitated with HA, followed by liquid chromatography with tandem mass spectrometry (LC–MS/MS) analysis. MS/MS spectrum for the identified acetylated peptides containing Lysine acetylation at position K19 and K211. Peak heights show the relative abundance of the corresponding fragmentation ions. The peptide sequences are shown at the top of the corresponding MS/MS spectrum with acetylated lysine residues highlighted in red. The identified fragmentation y (red color) and b (blue color) ions are indicated.
- Th2 cells were differentiated for 3 days and then treated with DMSO and JQ1 (250 nM) for 6, 24 and 48 h, followed by ChIP‐qPCR analysis of binding of Brd4 and EED on Foxp3 and Fbwx7.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All Western blotting data are representative of three independent experiments. All ChIP‐qPCR data represent mean ± SD and are representative of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
Brd4 has been shown to work with G9a (histone lysine methyltransferase for H3K9) to suppress autophagy genes through an unknown mechanism (Sakamaki et al, 2017). To determine how Brd4 represses the transcription of Foxp3 and Fbxw7 in Th2 cells, we performed ChIP‐qPCR and found that G9a binds to the Foxp3 gene locus, and G9a chromatin occupancy was reduced by JQ1 treatment (Fig EV5B). However, G9a binding to the Fbxw7 promoter is independent of Brd4 (Fig EV5B), suggesting a different mechanism attributing to Brd4's repressor function in gene transcription. We also noted that Brd4–G9a interaction is bromodomain‐independent, as JQ1 did not disrupt their interaction in Th2 cells (Fig EV5C). Importantly, we observed reduced H3K27me3 on the regulatory regions of Foxp3 and Fbxw7 in Th2 cells treated with JQ1 (Fig 3C), suggesting potential association of Brd4 with the PRC2 complex, known for H3K27me3‐mediated gene repression. Using Co‐IP, we found that Brd4 interacts with G9a and PRC2 subunits (EED, Suz12, H3K27 methyltransferase Ezh2) (Knutson et al, 2012) in Th2 cells (Fig 3D). To gain direct evidence of Brd4–PRC2 interaction and determine whether their interaction is functionally important, we characterized and observed enhanced Brd4, Ezh2, Suz12, and EED binding to Foxp3 and Fbxw7 gene loci in Th2 cells, as compared to Th0 cells (Fig 3E). JQ1 treatment of Th2 cells had no effects on mRNA expression of EED, Suz12, and Ezh2 (Fig EV5D), but resulted in loss of Brd4 binding, leading to reduced EED, Suz12, and Ezh2 binding to Foxp3 and Fbxw7 gene loci (Fig 3F). Interestingly, Brd4 recruitment of the PRC2 complex is only limited to repressed genes, but not Il4 and Il5 (Fig 3F). Collectively, we established that Brd4 represses Foxp3 and Fbxw7 through working with distinct transcriptional repressive complexes. Brd4 recruits both G9a and PRC2 to upregulate H3K9me3 and H3K27me3 levels on the Foxp3 while Brd4 recruits only PRC2 to upregulate H3K27me3 level on the Fbxw7 gene loci.
Brd4‐BD2 interacts with lysine‐acetylated‐EED for gene transcriptional repression
Using ectopic expression of PRC2 subunits EED, Ezh2, and Suz12, we showed that while they are all associated with Brd4, Brd4–EED interaction is the most robust (Fig 4A). EED is known to bind H3K27me3 through its WD40 domain at target gene loci, and the physical presence of EED and Suz12 is required for the catalytic activity of Ezh2 (van Mierlo et al, 2019). Importantly, we found that Brd4 interacts with EED through its BD2, as JQ1 or Brd4‐BD2 inhibitor ABBV‐744 (Faivre et al, 2020) but not Brd4‐BD1 inhibitor MS402 (Cheung et al, 2017a) disrupted the interaction (Fig 4B). Brd4–Ezh2 and Brd4–Suz12 interactions with ectopic expression appeared to be independent of bromodomains (Fig 4B), yet sensitive to benzonase treatment (Fig EV5E). Notably, Brd4–EED interaction is insensitive to benzonase treatment, suggesting that BRD4 interacts with PRC2 subunits through distinct mechanisms (Fig EV5E). In Th2 cells, ABBV‐744 disrupts Brd4–EED interaction but not Brd4–Suz12 and Brd4–Ezh2 interactions (Fig 4C). These data suggest that Brd4‐BD2 interacts with lysine‐acetylated‐EED, while Brd4–Ezh2 and Brd4–Suz12 interactions are possibly through DNA/RNA‐mediated mechanism. Notably, we found EED that binds Brd4‐BD2 to be lysine‐acetylated (Fig 4D). Since the acetylation sites of EED and their functions were never reported, we searched PhosphoSitePlus® (Hornbeck et al, 2019), a knowledge base of curated information on post‐translational modifications of proteins, and revealed that K19, K211, K250, K261 of EED as putative acetylation sites. We confirmed that EED‐K19 and ‐K211 were indeed acetylated in HEK293 cells using LC–MS/MS analysis (Fig EV5F). Importantly, we observed that K19‐acetylated EED interacts with Brd4, as an EED‐K19ac peptide, but not other acetylated peptides or nonacetylated EED‐K19 peptide, disrupts the Brd4–EED interaction (Fig 4E). In addition, using ectopic expression of Flag‐Brd4, HA‐EED, and HA‐EED(K19R) mutant, we confirmed that acetylation of K19 residue of EED is required for Brd4 interaction (Fig 4F). We introduced the EED(K19R) mutant into our established luciferase system to address the importance of this interaction for gene transcriptional repression. We showed that expression of BRD4 + EED synergistically repressed Fbxw7 promoter activity, which was rescued by EED(K19R) mutant (Fig 4G). Finally, we performed kinetic studies of the binding of BRD4 and EED to the regulatory regions of Foxp3 and Fbxw7. Th2 cells differentiated for 3 days were treated with BRD4 inhibitor JQ1, and the loss of BRD4 and EED binding to the regulatory regions of Foxp3 and Fbxw7 correlated with their de‐repression 6–24 h post‐treatments (Figs EV4F and G, and EV5G). Collectively, our results demonstrated that Brd4 through its BD2 binds to lysine‐acetylated‐EED at K19, maintaining an active PRC2 complex on target gene loci for transcriptional repression.
Figure 4. Brd4‐BD2 binds acetylated‐EED to repress Foxp3 and Fbxw7 in Th2 cells.
