

Abstract
Disruption of the transverse-axial tubule system (TATS) in diseases such as heart failure and atrial fibrillation occurs in combination with changes in the expression and distribution of key Ca2+-handling proteins. Together this ultrastructural and ionic remodeling is associated with aberrant Ca2+ cycling and electrophysiological instabilities that underly arrhythmic activity. However, due to the concurrent changes in TATs and Ca2+-handling protein expression and localization that occur in disease it is difficult to distinguish their individual contributions to the arrhythmogenic state. To investigate this, we applied our novel 3D human atrial myocyte model with spatially detailed Ca2+ diffusion and TATS to investigate the isolated and interactive effects of changes in expression and localization of key Ca2+-handling proteins and variable TATS density on Ca2+-handling abnormality driven membrane instabilities. We show that modulating the expression and distribution of the sodium-calcium exchanger, ryanodine receptors, and the sarcoplasmic reticulum (SR) Ca2+ buffer calsequestrin have varying pro and anti-arrhythmic effects depending on the balance of opposing influences on SR Ca2+ leak-load and Ca2+-voltage relationships. Interestingly, the impact of protein remodeling on Ca2+-driven proarrhythmic behavior varied dramatically depending on TATS density, with intermediately tubulated cells being more severely affected compared to detubulated and densely tubulated myocytes. This work provides novel mechanistic insight into the distinct and interactive consequences of TATS and Ca2+-handling protein remodeling that underlies dysfunctional Ca2+ cycling and electrophysiological instability in disease.
Keywords: Atria, Calcium, Arrhythmia, Transverse-axial tubule, Calcium-handling proteins
Graphical Abstract
Introduction
It is widely accepted that Ca2+ abnormalities can precipitate atrial fibrillation (AF), the most common cardiac arrhythmia. Both ionic remodeling and ultrastructural changes have been associated with aberrant Ca2+ signaling, excitation-contraction coupling, and AF (Nattel & Dobrev, 2012; Nattel & Harada, 2014; Dobrev & Wehrens, 2017; Denham et al., 2018). Disruption of the transverse-axial tubular system (TATS) is one hallmark of AF structural remodeling that likely occurs early in disease progression and contributes to the dysfunction observed. In a companion paper, we developed a three-dimensional model of the human atrial myocyte coupling electrophysiology, whole-cell and local Ca2+ handling, and subcellular ultrastructural details, to interrogate the mechanisms by which TATS variability and loss affect human atrial physiology (Zhang et al.). Our simulation predicted that TATS loss per se enhances vulnerability to proarrhythmic behaviors (i.e., spontaneous calcium releases, SCRs, delayed afterdepolarizations, DADs) by altering subcellular Ca2+ signaling. Nevertheless, SCRs and DADs occur more frequently in AF vs. sinus rhythm human atrial myocytes that both mostly lack TATS after isolation via enzymatic digestion (though a fairly robust TATS presence is seen in human atrial tissue (Richards et al., 2011)). Indeed, the reduced density and regularity of the TATS is one aspect of disease remodeling, and it occurs concomitantly with altered channel and transporter expression, regulatory state, and function, as well as subcellular redistribution of ion channels, transporters, and Ca2+ handling proteins.
Well established hallmarks of ionic remodeling in AF include reduced L-type Ca2+ channel (LCC) current (Lenaerts et al., 2009), increased inward rectifier K+ current (IK1) and constitutively activated acetylcholine-activated K+ current (IK,ACh) (Bosch et al., 1999), enhanced Na+/Ca2+ exchanger (NCX) activity (Lenaerts et al., 2009; Voigt et al., 2012), and hyperphosphorylated ryanodine receptors (RyRs) (Vest et al., 2005; Neef et al., 2010; Voigt et al., 2012). The basal subcellular localization of these proteins in the atria, and whether they are altered in disease, in not well understood. LCCs distribute equally in tubules and crest areas of the sarcolemma in control human atrial myocytes (Glukhov et al., 2015). Given atrial LCC current amplitude likely depends on TATS density as shown in rat ventricular myocyte experiments (Frisk et al., 2014), TATS loss in chronic AF might contribute to the observed reduction in current density. Recently, in human atrial myocytes from both normal sinus rhythm and chronic AF patients, RyR and the sarcoplasmic reticulum (SR) Ca2+ buffer calsequestrin (CSQ) were found more densely expressed near the cell periphery vs. the cell interior (Herraiz-Martínez et al., 2022). This is similar to what has previously been shown in rat atrial myocytes, where RyR-CSQ colocalization was reduced in the cell interior vs. periphery (Schulson et al., 2011). To our knowledge, NCX localization in human atrial myocytes has not yet been investigated, and while NCX is expressed at both the cell surface and in the TATS membrane, the precise quantitative distribution may vary in different species and cardiac regions (Melnyk et al., 2005; Scriven et al., 2010; Schulson et al., 2011).
The available data on ion channels and Ca2+ handling protein expression, localization, and function in human atrial myocytes is limited and may not be representative of the heterogeneous human atrial myocyte population in tissue. There are known regional differences across the atria, e.g., in right vs. left atrium (Arora et al., 2017), pulmonary vein vs. free atrial wall (Melnyk et al., 2005), and insight from isolated cells is also complicated by TATS damage from the enzymatic digestion (Chen et al., 2015). Furthermore, it remains unclear whether ionic and ultrastructural remodeling interacts to affect arrhythmogenic propensity. Indeed, because Ca2+ handling protein expression, localization, and regulatory states change simultaneously with TATs in disease (Brandenburg et al., 2016; Yue et al., 2017), their effects cannot easily be separated in experiments. To address this, we utilized our new 3D model of the human atrial myocyte with spatially detailed Ca2+ diffusion and TATS (Zhang et al.) to investigate the isolated and interactive effects of altered TATS and changes in expression and localization of key Ca2+-handling proteins (i.e., NCX, RyR, and CSQ) on Ca2+-driven membrane instabilities. We focused on these proteins because both 1) NCX and RyR are altered in AF and emerged as key mediators of TATS loss-induced diastolic instabilities in Ca2+ and membrane voltage (Vm), and 2) RyR and its regulator CSQ have been found to distribute non-uniformly between the cell periphery and interior. In this study, we found that TATS loss and Ca2+ handling protein remodeling collectively promote arrhythmia. Cells with intermediate TATS density were most sensitive to changes in protein expression and localization, whereby ultrastructural and Ca2+ handling remodeling synergistically contributed to the proarrhythmic outcomes. Conversely, when the TATS is depleted, changes in protein expression and distribution have little effect. Our study demonstrates the interactive contributions of TATS and Ca2+-handling protein expression and distribution on maintaining Ca2+ and membrane potential stability in human atrial myocytes and provides novel model-based mechanistic insight that may guide future therapeutic anti-AF strategies.
Methods
We simulated human atrial myocyte electrophysiology and Ca2+ handling using our recently developed model, integrating transmembrane voltage dynamics, spatially-detailed Ca2+ diffusion, and varying TATS structure (Zhang et al.). We modified the expression and subcellular distribution of various Ca2+ handling proteins (i.e., namely NCX, RyR, and CSQ), as detailed below. To investigate the interactive effects of altering protein expression and localization and varying tubular structures, we conducted simulations in cells with dense, median, and sparse TATS taken from the population of TATS structures described in our companion paper (Zhang et al.).
Varying subcellular distribution of NCX, RyR, and CSQ
To investigate how the heterogeneous subcellular localization of NCX, RyR, and CSQ impact electrophysiology and Ca2+ handling, we systematically varied the relative density of NCX, RyR or CSQ localized to surface Ca2+ release units (CRUs, i.e., the 2 outer CRU layers near cell surface) vs. those localized to inner (i.e., non-surface) CRUs while keeping the whole-cell total expression unchanged (Fig. 1). Specifically, we varied the ratio of surface/inner CRU protein density between 0.5 and 2 to reflect the surface-to-central gradient in the subcellular distribution of RyR and CSQ observed in experiments (Herraiz-Martínez et al., 2022).
Figure 1 – Varying Ca2+ handling proteins (Na+-Ca2+ exchanger, NCX, Ryanodine receptor, RyR, and calsequestrin, CSQ) distribution.
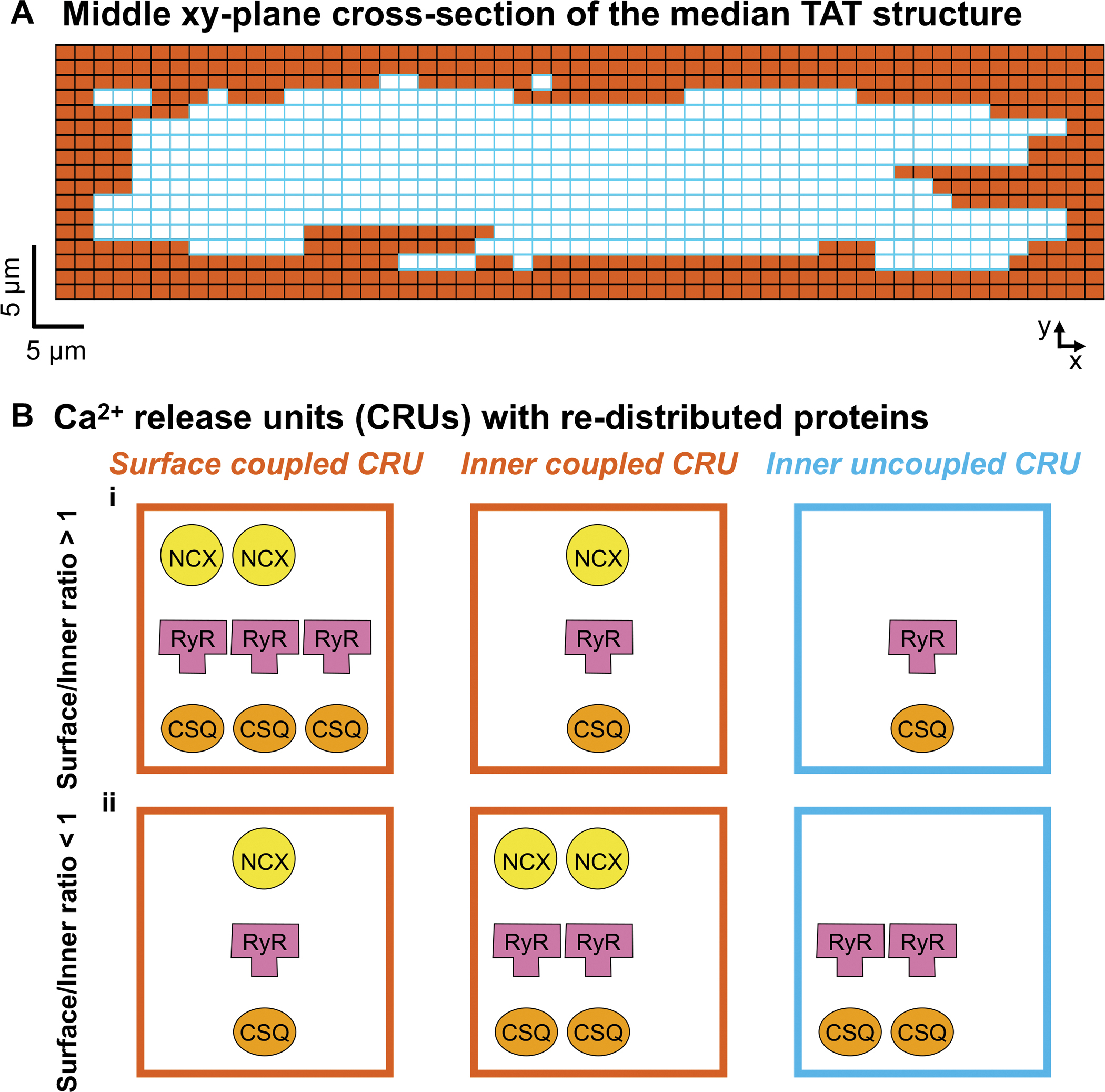
A) Two-dimensional section of a middle XY plane in the intermediate TAT structure shown in the companion paper (Zhang et al.). Surface coupled CRUs and inner coupled CRUs are in orange, and uncoupled CRUs are in light blue. B) Schematic of surface coupled (left), central coupled (middle), and uncoupled (right) CRUs when surface/inner CRU protein expression ratio > 1 (i) or < 1 (ii). When varying the NCX (yellow) distribution, we scale the NCX current density in inner coupled and surface coupled CRUs accordingly. When varying the RyR distribution, we scale the relative expression of RyRs (pink) in the inner (coupled and uncoupled) CRUs vs. surface coupled CRUs. When varying the CSQ (orange) distribution, we scale the relative CSQ expression in the inner (coupled and uncoupled) CRUs vs. surface coupled CRUs.
Varying the whole-cell expression of NCX, RyR, and CSQ
The whole-cell expression (i.e., NCX maximal transport rate, number of RyRs per CRU, and CSQ maximum buffering capacity, BCSQ) was scaled to explore the effects of experimentally-determined variation ranges of NCX (Schotten et al., 2002; El-Armouche et al., 2006; Lenaerts et al., 2009; Voigt et al., 2012; Yue et al., 2017), RyR (Ohkusa et al., 1999; Neef et al., 2010; Voigt et al., 2014; Greiser et al., 2014), and CSQ (Lüss et al., 1999; Faggioni et al., 2014). Specifically, NCX was varied between 10% and 200%, RyR was varied between 10% and 140%, and CSQ was varied between 50% and 140% of the baseline expression outlined in Zhang et al.
Since CSQ is not only a luminal SR Ca2+ buffer but also a regulator of RyR function (Terentyev et al., 2003; Györke et al., 2004; Knollmann et al., 2006; Restrepo et al., 2008), to dissect the relative and combined contributions of these processes we investigated three scenarios when performing simulations to study the impacts of varying CSQ: 1) only luminal SR Ca2+ buffering is affected; 2) only RyR regulation is affected; or 3) both SR buffering and RyR regulation are affected. To assess this, changes in Ca2+ buffering CSQ were implemented by modifying the BCSQ in the equations of junctional SR (JSR) Ca2+ rapid equilibrium approximation, i.e., β([Ca2+]JSR) in the companion paper by Zhang et al; whereas changes in RyR gating were implemented by modifying the BCSQ in RyR luminal-Ca2+-dependent transient rate equations, i.e., k14 and k23 in Table 5 of the companion paper by Zhang et al.
Numerical methods and simulation protocols
Simulation details are as in the companion paper (Zhang et al.). Briefly, our human atrial myocyte model was implemented in C++ and parallelized using OpenMP 5.1 (Dagum & Menon, 1998). The ordinary differential equations (ODEs) and subcellular Ca2+ diffusion were solved using an explicit Forward Euler method except that LCC current (ICa) and RyR gating behaviors were described stochastically as in (Restrepo et al., 2008; Sato & Bers, 2011), and that ODEs of fast Na+ current (INa) were solved using the Rush-Larsen scheme (Rush & Larsen, 1978). The time step was 0.01 ms.
Measurement of Ca2+-handling abnormalities, Vm instabilities and differences in Ca2+ release between CRUs
To compare Ca2+ signaling in surface CRUs, inner coupled CRUs (those CRUs located away from the cell surface but coupled to tubules), and inner uncoupled CRUs, the [Ca2+]Cyto within each group of CRUs throughout the cell was averaged. Ca2+-handling abnormalities and Vm instabilities associated with changes in protein expression and distribution were examined for SCRs, DADs, and spontaneous action potentials (SAPs). Detailed descriptions of SCR and DAD measurements are provided in our companion paper (Zhang et al.). Briefly, cells were paced to steady state with the stimulation then stopped and the subsequent diastolic period used for analysis in MATLAB R2021. SCRs were defined as Ca2+-release events with an amplitude > 0.3 μM, with DADs defined as membrane depolarizations > 10 mV. SAPs were deemed present when the amplitude of a DAD exceeded 70 mV.
Measurements of alternans
To understand the effects of varying expression and distribution of Ca2+ handling proteins on the regularity of Ca2+ release, the occurrence of pacing-induced alternans in [Ca2+]Cyto and Vm traces were analyzed as done in experiments (Myles et al., 2011; Hammer et al., 2015). Specifically, Ca2+ alternans were distinguished when the calculated index (r) that compares the alternating averaged smaller (S) and larger (L) Ca2+ transient amplitudes (i.e., r = 1 – S/L) was over 0.08. APD alternans were identified when the difference between the averaged longer APD90 and the averaged shorter APD90 (ΔAPD90) was over 10 ms.
Results
Lowering NCX promotes SCRs, but has biphasic effects on DADs and SAPs, depending on the balance between increased SCRs and reduced ΔVm/ΔCa2+ gain
To reveal how NCX expression and TATS changes affect arrhythmic biomarkers (i.e., SCRs, DADs, and SAPs), we varied NCX expression in the human atrial myocyte models with sparse, intermediate, and dense TATS. We found that the SCR rate threshold monotonically decreases with reduced NCX expression and increased NCX associated with fewer SCRs, especially in cells with sparse TATS (Fig. 2Ai). While low NCX expression is associated with decreased SCR rate threshold, it conversely increases the rate thresholds of both DADs and SAPs when below 20%–40% of its baseline value. Interestingly, at low expression levels above 40% of baseline, DAD and SAP rate threshold is decreased matching SCR-NCX expression dependence (Fig. 2Aii–iii). This is demonstrated in the representative voltage (i) and [Ca2+]Cyto (ii) traces in (Fig. 2B) where the biphasic effects of NCX reduction on DADs and SAPs (but not SCRs) are highlighted by the occurrence of SCRs at 0.2 and 0.5 NCX expression but DADs and SAPs solely occurring at 0.5. This result suggests that [Ca2+]Cyto-Vm coupling changes with low vs. high NCX expression. Interestingly, while the SCR pacing threshold is lower in cells with sparser TATS, SAPs occur at lower pacing rates in cells with intermediate and dense TATS, suggesting that NCX expression changes have the greatest impact on [Ca2+]Cyto-Vm coupling in cells with relatively intact TATS. We further analyzed the model to understand the mechanism by which reducing NCX promotes SCRs but has biphasic effects on DADs and SAPs. We found that lowering NCX expression reduced NCX Ca2+ extrusion (Fig. 2Ci), leading to elevated diastolic cleft Ca2+ concentration, augmented diastolic RyR open probability (PO) and leak, and increased SCRs and DADs. This mechanism resembles that suggested to be responsible for elevated SCR and DAD propensity in cells with sparse TATS in our companion paper (Zhang et al.). Conversely, less NCX reduced SCR-induced ΔVm, especially in cells with intermediate and dense TATS (Fig. 2Cii), leading to decoupling between SCR and DAD/SAPs and subsequent biphasic changes of DADs and SAPs.
Figure 2 – Inhibition of NCX promotes SCRs but has biphasic effects on DADs and spontaneous APs (SAP).
