

Abstract
A role for hypoxia‐inducible factors (HIFs) in hypoxia‐dependent regulation of tumor cell metabolism has been thoroughly investigated and covered in reviews. However, there is limited information available regarding HIF‐dependent regulation of nutrient fates in tumor and stromal cells. Tumor and stromal cells may generate nutrients necessary for function (metabolic symbiosis) or deplete nutrients resulting in possible competition between tumor cells and immune cells, a result of altered nutrient fates. HIF and nutrients in the tumor microenvironment (TME) affect stromal and immune cell metabolism in addition to intrinsic tumor cell metabolism. HIF‐dependent metabolic regulation will inevitably result in the accumulation or depletion of essential metabolites in the TME. In response, various cell types in the TME will respond to these hypoxia‐dependent alterations by activating HIF‐dependent transcription to alter nutrient import, export, and utilization. In recent years, the concept of metabolic competition has been proposed for critical substrates, including glucose, lactate, glutamine, arginine, and tryptophan. In this review, we discuss how HIF‐mediated mechanisms control nutrient sensing and availability in the TME, the competition for nutrients, and the metabolic cross‐talk between tumor and stromal cells.
Keywords: HIF, tumor metabolism, tumor microenvironment
Subject Categories: Cancer, Metabolism
This review summarizes emerging views on how hypoxia‐inducible factors control nutrient utilization in the tumor context, mediating stress responses and metabolic competition between cancer and stromal cells.

Introduction
Rapid tissue growth invariably requires a sufficient supply of oxygen and nutrients (Keith & Simon, 2015). Solid tumors greater than 2 mm in diameter are associated with oxygen and nutrient diffusion limits thereby requiring a vascular system (Keith & Simon, 2015). Cancer cells adapt to this stressful environment by inducing angiogenesis and reprograming cellular metabolism to promote survival and proliferation. However, rapid growth and dysfunctional blood flow contribute to hypoxia and nutrient deprivation in many tumors.
Given the importance of oxygen (O2) as an electron acceptor in a large number of biochemical reactions, including ATP generation via oxidative phosphorylation (OXPHOS), it is critical that cells rapidly respond to hypoxia. Similar to low O2, nutrient depletion can be disastrous to cancer cells. Therefore, a complex cross‐talk between cellular hypoxic responses and metabolic requirements is necessary. Many cancers increase glucose oxidation to maintain cell growth and proliferation. However, low glucose concentrations are correlated with poor patient survival suggesting the existence of biochemical and molecular adaptations that allow cells to respond to different nutrient conditions (Moscat et al, 2015).
Hypoxia‐mediated metabolic changes in tumor cells and stromal components (immune cells, blood vessels, fibroblasts, etc.) contribute to the metabolic blueprint of tumor microenvironments (TME). Notably, hypoxia increases expression of glycolytic enzymes, the pyruvate‐depleting enzyme indoleamine 2,3‐dioxygenase, and lactate, glutamine, and L‐arginine transporters. These changes lead to depletion of several substrates which can have detrimental effects on both tumor cell fitness and immune cell function. Tumor growth leads to nutrient deprivation and lactate accumulation in the TME resulting in immune cell deficiency. Recent reports demonstrate that nutrient availability and dependency is strongly tumor type‐dependent; however, even diverse cancers may show similar reliance on specific nutrients (Neuman & Mccoy, 1956; Krall et al, 2016; Pavlova et al, 2018; Garcia‐Bermudez et al, 2020). For example, several distinct cancers such as hepatocellular carcinoma, melanoma, acute myeloid leukemia, and colorectal tumors have urea cycle defects and rely on extracellular arginine (Garcia‐Bermudez et al, 2020). Others such as glioblastoma, pancreas, prostate, and breast cancer are dependent on extracellular cyst(e)ine. Furthermore, glutamine, proline, serine, lactate, and cholesterol are also subjected to cancer‐specific uptake (Garcia‐Bermudez et al, 2020). These dependencies on specific extracellular nutrient pools shape the TME in a tumor type‐specific manner, thus extensive nutrient uptake could lead to extracellular nutrient depletion. Interestingly, Reinfeld et al (2021) demonstrate that glucose and glutamine deprivation is not a common feature of the TME in human clear cell renal cell carcinoma (ccRCC). In fact, interstitial glucose concentrations were higher in ccRCC, a notoriously glucose avid cancer, (Reinfeld et al, 2021) compared to adjacent kidney tissue. In murine pancreatic cancers, lactate and glutamine concentrations were unchanged while glucose was depleted compared to plasma (Sullivan et al, 2019). In this review, we provide a brief overview of HIF‐mediated metabolic cross‐talk in cells and its implications on nutrient availability, stress response, and immune cell function in the TME.
Hypoxia stress response in tumor cells
Various molecular and biochemical processes rely on molecular oxygen (O2), the major electron acceptor for mitochondrial ATP production. The availability of O2 is therefore essential for the survival of aerobic cells, including cancer cells (Nakazawa et al, 2016). When oxygen levels decrease, cells attempt to adapt and match O2 demand with available supply. The decrease of O2 tension rapidly results in the induction of hypoxia‐inducible responses. Many evolutionarily conserved processes including those mediated by hypoxia‐inducible factors (HIFs), ER stress, autophagy, mechanistic target of rapamycin (mTOR), and other O2 sensing mechanisms become activated upon oxygen decrease (Nakazawa et al, 2016). Blood vessel growth commences through guided stimulation of angiogenesis leading to changes in respiration and cardiac function. Subsequently, cells reprogram various metabolic processes (Messmer‐Blust et al, 2009).
The relatively rapid proliferation rate of solid malignancies results in an environment in which tumors outgrow O2 diffusion capacity leading to intratumoral hypoxic regions. When challenged with hypoxia, cancer cells utilize multiple O2 sensing biochemical pathways to adapt to this environmental stress. Hypoxia induces multiple transcription factors including nuclear factor‐κB and HIFs, and the majority of O2‐sensitive genes are direct targets of HIF transcription factors (Fitzpatrick et al, 2011). HIF is a heterodimer containing an O2‐sensitive HIF‐α subunit and O2‐independent, constitutively expressed aryl hydrocarbon receptor nuclear translocator (ARNT), also called HIF‐β. Three HIF‐α subunits have been identified, HIF‐1α, HIF‐2α, and HIF‐3α. HIF‐1α is the most ubiquitously expressed, while HIF‐2α is primarily expressed in kidneys, brain, lungs, liver, gastrointestinal tract, pancreas, and heart (Belisario et al, 2020; Lee et al, 2020). While the regulation and stability of HIF‐1α and HIF‐2α is dependent on intracellular O2 levels, HIF‐3α is suspected to negatively regulate HIF‐1α and HIF‐2α hypoxic responses (Maynard et al, 2005; Dunwoodie, 2009). Under normoxic conditions, HIF‐α subunits are hydroxylated by prolyl hydroxylase domain (PHD)‐containing proteins on two proline residues within the O2‐dependent degradation (ODD) domain (Dunwoodie, 2009; Fig 1). Upon hydroxylation, HIF‐α is primed for polyubiquitination by the Von Hippel–Lindau (VHL) complex and targeted for proteasomal degradation (Kaelin, 2017). Hypoxia prevents HIF‐α hydroxylation and maintains HIF‐α transcriptional activity.
Figure 1. Hypoxia‐Induced Factor (HIF) regulation under normoxia and hypoxia.

In the presence of oxygen, HIF‐1α and/or HIF‐2α subunits are hydroxylated on proline‐402 and proline‐564 by HIF Prolyl Hydroxylases (PHD). Hydroxylated HIF is bound by von Hippel–Lindau (VHL) and subsequently ubiquitinated marking HIF for proteasomal degradation. In the absence of oxygen, HIF is not hydroxylated and translocated into the nucleus. In the nucleus, HIF forms a complex with aryl hydrocarbon receptor nuclear translocator (ARNT) and subsequently binds to the hypoxia response elements (HRE) in the regulatory regions of genes.
While HIF target genes usually promote adaptation to hypoxia (Carmeliet et al, 1998), chronic hypoxia or failure in adaption can result in HIF‐mediated apoptosis via induction of the pro‐death factors BNIP3 (Kothari et al, 2003) or NIX (Sowter et al, 2001). These findings demonstrate that HIF‐mediated hypoxia responses have important consequences and when dysregulated affect cell function. Importantly, several cancer types and diseases are associated with dysregulated hypoxia response mechanisms (Kim & Kaelin, 2004; Qiu et al, 2016; Hoefflin et al, 2020). Germline inactivation of the VHL‐tumor suppressor gene causes the von Hippel–Lindau hereditary cancer syndrome, characterized by retinal hemangiomas, clear cell renal cell carcinomas, and pheochromocytomas (Kim & Kaelin, 2004). Moreover, biallelic VHL inactivation is present in a vast majority of cases of clear cell renal cell carcinoma (ccRCC; Qiu et al, 2016; Hoefflin et al, 2020). The tumorigenic effect of VHL mutations results from constitutive activation of the HIF pathway and transcription of genes involved in angiogenesis, epithelial‐mesenchymal transition, invasion, metastasis, and metabolism (Shenoy & Pagliaro, 2016).
HIFs bind to hypoxia response elements (HREs) in the regulatory regions of a large number of targets (Fig 1). The list of confirmed HIF target genes is growing constantly and comprises various subclasses (Bruick, 2003). Many HIF target genes promote oxygen transport to hypoxic areas by stimulating angiogenesis (VEGF and endothelin‐1) or blood cell maturation (transferrin, Erythropoietin; Pezzuto & Carico, 2018; Belisario et al, 2020). Other HIF targets mediate cellular adaptations to reduce O2 consuming metabolic pathways and promote O2‐independent pathways, i.e. glycolysis (Robey et al, 2005). Although the role of HIF in hypoxia‐mediated induction of glycolysis is extensively investigated, several reports demonstrate the involvement of HIF signaling in the regulation of nutrient uptake mechanisms, counteracting hypoxia‐induced nutrient deprivation (see below).
Cross‐talk between hypoxia sensing and nutrient sensing
Hypoxic responses and induction of other stress pathways are strongly associated and interconnected. Oxygen scarcity can lead to oxidative stress, nutrient deprivation, signaling disruption, and protein misfolding leading to endoplasmic reticulum (ER) stress. Cells undergoing hypoxia‐related stress minimize O2 and energy consumption, and it is achieved by suppressing highly ATP‐dependent processes, such as global protein synthesis preventing the accumulation of unfolded/ misfolded proteins. Mechanistically, this occurs at the level of translation initiation, which is normally accomplished by the recruitment of 40 S ribosome subunits and initiation tRNAs at the mRNA AUG start codon (Lee et al, 2020). In hypoxic conditions, this suppression is mainly regulated by two pathways: downstream of Protein kinase R (PKR)‐like ER kinase (PERK), and mechanistic target of rapamycin (mTOR) complex 1 (mTORC1; Fig 2; Lee et al, 2020).
Figure 2. HIF employs multiple strategies to activate cellular stress responses.
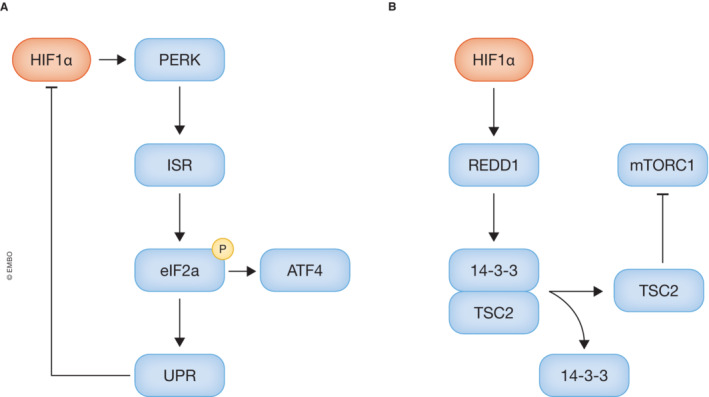
- HIF activates protein kinase R‐like ER kinase (PERK), subsequently activating the integrated stress response (ISR). Activating of the ISR results in phosphorylation of eukaryotic initiation factor 2α (eIF2a), which plays a role in the unfolded protein response (UPR) that decreases global translation including HIF‐1a translation. However, phosphorylation of EIF2a does promote cytoprotective genes regulated by activating transcription factor 4 (ATF4).
- Mammalian target of rapamycin complex 1 (mTORC1) responds to cellular stress responses via both a HIF‐dependent and HIF‐independent mechanism. In the HIF‐dependent mechanism, HIF induces expression of regulated in development and DNA damage response 1 (REDD1), which promotes the release of tuberous sclerosis complex 2 (TSC2) from 14‐3‐3 protein.
PERK is a known activator of the integrated stress response (ISR). The ISR is an evolutionarily conserved mechanism that functions through phosphorylation of eukaryotic initiation factor 2α (eIF2α) and plays a role in the unfolded protein response (UPR) decreasing global mRNA translation (Fig 2A). A recent study from Ivanova et al (2018) demonstrated that activation of the UPR prevents efficient HIF‐1α translation by reducing interactions between the mRNA binding protein YB‐1 and the 5′‐UTR of HIF‐1α mRNA. HIF pathway impairment by UPR activation results in a reduction of cell viability in low O2, suggesting that targeting the UPR may be a strategy to eliminate hypoxic malignant tumor cells (Ivanova et al, 2018).
Interestingly, not all mRNA translation is halted through eIF2α phosphorylation. Several cytoprotective genes are regulated through Activating Transcription Factor 4 (ATF4; Koumenis et al, 2002), and are essential to overcome the repressive nature of hypoxia on protein synthesis to mediate adaptive responses. Control of HIF‐1α expression and mRNA translation is clearly PERK‐dependent. Moreover, 40–50% of all increases in HIF‐1α protein levels are dependent on hypoxic stress response mediated HIF‐1α mRNA translation (Ivanova et al, 2018). Conversely, activation of PERK has been observed in several cancer cell types and is suspected to be HIF‐1α dependent (Bartoszewska & Collawn, 2020). In addition to PERK, three other kinases have also been reported to inhibit general protein translation through phosphorylation of eIF2α: double‐stranded RNA‐dependent protein kinase (PKR), the hemin‐regulated inhibitor (HRI), and amino acid (AA) starvation–dependent general control of AA biosynthesis kinase general control nonderepressible 2 (GCN2; Liu et al, 2010). Importantly, several of these kinases have been demonstrated to respond to hypoxia (Liu et al, 2010; Zhu et al, 2015).
Hypoxia can inhibit mTORC1 activity through both HIF‐dependent and HIF‐independent mechanisms. A previous report demonstrates that hypoxia induces rapid dephosphorylation of mTOR and its downstream effectors 4 E‐BP1, p70, rpS6, and eIF4G (Arsham et al, 2003). These effects are independent of HIF‐1 and do not correlate with changes in cellular ATP levels or AMPK signaling, demonstrating that O2 regulation of the mTOR pathway is not secondary to hypoxic effects on HIF‐1 or cellular energetics (Arsham et al, 2003). In addition, HIF‐1α regulates chronic mTORC1 activity by inducing expression of REDD1 (Sofer et al, 2005), which represses mTORC1 by promoting the release of sequestered TSC2, a negative regulator of mTORC1, from 14–3‐3 protein (Fig 2B; Deyoung et al, 2008). This inhibition may protect cells from hypoxic stress by reducing ATP‐consuming protein synthesis while increasing autophagy (Yuan et al, 2013). mTORC1 regulated initiation of peptide synthesis requires binding of the eukaryotic initiation factors 4 F4 (eIF4F) transcriptional complex (Lee et al, 2020). Hypoxia affects the composition of the eIF4F transcriptional complex, whereby under hypoxia eIF4F was shown to effectively recruit HIF‐dependent class III mRNAs to the ribosomes for translation. This was achieved most likely through the presence of HRE in their promotor regions, and via this process induce the hypoxia stress response (Ho et al, 2016).