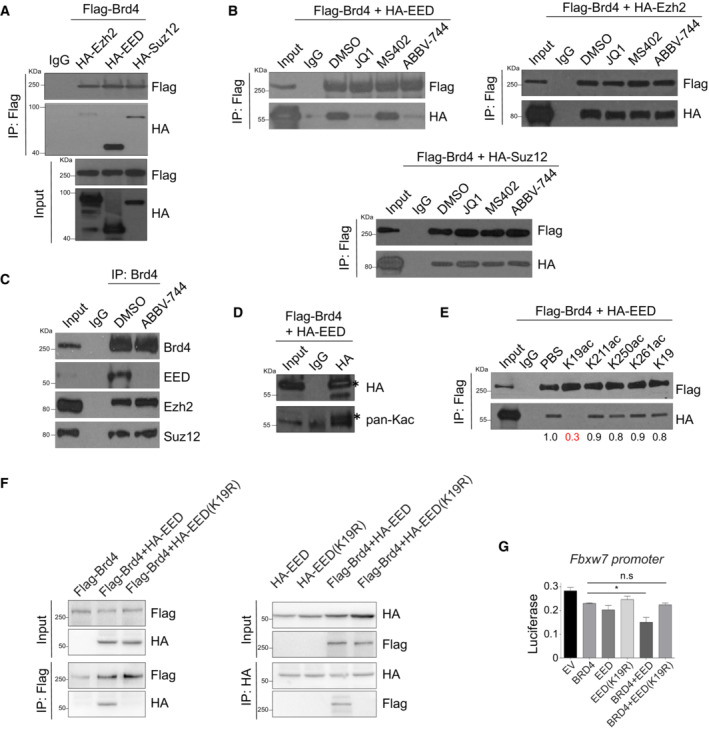
- Immunoprecipitation of Flag‐tagged Brd4, followed by Western blotting of HA to detect HA‐Ezh2, HA‐EED, and HA‐Suz12 in HEK293T cells transfected with Flag‐Brd4 and/or HA‐Ezh2, HA‐EED, and HA‐Suz12 plasmids.
- HEK293T cells are transfected with Flag‐Brd4 and HA‐EED, HA‐Ezh2, and Suz12 plasmids. Lysates are immunoprecipitated with Flag‐tagged Brd4, treated in vitro with JQ1 (5 μM), MS402 (5 μM), and ABBV‐744 (5 μM), followed by Western blotting of HA.
- Th2 cells were differentiated for 6 days. Lysates are immunoprecipitated with Brd4, treated in‐vitro with ABBV‐744 (5 μM), followed by Western blotting of EED, Ezh2, and Suz12.
- HEK293T cells are transfected with Flag‐Brd4 and HA‐EED plasmids. Lysates are immunoprecipitated with HA, followed by Western blotting of HA and pan‐acetylation (pan‐Ac). * denotes specific bands.
- HEK293T cells are transfected with Flag‐Brd4 and HA‐EED plasmids. Lysates are immunoprecipitated with Flag‐tagged Brd4, treated in vitro with 10 μM acetylated EED peptides (K19, K211, K250, K261) and nonacetylated EED‐K19 peptide, followed by Western blotting of HA to detect HA‐EED.
- HEK293T cells are transfected with Flag‐Brd4, HA‐EED, and HA‐EED(K19R) plasmids. Lysates are immunoprecipitated with Flag followed by Western blotting of HA, and vice versa.
- Dual‐luciferase reporter assay of HEK293T cells transfected with Fbxw7‐promoter, Flag‐Brd4, HA‐EED, and HA‐EED(K19R) plasmids.
Data information: All Western blotting data are representative of three independent experiments. All data represent mean ± SD and are representative of three independent experiments. Data are analyzed by Paired t test. *P < 0.05.
Source data are available online for this figure.
Brd4 functions through its BD2 to regulate gene transcription in Th2 cells
Our results suggest Th2 cell development is dependent on BRD4‐BD2 domain, offering therapeutic strategies to more selectively target Th2‐related inflammatory disorders. To determine the specific role of its individual BrDs in Brd4‐mediated gene transcriptional control in Th2 cells, we compared pan‐BET inhibitor JQ1, selective Brd4‐BD1 inhibitors MS611 (Gacias et al, 2014) and MS402 (Cheung et al, 2017a), and Brd4‐BD2 inhibitor ABBV‐744 (Faivre et al, 2020). Note that JQ1 but not MS402 treatment, leads to reduced mRNA expression of Il4, Il5, and Il13 as a result of diminished Brd4 binding to the cis‐regulatory regions of the genes (Fig EV6A–C). Consistently, we found that inhibition of Brd4 BD2 but not BD1 resulted in abolishment of IL‐4 expression during Th2 cell differentiation (Fig 5A).
Figure EV6. ChIP‐qPCR analysis of Brd2 and Brd4 on Il4/Il5/Il13 promoter treated with or without JQ1 and MS402.
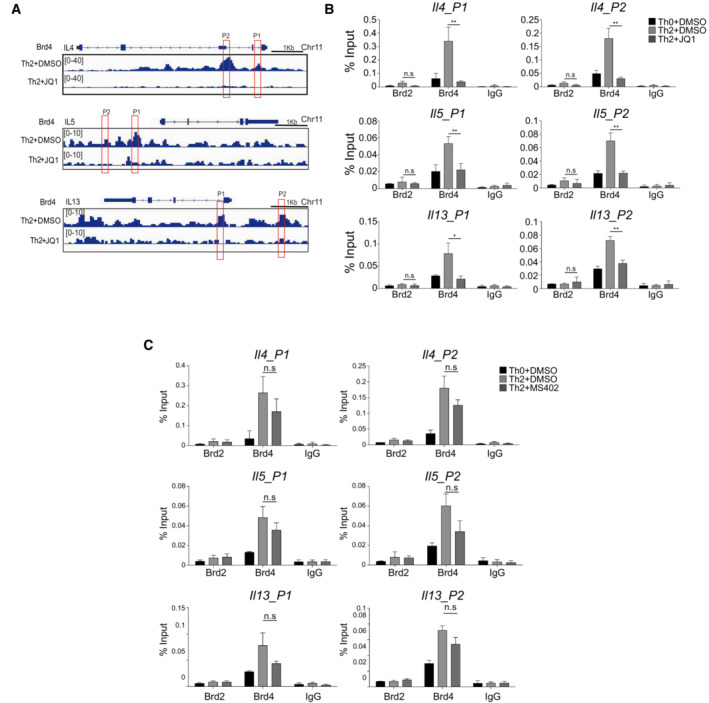
- ChIP‐seq tracks of Brd4 occupancy at the promoters of Il4, Il5, and Il13 in mouse Th2 cells treated with or without JQ1 (250 nM).
- ChIP‐qPCR analysis of Brd2 and Brd4 on Il4/Il5/Il13 promoter treated with or without JQ1 (250 nM).
- ChIP‐qPCR analysis of Brd2 and Brd4 on Il4/Il5/Il13 promoter treated with or without MS402 (3 μM).