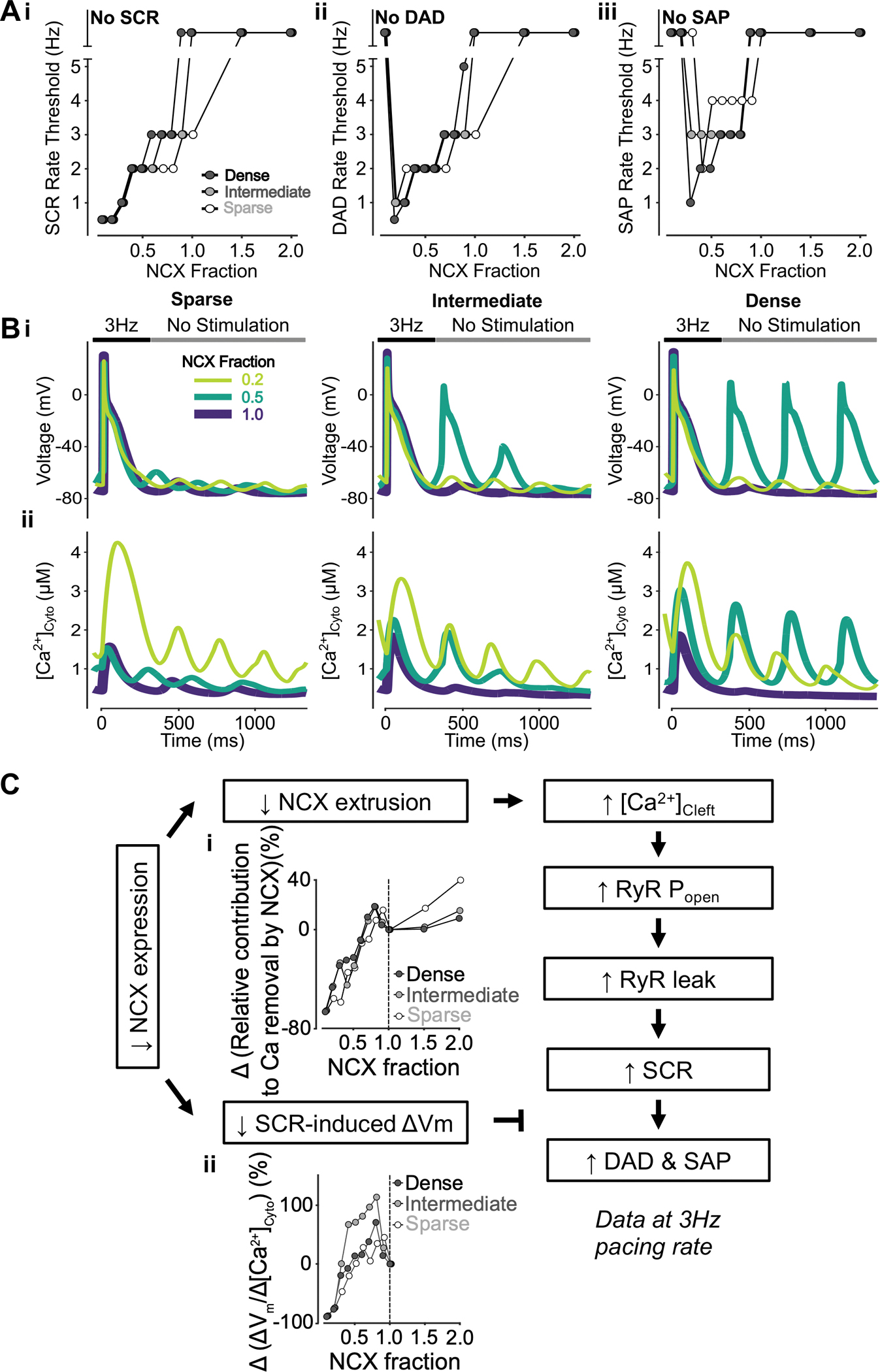
A) Reducing NCX monotonically decreases the rate threshold of SCRs (i), whereas the rate thresholds of DADs (ii) and SAP, defined as DADs with amplitudes over 70 mV, (iii) display biphasic dependence. B) Effect of altered NCX fraction on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs, SAPs and SCRs. C) Mechanism underlying NCX inhibition promoting SCRs but having biphasic effects on DADs and SAP. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained NCX fraction of 1.0. Lower NCX expression is associated with reduced NCX contribution to Ca2+ extrusion (i) and smaller SCR-induced voltage changes/SCR amplitude ratio (ii). Less NCX extrusion results in elevated cleft Ca2+ concentration, augmented RyR Po and leak leading to increased SCRs. While SCRs lead to DADs and SAP this transition is abated by the reduced SCR-induced changes in Vm, thus explaining the biphasic effects of reduced NCX.
We further sought to determine whether regional variations exist in the effects of varying NCX expression and TATS density on the pacing threshold for SCR. To do so, we analyzed the averaged Ca2+ transient of surface CRUs, inner CRUs coupled to TATS (inner coupled), and inner CRUs that are not coupled to TATS (inner uncoupled) in each cell in the population with varying NCX expression and TATS density and quantified the biomarkers of local Ca2+ transient. As expected, reduced NCX lowered the rate threshold for SCRs in all CRUs (Fig. 3Ai). The representative traces of average cytosolic Ca2+ concentration following pacing at 3 Hz in cells with dense (Fig. 3Bi), intermediate (Fig. 3Bii), and sparse TATS (Fig. 3Biii) also show larger SCRs with lower NCX expression. Interestingly, when NCX was severely reduced (scaling factor of 0.2 of baseline expression), Ca2+ alternans were observed in all CRUs from cells with intermediate and dense TATS (Fig. 3Bi–ii) indicating enhanced arrhythmic effect of lower NCX expression in tubulated cells. Overall, our simulations suggest that NCX inhibition shifts the balance between increased SCRs and reduced ΔVm/ΔCa2+ gain. While cells with sparse TATS consistently display a lower SCR pacing threshold, when SCRs occur in more densely tubulated cells they are greater in magnitude and the SAP pacing threshold appears more sensitive to variations in NCX levels compared to sparsely tubulated cells.
Figure 3 – Inhibition of NCX promotes simultaneous SCRs in all CRUs.
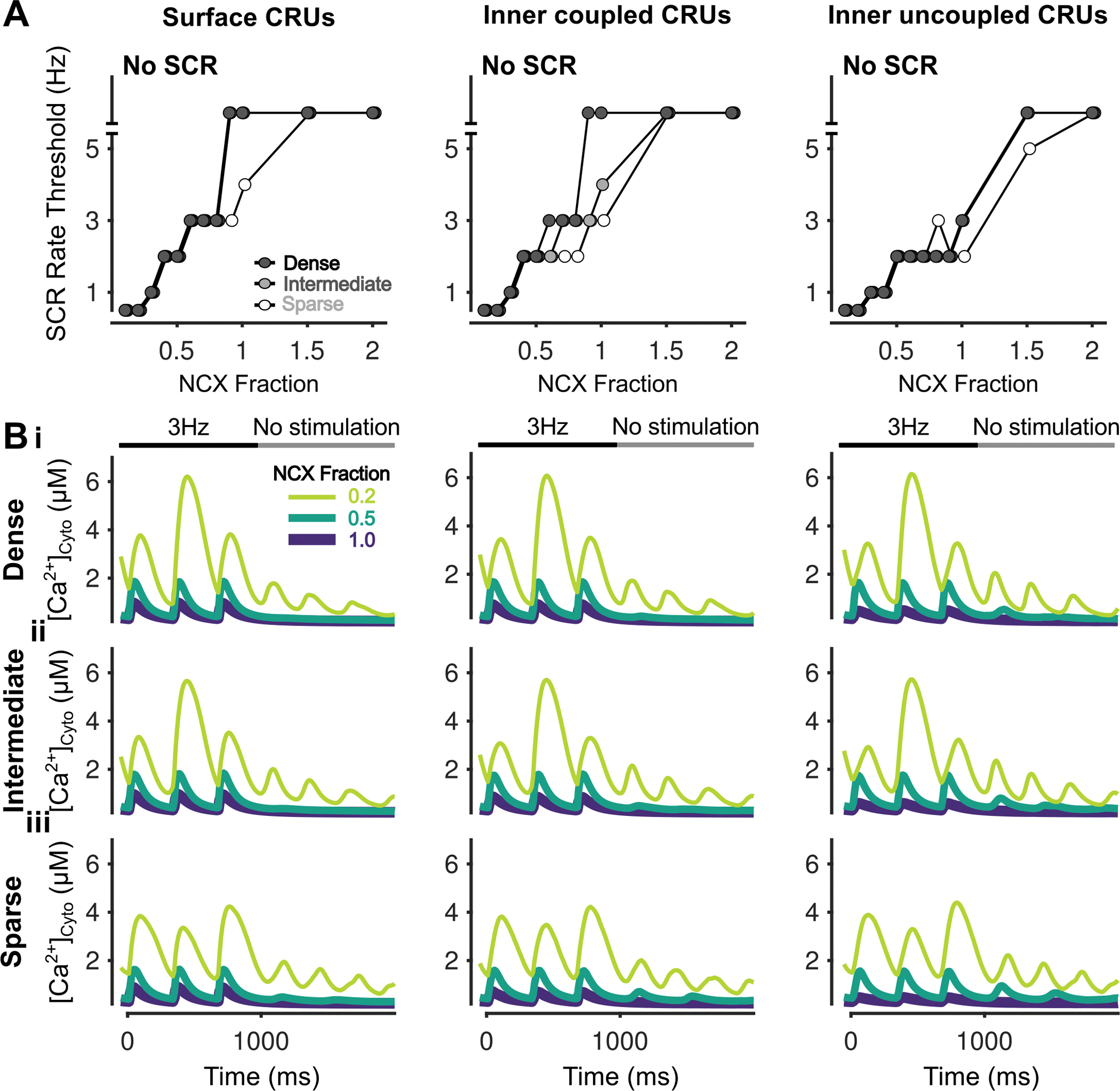
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in dense, intermediate and sparsely tubulated cells. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing SCRs and alternans for all CRUs at NCX Fraction of 0.2.
Increasing surface/inner CRU NCX expression ratio promotes SCRs and DADs by elevating inner [Ca2+]Cleft and RyR leak
After characterizing the impact of changes to global NCX expression on SCRs, DAD, and SAPs, we sought to investigate the effects of varying the spatial distribution of NCX and TATS density. To do this we altered the ratio of NCX in surface/inner CRUs in the human atrial myocyte models with sparse, intermediate, and dense TATS. Our simulations indicate that increasing the ratio of surface to inner CRU NCX expression decreases the rate threshold of SCRs (Fig. 4Ai) and DADs (Fig. 4Aii), without triggering SAPs (Fig. 4Aiii). This effect is more pronounced in cells with dense and intermediate TATS (Fig. 4Ai–ii), as also shown in the representative traces of Vm (Fig. 4Bi) and [Ca2+]Cyto (Fig. 2Bii). We found that increasing the surface-to-inner CRU NCX expression ratio decreased inner NCX Ca2+ extrusion but enhanced NCX Ca2+ extrusion at the surface. While [Ca2+]Cleft in surface CRUs remained unaffected (Fig. 4Ci), lower NCX expression in the inner CRUs lead to increased [Ca2+]Cleft in both inner coupled (Fig. 4Cii) and uncoupled (Fig. 4Ciii) CRUs, thus causing elevated RyR PO and leak, which promotes SCRs and DADs. The consequences of heterogeneous NCX distribution were especially pronounced in inner coupled CRUs in cells with dense and intermediate TATS (Fig. 5A–B). Overall, we found that increasing the surface/inner CRU NCX expression ratio promotes SCRs, especially in inner coupled CRUs, and DADs, but does not affect SAPs. These effects are marked in cells with intermediate and dense TATS, suggesting that loss of TATS and reduced NCX expression in inner CRUs favors SCRs and DADs via a similar mechanism, i.e., the elevation of inner [Ca2+]Cleft and RyR leak.
Figure 4 – Increasing surface/inner CRU NCX expression ratio inhibits SCRs and DADs but does not affect SAPs.
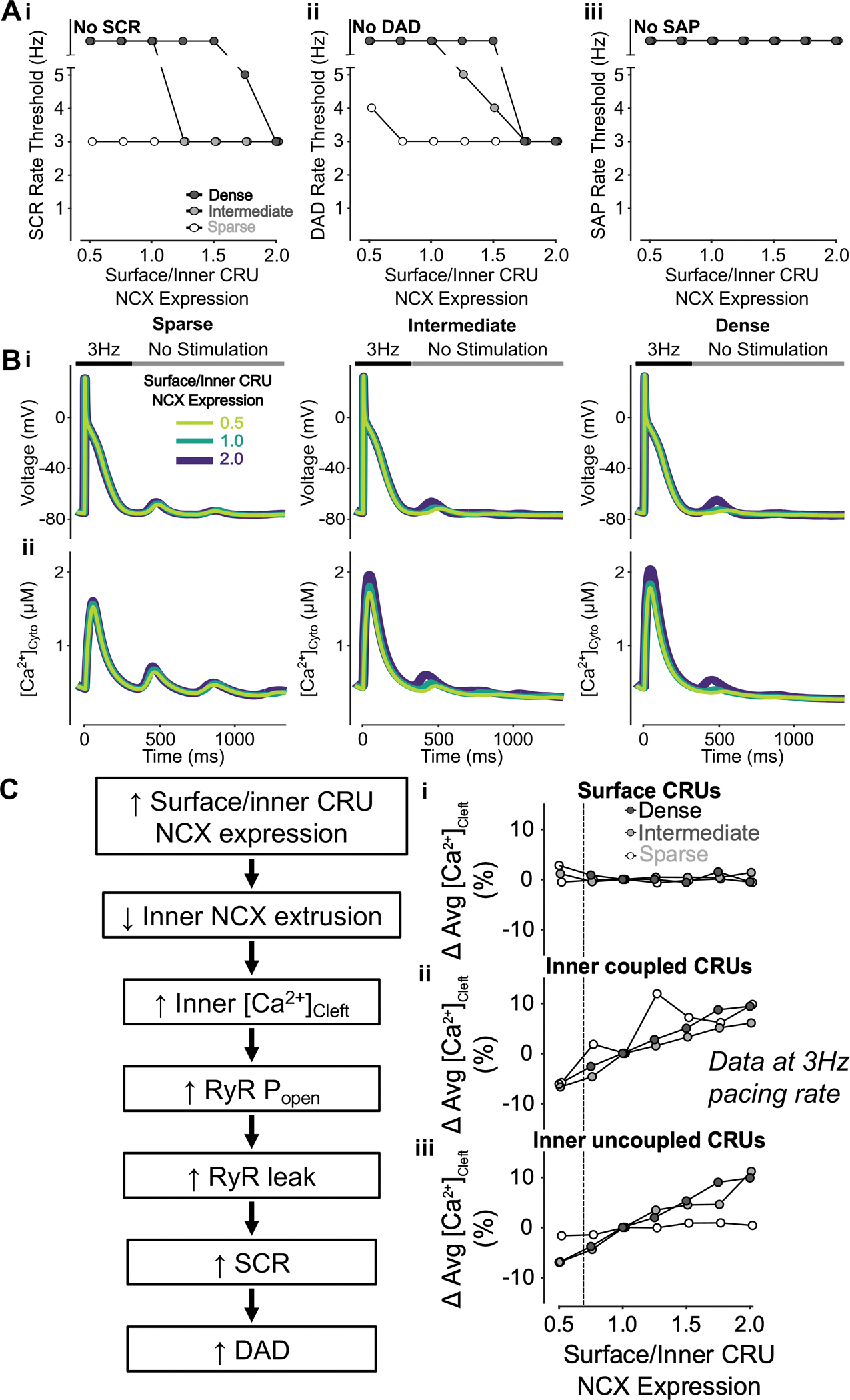
A) Increasing surface/inner CRU NCX expression ratio (i.e., increasing NCX density in surface CRUs without changing whole-cell NCX expression) monotonically decreases the rate threshold of SCRs (i) and DADs (ii), whereas SAP remains absent in all conditions. This effect is greater in cells with dense and intermediate tubular structures vs cells with sparse tubules. B) Effect of altered surface/inner CRU NCX expression ratio on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs. C) Mechanism underlying increasing surface/inner CRU NCX expression ratio inhibiting SCRs and DADs. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained surface/inner CRU NCX expression ratio of 1.0. Higher surface/inner CRU NCX expression ratio is associated with reduced NCX contribution to Ca2+ extrusion by inner coupled CRUs, resulting in higher cleft Ca2+ concentration in inner CRUs with no visible effect in surface CRUs. Higher inner cleft Ca2+ concentration in inner CRUs causes increased RyR Po and RyR leak, leading to increased SCRs and DADs. Since fewer inner CRUs containing NCX exist in sparsely tubulated cells to begin with, the impact of varying NCX distribution is more pronounced in cells with tubules. As such, the consequence of changing cleft Ca2+ concentration is greater in cells with dense and intermediate tubules rather than those with sparse tubules, explaining the observed differences in SCRs and DADs between cells with varying tubule densities.
Figure 5 – Increasing surface/inner CRU NCX expression ratio inhibits simultaneous SCRs in inner coupled CRUs.
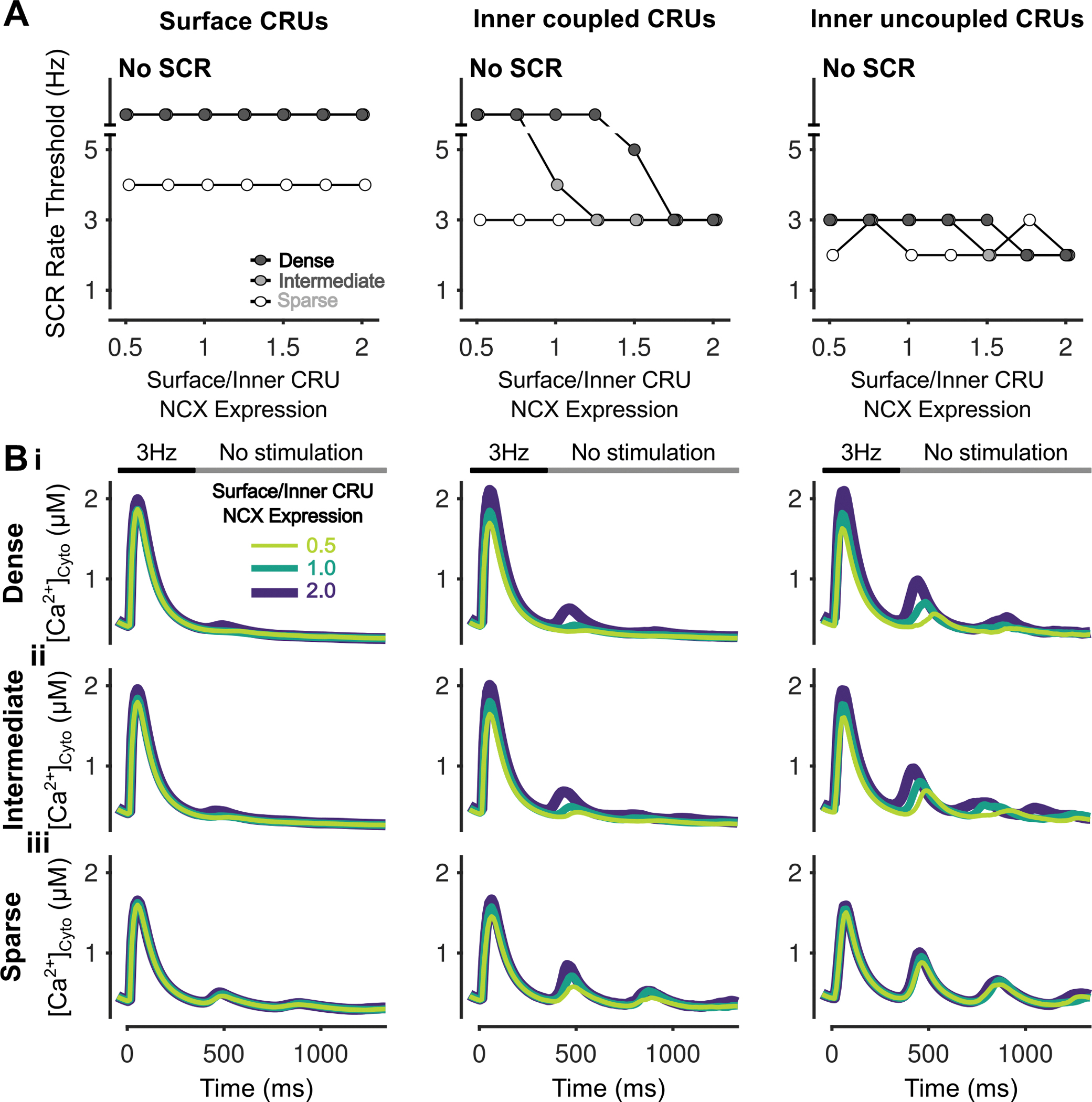
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered surface/inner NCX expression ratio greatest in inner coupled CRUs of cells with dense and intermediate tubules. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying surface/inner CRU NCX expression ratios.