Nutrient stress modulates hypoxia stress responses
Proliferating cells require large amounts of molecular oxygen for active biosynthesis and biomass production. Several growth factors activate major signaling pathways such as mitogen‐activated protein kinase (MAPK) and insulin‐stimulated PI3K pathways for cell growth. AMPK also has direct nutrient‐sensing capacities by monitoring cellular energy status via either AMP:ATP and ADP:ATP ratios, or other signals including oxidative stress (Hardie et al, 2012). Although there is substantial evidence of a reciprocal regulation between AMPK and HIF‐1α, the connections are highly context‐dependent. As previously stated, HIF‐1α induces glycolytic enzyme gene expression; however, this is counteracted by AMPK activation. Loss of AMPK signaling enhances HIF‐1α expression and increased aerobic glycolysis in mouse embryonic fibroblasts (Faubert et al, 2013). Importantly, AMPK inhibits ribosomal RNA synthesis, which represses the HIF‐1α mediated hypoxia stress response (Hoppe et al, 2009). On the other hand, AMPK signaling induced nuclear accumulation of HIF‐1α in several cancer types, which is a fundamental aspect of HIF‐1α activation (Chen et al, 2015b).
Cancer cells often exhibit dysregulation of key regulators that transmit growth signals to translational machinery (Chen & Sang, 2016). mTOR as an integrator of growth signals and nutrient status stimulates protein synthesis by phosphorylating ribosomal protein S6 kinase (S6K), eukaryotic elongation factor 2 kinase (EEF2K), and eukaryotic translation initiation factor 4 E (EIF4E)‐binding protein 1 (4EBP1), leading to increased mRNA translation (Düvel et al, 2010). Notably, mTORC1 modulates stress responses and increases HIF‐1α protein levels by promoting mRNA translation from its 5′UTR in a 4 E‐BP1 dependent manner (Düvel et al, 2010). Moreover, a previous report demonstrated mTORC1‐dependent increases in HIF‐1α and HIF‐2α transcript levels, which could be either mTORC1 regulated or resulting from HIF autoregulatory mechanisms (Chen & Sang, 2016).
The hexosamine biosynthetic pathway (HBP) has been proposed as a key player in intracellular nutrient sensing (see separate info box). HBP consumes glucose, glutamine, acetyl‐coenzyme A, and uridine‐5′‐triphosphate (UTP) to produce the amino sugar UDP‐N‐acetylglucosamine, a critical substrate for protein glycosylation (Ferrer et al, 2014; Akella et al, 2019). O‐GlcNAc transferase (OGT), responsible for O‐GlcNAcylation, prevents HIF‐1α hydroxylation impeding interaction with pVHL and consequently increasing HIF‐mediated metabolic adaptations. Therefore, O‐GlcNAcylation serves as a critical link between nutrient sensing and metabolic pathways critical for cancer cell survival via regulation of HIF‐1α hydroxylation. Importantly, decreased expression of O‐GlcNAcase (OGA), normally reducing O‐GlcNAcylation, correlates with poor patient survival due to increased O‐GlcNAcylation mediated stress responses (Ferrer et al, 2014).
In addition to O‐GlcNAcylation, acetylation is another nutrient‐dependent posttranslational modification of HIF that can affect its activity. HIF‐2α activity requires acetylation, which is regulated by coupled actions of a specific coactivator/lysine acetyltransferase, Creb binding protein (CBP) and a specific deacetylase, Sirtuin 1 (SIRT1; Chen et al, 2015a). Acetylation‐dependent HIF‐2α activation requires the specific acetyl CoA generator, acetate‐dependent acetyl CoA synthetase 2 (ACSS2). Acetate availability therefore dictates HIF‐2α activity; however, acetate stimulates tumor growth and metastasis in mice in an ACSS2‐ and HIF‐2α‐dependent manner, linking nutrient sensing and stress signaling with tumor growth (Chen et al, 2015a).
Mechanisms of nutrient sensing
The ability to sense nutrient levels and adapt to fluctuations is fundamental to cells. Nutrient availability is a selective pressure that shaped the evolution of most cellular processes and organisms. Different mechanisms have been developed to enable nutrient sensing of intracellular and extracellular levels of sugars, lipids, amino acids, etc. The ability to sense nutrients enables cells to adapt homeostatic processes, induce proliferation and cell growth, and induce protective stress responses and mobilization of internal stores through autophagy (Efeyan et al, 2015; Sanchez‐Garrido & Shenoy, 2020).
Cells within multicellular organisms are not directly exposed to environmental nutrient fluctuation, and homeostatic responses aim to maintain circulating nutrient levels within a narrow range. However, in pathological conditions, metabolic requirements and availability are different therefore nutrient sensing mechanisms and adaptations are essential for survival.
Cancer cells undergo metabolic adaptations in order to proliferate and survive and rely on proteins, nucleic acids, lipids, and glucose for biomass production (DeBerardinis & Chandel, 2016). To satisfy their needs, tumor cells must monitor nutrient availability and coordinate metabolic responses to nutrient supply (Torrence & Manning, 2018). However, how cancer cells use nutrient sensing mechanisms to influence growth, survival, and metastatic potential, remains poorly understood. Below, we provide an overview of intracellular and extracellular nutrient sensing mechanisms that are important but frequently dysregulated in tumor cells.
The hexosamine biosynthetic pathway (HBP) has been proposed to be key to intracellular nutrient sensing (Chaveroux et al, 2016). The HBP produces the activated amino sugar UDP‐N‐acetylglucosamine, a critical substrate for protein glycosylation. Since the HBP utilizes major metabolites such as glucose, glutamine, acetyl‐coenzyme A, and uridine‐5′‐triphosphate (UTP) to produce UDP‐GlcNAc, cells may use it as a ‘sensor’ of energy availability which can affect cellular stress responses through posttranslational addition of N‐ and O‐linked β‐N‐acetylglucosamine on intracellular proteins (Ferrer et al, 2014; Akella et al, 2019). It is well established that hyper glycosylation is a hallmark of cancer confirming that the HBP and nutrient sensing is critical in tumorigenesis and tumor progression (Muniz de Queiroz et al, 2019).
Amino acids: Nutrient auxotrophy of amino acids such as arginine, glutamine, proline, glycine, and cysteine is cancer type‐dependent. Typically, de novo synthesis of these semi‐essential amino acids is sufficient under physiologic conditions. However, during pathological (tumor cell growth) and physiological (immune cell activation) events, metabolic needs change and these amino acids must be obtained exogenously (DeBerardinis & Chandel, 2016).
Amino acids, including glutamine, methionine, leucine, arginine, methionine, histidine, or their respective downstream products, signal through specific receptors activating mTORC1. Leucine levels are detected by Sestrin2 and leucyl‐tRNA synthetase 1, arginine is detected by CASTOR1 and the lysosomal amino acid transporter SLC38A9, acetyl‐CoA (derived from leucine) is detected by mTORC1, S‐adenosyl methionine is sensed by BMT2/SAMTOR, alpha‐ketoglutarate produced through glutaminolysis acting via PHDs and ADP ribosylation factor 1.
In contrast, amino acid withdrawal results in the accumulation of uncharged tRNAs and ribosomal stalling activating EIF2AK4/GCN2 kinase activity (Carroll et al, 2015). Amino acid‐specific aminoacyl tRNA synthetases catalyze the loading of amino acids to tRNAs. In order to prevent cellular damage, cells must detect amino acid depletion and activate protective mechanisms preventing translation initiation via GCN2, whereby GCN2 has a high affinity for uncharged tRNAs under amino acid starvation. Binding of uncharged tRNAs to GCN2 leads to kinase activity and inhibitory phosphorylation of eukaryotic translation initiator factor 2 α (eIF2α) and inhibition of general protein translation. GCN2 mediated inhibition of protein translation occurs in concert with other cellular responses to amino acid shortage, such as the inhibition of mTORC1 (Sanchez‐Garrido & Shenoy, 2020).
Glucose and glycolytic intermediates: Glucose uptake, storage, and catabolism are tightly regulated extracellularly and intracellularly. Glucose sensing is mediated by glucose transporters, such as GLUT2. The low affinity of GLUT2 allows for efficient transport of glucose across the cell membrane only when extracellular glucose levels are high, but not under low concentrations when other glucose transporters are active. Glucose is further sensed by AMPK, which is activated during energy stress and is a fundamental regulator of cellular metabolism. It is responsive to cellular energy levels, as a surrogate sensing mechanism for glucose abundance, specifically increased levels of AMP and ADP directly activate AMPK. Although glucose metabolism and energy status are often tightly linked, AMPK can be activated upon glucose deprivation independently of changes in AMP and ADP levels (Hawley et al, 2010). The glycolytic intermediate fructose‐1,6‐bisphosphate (FBP) is sensed by the glycolytic enzyme aldolase. In the absence of FBP, interactions between aldolase and lysosomal v‐ATPase are altered, allowing an AXIN‐based AMPK activation complex priming cells for potential constant energy stress (Lin & Hardie, 2018).
The regulation of mTORC1 through Rag‐GTPases is not restricted to amino acids, as cellular glucose levels also affect the activity of Rag‐GTPases (Efeyan et al, 2013). It remains unknown how glucose affects Rag GTPases, but it is very likely that amino acid sensing and glucose sensing mechanisms converge upstream of mTORC1 through involvement of v‐ATPases (Muniz de Queiroz et al, 2019). Lactate, a glycolytic end‐product not only regulates intracellular and environmental pH levels, it represents a major fuel source for oxidative cancer cells and as a signaling agent (Payen et al, 2020). Regulation of lactate transport is essential for cellular function and survival. Several G protein‐coupled receptors (GPCRs; GPR4, GPR65, GPR68, GPR81 and GPR132) have been identified as putative lactate sensors (Justus et al, 2013). These receptors are activated via the protonation of several histidine residues in response to extracellular pH decreases. Acidity in tumor cells generated from lactate transmits intracellular signals through GPCRs such as adenylate cyclase (GPR4 and GPR65), phospholipase C (GPR68), or GPR 132 (Justus et al, 2013). In addition to tumor cells, stromal cells are also affected. Activation of GPR132 activates pro‐tumorigenic M2 macrophages, facilitating cancer cell migration, metastasis, and invasion (Chen et al, 2017).
Lipids: Lipids are detected through distinct sensors depending on their structure. Short chain fatty acids are detected by a family of GPCRs and histone deacetylases, such as GPR41 and GPR43, whereas butyrate is sensed by GPR109A and histone deacetylases (Alvarez‐Curto & Milligan, 2016). Long chain fatty acids, such as omega‐3 fatty acids are detected by GRP40 and GRP120. Cholesterol is sensed by sterol regulatory element‐binding protein (SREBP) and SREBP cleavage‐activating protein (SCAP). In the presence of cholesterol, the SCAP‐SREBP complex anchors to the membrane via INSIG. In the absence of cholesterol, SCAP‐SREBP does not anchor to the membrane and is trafficked to the Golgi, where proteolytic cleavage of SREBP subsequently activates cholesterol synthesis through activation of HMG‐CoA reductase (HMGCR). Triglycerides including low‐density and very‐low density lipoproteins are detected by the receptor CD36, which signals via non‐receptor tyrosine kinases (Silverstein & Febbraio, 2009). Key lipid sensors are the peroxisome proliferator activated receptors (PPARs), which are nuclear receptors regulating lipid metabolism, inflammation, and cellular growth. These receptors bind and are activated by a broad range of fatty acids and fatty acid derivatives (Cariello et al, 2021). PPARs directly regulate the expression of fatty acid transporters and control expression of principal enzymes of peroxisomal beta‐oxidation (Cariello et al, 2021). Moreover, phosphatidic acid (PA) interaction with the FK506‐binding protein–12‐rapamycin‐binding (FRB) domain of mTOR stabilizes both mTOR complexes: mTORC1 and mTORC2. Induction of mTOR by oleic acid is dependent on PA de novo synthesis, suggesting that mTOR senses lipids via production of PA, in addition to a glucose and amino acids (Menon et al, 2017).
Hypoxia induces HIF‐mediated metabolic responses
Metabolic alterations as a major feature of tumor cell metabolism was observed almost a century ago by the German physiologist Otto Warburg (Pavlova & Thompson, 2016). Canonically, glucose is converted to pyruvate and is further catabolized in the mitochondria through the TCA cycle generating NADH. Constant production of ATP as a result of mitochondrial OXPHOS inhibits the key glycolytic enzyme phosphofructokinase (PFK; Stine et al, 2014). In hypoxia, cells shift from O2‐dependent mitochondrial ATP production to O2‐independent production via glycolysis (Al Tameemi et al, 2019). In contrast, cancer cells preferentially utilize glycolysis over oxidative phosphorylation, even when O2 levels are adequate (Al Tameemi et al, 2019). There is an important balance between glycolytic ATP production and glucose import. Increased glucose uptake has been proposed as a hallmark of aggressive tumors (Schwartz et al, 2017) and is a feature commonly employed to detect malignancies using positron emission tomography (PET) imaging. PET determines glucose import activity using the non‐metabolized glucose analogue 2‐deoxy‐2‐[18F] fluoro‐D‐glucose ([18F]FDG; Patching, 2015).
From previous studies, it became clear that HIF‐mediated responses to hypoxia have strong influences on cell metabolism. HIF‐1α stimulates glycolysis and glucose metabolism by inducing expression of glucose transporters and numerous glycolytic protein isoforms (Fig 3; Semenza, 2003). Another glucose‐related metabolic pathway increased in cancer is the pentose phosphate pathway (PPP). The PPP branches off after the first step in glycolysis and consumes the glycolytic intermediate glucose 6‐phosphate to generate fructose 6‐phosphate and glyceraldehyde 3‐phosphate. The non‐oxidative PPP is responsible for nucleotide synthesis through production of ribose 5‐phosphate (R5P) and provides reducing power required for the synthesis of sterols, fatty acids, nucleotides, and AAs (Ge et al, 2020). Moreover, NADPH reducing power is needed for antioxidant defense mechanisms (Ge et al, 2020). HIF plays a significant role in mediating the PPP through regulation of GAPDH and induction of the direct HIF target glucose 6‐phosphate dehydrogenase (G6PD). G6PD is a key oxidative PPP enzyme which generates NADPH (Singh et al, 2017).
Figure 3. Regulation of glucose metabolism by HIF.