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All data represent mean ± SD and are representative of two independent experiments. *P < 0.05; and **P < 0.01.
Figure 5. Brd4 through its BD2 domain regulates transcription of Th2‐key genes and ‐negative regulators.
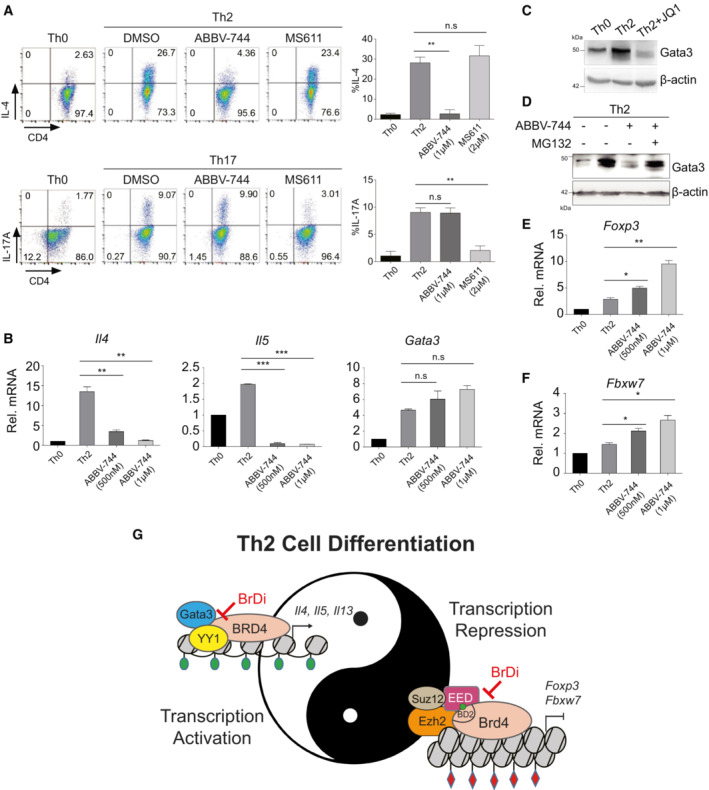
- Flow cytometric (left) and statistical analysis (right) of IL‐4 and IL‐17A in mouse Th2 and Th17 cells treated with BRD4 BD2‐selective inhibitor ABBV‐744 (1 μM) and BD1‐selective inhibitor MS611 (2 μM), respectively.
- qPCR analysis of Il4, Il5, Gata3 in mouse Th2 cells treated with ABBV‐744 at 500 nM and 1 μM.
- Western blotting of Gata3 in mouse Th2 cells treated with or without JQ1.
- Western blotting of Gata3 of mouse Th2 cells treated with ABBV‐744 (500 nM) and MG132 (20 μM).
- qPCR analysis of Foxp3 in mouse Th2 cells treated with ABBV‐744 at 500 nM and 1 μM.
- qPCR analysis of Fbxw7 in mouse Th2 cells treated with ABBV‐744 at 500 nM and 1 μM.
- Schematic diagram illustrating BRD4 functions in control of transcriptional activation and repression of key genes in chromatin that functionally cooperate with each other to regulate Th2 cell program. Histone lysine acetylation and methylation are depicted with color‐coded flags in green or red, respectively.
Data information: Mouse naïve CD4+ T cells were cultured in Th2 polarization condition and treated with or without inhibitors on Day 0 and were differentiated for 6 days before analysis, unless otherwise specified. All data represent mean ± SD and are representative of three independent experiments. Data are analyzed by Paired t test. *P < 0.05; **P < 0.01; and ***P < 0.001.
Source data are available online for this figure.
Notably, the mRNA levels of Th2 cytokines Il4 and Il5 were significantly reduced in Th2 cells treated with ABBV‐744 (Fig 5B). ABBV‐744 slightly induced the level of Gata3 mRNA, but not statistically significant (Fig 5B). In agreement with Th2 cells treated with JQ1, ABBV‐744 abolished Gata3 protein expression, which could be rescued by MG132 treatment (Fig 5C and D), and upregulated Foxp3 and E3‐ubiqutin ligase Fbxw7 expression (Fig 5E and F). Collectively, these results support the notion that Brd4‐BD2 is involved in and responsible for transcriptional activation of Th2 key genes Il4, Il5 as well as transcriptional repression of Foxp3 and Fbxw7 during Th2 cell differentiation. Our finding that Foxp3 was increased in Th2 cells by BD2 inhibition with JQ1 or ABBV‐744 highlights that Brd4 acts via its BD2 for transcriptional repression of Foxp3. Further study of how BRD4‐Foxp3 axis regulates Th2 differentiation will guide development of a new therapeutic strategy for Th2‐associated diseases such as allergies, asthma, and cancer.
Discussion
In this study, we uncovered previously unrecognized mechanistic insights into the multifaceted functions of BRD4 in gene transcriptional regulation of Th2 cell lineage‐specific differentiation. Unlike BRD4 in Th17 cells whose differentiation can be selectively blocked by BRD4 BD1‐specific inhibitor MS402 (Cheung et al, 2017a), we showed that BRD4 works with transcription factors Gata3 and YY1 to promote transcriptional activation of key Th2 cytokine genes Il4, Il5, and Il13, as well as recruits the PRC2 complex through its BD2 to exert transcriptional repression of Th2‐negative regulators Foxp3 and E3‐ubiquitin ligase Fbxw7, which in turn facilitates expression of Il4, Il5, and Il13 and ensures protein stability of Th2 transcription factor Gata3, respectively (Fig 5G). Thus, our study established that BRD4‐BD2 but not BD1 is required for Th2 cell differentiation, thereby highlighting the distinct functions of BD1 and BD2 in transcriptional regulation of different Th cells.
BRD4 is a key gene transcriptional co‐activator that promotes transcriptional elongation and is implicated in cancer and inflammatory disorders. However, how BRD4 directs gene transcriptional repression in chromatin in a cell type‐specific manner has remained a major outstanding question. We discovered that BRD4 recruits Polycomb repressor complex 2 (PRC2) to promote H3K27me3 and repress transcriptional expression of negative regulators of Gata3‐directed transcriptional program in a lineage‐specific differentiation of Th2 cells. Our discovery of BRD4–BD2 interaction with lysine‐acetylated‐EED and validation of the selective requirement of BRD4‐BD2 domain in Th2 cell development suggests a new therapeutic strategy to selectively treat Th2‐associated airway inflammation.
Importantly, our study presents a likely generalizable mechanism of how a major transcriptional regulator protein such as BRD4, through different molecules of the same protein, can perform distinct molecular functions to regulate different aspects of histone‐mediated gene transcriptional program to ensure a faithful cell transition from one state to another such as Th2 cell lineage‐specific differentiation from naïve CD4+ T cells.