Inhibiting RyR has biphasic effects on SCRs, DADs, and SAPs, depending on the balance between the reduced number of RyRs and increased PO
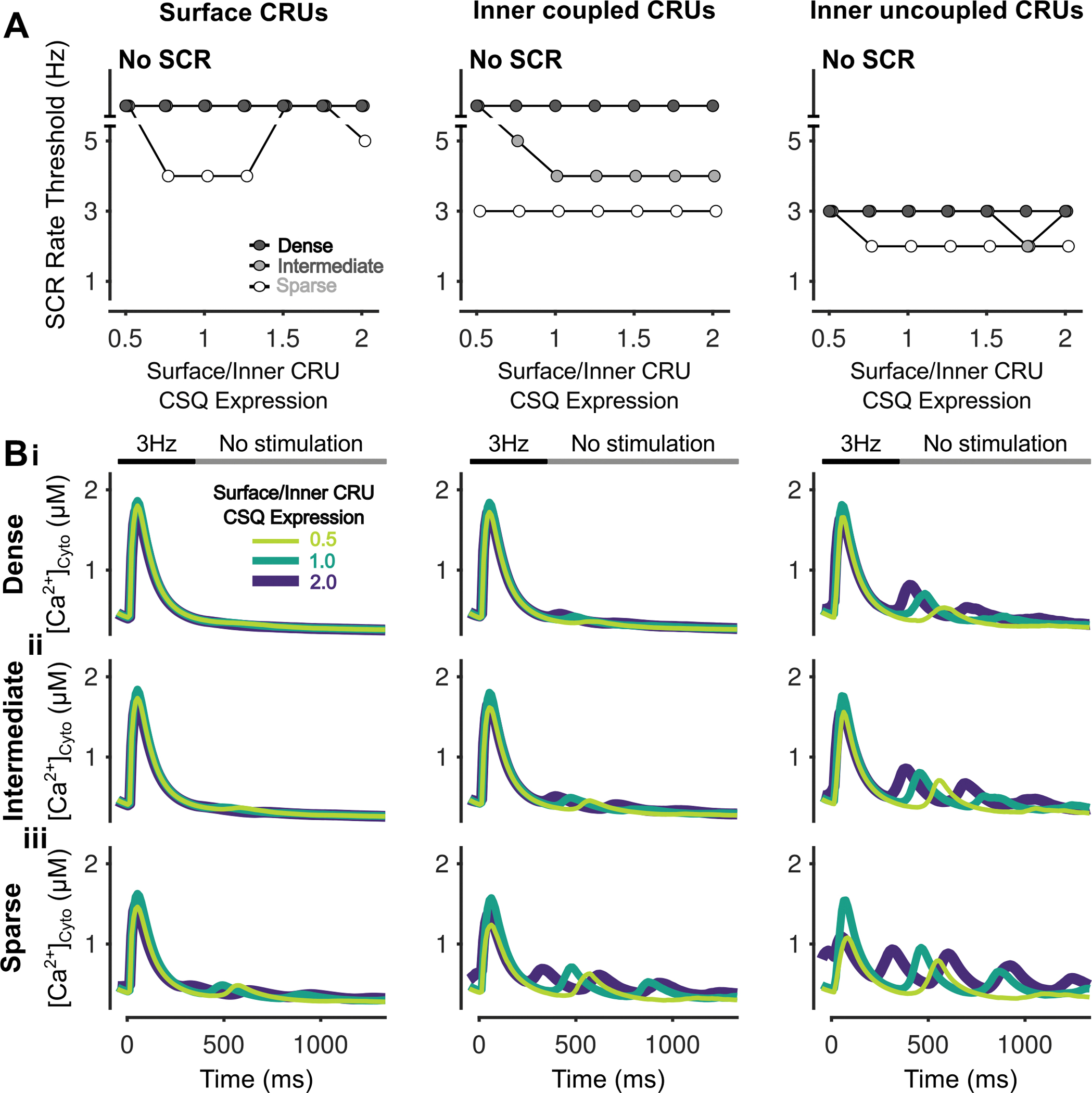
To understand the effects of varying RyR expression and TATS density on SCRs, DADs, and SAPs, we altered RyR expression in cells with sparse, intermediate, and dense TATS as described in the Methods. Slight lowering of RyR expression above 70% of its baseline value was proarrhythmic and reduced the rate thresholds of SCRs, DADs, and SAPs. RyR expression below 40–70% typically had the opposite effect with rate threshold increased (Fig. 6Ai–iii). Interestingly, in the cell with sparse TATs the SCR rate threshold was unaffected by varying RyR expression levels, despite the ability of SCRs to generate DADs and SAPs being altered in these cells. Indeed, DADs and SAPs occur at overall lower pacing rates in cells with sparse TATS vs. those with intermediate or dense TATS, suggesting that TATS loss further promotes arrhythmogenic behavior (due to larger SCRs, especially in inner uncoupled CRUs, Fig. 7B). The biphasic effects of RyR inhibition on SCRs, DADs, and SAPs are shown in representative traces of voltage (Fig. 6Bi) and [Ca2+]Cyto (Fig. 6Bii) following 4-Hz pacing. Our model analysis indicates that the biphasic effects of RyR inhibition are mediated by the balance between two contrasting processes: on one hand, reduction in RyR expression reduces SR Ca2+ release and increases the SR Ca2+ load (Fig. 6Ci), which elevates unitary RyR PO (Fig. 6Cii); on the other hand, the reduced RyR number limits the magnitude of diastolic Ca2+ release by affecting the number of unitary release events and thus their ability to recruit neighboring RyRs and CRUs. As with the impact on global Ca2+, subcellular Ca2+ signaling also shows biphasic changes in SCR rate thresholds, which are most remarkable in surface CRUs of the cell with sparse TATS and in inner coupled CRUs of cells with intermediate and dense TATS (Fig. 7A). Interestingly, the rate threshold of inner coupled SCRs is similar to the whole cell SCR rate threshold (Fig. 6Ai), suggesting that SCRs in inner coupled CRUs dominantly affect whole cell SCR behavior. Furthermore, the rate threshold of surface SCRs (Fig. 7A) is similar to the rate thresholds of DADs and SAPs (Fig. 6Aii–iii), suggesting that surface CRU SCRs are more likely associated with the occurrence of DADs/SAPs.
Figure 6 – Inhibition of RyR has biphasic effects on SCRs, DADs, and SAPs.
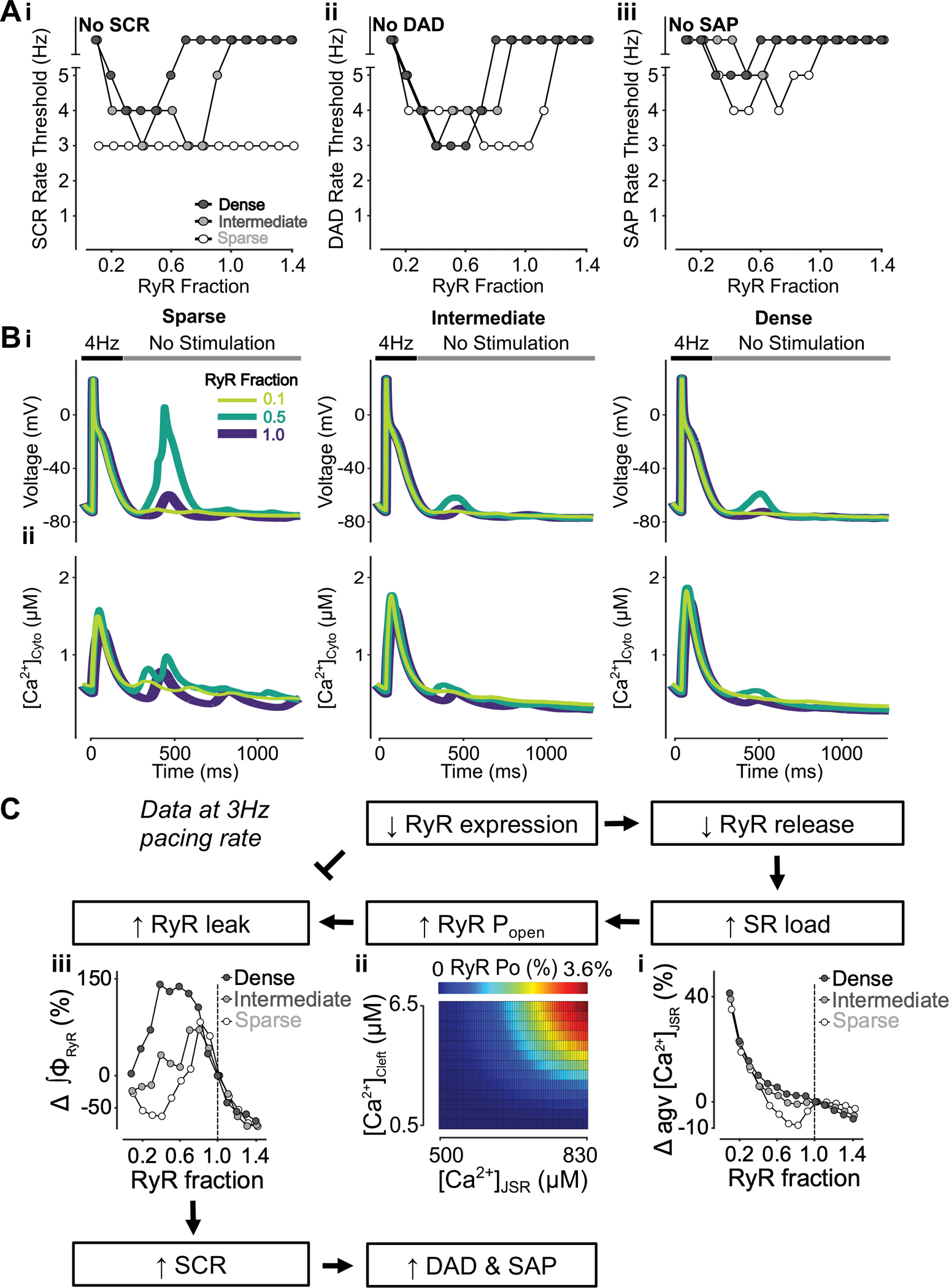
A) Rate threshold for SCRs (i), DADs (ii) and SAP (iii) in dense, intermediate and sparsely tubulated cells. Biphasic dependence on RyR inhibition is observed in all settings and cell types, with the exception of SCRs in sparsely tubulated cells. Here, the lowest observed baseline rate threshold for SCRs is unaltered by RyR inhibition. B) Effect of altered RyR fraction on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 4 Hz to examine the occurrence of DADs, SAPs and SCRs. C) Mechanism underlying RyR inhibition having biphasic effects on SCRs, DADs and SAP. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained RyR fraction of 1.0. Lower RyR expression is associated with reduced RyR release, leading to elevated SR load (i) and augmented RyR Po (ii). While higher RyR Po causes RyR leak leading to increased SCRs, lower RyR expression itself inhibits RyR leak. This results in the biphasic effect of RyR reduction on SCRs, DADs and SAPs.
Figure 7 – Inhibition of RyR has biphasic effects on SCRs in all CRUs.
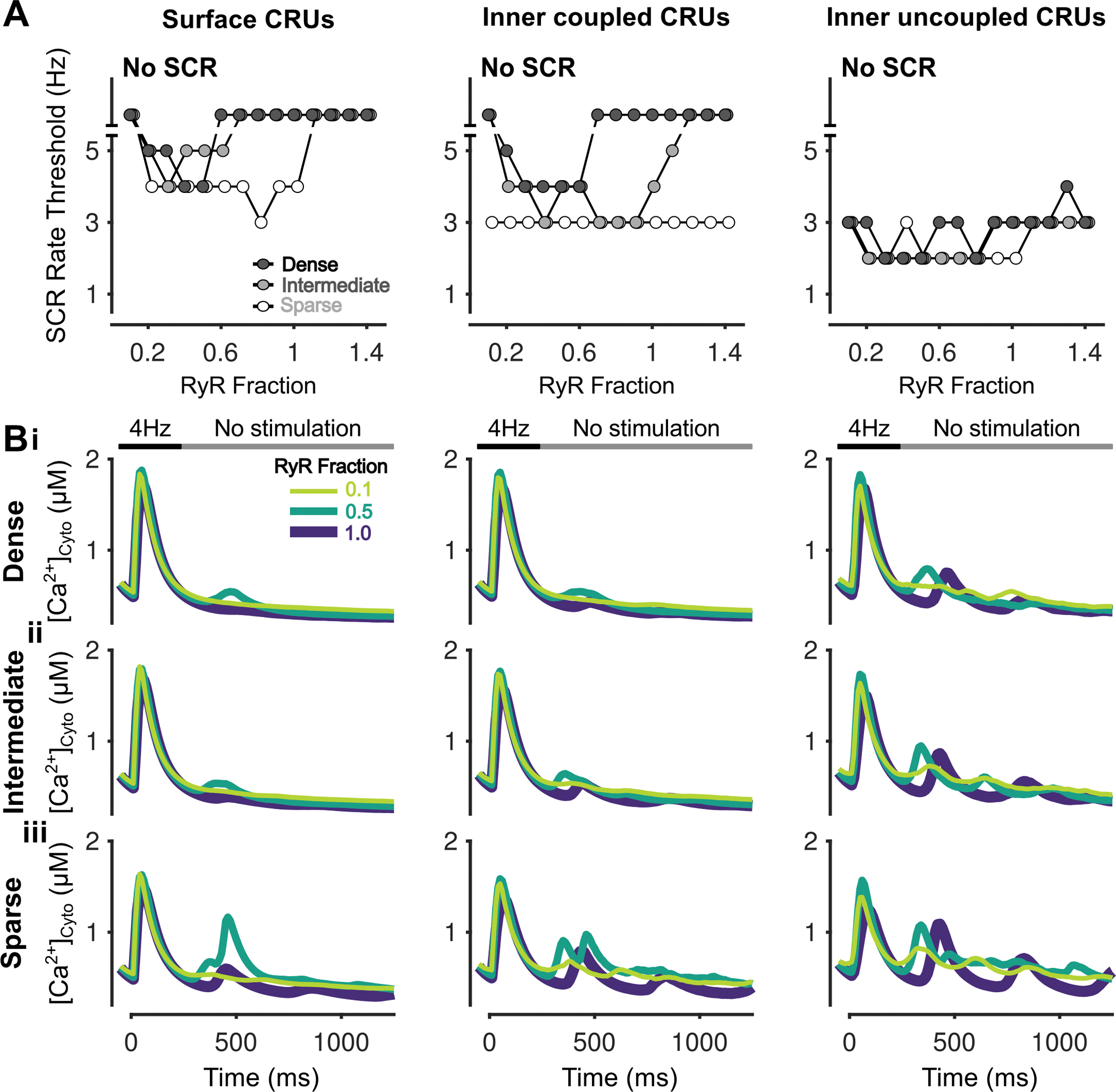
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered RyR expression ratio greatest in surface CRUs of all cells and inner coupled CRUs of cells with dense and intermediate tubules. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 4 Hz showing observed SCRs with varying RyR fraction.
Increasing RyR expression in surface vs. inner CRUs inhibits SCRs but has modest effects on DADs and SAPs
To reveal the effects of heterogenous CRU RyR localization and TATS on SCRs, DADs, and SAPs, we varied the surface-to-inner CRU RyR expression ratio in cells with sparse, intermediate, and dense TATS. The simulation results indicate that increasing the surface/inner CRU RyR expression ratio inhibits SCRs in the cell with sparse and intermediate TATS, with no changes observed in the densely tubulated cell (Fig. 8Ai). However, modest changes were seen in the DAD threshold and only for the most extreme changes (e.g., ratios of 0.5 and 1.75 – 2.0, Fig. 8Aii), while no changes were detected in the SAP rate threshold (Fig. 8Aiii). Both increasing and decreasing the RyR expression in surface vs. inner CRUs diminished DADs (i.e., reduced amplitude and increased pacing threshold) in the cell with sparse TATS, but enhanced DADs in the cell with intermediate TATS, as demonstrated in the representative traces (Fig. 8Bi). The representative traces of [Ca2+]Cyto also show that SCRs are larger in cells with sparse/intermediate vs. dense TATS (Fig. 8Bii) with varying surface/inner CRU RyR expression, as we show in the companion paper (Zhang et al.). The mechanistic analysis indicates increasing surface/inner CRU RyR expression enhances NCX-RyR coupling, thus leading to both larger NCX Ca2+ extrusion (Fig. 8Ci), with subsequent diminished SCRs that would attenuate DADs, and enhanced ΔVm/ΔCa2+ gain that would favor DADs (Fig. 8Cii). The balance of these two competing processes underlies the modest effects of varying RyR relative distribution in surface and inner CRUs on DADs. The analysis of subcellular Ca2+ signaling indicates that increasing surface RyR expression increases the SCR rate threshold in all CRUs, especially in inner CRUs, and that TATS loss exacerbates the effects of reducing RyR expression in inner CRUs (Fig. 9A–B).
Figure 8 – Increasing surface/inner CRU RyR expression ratio inhibits SCRs, has biphasic effects on DADs but does not affect SAP.