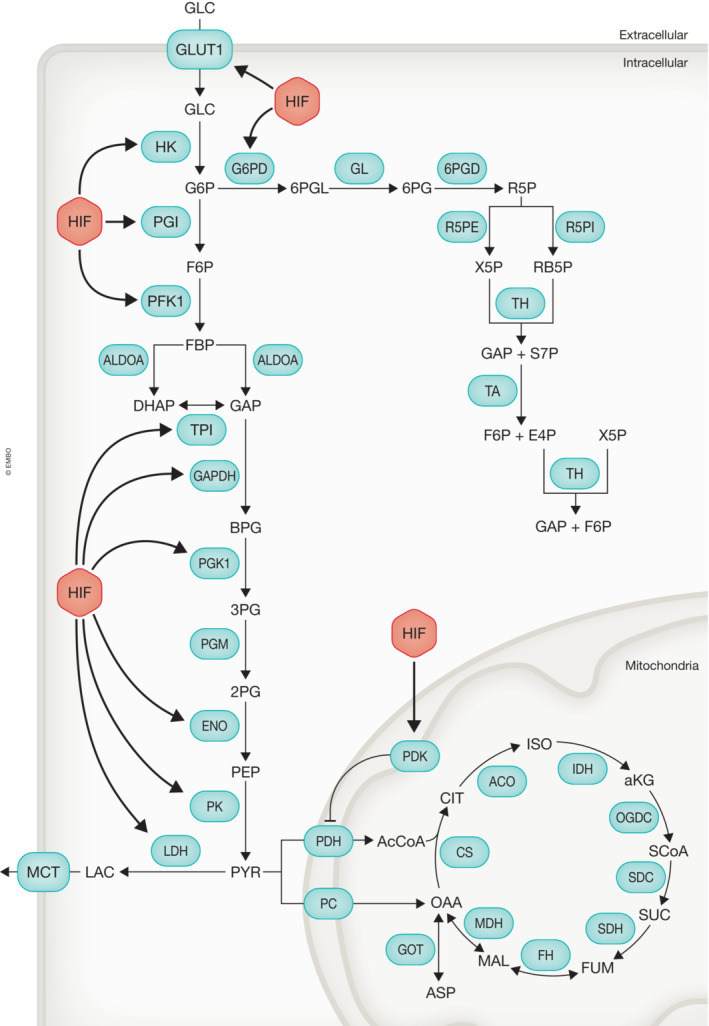
HIF increases the expression of a majority of genes involved in glucose import, glycolysis, and the pentose phosphate pathway (PPP). Additionally, HIF increases expression of pyruvate dehydrogenase kinase (PDK), which inhibits pyruvate dehydrogenase (PDH) activity subsequently reducing glucose entry into the mitochondria. Glycolysis: (enzymes) GLUT1, glucose transporter 1 or SLC2A1; HK, hexokinase; PGI, phosphoglucose isomerase; PFK1, phosphofructokinase 1; ALDOA, aldolase A; TPI, triosephosphate isomerase; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; PGK1, phosphoglycerate kinase 1; PGM, phosphoglucomutase 1; ENO, enolase; PK, pyruvate kinase; LDH, lactate dehydrogenase; (metabolites) GLC, glucose; G6P, glucose 6‐phosphate; F6P, fructose 6‐phosphate; FBP, fructose‐1,6‐bisphosphate; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde 3‐phosphate; BPG, 1,3‐bisphosphoglycerate; 3PG, 3‐phosphoglycerate; 2PG, 2‐phosphoglycerate; PEP, phosphoenol pyruvate; PYR, pyruvic acid; LAC, lactic acid. PPP: (enzymes) G6PD, glucose‐6‐phosphate dehydrogenase; GL, gluconolactonase; 6PGD, 6‐phosphogluconate dehydrogenase; R5PE, ribulose‐5‐phosphate 3‐epimerase; R5PI, ribulose‐5‐phosphate isomerase; TH, transketolase; TA, transaldolase; (metabolites) 6PGL, 6‐phosphogluconolactone; 6PG, 6‐phosphogluconate; R5P, ribulose‐5‐phosphate; X5P, xylulose‐5‐phosphate; RB5P, ribose‐5‐phosphate; S7P, sedoheptulose7‐phosphate; E4P, erythrose 4‐phosphate; Mitochondria: (enzymes) PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PC, pyruvate carboxylase; CS, citrate synthase; ACO, aconitase; IDH, isocitrate dehydrogenase; OGDC, oxoglutarate dehydrogenase complex; SCS, succinyl coenzyme A synthetase; SDH, succinate dehydrogenase; FH, fumarate hydratase; MDH, malate dehydrogenase; GOT, glutamic‐oxaloacetic transaminase; (metabolites) AcCoA, acetyl coenzyme A; CIT, citric acid; ISO, isocitric acid; aKG, alpha‐ketoglutarate; SUC, succinic acid; SCoA, succinyl coenzyme A; FUM, fumaric acid; MAL, malic acid; OAA, oxaloacetic acid; ASP, aspartic acid.
Pyruvate (the end product of glycolysis) enters the TCA cycle via pyruvate dehydrogenase, generating acetyl‐CoA and NADH or alternatively oxaloacetate through the anaplerotic enzyme, pyruvate carboxylase. Notably, hypoxia inhibits glucose carbon entry through the TCA cycle: HIF‐1α stimulates expression of pyruvate dehydrogenase kinase (PDK), which reduces acetyl‐CoA production through inhibitory phosphorylation of pyruvate dehydrogenase (Singh et al, 2017).
Effects of hypoxia on amino acid transport
Amino acid (AA) transport across plasma membranes is essential for supplying cells with nutrients needed for cellular metabolism and protein synthesis. Increased cancer cell proliferation results in increased consumption of AAs. As a result, cells adjust transport systems to optimize nutrient uptake and consequently modulate nutrient availability in the tumor microenvironment (Fig 4).
Figure 4. HIF regulates nutrient availability through plasma membrane transporters.
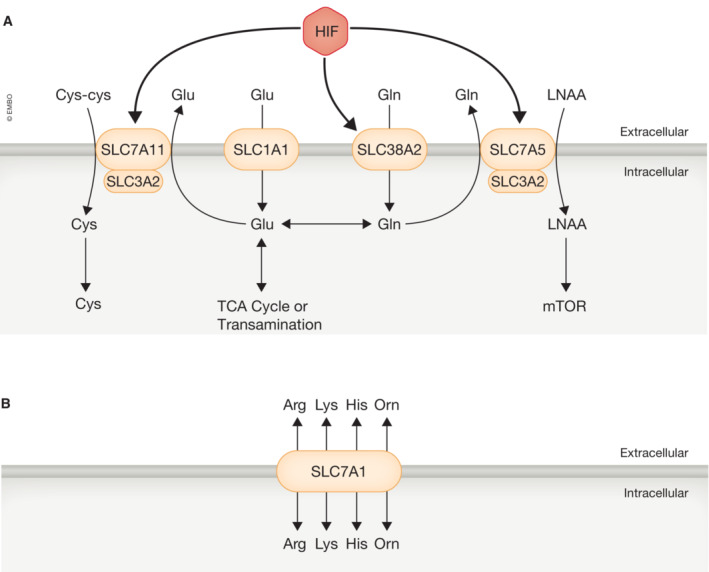
- HIF increases expression of the glutamine (gln) transporter (SLC38A2), which can be metabolized to glutamate (glu) or exported outside the cell in exchange from large neutral amino acids (LNAA) through SLC7A5. LNAAs can be used to promote protein synthesis through mammalian target of rapamycin (mTOR). Glutamate is also imported by SLC1A1 and is critical for the TCA cycle, transamination reactions, and glutathione (GSH) synthesis. Another critical component of GSH synthesis the cystine‐glutamate antiporter (SLC7A11). SLC3A2 forms a heterodimer with both SLC7A5 and SLC7A11.
- HIF also regulates transporters to either increase or decrease metabolite efflux from other metabolic reactions primarily through SLC7A1. arg, L‐arginine; lys, lysine; his, histidine; orn, ornithine.
Essential amino acids (EAAs) are critical for protein synthesis and cannot be de novo synthesized by cells. The branched‐chain EAAs (BCAA; leucine, isoleucine, and valine) are essential for in vivo lung (Mayers et al, 2016) and brain tumor (Tönjes et al, 2013) growth. Sustained levels of leucine are required to maintain mTORC1 activation, a master regulator of cell metabolism and growth (Parks et al, 2016). Various AA transporters have been linked with cancer progression, where the large neutral AA transporter (LAT1/ SLC7A5) is the primary example (Parks et al, 2016). SLC7A5 overexpression is associated with low overall survival in a wide range of tumor types including prostate, RCC, gliomas, and melanomas (Sakata et al, 2009; Kaira et al, 2010; Haining et al, 2012; Betsunoh et al, 2013; Shimizu et al, 2015). Several oncogenes activate SLC7A5 expression during carcinogenesis including Myc, Notch, and EZH2 (Salisbury & Arthur, 2018). In addition to oncogenic activation, adaptive pathways such as hypoxia can promote SLC7A5 expression in tumors.
Studies in neonatal rodents and pigs show increased levels of BCAAs in plasma and brain during hypoxia (Fitzgerald et al, 2021). Transport of BCAAs is mediated by SLC7A5, which functions in a heterodimer with SLC3A2. SLC7A5 and SLC3A2 expression is upregulated in multiple cell types in a HIF‐2α‐dependent manner (Elorza et al, 2012; Kucharzewska et al, 2015). Hypoxic induction of SLC38A2 mRNA and protein expression is significantly reduced by HIF‐1α siRNA, demonstrating HIF‐1α‐dependent regulation of AA transporters in hypoxic conditions (Morotti et al, 2019). Hypoxic upregulation of AA import may normalize intracellular levels of these key metabolic substrates supporting biomass production. Several reports demonstrate altered AA uptake in cells under hypoxic conditions (Kobayashi & Millhorn, 2001; Soh et al, 2007; Morotti et al, 2019; Belisario et al, 2020). Specifically, glioblastoma cells increase glutamine and leucine transport during hypoxia. Here, increased AA transport was completely blocked upon RNA or protein synthesis inhibition, demonstrating increased AA uptake is the result of elevated transporter expression without affecting their transport efficiency (Soh et al, 2007). Similarly, hypoxia induces expression of the glutamate transporters EAAC1 and GLT‐1 in rat pheochromocytoma cells (Kobayashi & Millhorn, 2001). RNA sequencing of various breast cancer cell lines revealed that 3 out of 25 AA transporters were upregulated in hypoxic conditions; SLC1A1, SLC7A5, and SLC38A2 (Morotti et al, 2019). Theoretically, these AA transporters combined can account for all AA precursors that can generate TCA cycle intermediates. SLC7A5 supports the uptake of ketogenic AAs, SLC1A1 the uptake of glutamate, and SLC38A2 the uptake of other neutral AAs such as glutamine, alanine, glycine, and serine (Fig 4A; Morotti et al, 2019).
The Na+‐independent cystine and glutamate antiporter (SLC7A11) is also upregulated under hypoxia (Fotiadis et al, 2013). Like SLC7A5, SLC7A11 forms a heterodimer with SLC3A2, which acts as a chaperone and promotes stabilization, trafficking, and insertion into plasma membranes. In cultured cells, SLC7A11 is the main provider of intracellular cysteine via import of cystine. Extracellular cysteine is readily oxidized to cystine, the substrate for SLC7A11 (Belisario et al, 2020). Reducing intracellular conditions favors the conversion of cystine to cysteine and its incorporation into the anti‐oxidant tripeptide glutathione (GSH). Previous studies in breast cancer cells demonstrated that knockdown and digoxin‐mediated inhibition of HIF‐1α, but not HIF‐2α decreased SLC7A11 expression in vitro and in vivo (Lu et al, 2015). Hypoxia increases SLC7A11 levels in triple‐negative breast cancer through HIF‐1α activity in cancer stem cell populations and plays a critical role in protection against oxidative stress (Lu et al, 2015). The importance of SLC7A11 in cysteine homeostasis is evident in vitro, however, its role in vivo is less clear as circulating cysteine and cystine concentrations are often similar. SLC7A11 is specific for cystine, while a number of other transporters (i.e., SLC1A5) can import cysteine.
Conversely, exposure to low O2 levels can decrease various AA transporters, potentially to prevent AA export since AA transporters can be bidirectional (Fig 4B; Nicklin et al, 2009). Hypoxia inhibits expression of system A amino acid transport of proline, 2‐aminoisobutyric acid (AIB), and 2‐ (methylamino) isobutyric acid (methyl‐AIB) and inhibited proline uptake in lung fibroblasts (Berk et al, 2000). It also reduces SLC7A1 expression in several cancer cell lines (HELA, P493‐6, HCT116, MCF7, RCC4; Lendahl et al, 2009). Interestingly, this cationic AA transporter is required for chronic lymphocytic leukemia (Werner et al, 2019) and colorectal cancer cell proliferation (Lu et al, 2013), but the rationale for this hypoxia‐mediated inhibition remains elusive. SLC7A1 is upregulated as a result of the integrated stress response. AA starvation results in uncharged tRNAs, activating GCN2 and ATF4, which in turn cause translation of the abundant SLC7A1 mRNA by a cap‐independent mechanism. A previous report demonstrates that loss of HIF‐1α induces ATF4 expression (Guimarães‐Camboa et al, 2015), potentially explaining how hypoxia and HIF‐1α inhibits SLC7A1 expression.
SLC7A5 overexpression is HIF‐2α dependent, and HIF‐2α also activates mTOR. As stated above, mTOR is a central sensor of cellular energy status and AA availability, and a stimulator of protein translation and cell proliferation (Sonenberg & Hinnebusch, 2009). This suggests that the HIF/AA transport axis is required within cancer cells to ensure uptake of essential AAs from the nutrient‐depleted tumor microenvironment for protein production necessary for survival and proliferation (Belisario et al, 2020).
Nutrient availability is strongly affected by hypoxia and mediates stromal cell activity
The tumor microenvironment is a complex and heterogeneous system where cancer cells and various stromal cell types including immune cells, fibroblasts, and endothelial cells reside (Hanahan & Coussens, 2012). Tumor cells alter their metabolism to promote biomass and proliferation relying on enhanced consumption of specific nutrients. Other cells surrounding the tumor parenchyma compete or cooperate with cancer cells causing metabolic antagonism or metabolic symbiosis, respectively (Wang et al, 2014). Depletion of specific nutrients in the TME will inevitably affect stromal cells relying on these factors. For instance, inhibition of the arginine‐producing urea cycle in ASS1‐deficient cancers prevents depletion of intracellular aspartate pools, a metabolite essential for de novo pyrimidine synthesis, but creates vulnerabilities by making cells dependent on extracellular arginine (Rabinovich et al, 2015). Therefore, the dependence of cancer cells on extracellular nutrients may rapidly deplete available nutrient pools. Longitudinal quantification of interstitial metabolites in a mouse model of pancreatic cancer demonstrates that arginine was the most significantly depleted metabolite relative to circulating concentrations (Sullivan et al, 2019). Pancreatic cancers are suggested to have a truncated urea cycle due to loss of ASS1 expression (Yang et al, 2021). Consequently, these tumor cells are arginine auxotrophs. Arginine depletion can negatively affect various immune cell populations that depend on exogenous arginine for their functionality such as macrophages (Murray, 2016) and T‐cells (Geiger et al, 2016). Thereby creating an environment where cancer cells and tumor‐residing immune cells compete for arginine availability. Similarly, enhanced aerobic glycolysis requires increased glucose import (Lin et al, 2020). These features result in depletion of extracellular nutrients which may significantly affect immune cell function. Alternatively, tumor cells can release several factors such as lactate and indoleamine 2,3‐dioxygenase (IDO), which have immunomodulatory properties (Rabinovich et al, 2015). Overall, potential metabolic competition between tumor and immune cells could promote immune suppression due to immune cells “exhaustion”. Interestingly, hypoxia sensing strongly affects cellular metabolism through HIF activity which consequently results in altered nutrient import and requirements, leading to modulation of extracellular nutrient pools in the tumor microenvironment and stromal cell activity.
Significant metabolic heterogeneity exists withing solid tumors creating O2 and nutrient gradients, partially due to changes in vascular integrity and proximal distance to the vasculature. As such, we assume that avascular areas, most likely located in the tumor core will be more hypoxic and more nutrient poor (Li & Simon, 2020). Indeed, a previous study demonstrated that tumor cores have much reduced levels of AAs including arginine, serine, glutamine and aspartate, compared to peripheral regions (Pan et al, 2016). Several major metabolites are subject to competition between tumor cells and immune cells, and their availability and necessity in hypoxic conditions will be described below (Table 1 and Fig 5).
Table 1.
