

Abstract
BACKGROUND:
SF3B1 mutations (SF3B1mut) in myelodysplastic syndromes (MDS) frequently involve codon K700E and have a favorable prognosis. The prognostic effect of non-K700E SF3B1mut is uncertain.
METHODS:
We analyzed the clinical-pathologic features and outcomes of a single-institutional series of 94 (19%) SF3B1mut and 415 SF3B1wt treatment-naïve MDS patients and explored the differences between K700E and non-K700E.
RESULTS:
Fifty-five (60%) patients carried K700E. Recurrent non-K700E mutations (39, 40%) included R625, H662 and K666. Compared to SF3B1mut-K700E, non-K700E patients had a lower median ANC (1·8 vs. 2·4, p=0·005) and were frequently “high” R-IPSS [19% vs. 4%, p=0·031]. Non-K700E MDS associated frequently with RUNX1 (26% vs. 7%, p=0·012) and exclusively with BCOR, IDH2, and SRSF2 mutations. Splicing analysis showed differential distribution of alternatively spliced events and gene expression profiles between K700 and non-K700E MDS patients. Majority (~80%) of SF3B1mut-K700E, SF3B1mut-non-K700E and SF3B1wt patients were treated with hypomethylating agents. Over a median follow-up of 16 months, SF3B1mut had superior overall survival (OS) than SF3B1wt in all MDS [not-reached vs. 25·2 months, p=0·0003], low-grade MDS, and MDS with ring sideroblasts (MDS-RS) patients. Compared to SF3B1wt, SF3B1mut-K700E had superior outcomes in all MDS (median OS, 25 months vs. not-reached, p=0·0001), low-grade MDS (median OS, 41.3 months vs. not-reached, p=0·0015) and MDS-RS (median OS, 22.3 months vs. not reached, p=0.0001), but no significant difference was seen between non-K700E and SF3B1wt MDS. By multivariable analysis, absence of SF3B1mut K700E mutations independently associated with prognosis.
CONCLUSIONS:
Our study highlights the importance of SF3B1 mutation subtype in MDS risk-assessment.
Keywords: SF3B1, ring sideroblasts, myelodysplastic syndromes, prognosis, K700E
Precis:
Myelodysplastic syndromes (MDS) with K700E and non-K700E SF3B1 mutations show distinct clinicopathological and genomic characteristics, with only SF3B1 K700E mutated MDS showing a significantly better OS compared to non-K700E mutations. SF3B1 mutation type is important for MDS risk-assessment.
Lay Summary
Myelodysplastic syndromes with SF3B1 mutations are regarded to have a favorable prognosis by both WHO and the International Working Group for Prognostication of MDS (IWG-PM). However, in this article, we show that only MDS with SF3B1 K700E mutations have a favorable prognosis, but not MDS with SF3B1 mutations involving other codons. This has important implications for refining future MDS sub-classification and risk assessment criteria.
INTRODUCTION
SF3B1 is one of the most frequently mutated genes in MDS.1 SF3B1 mutations are detected in a third of MDS patients and are present in up to 66% of MDS sub-categories with increased ring sideroblasts (RS).2–4 Prior studies have indicated that the presence of SF3B1 mutation (SF3B1mut) was an independently favorable prognostic factor for survival in MDS.2, 5–8 Based on the favorable outcome, a distinct gene expression profile, and association with ring sideroblasts (RS), the 2016 revisions to the World Health Organization (WHO) Classification of Myelodysplastic Syndromes recognized MDS-RS with single lineage (MDS-RS-SLD) and multilineage dysplasia (MDS-RS-MLD) as distinct sub-categories, and recommended testing for SF3B1 mutation in MDS patients with 5 and 15% ring sideroblasts in bone marrow (BM) aspirates.9 We have previously shown that the survival of MDS-RS-MLD patients (a majority of which had SF3B1 mutations) was significantly better compared to those with MDS-MLD without RS.10 Based on recent data suggesting sustained hematological responses in SF3B1mut MDS patients treated with luspatercept11, the International Working Group for the Prognosis of MDS (IWG-PM) has proposed that SF3B1mut MDS be considered a distinct entity with a favorable prognosis in the absence of >5% BM blasts or ≥1% peripheral blood (PB) blasts, absence of del(5q), monosomy 7, inv(3), abnormal 3q26 or complex karyotype (CK), mutations in RUNX1 and EZH2, and findings suggestive of other WHO-defined entities.6
Despite this body of information, the natural history of SF3B1 mutated MDS is heterogeneous. A high proportion of SF3B1 mutations occur within codon 700, causing a branch point recognition error of a cryptic splice site followed by nonsense decay due to aberrant splice junctions, ultimately resulting in large-scale downregulation of mRNA.12–14 The remainder of SF3B1 mutations occur outside of this hotspot and the downstream functional effects are not clear. Dalton, et al has recently suggested that SF3B1 K666N mutations had distinctive RNA splicing profiles and were associated with distinct clinico-pathologic features and worse outcomes. The authors suggested that these mutations may need aggressive management, even in the setting of a lower IPSS-R category.15 Beyond the SF3B1 mutation type, the clinical course can be potentially altered by other parameters, such as the variant allele frequency (VAF), presence of concomitant gene mutations and karyotype.
In this study, we investigated the clinico-pathologic and genetic features and outcomes in a single-institutional series of 94 SF3B1mut MDS patients and compared them to 415 with wild-type MDS (SF3B1wt). We also explored the differences between the SF3B1 K700E and non-K700E mutated MDS subgroups, and demonstrated distinct clinical and mutational profiles, with the K700E mutated subgroup showing a significantly better overall survival (OS) compared to the non-K700E subgroup. Further, only the SF3B1 K700E mutation subtype independently predicted for better OS in MDS.
MATERIALS AND METHODS
Patients and samples
We selected BM aspirates from all newly diagnosed previously untreated MDS patients presenting to The University of Texas MD Anderson Cancer Center (MDACC) from 2017–2019. Diagnoses were confirmed by BM examination and sub-classified using the 2016 WHO criteria.16 At least 20 metaphases were evaluated, and interpreted according to the 2016 International System for Human Cytogenetic Nomenclature. Risk stratification was done using the IPSS-R17 for patients with MDS. All patients underwent 81-gene panel next-generation sequencing (NGS) using BM aspirate samples for baseline mutation profiling. The study was approved by the MDACC Institutional Review Board and all samples were collected following institutional guidelines with informed consent in accord with the Declaration of Helsinki.
Targeted next-generation sequencing
All patient samples underwent comprehensive NGS-based mutation analysis using our 81-gene panel comprising the hotspots and whole coding regions of myeloid leukemia-related genes in a CLIA-certified Molecular Diagnostics Laboratory, as previously described.18 Briefly, genomic DNA was extracted from fresh BM aspirates using standard techniques, followed by library preparation and amplicon-based targeted NGS on a MiSeq sequencer. Sequences were aligned to the GRCh37/hg19 reference genome. With a minimum of 250X bidirectional coverage, a minimum variant call of 2% was considered as the limit of detection. The somatic nature of the variants was inferred based on the information in online SNP databases (i.e., Exome Aggregation Consortium [ExAC], dbSNP 137/138, 1000 Genomes) and the literature. FLT3-ITD mutations were assessed by PCR-based capillary electrophoresis.
Splicing analysis
We performed RNA-seq on CD34 positive cells from MDS patients 4 with K700E and 3 with non-K700E (1 E783K, 2 K666N) mutations, and compared these groups to healthy controls and to each other). We used replicate multivariate analysis of transcript splicing (rMATS) to assess the differential splicing.19 Aberrant splicing events associated with SF3B1 K700E, non-K700E and healthy controls were identified by comparison using false discovery rate [FDR] of <0.05 and inclusion level difference > PsiThresh of 0.1) followed by Gene Set Enrichment Analysis.
Statistical analysis
OS was calculated as the time from diagnosis to death or last follow-up date. Patients alive at their last follow-up were censored on that date. The Kaplan-Meier product limit method was used to estimate the median OS for each clinical/demographic factor. Univariate Cox proportional hazards regression analysis was used to identify any association with each of the variables and survival outcomes followed by multivariate analysis. Response assessment was performed following 2006 IWG criteria.20
RESULTS
Patient characteristics
A total of 509 treatment naïve MDS patients presented to our institution between 2017 and 2019, of which 94 (18·5%) had SF3B1 mutations. None of the patients had received prior therapy, including erythroid stimulating agents. The clinical characteristics of patients analyzed are shown in Table 1. The SF3B1mut MDS patients included 59 men and 35 women, with a median age of 74 (range, 39–92) years. The distribution of IPSS-R scores for this cohort were as follows: 14 (14·9%) very low; 37 (39·4%) low; 13 (13·8%) intermediate; 9 (9·6%) high; and 12 (12·8%) very high. Seventeen (18%) patients had received prior chemotherapy and/or radiation for an unrelated malignancy (therapy-related MDS, t-MDS). Among the remaining patients, 2016 WHO sub-classifications were as follows: 22 (23%) with MDS with single lineage dysplasia and ring sideroblasts (MDS-RS-SLD); 30 (32%) with MDS with ring sideroblasts and multilineage dysplasia (MDS-MLD-RS); 5 (5%) with MDS-MLD; 31 (33%) with MDS with excess blasts; 4 (4%) with MDS and isolated del(5q); and 2 (2%) with MDS-unclassifiable (MDS-U). Treatment data was available for 313 patients: 248 (79%) patients received hypomethylating agents (HMAs) alone or in combination with other agents and 35 (11·2%) received chemotherapy-based regimens.
Table 1.
