


Abstract
X chromosome inactivation (XCI) is an essential process, yet it initiates with remarkable diversity in various mammalian species. XIST, the main trigger of XCI, is controlled in the mouse by an interplay of lncRNA genes (LRGs), some of which evolved concomitantly to XIST and have orthologues across all placental mammals. Here, we addressed the functional conservation of human orthologues of two such LRGs, FTX and JPX. By combining analysis of single-cell RNA-seq data from early human embryogenesis with various functional assays in matched human and mouse pluripotent stem- or differentiated post-XCI cells, we demonstrate major functional differences for these orthologues between species, independently of primary sequence conservation. While the function of FTX is not conserved in humans, JPX stands as a major regulator of XIST expression in both species. However, we show that different entities of JPX control the production of XIST at various steps depending on the species. Altogether, our study highlights the functional versatility of LRGs across evolution, and reveals that functional conservation of orthologous LRGs may involve diversified mechanisms of action. These findings represent a striking example of how the evolvability of LRGs can provide adaptative flexibility to constrained gene regulatory networks.
Graphical Abstract
Graphical Abstract.
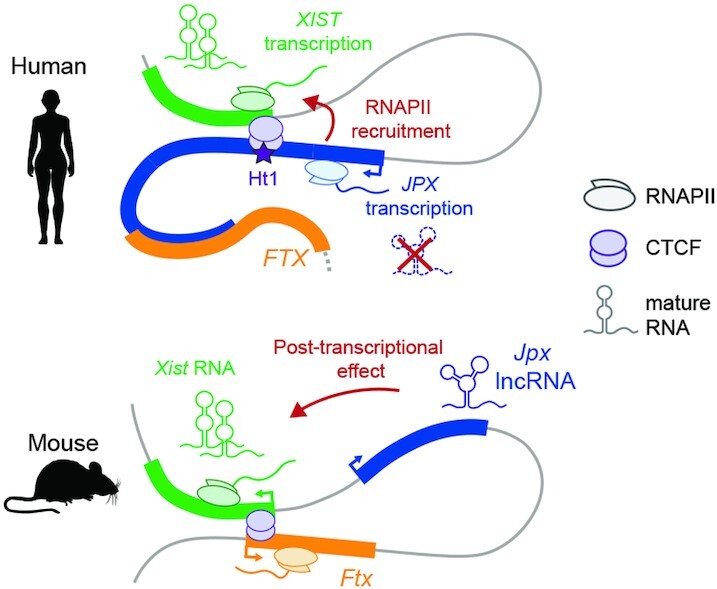
In contrast to the mouse, human JPX regulates XIST through mechanisms that are independent of JPX mature transcript, but which involve JPX transcription.
INTRODUCTION
Recent efforts in characterizing transcriptomes have revealed the abundance of long non-coding RNA (lncRNA) transcripts across tissues and species. The mechanistic dissection of lncRNA loci highlighted that their molecular function is not only mediated by the RNA molecule itself, but may also involve various entities such as the act of transcription or key regulatory elements embedded within their locus (1–4); defining these loci as lncRNA genes (LRGs) thus better depicts their mechanistic versatility.
From an evolutionary standpoint, one major challenge is that both LRGs functionality, if any, and their mechanism of action are hardly predictable based on the DNA or RNA sequence alone. For instance, it is known that syntenic LRGs often display strong primary sequence turnover during evolution, even among closely related species (5–8), whose impact on LRGs functional conservation is still poorly understood. The sharp contrast of sequence conservation between LRGs and protein-coding genes raised controversies on LRGs functionality, and experimental investigations are still lacking to provide a general understanding of the rules underlying LRGs functional conservation. X-chromosome inactivation (XCI) provides an interesting experimental paradigm to test this, since several LRGs, with orthologues conserved to various extent across placental mammals, have been shown to control this process in the mouse (9).
XCI is triggered early in development by the accumulation of the lncRNA XIST, which acts as a scaffold for multiple protein complexes involved, amongst others, in chromatin remodeling, nuclear organization and RNA modification (10). The concerted action of these ribonucleoprotein factors results in the conversion of one of the two X chromosomes in females into a compact and transcriptionally silent structure. XIST expression has to be tightly controlled in order to ensure female-restricted inactivation of a single X chromosome in a timely manner. However, it remains puzzling that such an essential process follows species-specific routes, in which the dynamics of XIST expression in early developmental stages differs markedly between mouse and human. For instance, XIST expression during human pre-implantation development initiates early on, in both male and female embryos, resulting in a biallelic XIST expression in females and a single XIST coated X chromosome in males at the early blastocyst stage. Mouse Xist expression seems to be more tightly regulated, coupled to XCI initiation, and mainly restricted to one X chromosome in females, although some cells do display transient bi-allelic Xist expression in blastocysts (11,12). The situation eventually homogenizes and Xist/XIST RNA coating becomes restricted to the sole inactivated X (Xi) in both mouse and human post-inactivation (post-XCI) cells (13). These observations raise questions regarding the functional conservation of XIST regulatory network across species.