Competition for metabolites between cell type functions in the tumor microenvironment.
| Metabolite | Cell type | Function | Cancer | References |
|---|---|---|---|---|
| Glucose | Tumor cells | Primarily use glycolysis for ATP and other biosynthetic substrates | Most solid tumors | Liberti and Locasale (2016) |
| Tumor inflitrating lymphocytes | Activated T cells shift metabolism to glycolysis with IFN‐y production high in glucose‐rich environments | a | Fox et al (2005) and Yin et al (2019) | |
| Macrophage | M1 macrophages utilize glycolysis while M2 use OXPHOS with glucose deprivation only affecting M1 | a | Venter et al (2014), Chang et al (2015), and Ho et al (2015) | |
| Lactate | Tumor cells | High lactate from glycolysis results in increased progression and mestastasis | Most solid tumors | Keenan and Chi (2015) |
| T cells | High TME lactate inhibits glycolysis and impairs IFN‐y production | a | Angelin et al (2017) and Chang et al (2015) | |
| Tregs | Alter metabolism to promote OXPHOS becoming resistant to high lactate to promote immunosupression | a | Angelin et al (2017) | |
| Tumor‐assoicated Macrophage | High lactate polarize to M2 phenotype | a | Colegio (2016) | |
| Tryptophan | Tumor cells | Generate kynurenine in the TME from tryptophan via IDO to manipulate immune cell function | Most solid tumors | Jovanovic et al (2020) |
| Dendritic cells | Kynurenine impairs cytokine induction via MHC class II | a | Gandhi et al (2010) and Rothhammer and Quintana (2019) | |
| T cells | Expression of FoxP3 to reprogram metabolism | a | Gandhi et al (2010) and Rothhammer and Quintana (2019) | |
| Glutamine | Tumor cells | Increase glutamine transport and metabolism (GLS) for proliferation | TNBC, NSCLC, GBM | Altman et al (2016) and Choi and Park (2018) |
| T cells | Require extracellular glutamine for activation | a | Liu et al (2017) | |
| Macrophage | Succinate generation from glutamine promotes M1 polarization, but may not be an essential factor | a | Tannahill et al (2013), Jha et al (2015), and Ren et al (2019) | |
| Arginine | Tumor cells | Most consumed metabolite from TME for growth following urea cycle deficiency | PDAC | |
| Macrophage | M1 macrophages consume arginine for NO production; M2 macrophages consume arginine to generate ornithine | a | Bronte and Zanovello (2005) and Peyssonnaux et al (2005) | |
| T cells | Arginine shifts T cell metabolism to OXPHOS, and arginase inhibition does not affect T cell proliferation | a | Geiger et al, (2016) |
A majority of the work on immune cell function has been performed after isolation of cells from a primary source and is under investigation in the TME.
Figure 5. The tumor microenvironment (TME) is composed of tumor and stromal cells that compete for nutrients.
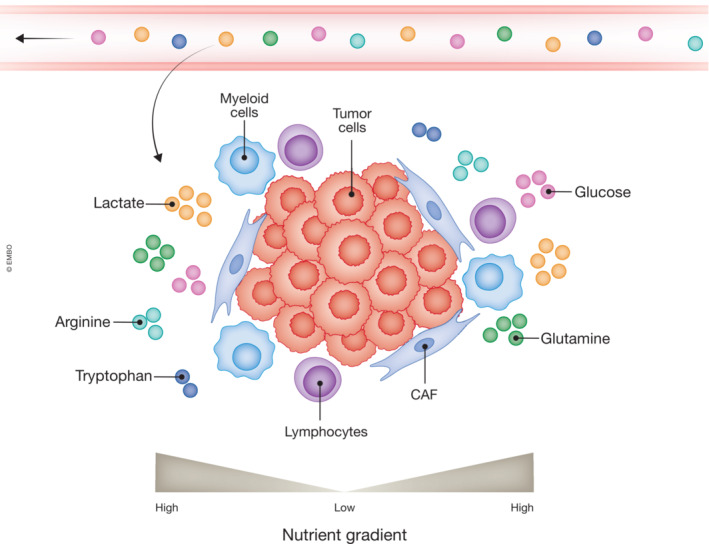
Schematic of cells and nutrients in the TME. Nutrients are circulated throughout the body, and the TME imports these nutrients from the blood. In the TME, stromal cells (immune cells, fibroblasts, endothelial cells, and more) and tumor cells have distinct metabolic requirements. Depending on the cellular composition, local metabolite concentration can be increased or decreased. The further into the TME (away from the circulation) the sparser nutrients and oxygen become resulting in cells competing for available nutrients.
Glucose
The principal glucose‐consuming pathway in tumor cells is glycolysis, and their glucose uptake and lactate production are significantly elevated (Liberti & Locasale, 2016). Glycolysis generates not only ATP, but also other substrates (serine, NADH, etc) required for tumor cell survival and proliferation (Liberti & Locasale, 2016; Lin et al, 2020). In cancer cells, glucose metabolism is regulated by both intrinsic signals (e.g. oncogenes) and environmental factors. Hypoxia strongly increases glucose uptake and glycolysis through HIF activity (Park et al, 2016; Huang et al, 2020). Various glycolytic enzymes and other glucose metabolic factors are transcriptionally regulated by HIF‐1α (Fig 3) including: pyruvate dehydrogenase kinase 1 (PDK1) hexokinase 1 and 2, enolase 1, autocrine motility factor (AMF/GPI), pyruvate kinase, 6‐phosphofructo‐2‐kinase/fructose‐2,6‐biphospathase‐3 (PFKFB3), glyceraldehyde‐3‐P‐dehydrogenase (GAPDH), and lactate dehydrogenase‐A (LDH‐A) (Semenza, 2003). Glucose import is mostly mediated by the glucose membrane transporter (GLUT) family. There are 14 known GLUT family members with GLUT1, GLUT3, and GLUT4 having a high affinity for glucose allowing its transport at a high rate (Barbosa & Martel, 2020); GLUT1 has received most attention in cancer research (Parks et al, 2016). Both GLUT1 and GLUT3 are HIF targets. HIF can promote glycolysis through elevated glucose import, and an increase in GLUT1 expression was observed in hypoxic colon cancer cells (Martinez et al, 2019). Increased glucose uptake by a large number of cancer cells could deplete extracellular glucose pools with detrimental consequences for tumor‐residing immune cells raising the possibility of competition among these cell types for glucose consumption in the TME (Ngwa et al, 2019). Several previous studies demonstrated that increased glycolytic activity in cancer cells and endothelial cells under hypoxia could restrict glucose availability for tumor‐infiltrating lymphocytes (TILs) resulting in T‐cell exhaustion and immune escape (Chang et al, 2015; Ho et al, 2015; Zhao et al, 2016; Wang et al, 2018a). However, a recent publication showed that metabolic competition for glucose and the immunosuppressive consequences are not a common feature in cancer. Reinfeld et al (2021) demonstrated that glucose is not broadly limiting, (Reinfeld et al, 2021) suggesting that diverse cell populations preferentially acquire distinct metabolites from a common pool of nutrients available in the TME. The authors propose a mechanism by which mTORC1‐driven glutamine uptake may suppress the expression of glycolytic genes in cancer cells allowing TME‐resident cells to increase glucose uptake when glutamine is limiting (Reinfeld et al, 2021).
It is well known that metabolic deregulation drives tumor progression, but it is still underappreciated how this may affect tumor‐infiltrating cells, such as TILs (Ho et al, 2015). Antigen‐specific naïve Tzcells proliferate extensively and acquire effector functions upon antigenic stimulation. Activated T‐cells adapt their metabolism to ensure sufficient energy generation to support cell biomass and proliferation (Fox et al, 2005). Quiescent naive T‐cells and regulatory T‐cells (Tregs) require little energy and nutrients and mostly utilize OXPHOS. While activated T‐cells consume large amounts of nutrients and reprogram their metabolism towards glycolysis for rapid ATP production (Yin et al, 2019). T‐cells produce significantly less IFNγ when cultured with tumor cells than when cultured alone. IFNγ production is restored by glucose addition, suggesting that competition with tumor cells for glucose suppresses T‐cell function (Chang et al, 2015). Moreover, TILs isolated from progressive sarcomas (exhibiting hyperglycolysis) are hyporesponsive, exhibit reduced glycolysis, and increase markers of nutrient deprivation. These TILs display impaired IFNγ and CD40L levels, two factors critical for maintaining an immunostimulatory microenvironment in Braf/PTEN melanomas, and increased expression of TGFβ and other genes associated with T‐cell anergy (Ho et al, 2014, 2015). Checkpoint blockade therapy increased cytokine production and glycolytic activity in TILs by directly inhibiting Akt/mTORC1 mediated glycolysis in tumor cells, thereby increasing glucose availability in the TME for tumor‐infiltrating cells (Ho et al, 2015). Overexpression of the gluconeogenic enzyme phosphoenolpyruvate carboxykinase 1 (PCK1) restores T‐cell activity, even under glucose poor conditions (Ho et al, 2015).
Macrophages, like T‐cells, are dependent on glucose availability and glucose metabolism. Macrophages are a heterogeneous population of immune cells with diverse functions. Rather than following a simple classification into two populations, macrophages form a continuum between two extremes, i.e. pro‐inflammatory M1 and anti‐inflammatory M2 phenotypes (Venter et al, 2014). These two populations are distinct in many ways and harbor different metabolic programs. M1 macrophages mainly rely on glycolysis and present TCA cycle dysfunction resulting in the accumulation of succinate. Excess succinate results in HIF‐1α stabilization and transcription of HIF‐1α‐dependent glycolytic genes, which sustains glycolytic metabolism. Macrophages stimulated with the M1 inducer LPS enhance glucose import through increased expression of the HIF‐1α target GLUT1 (Freemerman et al, 2014; Venter et al, 2014). HIF‐1α ‐ mediated PDK1 induction has been observed in severely hypoxic conditions, preventing entry of pyruvate into the TCA cycle. Notably, inhibition of the macrophage HIF‐1α‐PDK1 axis suppresses migration and inflammatory activity, demonstrating that adaptive responses in glucose metabolism contribute to macrophage function (Semba et al, 2016).
In contrast, M2 macrophages do not undergo such a dramatic metabolic rewiring and have metabolism comparable to that of unstimulated macrophages. They are more dependent on the TCA cycle to provide electrons for OXPHOS (Venter et al, 2014; Viola et al, 2019). 2‐DG treatment reduces glycolytic activity resulting in decreased, whole‐cell ATP concentrations and is associated with reduced JAK‐STAT6 pathway activity and M2 differentiation markers. Interestingly, although glucose deprivation dramatically reduced glycolytic flux it did not affect M2 polarization, OXPHOS, or ATP levels (Chang et al, 2015; Ho et al, 2015). This seems contradictory since an important role for pyruvate in macrophage polarization has been noted. However, more recent studies indicate that only 40% of pyruvate is derived from glycolysis, suggesting that glycolytic stimulation is not essential for M2 macrophage differentiation and activity, when OXPHOS remains intact (Hui et al, 2017; Wang et al, 2018b). Glucose deprivation will therefore mainly affect M1 function leading to an immune‐suppressive environment.
Lactate
L‐Lactate is produced in the cytosol via the rapid enzymatic conversion of pyruvate via lactate dehydrogenase A (LDH‐A), regenerating NAD+ supporting high rates of glycolysis. Lactate is subsequently transported across the plasma membrane through monocarboxylate transporters (MCTs) and accumulates in the extracellular space (Santos et al, 2019). Lactate normally circulates at levels of 1–2 mM, but rises to 30–40 mM in the tumor microenvironment (Santos et al, 2019). Accumulation of extracellular lactate and low pH, lactic acidosis, is a prominent microenvironmental stress found in most solid tumors (Keenan & Chi, 2015). High lactate levels are associated with severe tumor progression, increased metastasis, and poor clinical outcomes (Keenan & Chi, 2015).
Proton coupled lactate efflux is important for the maintenance of intratumoral pH homeostasis. In response to hypoxic conditions, cancer cells react by expressing various nutrient and ion transporters. HIF‐1α is associated with tumor lactogenesis by inducing expression of LDHA, MCT4, and membrane‐bound carbonic anhydrase IX (CAIX), leading to lactate accumulation in the TME (de la Cruz‐López et al, 2019). Lactate export prevents intracellular acidification and is primarily mediated by the monocarboxylate transporters MCT1 and MCT4, both of which carry lactate in co‐transport with H+ (Pérez‐Tomás & Pérez‐Guillén, 2020). MCT‐mediated H+ efflux increases extracellular acidification and supports the formation of a hostile environment favoring tumor growth (Jamali et al, 2015). Another key protein in the regulation of intracellular pH is CAIX, which serves to catalyze the reversible hydration of CO2 to HCO3 − + H+ (Noor et al, 2018). Importantly, CAIX‐dependent H+ shuttling is essential for MCT1/4‐mediated lactate efflux. Short interfering RNA targeting CAIX expression reduces MCT1‐mediated lactate transport in MCF7 breast cancer cells, and co‐expression of MCT1/4 with CAIX revealed that CAIX mediated MCT transport activity through the enzyme's intramolecular proton shuttle (Jamali et al, 2015; Noor et al, 2018). HIF‐dependent expression of lactate transporters varies between cancer types. Expression of HIF‐1α is strongly correlated with increased expression of CAIX. Cell base analyses indicate HIF‐1α mediated expression of CAIX in squamous cell carcinomas (Sáenz‐de‐Santa‐María et al, 2017) and MCT4 in HELA cells (Ullah et al, 2006). In addition to lactate transporters, various genes involved in lactate production are HIF regulated, including LDH‐A (Cui et al, 2017). HIF‐1α loss decreases LDH‐A expression in pancreatic cancer under hypoxic conditions, with a significant reduction in lactate production and glucose utilization (Cui et al, 2017). Similar observations were made in gastric cancer (Hao et al, 2019), ovarian cancer (Tang & Liu, 2016), colorectal cancer (Wang et al, 2015), and glioblastoma (Daniele et al, 2015).
There is increasing evidence that lactate secreted by tumor cells affects the behavior of both tumor and stromal cells. The mechanism (s) by which lactate impairs immune cell function are not fully understood, but it is thought that high concentrations of TME lactate block the export of lactate by glycolytic immune cells, and disrupting their function (Romero‐Garcia et al, 2016; Santos et al, 2019). However, lactate can be used by certain immune cells as an energy source, which impairs their glycolytic flux necessary for activation (Hargadon, 2017).
NAD+ generation is essential to sustain GAPDH activity and glycolysis in effector T‐cells. At a very low glucose concentration (20 mg per dL), effector T‐cells still are able to proliferate via glycolysis. However, influx of lactate and reversal of the LDH reaction consumes NAD+ and impairs glycolysis (Angelin et al, 2017). Loss of glycolysis is especially detrimental for effector T‐cells as it impairs intracellular calcium homeostasis and the ability to produce IFN‐γ (Chang et al, 2015; Ho et al, 2015). Moreover, lactate induces high IL‐7 production in CD4+ and loss of cytotoxic activity CD8+ T‐cells (Angelin et al, 2017). In contrast, Tregs have higher NAD+ levels to compensate and are less affected by reduced glycolysis (Beier et al, 2015; Gerriets et al, 2015). Tregs use the transcription factor FoxP3 to reprogram metabolism, which suppresses glycolysis, promotes OXPHOS, and causes Tregs to acquire resistance to lactate suppression (Angelin et al, 2017). Suggesting their role in immunosuppression and cancer cell evasion from effector T‐cells in the TME, and why lactate is correlated with poor patient survival. Whereas high expression of LDH1 and LDH5 and lactate transporters (MCT1 and MCT2) are inversely correlated with survival in colorectal cancer patients (Roseweir et al, 2019).
Tumor‐derived lactate induces expression of VEGF and arginase‐1 in tumor‐associated macrophages (TAMs), which are involved in suppressing T‐cell responses, and stimulate TAM polarization to an immunosuppressive and angiogenic phenotype (Colegio, 2016). Notably, induction of both VEGF and arginase‐1 by both hypoxia and tumor supernatant is abrogated in mice with HIF‐1α deficient macrophages. Moreover, lactate is sufficient to polarize macrophages to an M2 phenotype in a HIF‐1α‐dependent manner (Colegio, 2016).
Tryptophan
Access to nutrients is fundamental for proliferating cells; therefore, controlling nutrient availability is a strategy of tumor cells to manipulate immune cell function (Munn & Mellor, 2016). One approach is mediated by the enzyme indoleamine 2,3‐dioxygenase (IDO), which consumes tryptophan to produce kynurenine (Jovanovic et al, 2020). IDO affects immune cell functions by depleting extracellular tryptophan and generating kynurenine, a natural ligand for the aryl hydrocarbon receptor (AHR). This receptor, present on several immune cells has various effects on dendritic cells and T‐cells. In dendritic cells, AHR reduces MHC class II expression and impairs the induction of cytokines that promote polarization of cytotoxic T‐cells, while inducing expression of the regulatory T cell marker FoxP3 (Gandhi et al, 2010; Rothhammer & Quintana, 2019). Depletion of extracellular tryptophan pools can activate stress response kinase GCN2 which senses AA restriction. GCN2 activation in T‐cells will impair their proliferation, and promote differentiation of naïve CD4+ T‐cells to regulatory T‐cells (Munn & Mellor, 2013). Moreover, IDO‐dependent GCN2 activation inhibits macrophage‐dependent innate inflammatory responses to apoptotic cells (Ravishankara et al, 2015) and stimulates activity of immunosuppressive CD19+ dendritic cells (Manlapat et al, 2007). IDO expression in macrophages is strongly upregulated by tumor cell secreted CCL20 in hepatocellular carcinomas (HCCs). CCL20 is a chemokine that is transcriptionally activated by HIF‐1α in human HCC cells (Ye et al, 2016). Interestingly, a recent report demonstrated that hypoxia‐induced IDO expression in DCs is independent of HIF‐1α activity, while HIF‐1α signaling regulated IDO production in normoxic conditions (Song et al, 2018).
