

Abstract
The miniaturization of polymerase chain reaction (PCR) using drop-based microfluidics allows for amplification of single nucleic acids in aqueous picoliter-sized drops. Accurate data collection during PCR requires that drops remain stable to coalescence during thermocycling and drop contents are retained. Following systematic testing of known PCR additives, we identified an optimized formulation of 1% w/v Tween-20, 0.8 μg/μL bovine serum albumin, 1 M betaine in the aqueous phase, and 3 wt % (w/w) of the polyethylene glycol-perfluoropolyether2 surfactant in the oil phase of 50 μm diameter drops that maintains drop stability and prevents dye transport. This formulation enables a method we call off-chip drop reverse transcription quantitative PCR (OCD RT-qPCR) in which drops are thermocycled in a qPCR machine and sampled at various cycle numbers “off-chip”, or outside of a microfluidic chip. qPCR amplification curves constructed from hundreds of individual drops using OCD RT-qPCR and imaged using epifluorescence microscopy correlate with amplification curves of ≈300,000 drops thermocycled using a qPCR machine. To demonstrate the utility of OCD RT-qPCR, influenza A virus (IAV) RNA was detected down to a single viral genome copy per drop, or 0.320 cpd. This work was extended to perform multiplexed detection of IAV M gene RNA and cellular β-actin DNA in drops, and direct amplification of IAV genomes from infected cells without a separate RNA extraction step. The optimized additive formulation and the OCD-qPCR method allow for drop-based RT-qPCR without complex devices and demonstrate the ability to quantify individual or rare nucleic acid species within drops with minimal processing.
Graphical Abstract
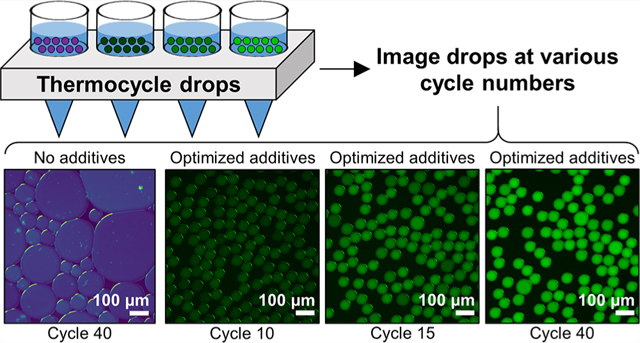
INTRODUCTION
Presymptomatic individuals or those with early viral infections as in the COVID-19 pandemic may not have obvious clinical symptoms.1,2 To determine positive confirmation of disease and viral loads in patient samples,3−5 polymerase chain reaction (PCR) is commonly used to amplify small segments of viral RNA or DNA.6,7 The application of PCR has been expanded using drop-based microfluidics, a method that increases the speed, sensitivity, and throughput of nucleic acid detection by reducing experimental volumes into aqueous microdrops separated by an immiscible oil phase and stabilized by surfactants.8−12
Two methods of performing PCR in drops include droplet digital PCR (ddPCR)8,13 and drop-based quantitative PCR (qPCR).9,10,14,15 The ddPCR method quantifies nucleic acids via limiting dilution, in which samples containing nucleic acids and PCR reagents are diluted to the extent where each drop contains either zero or at least one template. A PCR endpoint analysis that quantifies the number of fluorescent drops that contain a nucleic acid and dim empty drops are used to calculate the starting sample concentration through Poisson statistics.16 Currently, the majority of ddPCR applications use proprietary reagent mixtures for producing stable drops and a commercial device to detect drop fluorescence.8 Similarly, drop-based qPCR assays combine samples with PCR reagents to form picoliter-sized drops; however, limiting dilution is not required. Instead, fluorescent probes are released during nucleic acid amplification as thermocycling proceeds. Fluorescence intensities are converted to concentrations using a calibration curve generated from the cycle thresholds (Ct) of known starting nucleic acid concentrations.9,15
The production of stable aqueous drops for PCR is routinely achieved using biocompatible perfluorinated surfactants suspended in fluorinated oils. A commonly used perfluorinated surfactant is the tri-block copolymer polyethylene glycol-perfluoropolyether2 (PEG-PFPE2). The PEG group extends into the aqueous drop, making the surfactant biocompatible.17 However, drops made with PEG-PFPE2 have been shown to destabilize during PCR thermocycling where temperatures cycle between 60 and 95 °C.18,19 Additionally, the transport of fluorophores into the surrounding oil or neighboring drops has been shown to occur at time scales relevant to thermocycling.20−22 As drop-based qPCR assays rely on the detection of qPCR probes and reference dyes, drop stability and dye retention are critical for the detection and precise quantification of the genomic content in drops.
Approaches for maintaining drop stability and dye retention in droplet PCR assays include utilizing high surfactant concentrations (2−5 wt %),19,23,24 small drop sizes (typically <40 μm diameter), and/or incorporating additives. Common additives include PEG, Tween-20, bovine serum albumin (BSA), and betaine. Yet, systematic testing of these additives is critically lacking to determine how they improve drop stability and limit transport of small molecules. Though complicated microfluidic chips have been engineered to prevent transport between drops and coalescence by keeping drops completely separated,25,26 increasing the stability of drop interfaces using additives can enable thermocycling of a large collection of drops in a tube and subsequent downstream analysis. This would allow for stabilized drops to be sampled at various cycle numbers and analyzed “off-chip”, or outside of a microfluidic chip, using common imaging techniques. In addition, while drops are routinely thermocycled27,28 and endpoint analysis is usually implemented,23,29 sampling of drops undergoing qPCR has not yet been performed and correlated with real-time amplification curves. This can allow for nucleic acid quantification in drops, without performing limiting dilution as in ddPCR, and without the need for specialized microfluidic devices that perform qPCR on chip.9,15 The correlation of drop fluorescence intensities with qPCR amplification curves would validate future work in miniaturizing qPCR and yielding quantitative template numbers at the droplet level, simply by using a basic PCR thermocycler and a fluorescent microscope common in most laboratories and bypassing the need for extensive engineering expertise.