In summary, our new mechanistic findings provide much needed knowledge to illustrate previously underappreciated BRD4's role in gene transcriptional repression (Barrow et al, 2016; Sakamaki et al, 2017), and also appreciate the functional versatility to BRD4‐BD2‐dependent interactions with transcription factors or transcription repressors in control of gene transcription in chromatin. Our study further highlights the sophisticated molecular functions of BRD4 in the complex regulation of coordinated, cell type‐dependent transcriptional activation of positive factors and repression of negative regulators that together ensures faithful lineage‐specific differentiation of Th2 cells.
Materials and Methods
Reagents and Tools table
| Reagent/Resource | Reference or Source | Identifier or Catalog Number |
|---|---|---|
| Antibodies | ||
| PE anti‐mouse FOXP3 antibody | eBioscience | 12‐5773‐82 |
| APC anti‐mouse CD4 antibody | Biolegend | 100412 |
| PE anti‐mouse CD4 antibody | Biolegend | 100408 |
| BV421‐GATA3 antibody | Biolegend | 653814 |
| FITC anti‐mouse CD4 antibody | Biolegend | 100406 |
| APC anti‐mouse IFN‐γ antibody | Biolegend | 505810 |
| APC‐anti‐mouse IL‐17A antibody | Biolegend | 506916 |
| APC anti‐mouse CD25 antibody | Biolegend | 101910 |
| APC anti‐mouse IL‐4 antibody | Biolegend | 504106 |
| Anti‐BRD2 antibody | Bethyl | A302‐583A |
| Anti‐BRD3 antibody | Bethyl | A302‐368A |
| Anti‐BRD4 antibody | Bethyl | A301‐985A100 |
| Anti‐acetyl‐histone H4 antibody | Millipore | 06‐886 |
| Anti‐acetylated histone H3 antibody | Millipore | 06‐599 |
| Anti‐H3K27ac antibody | Abcam | ab4729 |
| Anti‐acetyl lysine antibody | Abcam | ab22550 |
| Anti‐acetyl‐lysine antibody | Cell Signaling | 9441 |
| Anti‐histone H3 (tri methyl K9) antibody | Abcam | ab8898 |
| Anti‐histone H3 (di methyl K9) antibody | Abcam | ab1220 |
| Anti‐histone H3 (mono methyl K9) antibody | Abcam | ab8896 |
| Anti‐histone H3 (acetyl K9) antibody | Abcam | ab4441 |
| Anti‐histone H3 (tri methyl K4) antibody | Abcam | ab8580 |
| Anti‐histone H3 (tri methyl K27) antibody | Abcam | ab6002 |
| Anti‐Foxp3 antibody | Abcam | ab215206 |
| Anti‐Fbxw7 anribody | Abcam | ab109617 |
| Anti‐Gata3 antibody | Abcam | ab282110 |
| Anti‐Ubiquitin antibody | Abcam | ab7780 |
| Anti‐EED antibody | Thermo | MA5‐35927 |
| Anti‐EED antibody | Abcam | ab240650 |
| Anti‐EED antibody | Cell Signaling | 51673 |
| Anti‐SUZ12 antibody | Abcam | ab175187 |
| Anti‐SUZ12 antibody | Cell Signaling | 3737 |
| Anti‐Ezh2 antibody | Abcam | ab191250 |
| Anti‐Ezh2 antibody | Cell Signaling | 5246 |
| Anti‐G9a antibody | Abcam | ab185050 |
| Anti‐Myc antibody | CMCTAG | AT0023 |
| Anti‐Myc antibody | Cell signaling | 2276 |
| Anti‐Flag antibody | CMCTAG | AT0022 |
| Anti‐Flag antibody | Sigma | F3165 |
| Anti‐HA antibody | Engibody | AT0024 |
| Anti‐HA antibody | Cell Signaling | 3724 |
| Anti‐β‐Actin antibody | CMCTAG | AT0001 |
| Anti‐β‐Actin antibody | Santa Cruz | sc‐47778 |
| Oligonucleotides and sequence‐based reagents | ||
| mIl4 | Antisense | CCATATCCACGGATGCGACA |
| Sense | CGTTGCTGTGAGGACGTTTG | |
| mIl5 | Sense | TGAGGCTTCCTGTCCCTACT |
| Antisense | CCCCCACGGACAGTTTGATT | |
| mIl13 | Sense | CTGTGTCTCTCCCTCTGACC |
| Antisense | CTGGGTCCTGTAGATGGCAT | |
| mFbxw7 | Sense | ATCGACTGACCGGACTCTCA |
| Antisense | CGAGAACCGCTTACAACCCT | |
| mNr4a2 | Sense | TGCGCTTAGCATACAGGTCC |
| Antisense | GCATTGCAACCTGTGCAAGA | |
| mGata3 | Antisense | AAAGAAGGCATCCAGACCCG |
| Sense | TTGAAGGAGCTGCTCTTGGG | |
| mFoxp3 | Sense | TTCTCCAGGACAGACCACACT |
| Antisense | GACCCCAGTGGCAGCAGAA | |
| mGapdh | Sense | AACTTTGGCATTGTGGAAGG |
| Antisense | GGATGCAGGGATGATGTTCT | |
| P‐mIL‐4 (‐971) | Sense | GGGCAATGAGTACCTCGACA |
| Antisense | TCTCACCCTCTCAGTCAGCA | |
| P‐mIL‐4 (‐1958) | Sense | CGAAGGATCAGCTTCAGAGC |
| Antisense | GGCTTGGTTTCCCTCTTCCA | |
| P‐mIL‐5 (‐18bp) | Sense | TTTCCTCAGAGAGAGAATAAATTGCTT |
| Antisense | GCTGGCCTTCAGCAAAGG | |
| P‐mIL‐5 (‐1004) | Sense | AAATGCGAACCTGCTGTGTG |
| Antisense | GTGTATGAGGGGTGCTCCAG | |
| P‐mIL‐13 (‐206) | Sense | ACCCAGAACCTGGAAACCCT |
| Antisense | GTGGCCGCTAAAGGAAAGAGT | |
| P‐mIL‐13 (‐1132) | Sense | GAAACGGGTGGGGAAAGAGT |
| Antisense | GATGCACCCTCACCCACTTC | |
| mus Fbxw7 Pomoter p1 | Sense | GCCACTCTCCTCCCGCCCCTCTGT |
| Antisense | CACTCACTCGCTCTCACGCTCTCC | |
| mus Fbxw7 Promoter p2 | Sense | CCCAGCAGCGCGGAAGAGACC |
| Antisense | CTGGCTAGGCGCGGAACTGAGGAA | |
| mus Foxp3 Promoter p1 | Sense | GGGAACGGGGCACGGCGGAGAG |
| Sense‐3 | CGGCGGTGGCGGGGCTGATG | |
| mus Foxp3 Promoter p2 | Sense | CGGGAGTGACGTACAGGTGATTGA |
| Antisense | AGGCCTCTCCATTGATTCCGTTAT | |
| sh‐Brd2 | GGATTCGAAAGCCTTCTCTGC | |
| sh‐Brd3 | GCGTGTGGTGCACATCATTCA | |
| sh‐Brd4 | GGATGAAGATGCGCTGGAACA | |
| sh‐Foxp3 | CCACCTGGAAGAATGCCAT | |
| sh‐Fbxw7 | GCAGCAAGCGAGACATCAA | |
| sh‐Control | TTCTCCGAACGTGTCACGT | |
Methods and Protocols
Mice
C57BL/6 mice were bought from Beijing Vital River. Rag1−/− mice were obtained from Model Animal Research Center of Nanjing University. All animals were housed and maintained in a conventional pathogen‐free facility at the 1st Hospital of Jilin University.