In addition, tryptophan can also be oxidized to kynurenine by tryptophan‐2,3‐dioxygenase (TDO2). This enzyme is closely related to IDO and is the second most downregulated enzyme in hypoxic glioblastoma cells (Mohapatra et al, 2019). TDO2 expression in hypoxia is controlled by HIF‐1α. While stabilization of HIF‐1α through dimethyloxalylglycine (DMOG) and cobalt chloride treatment reduced TDO2 expression, knockdown of HIF‐1α rescued TDO2 levels. In normoxic conditions, TDO2 expressing glioblastoma cells suppress T‐cell proliferation, while hypoxia restored the immune response, most likely through the reduced production of kynurenine and increased availability of tryptophan (Mohapatra et al, 2019).
Glutamine
Extracellular glutamine is another essential AA critically required by several cancer types. Triple‐negative breast cancer (TNBC) cells (which lack estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2) are glutamine addicted compared to other breast cancer subtypes (Altman et al, 2016). TNBC cells often express high levels of GLS, a rate‐limiting enzyme in glutaminolysis converting glutamine to glutamate, and ASCT2 (SLC1A5), a glutamine transporter (Edwards et al, 2021). Similar observations were made using non‐small cell lung cancer (NSCLC), breast cancer, and brain tumor cells (Choi & Park, 2018). They all exhibit a high dependency on glutamine for growth and survival with upregulated SLC1A5 expression (Choi & Park, 2018). Like tumor cells, activation of T‐cells depends on extracellular glutamine pools. T‐cells increase glutamine uptake and metabolism to support redox homeostasis, nucleotide synthesis, and AA production (Buck et al, 2015). T‐cell activation leads to increased expression of glutamine transporters such as SNAT1 (SLC38A1), SNAT2 (SLC38A2), and SLC1A5 (Edwards et al, 2021). Several of these are upregulated in hypoxia through HIF activity: e.g. SNAT1 and SNAT2 are upregulated in brown adipose tissue and MCF7 breast cancer cells, respectively (Horie et al, 2017; Morotti et al, 2019); and SLC1A5 is HIF‐2α dependent in pancreatic carcinoma cells (Yoo et al, 2020; Nachef et al, 2021).
Reduced extracellular glutamine levels significantly impair T‐cell proliferation and cytokine production. Importantly, Tregs maintain reserve respiratory capacity in low glutamine levels and even thrive in its absence (Klysz et al, 2015). Interestingly, when compared to other leukocytes such as lymphocytes and macrophages, neutrophils consume glutamine at the highest rates (Cruzat et al, 2018). Neutrophils play a major role linking inflammation and cancer and are actively involved in progression and metastasis (Wu et al, 2019). They possess unique structures called neutrophilic extracellular traps (NETs) composed of uncondensed chromatin and antimicrobial factors. NET activity requires ROS generation, which is highly dependent on glutamine (Branzk et al, 2014).
Glutamine also functions in modulating specific macrophage properties. For example, glutamine is especially effective in M2 polarization in mice. The production of α‐ketoglutarate and induction of glutamine‐dependent HBP is essential for GlcNAc synthesis, and critical for immune suppressive M2 macrophage polarization because it is responsible for glycosylation of the M2 marker proteins: macrophage mannose receptor and macrophage galactose binding lectin (Ren et al, 2019). Accordingly withdrawal of glutamine reduces expression of M2 markers: Arginase 1, Ym1 (Chitinase‐like 3), Retnla (Resistin‐like alpha 1), and Mrc1 (Mannose receptor C type 1; Liu et al, 2017). Glutamine is also essential for the polarization of immune‐stimulating M1 macrophages. Succinate produced by the γ‐aminobutyric acid (GABA) shunt, or glutamine‐dependent anaplerosis promotes M1 polarization. Interestingly, succinate promotes HIF‐1α stabilization through inhibition of the enzymatic activity of PHDs (Tannahill et al, 2013; Ren et al, 2019). Moreover, succinate indirectly promotes additional HIF‐1α stabilization through the production of reactive oxygen species (Mills et al, 2016). In contrast with the notion that glutamine can stimulate M1 polarization, a previous report stated that removal of glutamine did not affect the capacity for M1 polarization as measured by NOS2 (Jha et al, 2015). This suggests that although glutamine could stimulate M1 polarization, it is not an essential factor.
L‐Arginine
A recent report demonstrated that L‐arginine is the most depleted metabolite in tumor interstitial fluid in pancreatic ductal adenocarcinoma (PDAC; Sullivan et al, 2019). Similarly, several cancers suppress their urea cycle through epigenetic regulation of one or more key urea cycle enzymes (Keshet et al, 2018, 2020). Suppression of urea cycle activity in argininosuccinate synthase 1 (ASS1)‐deficient cancers is essential for the tumorigenic features of these cells (Rabinovich et al, 2015). ASS1 converts citrulline into argininosuccinate at the expense of intracellular aspartate, an essential factor for de novo pyrimidine synthesis (Rabinovich et al, 2015). Suppression of the urea cycle, a predominant source of L‐arginine in these cancers, results in L‐arginine auxotrophy (Alexandrou et al, 2018). PDAC, a highly hypoxic cancer, maintains a functional urea cycle suggesting the observed L‐arginine depletion could result from infiltration of L‐arginine consuming stromal cells. Macrophages are often present in hypoxic tissues, which strongly affects macrophage function. Moreover, most transcriptional responses are mediated by HIF‐1α (Takeda et al, 2010). Among HIF‐1α targets is inducible nitric oxide synthase (iNOS), which is primarily expressed in M1 polarized macrophages (Peyssonnaux et al, 2005). iNOS produces NO by metabolizing its substrate, L‐arginine. M2 macrophages primarily express arginase 1, which generates ornithine and urea at the expense of L‐arginine (Bronte & Zanovello, 2005). Arginase 1 could therefore compete with iNOS and regulate NO production by limiting L‐arginine availability in the extracellular environment (Takeda et al, 2010). High consumption rates of extracellular L‐arginine pools could strongly reduce L‐arginine availability in the tumor microenvironment.
In addition to macrophages, T‐cells also rely on exogenous L‐arginine for functionality (Geiger et al, 2016). Antigen‐specific naïve T‐cells proliferate extensively and acquire different types of effector functions. To support proliferation and survival, activated T‐cells adapt their metabolism to ensure sufficient energy and biomass production. Therefore, activated T‐cells consume large amounts of L‐arginine and it is converted into downstream metabolites (Geiger et al, 2016). Moreover, addition of L‐arginine significantly increases intracellular levels of several other metabolites (e.g. intermediates of the urea cycle, nucleotides, and amino acids) and induces a metabolic switch from glycolysis to OXPHOS (Geiger et al, 2016). Furthermore, arginase inhibition in human T‐cells does not affect T‐cell proliferation suggesting that the downstream fate of L‐arginine is less important in T cells than the levels of free L‐arginine. Interestingly only long‐term L‐arginine depletion affects T‐cell proliferation, while short‐term L‐arginine withdrawal has a negative impact on T‐cell survival instead, without affecting proliferation (Geiger et al, 2016).
Clinical implications of HIF inhibition and future perspectives
As stated above, HIF transcription factors play essential roles in tumorigenesis. Numerous studies have evaluated HIF expression and patient outcome. Most results showed that high HIF‐1α or HIF‐2α expression is associated with poor patient prognosis. Overexpression of HIF‐2α in oral squamous cell carcinoma is associated with early recurrence within 2 years of diagnosis (Lim et al, 2017). In non‐small cell lung cancer patients, HIF‐2α expression is associated with tumor progression and larger tumor sizes (Gao et al, 2017); and medullary cancer patients with high HIF‐1α levels show reduced 5‐year overall survival rates compared to patients with low HIF‐1α levels (Lodewijk et al, 2017).
These clinical findings suggest that targeting members of the HIF signaling pathway could have significant therapeutic benefits, especially the subunits HIF‐1α and HIF‐2α. It is therefore unsurprising that extensive research has been performed to investigate HIF inhibiting strategies (Albadari et al, 2019). Several HIF inhibitors have been proposed and evaluated in pre‐clinical and clinical models (Table 2; Samec et al, 2021). These therapeutic agents can be classified, based on their mechanism of HIF repression: as agents regulating gene expression, those modulating cascades associated with protein dimerization and accumulation, and molecules inhibiting transcriptional activity of HIF or DNA binding (Samec et al, 2021). The topoisomerase 1 inhibitor topotecan blocking HIF‐1α mRNA expression, and EZN‐2968, an antisense oligonucleotide targeting HIF‐1α mRNA suppresses HIF‐1α expression, are examples for the first classification. In addition, 2‐methoxyestrdiol and KC7F2 which affect protein synthesis markedly suppresses HIF‐1α in lung cancer and HIF‐2α protein synthesis in glioma, prostate, and breast cancer (Narita et al, 2009; Womeldorff et al, 2014; Aquino‐Gálvez et al, 2016). Moreover, histone deacetylase inhibitors, such as vorinostat induce anti‐hypoxic activity by degrading the HIF‐1α subunit in liver cancer‐derived cells (Hutt et al, 2014). An upcoming promising way to inhibit HIF activity is the disruption of HIF‐α and HIF‐β dimerization. Recent reports showed an essential role for a cyclic peptide (cyclo‐CLLFVY) via selective inhibition of HIF‐1α dimerization in various cell types, without affecting HIF‐2α activity (Miranda et al, 2013). Similarly, acriflavine, another compound disrupting HIF‐1α dimerization, targeted HIF‐1α mediated pathways and induced cell death and apoptosis in several glioma cell lines (Mangraviti et al, 2017). Notably, well‐known chemotherapeutics such as daunorubicin and doxorubicin, also exerts anti‐hypoxic activity through inhibition of HIF‐1 biding to hypoxia response elements (HREs). Chetomin inhibited the transcriptional activity of HIF‐1α by targeting the HIF‐1α/p300 complex in multiple myeloma cell lines (Viziteu et al, 2016). It is important to emphasize that only a subset of HIF inhibitors directly disrupt HIF expression, while others decrease HIF expression through indirect means. Nevertheless, these inhibitors have been investigated to alter HIF expression, and likely will disrupt metabolic cross‐talk as mentioned above.
Table 2.
Current HIF inhibitors in pre‐clinical and clinical studies.
| Inhibitor | Target | Mechanism of action | Status | References |
|---|---|---|---|---|
| Topotecan | HIF‐1α | Topoisomerase mediated prevention of HIF‐1α protein accumulation | Marketed | Beppu et al (2005) |
| EZN‐2968 | HIF‐1α | HIF‐1α RNA antagonist | Phase I | Jeong et al (2014) |
| 2‐methoxyestradiol | HIF‐1α | Small molecule impairs HIF‐1α nuclear translocation by binding to a buried pocket in the (PAS)‐B domain | Phase II/Suspended | Aquino‐Gálvez et al (2016) |
| KC7F2 | HIF‐1α | Inhibitor of HIF‐1α translation | Preclinical | Narita et al (2009) |
| vorinostat | HIF‐1α | Histone deacetylase inhibitor | FDA Approved | Krug et al (2011) and Hutt et al (2014) |
| cyclo‐CLLFVY | HIF‐1α | Binds to PAS‐B domain of HIF‐1α inhibiting dimerization | Preclinical | Miranda et al (2013) |
| acriflavine | HIF‐1α | Prevents dimerization of HIF | Preclinical | Mangraviti et al (2017) |
| daunorubicin | HIF‐1α | Topoisomerase mediated prevention of HIF‐1α protein accumulation | FDA Approved | Lee et al (2009) |
| chetomin | HIF‐1α | Inhibits HIF‐1α/p300 interaction | Preclinical | Viziteu et al (2016) |
| 2ME2 NCD (panzem) | HIF‐1α | Estradiol mimetic inhibiting HIF translation | Suspended/discontinued | Rajkumar et al (2007), Kulke et al (2011), Bruce et al (2012), and Fallah & Rini (2019) |
| 17‐AAG (tanespimycin) | HIF‐1α | Induction of proteasomal degradation of HIF‐1α | Suspended/discontinued | Ronnen et al (2006), Heath et al (2008), and Oki et al (2012) |
| IDF‐11774 | HIF‐1α | HSP70 inhibitor suppressing HIF‐1α accumulation | Phase I | Ban et al (2017) |
| PT2399 | HIF‐2α | Disrupts HIF‐2α/ ARNT binding | Preclinical | Chen et al (2016) and Cho et al (2016) |
| Belzutifan (MK‐6482) | HIF‐2α | Disrupts HIF‐2α/ ARNT binding | FDA Approved | Jonasch et al (2021) |
Several inhibitors are currently under investigation in clinical trials. 2ME2 NCD (panzem), an endogenous metabolite of estradiol which inhibits HIF‐1α protein synthesis and transcriptional activity, has been investigated in several phase II clinical trials including trials in patients with multiple myeloma (Rajkumar et al, 2007), metastatic renal cell carcinoma (Bruce et al, 2012), and locally unresectable or metastatic carcinoid neuroendocrine tumor (Kulke et al, 2011). However, responses varied based on tumor types and some trials were terminated due to toxicity events (Fallah & Rini, 2019). 17‐AAG (tanespimycin), a potent inhibitor of heat shock protein‐90 which increases degradation of HIF‐1α, has been evaluated in phase II trial with patients suffering from anaplastic large cell lymphoma, classical Hodgkin lymphoma, mantle cell lymphoma (Oki et al, 2012), metastatic renal cell carcinoma (Ronnen et al, 2006), and castration‐resistant metastatic prostate cancer (Heath et al, 2008) and demonstrated tumor size reduction (39%) in relapsed lymphoma patients (Oki et al, 2012). Another inhibitor that reached clinical trials is Vorinostat, a synthetic hydroxamic acid derivative which inhibits histone deacetylase (HDAC), and thereby increased degradation of HIF‐1α. This inhibitor showed promising results and has been advanced to phase III clinical trials for patients suffering from advanced malignant pleural mesothelioma (Krug et al, 2011).
The efficacy of HIF‐2α inhibitors was demonstrated by two studies with the HIF‐2α specific inhibitor, PT2399. They show that PT2399 causes tumor regression in patient derived xenograft tumor model of primary and metastatic pVHL‐defective clear cell renal cell carcinoma (Chen et al, 2016; Cho et al, 2016). The FDA has granted Breakthrough Therapy designation to the HIF‐2α inhibitor MK‐6482 (belzutifan) for the treatment of patients with RCC with nonmetastatic RCC tumors less than three centimeters in size, unless immediate surgery is required. The FDA also granted orphan drug designation to MK‐6482 for VHL disease.
More HIF inhibitors are being investigated, but despite many efforts to bring HIF inhibitors into the clinic, there is currently only one FDA‐approved drug that directly targets HIF‐dependent pathways, which is Merck's HIF‐2α inhibitor belzutifan for the treatment of patients with Von Hippel–Lindau (VHL) disease‐associated tumors. The difficulty bringing HIF inhibitors to the clinic could be at least in part attributed to the ability for HIF to control a vast network of essential signaling pathways and mechanisms with overlapping functions in other cells in the tumor microenvironment (Noman et al, 2019).