Here, we screen additive formulations of PEG, Tween-20, BSA, and betaine for their ability to stabilize 50 μm diameter drops during RT-qPCR thermocycling. An additive combination of 1% w/v Tween-20, 0.8 μg/μL BSA, and 1 M betaine in the aqueous phase and 3 wt % (w/w) of the PEG-PFPE2 surfactant in the oil phase provided stable drops and dye retention during droplet thermocycling. This result enables a method we call off-chip drop reverse transcription quantitative PCR (OCD RT-qPCR). In this method, drops containing unique qPCR additive formulations are created using a microfluidic drop maker, placed in several tubes within a standard qPCR machine, thermocycled, and removed at various cycle numbers (Figure 1). Drops are sampled “off-chip” at various cycle numbers and the fluorescence intensities obtained from sampled drops using epifluorescence microscopy are used to create PCR amplification curves that closely follow real-time amplification curves obtained using the standard qPCR machine. To our knowledge, this correlation between a sample of individual drops at various cycle numbers and a bulk measurement has never been systematically demonstrated.
Figure 1.

OCD RT-qPCR. (A) Drops containing PCR mix are placed in a standard qPCR machine. (B) Fluorescent images of drops are taken at various thermocycles. (C) Real-time amplification curves are read using the standard qPCR machine over 40 cycles (solid red line, pseudocontinuous). Amplification curves are constructed from drops imaged at various cycles (dashed blue line, discontinuous).
To demonstrate the utility of OCD RT-qPCR for use in studying single-cell infections within drops, we thermocycled drops containing low (101 copies per drop, cpd) and high (104 cpd) viral RNA concentrations. For both of these RNA concentrations, the qPCR amplification curves generated from epifluorescence measurements of sampled drops using OCD RT-qPCR correlate with qPCR amplification curves generated from measurements of thermocycled drops using a standard qPCR machine. We extend our findings to perform multiplexed detection of two target genomes, the influenza A virus (IAV) M gene and cellular β-actin. Finally, we demonstrate the ability to perform direct amplification of IAV genomes from infected cells without a separate RNA extraction and quantify IAV down to a single viral genome per drop. Our work can be extended to sampling of drops undergoing PCR thermocycling for the analysis of RNA and DNA nucleic acids of interest and for performing highly quantitative studies of other viruses. Furthermore, our ability to detect viral genomes without nucleic acid extraction is a powerful application of this technology that could be extended to other drop-based qPCR diagnostic approaches and for the study of viral infection at a single-cell level.
EXPERIMENTAL SECTION
Virus Strains and Cell Lines.
IAV A/California/07/2009 (H1N1) stocks were propagated on Madin-Darby canine kidney (MDCK) cells in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (FBS) and 1×Penicillin/Streptomycin (Pen/Strep). All experimental infections were performed on human alveolar epithelial A549 cells propagated in Ham’s F-12 media supplemented with 10% FBS and 1× Pen/Strep.
PCR Sequences and Reaction Concentrations.
All primer and probe sequences utilized in this study have been previously published. The sequences of qPCR amplification primers for the IAV Matrix gene (M gene) were designed for use by the CDC: M gene forward primer 5′-GACCRATCCTGTCACCTCTGAC-3′, M gene reverse primer 5′-AGGGCATTCTGGACAAATCGTCTA-3′.30 The sequence of the M gene TaqMan probe was: 5′-/FAM/TGCAGTCCTCGCTCACTGGGCACG/BHQ1/−3′. For the multiplexing assay, the primers targeting the β-actin plasmid (pCAG-mGFP-Actin, Addgene #21948) were: β-actin forward primer 5′-GTGTGGATCGGCGGCTCCATC-3′, β-actin reverse primer 5′-GACTCGTCATACTCCTGCTTG-3′, and β-actin TaqMan probe 5′-/Cy5/ACCTTCCAGCAGATGTGGATC/BHQ2/−3′.31 Samples were amplified using the SuperScript III Platinum One-Step qRT-PCR kit (Invitrogen 11732–020) with a final reaction volume of 25 μL. SUPERase RNase Inhibitor was added at 0.32 U/μL (Invitrogen AM2694). Tested additives were added at the following concentrations: 1.0% w/v Tween-20, 0.8 μg/μL BSA, 2.5% w/v PEG-6K, and 1.0 M betaine. Thermocycling was performed in a real-time qPCR machine (QuantStudio 3, Applied Biosystems): one cycle for 30 min at 60 °C, one cycle for 2 min at 95 °C, and 40 cycles between 15 s at 95 °C and 1 min at 60 °C.
In Vitro Transcribed RNA.
Standard curves were generated using serial dilutions of in vitro transcribed IAV M gene.32 To generate the in vitro transcribed RNA, a gBlock containing a T7 promoter (underlined), forward and reverse primer sites (italicized), and a probe sequence (bold) for the M gene was ordered from IDT: 5′-GTCTAATACGACTCACTATAG GACCAATCCTGTCACCTCTGACTGCAGTCCTCGCTCACTGGGCACGTGCTTCATCGCGAACTGCTTCGCGGATGCCATCGTCATGGCCACGAGGATATGTAAGAGTTAGACGATTTGTCCAGAATGCCCT-3’. The M gene was transcribed in vitro using a MEGAscript T7 RNA Synthesis Kit (Ambion, AM1333) following the manufacturer’s instructions that include a DNAse step prior to being purified over a GE Illustra Sephadex G-50 NICK column. RNA concentration was quantified using a NanoDrop spectrophotometer to determine the copy number per μL. The corresponding concentration in cpd is calculated by multiplying the copies/μL value by the volume of a 50 μm diameter drop (6.54 × 10−5 μL/drop).
Drop Encapsulation.
Flow-focusing drop-making devices were used to make 50 μm diameter drops for the drop stability and PCR dilution experiments and 100 μm diameter drops for infected supernatant experiments. The continuous phase consisted of a 3.0 wt % (w/w) solution of the PEG-PFPE2-based surfactant (RAN Biotechnologies, 008-FluoroSurfactant) in fluorinated HFE-7500 oil (3M). The dispersed and continuous phases were loaded in 1 mL syringes and injected into the drop-making microfluidic devices at a flow rate of 800 μL/h and 1600 μL/h, respectively, for 50 μm diameter drops, and 1000 μL/h and 2000 μL/h, respectively, for 100 μm diameter drops (New Era NE-1000 syringe pumps). Drops were collected in 100 μL PCR tubes for 90 s for 50 μm drops and 72 s for 100 μm drops, corresponding to a total volume of 60 μL composed of approximately 20 μL of drops and 40 μL of oil. While the microfluidic device was operated at room temperature, the PCR tubes were kept on a cooling block at approximately −20 °C until placed in the qPCR machine.