Baseline characteristics of the study group: all newly diagnosed previously untreated MDS samples that underwent mutation analysis using 81-gene next-generation sequencing panel.
| Variable | SF3B1 mutated (n=94) | SF3B1 wild-type (n=415) | p-value |
|---|---|---|---|
| Age (years) | 74 (39–92) | 70 (23–93) | 0.0002 |
| Gender (F:M) | 35:59 | 142:273 | 0.63 |
| BM blasts% | 2 (0–16) | 4 (0–19) | 0.003 |
| Hemoglobin (g/dL) | 9.2 (6.9–12.9) | 9.4 (5.2–16.4) | 0.14 |
| MCV fL | 105 (85–117) | 96 (66–122) | <0.0001 |
| Platelet count K/uL | 188 (8–628) | 78 (7–692) | <0.0001 |
| ANC | 2.1 (0.2–6.5) | 1.3 (0–112.9) | 0.76 |
| Chemotherapy | 9 (9.5%) | 122 (29.4%) | 0.0001 |
| Radiation | 11 (11.7%) | 79 (19.0%) | 0.10 |
| Therapy-related | 17 (18%) | 142 (34%) | 0.002 |
| Serum EPO (mIU/mL) | 108 (9–1694) | 56.9 (5.4–5650) | 0.70 |
| Serum Ferritin (ng/mL) | 462 (37–3017) | 462 (24–7261) | 0.47 |
| Morphologic findings | |||
| Ring sideroblasts (%) | 0.4 (0–0.9) | 0.07 (0–0.76) | <0.0001 |
| WHO 2016 sub-classification | |||
| Isolated del(5q) | 4 (4.3%) | 3 (0.7%) | <0.0001 |
| MDS-SLD-RS | 22 (23.4%) | 5 (1.2%) | <0.0001 |
| MDS-MLD | 5 (5.3%) | 137 (33%) | <0.0001 |
| MDS-MLD-RS | 30 (31.9%) | 28 (6.7%) | <0.0001 |
| MDS-EB | 31 (33%) | 201 (48.4%) | <0.0001 |
| MDS-U | 2 (2.1%) | 41 (9.9%) | 0.012 |
| R-IPSS risk categories | |||
| Very High | 12 (12.8%) | 107 (28.8%) | 0.0014 |
| High | 9 (9.6%) | 80 (21.5%) | 0.0079 |
| Intermediate | 13 (13.8%) | 75 (20.2%) | 0.19 |
| Low | 37 (39.4%) | 95 (25.5%) | 0.010 |
| Very Low | 14 (14.9%) | 15 (4%) | 0.0004 |
| IPSS risk categories | |||
| Low | 38 (45.2%) | 69 (18.6%) | <0.0001 |
| Int-1 | 28 (33.3%) | 174 (46.9%) | 0.028 |
| Int-2 | 16 (19%) | 103 (27.8%) | 0.13 |
| High | 2 (2.4%) | 25 (6.7%) | 0.20 |
| Karyotype | |||
| Diploid | 46 (52%) | 136 (35%) | 0.004 |
| Complex karyotype (≥3) | 9 (9.6%) | 133 (36%) | 0.47 |
Clinical and morphologic findings of SF3B1mut MDS
Compared to SF3B1wt, SF3B1mut MDS patients had a significantly higher median age (74 vs. 70, p=0·0008), higher median mean corpuscular volume (MCV) (105 vs. 96, p<0·0001), higher median platelet count (188 vs. 78, p<0·0001), and lower BM blast percentage (median, 2 vs. 4, p=0·003). There was no significant difference in median serum erythropoietin levels (108 vs. 57). Compared to SF3B1wt, SF3B1mut MDS patients were more likely to be in the IPSS-R very-low (4% vs. 14·9%, p=0·0004) and low (25·5% vs. 39·4%, p=0·010) categories and less likely to be in the IPSS-R-high (21.5% vs. 9·6%, p=0·008) and very-high (28·8% vs. 12·8%, p=0·001) categories. The median percentage of ring sideroblasts was higher in patients with SF3B1mut MDS (40% vs. 7%, p<0·0001) compared to the those with SF3B1wt; accordingly, the SF3B1mut MDS group was significantly enriched in WHO categories with ring sideroblasts [MDS-SLD-RS (23·4% vs. 1·2%, p<0·0001) and MDS-RS-MLD (31·.9% vs. 6·7%, p<0·0001)] and MDS with isolated del(5q) (4·3% vs. 0·7%, p<0·0001), while they were underrepresented in MDS-MLD (5·3% vs. 33%, p<0·0001), MDS-EB (33% vs. 48·4%, p<0·0001), and MDS-U (2·1% vs. 9·9%, p=0·012).
SF3B1mut MDS patients were less likely to be therapy-related [17 (18%) vs. 142 (34%); p=0·002]. However, within 24 t-MDS with >15% RS and <5% blasts, the most frequent mutation observed was in SF3B1 (n=13, 54%), 4 of which had concurrent TP53 mutations/CK. Six (25%) patients had TP53 mutations/CK without SF3B1 mutations, and 5 (20.8%) had neither SF3B1 nor TP53 mutations/CK.
Mutational landscape of SF3B1mut MDS (K700E and non-K700E subtypes)
BM aspirates from all patients underwent NGS analysis with an 81-gene panel at the time of diagnosis. The most frequent SF3B1 mutation, noted in ~60% of all patients, was the hotspot K700E. Among the remaining mutations observed, the most frequent involved the codons H662, K666, and R625, seen in 8 patients each (Figure 1 A). The mutational landscape is depicted in Figure 1 B. Only a third of the patients had an SF3B1 mutation as the sole driver of MDS, while the majority had concomitant mutations. The genes mutated in >10% of patients in decreasing order of frequency included TET2 (25%), DNMT3A (21%), RUNX1 (15%), TP53 (10%), ASXL1 (7%), BCOR (4%), IDH1/2 (4%), SRSF2 (3%), NRAS (3%), and EZH2 (3%). Conventional cytogenetic analysis showed a higher frequency of normal karyotype and lower frequency of CK in SF3B1mut compared to SF3B1wt cases. Three SF3B1mut patients had EVI1 gene rearrangements, but the frequency was not significantly different compared to the SF3B1wt group. Three SF3B1mut patients had t(1;3)(p36;q21) RPN1/PRDM16, which was absent in the SF3B1wt cohort.
Figure 1.
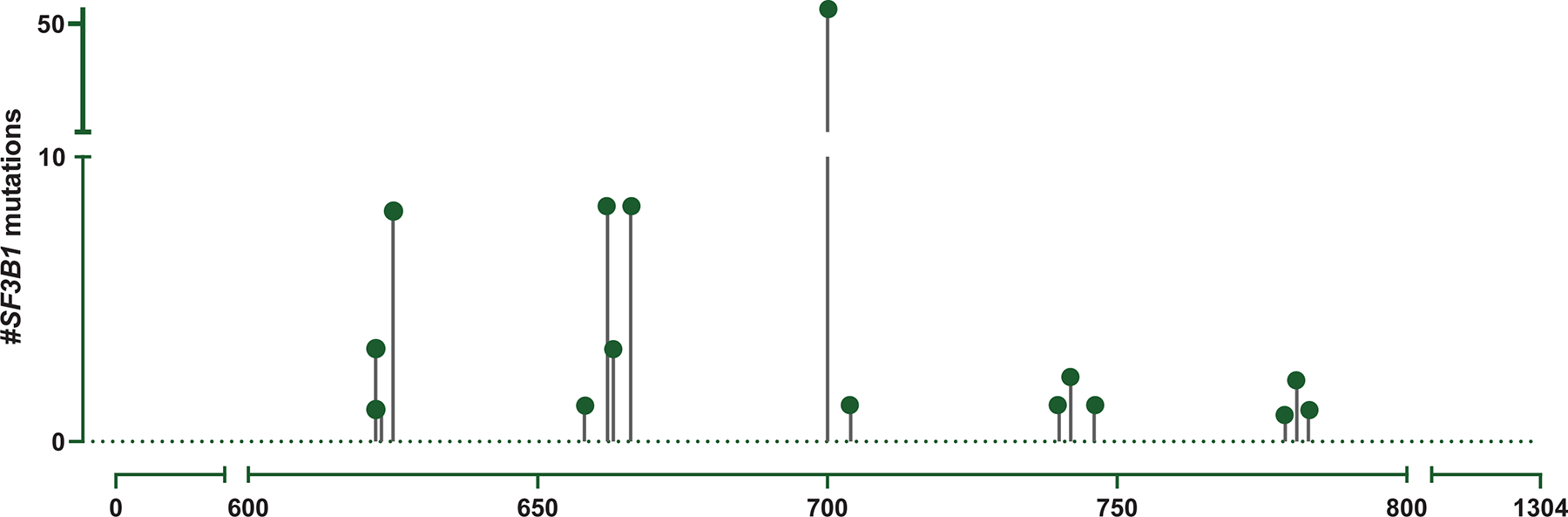
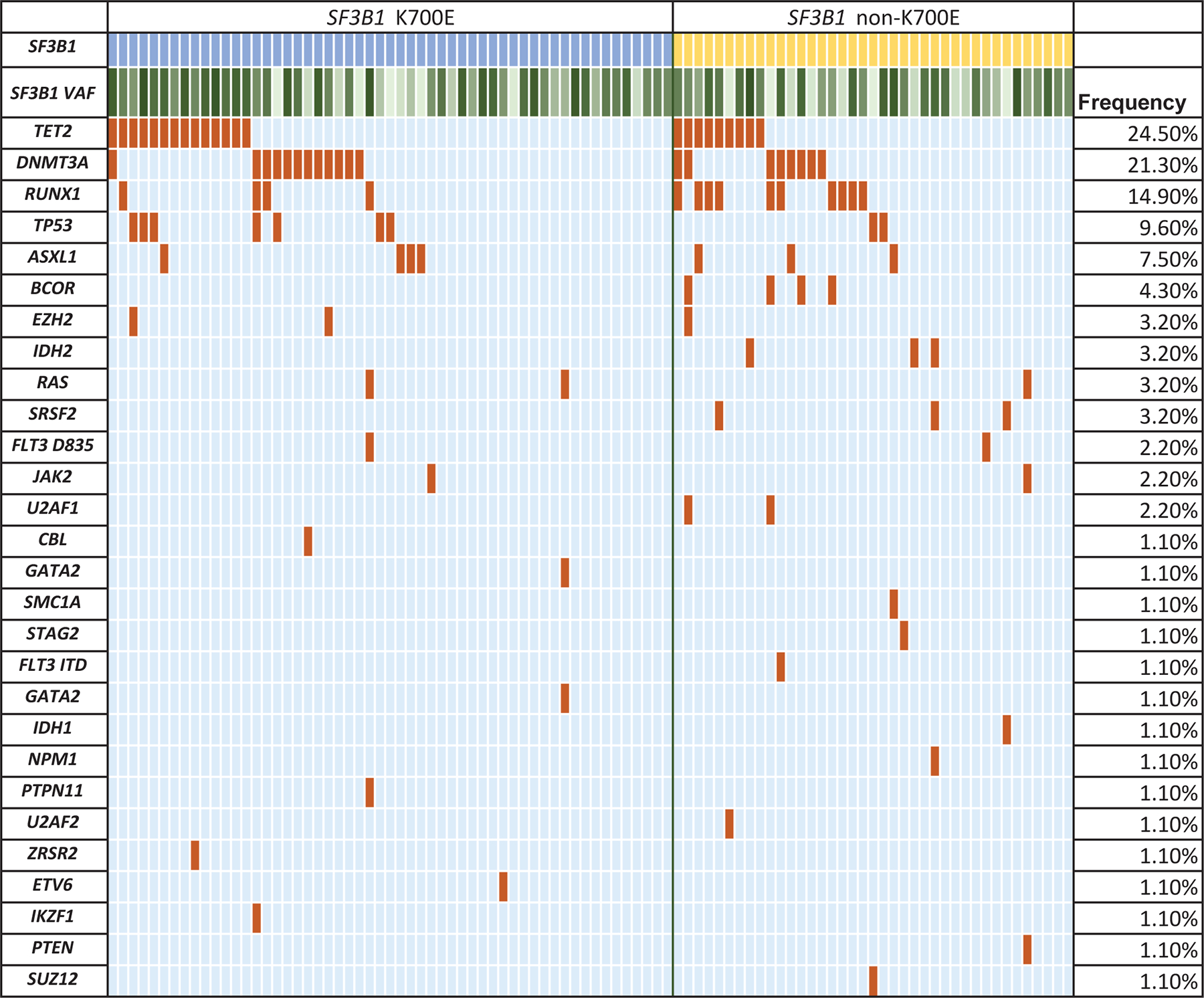
Mutational characteristics. (A) Spectrum of SF3B1 mutations within the coding region of the gene seen in the study group generated using MutationMapper from cbioportal. About ~60% of mutations involved the hotspot K700E, while ~40% involved the non-K700E regions. (B) Mutational landscape of SF3B1 K700E mutated MDS. Comutation plot of 94 SF3B1 mutated MDS patients showing nonsynonymous gene mutations (left), separated by the mutation subtype: K700E (blue) and non-K700E (yellow). Each column represents a patient, and mutations are represented by color. The frequency of gene mutations is shown on the right. Color code: Maroon indicates mutation while blue indicates wild-type. Variant allele frequency of SF3B1 is represented as a color gradient, darker color corresponding to higher VAF.