Recent studies investigated the effect of HIF inhibition on tumor metabolism and potential of targeting HIF‐dependent metabolic changes an anti‐cancer therapy. Ban et al (2017) demonstrated that IDF‐11774 (a HIF‐1α inhibitor) treatment reduced colorectal carcinoma HCT116 cell growth both in vitro and in vivo. HIF‐1α inhibition decreased the extracellular acidification rate, glucose uptake, and lactate secretion, which suggest that this could strongly affect conditions in the tumor microenvironment (Ban et al, 2017). Targeting hypoxia responses in the tumor microenvironment has been proposed as a novel therapeutic strategy to improve anti‐cancer immunotherapy (Wang et al, 2021). For instance, increased glycolysis in cancer cells can deplete extracellular glucose pools in the tumor microenvironment. Glucose consumption by murine sarcoma cells impaired T‐cell activity and their effector functions, which translated into immune resistance to adoptive T‐cell therapy in melanoma. According, inhibition of glycolysis in melanoma cells enhanced T‐cell mediated antitumor immunity (Cascone et al, 2018). HIF inhibitors such as IDF‐11774, could therefore increase immune responses in the tumor.
Increased glucose availability and reduced levels of the immunosuppressive metabolite lactate could potentially stimulate immune responses. Further research on this subject may benefit therapeutic strategy design and mediate immune responses to immune checkpoint inhibitors. HIF activity has been reported to increase protein death ligand 1 (PD‐L1) expression in various tumors (Bocanegra et al, 2019; Chen et al, 2019; Ferrata et al, 2019). PD‐L1 expressed by tumors binds to PD‐1 receptors on activated T‐ and other immune cells, leading to suppression of immune cells activity (Sharma & Allison, 2015). In addition, multiple HRE's were found at the promotor of HLA‐G (Yaghi et al, 2016), which is a non‐classical MHC‐I molecule expressed by several tumor types including melanoma, glioblastoma, colorectal, ovarian and cervical tumors (Yi et al, 2020). This molecule has immune suppressive properties by its ability to bind to specific receptors expressed by several immune cells, including B cells, T cells, NK, myelomonocytic cells, dendritic cells, monocytes, and macrophages (Noman et al, 2019).
These recent discoveries showing that hypoxia negatively impact tumor immune responses provide a major opportunity for innovative combination approaches. Moreover, anti‐HIF and immune‐checkpoint inhibitor combinations could not only benefits patients through the impact on expression of immune checkpoints (e.g., PD‐L1, PD‐1, HLA‐G), but HIF inhibition might modulate TME nutrient compositions favoring immune responses which could by further stimulated by immune check‐point inhibition. Further research is needed to address these hypotheses and could potentially lead to novel successful therapeutic approaches.
Concluding remarks
Since Gregg Semenza first identified the genes that encode HIF‐1α and HIF‐β (ARNT) in 1995, many advances have been made and HIF‐mediated metabolic responses to hypoxia have been well characterized on cellular level, its effect on metabolic properties in a more complex system such as the tumor microenvironment is far from fully elucidated. Recent reports show that HIF‐mediated metabolic changes have important consequences for tumor cell glucose availability and lactate levels. Moreover, many AA transporters and other major metabolites are regulated by HIF activity. All these nutrients have immunomodulating properties and it seems that HIF‐mediated metabolic alterations mainly induce an immunosuppressive tumor microenvironment. Future endeavors to investigate the effect of HIF activity in both tumor cells and tumor infiltrating immune cells on the nutrient composition of the TME, will provide essential information which could have substantial consequences for therapeutic design and could improve our understanding of the metabolic competition and symbiosis that exist in hypoxic areas of the tumor.
Author contributions
M Celeste Simon: Conceptualization. Rindert Missiaen: Conceptualization; writing—original draft. Nicholas P Lesner: Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
RM, NPL, MCS wrote and reviewed the manuscript.
Disclosure and competing interests statement
The authors declare no competing interests.
Acknowledgements
The authors thank the entire Simon laboratory for comments and discussion on the manuscript. This work was supported by the Belgian American Educational Foundation (B.A.E.F.) to R.M., and National Cancer Institute (NCI) grants T32CA009140 to N.P.L, and P01 CA104838 and R35 CA197602 to M.C.S.
The EMBO Journal (2023) 42: e112067
This review is part of the Cancer Review Series.
References
- Akella NM, Ciraku L, Reginato MJ (2019) Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol 17: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Tameemi W, Dale TP, Al‐Jumaily RMK, Forsyth NR (2019) Hypoxia‐modified cancer cell metabolism. Front Cell Dev Biol 7: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albadari N, Deng S, Li W (2019) The transcriptional factors HIF‐1 and HIF‐2 and their novel inhibitors in cancer therapy. Expert Opin Drug Discov 14: 667–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrou C, Al‐Aqbi SS, Higgins JA, Boyle W, Karmokar A, Andreadi C, Luo JL, Moore DA, Viskaduraki M, Blades M et al (2018) Sensitivity of colorectal cancer to arginine deprivation therapy is shaped by differential expression of urea cycle enzymes. Sci Rep 8: 12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman BJ, Stine ZE, Dang CV (2016) From Krebs to clinic: glutamine metabolism to cancer therapy. Nat Rev Cancer 16: 619–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Curto E, Milligan G (2016) Metabolism meets immunity: the role of free fatty acid receptors in the immune system. Biochem Pharmacol 114: 3–13 [DOI] [PubMed] [Google Scholar]
- Angelin A, Gil‐de‐Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ, Kopinski PK, Wang L et al (2017) Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab 25: 1282–1293.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquino‐Gálvez A, González‐Ávila G, Delgado‐Tello J, Castillejos‐López M, Mendoza‐Milla C, Zúñiga J, Checa M, Maldonado‐Martínez HA, Trinidad‐López A, Cisneros J et al (2016) Effects of 2‐methoxyestradiol on apoptosis and HIF‐1α and HIF‐2α expression in lung cancer cells under normoxia and hypoxia. Oncol Rep 35: 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsham AM, Howell JJ, Simon MC (2003) A novel hypoxia‐inducible factor‐independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem 278: 29655–29660 [DOI] [PubMed] [Google Scholar]
- Ban HS, Kim BK, Lee H, Kim HM, Harmalkar D, Nam M, Park SK, Lee K, Park JT, Kim I et al (2017) The novel hypoxia‐inducible factor‐1α inhibitor IDF‐11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis 8: e2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa AM, Martel F (2020) Targeting glucose transporters for breast cancer therapy: the effect of natural and synthetic compounds. Cancers 12: 154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoszewska S, Collawn JF (2020) Unfolded protein response (UPR) integrated signaling networks determine cell fate during hypoxia. Cell Mol Biol Lett 25: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier UH, Angelin A, Akimova T, Wang L, Liu Y, Xiao H, Koike MA, Hancock SA, Bhatti TR, Han R et al (2015) Essential role of mitochondrial energy metabolism in Foxp3+ T‐regulatory cell function and allograft survival. FASEB J 29: 2315–2326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belisario DC, Kopecka J, Pasino M, Akman M, De Smaele E, Donadelli M, Riganti C (2020) Hypoxia dictates metabolic rewiring of tumors: implications for Chemoresistance. Cell 9: 2598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beppu K, Nakamura K, Linehan WM, Rapisarda A, Thiele CJ (2005) Topotecan blocks hypoxia‐inducible factor‐1α and vascular endothelial growth factor expression induced by insulin‐like growth factor‐I in Neuroblastoma cells. Cancer Res 65: 4775–4781 [DOI] [PubMed] [Google Scholar]
- Berk JL, Hatch CA, Goldstein RH (2000) Hypoxia inhibits amino acid uptake in human lung fibroblasts. J Appl Physiol 89: 1425–1431 [DOI] [PubMed] [Google Scholar]
- Betsunoh H, Fukuda T, Anzai N, Nishihara D, Mizuno T, Yuki H, Masuda A, Yamaguchi Y, Abe H, Yashi M et al (2013) Increased expression of system large amino acid transporter (LAT)‐1 mRNA is associated with invasive potential and unfavorable prognosis of human clear cell renal cell carcinoma. BMC Cancer 13: 509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocanegra A, Fernandez‐Hinojal G, Zuazo‐Ibarra M, Arasanz H, Garcia‐Granda MJ, Hernandez C, Ibañez M, Hernandez‐Marin B, Martinez‐Aguillo M, Lecumberri MJ et al (2019) PD‐L1 expression in systemic immune cell populations as a potential predictive biomarker of responses to PD‐L1/PD‐1 blockade therapy in lung cancer. Int J Mol Sci 20: 1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzk N, Lubojemska A, Hardison SE, Wang Q, Gutierrez MG, Brown GD, Papayannopoulos V (2014) Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat Immunol 15: 1017–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronte V, Zanovello P (2005) Regulation of immune responses by L‐arginine metabolism. Nat Rev Immunol 5: 641–654 [DOI] [PubMed] [Google Scholar]
- Bruce JY, Eickhoff J, Pili R, Logan T, Carducci M, Arnott J, Treston A, Wilding G, Liu G (2012) A phase II study of 2‐methoxyestradiol nanocrystal colloidal dispersion alone and in combination with sunitinib malate in patients with metastatic renal cell carcinoma progressing on sunitinib malate. Invest New Drugs 30: 794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK (2003) Oxygen sensing in the hypoxic response pathway: regulation of the hypoxia‐inducible transcription factor. Genes Dev 17: 2614–2623 [DOI] [PubMed] [Google Scholar]
- Buck MD, O'Sullivan D, Pearce EL (2015) T cell metabolism drives immunity. J Exp Med 212: 1345–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cariello M, Piccinin E, Moschetta A (2021) Transcriptional regulation of metabolic pathways via lipid‐sensing nuclear receptors PPARs, FXR, and LXR in NASH. Cell Mol Gastroenterol Hepatol 11: 1519–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P et al (1998) Role of HIF‐1α in hypoxia‐mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394: 485–490 [DOI] [PubMed] [Google Scholar]
- Carroll B, Korolchuk VI, Sarkar S (2015) Amino acids and autophagy: cross‐talk and co‐operation to control cellular homeostasis. Amino Acids 47: 2065–2088 [DOI] [PubMed] [Google Scholar]
- Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, Williams LJ, Wang Z, Bristow CA, Carugo A et al (2018) Increased tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab 27: 977–987.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, Van Der Windt GJW et al (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162: 1229–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaveroux C, Sarcinelli C, Barbet V, Belfeki S, Barthelaix A, Ferraro‐Peyret C, Lebecque S, Renno T, Bruhat A, Fafournoux P et al (2016) Nutrient shortage triggers the hexosamine biosynthetic pathway via the GCN2‐ATF4 signalling pathway. Sci Rep 6: 27278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Sang N (2016) Hypoxia‐inducible Factor‐1: a critical player in the survival strategy of stressed cells. J Cell Biochem 117: 267–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Xu M, Nagati JS, Hogg RT, Das A, Gerard RD, Garcia JA (2015a) The acetate/ACSS2 switch regulates HIF‐2 stress signaling in the tumor cell microenvironment. PLoS One 10: e0116515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yin C, Lao T, Liang D, He D, Wang C, Sang N (2015b) AMPK‐HDAC5 pathway facilitates nuclear accumulation of HIF‐1α and functional activation of HIF‐1 by deacetylating Hsp70 in the cytosol. Cell Cycle 14: 2520–2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia‐Jimenez A, Homayoun F, Ma Y, Patel N, Yell P et al (2016) Targeting renal cell carcinoma with a HIF‐2 antagonist. Nature 539: 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, Siegwart DJ, Wan Y (2017) Gpr132 sensing of lactate mediates tumor‐macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA 114: 580–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Crabill GA, Pritchard TS, McMiller TL, Wei P, Pardoll DM, Pan F, Topalian SL (2019) Mechanisms regulating PD‐L1 expression on tumor and immune cells. J Immunother Cancer 7: 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, Carvo I, Signoretti S, Bruick RK, Josey JA et al (2016) On‐target efficacy of a HIF‐2α antagonist in preclinical kidney cancer models. Nature 539: 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YK, Park KG (2018) Targeting glutamine metabolism for cancer treatment. Biomol Ther 26: 19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegio OR (2016) Lactic acid polarizes macrophages to a tumor‐promoting state. Oncoimmunology 5: e1014774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruzat V, Rogero MM, Keane KN, Curi R, Newsholme P (2018) Glutamine: metabolism and immune function, supplementation and clinical translation. Nutrients 10: 1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Cruz‐López KG, Castro‐Muñoz LJ, Reyes‐Hernández DO, García‐Carrancá A, Manzo‐Merino J (2019) Lactate in the regulation of tumor microenvironment and therapeutic approaches. Front Oncol 9: 1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui XG, Han ZT, He SH, Wu X, Chen TR, Shao CH, Chen DL, Su N, Chen YM, Wang T et al (2017) HIF1/2α mediates hypoxia‐induced LDHA expression in human pancreatic cancer cells. Oncotarget 8: 24840–24852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele S, Giacomelli C, Zappelli E, Granchi C, Trincavelli ML, Minutolo F, Martini C (2015) Lactate dehydrogenase‐a inhibition induces human glioblastoma multiforme stem cell differentiation and death. Sci Rep 5: 15556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Chandel NS (2016) Fundamentals of cancer metabolism. Sci Adv 2: e1600200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW (2008) Hypoxia regulates TSC1/2‐mTOR signaling and tumor suppression through REDD1‐mediated 14‐3‐3 shuttling. Genes Dev 22: 239–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie SL (2009) The role of hypoxia in development of the mammalian embryo. Dev Cell 17: 755–773 [DOI] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S et al (2010) Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell 39: 171–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S et al (2021) Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple‐negative breast cancer. J Clin Invest 131: e140100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Zoncu R, Chang S, Gumper I, Snitkin H, Wolfson RL, Kirak O, Sabatini DD, Sabatini DM (2013) Regulation of mTORC1 by the rag GTPases is necessary for neonatal autophagy and survival. Nature 493: 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efeyan A, Comb WC, Sabatini DM (2015) Nutrient‐sensing mechanisms and pathways. Nature 517: 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elorza A, Soro‐Arnáiz I, Meléndez‐Rodríguez F, Rodríguez‐Vaello V, Marsboom G, de Cárcer G, Acosta‐Iborra B, Albacete‐Albacete L, Ordóñez A, Serrano‐Oviedo L et al (2012) HIF2α acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 48: 681–691 [DOI] [PubMed] [Google Scholar]
- Fallah J, Rini BI (2019) HIF inhibitors: status of current clinical development. Curr Oncol Rep 21: 6 [DOI] [PubMed] [Google Scholar]
- Faubert B, Boily G, Izreig S, Griss T, Samborska B, Dong Z, Dupuy F, Chambers C, Fuerth BJ, Viollet B et al (2013) AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab 17: 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrata M, Schad A, Zimmer S, Musholt TJ, Bahr K, Kuenzel J, Becker S, Springer E, Roth W, Weber MM et al (2019) PD‐L1 expression and immune cell infiltration in gastroenteropancreatic (GEP) and non‐GEP neuroendocrine neoplasms with high proliferative activity. Front Oncol 9: 343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer CM, Lynch TP, Sodi VL, Falcone JN, Schwab LP, Peacock DL, Vocadlo DJ, Seagroves TN, Reginato MJ (2014) O‐GlcNAcylation regulates cancer metabolism and survival stress signaling via regulation of the HIF‐1 pathway. Mol Cell 54: 820–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald E, Roberts J, Tennant DA, Boardman JP, Drake AJ (2021) Metabolic adaptations to hypoxia in the neonatal mouse forebrain can occur independently of the transporters SLC7A5 and SLC3A2. Sci Rep 11: 9092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick SF, Tambuwala MM, Bruning U, Schaible B, Scholz CC, Byrne A, O'Connor A, Gallagher WM, Lenihan CR, Garvey JF et al (2011) An intact canonical NF‐κB pathway is required for inflammatory gene expression in response to hypoxia. J Immunol 186: 1091–1096 [DOI] [PubMed] [Google Scholar]