Imaging of Drops.
Thermocycled drops were imaged with a 10× objective. Brightfield and fluorescence images of FAM TaqMan probe for M gene, Cy5 TaqMan probe for β-actin, and ROX reference dye were captured on an inverted epifluorescence microscope (Leica DMi8) with a 10× objective. Three fields of view (FOV) of drops were imaged for each M gene and supernatant dilution. For multiplexed drops, five FOV images of drops were taken.
Analysis of Epifluorescence Images of Drops during OCD RT-qPCR Thermocycling.
FAM fluorescence intensities were normalized as ΔRN/RN,0, where the qPCR-normalized reporter value RN is defined as RN = IFAM/IROX, ΔRN = RN − RN,baseline, Ii is the respective intensity of FAM or ROX at each cycle, RN,baseline is the average RN value of the first three cycles, and RN,0 is the initial RN value.
Analysis of Epifluorescence Images of Endpoint OCD RT-qPCR Drops.
FAM and Cy5 fluorescence intensities were normalized by the ROX intensity in each drop and denoted as RN,FAM and RN,Cy5. For Poisson estimates, fluorescence thresholds were set with Otsu’s method33 to distinguish positive and negative drops. This method was applied to the image analysis presented in Figures 3E and 5D.
Figure 3.

Dilution series of in vitro transcribed IAV M gene in bulk and in drops. (A) Amplification curves of six 10-fold dilutions of M gene RNA amplified in bulk (solid line) ranging from 2.62 × [109 to 104] copies/μL and drops (dashed lines) ranging from 1.71 × [104 to 10−1] copies per drop (cpd). No template controls (NTC) are included for bulk and drop conditions. (B) Ct standard curves for the bulk (red, y = −3.52x + 41.4, R2 = 0.999) and drop (blue, y = −3.56x + 41.2, R2 = 0.999) amplification curves. The PCR efficiency in drops was 91.0% and 92.3% for bulk. Representative epifluorescence images of the FAM (reporter) and ROX (reference) channels of drops containing (C) 10−1 cpd and (D) 104 cpd after 40 thermocycles. (E) Percentage of amplified drops as a function of RNA concentration CRNA. The total percentage of bright drops (red circles) increases as a function of RNA concentration and closely follows the Poisson estimate with R2 = 0.929 (blue-dotted line).
Figure 5.

Multiplexed endpoint OCD RT-qPCR detection of target genomes. Representative epifluorescence images of drops containing (A) M gene in the FAM channel and (B) β-actin in the Cy5 channel. Circled drops contain M gene only (yellow circle), β-actin only (pink circle), and both M gene and β-actin (orange circle). (C) Image analysis of drops (N = 1127) yields a scatter plot of drops containing only β-actin, both β-actin and M gene, only M gene, and no template. (D) IAV from infected cells analyzed using endpoint OCD RT-qPCR provides N+/Ntotal as a function of dilution factor of the infected viral supernatant (blue circles). Background amplification level is determined using the mock-infected cell supernatant (dashed red line). Poisson estimate fit of N+/Ntotal values above the background, R2 = 0.950 (dashed green line).
Imaging of Multiplexed OCD-qPCR Drops.
M gene (2.62 × 105 copies/μL) and β-actin (6.98 × 104 copies/μL) were encapsulated with qPCR mix in 50 μm diameter drops and thermocycled. Drops were imaged using an epifluorescence microscope to quantify fluorescence from the FAM (6-carboxyfluorescein) dye-labeled TaqMan probe for M gene, Cy5 (Cyanine5 NHS ester, ex. 649 nm, em. 666 nm) dyelabeled TaqMan probe for β-actin, and the ROX (6-carboxy-X-rhodamine) reference dye. Five FOVs were captured of drops on the epifluorescence microscope for a total of 1127 drops after pooling from all FOV.
Virus Infections.
A549 cells were seeded into 6-well plates with 1 × 106 cells per well. The cells were infected with the H1N1 virus at an MOI of 0.1 in infection media consisting of Ham’s F-12 supplemented with 1 mM N-(2-hydroxyethyl)-piperazine-N′-ethanesulfonic acid, 1× Pen/Strep, and 0.1% BSA. Infection with the H1N1 virus was performed in the presence of 1 μg TPCK-trypsin/mL.34 Cells were washed with 1× phosphate buffered saline and then incubated with virus for 1 h. The inoculum was removed and replaced with fresh infection media and the supernatant was collected at 24 hours post infection (hpi). The resulting supernatant, which contains infectious virus released from the cells, was serial diluted in infection media. The diluted supernatant was then added directly to the optimized OCD qRT-PCR master mix before encapsulation into drops as described above. The drops were thermocycled and three images of each dilution were used to calculate the ratio of “positive” drops, based on FAM fluorescence, to the number of total drops.
RESULTS AND DISCUSSION
OCD RT-qPCR.
A schematic of the OCD RT-qPCR method is outlined in Figure 1A–C. In this method, drops containing our optimized qPCR additive formulation are created using a microfluidic drop maker and placed in a standard qPCR machine (Figure 1A). The qPCR machine is stopped at various cycle numbers, at which a tube is removed, and drops are sampled for epifluorescence imaging (Figure 1B). Real-time amplification curves are read using the standard qPCR machine over 40 cycles (solid red line, pseudocontinuous, Figure 1C) and compared to amplification curves constructed using drops imaged at various cycle numbers (dashed blue line, discontinuous, Figure 1C).
Screening Additive Formulations.