In vitro differentiation of CD4 + T cells
Single‐cell suspensions were prepared from lymph nodes and spleens of the C57BL/6 mice. Naïve CD4+ T cells were purified with naïve CD4+ T Cell Isolation Kit (Miltenyi Biotec, 130‐104‐453) and cultured in RPMI‐1640 medium (Gibco) supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 U/ml of streptomycin, 10 mM HEPES buffer, and 50 μM β‐mercaptoethanol. Cells were activated by plate bound anti‐CD3 5 μg/ml (2c11, Biolegend), and soluble anti‐CD28 1 μg/ml (37.51, Biolegend). Th0 cells were cultured in the presence of 20 ng/ml IL‐2 (Peprotec). Th1 cells were cultured with IL‐12 (20 ng/ml) and anti‐IL4 antibody. Th2 cells were cultured with IL‐2 (20 ng/ml), IL‐4 (50 ng/ml) and anti‐IFNγ antibody. Th17 cells were cultured with IL‐6 (20 ng/ml), TGF‐β (2.5 ng/ml), anti‐IFNγ, and anti‐IL‐4 antibodies. Treg cells were cultured with TGF‐β (10 ng/ml) and IL‐2 (20 ng/ml). Th2 cells were induced for 6 days, and other Th cells were induced for 3 days.
Cell culture, plasmid construction, transfection, and retroviral transduction
HEK293T cells were cultured in DMEM (Gibco) supplemented with 10% fetal calf serum, 100 U/ml of penicillin, 100 U/ml of streptomycin. Flag‐Brd4, HA‐Ezh2, HA‐EED, and HA‐Suz12 plasmids were purchased from Addgene. The cDNA encoding Foxp3 was cloned by PCR and ligated to pWPXLd expression vector. Lentiviral vectors encoding murine Brd2, Brd4, Foxp3, Fbxw7, or negative control shRNA were ligated with BamHI and EcoRI into the pLVshRNA‐EGFP‐Puro. PEI were used for transfection in HEK293T cells. For lentivirus‐mediated gene transfer to naïve CD4+ T cells, HEK293T cells were transfected with lentiviral plasmids. After 6 h, culture supernatant containing lentivirus was collected 48 and 72 h after transfection. The supernatant was filtrated and concentrated, supplemented with 5 μg/ml polybrene and were used to spin‐infect naïve CD4+ T cells activated with anti‐CD3 and anti‐CD28. Spin infection was performed at 500 g for 90 min at room temperature. Cells were then cultured at 37°C for 4 h, and cytokines were added for polarization.
Chromatin immunoprecipitation
Cells were chemically cross‐linked with 1% formaldehyde solution for 10 min at room temperature followed by the addition of 2.5 M glycine (to a final concentration of 125 mM) for 5 min. Cells were rinsed twice with cold 1×PBS and then lysed in Szak's RIPA buffer (150 mM NaCl, 1% Nonidet P‐40, 0.5% deoxycholate, 0.1% SDS, 50 mM Tris–HCl pH 8, 5 mM EDTA, Protease Inhibitor Cocktail (Roche), 10 mM PMSF). Cells were then sonicated using a sonicator (Thermo) for 15 pulses of 15 s at a power of 30%, followed by 1 min rest on ice. Sonicated chromatin was cleared by centrifugation. The resulting chromatin extract was incubated overnight at 4°C with appropriate primary antibodies and 20 μl of Protein G/A magnetic beads (Dynabeads, Life Technologies). Beads were washed two times with incomplete Szak's RIPA buffer (without PMSF and Protease Inhibitor cocktail), four times with Szak's IP Wash Buffer (100 mM Tris HCl pH 8.5, 500 mM LiCl, 1% Nonidet P‐40, 1% deoxycholate), then twice again with incomplete RIPA buffer and twice with cold 1× TE. Complexes were eluted from beads in Talianidis Elution Buffer by heating at 65°C for 30 min and then by adding NaCl to a final concentration of 200 mM and reverse crosslinking was performed overnight at 65°C. Input DNA was concurrently treated for crosslink reversal. Samples were then treated with RNaseA and Proteinase K for an hour, extracted with Phenol/Chloroform and ethanol precipitated. The pellet was resuspended in water and used for subsequent ChIP‐seq library preparation or analyzed by qPCR. For re‐ChIP, the elute from the first ChIP were incubated in 200 μl TE (with 10 mmol/L DTT) at 37°C for 30 min and then diluted with RIPA buffer, followed by re‐ChIP with the second antibody, washes, elution, and DNA isolation.
Analysis of RNA‐Seq, ChIP‐Seq, and ATAC‐seq
RNA was extracted from Th2 cells using RNeasy Kits (Qiagen), followed by polyA selection, library preparation and Bioanalyzer Quality Control (QC). Libraries were sequenced with the average depth of 20 million paired end reads per sample. Adaptor was trimmed with Cutadapt and low‐quality reads (Q30) were filtered out before aligning with STAR to the reference genome mm10. Raw counts were calculated with featureCounts. DEseq2 was used to calculate differential expression genes, Gene‐ontology over‐representation analysis on DEGs (P < 0.05) was conducted using clusterProfiler. SimpleChIP Kit (Cell Signaling, #CST 9003 S) was used to generate Chip libraries, followed by Bioanalyzer QC, sequencing with Novaseq6000, and 40million pair‐end reads per sample were generated. Encode ChIP‐seq processing pipeline (chip‐seq‐pipeline2 v1.3.5.1) was used for data processing. In brief, raw read was trimmed and low‐quality reads (Q30) were filtered out along with mitochondrial reads and alignment was done with bowtie2 using mm10 reference genome. MACS2 for histone ChIP raw peaks and SPP for transcription factor ChIP raw peak at P‐value threshold of 0.01 were used. Irreproducible Discovery Rate (IDR) peak set (IDR P‐value threshold 0.05) and IGV track files were generated and pooled with idr and BEDTools. Annotation of genomic location of binding sites was achieved with R package ChIPseeker. Motif analysis was performed with HOMER package and peak comparison was done with deepTools package computeMatrix. Publicly available ATAC‐seq data were downloaded and processed with ENCODE ATAC pipeline (version 2.2.0) using standard approaches. ChIP‐seq and RNA‐seq data in this study are deposited in SRA database with accession number PRJNA798535. Gata3 ChIP‐seq data used in this study can be found in GEO dataset with accession number GSE28292. ATAC‐seq data used in this study can be found in GEO dataset with accession number GSE157597.
