

Summary
Colonic goblet cells are specialized epithelial cells that secrete mucus to physically separate the host and its microbiota, thus preventing bacterial invasion and inflammation. How goblet cells control the amount of mucus they secrete is unclear. We found that constitutive activation of autophagy in mice via Beclin 1 enables the production of a thicker and less penetrable mucus layer by reducing endoplasmic reticulum (ER) stress. Accordingly, genetically inhibiting Beclin 1-induced autophagy impairs mucus secretion, while pharmacologically alleviating ER stress results in excessive mucus production. This ER-stress-mediated regulation of mucus secretion is microbiota dependent and requires the Crohn’s-disease-risk gene Nod2. Overproduction of mucus alters the gut microbiome, specifically expanding mucus-utilizing bacteria, such as Akkermansia muciniphila, and protects against chemical and microbial-driven intestinal inflammation. Thus, ER stress is a cell-intrinsic switch that limits mucus secretion, whereas autophagy maintains intestinal homeostasis by relieving ER stress.
Keywords: mucus, goblet cell, autophagy, ER stress, inflammatory bowel diseases, microbiota, colitis, unfolded protein response, Beclin 1, Nod2
Graphical abstract
Highlights
-
•
ER stress controls mucus secretion from colonic goblet cells
-
•
Autophagy relieves ER stress to facilitate proper mucus secretion
-
•
ER-stress-mediated control of mucus secretion is dependent on the microbiota and NOD2
-
•
Excess mucus secretion reshapes the microbiota and protects from colitis
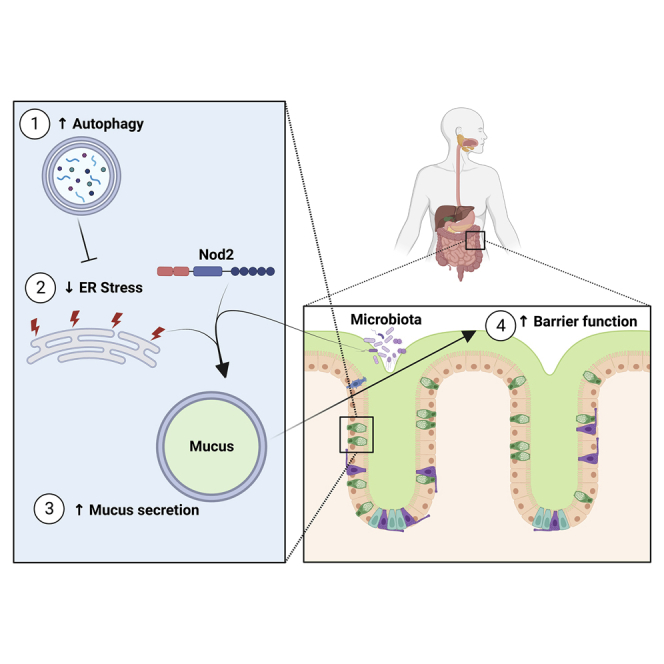
Mutations in autophagy genes and NOD2 predispose individuals to the development of inflammatory bowel diseases; yet, the reason(s) is not clear. Naama et al. show that autophagy regulates mucus secretion from goblet cells and offers protection from colitis by alleviating ER stress in a microbiota- and Nod2-dependent manner.
Introduction
Goblet cells are intestinal epithelial cells that secrete mucus and antimicrobial proteins, creating a chemical barrier between the host and its microbiota. This mucus barrier is crucial for symbiosis between the host and its microbiota as it limits microbial contact with the host epithelium and subsequent proinflammatory responses.1 Breakdown of this barrier and penetration of bacteria into the inner mucus layer is a hallmark, and perhaps the cause, of inflammatory bowel diseases (IBD) and enteric infections.2,3 Yet, despite their importance, how goblet cells control the amount of mucus they secrete into the gut lumen is unclear.
Macroautophagy (hereafter autophagy) is a cell-intrinsic recycling mechanism crucial for cellular homeostasis, which is activated in times of stress such as starvation, infection, and accumulation of unfolded proteins in the endoplasmic reticulum (ER). Intestinal secretory cells, such as Paneth and goblet cells, are highly sensitive to perturbations in the autophagy or the ER stress response pathways. Indeed, mutations in autophagy- and ER-stress-related genes are a risk factor for the breakdown of the intestinal barrier and the development of IBD. Yet, the exact reason for this is not fully clear.4
We wanted to understand how goblet cells control the amount of mucus they secrete and why autophagy is needed for proper goblet cell function. Here, we show that autophagy relieves ER stress in goblet cells to facilitate mucus secretion. We found that the genetic activation of autophagy in mice via the Beclin 1 protein led to the suppression of intestinal ER stress, which facilitated excess mucus secretion. This over-production of mucus could be achieved without manipulation of the autophagy process, by pharmacologically reducing ER stress or by activating specific arms of the unfolded protein response (UPR). Moreover, this mucus over-production was microbiota dependent and required the nucleotide-binding oligomerization-domain-containing protein 2 (Nod2). Excess mucus secretion led to expansion of mucus-utilizing bacteria and protection from colitis. Thus, ER stress is a cell-intrinsic switch that limits mucus secretion, while autophagy maintains proper mucus secretion and intestinal homeostasis by relieving ER stress.
Results
Activation of autophagy via Beclin 1 leads to production of a thicker and less penetrable colonic mucus layer
During steady state, the autophagy-initiating protein Beclin 1 is sequestered and inactivated by binding to the B cell lymphoma 2 protein (Bcl-2). To trigger autophagy, Bcl-2 must be phosphorylated to release Beclin 1, which goes on to initiate the synthesis of autophagosomes (Figure 1A).5 As functional autophagy is vital for proper goblet cell function,6 we hypothesized that constitutive activation of the autophagy process will improve goblet cell function and mucus secretion. We thus inspected knockin mice in which the phenylalanine residue at position 121 of Beclin 1 is mutated to alanine (Becn1F121A mice), preventing the binding of Beclin 1 to its inhibitor, Bcl-2, leading to the constitutive activation of autophagy (Figure 1A).5 We found that the colonic mucus layer in Becn1F121A mice was 50% thicker than that in wild-type mice (Figures 1B and 1C). To determine whether the thicker mucus layer in Becn1F121A mice improved barrier function, we measured the levels of luminal antigens in the sera of mice. Presence of microbial antigens in the blood is directly linked to intestinal barrier function.7 We found lower levels of agonists, which activate NOD1, NOD2, and Toll-like receptors 4 (TLR4) and TLR5 in the sera of Becn1F121A mice (Figures 1D, 1E, and S1A–S1C). This implied that the thicker mucus layer of these mice limited the translocation of bacterial antigens into host tissues.
Figure 1.

Becn1F121A mice produce a thicker and less penetrable colonic mucus layer and mount a dampened immune response in the colonic tissue
(A) Scheme depicting regulation of autophagy activation via Bcl-2 phosphorylation and its alteration in Becn1F121A mice.
(B) Alcian blue staining of Carnoy’s-fixed colonic tissue. The mucus layer is defined by the dashed line. Scale bars, 50 μm.
(C) Measurements of mucus thickness as shown in (B).
(D and E) (D) Detection of TLR4 and (E) TLR5 agonist in mouse serum using reporter cell lines.
(F) Principal coordinates analysis (PCoA) plot of RNA sequencing performed on colonic tissues.
(G) Volcano plot of transcripts from RNA sequencing. Genes in blue were down-regulated in Becn1F121A mice compared with wild-type mice and genes in red were up-regulated.
(H) Pathway analysis of transcripts, which are down-regulated in Becn1F121A mice according to GO biological function. Bars represent −log (p value) and dots represent number of genes in pathway.
(I) Heatmap depicting differentially expressed innate immune genes with an FDR < 0.01. Each column represents a mouse and each row a gene.
(C–F) Each dot represents a mouse. (G) Each dot represents a gene. ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; Student’s t test. WT, wild type; F121A, Becn1F121A; a.u., arbitrary units.