Though systematic testing of additives, to our knowledge, has never been performed, the additives PEG-6K, BSA, Tween-20, and betaine have been utilized in prior drop PCR assays.23,24,29,35−38 These additives are thought to enhance reactions and/or the stability of drops under PCR thermocycling. The polyether PEG improves the stability of drops containing a high salt content.18,39 Additionally, PEG enhances PCR reactions through macromolecular crowding, increasing DNA polymerase activity.40,41 The protein BSA has been shown to limit dye diffusion between drops and enhance PCR yields.21,42 Tween20 has been utilized as a cosurfactant for aqueous drops in fluorinated oils23,29,37 to reduce surface tension. Finally, the common PCR enhancer betaine improves PCR amplification of GC-rich regions by reducing the formation of a nucleic acid secondary structure.43,44
To probe how these PCR additives impact drop stability and dye retention during thermocycling, we measured the effect of Tween-20, BSA, PEG-6K, and betaine individually and in various formulations on the final drop diameter and fluorescence using 3.0 wt% of PEG-PFPE2 as the primary surfactant. All drops contain a TaqMan-based qPCR assay to detect and quantify in vitro transcribed M gene IAV RNA. The M gene is highly conserved across different IAV species,30,45 making it a frequently used standard target for detection. As a no-additive control, drops were produced from a solution of qPCR mix containing 107 copies/μL (≈171 cpd) of M gene. To test additive formulations, the reaction mixture was supplemented with: Tween-20/PEG-6K; Tween-20/BSA; and Tween-20/BSA/betaine. Drops were imaged on an epifluorescence microscope following thermocycling to assess drop stability from drop diameter measurements and retention of the ROX reference dye from fluorescence intensity measurements.
Drops that contained no additives, BSA, PEG-6K, or betaine alone showed evidence of extensive coalescence (Figure 2A). Post-thermocycling drop diameters D were compared to initial D0 with no PCR additives (D0 = 61.1 μm ± 0.8 μm). Normalized diameters (D − D0)/D0 ≈ 0 indicate stability. Values of (D − D0)/D0 in Figure 2B demonstrate that drops containing no additives or only one additive of BSA, PEG-6K, or betaine deviated highly from zero, had a wide distribution of diameters, and produced a large number of outliers indicating coalescence. The addition of Tween-20 alone to the qPCR mix resulted in a narrow distribution of drop diameters with a slight decrease in size ((D − D0)/D0 = −0.12, Figure 2B, red box). As the addition of Tween-20 prevented coalescence, we tested PEG-6K, BSA, and both BSA and betaine in combination with Tween-20. This resulted in (D − D0)/D0 < 0 and lowered coefficients of variation (CV) more effectively than with no additives and with each of the additives alone (Figure 2B, Supporting Information Appendix, Table S1). The formulation of Tween-20/BSA/betaine resulted in (D − D0)/D0 = −0.04 with very few outliers, indicating these drops were the most stable to coalescence upon thermocycling (Figure 2B, orange box).
Figure 2.

Screening of drop additives for PCR. (A) Representative epifluorescence images of drops after thermocycling. Fluorescence intensity I of the ROX dye is quantified from 8-bit pixel values and ranges from 0 (low) to 255 (high). (B) Effects of additives on the drop size. Boxplots represent values of normalized diameters, (D − D0)/D0. (C) Effects of additives on dye retention within drops. Boxplots represent values of normalized mean ROX fluorescence intensity within drops, I − IB.
We investigated the retention of the ROX reference dye within drops after thermocycling. The ROX fluorescence intensity I of each drop was normalized by subtracting the background signal IB. The value I − IB of each drop quantified retention of the reference dye (Figure 2C). A lower value of I − IB indicates that ROX diffused out of the drops into the oil and not between neighboring drops. To verify this, we compared IB for all additive conditions and found that the background fluorescence intensity was the highest for the noadditive condition (Supporting Information Appendix, Figure S2), indicating that all additives had a positive effect on ROX retention in drops.
A smaller CV of I − IB within drops indicates less variability in ROX measurements, ideal for normalizing the TaqMan probe signal that increases during qPCR. An ideal additive formulation for dye retention has high I − IB and small CV (Figure 2C). With no additives, there is significant ROX transport out of the drops and I − IB = 33.6 a.u., the lowest of all eight conditions tested. ROX retention is improved with Tween-20 alone and Tween-20/BSA/betaine. Tween-20 had the highest I − IB at 99.6 a.u (Figure 2C, red box). compared to the Tween-20/BSA/betaine formulation with I − IB = 89.7 a.u (Figure 2C, orange box). However, the Tween-20/BSA/betaine formulation resulted in a smaller CV (15.9%) of I − IB compared to the Tween-20 only condition (18.5%). Based on our results, the additive formulation of Tween-20/BSA/betaine had the lowest (D − D0)/D0 with a relatively high overall ROX fluorescence intensity and the lowest CV of fluorescence intensity. Thus, we apply this optimized additive formulation in subsequent experiments. Numbers of drops measured and statistical information from Figure 2B,C can be found in Supporting Information Appendix, Table S1.
Dilution Series Verifies Efficiency of Drops versus Bulk.
To evaluate the reaction efficiency of PCR amplification in drops, we performed a drop dilution series of in vitro transcribed RNA compared to conventional bulk dilutions. Standard curves relating cycle threshold (Ct) values to log10 dilutions of M gene RNA concentrations were generated for drop and bulk reactions. Serial dilutions for the bulk reactions ranged from 2.62 × [104 to 109] copies/μL. When loaded into 50 μm diameter drops, with a volume of 65 pL, this corresponds to 1.71 × [10−1 to 104] cpd. Amplification curves obtained using qPCR in conventional bulk reactions (Figure 3A, solid lines) follow the same trend as in drops (Figure 3A, dotted lines).
The Ct values of drops and bulk dilutions were used to generate standard curves. The slopes of the curves yielded an amplification efficiency of 91.0% in drops and 92.3% in bulk (Figure 3B). Both fall within typical efficiencies of 90−110%,46,47 where 100% indicates all cDNA is multiplied by a factor of two upon completion of every cycle. Reactions with PCR efficiencies outside this range will limit the assay dynamic range and sensitivity. We hypothesize that the lower fluorescence plateau of ΔRN in drops (Figure 3A) is because of the sequestration of reagents and an earlier exhaustion of reagents compared to bulk. Nonetheless, there is still a notable dynamic range and sensitivity of the assay in drops down to 1 cpd. Below this, at 10−1 cpd, the drops overlap with the NTC; however, this measurement is acquired from drops thermocycled in the qPCR machine. When drops are sampled at the endpoint using epifluorescence, bright and dark drops can be resolved at 10−1 cpd (Figure 3C).