Comparison between SF3B1 K700E and non-K700E mutated MDS
We compared the clinico-pathologic features of 55 K700E vs. 39 non-K700E treatment naïve SF3B1mut MDS patients (Table 2). MDS with SF3B1 K700E mutations had a higher percentage of ring sideroblasts (median 50% vs. 34%; p=0·038), higher ANC (2·4 vs. 1·8, p=0·005), and a trend of higher platelet count (196 vs. 124, p=0·05). Among IPSS-R categories, SF3B1mut MDS patients with non-K700E mutations had a significantly higher representation in IPSS-R high category [7(19%) vs. 2(4%), p=0·031], but the distribution was not significantly different among other categories. Likewise, within 2016 WHO categories, SF3B1mut MDS patients with K700E mutations were less likely to be classified as MDS-EB than non-K700E SF3B1 mutated patients (22% vs. 49%, p=0·008). All 4 patients that fit the criteria for MDS with isolated del(5q) had K700E mutations.
Table 2.
Comparison of baseline characteristics of all newly diagnosed untreated SF3B1 mutated MDS segregated by the mutation type, K700E vs. non-K700E.
| Clinical Variable |
SF3B1 K700E (n=55, 59%) |
SF3B1 Non-K700E (n=39, 41%) |
p-value |
|---|---|---|---|
| Age (years) | 74 (39–92) | 74 (51–86) | 0.946 |
| BM blasts% | 2 (0–16) | 3 (0–16) | 0.122 |
| Gender (F:M) | 19;36 | 16;23 | |
| Ring sideroblasts (%) | 0.5 (0–0.91) | 0.34 (0–0.77) | 0.038 |
| Hemoglobin (g/dL) | 9.35 (7–12.9) | 9.15 (6.9–12.5) | 0.675 |
| MCV fL | 106.5 (86–116) | 103.5 (85–117) | 0.248 |
| Platelet count K/uL | 195.5 (12–628) | 123.5 (8–421) | 0.054 |
| ANC | 2.41 (0.48–6.46) | 1.78 (0.22–4.46) | 0.005 |
| Serum EPO (mIU/mL) | 102 (9–1694) | 114.4 (14.2–1286.4) | 0.446 |
| Serum Ferritin (ng/mL) | 527 (72–3017) | 373 (37–1725) | 0.157 |
| Chemoradiation | 10 (18.1%) | 7 (17.9%) | 1.00 |
| R-IPSS | |||
| Very-high | 5 (10%) | 7 (19%) | 0.226 |
| High | 2 (4%) | 7 (19%) | 0.031 |
| Intermediate | 5 (10%) | 8 (22%) | 0.137 |
| Low | 26 (53%) | 11 (31%) | 0.087 |
| Very low | 11 (22%) | 3 (8%) | 0.143 |
| 2016 WHO sub-classification | |||
| MDS-SLD-RS, MDS-MLD-RS | 35 (64%) | 17 (44%) | 0.06 |
| MDS-MLD | 3 (5%) | 2 (5%) | 1 |
| MDS-EB | 12 (22%) | 19 (49%) | 0.008 |
| MDS-U | 1 (2%) | 0 | 1 |
| MDS with isolated del(5q) | 4 (7%) | 0 | 0.139 |
| Cytogenetics | n=49 | n=36 | |
| Diploid karyotype | 28 (57%) | 18 (50%) | 1.0 |
| Complex karyotype | 3 (6%) | 6 (16.7%) | 0.159 |
| Somatic mutation frequencies | |||
| TET2 | 14 (25.5%) | 9 (23.1%) | 0.691 |
| DNMT3A | 12 (21.8%) | 8 (20.5%) | 0.655 |
| TP53 | 7 (12.7%) | 2 (5.1%) | 0.949 |
| RUNX1 | 4 (7.3%) | 10 (25.6%) | 0.015 |
| ASXL1 | 4 (7.3%) | 3 (7.7%) | 0.618 |
| EZH2 | 2 (3.6%) | 1 (2.6%) | 0.804 |
| RAS | 2 (3.6%) | 1 (2.6%) | 0.804 |
| FLT3 D835 | 1 (1.8%) | 1 (2.6%) | 0.660 |
| JAK2 | 1 (1.8%) | 1 (2.6%) | 0.660 |
| BCOR | 0 | 4 (10.3%) | 0.027 |
| IDH2 | 0 | 3 (7.7%) | 0.068 |
| SRSF2 | 0 | 3 (7.7%) | 0.068 |
| U2AF1 | 0 | 2 (5.1%) | 0.170 |
| Variant allele frequency | |||
| SF3B1 | 33.5 (1.1–46.8) | 29.5 (1.1–45) | 0.18 |
There was no difference in the median SF3B1 variant allele frequency (VAF) between the 2 groups. The frequency of RUNX1 mutation was significantly higher in non-K700E cases (26% vs. 7·3%, p=0·012), and mutations in BCOR (p=0·02), IDH2 (p=0·07), and SRSF2 (p=0·07) were exclusively observed in non-K700E cases (Figure 2A). No significant differences were noted in the frequencies of TP53 or clonal hematopoiesis-associated mutations, including DNMT3A, ASXL1, and TET2.
Figure 2.