To determine how the improved barrier in Becn1F121A mice affected the colonic tissue, we performed a transcriptional analysis. RNA sequencing of colonic tissues from wild-type and Becn1F121A mice presented a clear difference in transcriptional patterns between the two groups of mice (Figures 1F and 1G). Unbiased pathway analysis of RNA sequencing data revealed that the expression of genes involved in immune regulation and response to bacteria was dampened in Becn1F121A mice (Figure 1H). Specifically, levels of transcripts encoding innate immune cytokines, antimicrobial proteins, and acute response proteins were diminished in Becn1F121A mice (Figure 1I). Thus, constitutive activation of autophagy leads to the excess secretion of mucus from colonic goblet cells. This excess mucus forms a barrier that better limits the translocation of luminal antigens into the host serum as well as limiting immune responses in the colon.
Autophagy reduces ER stress to promote mucus secretion from goblet cells
Next, we wanted to determine the mechanism by which autophagy activation promotes mucus secretion. One possibility is that the constitutive activation of autophagy leads to the expansion of goblet cell numbers. However, we found only a slight reduction in the number of goblet cells in Becn1F121A mice compared with wild-type mice (Figure 2A) and no difference in the mucus-filled cytoplasmatic (goblet cell theca) area (Figure 2B). Additionally, transcript levels of transcription factors that mark the goblet cell lineage, Atoh1 and Spdef,8 were similar in the two groups of mice (Figure S2A). Another possibility is that constitutive autophagy promotes mucus secretion by regulating transcription of mucus-forming genes, yet we found no significant differences in transcript levels of these genes when comparing wild-type and Becn1F121A mice (Figures 2C and S2A). A further possible explanation is changes in reactive oxygen species (ROS) levels in goblet cells. ROS have been previously shown to be affected by defective autophagy and thus impact mucus secretion.6 However, we found no difference in goblet cells’ levels of the ROS-indicator 4 hydroxynonenal (Figure S2B). Thus, expansion of goblet cells, overexpression of mucus-forming genes, or ROS levels could not explain the reason behind the thicker mucus layer in Becn1F121A mice.
Figure 2.

Autophagy relieves ER stress to facilitate mucus secretion from goblet cells
(A) Colonic goblet cell numbers per crypt.
(B) Quantification of the mucus-filled cytoplasmatic area of colonic goblet cells.
(C) Expression levels of Muc2, which encodes the mucus-forming protein MUC2 in the colons of mice via RNA sequencing.
(D) Heatmap depicting differentially expressed ER stress response genes with an FDR < 0.01. Each column represents a mouse and each row a gene.
(E) Representative western blot of mouse colons detected with the indicated antibodies.
(F) Densitometry analysis of western blots as in (E).
(G) qPCR analysis of Hspa5 mRNA in goblet cells isolated from colonic tissue via fluorescence-activated cell sorting.
(H) Quantification of protein levels of CHOP, specifically in colonic goblet cells via immunohistochemistry.
(I) Scheme depicting ER stress activation via thapsigargin.
(J and K) Measurements of mucus thickness in colonic sections from mice treated with thapsigargin as indicated, and stained with Alcian blue. Scale bars, 50 μm.
(L) Scheme depicting regulation of autophagy activation via Bcl-2 phosphorylation and its suppression in Bcl2AAA mice.
(A–C, F–H, and K) Each dot represents a mouse. ns, not significant; ∗p < 0.05; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. (A–C and F–H) Student’s t test; (K) one-way ANOVA; WT, wild type; F121A, Becn1F121A; AAA, Bcl2AAA; AB, Alcian blue; RPKM, reads per kilobase per million mapped reads; MFI, mean fluorescent intensity; RQ, relative quantity.
We then reanalyzed the RNA sequencing data using Gene Ontology (GO) cellular components analysis and found that many of the differentially transcribed genes in the colon of Becn1F121A mice encode proteins that are localized to the ER (Figure S2C). As a mechanism to relieve ER stress, cells activate the autophagy process,9,10 while impaired autophagy leads to increased ER stress.11 Of note, impairment of the ER stress response is linked to the dysfunction of goblet cells.12 Indeed, we found that levels of transcripts that are expressed in response to ER stress were reduced in the colonic tissue of Becn1F121A mice (Figure 2D). Accordingly, protein levels of the ER stress marker C/EBP homologous protein (CHOP) were reduced in the colons of Becn1F121A mice (Figures 2E and 2F). To verify that ER stress levels are reduced specifically in goblet cells, we isolated colonic goblet cells from wild-type and Becn1F121A mice using fluorescence-activated cell sorting and analyzed the expression of the ER stress response gene Hspa5. In line with our findings in whole colonic tissues, we found that mRNA levels of Hspa5 were reduced in sorted goblet cells from Becn1F121A mice (Figure 2G). We then validated this reduction of ER stress in goblet cells at the protein level by immunohistochemical analysis of goblet cells in a colonic section. We found a reduction in the levels of the ER stress protein CHOP specifically in the goblet cells of Becn1F121A mice (Figure 2H). Thus, constitutive activation of autophagy reduces ER stress in colonic goblet cells.
This led us to hypothesize that constitutive activation of autophagy in Becn1F121A mice promotes mucus secretion by reducing ER stress in goblet cells. To test this hypothesis, we treated Becn1F121A mice with the ER stress inducer thapsigargin (Figure 2I).13 In line with our hypothesis, we found that the mucus layer in thapsigargin-treated Becn1F121A mice was decreased compared with that in vehicle-treated Becn1F121A mice and was indistinguishable from the mucus layer in wild-type mice (Figures 2J and 2K). To further test the concept that autophagy facilitates mucus secretion by reducing ER stress, we tested whether preventing the activation of autophagy via Beclin 1, thus leaving goblet cells unable to alleviate the ER stress via autophagy, impairs mucus secretion. We used knockin mice in which the three specific residues of Bcl-2, which are phosphorylated to allow dissociation from Beclin 1, are mutated to alanine (Figure 2L). In these Bcl2AAA mice, Beclin 1 is constantly bound to Bcl-2 and is thus inactive.14 We found that the mucus layer in Bcl2AAA mice was thinner than that in wild-type mice—and in some colonic regions completely absent (Figures 2J and 2K). Thus, autophagy facilitates mucus secretion via Beclin 1 by alleviating ER stress in goblet cells.
Pharmacologically reducing ER stress leads to sustainable excess secretion of mucus
Our observation that the activation of autophagy can alleviate ER stress in a manner which results in elevated mucus secretion implies that ER stress might be an intracellular cue that limits mucus secretion. To test this hypothesis, we treated wild-type mice with the bile acid tauroursodeoxycholic acid (TUDCA), a chemical chaperone that reduces ER stress (Figure 3A).13 We found that TUDCA treatment resulted in the secretion of a thicker mucus layer, which resembled the mucus layer in Becn1F121A mice (Figures 3B and 3C). In 20% of TUDCA-treated wild-type mice, we found that the colonic luminal cavity was filled with mucus (Figure 3D).
Figure 3.

ER stress is a rate-limiting switch controlling mucus secretion
(A) Scheme depicting ER stress reduction by inhibiting eIF2α dephosphorylation via salubrinal, by TUDCA, or by activating ATF6 via AA147.
(B–D) Measurements of mucus thickness in colonic sections from mice treated with TUDCA or salubrinal as indicated, and stained with Alcian blue. Scale bars, 50 μm. Each dot represents a mouse.
(E) Measurements of mucus thickness in colonic sections from mice treated with the ATF6 activator AA147 and stained with Alcian blue. Each dot represents a mouse.
(F) Scheme depicting mucus growth rate measurements. Created with BioRender.com.
(G) Mucus growth over time ± SEM.
(H) Mucus growth rate. Lines connect tissues from the same mouse.
∗p < 0.05, ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; (C) one-way ANOVA; (E) Student’s t test; (G) nonlinear regression; (H) paired t test. WT, wild type; F121A, Becn1F121A; TUDCA, tauroursodeoxycholic acid.