We demonstrate successful implementation of the optimized additive formulation over five orders of magnitude of RNA in drops. Drops from the dilution series in Figure 3A were imaged at the endpoint. Representative images of the FAM and ROX signals in drops containing 10−1 (Figure 3C) and 104 (Figure 3D) RNA cpd show excellent drop stability and dye retention. The 104 cpd case resulted in all bright drops (N = 2051) because of the high concentration of RNA in each drop, while the 10−1 cpd case resulted in a combination of bright and dark drops (N = 2298) because of not every drop containing template at this limiting dilution of RNA.
We performed endpoint analysis by counting the number of bright (positive) and dim (empty) drops from the endpoint reactions of the dilution series (Supporting Information Appendix, Table S2). Poisson statistics was used to fit the fraction of positive drops from the dilution series images, following eq 1
| (1) |
where λ represents the average copy numbers of target RNA (cpd) and p is the fraction of endpoint positive drops.8 The percentage of amplified drops demonstrated good agreement with Poisson statistics yielding R2 = 0.929 (Figure 3E and Supporting Information Appendix, Table S2).
Constructing Amplification Curves Using OCD RTqPCR.
Real-time qPCR amplification curves were constructed from drops using OCD RT-qPCR. Here, we performed a high-resolution investigation of two concentrations of M gene in drops. We compared drops containing low (101 cpd) and high (104 cpd) viral RNA by sampling drops at various cycle numbers on the qPCR machine and imaging them using an epifluorescence microscope.
The qPCR machine tracks fluorescence amplification at each cycle number without interruption. We amplify RNA within the ≈300,000 drops contained in a sample tube using qPCR, which we call the pseudocontinuous data. Hundreds of drops at intermittent thermocycles were sampled from individual tubes, which we call the discontinuous data. This allows construction of discontinuous amplification curves (Figure 4A, blue-dashed line, Figure 4B, red-dashed line) that correlate with the real-time pseudocontinuous measurements acquired using the qPCR machine (Figure 4A, green solid line and Figure 4B, orange solid line).
Figure 4.

OCD RT-qPCR amplification curves compared to standard qPCR curves. Drops containing (A) 1.71 × 101 cpd of M gene RNA (low). The solid green line represents real-time pseudocontinuous fluorescence measurements of drops at each cycle using qPCR. The blue-dashed line follows fluorescence measurements of sampled individual drops. Shaded error bars represent one standard deviation. Drops containing (B) 1.71 × 104 cpd of M gene RNA (high). The solid orange line represents pseudocontinuous fluorescence measurements of drops at each cycle using qPCR. The red-dashed line follows fluorescence measurements of sampled individual drops. (C) Histograms of individual drop fluorescence values from epifluorescence images for 1.71 × 101 cpd of M gene (low). N represents the number of drops measured. The arrow in cycle 40 indicates unamplified drops. (D) Histograms of individual drop fluorescence values from epifluorescence images for 1.71 × 104 cpd of M gene RNA (high). (E) Changes in fluorescence are observed from 1.71 × 101 or 104 cpd of M gene RNA at cycles 22 and 28 and cycles 10 and 15, respectively. Yellow circles indicate dark drops containing no template at cycle 28.
Drops from the low RNA loading conditions (101 cpd) were analyzed across eight cycles (Figure 4A). The distributions of drop fluorescence at the various cycle numbers for the low RNA loading condition are presented in Figure 4C. As amplification starts to increase exponentially at cycle 20, the distribution of drops shifts to the right as the fluorescence increases. From cycle 20−30, we observe that a majority of the drops began to amplify while a small population does not (Figure 4C, arrow, cycle 40), forming a bimodal distribution. We hypothesize that this small population of nonamplifying drops did not contain template RNA, which is confirmed by the presence of dark drops seen under epifluorescence imaging (Figure 4E, yellow circles at cycle 28). This would indicate that the 1.71 × 101 cpd loading was an overestimate as empty drops are not expected at this concentration. In comparison, the high RNA loading condition (1.71 × 104 cpd) should result in every drop containing RNA; all drops should amplify in unison without a bimodal distribution. To test this, drops containing 1.71 × 104 RNA cpd were analyzed across four cycles (Figure 4B). The distributions of drop fluorescence at each cycle for the high RNA loading conditions (Figure 4D) show that all drops amplify as expected.
Discrete particles such as RNA are encapsulated into drops following a Poisson distribution.48 The high RNA loading condition is expected to create a tight distribution while the low RNA loading condition is expected to create a wide distribution. As predicted, the fluorescence intensities of the high RNA loading condition showed tighter distributions at all cycles compared to the low RNA loading condition. Representative images of drops for both low and high RNA loading at early and late cycles provide a visual reference for the distributions in the histograms (Figure 4E). OCD RT-qPCR demonstrates that real-time PCR amplification curves can be created from aggregated fluorescence measurements of individual drops that correlate with measurements taken using a standard qPCR machine. This suggests that quantification of nucleic acid concentrations from a large population of drops can be performed without the need for custom qPCR microfluidic devices.8,48
Multiplexed OCD RT-qPCR Drop Detection of Target Genomes.