FACS staining
For cytokine analysis, cells were incubated for 6 h with PMA (50 ng/ml; Sigma), ionomycin (500 ng/ml; Sigma), and BFA (Biolegend). Intracellular cytokine staining was performed according to the manufacturer's protocol (FoxP3 staining buffer set from eBioscience). LSR II flow cytometer (BD Biosciences) and FlowJo (Tree Star) software were used for flow cytometry and analysis. Dead cells were excluded using the Live/Dead fixable aqua dead cell stain kit (Invitrogen).
Co‐immunoprecipitation
Total cell extraction was incubated overnight at 4°C with appropriate primary antibodies and 20 μl of Protein G/A magnetic beads (Dynabeads, Life Technologies). Beads were washed three times with Buffer A (20 mM Tris PH 8.0, 10 mM NaCl, 1 mM EDTA, 0.5% NP‐40). The precipitates were then subjected to SDS–PAGE followed by transfer onto a PVDF membrane and incubation with antibody. Samples were detected using the Enhanced ECL Chemiluminescent Substrate Kit (Yeasen) detection method in accordance with the manufacturer's instructions.
Western blot analysis
Cells were scraped and lyzed with RIPA lysis buffer (150 mM NaCl, 0.5% Triton X‐100, 50 mM Tris–HCl, pH 7.4, 25 mM NaF, 20 mM EGTA, 1 mM DTT, 1 mM Na3VO4, with protease inhibitor cocktail tablet) for 30 min on ice followed by centrifugation at 14,800 g for 15 min. The protein concentration of the supernatant was measured by using the BCA reagent. Protein (20 μg) was loaded on 4–15% Tris‐Glycine gels and transferred onto polyvinylidenedifluoride (PVDF) membrane in Tris–glycine buffer (pH 8.4) containing 20% methanol. The membrane was then blocked in 5% fat‐free dry milk in phosphate‐buffered saline with 0.1% Tween‐20 (PBS‐T) for 1 h and then probed with primary antibodies and horseradish peroxidase‐conjugated secondary antibody by standard Western blotting procedures. Densitometry analyses were analyzed with ImageJ.
Enzyme‐linked immunosorbent assay (ELISA)
All ELISA kits were purchased from eBioscience and experiments were performed according to protocols provided by the manufacturer. Briefly, supernatants of samples were incubated in plates coated with capture‐antibody. Detection antibody was added after a total of five washes. Avidin‐HRP was then added after a total of five washes. Plates were read at 450 nm after addition of substrate solution and stop solution. Concentration of the cytokines in samples was calculated with reference to the absorbance value obtained from the standard curve.
In‐gel digestion and mass spectrometry analysis
The corresponding protein bands containing EED proteins on the SDS–PAGE gel were excised, chopped into small pieces and transferred to 1.7 ml Maximum recovery microcentrifuge tubes and subjected to in‐gel trypsin digestion. In brief, the gel pieces were de‐stained by 25 mM ammonium bicarbonate in 50% acetonitrile. The proteins in the gel pieces were reduced by dithiothreitol (DTT), alkylated by iodoacetamide (IAA), and then subjected to overnight trypsin (working concentration 10 ng/ml) digestion at 37°C. Tryptic peptides were extracted, lyophilized, resuspended in 40 μl of 5% formic acid, and further processed on C18 resin, using handmade StageTips. Peptides were eluted with 5% formic acid, 50% acetonitrile, lyophilized with a speed‐vac, reconstituted in 0.1% trifluoroacetic acid, 2% acetonitrile, and subjected to LC–MS/MS analysis. LC/MS/MS analyses were performed on an Orbitrap Eclipse™ Tribrid™ Mass Spectrometer (Thermo Scientific). Peptides were injected onto a 75 mm 6,150 mm BEH C18 column (particle size 1.7 mm, Waters) and separated using a Waters nanoACQUITY Ultra Performance LCTM (UPLCTM) System (Waters, Milford, MA). The Orbitrap Eclipse™ Tribrid™ Mass Spectrometer was operated in the data‐dependent mode using the TOP20 strategy. Selected ions were dynamically excluded for 30 s. Singly charged ions were excluded from MS/MS analysis. MS/MS spectra were subjected to SEQUEST search analysis against a small database containing the human EED protein sequence and its reverse sequence. Search parameters allowed two missed tryptic cleavages, a mass tolerance of < 10 ppm for precursor ion, and a mass tolerance of < 0.8 Da for product ions, a static modification of 57.02146 Da (carboxyamidomethylation) on cysteine, and a dynamic modification of 42.01056 (acetylation) on lysine or 15.99491 Da (oxidation) on methionine, respectively. Search results were manually verified to check if most of the major peaks in the MS/MS spectra can be accounted for and annotated.
Induction of acute allergic lung inflammation model
Th2 cells were started to prepare 1 week before the induction of the acute allergic lung inflammation model. Naïve CD4+ T cells from 6 to 8 weeks C57BL/6 mice were purified and activated with ConA (Sigma) for 1 day. On the second day, cells were washed with PBS for one time, and then infected with sh‐Ctrl and sh‐Brd4 lentivirus (50ul virus every 1 × 106 cells, and added 6 μl polybrene), incubate at 37°C for 4 h, followed by the addition of IL‐2 20 ng/ml and IL‐4 50 ng/ml, ConA 5ug/ml to induce Th2 cells. New medium and cytokines were replenished on the 5th day and the cells were cultured for three more days. Th2 cells infected with sh‐Ctrl and sh‐Brd4 were sorted with flow cytometry to purify cells expressing GFP indicating infection with virus. Cells were diluted in 2 × 106/ml in PBS and injected via the tail vein into Rag1−/− female mice (2 × 106/mice). Starting from the second day, Rag1−/− mice were challenged for 30 min daily for 6 days with aerosolized 5% OVA (Sigma) via the airways. All the mice were sacrificed for analyses upon final challenge. BAL lavage was collected using a 1 ml injection syringe to cannulas from the trachea with PBS. Cells in the BAL were counted and stained with Diff‐Quick staining kit (Yeasen) and OVA‐specific IgE was measured using ELISA kits (eBiosciecne). Lungs of OVA‐stimulated mice were fixed in 4% paraformaldehyde and analyzed with H&E and PAS staining.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
PDF+
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Acknowledgments
This work was supported by research grants from the Crohn's and Colitis Foundation (579304; KLC) and National Natural Science Foundation of China and Science Foundation of Jilin Province (81601409, 20180520135JH; LZ).