TUDCA affects all three arms of the unfolded protein response (UPR) in an unspecific manner.15 The UPR is a highly regulated cellular program which relieves ER stress to maintain cell function and avoid apoptosis.12 Thus, we next wanted to determine whether relieving ER stress via activation of specific arms of the UPR is sufficient for the induction of mucus secretion. To test this, we treated wild-type and Becn1F121A mice with salubrinal, a specific inhibitor of eukaryotic translation initiation factor 2 alpha (eIF2α) dephosphorylation (Figure 3A).16 The pancreatic ER kinase (PERK) arm of the UPR is activated when the ER stress-sensor PERK phosphorylates eIF2α. When eIF2α is phosphorylated, it inhibits the translation of certain transcripts, while promoting the translation of transcripts encoding chaperons and other proteins needed to resolve ER stress.12 We found that the activation of eIF2α in wild-type mice via salubrinal treatment led to the excessive secretion of mucus from goblet cells, as was seen in TUDCA-treated mice (Figures 3B and 3C). Additionally, the mucus layer in salubrinal-treated Becn1F121A mice did not differ from that of vehicle-treated Becn1F121A mice (Figures 3B and 3C), indicating that autophagy activation via Beclin 1 and eIF2α activation converge on the same pathway to induce mucus secretion. Moreover, pharmacological activation of activating transcription factor 6 (ATF6), a transcription factor that transcribes ER chaperone genes in response to ER stress,17 also led to an increase in mucus secretion in wild-type mice (Figure 3E). Thus, relieving ER stress via the activation of eIF2α or ATF6 is sufficient to increase mucus secretion from goblet cells.
We then wanted to determine the kinetics of the elevated mucus secretion that is induced by the reduction of ER stress. We removed two adjacent colon sections from each wild-type mouse and placed them in two chambers that allow the measurement of mucus secretion from live, unfixed tissues under physiological conditions. Only one chamber was infused with TUDCA, allowing us to compare different conditions in tissues that originated from the same mouse (Figure 3F). We found that TUDCA treatment significantly increased mucus secretion rates by 2-fold (Figures 3G and 3H). Additionally, unlike inducing rapid goblet cell degranulation with the cholinergic agonist carbachol, which leads to rapid mucus secretion over 15 min followed by a reduction in the secretion rate,18 TUDCA treatment led to a sustained increase in mucus secretion rates over the entire measurement period of 45 min (Figure S3A). This increase in secretion rates did not impact mucus permeability as compared with the control group (Figure S3B). Thus, ER stress is a cell-intrinsic switch that limits mucus secretion from goblet cells.
ER stress control over mucus secretion rate is regulated by the microbiota and Nod2
Crosstalk between the host immune system and the gut microbiota regulates multiple components which form the intestinal barrier.19 Thus, we next wanted to examine whether these immune-microbe interactions affect the control of ER stress on mucus secretion. First, we treated germ-free mice with TUDCA to reduce ER stress. Unlike conventionally colonized mice, we found that TUDCA treatment did not affect the mucus layer thickness of wild-type germ-free mice (Figure 4A). We then tested whether microbiota depletion also affects mucus secretion rates. We treated wild-type mice with a cocktail of antibiotics in drinking water, after which we removed two adjacent colon sections and placed them in two chambers to measure mucus secretion rates (as in Figure 3). Although TUDCA treatment of the colons from microbiota-depleted mice still led to increased mucus secretion, the stimulatory effect of the infusion was significantly lower as compared with the colonized mice (Figure 4B). Furthermore, we found in histological sections that the thickness of the colonic mucus layer in antibiotic-treated mice did not expand in response to TUDCA treatment (Figure 4C). Thus, presence of the gut microbiota is needed for ER stress control of mucus secretion.
Figure 4.

ER stress-controlled mucus secretion is regulated by the microbiota and Nod2
(A) Measurements of mucus thickness in colonic section from wild-type germ-free mice treated with TUDCA.
(B) % change in mucus secretion rates in response to TUDCA of colonic tissues from mice untreated or treated with antibiotics.
(C) Measurements of mucus thickness in colonic sections from wild-type mice treated with combinations of antibiotics, TUDCA, and IL-22.
(D) Measurements of mucus thickness in colonic sections from Rag1−/− mice treated with TUDCA.
(E) Measurements of mucus thickness in colonic sections from MyD88fl/fl and MyD88ΔIEC mice treated with TUDCA.
(F) Measurements of mucus thickness in colonic sections from Nod2−/− mice treated with TUDCA.
(A–F) Each dot represents a mouse. ns, not significant; ∗p < 0.05; ∗∗∗p < 0.001. (A, B, D, and F) Student’s t test; (C and E) one-way ANOVA; WT, wild type; TUDCA, tauroursodeoxycholic acid; ABX, antibiotics; IL-22, interleukin 22; fl, flox; IEC, intestinal epithelial cell.
Next, we wanted to determine how the microbiota is sensed by the host to allow excess mucus secretion. Multiple aspects of intestinal barrier function are controlled by the secretion of interleukin 22 (IL-22) from T helper cells or innate lymphoid cells, which then induces an antimicrobial response in intestinal epithelial cells. Thus, we treated microbiota-depleted mice with IL-22 and TUDCA. We found that IL-22 was not sufficient to induce mucus secretion in response to reduced ER stress in these microbiota-depleted mice (Figure 4C). We next tested whether the adaptive arm of the immune system senses the microbiota to allow excess mucus secretion. To do so, we treated Rag1-deficient mice, which lack mature T and B cells, with TUDCA. We found that this treatment expanded the mucus layer (Figure 4D), indicating that T and B cells are not the components that regulate excess mucus secretion in response to microbiota presence. Next, we tested whether direct microbiota sensing by intestinal epithelial cells through TLRs is needed to facilitate excess mucus secretion. We found that mice lacking the adaptor protein MyD88, which is needed for TLR-induced signaling, also responded to TUDCA treatment with excess mucus secretion (Figure 4E). Finally, we tested whether the intracellular sensor of bacteria, Nod2, was needed to facilitate excess mucus secretion. We found that mice lacking Nod2 did not respond to TUDCA treatment with excess mucus secretion (Figure 4F). Thus, functional Nod2 is crucial for excess mucus secretion in response to reduced ER stress.
Excess mucus secretion in Becn1F121A mice alters the gut microbiota
The colonic mucus layer also serves as a nutrient source for the microbiota by providing glycans for energy production by bacteria.20 Thus, we next examined whether the constitutive activation of autophagy shapes the gut microbiota via modulation of the mucus layer. 16S rRNA gene sequencing of fecal samples from separately bred and housed mice revealed that Becn1F121A mice harbor a distinct gut microbiota compared with wild-type mice (Figures 5A, 5B, and S4A), characterized by higher microbial diversity (Figure 5C). Relative abundance analysis revealed that bacteria from the genus Akkermansia were enriched in Becn1F121A mice (Figure 5D). Linear discriminant analysis effect size (LEfSe) analysis further confirmed that the mucus-utilizing bacterium Akkermansia muciniphila was abnormally over-abundant in Becn1F121A mice (Figures 5E, 5F, and S4B).
Figure 5.

Becn1F121A mice contain an altered gut microbiota
16S rRNA sequencing was performed to characterize gut microbiota composition.
(A and B) (A) PCoA of fecal microbiota β diversity based on weighted UniFrac and (B) Bray-Curtis dissimilarity in mice housed at the Bar-Ilan University.
(C) α diversity comparison based on richness and evenness.
(D) Relative taxonomic composition.
(E) Cladogram depicting LEfSe analysis of differently abundant bacteria in wild-type and Becn1F121A mice.
(F) Relative abundance of Akkermansia muciniphila in mice housed at the Bar-Ilan University.
(G) Principal coordinates analysis (PCoA) of fecal microbiota β diversity based on weighted UniFrac on mice housed at UTSW.
(H) Relative abundance of Akkermansia muciniphila in mice housed at UTSW.
(A, B, and F–H) Each symbol represents a mouse. (D) Each column represents a mouse. ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; (C, F and H) Student’s t test. WT, wild type; F121A, Becn1F121A; UTSW, UT Southwestern.
Variations in microbiota composition are highly dependent on mouse housing conditions and diet.21 To test how robust the effects of constitutive activation of autophagy are on microbiota composition, we performed 16S rRNA gene sequencing on feces collected from wild-type and Becn1F121A mice housed at a different animal facility (UT Southwestern Medical Center), fed a diet from a different supplier. Again, we found a clear difference in microbial composition between wild-type and Becn1F121A mice and an abnormal over-abundance of Akkermansia muciniphila (Figures 5G and 5H). As Akkermansia muciniphila is a mucus-utilizing bacterium that can be cultured only on mucus,22 its high relative abundance in the colons of Becn1F121A mice further points to the over-abundance of mucus in these mice. Thus, constitutive activation of autophagy reproducibly and rigorously alters the gut microbiota with the expansion of mucus-utilizing bacteria.