- Fotiadis D, Kanai Y, Palacín M (2013) The SLC3 and SLC7 families of amino acid transporters. Mol Aspects Med 34: 139–158 [DOI] [PubMed] [Google Scholar]
- Fox CJ, Hammerman PS, Thompson CB (2005) Fuel feeds function: energy metabolism and the T‐cell response. Nat Rev Immunol 5: 844–852 [DOI] [PubMed] [Google Scholar]
- Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha‐Hicks P, Rathmell JC, Makowski L (2014) Metabolic reprogramming of macrophages. J Biol Chem 289: 7884–7896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, Kozoriz D, Weiner HL, Quintana FJ (2010) Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell‐like and Foxp3+ regulatory T cells. Nat Immunol 11: 846–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao ZJ, Wang Y, Yuan WD, Yuan JQ, Yuan K (2017) HIF‐2α not HIF‐1α overexpression confers poor prognosis in non‐small cell lung cancer. Tumour Biol 39: 101042831770963 [DOI] [PubMed] [Google Scholar]
- Garcia‐Bermudez J, Williams RT, Guarecuco R, Birsoy K (2020) Targeting extracellular nutrient dependencies of cancer cells. Mol Metab 33: 67–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge T, Yang J, Zhou S, Wang Y, Li Y, Tong X (2020) The role of the pentose phosphate pathway in diabetes and cancer. Front Endocrinol 11: 365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, Kogadeeva M, Picotti P, Meissner F, Mann M et al (2016) L‐arginine modulates T cell metabolism and enhances survival and anti‐tumor activity. Cell 167: 829–842.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerriets VA, Kishton RJ, Nichols AG, MacIntyre AN, Inoue M, Ilkayeva O, Winter PS, Liu X, Priyadharshini B, Slawinska ME et al (2015) Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J Clin Invest 125: 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimarães‐Camboa N, Stowe J, Aneas I, Sakabe N, Cattaneo P, Henderson L, Kilberg MS, Johnson RS, Chen J, McCulloch AD et al (2015) HIF1α represses cell stress pathways to allow proliferation of hypoxic fetal Cardiomyocytes. Dev Cell 33: 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haining Z, Kawai N, Miyake K, Okada M, Okubo S, Zhang X, Fei Z, Tamiya T (2012) Relation of LAT1/4F2hc expression with pathological grade, proliferation and angiogenesis in human gliomas. BMC Clin Pathol 12: 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Coussens LM (2012) Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell 21: 309–322 [DOI] [PubMed] [Google Scholar]
- Hao LS, Liu Q, Tian C, Zhang DX, Wang B, Zhou DX, Li ZP, Yuan ZX (2019) Correlation and expression analysis of hypoxia‐inducible factor 1α, glucose transporter 1 and lactate dehydrogenase 5 in human gastric cancer. Oncol Lett 18: 1431–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargadon KM (2017) Strategies to improve the efficacy of dendritic cell‐based immunotherapy for melanoma. Front Immunol 8: 1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley SA, Ross FA, Chevtzoff C, Green KA, Evans A, Fogarty S, Towler MC, Brown LJ, Ogunbayo OA, Evans AM et al (2010) Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11: 554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP et al (2008) A phase II trial of 17‐allylamino‐17‐demethoxygeldanamycin in patients with hormone‐refractory metastatic prostate cancer. Clin Cancer Res 14: 7940–7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Meeth KM, Tsui YC, Srivastava B, Bosenberg MW, Kaech SM (2014) Immune‐based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNγ. Cancer Res 74: 3205–3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, MacIntyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC et al (2015) Phosphoenolpyruvate is a metabolic checkpoint of anti‐tumor T cell responses. Cell 162: 1217–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho JJD, Wang M, Audas TE, Kwon D, Carlsson SK, Timpano S, Evagelou SL, Brothers S, Gonzalgo ML, Krieger JR et al (2016) Systemic reprogramming of translation efficiencies on oxygen stimulus. Cell Rep 14: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoefflin R, Harlander S, Schäfer S, Metzger P, Kuo F, Schönenberger D, Adlesic M, Peighambari A, Seidel P, Chen C et al (2020) HIF‐1α and HIF‐2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat Commun 11: 4111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppe S, Bierhoff H, Cado I, Weber A, Tiebe M, Grummt I, Voit R (2009) AMP‐activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci USA 106: 17781–17786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horie T, Fukasawa K, Iezaki T, Park G, Onishi Y, Ozaki K, Kanayama T, Hiraiwa M, Kitaguchi Y, Kaneda K et al (2017) Hypoxic stress upregulates the expression of Slc38a1 in brown adipocytes via hypoxia‐inducible factor‐1α. Pharmacology 101: 64–71 [DOI] [PubMed] [Google Scholar]
- Huang M, Yang L, Peng X, Wei S, Fan Q, Yang S, Li X, Li B, Jin H, Wu B et al (2020) Autonomous glucose metabolic reprogramming of tumour cells under hypoxia: opportunities for targeted therapy. J Exp Clin Cancer Res 39: 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Zhan L, Yanxiang Guo J et al (2017) Glucose feeds the TCA cycle via circulating lactate. Nature 551: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutt DM, Roth DM, Vignaud H, Cullin C, Bouchecareilh M (2014) The histone deacetylase inhibitor, vorinostat, represses hypoxia inducible factor 1 alpha expression through translational inhibition. PLoS One 9: e106224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanova IG, Park CV, Yemm AI, Kenneth NS (2018) PERK/eIF2α signaling inhibits HIF‐induced gene expression during the unfolded protein response via YB1‐dependent regulation of HIF1α translation. Nucleic Acids Res 46: 3878–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamali S, Klier M, Ames S, Felipe Barros L, McKenna R, Deitmer JW, Becker HM (2015) Hypoxia‐induced carbonic anhydrase IX facilitates lactate flux in human breast cancer cells by non‐catalytic function. Sci Rep 5: 13605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W, Rapisarda A, Park SR, Kinders RJ, Chen A, Melillo G, Turkbey B, Steinberg SM, Choyke P, Doroshow JH et al (2014) Pilot trial of EZN‐2968, an antisense oligonucleotide inhibitor of hypoxia‐inducible factor‐1 alpha (HIF‐1α), in patients with refractory solid tumors. Cancer Chemother Pharmacol 73: 343–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha AK, Huang SCC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B et al (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42: 419–430 [DOI] [PubMed] [Google Scholar]
- Jonasch E, Donskov F, Iliopoulos O, Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie JK, Welsh SJ et al (2021) Belzutifan for renal cell carcinoma in von Hippel‐Lindau disease. N Engl J Med 385: 2036–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic F, Candido KD, Knezevic NN (2020) The role of the kynurenine signaling pathway in different chronic pain conditions and potential use of therapeutic agents. Int J Mol Sci 21: 6045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justus CR, Dong L, Yang LV (2013) Acidic tumor microenvironment and pH‐sensing G protein‐coupled receptors. Front Physiol 4: 354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG (2017) The VHL tumor suppressor gene: insights into oxygen sensing and cancer. Trans Am Clin Climatol Assoc 128: 298–307 [PMC free article] [PubMed] [Google Scholar]
- Kaira K, Oriuchi N, Imai H, Shimizu K, Yanagitani N, Sunaga N, Hisada T, Kawashima O, Kamide Y, Ishizuka T et al (2010) Prognostic significance of L‐type amino acid transporter 1 (LAT1) and 4F2 heavy chain (CD98) expression in surgically resectable stage III non‐small cell lung cancer. Exp Ther Med 1: 799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keenan MM, Chi JT (2015) Alternative fuels for cancer cells. Cancer J 21: 49–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith B, Simon MC (2015) Tumor angiogenesis. In The molecular basis of cancer, Mendelsohn J, Howley PM, Israel MA, Gray JW, Thompson CB (eds), 4th edn, pp 257–268.e2. Philadelphia, PA: Elsevier; [Google Scholar]
- Keshet R, Szlosarek P, Carracedo A, Erez A (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18: 634–645 [DOI] [PubMed] [Google Scholar]
- Keshet R, Lee JS, Adler L, Iraqi M, Ariav Y, Lim LQJ, Lerner S, Rabinovich S, Oren R, Katzir R et al (2020) Targeting purine synthesis in ASS1‐expressing tumors enhances the response to immune checkpoint inhibitors. Nat Cancer 1: 894–908 [DOI] [PubMed] [Google Scholar]
- Kim WY, Kaelin WG (2004) Role of VHL gene mutation in human cancer. J Clin Oncol 22: 4991–5004 [DOI] [PubMed] [Google Scholar]
- Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, Mongellaz C, Floess S, Fritz V, Matias MI et al (2015) Glutamine‐dependent α‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8: ra97 [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Millhorn DE (2001) Hypoxia regulates glutamate metabolism and membrane transport in rat PC12 cells. J Neurochem 76: 1935–1948 [DOI] [PubMed] [Google Scholar]
- Kothari S, Cizeau J, McMillan‐Ward E, Israels SJ, Bailes M, Ens K, Kirshenbaum LA, Gibson SB (2003) BNIP3 plays a role in hypoxic cell death in human epithelial cells that is inhibited by growth factors EGF and IGF. Oncogene 22: 4734–4744 [DOI] [PubMed] [Google Scholar]
- Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG (2002) Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol 22: 7405–7416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, Christofk HR (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7: 11457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug LM, Kindler H, Calvert H, Manegold C, Tsao AS, Fennell D, Lubiniecki GM, Sun X, Smith M, Baas P (2011) VANTAGE 014: Vorinostat (V) in patients with advanced malignant pleural mesothelioma (MPM) who have failed prior Pemetrexed and either cisplatin or carboplatin therapy: a phase III, randomized, double‐blind, placebo‐controlled trial. Eur J Cancer 47: 2–3 [Google Scholar]
- Kucharzewska P, Christianson HC, Belting M (2015) Global profiling of metabolic adaptation to hypoxic stress in human glioblastoma cells. PLoS One 10: e0116740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulke MH, Chan JA, Meyerhardt JA, Zhu AX, Abrams TA, Blaszkowsky LS, Regan E, Sidor C, Fuchs CS (2011) A prospective phase II study of 2‐methoxyestradiol administered in combination with bevacizumab in patients with metastatic carcinoid tumors. Cancer Chemother Pharmacol 68: 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KA, Qian DZ, Rey S, Wei H, Liu JO, Semenza GL (2009) Anthracycline chemotherapy inhibits HIF‐1 transcriptional activity and tumor‐induced mobilization of circulating angiogenic cells. Proc Natl Acad Sci USA 106: 2353–2358 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee P, Chandel NS, Simon MC (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat Rev Mol Cell Biol 21: 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendahl U, Lee KL, Yang H, Poellinger L (2009) Generating specificity and diversity in the transcriptional response to hypoxia. Nat Rev Genet 10: 821–832 [DOI] [PubMed] [Google Scholar]
- Li F, Simon MC (2020) Cancer cells don't live alone: metabolic communication within tumor microenvironments. Dev Cell 54: 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, Locasale JW (2016) The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 41: 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim E, Kuo CC, Tu HF, Yang CC (2017) The prognosis outcome of oral squamous cell carcinoma using HIF‐2α. J Chin Med Assoc 80: 651–656 [DOI] [PubMed] [Google Scholar]
- Lin SC, Hardie DG (2018) AMPK: sensing glucose as well as cellular energy status. Cell Metab 27: 299–313 [DOI] [PubMed] [Google Scholar]
- Lin X, Xiao Z, Chen T, Liang SH, Guo H (2020) Glucose metabolism on tumor plasticity, diagnosis, and treatment. Front Oncol 10: 317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, László C, Liu Y, Liu W, Chen X, Evans SC, Wu S (2010) Regulation of G1 arrest and apoptosis in hypoxia by PERK and GCN2‐mediated eIF2α phosphorylation. Neoplasia 12: 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M et al (2017) α‐Ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol 18: 985–994 [DOI] [PubMed] [Google Scholar]
- Lodewijk L, van Diest P, van der Groep P, ter Hoeve N, Schepers A, Morreau J, Bonenkamp J, van Engen‐van Grunsven A, Kruijff S, van Hemel B et al (2017) Expression of HIF‐1α in medullary thyroid cancer identifies a subgroup with poor prognosis. Oncotarget 8: 28650–28659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wang W, Wang J, Yang C, Mao H, Fu X, Wu Y, Cai J, Han J, Xu Z et al (2013) Overexpression of arginine transporter CAT‐1 is associated with accumulation of L‐arginine and cell growth in human colorectal cancer tissue. PLoS One 8: e73866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Samanta D, Xiang L, Zhang H, Hu H, Chen I, Bullen JW, Semenza GL (2015) Chemotherapy triggers HIF‐1‐dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc Natl Acad Sci USA 112: E4600‐9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangraviti A, Raghavan T, Volpin F, Skuli N, Gullotti D, Zhou J, Asnaghi L, Sankey E, Liu A, Wang Y et al (2017) HIF‐1α‐ targeting Acriflavine provides long term survival and radiological tumor response in brain cancer therapy. Sci Rep 7: 14978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manlapat AK, Kahler DJ, Chandler PR, Munn DH, Mellor AL (2007) Cell‐autonomous control of interferon type I expression by indoleamine 2, 3‐dioxygenase in regulatory CD19 + dendritic cells. Eur J Immunol 37: 1064–1071 [DOI] [PubMed] [Google Scholar]
- Martinez CA, Kerr B, Jin C, Cistulli PA, Cook KM (2019) Obstructive sleep apnea activates HIF‐1 in a hypoxia dose‐dependent manner in HCT116 colorectal carcinoma cells. Int J Mol Sci 20: 445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM et al (2016) Tissue of origin dictates branched‐chain amino acid metabolism in mutant Kras‐driven cancers. Science 353: 1161–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard MA, Evans AJ, Hosomi T, Hara S, Jewett MA, Ohh M (2005) Human HIF‐3alpha4 is a dominant‐negative regulator of HIF‐1 and is down‐regulated in renal cell carcinoma. FASEB J 19: 1396–1406 [DOI] [PubMed] [Google Scholar]
- Menon D, Salloum D, Bernfeld E, Gorodetsky E, Akselrod A, Frias MA, Sudderth J, Chen PH, DeBerardinis R, Foster DA (2017) Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J Biol Chem 292: 6303–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messmer‐Blust A, An X, Li J (2009) Hypoxia‐regulated angiogenic inhibitors. Trends Cardiovasc Med 19: 252–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Däbritz JHM, Gottlieb E, Latorre I et al (2016) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167: 457–470.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, Court W, Fox KR, Townsend PA, Packham GK et al (2013) A cyclic peptide inhibitor of HIF‐1 heterodimerization that inhibits hypoxia signaling in cancer cells. J Am Chem Soc 135: 10418–10425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra SR, Sadik A, Tykocinski LO, Dietze J, Poschet G, Heiland I, Opitz CA (2019) Hypoxia inducible factor 1α inhibits the expression of immunosuppressive tryptophan‐2,3‐dioxygenase in glioblastoma. Front Immunol 10: 2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotti M, Bridges E, Valli A, Choudhry H, Sheldon H, Wigfield S, Gray N, Zois CE, Grimm F, Jones D et al (2019) Hypoxia‐induced switch in SNAT2/SLC38A2 regulation generates endocrine resistance in breast cancer. Proc Natl Acad Sci USA 116: 12452–12461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscat J, Richardson A, Diaz‐Meco MT (2015) Nutrient stress revamps cancer cell metabolism. Cell Res 25: 537–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muniz de Queiroz R, Oliveira IA, Piva B, Catão FB, Rodrigues BC, Pascoal AC, Diaz BL, Todeschini AR, Caarls MB, Dias WB (2019) Hexosamine biosynthetic pathway and glycosylation regulate cell migration in melanoma cells. Front Oncol 9: 116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL (2013) Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol 34: 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, Mellor AL (2016) IDO in the tumor microenvironment: inflammation, counter‐regulation, and tolerance. Trends Immunol 37: 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PJ (2016) Amino acid auxotrophy as a system of immunological control nodes. Nat Immunol 17: 132–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachef M, Ali AK, Almutairi SM, Lee SH (2021) Targeting SLC1A5 and SLC3A2/SLC7A5 as a potential strategy to strengthen anti‐tumor immunity in the tumor microenvironment. Front Immunol 12: 624324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa MS, Keith B, Simon MC (2016) Oxygen availability and metabolic adaptations. Nat Rev Cancer 16: 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita T, Yin S, Gelin CF, Moreno CS, Yepes M, Nicolaou KC, Van Meir EG (2009) Identification of a novel small molecule HIF‐1α translation inhibitor. Clin Cancer Res 15: 6128–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman RE, Mccoy TA (1956) Dual requirement of Walker carcinosarcoma 256 in vitro for asparagine and glutamine. Science 124: 124–125 [DOI] [PubMed] [Google Scholar]