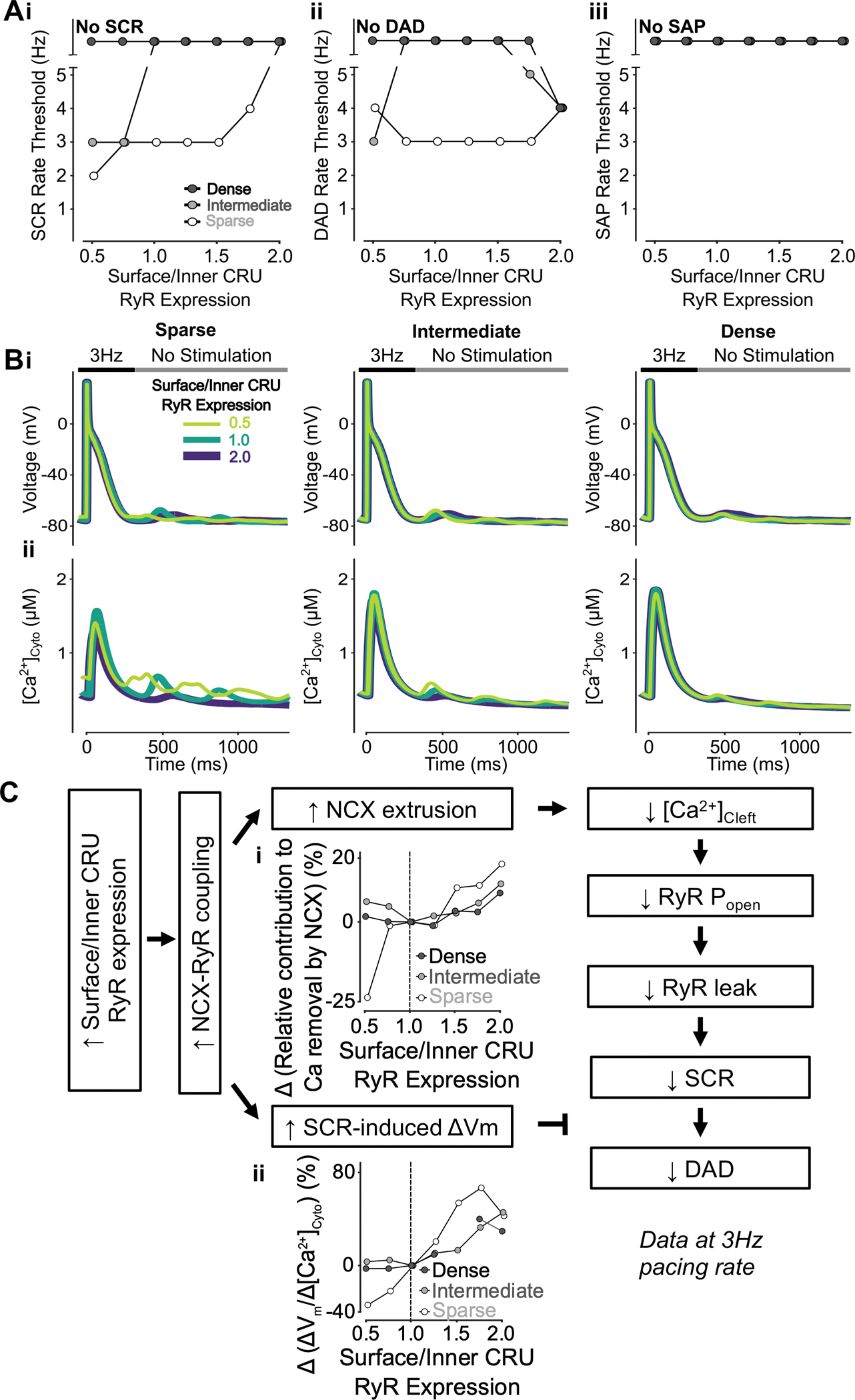
A) Increasing surface/inner CRU RyR expression ratio (i.e., increasing RyR density in surface CRUs without changing whole-cell RyR expression) monotonically increases the rate threshold of SCRs (i), whereas DADs (ii) display biphasic dependence and SAP (iii) remains absent in all conditions. The effect on SCRs is greater in cells with sparse and intermediate tubular structures vs cells with dense tubules, whereas the biphasic effect on DADs is less pronounced in cells with sparse tubular structures vs cells with intermediate and dense tubules. B) Effect of altered surface/inner CRU RyR expression ratio on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs. C) Mechanism underlying increasing surface/inner CRU RyR expression ratio inhibiting SCRs, having biphasic effects on DADs but not affecting SAP. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained surface/inner CRU RyR expression ratio of 1.0. Increasing surface/inner CRU RyR expression ratio means more RyRs are located closer to NCX, resulting in increased NCX-RyR coupling. This is associated with increased NCX contribution to Ca2+ extrusion (i) and higher SCR-induced voltage changes/SCR amplitude ratio (ii). Enhanced NCX extrusion results in lower cleft Ca2+ concentration, smaller RyR Po and leak leading to milder SCRs. While this in itself limits DADs, DAD likelihood is promoted by the increased SCR-induced changes in Vm, thus explaining the biphasic effects of increasing surface/inner CRU RyR expression ratio.
Figure 9 – Increasing surface/inner CRU RyR expression ratio inhibits simultaneous SCRs in all CRUs.
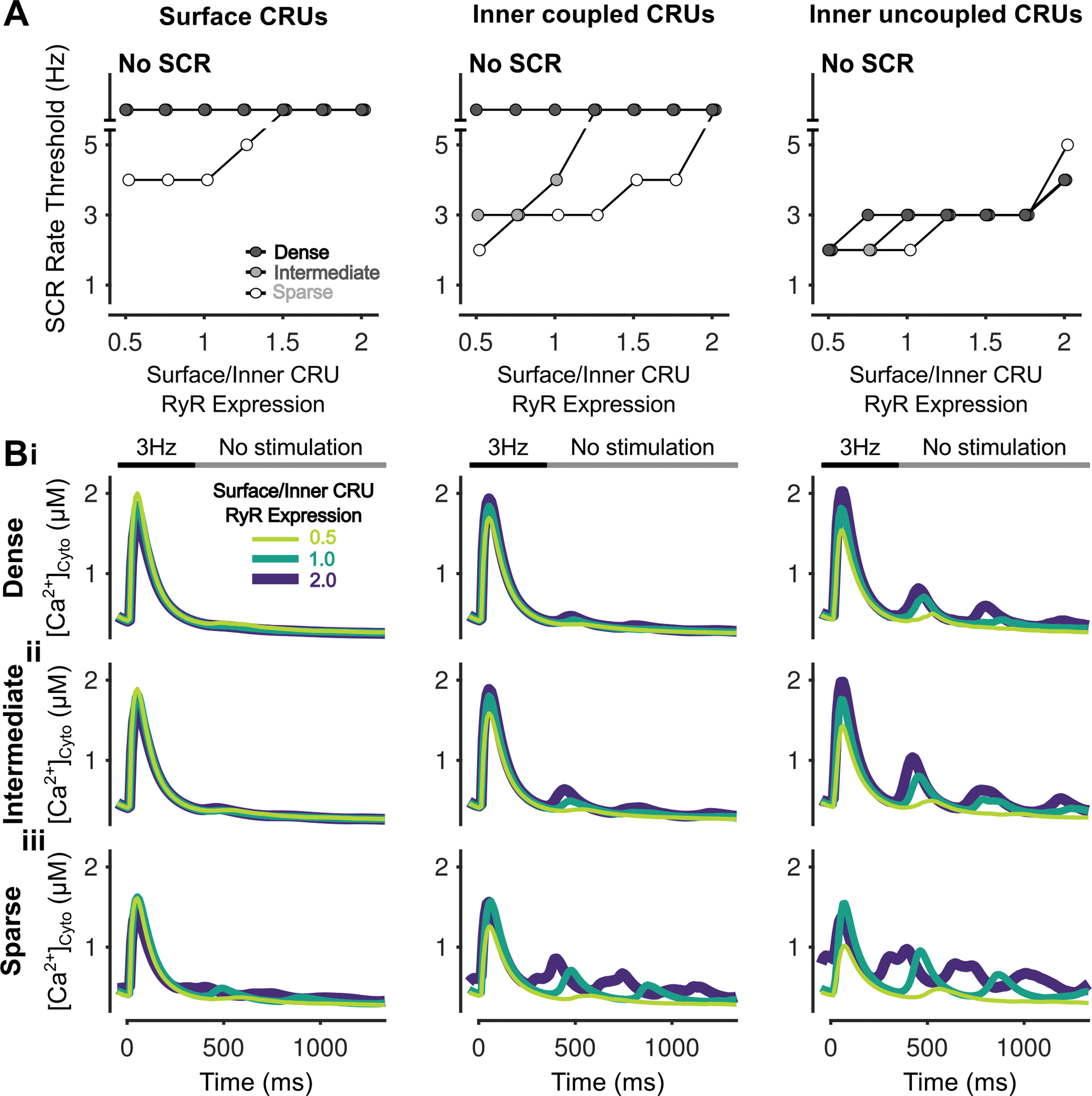
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered surface/inner RyR expression ratio greatest in inner coupled CRUs of cells with sparse and intermediate tubules. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying surface/inner CRU RyR expression ratios.
Loss of CSQ promotes SCRs and DADs, primarily through diminished CSQ Ca2+ buffering, with no effect on SAPs
The impact of CSQ expression levels and TATS density on SCRs, DADs, and SAPs was examined by varying the expression of CSQ in cells with sparse, intermediate, and dense TATS. Because CSQ both affects luminal Ca2+ buffering and RyR PO, we simulated the changes in CSQ expression level by 1) only changing the Ca2+ buffering parameters (Figs. 10–11), 2) only modifying the parameters associated with CSQ regulation of RyR gating (Figs. 11–12), and 3) modulating both processes (Figs. 13–14), as described in the Methods. Reducing CSQ-mediated luminal SR Ca2+ buffering promotes SCRs, DADs, and SAPs (Figs. 10A–B), and enhances subcellular SCRs in all CRUs (Fig. 11). However, inhibiting CSQ RyR-regulation, i.e., decreasing SR Ca2+ sensitivity of RyR PO, conversely increases pacing rate thresholds of SCRs, DADs, and SAPs, especially in cells with sparse TATS (Fig. 12A–B). Inhibiting CSQ RyR-regulation also reduces subcellular SCRs in all CRUs, especially in inner CRUs (Fig. 13A–B). When concomitantly simulating the changes in both luminal Ca2+ buffering and RyR regulation, we found that CSQ inhibition promotes SCRs and DADs, without changing the SAP rate threshold (Fig. 14A–B), i.e., similar to the effects of inhibiting CSQ Ca2+ buffering alone (Fig. 10A–B). Indeed, our mechanistic analysis suggests that luminal Ca2+ buffering by CSQ plays a more important role in affecting arrhythmic outcomes than its role on RyR gating. On one hand, inhibiting CSQ Ca2+ buffering (Fig. 14C, 1) increased the SR load (Fig. 14Ci) and RyR leak (Fig. 14Cii) to promote SCRs and DADs. On the other hand, inhibition of CSQ RyR-regulation (Fig. 14C, 2) elevated RyR systolic PO and enhanced SR release fraction, and decreased SR load (Fig. 14Ciii), leading to subsequent smaller RyR leak (Fig. 14Civ) and reduced SCRs and DADs, especially in inner coupled CRUs (Fig. 15). When combining the two opposing effects, we found an overall net increase in RyR leak (Fig. 14Cvi), which was dominant over the slight decrease in SR load (Fig. 14Cv), and promoted SCRs, DADs, and SAPs. In these settings, the rate thresholds of SCRs, DADs, and SAPs are consistently lower in the cell with sparse TATS (in agreement with results in the companion Zhang et al. paper), but cells with intermediate and dense TATS are more susceptible to changes in CSQ expression.
Figure 10 – Promotion of CSQ Ca2+ buffering effects inhibits SCRs, DADs and SAPs.
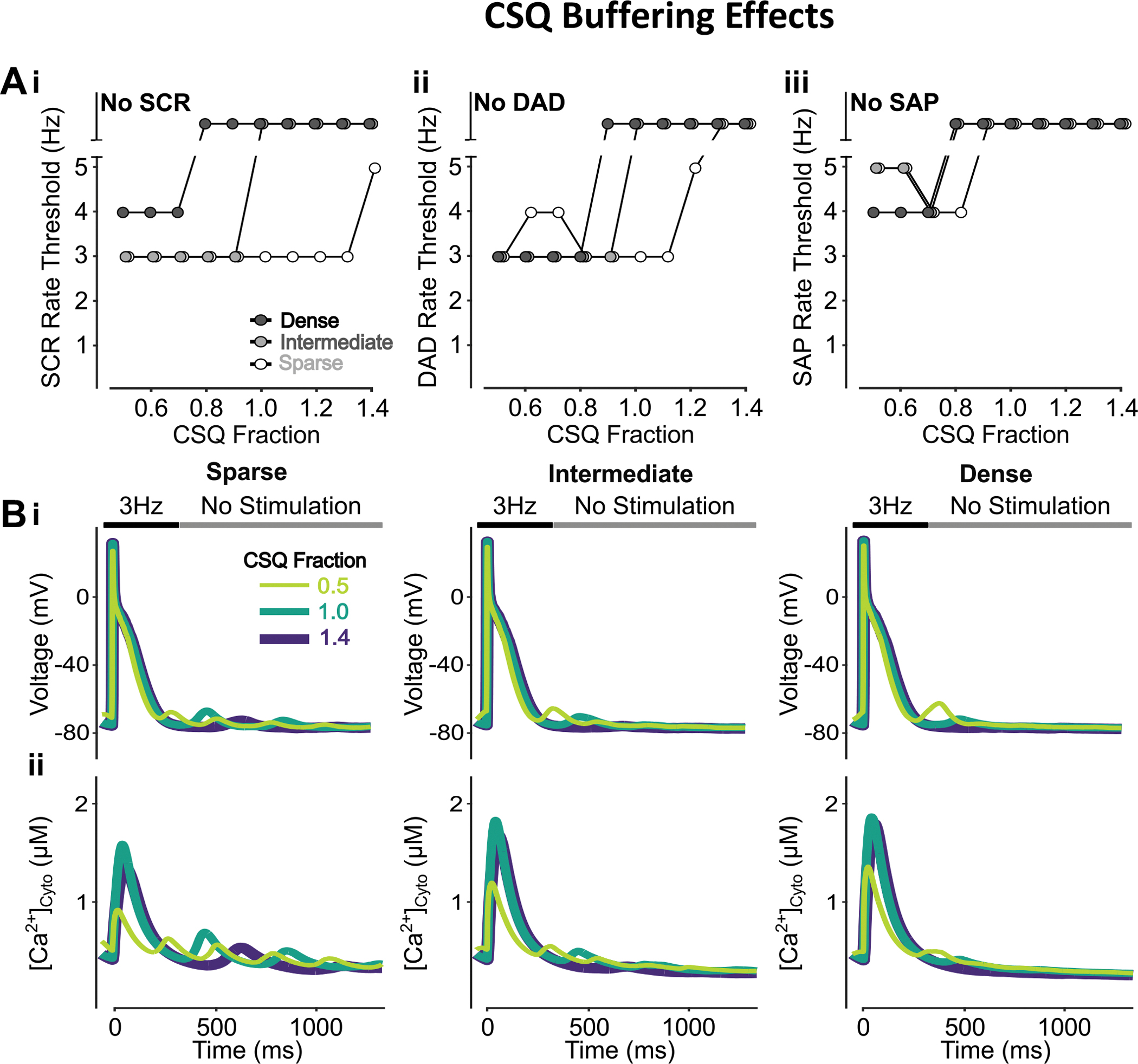
A) Promoting Ca2+ buffering by CSQ monotonically increases the rate threshold of SCRs (i), DADs (ii), and SAPs (iii). This effect is enhanced in cells with dense and intermediate tubular structures vs cells with sparse tubules. B) Effect of altered CSQ-mediated Ca2+ buffering on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs.
Figure 11 – Inhibition of CSQ Ca2+ buffering effects promotes SCRs in all CRUs.
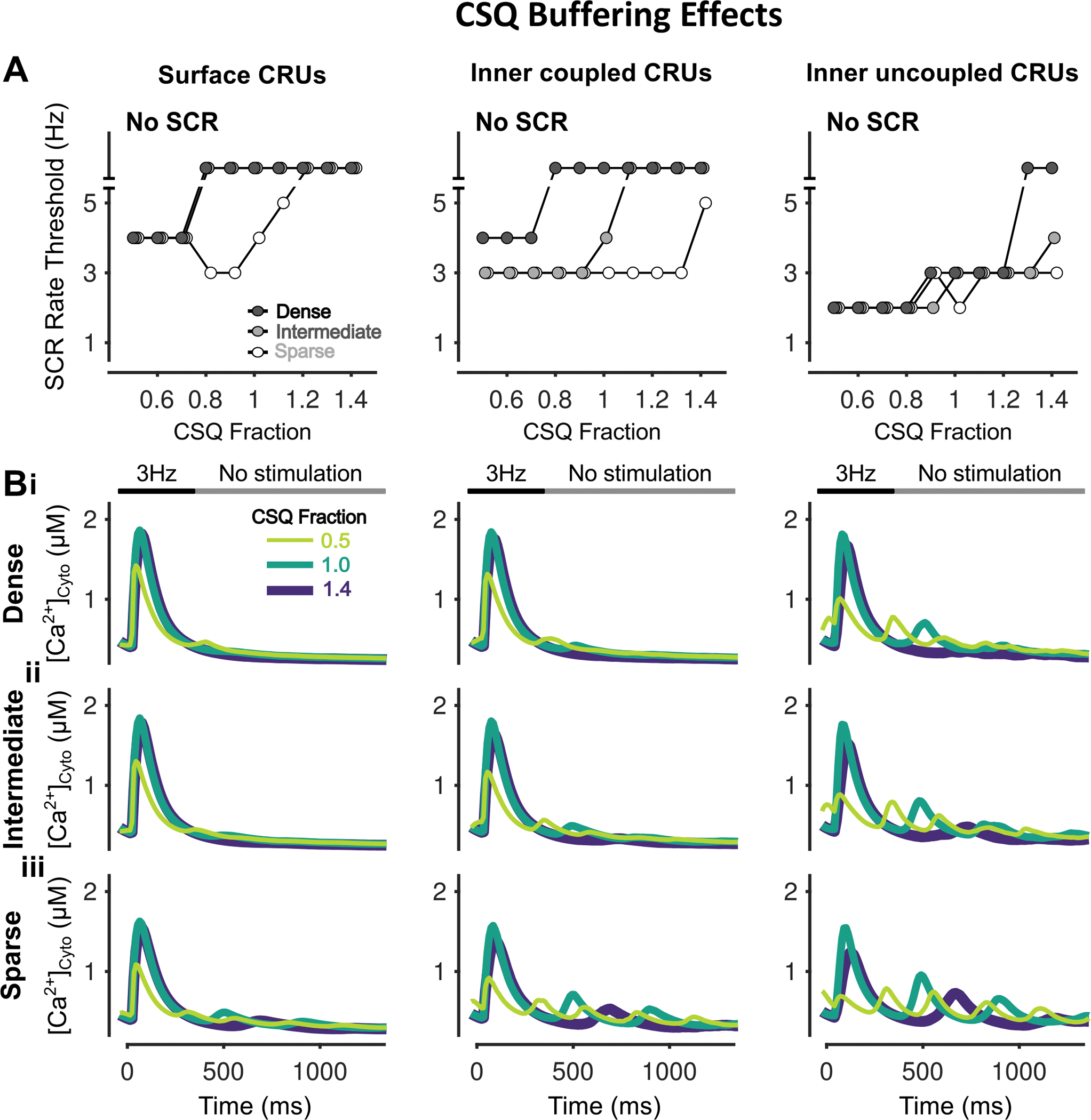
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered CSQ Ca2+ buffering effects greatest in inner coupled/uncoupled CRUs of cells with dense and intermediate tubules. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying fraction of CSQ Ca2+ buffering effects.
Figure 12 – Increasing CSQ-RyR regulation promotes SCRs, DADs and SAPs.