The microbiota of Becn1F121A mice confers protection from colitis
Next, we assessed how the altered mucus layer in Becn1F121A mice affects susceptibility to colonic inflammation. Penetrability of the colonic mucus layer is directly linked to colonic inflammation in both mice and humans.2,3 Thus, we hypothesized that Becn1F121A mice would be less susceptible to colitis. Dextran sulfate sodium (DSS) treatment instigates colonic inflammation through a direct toxic effect on the mucus barrier2 and thus was chosen as a suitable model to test the robustness of the mucus layer in Becn1F121A mice. When treating mice with 3% DSS for 5 and 7 days, we found that Becn1F121A mice lost less weight, displayed less signs of severe disease, suffered from reduced colon shortening, and had less histological damage as compared with wild-type mice (Figures 6A–6D and S5A–S5E). We further confirmed this in a model of moderate colitis by treating mice with 2% DSS for 7 days (Figures S5F–S5H). Additionally, we found that Becn1F121A mice still maintained better barrier function than wild-type mice following DSS treatment (Figure S5I).
Figure 6.

The microbiota of Becn1F121A mice confers protection from colitis
(A–D) Mice were treated with 3% DSS for 7 days. (A) Relative weight change ± SEM, (B) disease activity index ± SEM, (C) colon length, and (D) histological damage score.
(E) Scheme depicting FMT experiment. Created with BioRender.com.
(F and G) PCoA of fecal microbiota β diversity based on weighted UniFrac (F) and Bray-Curtis dissimilarity (G) of wild-type germ-free mice following FMT and separately housed wild-type and Becn1F121A mice fecal donors.
(H) α diversity comparison based on richness and evenness of wild-type germ-free mice following FMT.
(I–K) Mice were treated with 3% DSS for 7 days following FMT. (I) Disease activity index ± SEM, (J) colon length and histological damage score (K).
(C, D, F, G, J, and K) Each symbol represents a mouse. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗∗p < 0.0001; (C, D, J, and K) Student’s t test; (A, B, and I) multiple t tests corrected for false discovery rate; (H) one-way ANOVA. WT, wild type; F121A, Becn1F121A; FMT, fecal microbiota transplant.
The gut microbiota plays an important role in susceptibility to colitis.23 As the microbiota of separately bred Becn1F121A mice is altered in comparison with wild-type mice, we investigated whether this microbiota contributes to the reduced susceptibility of Becn1F121A mice to DSS-induced colitis. We colonized germ-free wild-type mice with microbiota from either wild-type or Becn1F121A mice (Figure 6E) and analyzed the effects of fecal microbiota transplant (FMT). Although FMT from wild-type donors into wild-type recipients was highly efficient, FMT from Becn1F121A donors into wild-type recipients was only partially efficient (Figures 6F and 6G). This implies that specific conditions present in the intestines of Becn1F121A mice are needed to support their microbiota structure. However, the microbiota of recipients from Becn1F121A mice was still highly diverse and different than the microbiota of recipients from wild-type mice (Figures 6F–6H). We then challenged these recipient mice with DSS and found that the microbiota of Becn1F121A mice did not prevent weight loss or colon shortening but did reduce histological damage (Figures 6I–6K). Thus, the microbiota of Becn1F121A mice provides partial protection from colitis.
Becn1F121A mice are protected from colitis in a microbiota-independent manner
We then investigated whether the distinct microbiota of Becn1F121A mice was essential to protect these mice from colitis. We bred heterozygous Becn1F121A mice together so that the wild-type and Becn1F121A offspring were exposed to the same microbiota throughout development and adulthood (Figure 7A). We found that the microbiota of littermate wild-type and Becn1F121A mice were similar to each other, and indistinguishable from the microbiota of wild-type mice bred separately (Figures 7B–7D). Yet, the mucus layer of Becn1F121A mice remained thicker than that of their wild-type littermates (Figure 7E), indicating that the unique Becn1F121A mice microbiota did not play an essential role in promoting excessive mucus secretion in Becn1F121A mice. We then challenged these littermate mice, and separately bred wild-type and Becn1F121A mice, with DSS. We found that Becn1F121A mice were still protected from acute colitis compared with their littermate wild-type mice, though to a lesser extent than separately bred Becn1F121A mice (Figures 7F and 7G). Thus, the altered microbiota is not needed for protection against colitis in Becn1F121A mice.
Figure 7.

Becn1F121A mice are protected from DSS- and AIEC-induced colitis in a microbiota-independent manner
(A) Scheme depicting littermate (L.M.) experimental design. Created with BioRender.com.
(B and C) (B) PCoA of fecal microbiota β diversity based on weighted UniFrac and (C) Bray-Curtis dissimilarity of separately bred and littermate wild-type and Becn1F121A mice.
(D) α diversity comparison based on richness and evenness.
(E) Measurements of mucus thickness in colonic section from littermate mice stained with Alcian blue.
(F and G) Disease activity index ± SEM (F), and histological damage score (G) of mice treated with 3% DSS for 7 days.
(H–K) Survival curve (H), stool lipocalin 2 levels (I), colon length (J), and histological damage score (K) of mice pretreated with 20 mg streptomycin and infected with 108 CFU AIEC for 10 weeks.
(L and M) Colon length (L) and histological score (M) of mice pretreated with 20 mg streptomycin and infected with 108 CFU AIEC for 14 days.
(B, C, E, G, and I–M) Each symbol represents a mouse. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; (D and G) one-way ANOVA; (E and I–M) Student’s t test. WT, wild type; F121A, Becn1F121A; L.M., littermates; a.u., arbitrary units.
Becn1F121A mice are protected from infection-driven colitis
Finally, we tested whether Becn1F121A mice are also protected from a clinically relevant model of intestinal inflammation by infecting mice with an adherent-invasive E. coli (AIEC).24 AIEC infection is strongly associated with IBD and is thought to be involved in the early stages of IBD development.25 Microbiota differences are negligible in this model as mice are pretreated with antibiotics.24 We found that, unlike wild-type mice, Becn1F121A mice did not develop chronic intestinal inflammation or suffer from colon shortening 10 weeks after being infected with AIEC (Figures 7H–7K). We further validated these results in an acute infection model (14 days) as well (Figures 7L and 7M). Thus, constitutive activation of autophagy also protects mice from infection-driven chronic colitis.
Discussion
Goblet cells are tasked with producing mucus to protect the intestine from microbial invasion.26 Yet, how these cells control the exact amount of mucus they need to secrete was not clear. Our results illuminate how mucus secretion from intestinal goblet cells is controlled by ER stress and autophagy. As secretory cells that produce large amounts of proteins, goblet cells are inherently susceptible to the accumulation of ER stress.12 Indeed, the loss of genes involved in the UPR leads to apoptosis of goblet cells in vivo.27 Additionally, single-cell analysis has revealed that genes involved in relieving ER stress are specifically enriched in the goblet cells population in wild-type mice during steady state.8 This observation suggests that, compared with other intestinal epithelial cells, goblet cells carry out a transcriptional program that is meant to relieve ER stress during steady state in an attempt by these goblet cells to preserve proteostasis. Thus, coupling ER stress to mucus secretion might be a mechanism by which goblet cells preserve viability by reducing mucus secretion when cells are pushed to their limits. As mucus is released and passes to the outside of the body, its concentration cannot be used as feedback to adjust secretion rates. However, the mechanism of regulation via ER stress that we describe here can provide cell-intrinsic control over the amount of mucus that is secreted.