- Ngwa VM, Edwards DN, Philip M, Chen J (2019) Microenvironmental metabolism regulates antitumor immunity. Cancer Res 79: 4003–4008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C et al (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136: 521–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noman MZ, Hasmim M, Lequeux A, Xiao M, Duhem C, Chouaib S, Berchem G, Janji B (2019) Improving cancer immunotherapy by targeting the hypoxic tumor microenvironment: new opportunities and challenges. Cell 8: 1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor SI, Jamali S, Ames S, Langer S, Deitmer JW, Becker HM (2018) A surface proton antenna in carbonic anhydrase II supports lactate transport in cancer cells. Elife 7: e35176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oki Y, Copeland A, Romaguera J, Fayad L, Fanale M, Faria SDC, Medeiros LJ, Ivy P, Younes A (2012) Clinical experience with the heat shock protein‐90 inhibitor, tanespimycin, in patients with relapsed lymphoma. Leuk Lymphoma 53: 990–992 [DOI] [PubMed] [Google Scholar]
- Pan M, Reid MA, Lowman XH, Kulkarni RP, Tran TQ, Liu X, Yang Y, Hernandez‐Davies JE, Rosales KK, Li H et al (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18: 1090–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Kim JH, Sun BK, Song SU, Suh W, Sung JH (2016) Hypoxia induces glucose uptake and metabolism of adipose‐derived stem cells. Mol Med Rep 14: 4706–4714 [DOI] [PubMed] [Google Scholar]
- Parks SK, Cormerais Y, Marchiq I, Pouyssegur J (2016) Hypoxia optimises tumour growth by controlling nutrient import and acidic metabolite export. Mol Aspects Med 47‐48: 3–14 [DOI] [PubMed] [Google Scholar]
- Patching S (2015) Roles of facilitative glucose transporter GLUT1 in [18F]FDG positron emission tomography (PET) imaging of human diseases. J Diagn Imaging Ther 2: 30–102 [Google Scholar]
- Pavlova NN, Thompson CB (2016) The emerging hallmarks of cancer metabolism. Cell Metab 23: 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, Rabinowitz JD, Thompson CB, Zhang J (2018) As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab 27: 428–438.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payen VL, Mina E, Van Hée VF, Porporato PE, Sonveaux P (2020) Monocarboxylate transporters in cancer. Mol Metab 33: 48–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Tomás R, Pérez‐Guillén I (2020) Lactate in the tumor microenvironment: An essential molecule in cancer progression and treatment. Cancers 12: 3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado‐Ziola N, Nizet V, Johnson RS (2005) HIF‐1α expression regulates the bactericidal capacity of phagocytes. J Clin Investig 115: 1806–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzuto A, Carico E (2018) Role of HIF‐1 in cancer progression: novel insights. A review. Curr Mol Med 18: 343–351 [DOI] [PubMed] [Google Scholar]
- Qiu B, Ackerman D, Sanchez DJ, Li B, Ochocki JD, Grazioli A, Bobrovnikova‐Marjon E, Alan Diehl J, Keith B, Celeste Simon M (2016) HIF2α‐dependent lipid storage promotes endoplasmic reticulum homeostasis in clear‐cell renal cell carcinoma. Cancer Discov 5: 652–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D et al (2015) Diversion of aspartate in ASS1‐deficient tumours fosters de novo pyrimidine synthesis. Nature 527: 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, Richardson PG, Lacy MQ, Dispenzieri A, Greipp PR, Witzig TE, Schlossman R, Sidor CF, Anderson KC, Gertz MA (2007) Novel therapy with 2‐methoxyestradiol for the treatment of relapsed and plateau phase multiple myeloma. Clin Cancer Res 13: 6162–6167 [DOI] [PubMed] [Google Scholar]
- Ravishankara B, Liua H, Shindea R, Chaudharya K, Xiaoa W, Bradleya J, Koritzinsky M, Madaio MP, McGaha TL (2015) The amino acid sensor GCN2 inhibits inflammatory responses to apoptotic cells promoting tolerance and suppressing systemic autoimmunity. Proc Natl Acad Sci USA 112: 10774–10779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT et al (2021) Cell‐programmed nutrient partitioning in the tumour microenvironment. Nature 593: 282–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren W, Xia Y, Chen S, Wu G, Bazer FW, Zhou B, Tan B, Zhu G, Deng J, Yin Y (2019) Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv Nutr 10: 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robey IF, Lien AD, Welsh SJ, Baggett BK, Gillies RJ (2005) Hypoxia‐inducible factor‐1α and the glycolytic phenotype in tumors. Neoplasia 7: 324–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero‐Garcia S, Moreno‐Altamirano MMB, Prado‐Garcia H, Sánchez‐García FJ (2016) Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front Immunol 7: 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnen EA, Kondagunta GV, Ishill N, Sweeney SM, DeLuca JK, Schwartz L, Bacik J, Motzer RJ (2006) A phase II trial of 17‐ (Allylamino)‐17‐demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs 24: 543–546 [DOI] [PubMed] [Google Scholar]
- Roseweir AK, Clark J, McSorley ST, vanWyk HC, Quinn JA, Horgan PG, McMillan DC, Park JH, Edwards J (2019) The association between markers of tumour cell metabolism, the tumour microenvironment and outcomes in patients with colorectal cancer. Int J Cancer 144: 2320–2329 [DOI] [PubMed] [Google Scholar]
- Rothhammer V, Quintana FJ (2019) The aryl hydrocarbon receptor: an environmental sensor integrating immune responses in health and disease. Nat Rev Immunol 19: 184–197 [DOI] [PubMed] [Google Scholar]
- Sáenz‐de‐Santa‐María I, Bernardo‐Castiñeira C, Secades P, Bernaldo‐de‐Quirós S, Rodrigo JP, Astudillo A, Chiara MD (2017) Clinically relevant HIF‐1α‐dependent metabolic reprogramming in oropharyngeal squamous cell carcinomas includes coordinated activation of CAIX and the miR‐210/ISCU signaling axis, but not MCT1 and MCT4 upregulation. Oncotarget 8: 13730–13746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata T, Ferdous G, Tsuruta T, Satoh T, Baba S, Muto T, Ueno A, Kanai Y, Endou H, Okayasu I (2009) L‐type amino‐acid transporter 1 as a novel biomarker for high‐grade malignancy in prostate cancer. Pathol Int 59: 7–18 [DOI] [PubMed] [Google Scholar]
- Salisbury TB, Arthur S (2018) The regulation and function of the L‐type amino acid transporter 1 (LAT1) in cancer. Int J Mol Sci 19: 2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samec M, Liskova A, Koklesova L, Mersakova S, Strnadel J, Kajo K, Pec M, Zhai K, Smejkal K, Mirzaei S et al (2021) Flavonoids targeting HIF‐1: implications on cancer metabolism. Cancers 13: 130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Garrido J, Shenoy AR (2020) Regulation and repurposing of nutrient sensing and autophagy in innate immunity. Autophagy 17: 1571–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos N, Pereira‐Nunes A, Baltazar F, Granja S (2019) Lactate as a regulator of cancer inflammation and immunity. Immunometabolism 1: e190015 [Google Scholar]
- Schwartz L, Supuran C, Alfarouk K (2017) The Warburg effect and the hallmarks of cancer. Anticancer Agents Med Chem 17: 164–170 [DOI] [PubMed] [Google Scholar]
- Semba H, Takeda N, Isagawa T, Sugiura Y, Honda K, Wake M, Miyazawa H, Yamaguchi Y, Miura M, Jenkins DMR et al (2016) HIF‐1α‐PDK1 axis‐induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun 7: 11635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL (2003) Targeting HIF‐1 for cancer therapy. Nat Rev Cancer 3: 721–732 [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP (2015) The future of immune checkpoint therapy. Science 348: 56–61 [DOI] [PubMed] [Google Scholar]
- Shenoy N, Pagliaro L (2016) Sequential pathogenesis of metastatic VHL mutant clear cell renal cell carcinoma: putting it together with a translational perspective. Ann Oncol 27: 1685–1695 [DOI] [PubMed] [Google Scholar]
- Shimizu A, Kaira K, Kato M, Yasuda M, Takahashi A, Tominaga H, Oriuchi N, Nagamori S, Kanai Y, Oyama T et al (2015) Prognostic significance of l‐type amino acid transporter 1 (LAT1) expression in cutaneous melanoma. Melanoma Res 25: 399–405 [DOI] [PubMed] [Google Scholar]
- Silverstein RL, Febbraio M (2009) CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal 2: re3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh D, Arora R, Kaur P, Singh B, Mannan R, Arora S (2017) Overexpression of hypoxia‐inducible factor and metabolic pathways: possible targets of cancer. Cell Biosci 7: 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofer A, Lei K, Johannessen CM, Ellisen LW (2005) Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol 25: 5834–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh H, Wasa M, Fukuzawa M (2007) Hypoxia upregulates amino acid transport in a human neuroblastoma cell line. J Pediatr Surg 42: 608–612 [DOI] [PubMed] [Google Scholar]
- Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136: 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Zhang Y, Zhang L, Song W, Shi L (2018) Hypoxia enhances indoleamine 2,3‐dioxygenase production in dendritic cells. Oncotarget 9: 11572–11580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowter HM, Ratcliffe PJ, Watson P, Greenberg AH, Harris AL (2001) HIF‐1‐dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res 61: 6669–6673 [PubMed] [Google Scholar]
- Stine ZE, Altman BJ, Hsieh AL, Gouw AM, Dang CV (2014) Deregulation of the cellular energetics of cancer cells. In Pathobiology of human disease: a dynamic encyclopedia of disease mechanisms, McManus LM, Mitchell RN (eds). Amsterdam: Academic Press; [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Heiden MGV, Muir A (2019) Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8: e44235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda N, O'Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS (2010) Differential activation and antagonistic function of HIF‐α isoforms in macrophages are essential for NO homeostasis. Genes Dev 24: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Liu W (2016) LDHA is a feedback activator of hypoxia inducible factor 1‐alpha in ovarian cancer. Int J Clin Exp Pathol 9: 10437–10443 [Google Scholar]
- Tannahill GM, Curtis AM, Adamik J, Palsson‐Mcdermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH et al (2013) Succinate is an inflammatory signal that induces IL‐1β through HIF‐1α. Nature 496: 238–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tönjes M, Barbus S, Park YJ, Wang W, Schlotter M, Lindroth AM, Pleier SV, Bai AHC, Karra D, Piro RM et al (2013) BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild‐type IDH1. Nat Med 19: 901–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrence ME, Manning BD (2018) Nutrient sensing in cancer. Annu Rev Cancer Biol 2: 251–269 [Google Scholar]
- Ullah MS, Davies AJ, Halestrap AP (2006) The plasma membrane lactate transporter MCT4, but not MCT1, is up‐regulated by hypoxia through a HIF‐1α‐dependent mechanism. J Biol Chem 281: 9030–9037 [DOI] [PubMed] [Google Scholar]
- Venter G, Oerlemans FTJJ, Wijers M, Willemse M, Fransen JAM, Wieringa B (2014) Glucose controls morphodynamics of LPS‐stimulated macrophages. PLoS One 9: e96786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola A, Munari F, Sánchez‐Rodríguez R, Scolaro T, Castegna A (2019) The metabolic signature of macrophage responses. Front Immunol 10: 1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viziteu E, Grandmougin C, Goldschmidt H, Seckinger A, Hose D, Klein B, Moreaux J (2016) Chetomin, targeting HIF‐1α/p300 complex, exhibits antitumour activity in multiple myeloma. Br J Cancer 114: 519–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Liu G, Wang R (2014) The intercellular metabolic interplay between tumor and immune cells. Front Immunol 5: 358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang H, Liu A, Fang C, Hao J, Wang Z (2015) Lactate dehydrogenase a negatively regulated by miRNAs promotes aerobic glycolysis and is increased in colorectal cancer. Oncotarget 6: 19456–19468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wang Q, Yan G, Qiao Y, Zhu B, Liu B, Tang C (2018a) Hypoxia induces lactate secretion and glycolytic efflux by downregulating mitochondrial pyruvate carrier levels in human umbilical vein endothelial cells. Mol Med Rep 18: 1710–1717 [DOI] [PubMed] [Google Scholar]
- Wang F, Zhang S, Vuckovic I, Jeon R, Lerman A, Folmes CD, Dzeja PP, Herrmann J (2018b) Glycolytic stimulation is not a requirement for M2 macrophage differentiation. Cell Metab 28: 463–475.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhao Q, Zhang Y, Liu Z, Zheng Z, Liu S, Meng L, Xin Y, Jiang X (2021) Targeting hypoxia in the tumor microenvironment: a potential strategy to improve cancer immunotherapy. J Exp Clin Cancer Res 40: 24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner A, Pieh D, Echchannaoui H, Rupp J, Rajalingam K, Theobald M, Closs EI, Munder M (2019) Cationic amino acid Transporter‐1‐mediated arginine uptake is essential for chronic lymphocytic leukemia cell proliferation and viability. Front Oncol 9: 1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womeldorff M, Gillespie D, Jensen RL (2014) Hypoxia‐inducible factor‐1 and associated upstream and downstream proteins in the pathophysiology and management of glioblastoma. Neurosurg Focus 37: E8 [DOI] [PubMed] [Google Scholar]
- Wu L, Saxena S, Awaji M, Singh RK (2019) Tumor‐associated neutrophils in cancer: going pro. Cancers 11: 564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaghi L, Poras I, Simoes RT, Donadi EA, Tost J, Daunay A, de Almeida BS, Carosella ED, Moreau P (2016) Hypoxia inducible factor‐1 mediates the expression of the immune checkpoint HLA‐G in glioma cells through hypoxia response element located in exon 2. Oncotarget 7: 63690–63707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J‐S, Wang C‐C, Qiu J‐D, Ren B, You L (2021) Arginine metabolism: a potential target in pancreatic cancer therapy. Chin Med J 134: 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye LY, Chen W, Bai XL, Xu XY, Zhang Q, Xia XF, Sun X, Li GG, Hu Q, Fu QH et al (2016) Hypoxia‐induced epithelial‐to‐mesenchymal transition in hepatocellular carcinoma induces an immunosuppressive tumor microenvironment to promote metastasis. Cancer Res 76: 818–830 [DOI] [PubMed] [Google Scholar]
- Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K (2020) The role of cancer‐derived microRNAs in cancer immune escape. J Hematol Oncol 13: 25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Bai L, Li W, Zeng T, Tian H, Cui J (2019) Targeting T cell metabolism in the tumor microenvironment: an anti‐cancer therapeutic strategy. J Exp Clin Cancer Res 38: 403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo HC, Park SJ, Nam M, Kang J, Kim K, Yeo JH, Kim JK, Heo Y, Lee HS, Lee MY et al (2020) A variant of SLC1A5 is a mitochondrial glutamine transporter for metabolic reprogramming in cancer cells. Cell Metab 31: 267–283.e12 [DOI] [PubMed] [Google Scholar]
- Yuan HX, Xiong Y, Guan KL (2013) Nutrient sensing, metabolism, and cell growth control. Mol Cell 49: 379–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao E, Maj T, Kryczek I, Li W, Wu K, Zhao L, Wei S, Crespo J, Wan S, Vatan L et al (2016) Cancer mediates effector T cell dysfunction by targeting microRNAs and EZH2 via glycolysis restriction. Nat Immunol 17: 95–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhong H, Zhou Q, Hu X, Chen D, Wang J, Wu J, Cai J, Zhou S, Chen AF (2015) Inhibition of PKR impairs angiogenesis through a VEGF pathway. Am J Physiol Endocrinol Metab 308: E518–E524 [DOI] [PubMed] [Google Scholar]