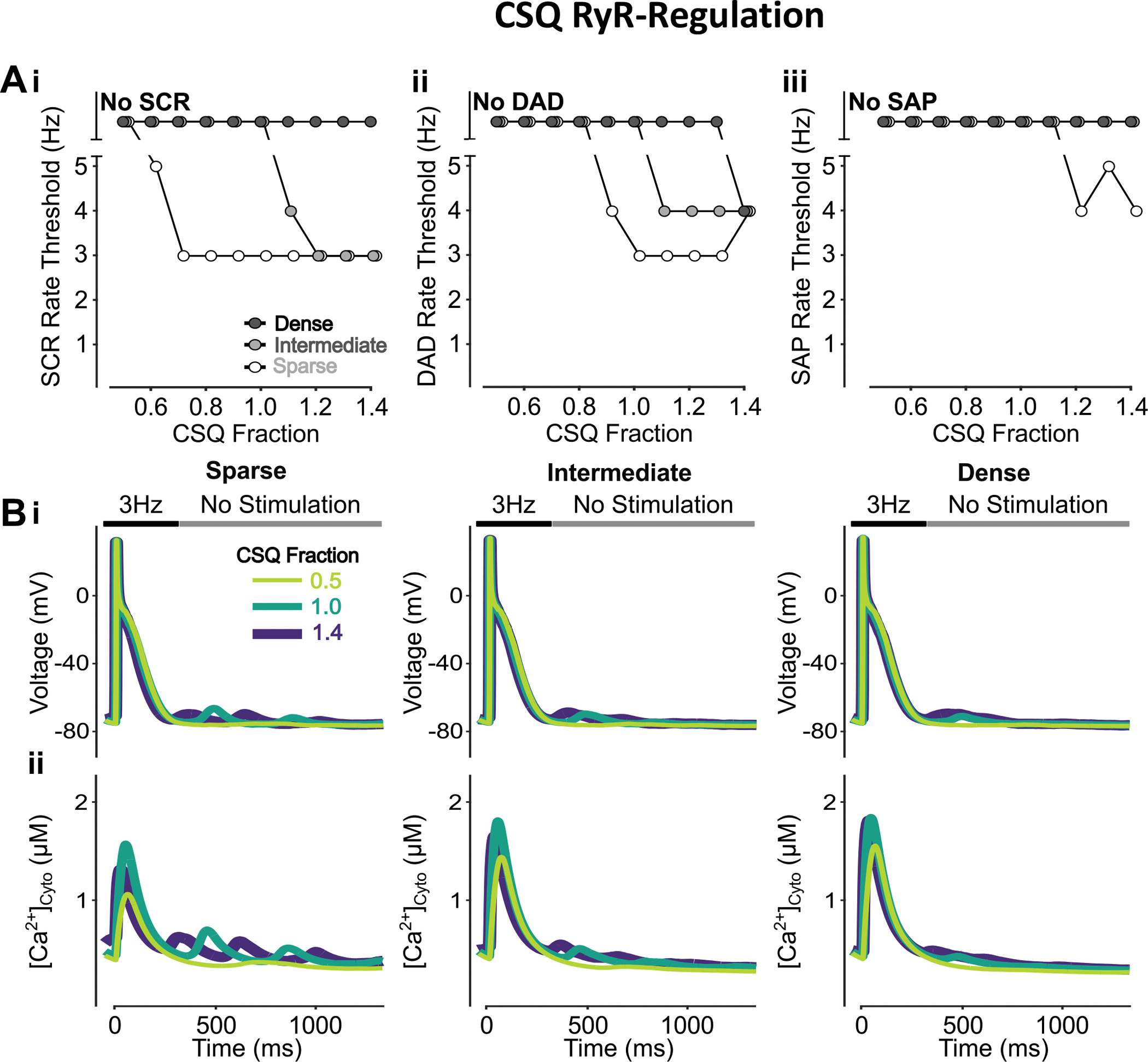
A) Increasing CSQ-RyR regulation monotonically decreases the rate threshold of SCRs (i), DADs (ii), and SAPs (iii) with the effect greater in cells with sparse and intermediate tubular structures vs cells with dense tubules. B) Effect of altered CSQ-RyR regulation on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs.
Figure 13 – Inhibition of CSQ-RyR regulation suppresses SCRs in all CRUs.
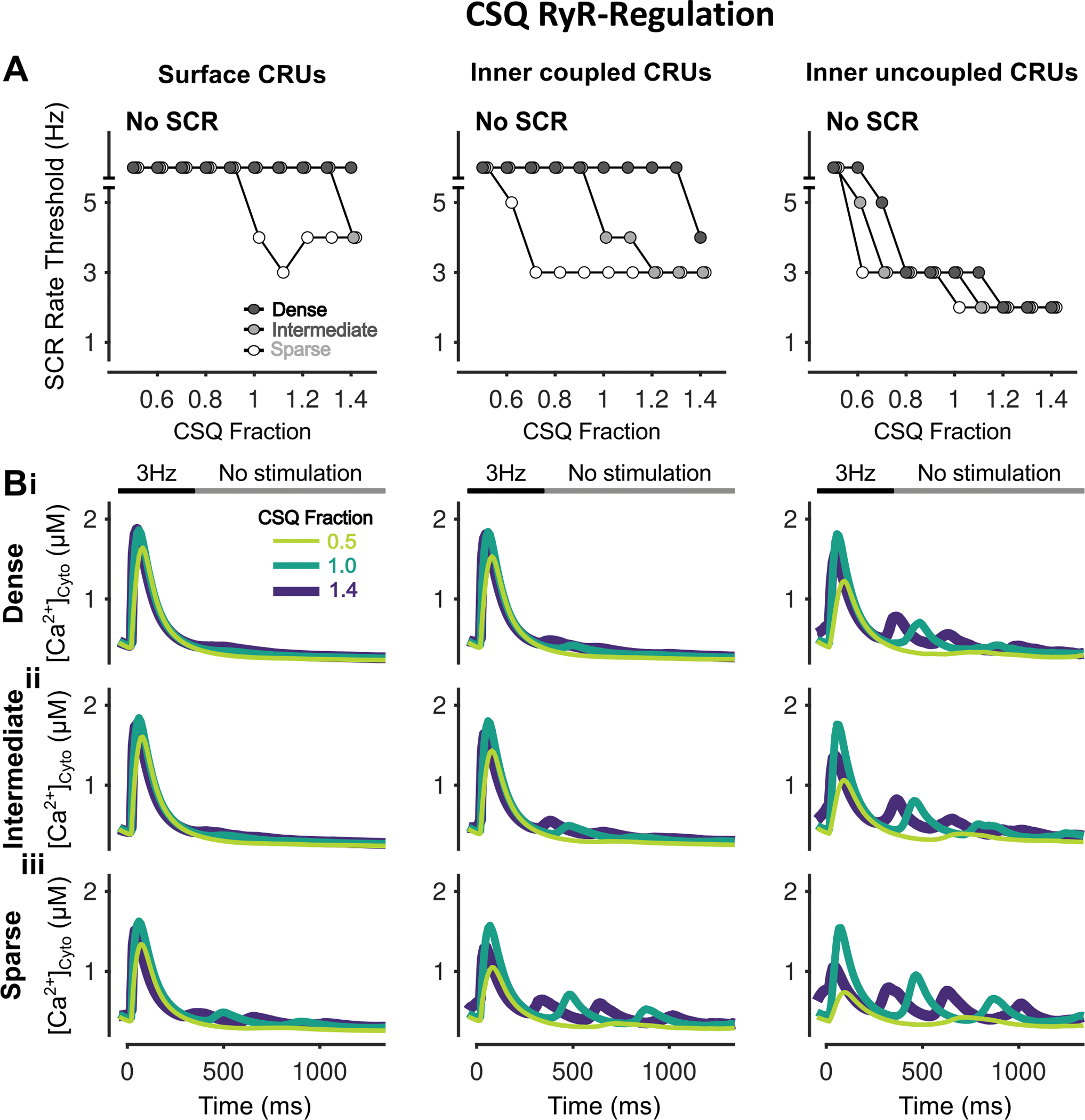
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered CSQ-RyR regulation greatest in inner coupled/uncoupled CRUs. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying fraction of CSQ RyR-regulation.
Figure 14 – Increasing CSQ fraction inhibits SCRs and DADs with no effect on SAPs.
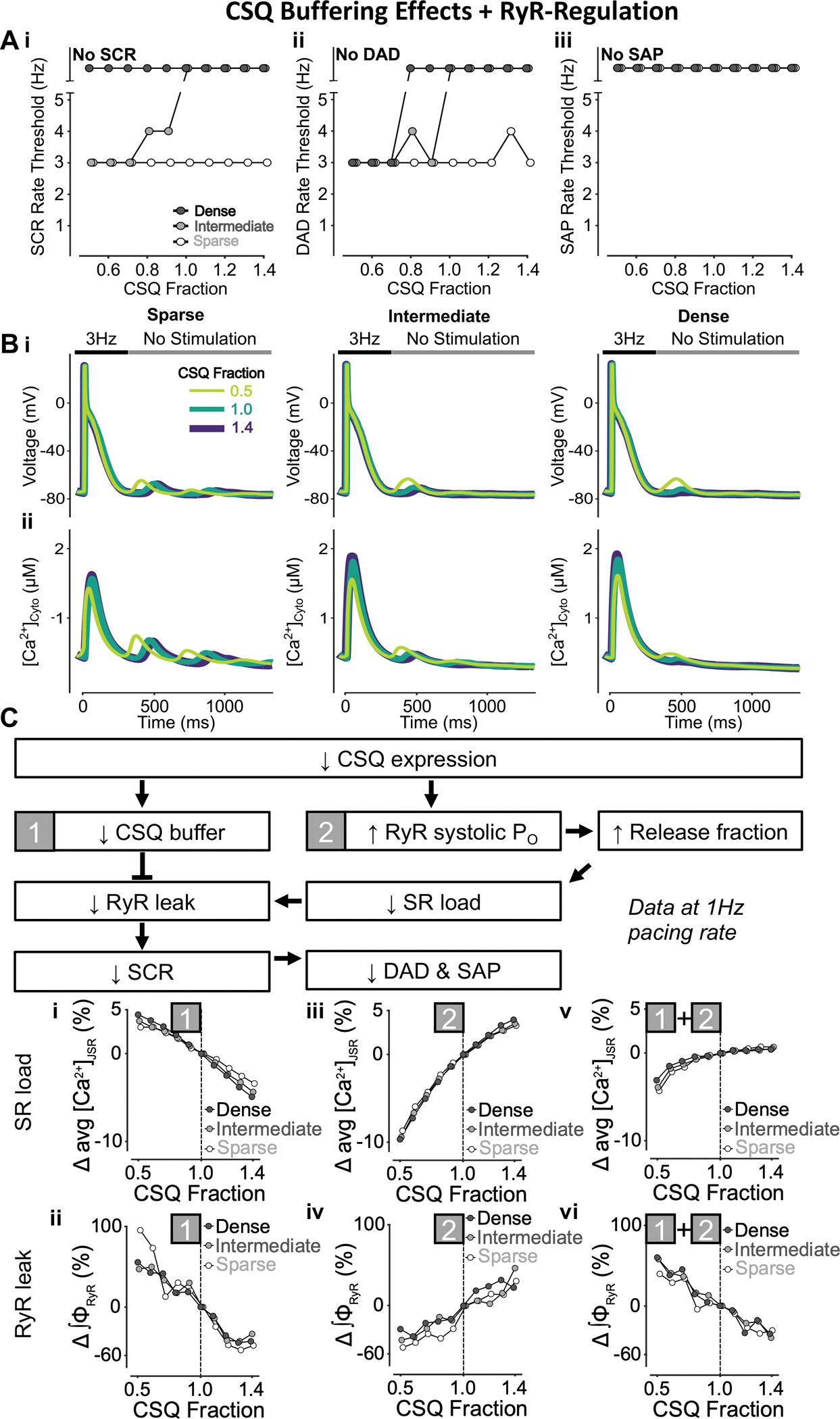
A) Increasing CSQ fraction (i.e., both CSQ-mediated Ca2+ buffering and RyR regulation) monotonically raises the rate threshold of SCRs (i) and DADs (ii), with SAP (iii) remaining absent in all conditions. While CSQ-mediated changes in SCRs are solely observed in cells with intermediate tubular structures, the effect of reduced CSQ fraction on DADs is greater in cells with dense and intermediate tubular structures vs cells with sparse tubules. B) Effect of altered CSQ fraction on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs. C) Mechanism underlying increased CSQ fraction inhibiting SCRs and DADs without affecting SAPs. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained CSQ fraction of 1.0. Since CSQ is a jSR Ca2+ buffer and regulates RyR Po, there is interplay between varying CSQ buffering effects (1), CSQ-RyR regulation (2) and CSQ fraction (1+2). Lower CSQ expression is associated with reduced CSQ-mediated jSR Ca2+ buffering (1) and increased RyR systolic Po (2). Reduced CSQ Ca2+ buffering (1) decreases SR load (i) but promotes RyR leak (ii), and thus SCRs. Conversely, enhanced systolic RyR Po (2) increases SR release fraction, which lowers SR load (iii) and diminishes RyR leak (iv), leading to reduced SCRs. Combining these two effects, although decreasing CSQ expression lowers down SR Ca2+ load (v), RyR leak is enhanced (vi), which leads to promotion of SCRs, DADs, and SAPs.
Figure 15 – Varying CSQ fraction has differing effects on SCR dependent on CRU location.
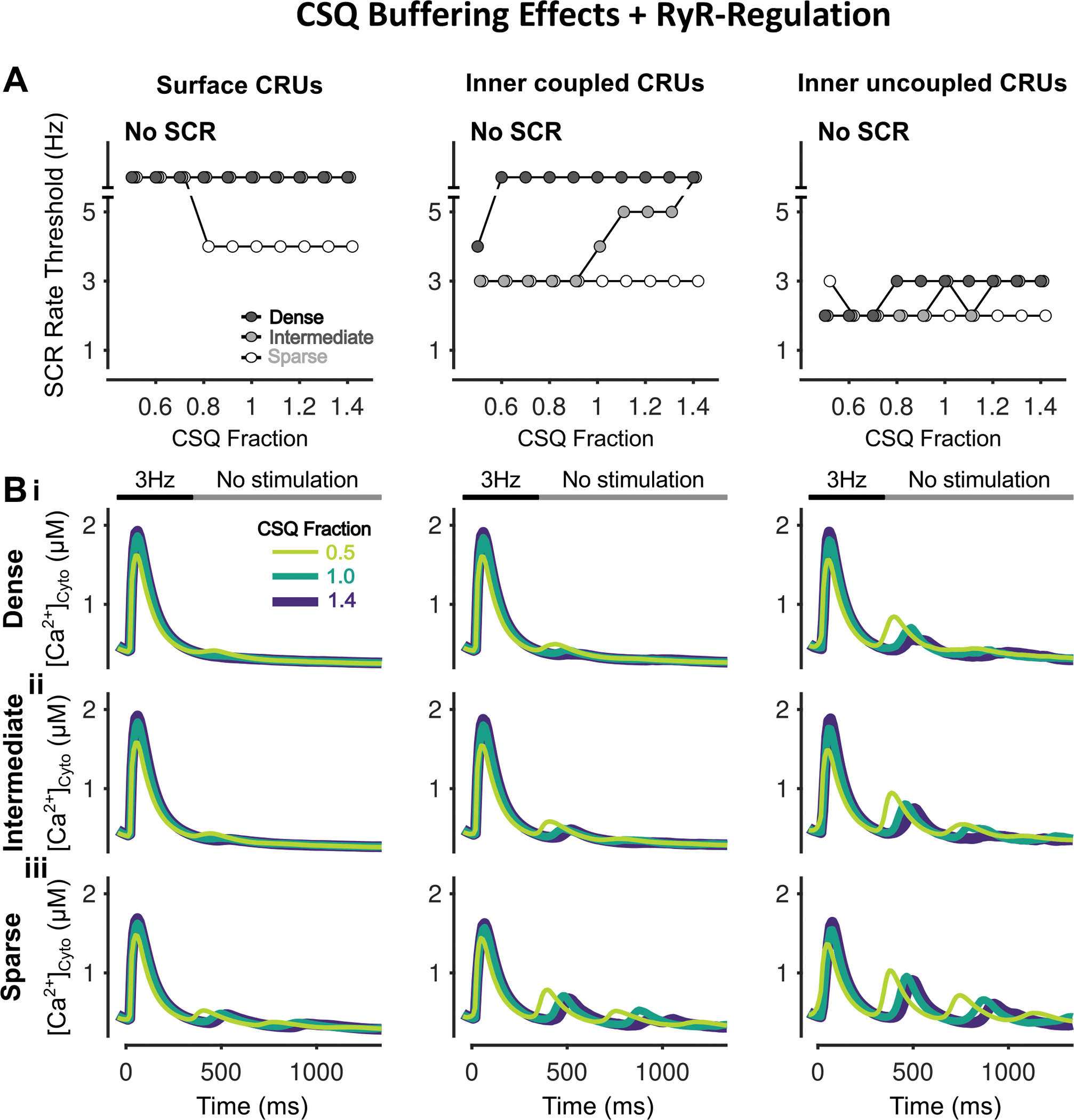
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells. Whereas CSQ downregulation raises the SCR rate threshold in cells with sparse tubules, it lowers the threshold in inner coupled/uncoupled CRUs of cells with intermediate and dense tubular structures. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying CSQ fraction.
Increasing surface vs. inner CRU CSQ expression promotes SCRs but inhibits DADs by elevating inner RyR leak but decreasing surface RyR leak
To reveal the effects of heterogeneous subcellular CSQ distribution and TATS density on SCRs, DADs, and SAPs, we simulated various surface/inner CRU CSQ expression ratios in cells with sparse, intermediate, and dense TATS. The simulation results indicate that increasing surface vs. inner CRU CSQ expression lowers the pacing threshold for SCRs (in cells with intermediate TATS, Fig. 16Ai), but increased the pacing threshold for DADs (in cells with sparse and intermediate TATS, Fig. 16Aii), without remarkable effects on SAP rate threshold (Fig. 16Aiii). These results are also illustrated by the representative traces of [Ca2+]Cyto and Vm (Fig. 16B). We found that increasing surface-to-inner CRU CSQ expression ratio changes RyR leak locally, i.e., decreasing RyR leak at the periphery and increasing it at the cell interior. A larger inner RyR leak favors SCRs and DADs (Fig. 16Ci), but a smaller RyR leak in the periphery area (Fig. 16Cii) reduces NCX Ca2+ extrusion (Fig. 16Ciii), ΔVm/ΔCa2+ gain (Fig. 16Civ) and DADs. The effects of varying surface-to-inner CRU CSQ expression ratio on ΔVm/ΔCa2+ gain are stronger in cells with sparse TATS (Fig. 16Civ), which is reflected in DAD rate threshold changes being more marked compared to intermediate/densely tubulated cells (Fig. 16Aii). Examination of the subcellular Ca2+ signals suggests that increasing surface vs. inner CSQ expression reduces the SCR rate threshold in inner coupled CRUs of the cell with intermediate TATS (Fig. 17A), matching the changes in SCR rate threshold (Fig. 16Ai), and in all cases, SCRs are favored in cells with sparse TATS (Fig. 17A–B).
Figure 16 – Increasing surface/inner CRU CSQ expression ratio promotes SCRs but inhibits DADs and does not affect SAP.