How Beclin 1 activation relieves ER stress in goblet cells is not clear at this point. Autophagy has been shown to alleviate ER stress in other cells by multiple mechanisms.10 Specifically in intestinal epithelial cells, autophagy has been shown to remove inositol-requiring enzyme 1 α (IRE1α) via Atg16L1-dependent autophagy, thus reducing ER stress and preserving goblet cell function.11 Yet another possibility is that autophagy removes misfolded proteins from goblet cells. Properly folding the mucus-forming protein MUC2 is a demanding task for goblet cells, and defects in this folding process have been linked to goblet cell dysfunction.28
Our discovery that autophagy is needed to sustain mucus production by relieving ER stress provides a possible explanation for the prevalence of mutations in key autophagy genes found in IBD patients.4 With the inability to relieve ER stress because of dysfunctional autophagy, goblet cells might have difficulties keeping up with mucus production demands. Additionally, our observation that Nod2 is needed to facilitate mucus secretion when ER stress is reduced provides a possible explanation for the high frequency of mutations in this gene found in IBD patients.29,30 NOD2 activates an inflammatory response when sensing bacterial-derived muramyl dipeptide (MDP). Thus, the observation that inactivating mutations in NOD2 predispose to diseases that are characterized by chronic inflammation is somewhat paradoxical. The role we discovered for Nod2 in mucus secretion could thus provide an explanation for the observation that the mucus layer becomes penetrable in IBD patients,3 leading to the constant contact of microbes with the host epithelium and subsequently to chronic inflammation.
Finally, our observation that goblet cells possess the ability to produce more mucus when autophagy is constitutively activated, in a way that protects from intestinal inflammation, raises the question of why evolution has shaped goblet cells to not do so in the wild? Mucus production is very energetically costly, as the MUC2 protein is highly glycosylated. Thus, it is possible that evolutionary constraints have shaped the mucus layer to fit the energy consumption of the animal. Excess mucus secretion can also be detrimental, for example, in cystic fibrosis, allergy, or asthma. Thus, it is possible that altering the regulation of mucus secretion in the intestine might also affect mucus secretion in other organs. Indeed, if an organism has a need to produce more mucus, it does so by increasing goblet cell numbers, as seen during infection by intestinal parasites and worms.31 Interestingly, the same signals that lead to the production of more goblet cells in the intestine (type 2 cytokines) also lead to an increase in goblet cells and mucus secretion in the airways.32
Limitations of the study
Here, we show that ER stress levels control mucus secretion from colonic goblet cells and that constitutively activated autophagy reduces ER stress to facilitate excess mucus secretion and protection from colitis. We acknowledge that there are several limitations to our study. First, we cannot rule out the possibility that the constitutive activation of autophagy or reduction of ER stress affects cells other than goblet cells and that these cells then affect mucus secretion, the microbiota, and susceptibility to colitis. For example, although Paneth cells are not present in the colon, the secretion of antimicrobial proteins from Paneth cells is highly dependent on both autophagy and ER stress levels,13,33 and these proteins have been shown to reach the colon.34 However, there are no mouse models currently available that allow the constitutive activation of autophagy—or reduction of ER stress—in goblet cells alone. Second, the biochemical changes to the mucus layer in Becn1F121A mice that lead to the expansion of mucin-utilizing bacteria are not clear. It would be valuable to characterize these changes to the mucus layer using sensitive glycomics and proteomics methods. Finally, understanding the exact molecular mechanism by which autophagy relieves ER stress, in goblet cells specifically, could provide a viable option for the development of mucus-targeted therapeutics.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-GADD153/CHOP | SCBT | sc-166682 |
| Anti-Tubulin | Abcam | ab176560 |
| Anti-EPCAM | BD Biosciences | 563214 |
| Anti-4 Hydroxynonenal | Abcam | ab48506 |
| Bacterial and virus strains | ||
| Adherent-invasive E. Coli pathotype (AIEC), NRG857c strain (O83:H1) | Small et al.24 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| tauroursodeoxycholic acid (TUDCA) | Sigma-Aldrich | T0266 |
| Ulex Europaeus Agglutinin I (UEA I), DyLight™ 649 | Vector Laboratories | DL-1068-1 |
| Fixable Viability Stain 510 | BD Biosciences | 564406 |
| Salubrinal | Enzo | ALX-270-428 |
| Thapsigargin | Sigma-Aldrich | T9033 |
| Dextran sulfate sodium colitis grade, 36,000 - 50,000 Da | MP Biomedicals | SKU: 02160110-CF |
| Compound 147 (AA147) | Abbexa | abx283245 |
| IL-22-Fc | Genentech | PRO312045 |
| Critical commercial assays | ||
| TLR4 HEK-Blue™ reporter cells | Invivogen | hkb-mtlr4 |
| TLR5 HEK-Blue™ reporter cells | Invivogen | hkb-mtlr5 |
| RNeasy Universal kit | Qiagen | 73404 |
| Purelink™ Microbiome DNA Purification Kit | Invitrogen | A29790 |
| AMPure magnetic beads | Beckman Coulter | A63880 |
| Lcn-2 ELISA kit | R&D systems | DY1857-05 |
| NOD1 HEK-Blue™ reporter cells | Invivogen | hkb-mnod1 |
| NOD2 HEK-Blue™ reporter cells | Invivogen | hkb-mnod2 |
| TLR9 HEK-Blue™ reporter cells | Invivogen | hkb-mtlr9 |
| Deposited data | ||
| RNA sequencing | This paper | GSE220457 |
| 16S rRNA gene sequencing | This paper | GSE220883 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6JOlaHsd | Envigo Israel | 2BL |
| Mouse: Becn1F121A (C57BL/6) | Fernández et al.5 | N/A |
| Mouse: Nod2flox/flox (C57BL/6) | Kim et al.35 | N/A |
| Mouse: B6.129X1(Cg)-Bcl2tm1.1Sjk/J | The Jackson Laboratory | #018430 |
| Mouse: Germ-free Mice/Swiss Webster | Taconic Biosciences | SW-F/SW-M |
| Mouse: B6.129S7-Rag1tm1Mom/J | The Jackson Laboratory | # 002216 |
| Mouse: B6.129P2(SJL)-Myd88tm1Defr/J | The Jackson Laboratory | # 008888 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | The Jackson Laboratory | # 006054 |
| Mouse: B6.Cg-Tg(Vil1-cre)997Gum/J | The Jackson Laboratory | # 004586 |
| Oligonucleotides | ||
| 515F - (barcode) 5′-AATGATACGGCGACCACCGAG ATCTACACGCTAGCCTTCGTCGCTATGGTAATTGT G TGYCAGCMGCCGCGGTAA-3′ |
DeSantis et al.36 | N/A |
| 806 R 5′- CAAGCAGAAGACGGCATACGAGATAG TCAGTCAGCCGGACTACHVGGGTWTCTAAT -3′ |
DeSantis et al.36 | N/A |
| Software and algorithms | ||
| Quantitative Insights into Microbial Ecology 2 (QIIME2) | Bolyen et al.37 | N/A |
| PRISM | Graphpad | N/A |
| FIJI | Schindelin et al.38 | N/A |
| Divisive Amplicon Denoising Algorithm (DADA2) | DeSantis et al.36 | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Shai Bel (shai.bel@biu.ac.il).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Mice (see key resources table) were separately bred, unless indicated otherwise, and maintained in the SPF barrier at the Azrieli Faculty of Medicine, Bar-Ilan University, Israel. MyD88ΔIEC mice were generated by breeding MyD88flox and Vil1-cre mice. Nod2-/- mice were generated by breeding Nod2flox (a kind gift from Philip Rosenstiel35) and CMV-cre mice. Germ-free Swiss Webster mice were bred and maintained in isolators as described.39 8–14-week-old mice were used for all experiments. All experiments were performed using protocols approved by the Institutional Animal Care and Use Committees (IACUC) of the Bar-Ilan University.
Mucus thickness measurements
Mice were euthanized by cervical dislocation, according to IACUC guidelines, and a 1cm long colon tissue sample containing a fecal pellet was removed. Tissues were immediately fixed in Carnoy’s fixative to preserve the mucus layer and processed for paraffin embedding.40 5μm thick sections were cut using a microtome and stained with Alcian blue stain. Stained sections were visualized using a Zeiss Axioimager M2 microscope and mucus layer thickness (the blue stained area engulfing the fecal pellet) was measured using Zeiss Zen software. 30 measurements were performed per section and the average calculated for each mouse. For measurements following antibiotic treatment, mice were treated with 1 mg/ml ampicillin, 0.5 mg/ml vancomycin, 1 mg/ml neomycin, 1 mg/ml streptomycin and 1 mg/ml metronidazole for 6 weeks.