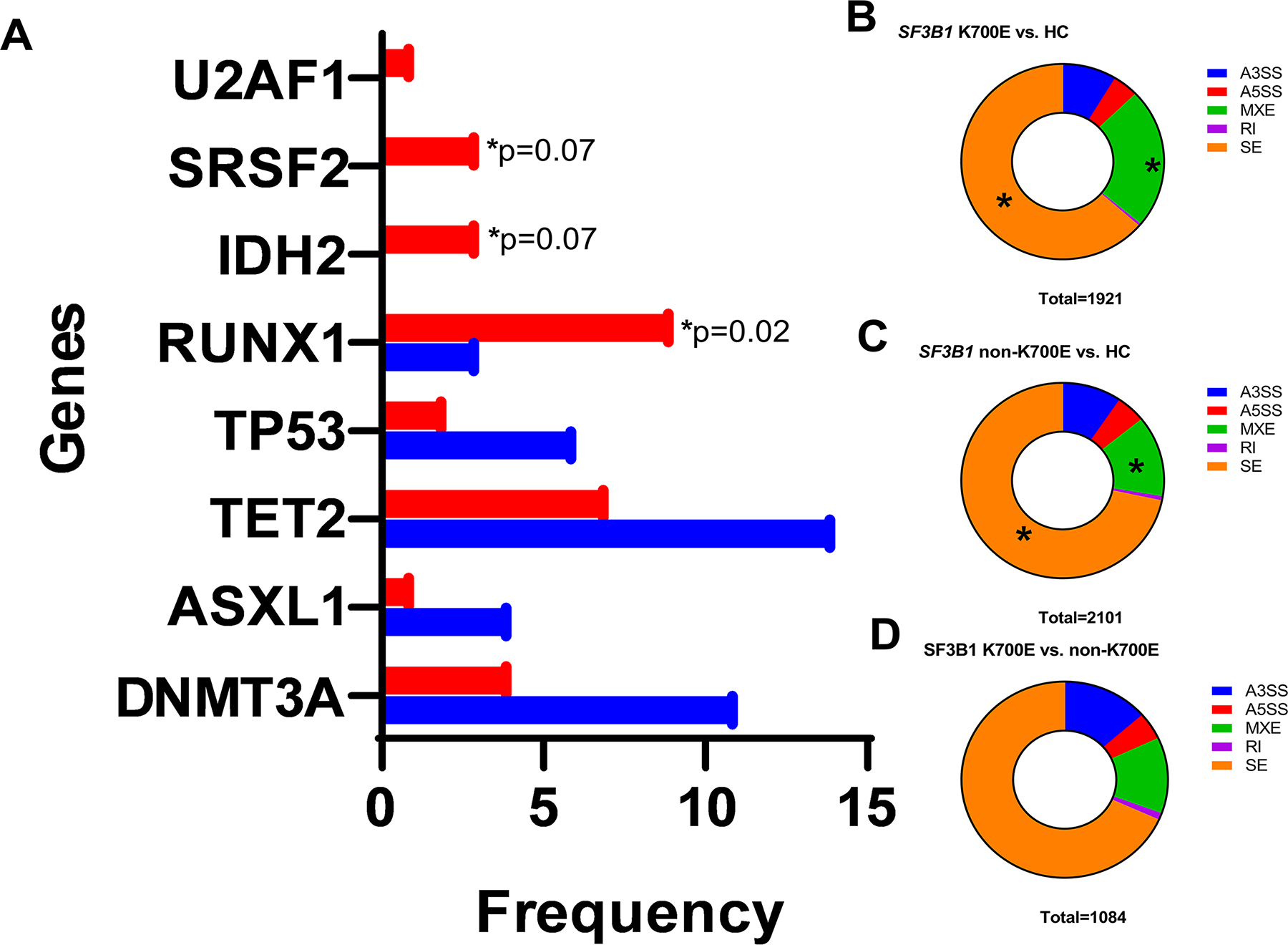
A. Within SF3B1 mutated MDS, the K700E mutant group has distinct clinico-pathologic and genetic characteristics compared to non-K700E mutant MDS. SF3B1 non-K700E mutated patients (red) had a significantly higher frequency of mutations in RUNX1, and a trend to higher frequencies of mutations in SRSF2 and IDH2 genes compared to SF3B1 K700E mutated MDS patients. B, C: Distribution of alternative splicing events when comparing SF3B1 K700E vs. healthy controls (panel B) and SF3B1 non-K700E vs. healthy controls (panel C) and K700E vs. non-K700E patients (panel D).
There were no significant differences in normal vs. complex karyotype. When cytogenetic aberrations were classified using the comprehensive cytogenetic scoring system (CCSS; scores from 0–5), SF3B1mut K700E mutated patients were more likely to have a lower CCSS scores (0/1) compared to non-K700E MDS patients [39 (80%) vs. 19 (53%); p=0·011].
Splicing analysis between K700E and non-K700E mutated MDS
Using rMATS, we identified the five most frequent types of alternative splicing events (alternative 5’ splice site, A5SS; alternative 3’ splice site, A3SS; mutually exclusive exon, MXE; retained intron, RI and skipped exon, SE). Comparing the SF3B1 K700E mutated MDS and healthy controls, we identified 1921 alternative splicing events in 852 genes. Similarly, there were 2101 events in 1457 genes when comparing SF3B1 non-K700E mutated MDS and healthy controls. The most frequent differentially spliced events in both SF3B1 K700E and non-K700E mutated MDS were SE (64%, 72%) and MXE (23%, 13%). Within SF3B1 K700E mutated MDS, differentially spliced MXE events were significantly more frequent (23% vs. 13%, p<0.0001) while SE events were less frequent (64% vs. 72%, p<0.0001) compared to non-K700E mutated MDS [Figure 2B, 2C]. There was no difference in the number of A3SS events (9% vs. 10%, p=0.38). Comparison between SF3B1 K700E and non-K700E mutated MDS showed the distribution of aberrant splicing events (n=1084): SE (68%) followed by A3SS (14%) and MXE (13%) [Figure 2D]. GSEA analysis demonstrated SF3B1 K700E and non-K700E MDS to have distinct profiles compared to healthy controls and to each other. When comparing K700E and non-K700E mutated MDS, there were 13 enriched pathways involving processing of capped intron-containing pre-mRNA, mRNA processing, splicing, metabolism, regulation and 3’ end processing, membrane trafficking, transport of transcripts to cytoplasm, immune cytokine signaling and RIG-I/MDA5 mediated induction of IFN-alpha/beta pathways.
Therapy and outcomes
The majority of patients in all 3 categories were treated with an HMA: 16/17 (94%) K700E mutated patients; 16/19 (84%) non-K700E mutated patients; and 217/277 (78%) SF3B1wt patients. The treatment details are provided in Supplementary Table 1. The median onset of HMA from the time of diagnosis was 10.1 months. With a median follow-up of 15·6 months, SF3B1mut MDS patients had a significantly better OS than SF3B1wt patients. This was true in the entire cohort (not reached vs. 25·2 months, p=0·0003), low-grade MDS categories (not reached vs. 41·3 months, p=0·0015), and low-grade MDS-RS categories (not reached vs. 22·3 months, p=0·0004). When segregated based on SF3B1 mutation types, the outcomes of K700E SF3B1mut MDS patients were significantly better compared to SF3B1wt within the entire cohort (median OS, not reached vs. 25.2 months, p=0.0001). We focused on the low-grade MDS patients with <5% blasts, and found similar findings: within the low-grade MDS (median OS, not reached vs. 41.3 months, p=0·0015) and with low-grade MDS-RS categories (median OS, not reached vs. 22.3 months, p=0.0001). In contrast, the outcomes of non-K700E SF3B1mut MDS patients were similar to SF3B1wt MDS patients, within the entire cohort (median OS, not reached vs. 25·2 months, p=0·2314), and within the low-grade MDS (median OS, not reached vs. 41·3 months, p=0·2598), and MDS-RS categories (median OS, not reached vs. 22·3 months, p=0·2327).
Within the MDS-EB categories, compared to SF3B1wt, there were no significant differences in OS of K700E SF3B1mut (median OS, not reached vs. 17·7 month, p=0·355) and non-K700E SF3B1mut (median OS, 20·5 vs. 17·7 months; p=0·5477) patients (Figure 3A–D).
Figure 3.
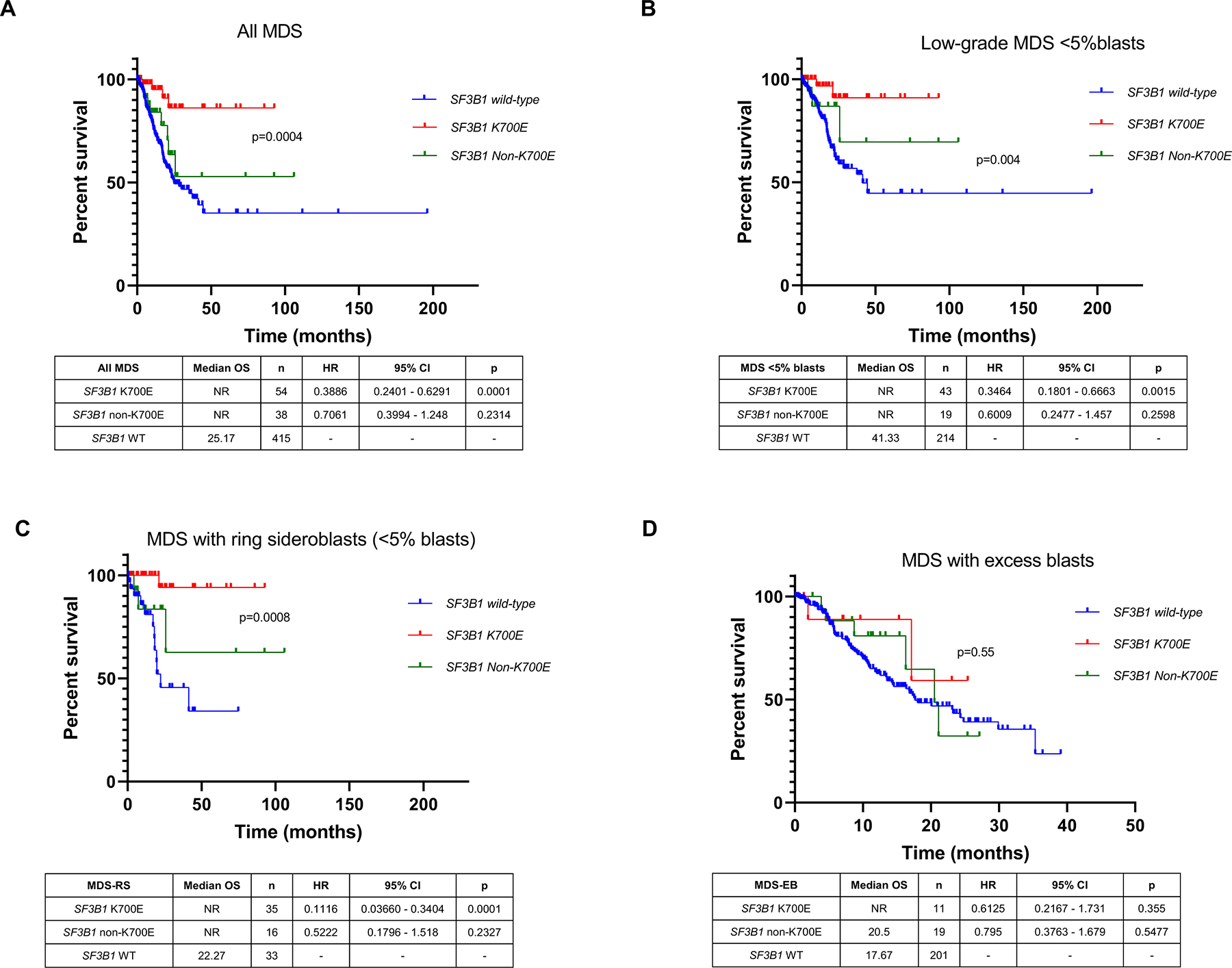
Overall survival (OS) of SF3B1 mutated MDS subtypes compared to SF3B1 wild-type MDS patients. OS of SF3B1 mutated MDS patients was significantly better than wild-type patients. When segregated based on SF3B1 mutation types, the outcome of SF3B1 non-K700E patients was similar to wild-type MDS, within the entire cohort (A, median OS, not reached for both; p=0·02), within low-grade MDS (B), and MDS-RS (C) categories, while there were no significant differences in OS within MDS-EB categories (D). The outcome of SF3B1 K700E patients was superior in all the categories. Kaplan-Meier curves show patients with SF3B1 K700E MDS mutations (red), SF3B1 non-K700E mutation MDS patients (green), and SF3B1 wild-type MDS patients (blue).