The optimized additive formulation enables endpoint OCD RT-qPCR detection of multiplexed fluorophores within a single sample volume. We perform multiplexed detection of two different nucleic acid targets, a FAM probe for IAV M gene RNA and a Cy5 probe for plasmid-based cellular β-actin DNA. Cellular β-actin DNA is a commonly used eukaryotic “housekeeping” gene for qPCR normalization.31 Selected M gene and β-actin concentrations were added to the optimized qPCR mix and thermocycled in 50 μm diameter drops. A representative image showing two fluorescence channels of M gene (FAM, Figure 5A) and β-actin (Cy5, Figure 5B) demonstrates that some drops contain M gene only (Figure 5A,B, yellow circle), β-actin only (Figure 5A,B, pink circle), and both M gene and β-actin (Figure 5A,B, orange circle). Analysis of drops (N = 1127) using endpoint OCD RT-qPCR analysis showed that 28.4% of drops contain M gene only, 7.3% of drops contain β-actin only, 2.7% of drops contain both, and 61.7% of drops were empty (Figure 5C). Multiplexing of qPCR probes allows measurements of multiple gene targets within a population of drops, demonstrating that multiplexing is possible off-chip without a decrease in drop stability or preferential loss of one or both reporter probes significantly increases the scope and applicability of OCD RT-qPCR.
Quantification of Influenza A Virus Genomes from Infected Cells.
The optimized additive formulation enabled endpoint OCD RT-qPCR detection of IAV down to a single viral genome per drop. We infected A549 cells, a human alveolar epithelial cell line, with A/California/07/2009 (H1N1) IAV at a 0.1 multiplicity of infection. At 24 h post infection, we prepared six dilutions of supernatant from infected cells (undiluted 100 down to 10−5 dilution). Supernatant from mock-infected cells was included as a control to determine the level of background amplification and viral detection limit. Supernatant solutions were added to the optimized qPCR mix and drops were produced, thermocycled, and imaged. The virus was heat-lysed during the reverse transcription step at 60 °C for 30 min and amplified without an RNA extraction step. This direct PCR method allowed us to quantify viral RNA directly from the supernatant of infected cells. The fraction of positive drops to total drops (N+/Ntotal) was plotted as a function of the dilution factor (Figure 5D, blue circles, Supporting Information Appendix, Table S2). N+/Ntotal, also defined as p in eq 1, can be used to calculate λ, the average cpd. The background amplification level of the mock-infected supernatant from the measured N+/Ntotal was determined to be 0.086 cpd (Figure 5D, red-dashed line, Ntotal = 208). Thus, infected supernatant dilutions below 10−3 were below the background level and excluded from analysis. A value of λ = 175 cpd for the uninfected supernatant was calculated based upon a Poisson estimate fit (R2 = 0.950) to the measured N+/Ntotal of the four infected supernatant dilutions above the background limit of 10−3 (Figure 5D, green-dashed line). The calculated λ corresponds to 3.34 × 106 copies/μL for the undiluted infected supernatant. The standard curve in Figure 3B was used to estimate the concentration of the undiluted infected supernatant based upon its measured Ct value. This yielded an estimated starting concentration of 1.96 × 106 copies/μL, in close agreement with 3.34 × 106 copies/μL determined from λ. The lowest level of detection at 10−3 dilution corresponds to 0.320 cpd (Ntotal = 208). Thus, we demonstrate the ability of our optimized endpoint OCD RT-qPCR assay to detect down to single viral genomes encapsulated within drops. Notably, we demonstrate the ability to amplify IAV RNA directly from infected cells.
CONCLUSIONS
OCD RT-qPCR is a versatile off-chip method for constructing qPCR amplification curves from individual drops that is enabled by a systematic screening of additive formulations, which has been critically lacking and previously unreported. Performing RT-qPCR in picoliter-sized drops offers distinct advantages for speed, sensitivity, and throughput. However, the results can be misleading if drops merge or leak. Furthermore, heating cycles are known to destabilize drops. Complex microfluidic devices25 have been developed to keep drops separated, preventing leakage of contents and coalescence. Yet, these devices are often impractical for many researchers. Other strategies have included replacing liquid drops with hydrogel beads49 or stabilizing drops using solid particles.50 These changes can limit downstream options as drops may no longer be easily subdivided by splitting or merged with additional reaction components. Instead, it is more desirable to increase the stability of droplet interfaces while making no irreversible changes to the liquid phase or structure of the droplet itself. We, therefore, screened a number of PCR additives and established optimal conditions that protect drops during PCR thermocycling. The optimized formulation of Tween-20/BSA/betaine maintained drop stability and limited dye transport during thermocycling.
Using OCD RT-qPCR, we also demonstrate for the first time that sampled drops imaged using epifluorescence correlates with real-time amplification curves obtained using a standard qPCR machine. Drop thermocycling and analysis is performed entirely off-chip, or outside of a microfluidic device. Previous drop-based qPCR methods relied on endpoint fluorescence measurements to confirm the presence or absence of specific nucleic acid species,23 or required complex devices to measure drop fluorescence following each thermocycle number.9,10,15 These approaches are either unattainable for laboratories without extensive engineering expertise or fail to provide any quantification of specific nucleic acid species. The OCD RT-qPCR method addresses these pitfalls by demonstrating that fluorescence measurements can be obtained using a standard epifluorescence microscope and sampling drops at various cycles allows for the construction of amplification curves. This combination can be used to quantify specific nucleic acid species of interest within individual drops with high accuracy.
Optimized drop stability can greatly improve single-cell studies, where drops must resist coalescence while containing destabilizing molecules and proteins such as those present in growth media or expressed during cellular metabolism.51−53 For example, we have confirmed the ability to detect down to a single genome in a drop, or 0.320 cpd, demonstrating the high sensitivity achievable using OCD RT-qPCR. A variety of downstream processing options become available to drops stable enough to be handled for off-chip analysis and eliminates the need for complicated custom microfluidic thermocycling devices.9,10 Fluorescence measurements of tens of thousands of drops may be accomplished by replacing epifluorescence microscopy measurements with flow-based drop fluorescence detection.48 Furthermore, there is potential to measure the viral output of infected single cells using OCD RT-qPCR, significantly increasing the number of cells that can be assayed compared to common serial dilution approaches performed using standard well plates.54 Importantly, our work establishes the possibility of directly amplifying viral RNA without the need for RNA extraction, which is advantageous in resource-limited situations such as the COVID-19 pandemic for performing massive testing of SARS-CoV-2.55
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Defense Advanced Research Projects Agency (DARPA) grant W911NF-17-2-0034 and by National Science Foundation (NSF) CAREER grant 1753352. We thank C. Brooke at the University of Illinois at Urbana-Champaign for providing H1N1 A/California/07/2009 and human alveolar epithelial A549 cells and A. Rynda-Apple at Montana State University for providing MDCK cells.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.analchem.0c03455.