Barrier function analysis
Blood was collected post-mortem via cardiac puncture and incubated at room temperature for 30 minutes in 1.5ml tubes to allow clotting. Samples were then centrifuged at 1,500g for 20 minutes at 4°C after which serum was collected to new tubes. 20μl of serum was added to wells containing InvivoGen HEK-Blue™ reporter cells, and luminal antigens were detected following manufacturer’s instructions.
In vivo tauroursodeoxycholic acid, salubrinal, thapsigargin, AA147 and IL-22 treatment
Tauroursodeoxycholic acid (TUDCA; Sigma T0266), was dissolved in PBS and administered at 250 mg/kg body weight. TUDCA was administered to mice via intraperitoneal injection twice a day for 3 days. On the 4th day, mice were treated in the morning and euthanized 4 hours after the last treatment. Control mice were treated with PBS. Salubrinal (Enzo ALX-270-428) was dissolved in dimethyl sulfoxide (DMSO) at 50 mg/ml and diluted 1:400 in PBS. Mice were treated with 1 mg Salubrinal/kg body weight via intraperitoneal injection once a day for 3 days before being euthanized. Thapsigargin (Sigma T9033), was dissolved in ethanol at 10 mg/ml and then diluted 1:40 in PBS. Thapsigargin (2.5 mg/kg body weight) was administered to mice via intraperitoneal injection once a day for 2 days and mice were euthanized 4 hours after the last treatment. Control mice were treated with ethanol diluted 1:40 in PBS. AA147 (2 mg/kg body weight) was administered to mice via intraperitoneal injection twice a day for 3 days and mice were euthanized 4 hours after the last treatment. IL-22-Fc (Genentech, 100ug per dose) was administered once a day for 4 days.
Chemical-induced colitis model
Mice were treated with dextran sulfate sodium (DSS, colitis grade, 36,000–50,000 Da, MP) in drinking water at the concentrations and durations indicated in the figure legends. Fresh DSS was prepared daily. Disease activity index (DAI) was measured daily, based on weight loss, stool consistency and rectal bleeding as previously described.41 Briefly, weight loss relative to initial weight, stool consistency (solid, loose or diarrhea) and rectal bleeding were each individually scored on a 0-4 scale and summed for each mouse at the indicated timepoints. Barrier function using FITC-dextran was performed on the last day as previously described.41 Briefly, mice were orally treated with 200 μl of 3-5 kDa FITC-dextran via gavage 4 hours prior to termination. Blood was then collected and serum separated as above. FITC-dextran was detected using a plate reader at 530 nm with excitation at 485 nm.
Infection-induced colitis model
Mice were infected with adherent-invasive E. Coli pathotype (AIEC), NRG857c strain (O83:H1) (a kind gift from Brian Coombes24). NRG857c was cultured in Luria broth (LB) containing chloramphenicol (34 μg∗ml-1) and ampicillin (100 μg∗ml-1). 24h before infection, mice were treated with 20mg streptomycin and then infected with 108 colony forming units (CFU) AIEC. After either 14 days or 10 weeks mice were euthanized, and intestines were fixed in 4% PFA. For quantification of Lipocalin-2 levels, feces were collected and immediately froze in liquid nitrogen and stored at -80°C. Then, samples were reconstituted in PBS containing 0.1% Tween 20, vortexed for 20 min, and centrifuged at 21,000g for 20 min at 4°C. Fecal Lcn-2 levels were determined by the Lcn-2 ELISA kit (R&D systems, DY1857-05) according to the manufacturer’s instructions. Results were calculated by OD and feces weight ratio.
Fecal microbiota transplant
Feces collected from mice housed at the SPF barrier facility were immediately transferred into an anaerobic chamber. Feces were vortexed in sterile PBS, debris allowed to settle by gravity, and supernatant transferred to new tubes. Sealed tubes were then removed from the anaerobic chamber and orally administered to germ-free Swiss Webster mice via gavage. Feces from each SPF-housed mouse were used to inoculate two germ-free mice. Inoculated mice were then transferred to Tecniplast IsoCages to prevent outside contamination for two weeks, after which mice were challenged with DSS as above.
Method details
RNA sequencing and analysis
RNA from frozen colonic tissues was extracted using Qiagen RNeasy Universal kit. Integrity of the isolated RNA was analysed using the Agilent TS HS RNA Kit and TapeStation 4200 at the Genome Technology Center at the Azrieli Faculty of Medicine, Bar-Ilan University. 1000ng of total RNA was treated with the NEBNext poly (A) mRNA Magnetic Isolation Module (NEB, #E7490L). RNA sequencing libraries were produced by using the NEBNext Ultra II RNA Library Prep Kit for Illumina (NEB #E7770L). Quantification of the library was performed using a dsDNA HS Assay Kit and Qubit (Molecular Probes, Life Technologies) and qualification was done using the Agilent TS D1000 kit and TapeStation 4200. 250nM of each library was pooled together and diluted to 4nM according to the NextSeq manufacturer’s instructions. 1.6pM was loaded onto the Flow Cell with 1% PhiX library control. Libraries were sequenced with the Illumina NextSeq 550 platform with single-end reads of 75 cycles according to the manufacturer's instructions. Sequencing data was aligned and normalized (reads per kilo base per million mapped reads) using Partek bioinformatics software. Pathway analysis was performed using the ShinyGO webtool.42 Heat maps, principal component analysis (PCA) plots and volcano plots were generated using GraphPad Prism software.
Isolation of goblet cells for qPCR analysis
Colonic tissues were washed with PBS, cut into 3 pieces, and washed with HBSS buffer supplemented with 5% FBS and 1% penicillin and streptomycin on ice. Cut tissues were then transferred to HBSS buffer supplemented with 10mM EDTA, 1mM DTT, 5% FBS and 1% penicillin and streptomycin and incubated at 37°C with shaking at 200 rpm for 10 minutes. Tissues were then vortexed, passed through a 70μm strainer and further dissociated with a syringe. Dissociated cells were then collected via centrifugation at 500g for 10 minutes at 4°C. Isolated cells were resuspended in PBS containing 5% FBS, incubated with BD Horizon™ Fixable Viability Stain 510, anti-EPCAM antibody and UEA-1-DyLight 649 and sorted using a BD FACSAria™ Cell Sorter. RNA was extracted from live EPCAM+ UEA-1+ cells were using Qiagen RNeasy Universal kit and cDNA prepared using Thermo High-Capacity cDNA Reverse Transcription Kit. mRNA levels of Hspa5 were analyzed using TaqMan probes and normalized to 18s levels.
Immunoblot
Total proteins were extracted from colonic tissue samples by homogenizing in RIPA buffer (Thermo Fisher 89900). 50μg of total protein (as determined by Bradford assay) were loaded onto a 4-20% gradient SDS-PAGE and subsequently transferred to a PVDF membrane. Membranes were blocked with 5% nonfat dry milk in PBS with 0.1% Tween-20. The membranes were incubated at 40C overnight with the following primary antibodies: α-GADD153/CHOP (SCBT sc-166682) and α-Tubulin (Abcam ab176560). After washing, membranes were incubated with the species-appropriate HRP-conjugated secondary antibodies. Membranes were visualized using an Alliance Q9 system and band density was quantified using FIJI software.38
Ex vivo mucus measurements and tauroursodeoxycholic acid treatment
Mucus growth rate in colonic tissue explant was measured as previously described18 with some modifications. Briefly, distal colon tissue from 8-12 week-old wild type mice was collected and washed with 4-5 ml of Kreb’s transport buffer. After muscle removal, tissue was separated into 2 pieces. The tissues were mounted in a heated chamber (37oC). One piece of colon was incubated with RPMI supplemented with 6 mg/ml tauroursodeoxycholic acid (TUDCA), while another was mounted and incubated with RPMI as a control. Mounted tissues were covered by Kreb’s mannitol buffer, the mucus was overlaid with 10 μm-sized beads and mucus thickness was measured repeatedly with a micromanipulator-connected glass needle. Mucus growth measurement was performed for 45 min. For measurements following antibiotic treatment, mice were treated with 1 mg/ml of Ampicillin, 0.5 mg/ml of Cefoperazone and 1 ml/ml of Clindamycin in sterile drinking water for 5 days. Antibiotics were then removed for 2 days (washout) and mice were then treated for an additional 5 days with 1 mg/ml of Streptomycin, 1mg/ml of Neomycin, and 0.5 mg/ml of Vancomycin. Antibiotics were then replaced with drinking water 12 hours prior to mucus measurements as above.