Between K700E vs. non-K700E SF3B1mut MDS categories, 9 of 39 (23%) K700E SF3B1mut MDS patients died compared to 4 of 55 (7%; p=0.02) non-K700E patients; findings were similar in low-grade MDS patients (16% vs. 3%, p=0.04). Within low-grade MDS, while the median OS of both K700E and non-K700E SF3B1mut were not reached in both categories, K700E SF3B1mut MDS patients had a significantly improved OS compared to non-K700E SF3B1mut MDS (p=0.02).
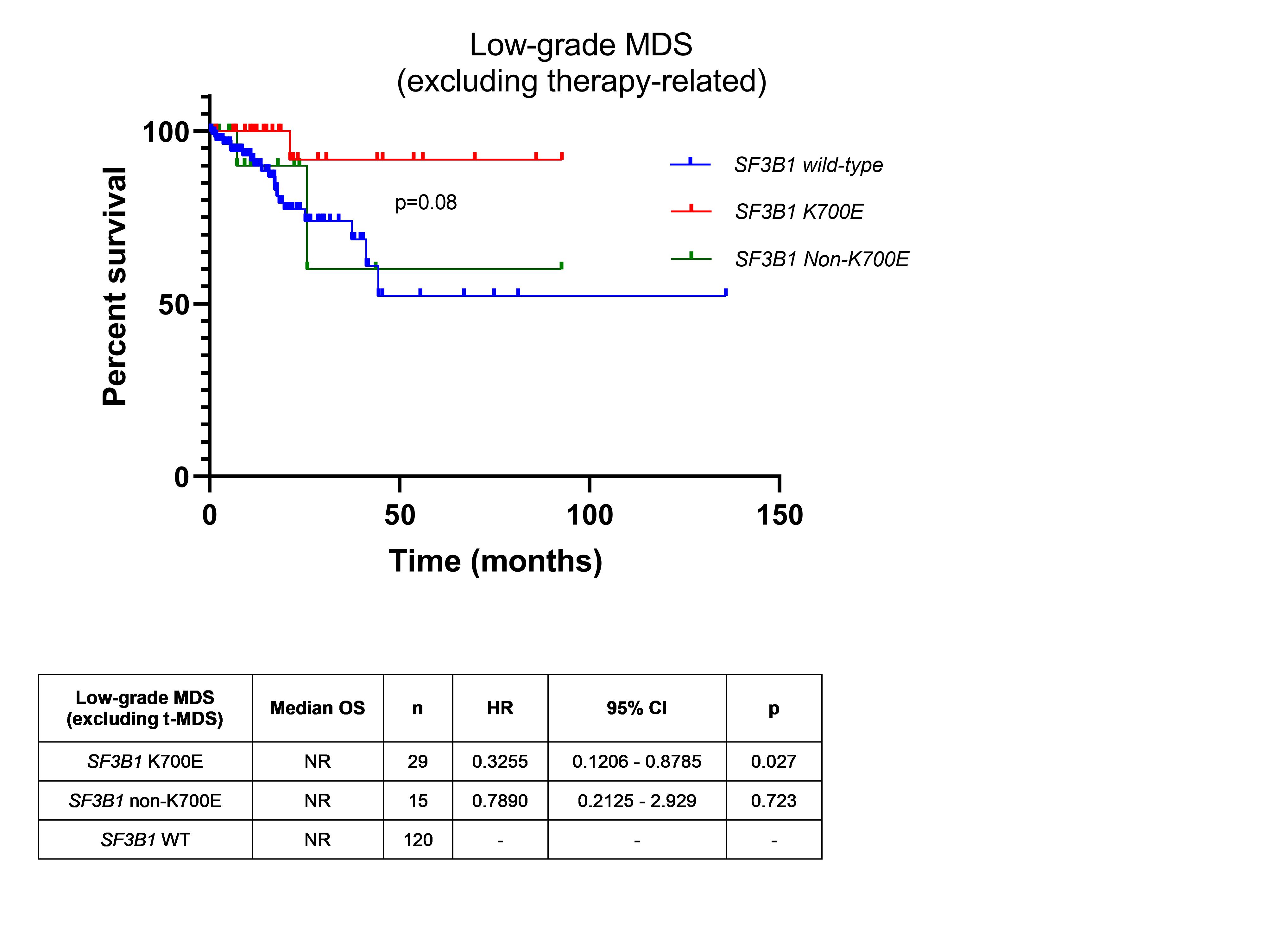
Therapy-related MDS cases were distributed equally between K700E and non-K700E groups. Within low-grade MDS (MDS-SLD, MDS-MLD and MDS-RS), we excluded therapy-related MDS cases due to a relatively higher representation of therapy-related MDS in our institutional cohort. Compared to SF3B1wt MDS, K700E SF3B1mut MDS had a significantly better OS (p=0.03) but not non-K700E SF3B1mut MDS (p=0.72) (Supplementary Figure 1). Compared to non-K700E SF3B1mut MDS, K700E SF3B1mut MDS showed a trend for better overall survival (p=0.08, median OS undefined for both).
As a next step, among low-grade MDS (<5% BM blasts and <1% PB blasts) with therapy-related cases excluded, we explored to see if the differences persisted between K700E and non-K700E SF3B1mut MDS groups even after applying the proposed 2020 IWG-PM criteria for SF3B1 mutant MDS. Based on these criteria, we excluded 2 cases of K700E SF3B1mut MDS (one with complex karyotype, other with EZH2 mutation) and 3 cases of non-K700E SF3B1mut MDS (one with EVI1 rearrangement and two with RUNX1 mutations). None met the criteria for other WHO entities (MDS/MPN), and none included isolated del(5q) or monosomy 7. Although numbers were small, K700E SF3B1mut MDS showed a trend for longer OS than non-K700E SF3B1mut MDS even in this cohort (undefined vs. 25.7 months; p=0.068; Supplementary Figure 2).
Within the entire MDS cohort, by univariate analysis (Supplementary Table 2), the following parameters were associated with worse outcomes: higher BM blasts percentage; lower hemoglobin, platelet and MCV; prior history of chemo-radiation (t-MDS); presence of CK; higher IPSS-R risk category; WHO MDS-EB category; absence of mutations in SF3B1 K700E, TET2, and U2AF1; and presence of a TP53 mutation. The outcome was superior when the SF3B1 mutation VAF was higher [p=0·026; OR, 0·96 (0·924–0·995)]. Non-K700E SF3B1 mutations did not associate with OS. By multivariable analysis (using a p-value cut-off of 0·200), lower hemoglobin, higher IPSS-R category, absence of SF3B1 K700E, and presence of TP53 mutation were independent predictors of worse OS. Within the MDS-RS categories (MDS-RS-SLD and MDS-RS-MLD), independent prognostic factors of worse OS included lower platelet count and presence of mutations in SF3B1 (non-K700E), ASXL1, SRSF2, and TP53 (Table 3).
Table 3.
Multivariate analysis for overall survival in all newly diagnosed untreated MDS patients (p-value cutoff of 0.200 from univariate analysis) SF3B1 mutations were segregated by subtype.
| p-value | Hazard Ratio | 95% CI | |
|---|---|---|---|
| Hemoglobin | <0.001 | 0.805 | 0.713–0.909 |
| R-IPSS | |||
| Very Low | 0.282 | 0.326 | 0.042–2.512 |
| Low | - | - | - |
| Intermediate | 0.016 | 2.288 | 1.166–4.487 |
| High | 0.01 | 2.436 | 1.237–4.798 |
| Very High | <0.001 | 4.143 | 2.076–8.467 |
| SF3B1 | |||
| Wild type | - | - | - |
| K700E | 0.041 | 0.288 | 0.087–0.951 |
| Non-K700E | 0.804 | 0.913 | 0.445–1.873 |
| BCOR | 0.154 | 0.234 | 0.032–1.724 |
| TP53 | <0.001 | 2.374 | 1.539–3.662 |
| U2AF1 | 0.082 | 0.513 | 0.242–1.089 |
Given the known independent prognostic influence of both SF3B1 and TP53 mutations, we assessed the co-mutation pattern and outcomes of SF3B1 mutations with TP53 mutations and CK. A total of 14 SF3B1mut patients had concurrent TP53 mutations [(n=9; median VAF 25·8 (range, 2–68%)] or CK (n=5). We divided the patients into 4 categories: (1) SF3B1wt MDS without TP53mut or CK (n=219); (2) SF3B1mut MDS without TP53mut or CK (n=71); (3) SF3B1mut MDS with TP53mut or CK (n=14); and (4) SF3B1wt MDS with TP53mut or CK (n=153). No survival differences were noted between patients with SF3B1mut MDS with or without mutated TP53 and/or CK (median OS, not reached) and patients with SF3B1wt MDS without TP53 mutations/CK (44·3 months). But MDS patients with TP53 mutations or CK had a significantly worse outcomes (median OS, 12·9 months, HR 1·46, p=0·001) in the absence of SF3B1 mutations. Similar findings were noted within the low-grade MDS and MDS-RS categories, where worse OS was associated with TP53 mutations or CK when SF3B1 was wild-type. This suggests that SF3B1 mutation may negate the poor prognostic effect of TP53 mutation or CK (Figure 4).
Figure 4.