Drop measurements and additional experimental details for microfluidic device fabrication, PCR mix formulation, image acquisition, and image analysis (PDF)
Contributor Information
Emma Kate Loveday, Center for Biofilm Engineering and the Department of Chemical and Biological Engineering, Montana State University, Bozeman, Montana 59717, United States.
Geoffrey K. Zath, Center for Biofilm Engineering and the Department of Chemical and Biological Engineering, Montana State University, Bozeman, Montana 59717, United States.
Dimitri A. Bikos, Center for Biofilm Engineering and the Department of Chemical and Biological Engineering, Montana State University, Bozeman, Montana 59717, United States
Zackary J. Jay, Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United States
Connie B. Chang, Center for Biofilm Engineering and the Department of Chemical and Biological Engineering, Montana State University, Bozeman, Montana 59717, United States
REFERENCES
- (1).He X; Lau EHY; Wu P; Deng X; Wang J; Hao X; Lau YC; Wong JY; Guan Y; Tan X; Mo X; Chen Y; Liao B; Chen W; Hu F; Zhang Q; Zhong M; Wu Y; Zhao L; Zhang F; Cowling BJ; Li F; Leung GM Nat. Med. 2020, 26, 672–675. [DOI] [PubMed] [Google Scholar]
- (2).Furukawa NW; Brooks JT; Sobel J. Emerg. Infect. Dis. 2020, 26, e1–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Maignan M; Viglino D; Hablot M; Masson NT; Lebeugle A; Muret RC; Makele PM; Guglielmetti V; Morand P; Lupo J; Forget V; Landelle C; Larrat S. PLoS One 2019, 14, No. e0216308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pfefferle S; Reucher S; Norz D; Lutgehetmann M. Euro Surveill. 2020, 25, 2000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thirion L; Dubot-Peres A; Pezzi L; Corcostegui I; Touinssi M; de Lamballerie X; Charrel RN Viruses 2020, 12, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mackay IM; Arden KE; Nitsche A. Nucleic Acids Res. 2002, 30, 1292–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Clementi M; Menzo S; Bagnarelli P; Manzin A; Valenza A; Varaldo PE PCR Methods Appl. 1993, 2, 191–196. [DOI] [PubMed] [Google Scholar]
- (8).Hindson BJ; Ness KD; Masquelier DA; Belgrader P; Heredia NJ; Makarewicz AJ; Bright IJ; Lucero MY; Hiddessen AL; Legler TC; Kitano TK; Hodel MR; Petersen JF; Wyatt PW; Steenblock ER; Shah PH; Bousse LJ; Troup CB; Mellen JC; Wittmann DK; Erndt NG; Cauley TH; Koehler RT; So AP; Dube S; Rose KA; Montesclaros L; Wang S; Stumbo DP; Hodges SP; Romine S; Milanovich FP; White HE; Regan JF; Karlin-Neumann GA; Hindson CM; Saxonov S; Colston BW Anal. Chem. 2011, 83, 8604–8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Beer NR; Wheeler EK; Lee-Houghton L; Watkins N; Nasarabadi S; Hebert N; Leung P; Arnold DW; Bailey CG; Colston BW Anal. Chem. 2008, 80, 1854–1858. [DOI] [PubMed] [Google Scholar]
- (10).Kiss MM; Ortoleva-Donnelly L; Beer NR; Warner J; Bailey CG; Colston BW; Rothberg JM; Link DR; Leamon JH Anal. Chem. 2008, 80, 8975–8981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kim SC; Clark IC; Shahi P; Abate AR Anal. Chem. 2018, 90, 1273–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pekin D; Skhiri Y; Baret J-C; Le Corre D; Mazutis L; Ben Salem C; Millot F; El Harrak A; Hutchison JB; Larson JW; Link DR; Laurent-Puig P; Griffiths AD; Taly V. Lab Chip 2011, 11, 2156–2166. [DOI] [PubMed] [Google Scholar]
- (13).Hopken MW; Piaggio AJ; Pabilonia KL; Pierce J; Anderson T; Abdo ZJ Virol. Methods 2020, 276, 113777. [DOI] [PubMed] [Google Scholar]
- (14).Hajji I; Serra M; Geremie L; Ferrante I; Renault R; Viovy J-L; Descroix S; Ferraro D. Sens. Actuators, B 2020, 303, 127171. [Google Scholar]
- (15).Prakash R; Pabbaraju K; Wong S; Wong A; Tellier R; Kaler KV I. S. J. Electrochem. Soc. 2014, 161, B3083–B3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Basu AS SLAS Technol. 2017, 22, 369–386. [DOI] [PubMed] [Google Scholar]
- (17).Holtze C; Rowat AC; Agresti JJ; Hutchison JB; Angile FE; Schmitz CHJ; Köster S; Duan H; Humphry KJ; Scanga RA; Johnson JS; Pisignano D; Weitz DA Lab Chip 2008, 8, 1632–1639. [DOI] [PubMed] [Google Scholar]
- (18).Etienne G; Kessler M; Amstad E. Macromol. Chem. Phys. 2017, 218, 1600365. [Google Scholar]
- (19).Chowdhury MS; Zheng W; Kumari S; Heyman J; Zhang X; Dey P; Weitz DA; Haag R. Nat. Commun. 2019, 10, 4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Gruner P; Riechers B; Semin B; Lim J; Johnston A; Short K; Baret JC Nat. Commun. 2016, 7, 10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Skhiri Y; Gruner P; Semin B; Brosseau Q; Pekin D; Mazutis L; Goust V; Kleinschmidt F; El Harrak A; Hutchison JB; Mayot E; Bartolo J-F; Griffiths AD; Taly V; Baret J-C Soft Matter 2012, 8, 10618–10627. [Google Scholar]
- (22).Chen Y; Wijaya Gani A; Tang SK Y. Lab Chip 2012, 12, 5093–5103. [DOI] [PubMed] [Google Scholar]