Bacterial DNA extraction, amplification, and purification
DNA was extracted from fecal samples using the Invitrogen Purelink™ Microbiome DNA Purification Kit according to the manufacturer’s instructions, following two minutes of bead beating (Bio Spec). Following DNA extraction, the V4 variable region of the bacterial 16S rRNA gene was amplified by polymerase chain reaction (PCR) using the 515F and 806R primers, and each sample received a unique 515F barcoded primer. Primer sequences used were: 515F- (barcode) 5′-AATGATACGGCGACCACCGAGATCTACACGCTAGCCTTCGTCGCTATGGTAATTGTG TGYCAGCMGCCGCGGTAA-3′ and 806 R 5′- CAAGCAGAAGACGGCATACGAGATAGTCAGTCAGCCGGACTACHVGGGTWTCTAAT -3′36
PCR reactions were carried out with the Primestar taq polymerase (Takara) for 30 cycles of denaturation (95 °C), annealing (55 °C) and extension (72 °C), and a final elongation at 72 °C. Amplicons were purified using AMPure magnetic beads (Beckman Coulter), and subsequently quantified using Picogreen dsDNA quantitation kit (Invitrogen). Samples were pooled at equal concentrations (50 ng/μl), loaded on 2% E-Gel (Thermo Fisher), and purified using NucleoSpin Gel and PCR Clean-up (Macherey-Nagel).
16S rRNA gene sequence analysis
Purified products were sequenced using the Illumina MiSeq platform (Genome Technology center at the Azrieli Faculty of Medicine Bar-Ilan University). FASTQ data was processed and analyzed using Quantitative Insights into Microbial Ecology 2 (QIIME2) version 2019.4.37 Single-end sequences were demultiplexed, and reads were denoised and clustered using Divisive Amplicon Denoising Algorithm (DADA2).36 Primers were trimmed off and single end reads were truncated to ≥150 base pairs. A phylogenetic tree was constructed and features were assigned taxonomy using Greengenes reference database with 99% confidence.36
Analysis was performed on a rarefied feature table of 7340 reads per sample after filtration of mitochondria and chloroplast sequences. Alpha diversity was calculated using the Shannon index, referring to bacterial evenness within the sample.43 For between sample diversity (beta diversity), weighted and unweighted UniFrac distances were calculated.44 Over-represented and under-represented features were identified using linear discriminant analysis effect size (LEfSe).45
Histological scoring of colitis severity
Distal colon tissues were fixed in 4% paraformaldehyde, paraffin embedded, sectioned, and stained with hematoxylin and eosin. Histopathological analysis and semi-quantitative scoring were performed by a board-certified toxicological pathologist according to the scoring system described by Cooper et al.,46 taking into consideration the grades of extension (laterally, along the mucosa and deep into the mucosa, submucosa, and/or muscular layers) of the inflammation and ulceration. Analysis was performed in a blinded fashion.
Immunohistochemistry
Paraffin-embedded colonic tissues were sectioned and washed in xylene followed by re-hydration in decreasing concentrations of ethanol. Antigen retrieval was performed by boiling slides in 0.1 M trisodium citrate and washing in PBS. Slides were blocked with 1% BSA, 10% FBS, 1% Triton X-100 in PBS and then incubated with primary antibodies at 4oC overnight at 1:200 dilution. Secondary antibodies were diluted 1:400 and applied to slides for 1 hour at room temperature in the dark. Slides were then washed and mounted. Images were captured using a Zeiss AxioImager M2 microscope and analyzed with either Zeiss or FIJI software.38
Quantification and statistical analysis
Statistical analysis was performed using GraphPad PRISM as indicated in figure legends.
Acknowledgments
This study was performed in memory of Beth Levine, a pioneer and a kind mentor. We thank Lora V. Hooper for donating samples from mice housed at UT Southwestern Medical Center. Funding: The Azrieli Foundation Early Career Faculty Fellowship (S.B.); Israel Science Foundation (ISF) grants 925/19 and 1851/19 (S.B.); European Crohn’s and Colitis Organization (ECCO) grant (S.B.); Mizutani Foundation for Glycoscience (S.B.); European Research Council (ERC) starting grant GCMech 101039927 (S.B.); Swedish Research Council—grants 2018-02095 and 2021-06602 (B.O.S.)
Author contributions
Conceptualization, M.N., M.W., B.O.S., M.N.-O., and S.B.; investigation, M.N., S.Telpaz, A.A., S.B.-S., S.H.-S., S.M., E.R., J.S., L.Z., M.Z., N.A., S.Turjeman, S.W., R.F., A.N., and M.N.-O.; funding acquisition, B.O.S. and S.B.; supervision, S.B.; writing – original draft, S.B.; writing – review & editing, M.W., B.O.S., S.Turjeman, M.N.-O., and S.B.
Declaration of interests
The authors declare no competing interests.
Published: February 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.chom.2023.01.006.
Supplemental information
Data and code availability
RNA-seq data (GSE220457) and 16S rRNA gene sequencing data (GSE220883) have been deposited at NCBI Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.
References
- 1.Johansson M.E.V., Hansson G.C. Immunological aspects of intestinal mucus and mucins. Nat. Rev. Immunol. 2016;16:639–649. doi: 10.1038/nri.2016.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johansson M.E.V., Gustafsson J.K., Holmén-Larsson J., Jabbar K.S., Xia L., Xu H., Ghishan F.K., Carvalho F.A., Gewirtz A.T., Sjövall H., et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut. 2014;63:281–291. doi: 10.1136/GUTJNL-2012-303207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Post S., Jabbar K.S., Birchenough G., Arike L., Akhtar N., Sjovall H., Johansson M.E.V., Hansson G.C. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut. 2019;68:2142–2151. doi: 10.1136/GUTJNL-2018-317571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaser A., Blumberg R.S. The road to Crohn’s disease. Science. 2017;357:976–977. doi: 10.1126/science.aao4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernández Á.F., Sebti S., Wei Y., Zou Z., Shi M., McMillan K.L., He C., Ting T., Liu Y., Chiang W.-C., et al. Disruption of the beclin 1–BCL2 autophagy regulatory complex promotes longevity in mice. Nature. 2018;558:136–140. doi: 10.1038/s41586-018-0162-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Patel K.K., Miyoshi H., Beatty W.L., Head R.D., Malvin N.P., Cadwell K., Guan J.L., Saitoh T., Akira S., Seglen P.O., et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013;32:3130–3144. doi: 10.1038/emboj.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serradas P., Thaiss C.A., Gertler A., Aamar S., Blacher E., Halpern Z., Katz M.N., Geiger B., Grosheva I., Grosfeld A., et al. Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science. 2018;359:1376–1383. doi: 10.1126/science.aar3318. [DOI] [PubMed] [Google Scholar]
- 8.Nyström E.E.L., Martinez-Abad B., Arike L., Birchenough G.M.H., Nonnecke E.B., Castillo P.A., Svensson F., Bevins C.L., Hansson G.C., Johansson M.E.V. An intercrypt subpopulation of goblet cells is essential for colonic mucus barrier function. Science. 2021;372:eabb1590. doi: 10.1126/SCIENCE.ABB1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adolph T.E., Tomczak M.F., Niederreiter L., Ko H.-J., Böck J., Martinez-Naves E., Glickman J.N., Tschurtschenthaler M., Hartwig J., Hosomi S., et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rashid H.-O., Yadav R.K., Kim H.-R., Chae H.-J. ER stress: autophagy induction, inhibition and selection. Autophagy. 2015;11:1956–1977. doi: 10.1080/15548627.2015.1091141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tschurtschenthaler M., Adolph T.E., Ashcroft J.W., Niederreiter L., Bharti R., Saveljeva S., Bhattacharyya J., Flak M.B., Shih D.Q., Fuhler G.M., et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease–like ileitis. J. Exp. Med. 2017;214:401–422. doi: 10.1084/JEM.20160791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaser A., Blumberg R.S. Endoplasmic reticulum stress in the intestinal epithelium and inflammatory bowel disease. Semin. Immunol. 2009;21:156–163. doi: 10.1016/J.SMIM.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bel S., Pendse M., Wang Y., Li Y., Ruhn K.A.K.A., Hassell B., Leal T., Winter S.E.S.E., Xavier R.J., R.J.R.J.R.J., Hooper L.V.L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017;357:eaal4677. doi: 10.1126/science.aal4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He C., Bassik M.C., Moresi V., Sun K., Wei Y., Zou Z., An Z., Loh J., Fisher J., Sun Q., et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481:511–515. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Berger E., Haller D. Structure–function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2011;409:610–615. doi: 10.1016/J.BBRC.2011.05.043. [DOI] [PubMed] [Google Scholar]