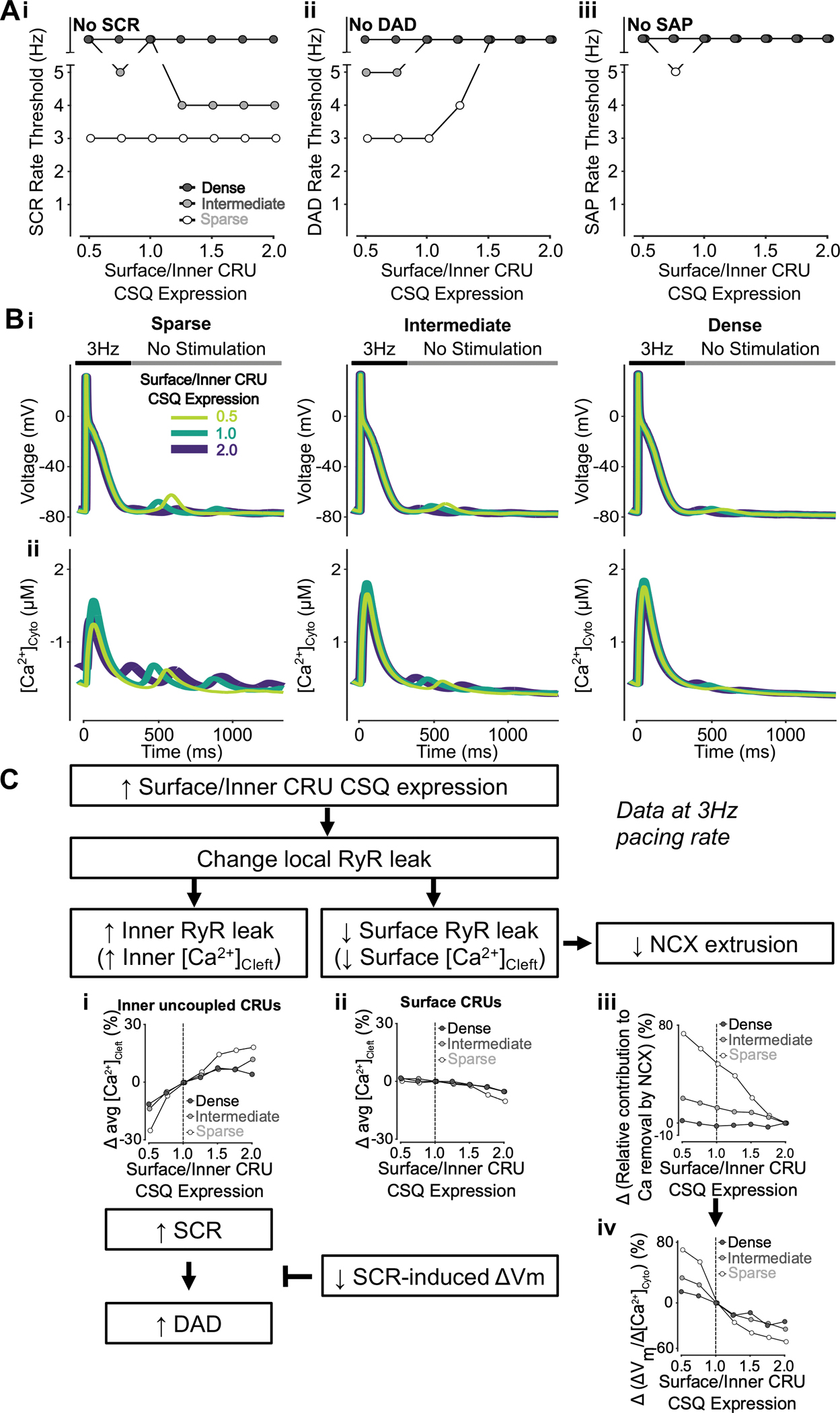
A) Increasing surface/inner CRU CSQ expression ratio (i.e., increasing CSQ density in surface CRUs without changing whole-cell CSQ expression) monotonically decreases the rate threshold of SCRs (i) and increases the rate threshold of DADs (ii), whereas SAPs (iii) only appear in cells with sparse tubules and surface/inner CRU CSQ expression ratio of 0.75. The effect on SCRs is greater in cells with intermediate tubular structures vs cells with sparse and dense tubules, whereas the effect on DADs is enhanced in cells with sparse and intermediate tubular structures vs cells with dense tubules. B) Effect of altered surface/inner CRU CSQ expression ratio on voltage (i) and global cytosolic Ca2+ concentration (ii) in cells with sparse (left), intermediate (middle), and dense (right) tubules following pacing at 3 Hz to examine the occurrence of DADs and SCRs. C) Mechanism underlying increasing surface/inner CRU CSQ expression ratio promoting SCRs, inhibiting DADs with no effect on SAPs. Biomarkers were determined from the first 100 ms of no-stimulation period and normalized to those of cells with a retained surface/inner CRU CSQ expression ratio of 1.0. As similarly described when altering overall CSQ expression (Figure 13), increasing surface/inner CRU CSQ expression ratio is associated with increased RyR leak in inner uncoupled CRUs (i) and decreased RyR leak in surface CRUs (ii). Enhanced RyR leak in inner uncoupled CRUs leads to stronger SCRs and DADs. However, less RyR leak in surface CRUs reduces NCX contribution to Ca2+ extrusion (iii) and lowers SCR-induced voltage changes/SCR amplitude ratio (iv). As such, while SCRs typically lead to DADs, this transition is prohibited by the decreased SCR-induced changes in Vm, thus explaining the opposing effects of increasing surface/inner CRU RyR expression ratio on SCRs and DADs.
Figure 17 – Increasing surface/inner CRU CSQ expression ratio promotes simultaneous SCRs in inner coupled CRUs of cells with intermediate tubules.
A) Rate threshold for local SCRs of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs in sparse, intermediate and densely tubulated cells with the impact of altered surface/inner CSQ expression ratio greatest in inner coupled CRUs of cells with intermediate tubules. B) Cytosolic Ca2+ concentration of surface (left), inner coupled (middle) and inner uncoupled (right) CRUs from cells with dense (i), intermediate (ii), and sparse (iii) tubules following pacing at 3 Hz showing observed SCRs with varying surface/inner CRU CSQ expression ratios.
Discussion
In this study, we utilized our integrative model of the human atrial myocyte (Zhang et al.) to predict and quantitatively explain how TATS loss interacts with varying expression and localization of key Ca2+ handling proteins to disrupt diastolic Ca2+ homeostasis and electrophysiological stability in human atrial cells. Specifically, we demonstrated that arrhythmogenic effects of Ca2+ handling protein remodeling are especially exaggerated in cells with intermediate TATS, with densely tubulated myocytes being more resilient, and detubulated myocytes being rather insensitive to the superimposed ionic remodeling. Recent studies have investigated t-tubule restoration as a therapeutic maneuver in cardiac disease, especially in HF, and suggest that therapeutic t-tubule protection and repair may benefit inotropy while inhibiting arrhythmia (Manfra et al., 2017). Analogously, several groups endeavored to optimize experimental conditions to produce human induced pluripotent derived cardiomyocytes with functional t-tubule networks (Parikh et al., 2017). Our findings provide mechanistic insight into the interactive contributions of TATS and Ca2+-handling protein expression and distribution on Ca2+-driven proarrhythmic behavior that underlie AF pathophysiology and may help to predict the effects of antiarrhythmic strategies at varying stages of ultrastructural remodeling.
NCX expression and distribution
In the healthy heart, NCX inhibition prevents Ca2+ extrusion, leading to Ca2+ overload (Bers, 2002) and thus enhancement of SCRs (Lotteau et al., 2021). This is seen in our simulations, whereby a decrease in NCX is associated with a reduction in the pacing threshold for SCRs. While increased SCRs are typically expected to promote Vm instabilities, drug-induced NCX block enhanced SCRs but reduced DADs in rabbit atrial myocyte experiments (Hohendanner et al., 2015). We also observed this in our simulations where inhibiting NCX (both globally or locally in the cell interior vs. periphery) varied the balance between increased SCRs and reduced ΔVm/ΔCa2+ gain that determines the net impact on SCRs, DADs, and SAPs (Fig. 2). As such, our model provides quantitative spatially-detailed insight into the interaction of these two opposing effects. Furthermore, our simulation results suggest that SCR is further enhanced with TATS loss, and the effects of inhibiting NCX on [Ca2+]Cyto-voltage coupling is stronger in cells with intermediate and dense TATS (Figs. 2 and 4). This suggests that with severe ultrastructural remodeling and TATS loss, additional changes in Ca2+ extrusion may not strongly modulate Ca2+ homeostasis and arrhythmogenesis. Interestingly, atrial myocytes from NCX knock-out mice also have reduced TATS density (Yue et al., 2017). It is reasonable to speculate that TATS loss in this setting may be a compensatory adaptation to Ca2+ overload, to limit Ca2+-Vm coupling and attenuate DAD and SAP risk. Indeed, this is supported by our sparsely tubulated myocyte model having a higher SAP rate threshold than the models with intermediate and dense TATS.
Interestingly, lower NCX promotes CaT alternans in cells with sparse and intermediate TATS but has biphasic effects on the rate threshold of CaT alternans in the densely tubulated cell (Fig. 18Ai), with the same effect observed in all CRU locations (Fig. 3Bi). Similarly, NCX inhibition has biphasic effects on the rate threshold of APD alternans (Fig. 18Aii), alongside changes in rate thresholds of DADs and SAPs (Fig. 2Aii–iii). Though these effects were observed when altering NCX expression, varying surface/inner CRU NCX localization had no effect on rate threshold of APD alternans besides initiating some CaT alternans at 5Hz (Fig. 18B). Taken together, these results suggest that atrial cells are more sensitive to whole-cell rather than subcellular changes in NCX-mediated Ca2+ extrusion.
Figure 18 – Inhibition of NCX has biphasic effects on CaT and APD alternans but increasing surface/inner CRU NCX expression ratio only exhibits CaT alternans at 5 Hz.
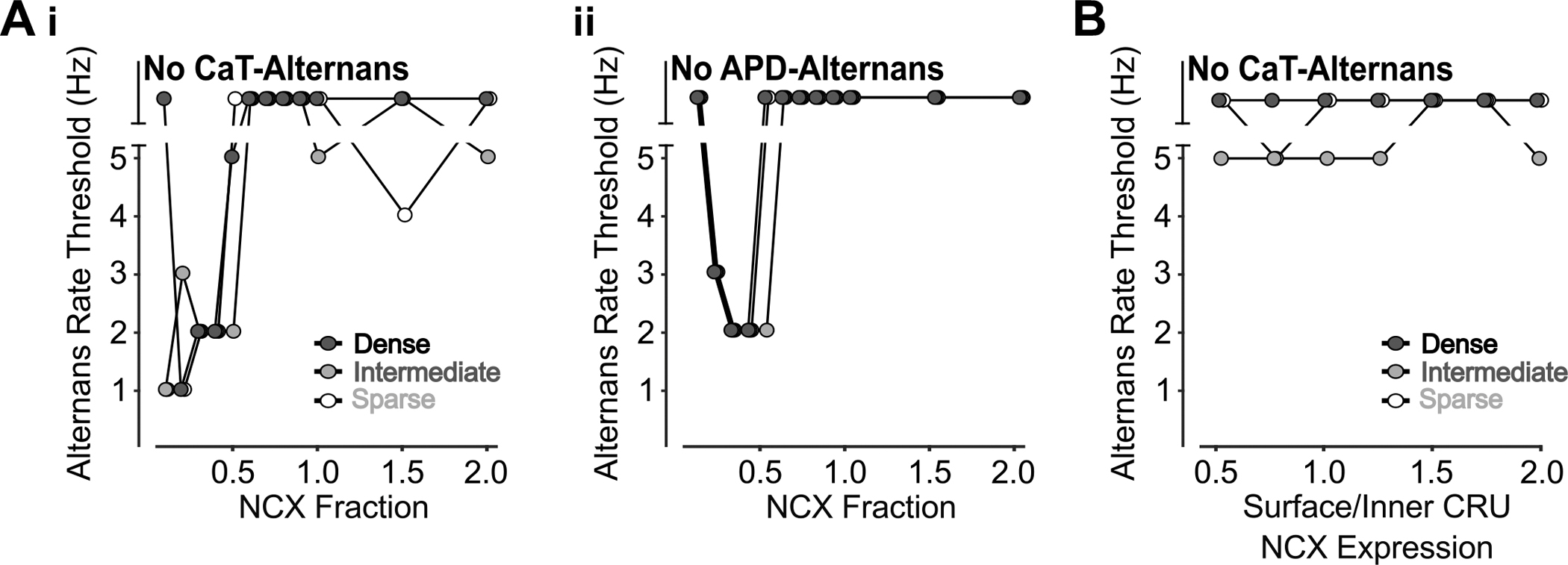
A) The rate thresholds of CaT alternans (i) and APD alternans (ii) display biphasic dependence on NCX reduction. B) Varying surface/inner CRU NCX expression ratio induces CaT alternans at 5 Hz, especially in cells with intermediate TATS.
As NCX is upregulated in disease (Voigt et al., 2012), we also considered the impact of increased, not just decreased, NCX expression on arrhythmogenesis. Based on our simulation results, enhanced NCX expression may be an adaptive ionic remodeling process to protect the cells against Ca2+ overload-induced arrhythmia, although this can also be proarrhythmic with increased Ca2+-Vm coupling. Indeed, in AF, enhanced NCX expression increases diastolic Ca2+-Vm coupling to augment the arrhythmogenic effects of enhanced SR Ca2+-leak, causing transient inward current and subsequent DADs (Hove-Madsen et al., 2004; Lenaerts et al., 2009; Neef et al., 2010; Voigt et al., 2012). Because of this, NCX inhibition is thought to be a suitable therapeutic AF treatment (Hobai & O’Rourke, 2004). Our simulation suggests that varying degrees of NCX inhibition could have opposite effects on arrhythmias with NCX inhibition treatment potentially having better effects in cells with intermediate and dense TATS than in cells with sparse TATS. In atrial myocytes, NCX likely distributes on both the cell surface and TATS (Melnyk et al., 2005; Scriven et al., 2010), with localization disrupted with TATS remodeling in disease. As such, altered NCX distribution alongside TATS loss promotes arrhythmia, while increasing NCX density within remaining TATS may preserve NCX-mediated Ca2+-extrusion and rescue the cells from Ca2+ overload. While NCX inhibition is thought to be more relevant to cardiac disease clinically, it is interesting to note that pharmacological NCX activators are emerging as promising strategies to ameliorate certain neurodegenerative diseases (Annunziato et al., 2020), such as stroke, neonatal hypoxia, multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer’s disease, Parkinson’s disease, and spinal muscular atrophy. In neuronal cells, various NCX isoforms work to maintain Na+ and Ca2+ homeostasis in the cytosol, endoplasmic reticulum, and mitochondria via both forward and reverse mode operation; and have been shown to increase survival of neuronal and glial-derived cells in pathophysiologic conditions (Annunziato et al., 2020; Pannaccione et al., 2020).
RyR expression and distribution
Previous computational studies using a ventricular myocyte model show that enhanced RyR expression per se increased SR Ca2+ leak, while the subsequent reduction SR Ca2+ load reduced RyR PO (Sato et al., 2016), similar to experimental data showing that SR Ca2+ leak reduces SR load (Bers, 2002), which can reduce the RyR PO (Sitsapesan & Williams, 1994). On the other hand, a recent experimental study found RyR loss-of-function mutations, which are associated with arrhythmia and sudden cardiac death, cause elevated SR Ca2+ load (Li et al., 2021), which can increase the RyR PO. These data suggest that increasing RyR expression might be pro- or anti-arrhythmic depending on the prevailing mechanism. Indeed, our simulations indicate that altering RyR expression shifts the balance between RyR number-associated release flux and opposite SR load changes that determine the net impact on RyR leak, and subsequent SCRs, DADs, and SAPs (Fig. 6). While RyR number does affect SCR in the cell with sparse TATS, DADs and SAPs are similarly affected in all cell types (Fig. 6A). Interestingly, these contrasting outcomes are similar to those reported following use of two different RyR-modulating drugs in ventricular myocytes from catecholaminergic polymorphic ventricular tachycardia (CPVT) mice. Here RyR inhibition by tetracaine decreased Ca2+ sparks and leak thus increasing SR Ca2+ load, whereas flecainide reduced Ca2+ spark mass and increased Ca2+ spark frequency with no effect on spark-mediated SR Ca2+ leak or content (Hilliard et al., 2010).
SCRs are increased in myocytes isolated from patients with paroxysmal AF lacking TATS likely because of increased RyR channel expression (Beavers et al., 2013; Voigt et al., 2014). Conversely, a reduction in RyR expression is seen in several animal models of atrial tachycardia remodeling as part of the underlying proarrhythmic substrate (Lenaerts et al., 2009; Wakili et al., 2010; Lugenbiel et al., 2015). Notably, despite reduced total RyR protein levels in the atria of patients with systolic HF, RyR function was found to be increased (Molina et al., 2018). As such, changes in RyR expression may not fully reflect changes in RyR regulation, as previously discussed (Zhang et al.), which may further affect the balance between adaptive and maladaptive responses. For example, mouse knock-in RyR mutations that result in increased diastolic Ca2+ leak are associated with CPVT and AF, which are inhibited with the RyR stabilizer S107 that reduces diastolic SR Ca2+ leak in atrial myocytes and decreases burst pacing-induced AF in vivo (Shan et al., 2012). Additionally, AF is associated with RyR hyperphosphorylation (Vest et al., 2005; Neef et al., 2010; Voigt et al., 2012) and thus larger diastolic SR leak and elevated Ca2+ levels (Neef et al., 2010).
In human atrial myocytes from both normal sinus rhythm and AF patient samples, RyR density is higher at the sarcolemma than in the cell interior (Herraiz-Martínez et al., 2022), but the physiological significance of heterogeneous subcellular RyR distribution is not understood. In our model, increasing RyR expression at the surface vs. inner CRUs enhances NCX-RyR coupling, which on one hand reduces SCRs due to NCX-mediated Ca2+ extrusion, but on the other hand, increases ΔVm/ΔCa2+ gain (Fig. 8). Our simulations suggest that the observed increase in peripheral RyR distribution might be a protective mechanism that allows achieving the optimal balance that guarantees stable Ca2+ and Vm homeostasis. Whilst these spatial differences exist in the healthy atria, complex remodeling occurs in AF involving RyR cluster fragmentation and redistribution to inter-z-line areas, with Ca2+ sparks increased in fragmented CRUs (Macquaide et al., 2015). This subcellular remodeling of RyR distribution in AF is outside the scope of this study but can be examined in future investigations. In the companion paper (Zhang et al), we highlighted that NCX-mediated Ca2+ extrusion inhibits SCRs in coupled CRUs, whereas SCRs persist in uncoupled CRUs due to Ca2+ released having to diffuse to coupled CRUs to be extruded by NCX. In line with this, spatial differences in subcellular Ca2+ dynamics may be differentially affected by RyR subcellular remodeling in coupled vs. uncoupled CRUs.