- (23).Eastburn DJ; Sciambi A; Abate AR Anal. Chem. 2013, 85, 8016–8021. [DOI] [PubMed] [Google Scholar]
- (24).Rane TD; Chen L; Zec HC; Wang T-H Lab Chip 2015, 15, 776–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Labanieh L; Nguyen T; Zhao W; Kang D-K Micromachines 2015, 6, 1469–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Pompano RR; Liu W; Du W; Ismagilov RF Annu. Rev. Anal. Chem. 2011, 4, 59–81. [DOI] [PubMed] [Google Scholar]
- (27).Nakano M; Komatsu J; Matsuura S.-i.; Takashima K; Katsura S; Mizuno AJ Biotechnol. 2003, 102, 117–124. [DOI] [PubMed] [Google Scholar]
- (28).Zhang F; Liao P; Sun Y; Chen Z; Pang Y; Huang Y. Lab Chip 2020, 20, 2328–2333. [DOI] [PubMed] [Google Scholar]
- (29).Tao Y; Rotem A; Zhang H; Chang CB; Basu A; Kolawole AO; Koehler SA; Ren Y; Lin JS; Pipas JM; Feldman AB; Wobus CE; Weitz DA Lab Chip 2015, 15, 3934–3940. [DOI] [PubMed] [Google Scholar]
- (30).Shu B; Wu K-H; Emery S; Villanueva J; Johnson R; Guthrie E; Berman L; Warnes C; Barnes N; Klimov A; Lindstrom SJ Clin. Microbiol. 2011, 49, 2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Piorkowski G; Baronti C; de Lamballerie X; de Fabritus L; Bichaud L; Pastorino BA; Bessaud MJ Virol. Methods 2014, 202, 101–105. [DOI] [PubMed] [Google Scholar]
- (32).Spackman E; Senne DA; Myers TJ; Bulaga LL; Garber LP; Perdue ML; Lohman K; Daum LT; Suarez DL J. Clin. Microbiol. 2002, 40, 3256–3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Otsu N. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [DOI] [PubMed] [Google Scholar]
- (34).Weingartl HM; Berhane Y; Hisanaga T; Neufeld J; Kehler H; Emburry-Hyatt C; Hooper-McGreevy K; Kasloff S; Dalman B; Bystrom J; Alexandersen S; Li Y; Pasick JJ Virol. 2010, 84, 2245–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lan F; Haliburton JR; Yuan A; Abate AR Nat. Commun. 2016, 7, 11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Azizi M; Zaferani M; Cheong SH; Abbaspourrad A. ACS Sens. 2019, 4, 841–848. [DOI] [PubMed] [Google Scholar]
- (37).Zhang H; Cockrell SK; Kolawole AO; Rotem A; Serohijos AWR; Chang CB; Tao Y; Mehoke TS; Han Y; Lin JS; Giacobbi NS; Feldman AB; Shakhnovich E; Weitz DA; Wobus CE; Pipas JM J. Virol. 2015, 89, 7722–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Kothekar SC; Ware AM; Waghmare JT; Momin SA J. Dispersion Sci. Technol. 2007, 28, 477–484. [Google Scholar]
- (39).Takei F; Akiyama M; Nobusawa K; Sabani NB; Han H; Nakatani K; Yamashita I. ChemistrySelect 2018, 3, 973–976. [Google Scholar]
- (40).Sasaki Y; Miyoshi D; Sugimoto N. Biotechnol. J. 2006, 1, 440–446. [DOI] [PubMed] [Google Scholar]
- (41).Tong Y; Lemieux B; Kong H. BMC Biotechnol. 2011, 11, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Farell EM; Alexandre G. BMC Res. Notes 2012, 5, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Zhang Z; Yang X; Meng L; Liu F; Shen C; Yang W. Biotechniques 2009, 47, 775–779. [DOI] [PubMed] [Google Scholar]
- (44).Henke W; Herdel K; Jung K; Schnorr D; Loening SA Nucleic Acids Res. 1997, 25, 3957–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hoffmann E; Stech J; Guan Y; Webster RG; Perez DR Arch. Virol. 2001, 146, 2275–2289. [DOI] [PubMed] [Google Scholar]
- (46).Pfaffl MW Quantification strategies in real-time PCR. In A-Z of Quantitative PCR; Bustin SA, Ed.; International University Line Press, 2004; pp 87–112. [Google Scholar]
- (47).Kubista M; Andrade JM; Bengtsson M; Forootan A; Jonák J; Lind K; Sindelka R; Sjöback R; Sjögreen B; Strömbom L; Stahlberg A; Zoric N. Mol. Aspects Med. 2006, 27, 95–125. [DOI] [PubMed] [Google Scholar]
- (48).Mazutis L; Gilbert J; Ung WL; Weitz DA; Griffiths AD; Heyman JA Nat. Protoc. 2013, 8, 870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li L; Yu L; Wu T. Direct PCR amplification and in situ imaging based on alginate droplets, 2017 IEEE 30th International Conference on Micro Electro Mechanical Systems (MEMS), 22−26 Jan. 2017, 2017; pp 1252–1255. [Google Scholar]
- (50).Yin K; Zeng X; Liu W; Xue Y; Li X; Wang W; Song Y; Zhu Z; Yang C. Anal. Chem. 2019, 91, 6003–6011. [DOI] [PubMed] [Google Scholar]
- (51).Pratt SL; Zath GK; Akiyama T; Williamson KS; Franklin MJ; Chang CB Front. Microbiol. 2019, 10, 2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Akiyama T; Williamson KS; Schaefer R; Pratt S; Chang CB; Franklin MJ Proc. Natl. Acad. Sci. U.S.A. 2017, 114, 3204–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fischer AE; Wu SK; Proescher JBG; Rotem A; Chang CB; Zhang H; Tao Y; Mehoke TS; Thielen PM; Kolawole AO; Smith TJ; Wobus CE; Weitz DA; Lin JS; Feldman AB; Wolfe JT J. Virol. Methods 2015, 213, 111–117. [DOI] [PubMed] [Google Scholar]
- (54).Heldt FS; Kupke SY; Dorl S; Reichl U; Frensing T. Nat. Commun. 2015, 6, 8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fomsgaard AS; Rosenstierne MW Euro Surveill. 2020, 25, 2000398. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.