- 16.Boyce M., Bryant K.F., Jousse C., Long K., Harding H.P., Scheuner D., Kaufman R.J., Ma D., Coen D.M., Ron D., et al. A selective inhibitor of elF2α dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/SCIENCE.1101902. [DOI] [PubMed] [Google Scholar]
- 17.Grandjean J.M.D., Wiseman R.L. Small molecule strategies to harness the unfolded protein response: where do we go from here? J. Biol. Chem. 2020;295:15692–15711. doi: 10.1074/jbc.REV120.010218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gustafsson J.K., Ermund A., Johansson M.E.V., Schütte A., Hansson G.C., Sjövall H. An ex vivo method for studying mucus formation, properties, and thickness in human colonic biopsies and mouse small and large intestinal explants. Am. J. Physiol. Gastrointest. Liver Physiol. 2012;302:G430–G438. doi: 10.1152/AJPGI.00405.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thaiss C.A., Zmora N., Levy M., Elinav E. The microbiome and innate immunity. Nature. 2016;535:65–74. doi: 10.1038/nature18847. [DOI] [PubMed] [Google Scholar]
- 20.Hansson G.C. 2020. Annual review of biochemistry mucins and the microbiome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stappenbeck T.S., Virgin H.W. Accounting for reciprocal host-microbiome interactions in experimental science. Nature. 2016;534:191–199. doi: 10.1038/nature18285. [DOI] [PubMed] [Google Scholar]
- 22.Desai M.S., Seekatz A.M., Koropatkin N.M., Kamada N., Hickey C.A., Wolter M., Pudlo N.A., Kitamoto S., Terrapon N., Muller A., et al. A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell. 2016;167:1339–1353.e21. doi: 10.1016/J.CELL.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bel S., Elkis Y., Elifantz H., Koren O., Ben-Hamo R., Lerer-Goldshtein T., Rahimi R., Horin S. ben, Nyska A., Shpungin S., et al. Reprogrammed and transmissible intestinal microbiota confer diminished susceptibility to induced colitis in TMF-/- mice. Proc. Natl. Acad. Sci. USA. 2014;111:4964–4969. doi: 10.1073/pnas.1319114111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Small C.L.N., Reid-Yu S.A., McPhee J.B., Coombes B.K. Persistent infection with Crohn’s disease-associated adherent-invasive Escherichia coli leads to chronic inflammation and intestinal fibrosis. Nat. Commun. 2013;4:1957. doi: 10.1038/ncomms2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buisson A., Sokol H., Hammoudi N., Nancey S., Treton X., Nachury M., Fumery M., Hébuterne X., Rodrigues M., Hugot J.-P., et al. Role of adherent and invasive Escherichia coli in Crohn’s disease: lessons from the postoperative recurrence model. Gut. 2022;72:39–48. doi: 10.1136/GUTJNL-2021-325971. [DOI] [PubMed] [Google Scholar]
- 26.Modilevsky S., Naama M., Bel S. Encyclopedia of Cell Biology. Elsevier; 2023. Goblet and Paneth cells: Producers of the Intestinal Barrier; pp. 66–71. [DOI] [Google Scholar]
- 27.Kaser A., Lee A.H., Franke A., Glickman J.N., Zeissig S., Tilg H., Nieuwenhuis E.E.S., Higgins D.E., Schreiber S., Glimcher L.H., et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell. 2008;134:743–756. doi: 10.1016/j.cell.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasnain S.Z., Tauro S., Das I., Tong H., Chen A.C., Jeffery P.L., McDonald V., Florin T.H., McGuckin M.A. IL-10 promotes production of intestinal mucus by suppressing protein misfolding and endoplasmic reticulum stress in goblet cells. Gastroenterology. 2013;144:357–368.e9. doi: 10.1053/j.gastro.2012.10.043. [DOI] [PubMed] [Google Scholar]
- 29.Balasubramanian I., Gao N. From sensing to shaping microbiota: insights into the role of NOD2 in intestinal homeostasis and progression of Crohn’s disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2017;313:G7–G13. doi: 10.1152/ajpgi.00330.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirkov M.U., Verstockt B., Cleynen I. Genetics of inflammatory bowel disease: beyond NOD2. Lancet Gastroenterol. Hepatol. 2017;2:224–234. doi: 10.1016/S2468-1253(16)30111-X. [DOI] [PubMed] [Google Scholar]
- 31.von Moltke J., Ji M., Liang H.E., Locksley R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature. 2016;529:221–225. doi: 10.1038/nature16161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jaramillo A.M., Azzegagh Z., Tuvim M.J., Dickey B.F. Airway mucin secretion. Ann. Am. Thorac. Soc. 2018;15:S164–S170. doi: 10.1513/AnnalsATS.201806-371AW. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bel S., Hooper L.V.L.V. Secretory autophagy of lysozyme in Paneth cells. Autophagy. 2018;14:719–721. doi: 10.1080/15548627.2018.1430462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mastroianni J.R., Ouellette A.J. Alpha-defensins in enteric innate immunity: functional Paneth cell alpha-defensins in mouse colonic lumen. J. Biol. Chem. 2009;284:27848–27856. doi: 10.1074/jbc.M109.050773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D., Kim Y.G., Seo S.U., Kim D.J., Kamada N., Prescott D., Chamaillard M., Philpott D.J., Rosenstiel P., Inohara N., et al. Nod2-mediated recognition of the microbiota is critical for mucosal adjuvant activity of cholera toxin. Nat. Med. 2016;22:524–530. doi: 10.1038/nm.4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeSantis T.Z., Hugenholtz P., Larsen N., Rojas M., Brodie E.L., Keller K., Huber T., Dalevi D., Hu P., Andersen G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006;72:5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolyen E., Rideout J.R., Dillon M.R., Bokulich N.A., Abnet C.C., Al-Ghalith G.A., Alexander H., Alm E.J., Arumugam M., Asnicar F., et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019;37:852–857. doi: 10.1038/S41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., et al. Fiji: an open source platform for biological image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cash H.L., Whitham C.V. v, Behrendt C.L., Hooper L.V. Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science. 2006;313:1126–1130. doi: 10.1126/science.1127119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson M.E. v, Hansson G.C. Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods Mol. Biol. 2012;842:229–235. doi: 10.1007/978-1-61779-513-8_13. [DOI] [PubMed] [Google Scholar]
- 41.Bel S., Elkis Y., Lerer-Goldstein T., Nyska A., Shpungin S., Nir U. Loss of TMF/ARA160 protein renders colonic mucus refractory to bacterial colonization and diminishes intestinal susceptibility to acute colitis. J. Biol. Chem. 2012;287:25631–25639. doi: 10.1074/jbc.M112.364786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ge S.X., Jung D., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/BIOINFORMATICS/BTZ931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pielou E.C. Species-diversity and pattern-diversity in the study of ecological succession. J. Theor. Biol. 1966;10:370–383. doi: 10.1016/0022-5193(66)90133-0. [DOI] [PubMed] [Google Scholar]
- 44.Lozupone C., Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/GB-2011-12-6-R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab. Invest. 1993;69:238–249. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data (GSE220457) and 16S rRNA gene sequencing data (GSE220883) have been deposited at NCBI Gene Expression Omnibus and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request.