

To the Editor:
The anti‐apoptotic protein B‐cell lymphoma 2 (BCL‐2) is a key regulator of the intrinsic apoptotic pathway, 1 and survival dependence on BCL‐2 has been demonstrated in chronic lymphocytic leukemia (CLL). 2 Venetoclax is a highly selective, potent, orally bioavailable BCL‐2 inhibitor 3 that has demonstrated efficacy as monotherapy for patients with previously treated CLL in four clinical studies. 4 , 5 , 6 , 7 , 8 Because venetoclax is the first approved BH3 mimetic and an approved treatment option for CLL, strong interest exists in learning about potential mechanisms of resistance.
To investigate resistance to venetoclax in vitro, we performed a genome‐wide CRISPR knockout screen. In the absence of a suitable CLL cell line, it was performed in SU‐DHL4, a diffuse large B‐cell lymphoma cell line. Upon venetoclax treatment, cells with potential genetic perturbations leading to drug resistance were enriched in the surviving population. Consistent with previously described functional genomic screens across multiple disease models, 9 genes encoding members of the pro‐apoptotic machinery, including BAX, BCL2L11 (BIM), and PMAIP1 (NOXA), were among the strongest hits associated with venetoclax resistance (Figure 1A). Loss of ID3 or NFKBIA was also associated with decreased venetoclax sensitivity, suggesting multiple possible mechanisms of resistance. 10
FIGURE 1.
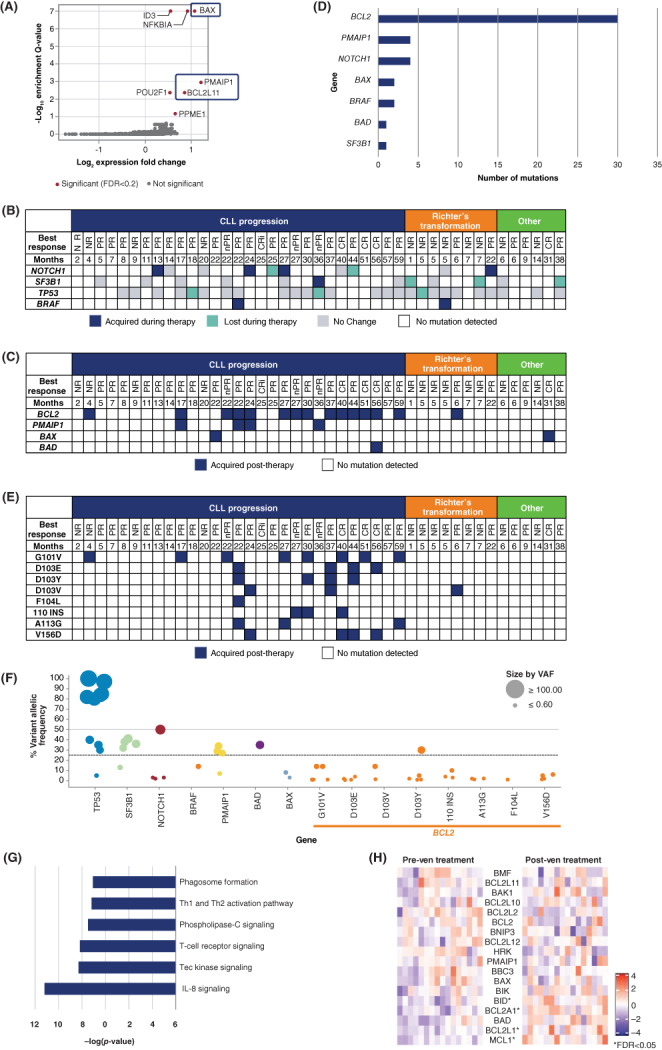
(A) Gene deletions identified from a pooled genome‐wide CRISPR screen in SU‐DHL4 cells that confer venetoclax resistance. (B) Tumor genomic changes in patients with CLL after venetoclax monotherapy. (C) Acquired mutations in apoptotic machinery after venetoclax monotherapy. (D) Frequency of acquired mutations after venetoclax monotherapy. (E) Acquired BCL2 mutations after venetoclax monotherapy. (F) Variant allele frequency of mutations. (G) Upregulation of genes in patients who relapsed on venetoclax monotherapy. (H) Changes in expression of BCL2 family genes pre‐ and post‐treatment. * represents significant expression changes (FDR < 0.05). CR, complete remission; CRi, complete remission with incomplete hematopoietic recovery; nPR, nodular partial remission; NR, nonresponder; PR, partial remission; VAF, variant allele frequency
To compare these results with potential mechanisms of resistance in patients treated with venetoclax, we performed whole‐exome and targeted sequencing of DNA extracted from CD19+ B cells collected before treatment initiation and/or upon treatment discontinuation from 43 patients with relapsed/refractory CLL or small lymphocytic lymphoma. All patients were enrolled on a venetoclax monotherapy study (NCT01328626, 5 n = 18 or NCT01889186, 6 n = 25) and received treatment until disease progression or unacceptable toxicity. Selection for these analyses was based on sample availability. Baseline characteristics are shown in Table S1.