CSQ expression and distribution
CSQ is a major luminal Ca2+ buffer and RyR regulator (Terentyev et al., 2003; Györke et al., 2004; Knollmann et al., 2006; Restrepo et al., 2008). Reduced atrial CSQ expression promotes arrhythmia in HF (Yeh et al., 2008), with CSQ loss enhancing SCRs and DADs to increase AF risk (Faggioni et al., 2014). Our model confirms these findings and demonstrates that diminished Ca2+ buffering associated with lower CSQ expression has a predominant effect promoting SCRs and DADs (Figs. 10 and 14), with associated changes to RyR gating having the opposing (and more modest) effect of decreasing SR load to limit SCRs and DADs (Figs. 12 and 14). Indeed, while impaired SR Ca2+ buffering is a well-accepted mechanism for CPVT-associated CSQ mutations (Wleklinski et al., 2020), CSQ-RyR interaction may also be altered by some of these mutations (Terentyev et al., 2006). Notably, despite SCRs and DADs being consistently greater in the cell with sparse TATS, cells with intermediate and dense TATS are more susceptible to changes in CSQ expression (Fig. 14).
In human atrial myocytes, CSQ is more abundant near the cell periphery vs. the inner area (Schulson et al., 2011; Herraiz-Martínez et al., 2022). Our simulations suggest that this baseline subcellular CSQ distribution gradient might be protective against arrhythmia, as increasing surface vs. inner CRU CSQ expression promotes SCRs but inhibits DADs (Fig. 16). The protection from DADs is stronger in cells with sparse and intermediate TATS (Fig. 16), and might be relevant for chronic AF, where a cell surface to interior gradient in CSQ expression is seen and there is TATS loss (Lenaerts et al., 2009).
Notably, when we simulated concomitant changes in surface/inner CRU expression ratios (ratio = 2) of both RyR and CSQ to mimic experimental findings (Herraiz-Martínez et al., 2022), we found that the pacing threshold for SCRs increased vs varying CSQ distribution only, and varying-RyR-induced DADs are ultimately prevented by concomitant changes (i.e., CSQ + RyR) (Fig. 19). This indicates that appropriate relative expression of RyR and CSQ through non-uniform surface/inner CRU expression ratios, as seen in human atrial myocyte experiments (Herraiz-Martínez et al., 2022), provides protection against arrhythmia. Of note, adaptive changes to CSQ deficiency in CPVT include increased RyR expression, which on one hand maintains excitation-contraction coupling but also increases RyR leakiness (Song et al., 2007).
Figure 19 – Effect of concomitantly increasing surface/inner CRU expression ratio of CSQ and RyR to 2.0.
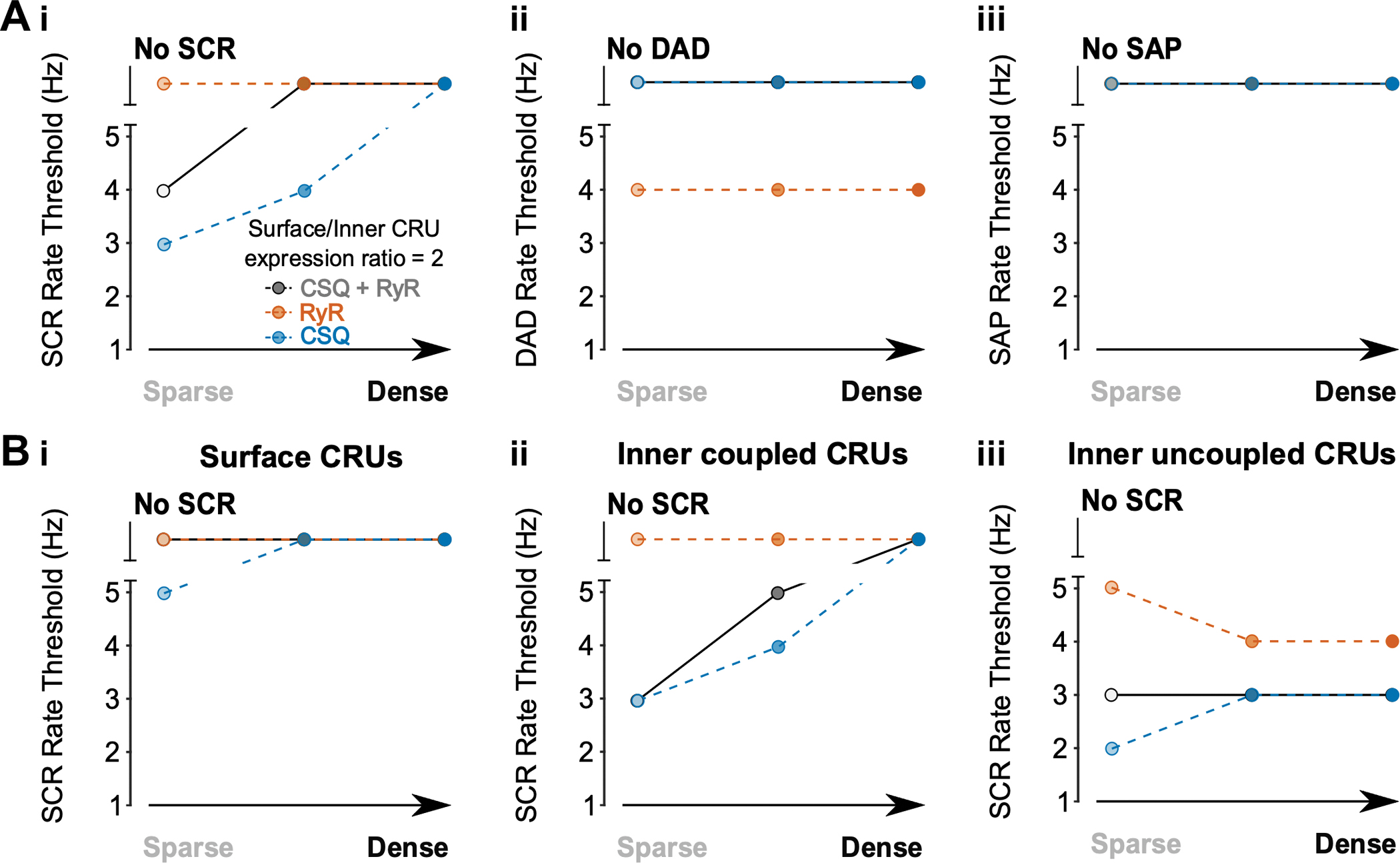
A) With increased CRU CSQ and RyR expression ratio, tubule loss decreases the rate threshold of SCRs (i), whereas DADs (ii) and SAPs (iii) remain absent in all conditions. The simulation results with surface/inner CRU expression ratio of RyR (orange) or CSQ (blue) at 2.0 are superimposed for comparison. B) Rate threshold for local SCRs from surface (i), inner coupled (ii) and inner uncoupled (iii) CRUs in sparse, intermediate, and densely tubulated cells. The impact of altered tubular density is greatest in inner coupled CRUs, with less change in SCR rate threshold associated with changes in tubule density in surface or inner uncoupled CRUs.
LCC expression and localization
In human atrial myocytes, LCCs are evenly distributed in TATS and surface membrane (Glukhov et al., 2015). While LCC current is decreased in AF (Christ T. et al., 2004), to our knowledge LCC subcellular distribution in AF is yet to be elucidated. In HF ventricular myocytes, altered colocalization between LCCs and RyRs may impair the ability of the LCC to trigger RyR Ca2+ release (Gómez et al., 1997). Indeed, computational atrial myocyte simulations indicate that disrupted LCC localization (i.e., removal of LCCs away from the dyadic cleft) changes subcellular Ca2+ dynamics to elevate SR load and enhance AP-evoked Ca2+ wave propagation (Shiferaw et al., 2020). In our preliminary simulations, we found that varying LCC expression and distribution per se did not change SCR and DAD properties (data not shown). Nevertheless, LCCs play a critical role in systolic Ca2+ signaling, as well as in setting the steady-state conditions for diastolic behavior (e.g., cellular Ca2+ loading and removal, which we take into account by simulating at various pacing frequencies).
Future Directions
The limitations of this model are discussed in our companion paper (Zhang et al). Taking those into account, our novel 3D human atrial myocyte model provides a valuable platform that can be further adapted for future investigations. These could include, but are not limited to, incorporating immunofluorescence microscopy-based protein distributions and localization information of other proteins, calibrating additional experimental results of SR Ca2+ dynamics, and integrating extracellular ion dynamics.
Estimation of protein localization
While spatial localization and activity of key Ca2+ handling proteins in the ventricle have been characterized experimentally using immunofluorescence microscopy and/or detubulation (Kawai et al., 1999; Despa et al., 2003; Gadeberg et al., 2017), little is currently known regarding the atria. However, various experimental data indicate heterogeneous Ca2+ handling proteins function, expression, and localization (Chen-Izu et al., 2006; Macquaide et al., 2015; Brandenburg et al., 2016; Galice et al., 2018; Herraiz-Martínez et al., 2022), and variable TATs (Trafford et al., 2013; Brandenburg et al., 2016, 2018) in atrial myocytes, which may destabilize Ca2+ homeostasis and the bidirectional interaction between electrical excitation and Ca2+ signaling. LCCs have been shown to distribute equally in tubules and crest areas of the sarcolemma (Glukhov et al., 2015), and rabbit atrial experiments confirm that LCCs are colocalized with RyRs as in the ventricle (Carl et al., 1995). Compared to the ventricle, however, atrial cells are much more diverse, with varied cell width, sarcomere spacing (Arora et al., 2017), and heterogeneity in the TATS between cells and across the atrium (Frisk et al., 2014; Gadeberg et al., 2016). Indeed, the subcellular distribution and expression of K+ ion channels, LCC, and NCX are heterogeneous in canine pulmonary veins vs. left atrium (Melnyk et al., 2005). While the distribution of these proteins in the healthy atria has begun to be identified, more work is needed, especially to provide insight into the remodeling that occurs in disease. Once experimental data on protein localization of human atrial myocytes becomes more readily available, this model has the potential to integrate all the spatial information to investigate how varying expression and subcellular structures may affect Ca2+ and voltage homeostasis.
Effect on extracellular ion concentrations
The TATS permits rapid AP propagation but slows ion (i.e., Na+, Ca2+, and K+) diffusion between the extracellular space and TATS lumen (Yao et al., 1997; Blatter & Niggli, 1998; Shepherd & McDonough, 1998; Swift et al., 2006). However, myocyte contraction and relaxation cause distortion of the TATS that aids pumping the fluid to accelerate the diffusion (Savio-Galimberti et al., 2008; Rog-Zielinska et al., 2021). A computational model of Ca2+ diffusion within a 10-μm long tubule suggests there is a high-to-low Ca2+ concentration dynamic gradient between peripheral, central and deep sections, which are regulated by tubule diameters, depth, and buffering effects, extracellular ion concentration and diffusion coefficient (Pásek et al., 2008b). Experimental observations and mathematical models indicate the difference between tubules and surface membrane on membrane space, specific membrane capacitance and distribution of membrane ion channels, transporters and pumps in rat ventricular myocytes (Pásek et al., 2008a). Computational models of tubules in rat (Pásek et al., 2006) and guinea-pig (Pásek et al., 2008c) ventricular myocytes both indicate that current-clamp pacing brings transient K+ accumulation but Ca2+ depletion in the TATS lumen, whereas fast pacing weakens K+ accumulation but increases Ca2+ depletion to decrease SR load and Ca2+ transient amplitude. These ion concentration gradients could also be integrated into our model to investigate not only intracellular but also extracellular local Ca2+ dynamics.
Conclusions
We utilized our novel 3D human atrial myocyte model to examine the arrhythmic effect of varying the expression and distribution of key Ca2+-handling proteins alongside changes in TATS density. We reveal a balance between the pro- and anti-arrhythmic effects of protein remodeling and that this is differentially impacted by TATS density. These findings mechanistically elucidate how remodeling underlies Ca2+-handling abnormalities and Vm instabilities that may precipitate AF.
Supplementary Material
Key Points.
In our companion paper we developed a 3D human atrial myocyte model, coupling electrophysiology and Ca2+ handling with subcellular spatial details governed by the transverse-axial tubule system (TATS).
Here we utilize this model to mechanistically examine the impact of TATS loss and changes in the expression and distribution of key Ca2+-handling proteins known to be remodeled in disease on Ca2+ homeostasis and electrophysiological stability.
We demonstrate that varying the expression and localization of these proteins has variable pro- and anti-arrhythmic effects with outcomes displaying dependence on TATS density.
Whereas detubulated myocytes typically appear unaffected and densely tubulated cells seem protected, the arrhythmogenic effects of Ca2+ handling protein remodeling are profound in intermediately tubulated cells.
Our work shows the interaction between TATS and Ca2+-handling protein remodeling that underlies the Ca2+-driven proarrhythmic behavior observed in AF and may help to predict the effects of antiarrhythmic strategies at varying stages of ultrastructural remodeling.
Funding
American Heart Association Predoctoral Fellowship 20PRE35120465 (X.Z.), Postdoctoral Fellowship 20POST35120462 (H.N.); NIH/NHLBI Grants R01HL131517 (E.G.), P01HL141084 (E.G.), R01HL141214 (E.G.), R00HL138160 (S.M.); NIH Stimulating Peripheral Activity to Relieve Conditions Grant 1OT2OD026580–01 (E.G.)
Abbreviations
- AF
Atrial fibrillation
- AP
Action potential
- APD90
Action Potential Duration at 90% repolarization
- BCSQ
CSQ maximum buffering capacity
- CaT
Ca2+ transient
- CPVT
Catecholaminergic polymorphic ventricular tachycardia
- CRU
Ca2+ release unit
- CSQ
Calsequestrin
- DAD
Delayed afterdepolarization
- ICa
L-type Ca2+ channel current
- IK1
Inward rectifier K+ current
- IK,ACh
Acetylcholine-activated K+ current
- INa
Fast Na+ current
- JSR
Junctional SR
- LCC
L-type Ca2+ channel
- NCX
Na+/Ca2+ exchanger
- NSR
Network SR
- PO
RyR open probability
- RyR
Ryanodine receptor
- SAP
Spontaneous action potential
- SCR
Spontaneous Ca2+ release
- SR
Sarcoplasmic reticulum
- TATS
Transverse-axial tubular system
- Vm
Membrane voltage
Biography

Xianwei Zhang received his BS in Physics from Zhejiang University in 2017. Currently, he is completing his PhD in Biophysics in the lab of Dr. Eleonora Grandi at the University of California Davis. The studies described in the present article are the focus of his PhD work and were carried out as part of an AHA pre-doctoral fellowship. Xianwei’s long-term research interests are to apply quantitative methods in physiology and pathophysiology to inform translational therapeutic strategies and benefit patient care.
Footnotes
Additional information
Competing Interests
No conflicts to disclose.
Data availability statement
The source code of our new 3D human atrial cell model can be accessed from http://elegrandi.wixsite.com/grandilab/downloads and https://github.com/drgrandilab.