Author contributions
Li Zhao: Conceptualization; writing – review and editing. Yiqi Wang: Data curation; formal analysis; validation. Anbalagan Jaganathan: Data curation; formal analysis; validation. Yifei Sun: Data curation; formal analysis; validation; methodology. Ning Ma: Data curation. Ning Li: Data curation. Xinye Han: Data curation. Xueying Sun: Data curation. Huanfa Yi: Data curation. Shibo Fu: Data curation. Fangbin Han: Data curation. Xue Li: Data curation. Kunhong Xiao: Data curation. Martin J Walsh: Data curation. Lei Zeng: Data curation; funding acquisition; validation. Ming‐Ming Zhou: Conceptualization; data curation; supervision; validation; methodology; writing – review and editing. Ka Lung Cheung: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing.
The EMBO Journal (2023) 42: e111473
Contributor Information
Li Zhao, Email: zhaol056@163.com.
Ming‐Ming Zhou, Email: ming-ming.zhou@mssm.edu.
Ka Lung Cheung, Email: kalung.cheung@mssm.edu.
Data availability
ChIP‐seq data: Sequence Read Archive PRJNA798535 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA798535).
RNA‐seq data: Sequence Read Archive PRJNA798535 (https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA798535).
References
- Abraham C, Medzhitov R (2011) Interactions between the host innate immune system and microbes in inflammatory bowel disease. Gastroenterology 140: 1729–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrow JJ, Balsa E, Verdeguer F, Tavares CD, Soustek MS, Hollingsworth LR, Jedrychowski M, Vogel R, Paulo JA, Smeitink J et al (2016) Bromodomain inhibitors correct bioenergetic deficiency caused by mitochondrial disease complex I mutations. Mol Cell 64: 163–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina AC, Nikolajczyk BS, Denis GV (2013) BET protein function is required for inflammation: Brd2 genetic disruption and BET inhibitor JQ1 impair mouse macrophage inflammatory responses. J Immunol 190: 3670–3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brusselle GG, Kips JC, Tavernier JH, van der Heyden JG, Cuvelier CA, Pauwels RA, Bluethmann H (1994) Attenuation of allergic airway inflammation in IL‐4 deficient mice. Clin Exp Allergy 24: 73–80 [DOI] [PubMed] [Google Scholar]
- Cheung K, Lu G, Sharma R, Vincek A, Zhang R, Plotnikov AN, Zhang F, Zhang Q, Ju Y, Hu Y et al (2017a) BET N‐terminal bromodomain inhibition selectively blocks Th17 cell differentiation and ameliorates colitis in mice. Proc Natl Acad Sci U S A 114: 2952–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KL, Zhang F, Jaganathan A, Sharma R, Zhang Q, Konuma T, Shen T, Lee JY, Ren C, Chen CH et al (2017b) Distinct roles of Brd2 and Brd4 in potentiating the transcriptional program for Th17 cell differentiation. Mol Cell 65: e1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn L, Elias JA, Chupp GL (2004) Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol 22: 789–815 [DOI] [PubMed] [Google Scholar]
- Conrad RJ, Fozouni P, Thomas S, Sy H, Zhang Q, Zhou MM, Ott M (2017) The short isoform of BRD4 promotes HIV‐1 latency by engaging repressive SWI/SNF chromatin‐remodeling complexes. Mol Cell 67: 1001–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou MM (1999) Structure and ligand of a histone acetyltransferase bromodomain. Nature 399: 491–496 [DOI] [PubMed] [Google Scholar]
- Faivre EJ, McDaniel KF, Albert DH, Mantena SR, Plotnik JP, Wilcox D, Zhang L, Bui MH, Sheppard GS, Wang L et al (2020) Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 578: 306–310 [DOI] [PubMed] [Google Scholar]
- Fang D, Cui K, Hu G, Gurram RK, Zhong C, Oler AJ, Yagi R, Zhao M, Sharma S, Liu P et al (2018) Bcl11b, a novel GATA3‐interacting protein, suppresses Th1 while limiting Th2 cell differentiation. J Exp Med 215: 1449–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I et al (2010) Selective inhibition of BET bromodomains. Nature 468: 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacias M, Gerona‐Navarro G, Plotnikov AN, Zhang G, Zeng L, Kaur J, Moy G, Rusinova E, Rodriguez Y, Matikainen B et al (2014) Selective chemical modulation of gene transcription favors oligodendrocyte lineage progression. Chem Biol 21: 841–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Lora JM (2016) Suppression of TH17‐mediated pathology through BET bromodomain inhibition. Drug Discov Today Technol 19: 39–44 [DOI] [PubMed] [Google Scholar]
- Guo J, Casolaro V, Seto E, Yang WM, Chang C, Seminario MC, Keen J, Georas SN (2001) Yin‐Yang 1 activates interleukin‐4 gene expression in T cells. J Biol Chem 276: 48871–48878 [DOI] [PubMed] [Google Scholar]
- Guo J, Lin X, Williams MA, Hamid Q, Georas SN (2008) Yin‐Yang 1 regulates effector cytokine gene expression and T(H)2 immune responses. J Allergy Clin Immunol 122: 195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammitzsch A, Tallant C, Fedorov O, O'Mahony A, Brennan PE, Hay DA, Martinez FO, Al‐Mossawi MH, de Wit J, Vecellio M et al (2015) CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci U S A 112: 10768–10773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho IC, Tai TS, Pai SY (2009) GATA3 and the T‐cell lineage: essential functions before and after T‐helper‐2‐cell differentiation. Nat Rev Immunol 9: 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Kornhauser JM, Latham V, Murray B, Nandhikonda V, Nord A, Skrzypek E, Wheeler T, Zhang B, Gnad F (2019) 15 years of PhosphoSitePlus(R): integrating post‐translationally modified sites, disease variants and isoforms. Nucleic Acids Res 47: D433–D441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosokawa H, Kimura MY, Shinnakasu R, Suzuki A, Miki T, Koseki H, van Lohuizen M, Yamashita M, Nakayama T (2006) Regulation of Th2 cell development by Polycomb group gene bmi‐1 through the stabilization of GATA3. J Immunol 177: 7656–7664 [DOI] [PubMed] [Google Scholar]