Median time on therapy was 20.1 months (range, 1.2–60.3) with a median duration follow‐up of 61.3 months (95% CI, 48.6–73.4). Venetoclax was nearly as efficacious in this cohort as it was in the overall cohort 5 , 6 (and pooled data of 347 patients across three venetoclax studies 11 ), with an overall response rate (ORR) of 69.8%, a complete remission/complete remission with incomplete hematopoietic recovery (CR/CRi) rate of 11.6%, and median progression‐free survival (PFS) of 21.9 months (Table S2). Although minimal residual disease (MRD) evaluation was not required for all patients, of the 12 patients with ≥ 1 MRD assessment, five achieved undetectable MRD at 10−4 in the peripheral blood, whereas seven had detectable MRD. Post‐treatment specimens were collected at study discontinuation due to CLL progression (n = 29), Richter's transformation (n = 8), or other reasons (n = 6) (Table S3).
To investigate changes in clonal architecture after venetoclax treatment in CLL, we focused on three of the most mutated genes in CLL: NOTCH1, SF3B1, and TP53 (Figure 1B). Although some gains and losses of mutations in these genes were observed, most mutations present before treatment initiation were detected in post‐venetoclax specimens. Gains and losses of NOTCH1 mutations were detected in post‐treatment samples of four and two patients, respectively. One patient's CLL gained an SF3B1 mutation at progression, whereas in three patients, SF3B1‐mutated clones present at commencement were no longer detected. We did not observe gains of TP53 mutations. For three patients, TP53‐mutant clones present at commencement were not detected subsequently, supporting the notion that venetoclax‐based treatments are efficacious in CLL patients with these mutations. Two patients gained BRAF V600E mutations, consistent with a recent report. 12 Overall, based on the markers evaluated, one‐third (n = 14/43) of patients exhibited changes in clonal architecture during treatment.
Previous studies have identified BCL2 mutations associated with relapse on venetoclax. 13 , 14 , 15 In our cohort, no BCL2 mutations were found at study entry, but BCL2 mutations were acquired in CLL cells from 15 of 43 patients (35%), 14 of whom experienced CLL progression (Figure 1C). Of 29 patients with CLL progression, BCL2 mutations were identified in 10 patients who were on treatment for ≥24 months. Overall, 30 distinct BCL2 mutations were identified, with multiple mutations identified in nine patients (Figure 1D,E). The most frequent mutations of any description observed were at residue D103 (11 occasions) and G101 (8 occasions). Additionally, mutations were identified at F104 (one occasion), A113 (three occasions), and V156 (four occasions). A 4–amino acid (RRYR) insertion at residue 110 was identified in samples from three patients. Except for the insertion, all BCL2 alterations were missense mutations. In agreement with previous findings, 13 , 14 most BCL2 mutations were found in minor clones (Figure 1F). Only 1 BCL2 mutation (D103Y) had a variant allele frequency (VAF) > 20%, whereas most SF3B1 and TP53 mutations were in larger clones (VAF > 20%).