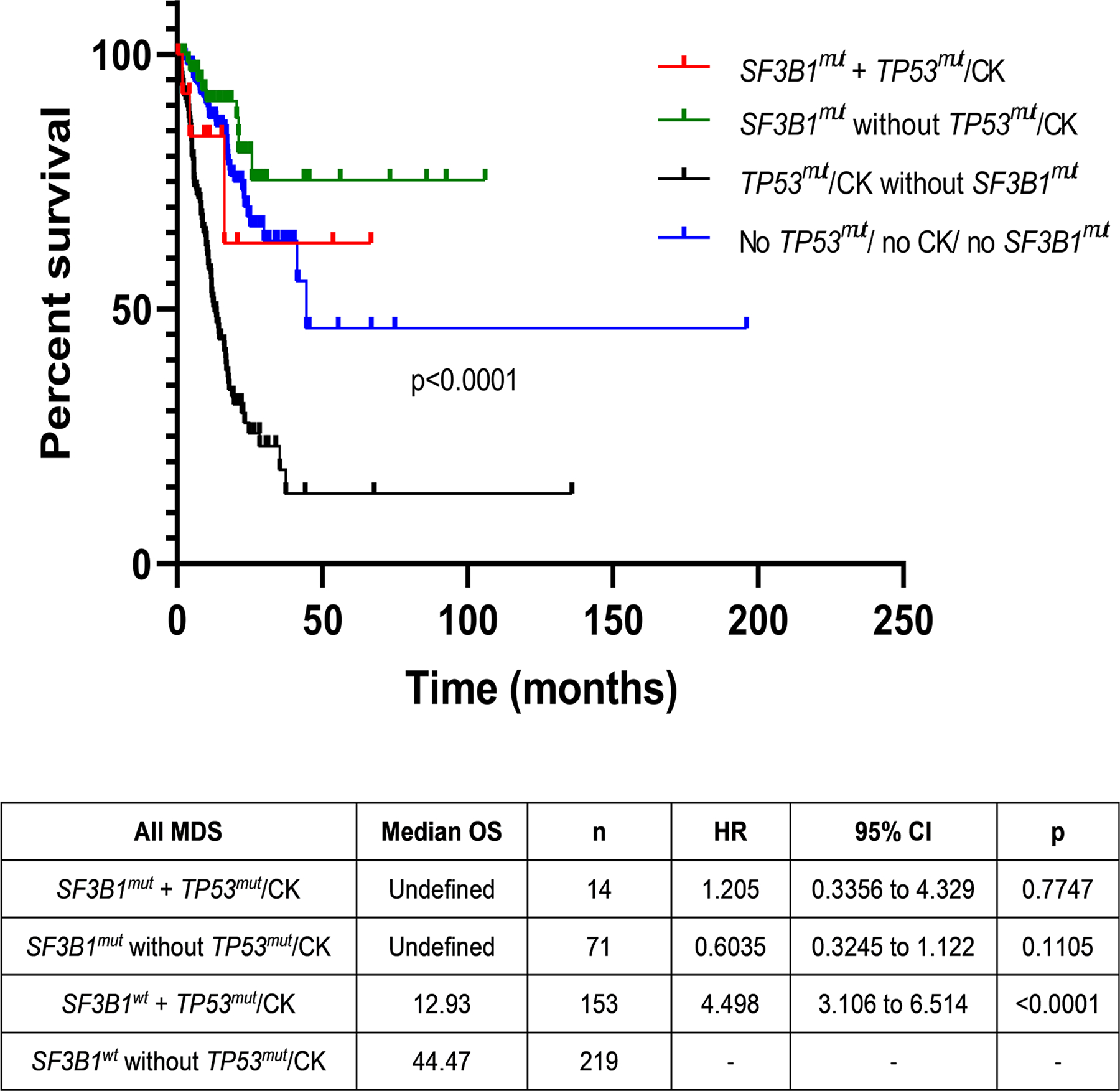
Survival differences in MDS based on concurrent SF3B1 and TP53 mutations and/or complex karyotype (CK). No overall survival differences were noted between SF3B1 mutated MDS patients with or without mutated TP53 and/or CK (median OS not reached) and SF3B1 wild-type MDS patients without TP53 mutations/CK (44·3 months). MDS patients with TP53 mutations or CK had significantly worse outcomes (median OS 12·9 months, HR 1·46, p=0·001) in the absence of SF3B1 mutations, suggesting that SF3B1 mutation may negate the poor prognostic effects of TP53 mutation or CK.
SF3B1 mutation acquired during disease course
SF3B1 mutation is considered a founder clone, however we observed 2 patients in which the mutation arose during disease evolution. The first patient was a 74-year-old man who was diagnosed with MDS-EB with trisomy 8 and mutations in BCOR, DNMT3A (x2), EZH2, TET2, and U2AF1 (33% VAF). BM morphology did not show any ring sideroblasts. Flow cytometry analysis showed 5% B-cells with a CLL/SLL-like immunophenotype. Following HMA treatment, a 6-month follow-up BM aspirate analysis showed persistent MDS with 2% blasts and clonal evolution with additional sub-clonal mutations in TET2 and SF3B1 R625C (VAF, 29·6%), expansion in the EZH2 clone, clearance of BCOR, and reduction in previously detected TET2 and U2AF1 mutations. As expected, given the acquisition of the SF3B1 mutation, BM morphology showed 18% ring sideroblasts (Figure 5A). The second case was a 68-year-old woman with pancytopenia. BM aspirate analysis showed MDS-EB with multi-lineage dysplasia and MF-3 fibrosis with an del(5q) abnormality in 16 of 20 metaphases. NGS analysis demonstrated a JAK2 V617 (<5% VAF) mutation at baseline. The patient underwent treatment with an HMA and PD-1/CTLA-4 blockers and transformed to AML over the next 4 months. Karyotype showed del(5q) abnormality. Additional mutations in SF3B1 K700E (29% VAF) and SETBP1 E862K (23·8% VAF) were acquired at transformation (Figure 5B). Both the patients underwent allogenic stem cell transplantation and have remained in morphologic and molecular remission. While these cases are rare, these interesting scenarios allow us to explore potential mechanisms of clonal evolution. Further evaluation in larger cohorts specifically evaluating this is warranted.
Figure 5.
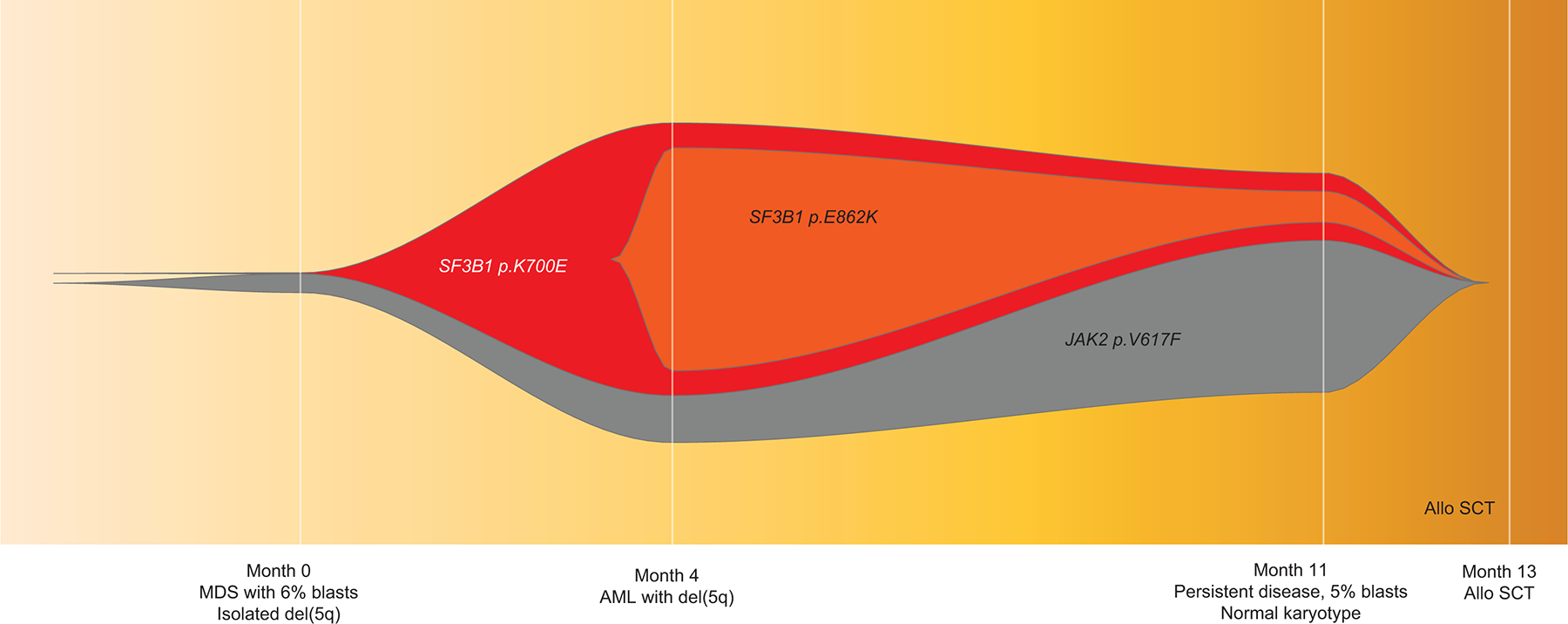
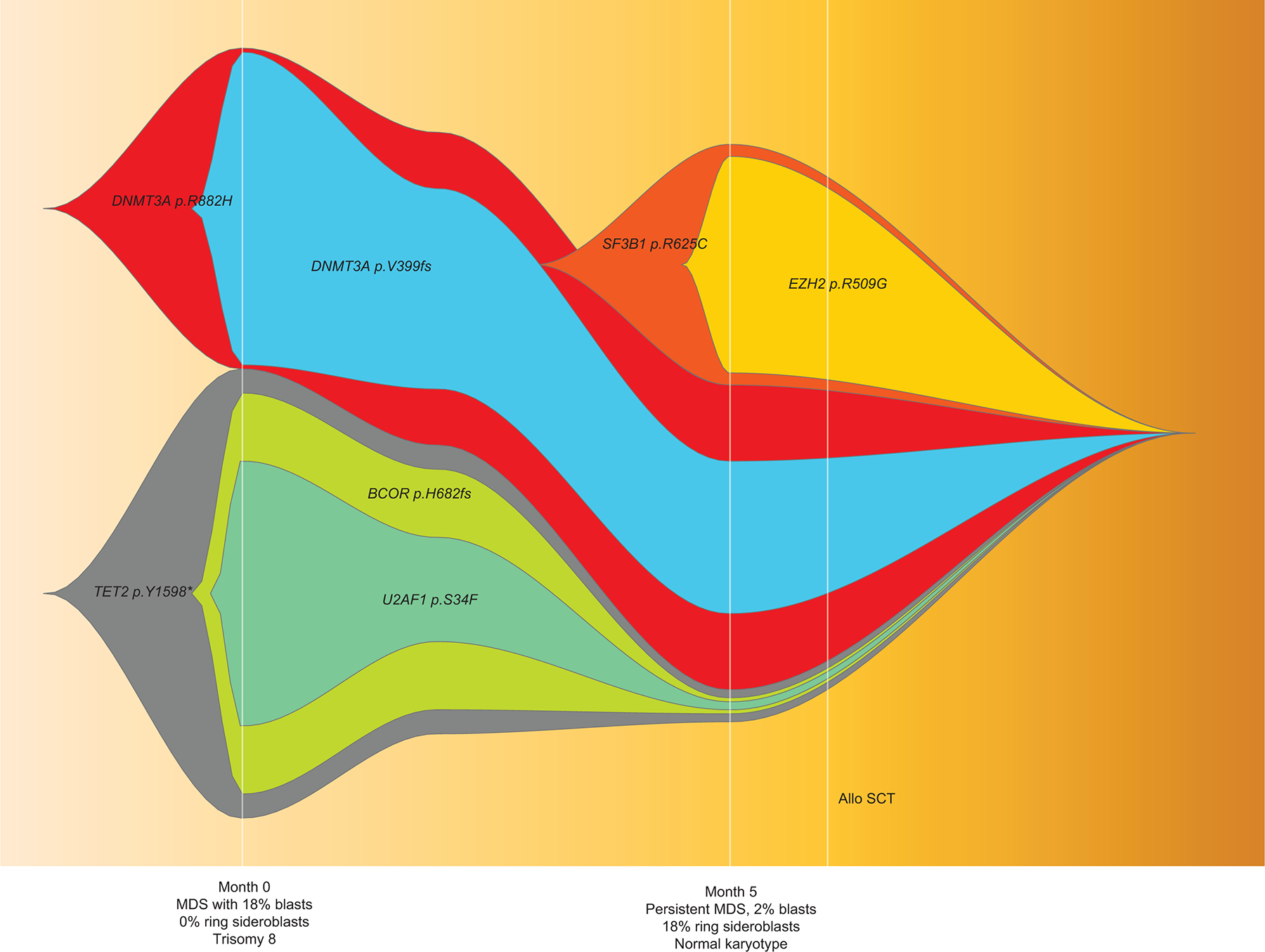
NGS analysis of serial of samples in two MDS patients who acquired SF3B1 mutations over the course of the disease: in patient #1, at the time of AML progression (A), and in patient #2, post HMA therapy at the time of morphologically persistent disease with genotype change corresponding to phenotypic acquisition of ring sideroblasts (B). Representative FISH plots from both cases showing clonal architecture at different time points. In both cases, mutations cleared following allogeneic stem cell transplantation.
DISCUSSION
The outcomes of SF3B1mut MDS, although regarded as a favorable prognostic biomarker, are variable. In this study, to understand the heterogeneity in clinico-pathologic features and outcomes of different types of SF3B1 mutations, we reviewed the entire spectrum of MDS, including low-grade (<5% blasts) through high-grade (≥5% blasts) MDS, and evaluated in detail within the low-grade MDS categories. Majority of these patients were primarily treated using hypomethylating agents.
SF3B1 mutations are observed in a third of MDS cases, are associated with ≥15% RS, and are overrepresented in the MDS-RS-SLD and MDS-RS-MLD WHO subtypes.2, 3, 21 Ring sideroblasts represent dysplastic erythroid precursors with abnormal iron-laden mitochondria encircling the nucleus.22, 23 Based on the distinct gene expression profile of SF3B1mut MDS and strong association with RS, the current WHO criteria classifies MDS with <5% BM blasts and 5–15% RS in the absence of isolated de(5q) with SF3B1 mutation as equivalent to MDS-RS-SLD or MDS-RS-MLD characterized by ≥15% RS.9 SF3B1 mutation is regarded as a favorable prognostic biomarker in MDS.2 The most frequent hotspot SF3B1 mutation is located on K700E, while the rest are located within exons 14–16.3, 4, 14
Consistent with the literature, our study found that SF3B1mut MDS had favorable clinical characteristics, with enrichment in lower IPSS-R categories and ring sideroblastic WHO subtypes. However, when segregated by the type of mutation, we demonstrated that only a subset of SF3B1mut MDS patients, those with K700E mutations, had favorable outcomes, while the outcomes of those with non-K700E mutations, noted in ~40% of all SF3B1 mutated patients, were similar to those with SF3B1wt MDS. This was true even in low-grade MDS, MDS with ring sideroblastic categories and in all MDS. The only significant adverse clinical feature in non-K700E mutated SF3B1 was lower ANC. Both K700E and non-K700E SF3B1mut MDS patient groups showed frequent RS (with a median RS percentage exceeding 15% in both), no significant difference