Reference
- Annunziato L, Secondo A, Pignataro G, Scorziello A & Molinaro P (2020). New perspectives for selective NCX activators in neurodegenerative diseases. Cell Calcium 87, 102170. [DOI] [PubMed] [Google Scholar]
- Arora R, Aistrup GL, Supple S, Frank C, Singh J, Tai S, Zhao A, Chicos L, Marszalec W, Guo A, Song L-S & Wasserstrom JA (2017). Regional distribution of T-tubule density in left and right atria in dogs. Heart Rhythm 14, 273–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beavers DL, Wang W, Ather S, Voigt N, Garbino A, Dixit SS, Landstrom AP, Li N, Wang Q, Olivotto I, Dobrev D, Ackerman MJ & Wehrens XHT (2013). Mutation E169K in Junctophilin-2 Causes Atrial Fibrillation Due to Impaired RyR2 Stabilization. Journal of the American College of Cardiology 62, 2010–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM (2002). Cardiac excitation–contraction coupling. Nature 415, 198–205. [DOI] [PubMed] [Google Scholar]
- Blatter LA & Niggli E (1998). Confocal nearmmembrane detection of calcium in cardiac myocytes. Cell Calcium 23, 269–279. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C & Kühlkamp V (1999). Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res 44, 121–131. [DOI] [PubMed] [Google Scholar]
- Brandenburg S, Kohl T, Williams GSB, Gusev K, Wagner E, Rog-Zielinska EA, Hebisch E, Dura M, Didié M, Gotthardt M, Nikolaev VO, Hasenfuss G, Kohl P, Ward CW, Lederer WJ & Lehnart SE (2016). Axial tubule junctions control rapid calcium signaling in atria. J Clin Invest 126, 3999–4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandenburg S, Pawlowitz J, Fakuade FE, Kownatzki-Danger D, Kohl T, Mitronova GY, Scardigli M, Neef J, Schmidt C, Wiedmann F, Pavone FS, Sacconi L, Kutschka I, Sossalla S, Moser T, Voigt N & Lehnart SE (2018). Axial Tubule Junctions Activate Atrial Ca2+ Release Across Species. Frontiers in Physiology 9, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carl SL, Felix K, Caswell AH, Brandt NR, Ball WJ, Vaghy PL, Meissner G & Ferguson DG (1995). Immunolocalization of sarcolemmal dihydropyridine receptor and sarcoplasmic reticular triadin and ryanodine receptor in rabbit ventricle and atrium. The Journal of Cell Biology 129, 673–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Zhang C, Guo A & Song L-S (2015). In situ single photon confocal imaging of cardiomyocyte T-tubule system from Langendorff-perfused hearts. Frontiers in Physiology 6, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Izu Y, McCulle SL, Ward CW, Soeller C, Allen BM, Rabang C, Cannell MB, Balke CW & Izu LT (2006). Three-Dimensional Distribution of Ryanodine Receptor Clusters in Cardiac Myocytes. Biophysical Journal 91, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christ T, Boknik P, Wöhrl S, Wettwer E, Graf EM, Bosch RF, Knaut M, Schmitz W, Ravens U, & Dobrev D (2004). L-Type Ca2+ Current Downregulation in Chronic Human Atrial Fibrillation Is Associated With Increased Activity of Protein Phosphatases. Circulation 110, 2651–2657. [DOI] [PubMed] [Google Scholar]
- Dagum L & Menon R (1998). OpenMP: An Industry-Standard API for Shared-Memory Programming. IEEE Comput Sci Eng 5, 46–55. [Google Scholar]
- Denham NC, Pearman CM, Caldwell JL, Madders GWP, Eisner DA, Trafford AW & Dibb KM (2018). Calcium in the Pathophysiology of Atrial Fibrillation and Heart Failure. Front Physiol 9, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Despa S, Brette F, Orchard CH & Bers DM (2003). Na/Ca Exchange and Na/K-ATPase Function Are Equally Concentrated in Transverse Tubules of Rat Ventricular Myocytes. Biophysical Journal 85, 3388–3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrev D & Wehrens XHT (2017). Calcium-mediated cellular triggered activity in atrial fibrillation. The Journal of Physiology 595, 4001–4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Armouche A, Boknik P, Eschenhagen T, Carrier L, Knaut M, Ravens U & Dobrev D (2006). Molecular Determinants of Altered Ca 2+ Handling in Human Chronic Atrial Fibrillation. Circulation 114, 670–680. [DOI] [PubMed] [Google Scholar]
- Faggioni M, Savio-Galimberti E, Venkataraman R, Hwang HS, Kannankeril PJ, Darbar D & Knollmann BC (2014). Suppression of Spontaneous Ca Elevations Prevents Atrial Fibrillation in Calsequestrin 2-Null Hearts. Circulation: Arrhythmia and Electrophysiology 7, 313–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisk M, Koivumäki JT, Norseng PA, Maleckar MM, Sejersted OM & Louch WE (2014). Variable t-tubule organization and Ca2+ homeostasis across the atria. American Journal of Physiology-Heart and Circulatory Physiology 307, H609–H620. [DOI] [PubMed] [Google Scholar]
- Gadeberg HC, Bond RC, Kong CHT, Chanoit GP, Ascione R, Cannell MB & James AF (2016). Heterogeneity of T-Tubules in Pig Hearts. PLOS ONE 11, e0156862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadeberg HC, Kong CHT, Bryant SM, James AF & Orchard CH (2017). Sarcolemmal distribution of I Ca and I NCX and Ca 2+ autoregulation in mouse ventricular myocytes. American Journal of Physiology-Heart and Circulatory Physiology 313, H190–H199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galice S, Xie Y, Yang Y, Sato D & Bers DM (2018). Size Matters: Ryanodine Receptor Cluster Size Affects Arrhythmogenic Sarcoplasmic Reticulum Calcium Release. Journal of the American Heart Association 7, e008724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glukhov AV, Balycheva M, Sanchez-Alonso JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, Diakonov I, Schobesberger S, Sikkel MB, Bhargava A, Faggian G, Punjabi PP, Houser SR & Gorelik J (2015). Direct Evidence for Microdomain-Specific Localization and Remodeling of Functional L-Type Calcium Channels in Rat and Human Atrial Myocytes. Circulation 132, 2372–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez AM, Valdivia HH, Cheng H, Lederer MR, Santana LF, Cannell MB, McCune SA, Altschuld RA & Lederer WJ (1997). Defective Excitation-Contraction Coupling in Experimental Cardiac Hypertrophy and Heart Failure. Science 276, 800–806. [DOI] [PubMed] [Google Scholar]
- Greiser M, Kerfant B-G, Williams GSB, Voigt N, Harks E, Dibb KM, Giese A, Meszaros J, Verheule S, Ravens U, Allessie MA, Gammie JS, van der Velden J, Lederer WJ, Dobrev D & Schotten U (2014). Tachycardia-induced silencing of subcellular Ca2+ signaling in atrial myocytes. J Clin Invest 124, 4759–4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Györke I, Hester N, Jones LR & Györke S (2004). The Role of Calsequestrin, Triadin, and Junctin in Conferring Cardiac Ryanodine Receptor Responsiveness to Luminal Calcium. Biophysical Journal 86, 2121–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer KP, Ljubojevic S, Ripplinger CM, Pieske BM & Bers DM (2015). Cardiac myocyte alternans in intact heart: Influence of cell–cell coupling and β-adrenergic stimulation. Journal of Molecular and Cellular Cardiology 84, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herraiz-Martínez A, Tarifa C, Jiménez-Sábado V, Llach A, Godoy-Marín H, Colino-Lage H, Nolla-Colomer C, Casabella-Ramon S, Izquierdo-Castro P, Benítez I, Benítez R, Roselló-Díez E, Rodríguez-Font E, Viñolas X, Ciruela F, Cinca J & Hove-Madsen L (2022). Influence of sex on intracellular calcium homoeostasis in patients with atrial fibrillation. Cardiovascular Research 118, 1033–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilliard FA, Steele DS, Laver D, Yang Z, Marchand SJL, Chopra N, Piston DW, Huke S & Knollmann BC (2010). Flecainide inhibits arrhythmogenic Ca2+ waves by open state block of ryanodine receptor Ca2+ release channels and reduction of Ca2+ spark mass. Journal of Molecular and Cellular Cardiology 48, 293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai IA & O’Rourke B (2004). The potential of Na+/Ca2+ exchange blockers in the treatment of cardiac disease. Expert Opinion on Investigational Drugs 13, 653–664. [DOI] [PubMed] [Google Scholar]
- Hohendanner F, Walther S, Maxwell JT, Kettlewell S, Awad S, Smith GL, Lonchyna VA & Blatter LA (2015). Inositol-1,4,5-trisphosphate induced Ca2+ release and excitation–contraction coupling in atrial myocytes from normal and failing hearts. The Journal of Physiology 593, 1459–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hove-Madsen L, Llach A, Bayes-Genís A, Roura S, Font ER, Arís A & Cinca J (2004). Atrial Fibrillation Is Associated With Increased Spontaneous Calcium Release From the Sarcoplasmic Reticulum in Human Atrial Myocytes. Circulation 110, 1358–1363. [DOI] [PubMed] [Google Scholar]
- Kawai M, Hussain M & Orchard CH (1999). Excitation-contraction coupling in rat ventricular myocytes after formamide-induced detubulation. American Journal of Physiology-Heart and Circulatory Physiology 277, H603–H609. [DOI] [PubMed] [Google Scholar]
- Knollmann BC, Chopra N, Hlaing T, Akin B, Yang T, Ettensohn K, Knollmann BEC, Horton KD, Weissman NJ, Holinstat I, Zhang W, Roden DM, Jones LR, Franzini-Armstrong C & Pfeifer K (2006). Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 116, 2510–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenaerts I, Bito V, Heinzel FR, Driesen RB, Holemans P, D’hooge J, Heidbüchel H, Sipido KR & Willems R (2009). Ultrastructural and Functional Remodeling of the Coupling Between Ca2+ Influx and Sarcoplasmic Reticulum Ca2+ Release in Right Atrial Myocytes From Experimental Persistent Atrial Fibrillation. Circulation Research 105, 876–885. [DOI] [PubMed] [Google Scholar]
- Li Y, Wei J, Guo W, Sun B, Estillore JP, Wang R, Yoruk A, Roston TM, Sanatani S, Wilde AAM, Gollob MH, Roberts JD, Tseng ZH, Jensen HK & Chen SRW (2021). Human RyR2 (Ryanodine Receptor 2) Loss-of-Function Mutations. Circulation: Arrhythmia and Electrophysiology 14, e010013. [DOI] [PubMed] [Google Scholar]
- Lotteau S, Zhang R, Hazan A, Grabar C, Gonzalez D, Aynaszyan S, Philipson KD, Ottolia M & Goldhaber JI (2021). Acute Genetic Ablation of Cardiac Sodium/Calcium Exchange in Adult Mice: Implications for Cardiomyocyte Calcium Regulation, Cardioprotection, and Arrhythmia. Journal of the American Heart Association 10, e019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugenbiel P, Wenz F, Govorov K, Schweizer PA, Katus HA & Thomas D (2015). Atrial Fibrillation Complicated by Heart Failure Induces Distinct Remodeling of Calcium Cycling Proteins. PLOS ONE 10, e0116395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüss I, Boknik P, Jones LR, Kirchhefer U, Knapp J, Linck B, Lüss H, Meissner A, Müller FU, Schmitz W, Vahlensieck U & Neumann J (1999). Expression of Cardiac Calcium Regulatory Proteins in Atrium v Ventricle in Different Species. Journal of Molecular and Cellular Cardiology 31, 1299–1314. [DOI] [PubMed] [Google Scholar]
- Macquaide N, Tuan H-TM, Hotta J, Sempels W, Lenaerts I, Holemans P, Hofkens J, Jafri MS, Willems R & Sipido KR (2015). Ryanodine receptor cluster fragmentation and redistribution in persistent atrial fibrillation enhance calcium release. Cardiovasc Res 108, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfra O, Frisk M & Louch WE (2017). Regulation of Cardiomyocyte T-Tubular Structure: Opportunities for Therapy. Curr Heart Fail Rep 14, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnyk P, Ehrlich JR, Pourrier M, Villeneuve L, Cha T-J & Nattel S (2005). Comparison of ion channel distribution and expression in cardiomyocytes of canine pulmonary veins versus left atrium. Cardiovascular Research 65, 104–116. [DOI] [PubMed] [Google Scholar]
- Molina CE, Abu-Taha IH, Wang Q, Roselló-Díez E, Kamler M, Nattel S, Ravens U, Wehrens XHT, Hove-Madsen L, Heijman J & Dobrev D (2018). Profibrotic, Electrical, and Calcium-Handling Remodeling of the Atria in Heart Failure Patients With and Without Atrial Fibrillation. Frontiers in Physiology 9, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myles RC, Burton FL, Cobbe SM & Smith GL (2011). Alternans of action potential duration and amplitude in rabbits with left ventricular dysfunction following myocardial infarction. Journal of Molecular and Cellular Cardiology 50, 510–521. [DOI] [PubMed] [Google Scholar]
- Nattel S & Dobrev D (2012). The multidimensional role of calcium in atrial fibrillation pathophysiology: mechanistic insights and therapeutic opportunities. Eur Heart J 33, 1870–1877. [DOI] [PubMed] [Google Scholar]
- Nattel S & Harada M (2014). Atrial Remodeling and Atrial Fibrillation: Recent Advances and Translational Perspectives. Journal of the American College of Cardiology 63, 2335–2345. [DOI] [PubMed] [Google Scholar]
- Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schöndube FA, Hasenfuss G & Maier LS (2010). CaMKII-Dependent Diastolic SR Ca2+ Leak and Elevated Diastolic Ca2+ Levels in Right Atrial Myocardium of Patients With Atrial Fibrillation. Circulation Research 106, 1134–1144. [DOI] [PubMed] [Google Scholar]
- Ohkusa T, Ueyama T, Yamada J, Yano M, Fujumura Y, Esato K & Matsuzaki M (1999). Alterations in cardiac sarcoplasmic reticulum Ca2+regulatory proteins in the atrial tissue of patients with chronic atrial fibrillation. Journal of the American College of Cardiology 34, 255–263. [DOI] [PubMed] [Google Scholar]
- Pannaccione A, Piccialli I, Secondo A, Ciccone R, Molinaro P, Boscia F & Annunziato L (2020). The Na+/Ca2+exchanger in Alzheimer’s disease. Cell Calcium 87, 102190. [DOI] [PubMed] [Google Scholar]
- Parikh SS, Blackwell DJ, Gomez-Hurtado N, Frisk M, Wang L, Kim K, Dahl CP, Fiane A, Tønnessen T, Kryshtal DO, Louch WE & Knollmann BC (2017). Thyroid and Glucocorticoid Hormones Promote Functional T-Tubule Development in Human-Induced Pluripotent Stem Cell–Derived Cardiomyocytes. Circulation Research 121, 1323–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pásek M, Brette F, Nelson A, Pearce C, Qaiser A, Christe G & Orchard CH (2008a). Quantification of t-tubule area and protein distribution in rat cardiac ventricular myocytes. Progress in Biophysics and Molecular Biology 96, 244–257. [DOI] [PubMed] [Google Scholar]
- Pásek M, Šimurda J & Christé G (2006). The functional role of cardiac T-tubules explored in a model of rat ventricular myocytes. Philosophical Transactions of the Royal Society A: Mathematical, Physical and Engineering Sciences 364, 1187–1206. [DOI] [PubMed] [Google Scholar]
- Pásek M, Šimurda J, Christé G & Orchard CH (2008b). Modelling the cardiac transverse-axial tubular system. Progress in Biophysics and Molecular Biology 96, 226–243. [DOI] [PubMed] [Google Scholar]
- Pásek M, Šimurda J, Orchard CH & Christé G (2008c). A model of the guinea-pig ventricular cardiac myocyte incorporating a transverse–axial tubular system. Progress in Biophysics and Molecular Biology 96, 258–280. [DOI] [PubMed] [Google Scholar]
- Restrepo JG, Weiss JN & Karma A (2008). Calsequestrin-Mediated Mechanism for Cellular Calcium Transient Alternans. Biophys J 95, 3767–3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MA, Clarke JD, Saravanan P, Voigt N, Dobrev D, Eisner DA, Trafford AW & Dibb KM (2011). Transverse tubules are a common feature in large mammalian atrial myocytes including human. American Journal of Physiology-Heart and Circulatory Physiology 301, H1996–H2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rog-Zielinska EA, Scardigli M, Peyronnet R, Zgierski-Johnston CM, Greiner J, Madl J, O’Toole ET, Morphew M, Hoenger A, Sacconi L & Kohl P (2021). Beat-by-Beat Cardiomyocyte T-Tubule Deformation Drives Tubular Content Exchange. Circulation Research 128, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush S & Larsen H (1978). A Practical Algorithm for Solving Dynamic Membrane Equations. IEEE Transactions on Biomedical Engineering BME-25, 389–392. [DOI] [PubMed] [Google Scholar]
- Sato D & Bers DM (2011). How Does Stochastic Ryanodine Receptor-Mediated Ca Leak Fail to Initiate a Ca Spark? Biophysical Journal 101, 2370–2379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato D, Shannon TR & Bers DM (2016). Sarcoplasmic Reticulum Structure and Functional Properties that Promote Long-Lasting Calcium Sparks. Biophys J 110, 382–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savio-Galimberti E, Frank J, Inoue M, Goldhaber JI, Cannell MB, Bridge JHB & Sachse FB (2008). Novel Features of the Rabbit Transverse Tubular System Revealed by Quantitative Analysis of Three-Dimensional Reconstructions from Confocal Images. Biophysical Journal 95, 2053–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotten U, Greiser M, Benke D, Buerkel K, Ehrenteidt B, Stellbrink C, Vazquez-Jimenez JF, Schoendube F, Hanrath P & Allessie M (2002). Atrial fibrillation-induced atrial contractile dysfunction: a tachycardiomyopathy of a different sort. Cardiovasc Res 53, 192–201. [DOI] [PubMed] [Google Scholar]
- Schulson MN, Scriven DRL, Fletcher P & Moore EDW (2011). Couplons in rat atria form distinct subgroups defined by their molecular partners. Journal of Cell Science 124, 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scriven DRL, Asghari P, Schulson MN & Moore EDW (2010). Analysis of Cav1.2 and Ryanodine Receptor Clusters in Rat Ventricular Myocytes. Biophys J 99, 3923–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan J, Xie W, Betzenhauser M, Reiken S, Chen B-X, Wronska A & Marks AR (2012). Calcium Leak Through Ryanodine Receptors Leads to Atrial Fibrillation in 3 Mouse Models of Catecholaminergic Polymorphic Ventricular Tachycardia. Circulation Research 111, 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd N & McDonough HB (1998). Ionic diffusion in transverse tubules of cardiac ventricular myocytes. American Journal of Physiology-Heart and Circulatory Physiology 275, H852–H860. [DOI] [PubMed] [Google Scholar]
- Shiferaw Y, Aistrup GL, Louch WE & Wasserstrom JA (2020). Remodeling Promotes Proarrhythmic Disruption of Calcium Homeostasis in Failing Atrial Myocytes. Biophysical Journal 118, 476–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sitsapesan R & Williams AJ (1994). Regulation of the gating of the sheep cardiac sarcoplasmic reticulum ca2+-release channel by luminal Ca2+. J Membarin Biol 137, 215–226. [DOI] [PubMed] [Google Scholar]
- Song L, Alcalai R, Arad M, Wolf CM, Toka O, Conner DA, Berul CI, Eldar M, Seidman CE & Seidman JG (2007). Calsequestrin 2 (CASQ2) mutations increase expression of calreticulin and ryanodine receptors, causing catecholaminergic polymorphic ventricular tachycardia. J Clin Invest 117, 1814–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swift F, Strømme TA, Amundsen B, Sejersted OM & Sjaastad I (2006). Slow diffusion of K+ in the T tubules of rat cardiomyocytes. Journal of Applied Physiology 101, 1170–1176. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Nori A, Santoro M, Viatchenko-Karpinski S, Kubalova Z, Gyorke I, Terentyeva R, Vedamoorthyrao S, Blom NA, Valle G, Napolitano C, Williams SC, Volpe P, Priori SG & Gyorke S (2006). Abnormal Interactions of Calsequestrin With the Ryanodine Receptor Calcium Release Channel Complex Linked to Exercise-Induced Sudden Cardiac Death. Circulation Research 98, 1151–1158. [DOI] [PubMed] [Google Scholar]
- Terentyev D, Viatchenko-Karpinski S, Györke I, Volpe P, Williams SC & Györke S (2003). Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proceedings of the National Academy of Sciences 100, 11759–11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafford AW, Clarke JD, Richards MA, Eisner DA & Dibb KM (2013). Calcium signalling microdomains and the t-tubular system in atrial mycoytes: potential roles in cardiac disease and arrhythmias. Cardiovasc Res 98, 192–203. [DOI] [PubMed] [Google Scholar]
- Vest JA, Wehrens XHT, Reiken SR, Lehnart SE, Dobrev D, Chandra P, Danilo P, Ravens U, Rosen MR & Marks AR (2005). Defective Cardiac Ryanodine Receptor Regulation During Atrial Fibrillation. Circulation 111, 2025–2032. [DOI] [PubMed] [Google Scholar]
- Voigt N, Heijman J, Wang Q, Chiang DY, Li N, Karck M, Wehrens XHT, Nattel S & Dobrev D (2014). Cellular and Molecular Mechanisms of Atrial Arrhythmogenesis in Patients With Paroxysmal Atrial Fibrillation. Circulation 129, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu-Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XHT & Dobrev D (2012). Enhanced Sarcoplasmic Reticulum Ca2+ Leak and Increased Na+-Ca2+ Exchanger Function Underlie Delayed Afterdepolarizations in Patients With Chronic Atrial Fibrillation. Circulation 125, 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakili R, Yeh Y-H, Yan Qi X, Greiser M, Chartier D, Nishida K, Maguy A, Villeneuve L-R, Boknik P, Voigt N, Krysiak J, Kääb S, Ravens U, Linke WA, Stienen GJM, Shi Y, Tardif J-C, Schotten U, Dobrev D & Nattel S (2010). Multiple Potential Molecular Contributors to Atrial Hypocontractility Caused by Atrial Tachycardia Remodeling in Dogs. Circulation: Arrhythmia and Electrophysiology 3, 530–541. [DOI] [PubMed] [Google Scholar]
- Wleklinski MJ, Kannankeril PJ & Knollmann BC (2020). Molecular and tissue mechanisms of catecholaminergic polymorphic ventricular tachycardia. The Journal of Physiology 598, 2817–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao A, Spitzer KW, Ito N, Zaniboni M, Lorell BH & Barry WH (1997). The restriction of diffusion of cations at the external surface of cardiac myocytes varies between species. Cell Calcium 22, 431–438. [DOI] [PubMed] [Google Scholar]
- Yeh Y-H, Wakili R, Qi X-Y, Chartier D, Boknik P, Kääb S, Ravens U, Coutu P, Dobrev D & Nattel S (2008). Calcium-Handling Abnormalities Underlying Atrial Arrhythmogenesis and Contractile Dysfunction in Dogs With Congestive Heart Failure. Circulation: Arrhythmia and Electrophysiology 1, 93–102. [DOI] [PubMed] [Google Scholar]
- Yue X, Zhang R, Kim B, Ma A, Philipson KD & Goldhaber JI (2017). Heterogeneity of transverse-axial tubule system in mouse atria: Remodeling in atrial-specific Na+–Ca2+ exchanger knockout mice. Journal of Molecular and Cellular Cardiology 108, 50–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ni H, Morotti S, Smith CER, Sato D, Louch WE, Edwards AG, Grandi E. Mechanisms of spontaneous Ca2+ release-mediated arrhythmia in a novel 3D human atrial myocyte model: I. Transverse-axial tubule variation. Submitted to The Journal of Physiology. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The source code of our new 3D human atrial cell model can be accessed from http://elegrandi.wixsite.com/grandilab/downloads and https://github.com/drgrandilab.