Mutations in apoptosis regulators other than BCL2 were identified, including PMAIP1 and BAX, two of the strongest hits identified in the CRISPR screen. All PMAIP1 mutations were loss‐of‐function, including three nonsense mutations and one mutation leading to loss of translation‐initiating methionine, M1. One BAX mutation was nonsense; the other mutation replaced leucine at amino acid 181 with histidine. Although both BAX mutations had a VAF < 10%, three of four PMAIP1 mutations had a VAF > 20%, suggesting that PMAIP1 mutations could perhaps be a key driver of venetoclax resistance, although further study is needed. In addition to a BCL2 mutation, one patient also had a mutation in the pro‐apoptotic gene BAD (G111R) with a VAF >25%; the functional relevance of this is unknown. Three PMAIP1 mutant patients also had BCL2 mutations present at the end of treatment, albeit at different allele frequencies, supporting the idea of multiple potential mechanisms of resistance within an individual patient.
Although initial clinical response to venetoclax‐based therapies are similar in CLL patients regardless of TP53 disruption via del(17p) and/or TP53 mutation, patients with del(17)p/TP53 mutation had shorter response duration. 11 , 16 , 17 In our cohort, 78% of patients (n = 29/37) with CLL progression or Richter's transformation had a del(17p)/TP53 mutation at baseline. Additionally, 87% of patients (n = 13/15) with BCL2 mutations had evidence of TP53 disruption.
To provide a deeper understanding of the characteristics of the patients available for these exploratory analyses, the outcomes are reported. Among the eight patients who progressed with Richter's transformation, the ORR was 25.0% with a median PFS of 5.3 months (Table S2). While two patients achieved partial remission (PR), most patients were refractory to venetoclax prior to diagnosis of Richter's transformation. One patient who acquired a BCL2 mutation achieved a PR but relapsed after 6 months. In contrast, the ORR for patients who eventually relapsed due to CLL progression was 86.2%, with seven achieving a deep remission (CR/CRi/nodular partial remission) and 18 achieving a PR. Median duration of response (DOR) was 23.7 months and median PFS was 21.9 months. Among patients who relapsed with CLL progression, median PFS for patients with a BCL2 mutation at relapse was 27.3 months but for patients without a BCL2 mutation median PFS was 10.8 months. Similar results were demonstrated for patients with a mutation in any apoptotic gene; median PFS for patients with versus without an acquired mutation in any apoptotic gene was 27.3 and 10.8 months, respectively. Lastly, for patients who acquired a mutation in BCL2 or other apoptotic genes, the DOR was longer compared with DOR in patients without a mutation (Table S2). Given the observation that acquisition of mutations in apoptotic genes did not portend a shorter response, we evaluated gene expression changes in the pre‐ and post‐progression specimens.
To investigate transcriptional changes that occurred, we performed RNA‐seq on 16 pairs of pre‐ and post‐venetoclax samples from patients with CLL progression. Comparison of samples identified changes in expression of over 800 genes (FDR < 0.05). Several genes that play a role in IL‐8, Tec kinase, and phospholipase C signaling were upregulated in patients at relapse (Figure 1G). Comparison of pre‐ and post‐treatment samples between patients who were on study for different durations of time did not yield any differentially expressed genes. We were interested in looking at changes in expression of BCL2 family genes because they are known to be able to affect venetoclax resistance in vitro (Figure 1H). A trend of decreased BCL2 expression in post‐venetoclax samples was observed, although the differences did not reach statistical significance. Concomitantly, we observed a statistically significant increase in expression of MCL1, BCL2L1 (BCL‐XL), and BCL2A1 (BFL‐1), three genes whose protein products can inhibit apoptosis when BCL‐2 is effectively targeted by venetoclax.