- Hwang SS, Kim YU, Lee S, Jang SW, Kim MK, Koh BH, Lee W, Kim J, Souabni A, Busslinger M et al (2013) Transcription factor YY1 is essential for regulation of the Th2 cytokine locus and for Th2 cell differentiation. Proc Natl Acad Sci U S A 110: 276–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R (2015) Control of adaptive immunity by the innate immune system. Nat Immunol 16: 343–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J et al (2012) A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol 8: 890–896 [DOI] [PubMed] [Google Scholar]
- Lambrecht BN, Hammad H (2015) The immunology of asthma. Nat Immunol 16: 45–56 [DOI] [PubMed] [Google Scholar]
- Lee GR, Fields PE, Flavell RA (2001) Regulation of IL‐4 gene expression by distal regulatory elements and GATA‐3 at the chromatin level. Immunity 14: 447–459 [DOI] [PubMed] [Google Scholar]
- Liu B, Liu X, Han L, Chen X, Wu X, Wu J, Yan D, Wang Y, Liu S, Shan L et al (2022) BRD4‐directed super‐enhancer organization of transcription repression programs links to chemotherapeutic efficacy in breast cancer. Proc Natl Acad Sci U S A 119: e2109133119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Mierlo G, Veenstra GJC, Vermeulen M, Marks H (2019) The complexity of PRC2 subcomplexes. Trends Cell Biol 29: 660–671 [DOI] [PubMed] [Google Scholar]
- Mosmann TR, Coffman RL (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7: 145–173 [DOI] [PubMed] [Google Scholar]
- Mujtaba S, Zeng L, Zhou MM (2007) Structure and acetyl‐lysine recognition of the bromodomain. Oncogene 26: 5521–5527 [DOI] [PubMed] [Google Scholar]
- Murphy KM, Reiner SL (2002) The lineage decisions of helper T cells. Nat Rev Immunol 2: 933–944 [DOI] [PubMed] [Google Scholar]
- Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, Murphy KM (2000) Stat6‐independent GATA‐3 autoactivation directs IL‐4‐independent Th2 development and commitment. Immunity 12: 27–37 [DOI] [PubMed] [Google Scholar]
- Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q et al (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6: 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul WE (2010) What determines Th2 differentiation, in vitro and in vivo? Immunol Cell Biol 88: 236–239 [DOI] [PubMed] [Google Scholar]
- Paul WE, Zhu J (2010) How are TH2‐type immune responses initiated and amplified? Nat Rev Immunol 10: 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puleston DJ, Baixauli F, Sanin DE, Edwards‐Hicks J, Villa M, Kabat AM, Kaminski MM, Stanckzak M, Weiss HJ, Grzes KM et al (2021) Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 184: 4186–4202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren C, Zhang G, Han F, Fu S, Cao Y, Zhang F, Zhang Q, Meslamani J, Xu Y, Ji D et al (2018) Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. Proc Natl Acad Sci U S A 115: 7949–7954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengarajan J, Szabo SJ, Glimcher LH (2000) Transcriptional regulation of Th1/Th2 polarization. Immunol Today 21: 479–483 [DOI] [PubMed] [Google Scholar]
- Sakamaki JI, Ryan KM (2017) Transcriptional regulation of autophagy and lysosomal function by bromodomain protein BRD4. Autophagy 13: 2006–2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamaki JI, Wilkinson S, Hahn M, Tasdemir N, O'Prey J, Clark W, Hedley A, Nixon C, Long JS, New M et al (2017) Bromodomain protein BRD4 is a transcriptional repressor of autophagy and lysosomal function. Mol Cell 66: 517–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder S, Cho S, Zeng L, Zhang Q, Kaehlcke K, Mak L, Lau J, Bisgrove D, Schnolzer M, Verdin E et al (2012) Two‐pronged binding with bromodomain‐containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J Biol Chem 287: 1090–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang Y, Zeng L, Wu Y, Deng J, Zhang Q, Lin Y, Li J, Kang T, Tao M et al (2014) Disrupting the interaction of BRD4 with Diacetylated twist suppresses tumorigenesis in basal‐like breast cancer. Cancer Cell 25: 210–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinnakasu R, Yamashita M, Kuwahara M, Hosokawa H, Hasegawa A, Motohashi S, Nakayama T (2008) Gfi1‐mediated stabilization of GATA3 protein is required for Th2 cell differentiation. J Biol Chem 283: 28216–28225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Cao C, Tang Y, Bi L, Jiang Y, Zhou Y, Song X, Liu L, Ge W (2018) The ubiquitin ligase SCF(FBXW7alpha) promotes GATA3 degradation. J Cell Physiol 233: 2366–2377 [DOI] [PubMed] [Google Scholar]
- Suehiro KI, Suto A, Suga K, Furuya H, Iwata A, Iwamoto T, Tanaka S, Kageyama T, Suzuki K, Hirose K et al (2020) Sox12 enhances Fbw7‐mediated ubiquitination and degradation of GATA3 in Th2 cells. Cell Mol Immunol 18: 1729–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Motomura Y, Suzuki Y, Yagi R, Inoue H, Miyatake S, Kubo M (2011) The enhancer HS2 critically regulates GATA‐3‐mediated Il4 transcription in T(H)2 cells. Nat Immunol 12: 77–85 [DOI] [PubMed] [Google Scholar]
- Yamashita M, Shinnakasu R, Asou H, Kimura M, Hasegawa A, Hashimoto K, Hatano N, Ogata M, Nakayama T (2005) Ras‐ERK MAPK cascade regulates GATA3 stability and Th2 differentiation through ubiquitin‐proteasome pathway. J Biol Chem 280: 29409–29419 [DOI] [PubMed] [Google Scholar]
- Zhang DH, Cohn L, Ray P, Bottomly K, Ray A (1997) Transcription factor GATA‐3 is differentially expressed in murine Th1 and Th2 cells and controls Th2‐specific expression of the interleukin‐5 gene. J Biol Chem 272: 21597–21603 [DOI] [PubMed] [Google Scholar]
- Zhang W, Prakash C, Sum C, Gong Y, Li Y, Kwok JJ, Thiessen N, Pettersson S, Jones SJ, Knapp S et al (2012) Bromodomain‐containing protein 4 (BRD4) regulates RNA polymerase II serine 2 phosphorylation in human CD4+ T cells. J Biol Chem 287: 43137–43155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng W, Flavell RA (1997) The transcription factor GATA‐3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell 89: 587–596 [DOI] [PubMed] [Google Scholar]
- Zhu J, Paul WE (2010) Peripheral CD4+ T‐cell differentiation regulated by networks of cytokines and transcription factors. Immunol Rev 238: 247–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Cote‐Sierra J, Guo L, Paul WE (2006) GATA‐3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell‐specific factors. Cell Res 16: 3–10 [DOI] [PubMed] [Google Scholar]