in median BM blast percentage, although non-K700E cases had a higher representation within MDS-EB and IPSS-R high groups. Survival analysis within low-grade MDS and within MDS with ring sideroblastic categories (MDS-RS-SLD and MDS-RS-MLD) also showed a significantly favorable outcome for SF3B1mut K700E MDS patients compared to non-K700E patients. Non-K700E SF3B1mut MDS had a higher frequency of concomitant mutations in RUNX1 (>25% cases) and BCOR. RUNX1 mutation is an independent predictor of poor survival in MDS.6, 24 RUNX1 has been reported to be recurrently mutated in SF3B1mut MDS (~5% frequency) following TET2, DMT3A, SRSF2, and ASXL1 and is known to be associated with worse OS and disease progression.2 The higher frequency of RUNX1 mutations in our cohort (~15%) may be due to tertiary referral bias; it was much more frequent in non-K700E mutated patients compared to K700E patients (26% vs. 7.3%, p=0.012). Presence of K700E SF3B1mut alone (not non-K700E mutations) was associated with favorable OS in both univariate and multivariate analysis, along with other prognostic factors including hemoglobin, IPSS-R category, and TP53 mutations. The latter finding was true within low-grade MDS (<5% blasts) and MDS–RS categories (also <5% blasts). Thus, recognition of SF3B1 mutation type by sequencing the coding region that includes at least exons 14–16, is important and has bearing on clinical management. Hence, incorporation of the type of SF3B1 mutation rather than simply the presence of mutation and ring sideroblast percentage will be helpful for future WHO sub-classification. To support the biological distinction of these subtypes, we also performed exploratory splicing analysis on a small number of K700E and non-K700E SF3B1mut MDS cases and found differences in the distribution of aberrant splicing events, and gene expression profiles. But, the small number of patients precludes definitive conclusions, and does not rule out the possibility of co-mutated genes affecting these changes.
We evaluated clinicopathologic associations between SF3B1mut and other co-mutations. SF3B1 mutation is an MDS initiating clone arising in hematopoietic stem cells of lymphomyeloid origin, with the mutation often being the sole driver with a near-heterozygous VAF.25, 26 Within our MDS cohort, >20% of SF3B1mut MDS patients had common CHIP-associated gene mutations in TET2 and DNMT3A. The presence of these additional gene mutations was not associated with any particular clinical or pathologic characteristics.
TP53 mutations were noted in >10% of SF3B1mut MDS patients. Since TP53 mutations associated with CK, as observed in ~10% of MDS patients, we assessed the interplay of these 3 parameters on outcomes. Our findings showed that SF3B1 mutations negate the poor prognostic effects of TP53 mutations and/or CK. The frequency of concurrent TP53 mutations in SF3B1mut MDS was higher than expected in our cohort due to a high frequency of therapy-related cases. About ~55% of SF3B1mut MDS with concurrent TP53 mutations were therapy-related. Within all t-MDS cases (n=159), 17 (11%) were SF3B1mut MDS, 5 (29%) of which had TP53 mutations, and majority 142 (89%) were SF3B1 wild-type, 64 (45%) of which had TP53 mutations. Interestingly, SF3B1 mutations were enriched in t-MDS with ring sideroblasts, more frequent than TP53 mutations.
In addition to CK, other poor prognostic cytogenetic features in a subset of SF3B1mut MDS included del(7q)/-7 and EVI1/MECOM rearrangement. Studies have shown enrichment of SF3B1 mutations in AML with inv(3) or t(3;3) and overexpression of EVI1 in SF3B1mut MDS that transform to AML.27, 28 But, in our cohort, the frequency of EVI1 rearrangement was not significantly different from the wild-type cases. There were also 3 SF3B1mut patients with t(1;3)(p36;q21), a rare but recurrent rearrangement in MDS/AML involving PRDM16 (PR/SET domain containing 16) or MEL1 (located on 1p36) and the enhancer element of GATA2 (RPN1), located on 3q21. PRDM16 is zinc finger transcriptional regulator structurally similar to MECOM (MDS1 and EVI1 complex). Rearrangements of PRDM16 in t(1;3)(p36;q21) and MECOM in inv(3)(q21q26.2) or t(3;3)(q21;q26.2) to the GATA2/RPN1 locus lead to overexpression of the aberrant oncogenic short-forms of PRDM16s and EVI1 that lack the PR/SET domain. Aberrant Prdm16s has been implicated in leukemogenesis or progression by transformation of megakaryocyte-erythroid progenitors to myeloid leukemia stem cells.29, 30
Within this single institution large dataset, we were able to evaluate SF3B1 mutations in unique clinical scenarios, including isolated del(5q) (all of which were associated with K700E mutations), SF3B1 mutations acquired during the disease course and t-MDS. Within SF3B1mut MDS, a high proportion of t-MDS cases had concurrent TP53 mutations and/or CK (57%), while fewer had SF3B1 mutations without a TP53 mutation or CK (19%). There were no differences in outcome based on therapy-related disease [HR 2·11, 95% CI 0·78–5·66, p=0·140].