In addition to previously published mutations in BCL2, we identified novel mutations in PMAIP1, BAX, and BAD in CLL patients who developed resistance to venetoclax monotherapy. This is the first confirmation of the top hits from an in vitro screen with patient samples from clinical trials. Our findings suggest that inactivation of BAX, BAD, or PMAIP1 may be sufficient to render CLL cells resistant to venetoclax treatment, identifying these factors as potential critical regulators of venetoclax response. This is the first report to evaluate mRNA expression differences in this patient population. Observed gene expression changes indicated that tumor cells may upregulate anti‐apoptotic BCL‐2 proteins other than BCL‐2 to avoid apoptosis induction by venetoclax. Overall, our data suggest that resistance to venetoclax monotherapy in CLL patients is multifactorial and highly dependent on mutational and gene expression changes that alter the balance between the pro‐ and anti‐apoptotic machinery. Additional studies are needed to determine the clinical relevance of minor BCL2 mutant clones in venetoclax resistance and whether similar mechanisms of resistance arise in patients treated with fixed‐duration venetoclax combination therapies.
CONFLICT OF INTEREST
R.P., F.D., C.L., K.R., D.Q., S.E.W., N.M., A.J.S., J.F.W., J.A.R., and B.J.C. are employees of AbbVie and may hold stock or other options; J.P. is a former employee of AbbVie, current employee of GlaxoSmithKline, and may hold stock or other options with AbbVie; M.S.D. received institutional research grants from AbbVie, Ascentage Pharma, AstraZeneca, Genentech, MEI Pharma, Novartis, Surface Oncology, TG Therapeutics, and Verastem; and personal fees from AbbVie, Adaptive Biotechnologies, Ascentage Pharma, AstraZeneca, BeiGene, Bristol Myers Squibb, Celgene, Eli Lilly, Genentech, Janssen, MEI Pharma, Merck, Novartis, Research to Practice, Takeda, TG Therapeutics, and Verastem; E.T. received fees for consulting and research support from AbbVie, Janssen, and Roche; S.S. received fees for consulting, drug/equipment, funding, and writing assistance from AbbVie, AstraZeneca, Celgene, Gilead, GlaxoSmithKline, Hoffman La‐Roche, Janssen, Novartis, Pharmacyclics, and Sunesis; J.D.L. is a former employee of AbbVie, current employee of Pfizer, and may hold stock or other options with AbbVie; S.Y.K. is a former employee of AbbVie, current employee of Nurix Therapeutics, and may hold stock or other options with AbbVie.
AUTHOR CONTRIBUTIONS
Relja Popovic, Kenneth Robinson, Scott E. Warder, Andrew J. Souers, Jeffrey F. Waring, Matthew S. Davids, Stephan Stilgenbauer, Eugen Tausch, Jeremy A. Ross, Joel D. Leverson, Su Young Kim, and Brenda J. Chyla conceived study. Relja Popovic, Kenneth Robinson, Su Young Kim, and Brenda J. Chyla contributed to data collection. Relja Popovic, Fengjiao Dunbar, Charles Lu, Danjuma Quarless, Nabanita Mukherjee, John Pesko, and Brenda J. Chyla contributed to data analysis. All authors contributed to manuscript drafting.
Supporting information
Table S1 Patient demographics and disease characteristics at baseline.
Table S2 Summary of efficacy parameters.
Table S3 Post‐treatment specimens.
ACKNOWLEDGMENTS
Venetoclax is being developed in collaboration between Genentech Inc. and AbbVie. AbbVie funded this study and participated in the study design, research, analysis, data collection, interpretation of data, as well as the writing, review, and approval of the publication. All authors had access to relevant data and participated in the drafting, review, and approval of this publication. No honoraria or payments were made for authorship. E.T. and S.S. were supported by Deutsche Forschungsgemeinschaft (DFG) (SFB1074, subprojects B1 and B2). Medical writing support was provided by Grace Lewis, PharmD, of BioConnections, LLC and funded by AbbVie.
Funding information AbbVie
[Correction added on January 29, 2022, after first online publication: Copyright was changed.]
DATA AVAILABILITY STATEMENT
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.
REFERENCES
- 1. Cory S, Roberts AW, Colman PM, Adams JM. Targeting BCL‐2‐like proteins to kill cancer cells. Trends Cancer. 2016;2(8):443‐460. doi: 10.1016/j.trecan.2016.07.001 [DOI] [PubMed] [Google Scholar]
- 2. Del Gaizo MV, Brown JR, Certo M, Love TM, Novina CD, Letai A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT‐737. J Clin Invest. 2007;117(1):112‐121. doi: 10.1172/jci28281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Souers AJ, Leverson JD, Boghaert ER, et al. ABT‐199, a potent and selective BCL‐2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013;19(2):202‐208. doi: 10.1038/nm.3048 [DOI] [PubMed] [Google Scholar]
- 4. Kater AP, Arslan O, Demikran F, et al. Efficacy of Venetoclax in patients with relapsed/refractory chronic lymphocytic leukemia: primary endpoint analysis of the international phase 3b trial (VENICE I). HemaSphere. 2020;4:S156. [Google Scholar]
- 5. Roberts AW, Davids MS, Pagel JM, et al. Targeting BCL2 with Venetoclax in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):311‐322. doi: 10.1056/NEJMoa1513257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stilgenbauer S, Eichhorst B, Schetelig J, et al. Venetoclax for patients with chronic lymphocytic leukemia with 17p deletion: results from the full population of a phase II pivotal trial. J Clin Oncol. 2018;36(19):1973‐1980. doi: 10.1200/JCO.2017.76.6840 [DOI] [PubMed] [Google Scholar]
- 7. Coutre S, Choi M, Furman RR, et al. Venetoclax for patients with chronic lymphocytic leukemia who progressed during or after idelalisib therapy. Blood. 2018;131(15):1704‐1711. doi: 10.1182/blood-2017-06-788133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jones JA, Mato AR, Wierda WG, et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open‐label, phase 2 trial. Lancet Oncol. 2018;19(1):65‐75. doi: 10.1016/S1470-2045(17)30909-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nechiporuk T, Kurtz SE, Nikolova O, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov. 2019;9(7):910‐925. doi: 10.1158/2159-8290.Cd-19-0125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guièze R, Liu VM, Rosebrock D, et al. Mitochondrial reprogramming underlies resistance to BCL‐2 inhibition in lymphoid malignancies. Cancer Cell. 2019;36(4):369‐384.e13. doi: 10.1016/j.ccell.2019.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Roberts AW, Ma S, Kipps TJ, et al. Efficacy of venetoclax in relapsed chronic lymphocytic leukemia is influenced by disease and response variables. Blood. 2019;134(2):111‐122. doi: 10.1182/blood.2018882555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Herling CD, Abedpour N, Weiss J, et al. Clonal dynamics towards the development of venetoclax resistance in chronic lymphocytic leukemia. Nat Commun. 2018;9(1):727. doi: 10.1038/s41467-018-03170-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blombery P, Anderson MA, Gong JN, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to Venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov. 2019;9(3):342‐353. doi: 10.1158/2159-8290.Cd-18-1119 [DOI] [PubMed] [Google Scholar]
- 14. Tausch E, Close W, Dolnik A, et al. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica. 2019;104(9):e434‐e437. doi: 10.3324/haematol.2019.222588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blombery P, Thompson ER, Nguyen T, et al. Multiple BCL2 mutations cooccurring with Gly101Val emerge in chronic lymphocytic leukemia progression on venetoclax. Blood. 2020;135(10):773‐777. doi: 10.1182/blood.2019004205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kater AP, Wu JQ, Kipps T, et al. Venetoclax plus rituximab in relapsed chronic lymphocytic leukemia: 4‐year results and evaluation of impact of genomic complexity and gene mutations from the MURANO phase III study. J Clin Oncol. 2020;38(34):4042‐4054. doi: 10.1200/jco.20.00948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tausch E, Schneider C, Robrecht S, et al. Prognostic and predictive impact of genetic markers in patients with CLL treated with obinutuzumab and venetoclax. Blood. 2020;135(26):2402‐2412. doi: 10.1182/blood.2019004492 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Patient demographics and disease characteristics at baseline.
Table S2 Summary of efficacy parameters.
Table S3 Post‐treatment specimens.
Data Availability Statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial‐level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications. This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our‐science/clinical‐trials/clinical‐trials‐data‐and‐information‐sharing/data‐and‐information‐sharing‐with‐qualified‐researchers.html.