Our study is a retrospective analysis of a large single-institutional series of MDS patients primarily treated using hypomethylating agents, with limited numbers of K700E and non-K700E SF3B1mut mutated MDS cases, and enrichment of therapy-related cases due to referral bias etc. and relatively short duration of follow-up. Due to the heterogeneity in MDS, large number of patients are needed to define the clinical and prognostic effect of somatic mutations in MDS. Hence, these findings are largely descriptive, and validation of these findings in a large independent cohort, as well as prospective analysis of the outcome differences between K700E and non-K700E SF3B1mut MDS patients, in the context of newer therapies such as luspatercept, are needed.
In summary, we show that ~40% of SF3B1mut MDS show non-K700E mutations. Hotspot K700E and non-K700E SF3B1mut MDS show distinct clinical and mutational profiles, with K700E showing a significantly better OS compared to non-K700E and SF3B1wt. Only absence of SF3B1mut K700E mutation independently predicted for worse OS in MDS. Hence, identification of the SF3B1 mutation type is important for risk stratification.
Supplementary Material
Supplementary Figure 2. When SF3B1 mutated MDS cases were selected based on the proposed 2020 IWG-PM criteria, K700E SF3B1mut MDS showed a trend for longer OS than non-K700E SF3B1mut MDS although the numbers were small (undefined vs. 25.7 months; p=0.068).
{kind=link}
Supplementary Figure 1. Even after excluding therapy-related MDS cases, K700E SF3B1mut MDS had a significantly better OS (p=0.03) compared to SF3B1wt MDS, but not non-K700E SF3B1mut MDS (p=0.72). Compared to non-K700E SF3B1mut MDS, K700E SF3B1mut MDS showed a trend for better overall survival (p=0.08).
{kind=link}
Acknowledgements:
This work was supported in part by grants from the Ladies Leukemia League and leukemia SPORE career enhancement award (to Rashmi Kanagal-Shamanna), the National Cancer Institute of the National Institutes of Health (University of Texas MD Anderson Cancer Center Support Award CA016672 [all authors]), and the University of Texas MD Anderson MDS/AML Moon Shot (to Yue Wei, Kelly A. Soltysiak, Faezeh Darbaniyan, Hagop Kantarjian, and Guillermo Garcia-Manero).
Funding:
This work was supported by grants from the Ladies Leukemia League, the National Institutes of Health (CA016672) and the MD Anderson MDS/AML Moon Shot.
Author Disclosures:
Rashmi Kanagal-Shamanna: This author declares no conflict of interest.
Guillermo Montalban-Bravo: This author declares no conflict of interest.
Koji Sasaki: This author declares an advisory role with Pfizer Japan.
Faezeh Darbaniyan: This author declares no conflict of interest.
Elias Jabbour: This author declares research support and an advisory role with Adaptive, AbbVie, Amgen, Pfizer, Cyclacel LTD, Takeda, Bristol Myers Squibb.
Carlos Bueso-Ramos: This author declares no conflict of interest.
Yue Wei: This author declares no conflict of interest.
Kelly Chien: This author declares no conflict of interest.
Tapan Kadia: This author declares no conflict of interest.
Farhad Ravandi: This author declares no conflict of interest.
Gautam Borthakur: This author declares no conflict of interest.
Kelly A. Soltysiak: This author declares no conflict of interest.
Mark Routbort: This author declares no conflict of interest.
Keyur Patel: This author declares no conflict of interest.
Sherry Pierce: This author declares no conflict of interest.
L. Jeffrey Medeiros: This author declares no conflict of interest.
Hagop Kantarjian: This author declares research support and an advisory role with Actinium, and research support from AbbVie, Agio, Amgen, Ariad, Astex, BMS, Cyclacel, Daiichi-Sankyo, Immunogen, Jazz Pharma, Novartis, and Pfizer.
Guillermo Garcia-Manero: This author declares research support and an advisory role with Bristol Myers Squibb, Astex, and Helsinn, and research support from Amphivena, Novartis, AbbVie, H3 Biomedicine, Onconova, and Merck.
Footnotes
Competing Interests: The authors declare no competing interests.
REFERENCES
- 1.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122: 3616–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Malcovati L, Karimi M, Papaemmanuil E, et al. SF3B1 mutation identifies a distinct subset of myelodysplastic syndrome with ring sideroblasts. Blood. 2015;126: 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papaemmanuil E, Cazzola M, Boultwood J, et al. Somatic SF3B1 mutation in myelodysplasia with ring sideroblasts. N Engl J Med. 2011;365: 1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478: 64–69. [DOI] [PubMed] [Google Scholar]
- 5.Chen J, Kao YR, Sun D, et al. Myelodysplastic syndrome progression to acute myeloid leukemia at the stem cell level. Nat Med. 2019;25: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malcovati L, Stevenson K, Papaemmanuil E, et al. SF3B1-mutant myelodysplastic syndrome as a distinct disease subtype-A Proposal of the International Working Group for the Prognosis of Myelodysplastic Syndromes (IWG-PM). Blood. 2020;136: 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Migdady Y, Barnard J, Al Ali N, et al. Clinical outcomes with ring sideroblasts and SF3B1 mutations in myelodysplastic syndromes: MDS clinical research consortium analysis. Clin Lymphoma Myeloma Leuk. 2018;18: 528–532. [DOI] [PubMed] [Google Scholar]
- 8.Montalban-Bravo G, Garcia-Manero G. Myelodysplastic syndromes: 2018 update on diagnosis, risk-stratification and management. Amer J Hematol. 2018;93: 129–147. [DOI] [PubMed] [Google Scholar]
- 9.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127: 2391–2405. [DOI] [PubMed] [Google Scholar]
- 10.Kanagal-Shamanna R, Hidalgo Lopez JE, Milton DR, et al. Validation of the 2016 revisions to the WHO classification in lower-risk myelodysplastic syndrome. Am J Hematol. 2017;92: E168–E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fenaux P, Platzbecker U, Mufti GJ, et al. Luspatercept in Patients with Lower-Risk Myelodysplastic Syndromes. N Engl J Med. 2020;382: 140–151. [DOI] [PubMed] [Google Scholar]
- 12.Alsafadi S, Houy A, Battistella A, et al. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun. 2016;7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darman RB, Seiler M, Agrawal AA, et al. Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3’ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 2015;13: 1033–1045. [DOI] [PubMed] [Google Scholar]
- 14.Obeng EA, Chappell RJ, Seiler M, et al. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell. 2016;30: 404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalton WB, Helmenstine E, Pieterse L, et al. The K666N mutation in SF3B1 is associated with increased progression of MDS and distinct RNA splicing. Blood Adv. 2020;4: 1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia. Blood. 2016;127: 2391–405. [DOI] [PubMed] [Google Scholar]
- 17.Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120: 2454–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanagal-Shamanna R, Luthra R, Yin CC, et al. Myeloid neoplasms with isolated isochromosome 17q demonstrate a high frequency of mutations in SETBP1, SRSF2, ASXL1 and NRAS. Oncotarget. 2016;7: 14251–14258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shen S, Park JW, Lu Z-x, et al. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci. 2014;111: E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108: 419–425. [DOI] [PubMed] [Google Scholar]
- 21.Malcovati L, Cazzola M. Recent advances in the understanding of myelodysplastic syndromes with ring sideroblasts. Br J Haematol. 2016;174: 847–858. [DOI] [PubMed] [Google Scholar]
- 22.Della Porta MG, Travaglino E, Boveri E, et al. Minimal morphological criteria for defining bone marrow dysplasia: a basis for clinical implementation of WHO classification of myelodysplastic syndromes. Leukemia. 2015;29: 66–75. [DOI] [PubMed] [Google Scholar]
- 23.Cazzola M, Invernizzi R. Ring sideroblasts and sideroblastic anemias. Haematologica. 2011;96: 789–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bejar R, Stevenson K, Abdel-Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mortera-Blanco T, Dimitriou M, Woll PS, et al. SF3B1-initiating mutations in MDS-RSs target lymphomyeloid hematopoietic stem cells. Blood. 2017;130: 881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mian SA, Rouault-Pierre K, Smith AE, et al. SF3B1 mutant MDS-initiating cells may arise from the haematopoietic stem cell compartment. Nat Commun. 2015;6: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiozawa Y, Malcovati L, Gallì A, et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood. 2017;130: 2642–2653. [DOI] [PubMed] [Google Scholar]
- 28.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374: 2209–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hu T, Morita K, Hill MC, et al. PRDM16s transforms megakaryocyte-erythroid progenitors into myeloid leukemia–initiating cells. Blood. 2019;134: 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mochizuki N, Shimizu S, Nagasawa T, et al. A novel gene, MEL1, mapped to 1p36. 3 is highly homologous to the MDS1/EVI1 gene and is transcriptionally activated in t (1; 3)(p36; q21)-positive leukemia cells. Blood. 2000;96: 3209–3214. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 2. When SF3B1 mutated MDS cases were selected based on the proposed 2020 IWG-PM criteria, K700E SF3B1mut MDS showed a trend for longer OS than non-K700E SF3B1mut MDS although the numbers were small (undefined vs. 25.7 months; p=0.068).
Supplementary Figure 1. Even after excluding therapy-related MDS cases, K700E SF3B1mut MDS had a significantly better OS (p=0.03) compared to SF3B1wt MDS, but not non-K700E SF3B1mut MDS (p=0.72). Compared to non-K700E SF3B1mut MDS, K700E SF3B1mut MDS showed a trend for better overall survival (p=0.08).