We previously identified the transcription factor YY1 as a potent trans-activator of XIST in mouse and human (14), but the activity of non-coding regulatory elements from the genomic region surrounding XIST, known as the X-inactivation center (XIC), has never been addressed in species other than the mouse. Of note, all LRGs hosted within the mouse Xic were shown to intervene, through various mechanisms, in the regulation of Xist expression: Tsix and Linx act as major repressors while Jpx and Ftx act as positive regulators (15). The antagonistic action of the Xic-linked LRGs is likely facilitated by the spatial segregation of the Xist- and Tsix-associated regulators in two adjacent and oppositely regulated topologically associated domains (TADs) (16). These TADs also delimit internal long-range interactions to ensure contacts between regulatory elements and their target genes; intra-TAD interactions have been described between the Xist and Ftx LRGs and between Tsix promoter and Linx LRG (3,16,17). XIST, JPX and FTX emerged by pseudogenization of protein-coding genes after the divergence between eutherians and marsupials (18) and are conserved across all placental mammals. Whether their function is similar in all species deserves urgent attention, as a framework to understand the evolution of XCI strategies across mammals, and to gain insights into the principles of LRGs functional evolution at large.
Here, we investigated the regulatory network involved in the initial steps of XIST expression during human early embryogenesis. The analysis of single-cell RNA-seq data from early human embryos designated JPX, but not FTX, as a potent candidate for XIST regulation in human. Using a panel of functional approaches to target various modules of the JPX LRG, we could show that, while mature JPX transcripts are dispensable for XIST to be expressed, transcription of the JPX locus and/or nascent JPX RNA is essential to sustain XIST transcription in human post-XCI cells. This process is fostered within a sub-TAD domain that involves RNA polymerase II-mediated 3D interactions. We also re-addressed the role of mouse Jpx RNA, matching cellular models and functional approaches between human and mouse. We could identify that spliced Jpx RNA acts as a positive regulator of Xist in mouse post-XCI cells and our findings suggest that Jpx regulates Xist accumulation in a post-transcriptional manner. These findings provide a striking demonstration that the conservation of LRGs regulatory function may involve divergent mechanisms, which might confer additional plasticity to gene regulatory networks and provide species-specific opportunities to shape the regulome of target gene(s).
MATERIALS AND METHODS
Cell culture
Primary mouse embryonic fibroblasts (pMEFs) and primary fetal lung fibroblast (IMR90, ATCC CCL-186) were cultured in Dulbecco′s modified Eagle's medium (DMEM, Gibco) supplemented with 10% of heat-inactivated fetal bovine serum (FBS, Gibco), 100 U/ml of penicillin and 100 μg/ml of streptomycin (Thermo Fisher Scientific). Cells were routinely passaged 0.05% Trypsin–EDTA (Thermo Fisher Scientific) and cultured in 20% O2 and 8% CO2 at 37°C.
Female EpiSCs (gift from Alice Jouneau) were cultured using chemically defined medium (CDM) as previously defined (19), supplemented with Activin A (20ng/ml, Cell Guidance System) and Fgf2 (12 ng/ml, Cell Guidance System). EpiSCs were passaged using 4 mg/ml Collagenase II (Sigma) and then plated into plates pre-coated with fetal bovine serum.
Research on human embryonic stem cells has been approved by Agence de la Biomédecine and informed consent was obtained from all subjects. Primed hESC lines H9 (20), obtained from the WiCell Research Institute, and WIBR2 (21), obtained from the Whitehead Institute for Biomedical Research, were cultured on Matrigel-coated culture dishes (BD Biosciences) in mTeSR™1 media (Stemcells technologies) according to the manufacturer instructions, in 20% O2 and 5% CO2 at 37°C. Primed hESCs were routinely passaged in clumps using a 0.5 mM EDTA solution as previously described (22). For experiments requiring single-cell suspension, cells were incubated with Accutase (Stemcells technologies) and plated in fresh mTeSR™1 media supplemented with 10 μM of Y-27632 (Stemcells Technologies).
Derivation of primary MEFs
pMEFs were derived from 13.5 days post-coitum embryos obtained from crosses of CD1 and Crl:CD1(ICR) mice (Charles River). Embryos were manually cut, further dissociated in 0.05% Trypsin-EDTA (Thermo Fisher Scientific) and plated on gelatin-coated dishes. At confluence, pMEFs were frozen until further use (passage 1) and all experiments were performed between passage 1 and 4, from at least three independent female embryos.
LNA GapmRs and siRNAs lipofection
All LNA Gapmers (LGs) were designed using the Exiqon online tool (https://www.exiqon.com/) and the siRNA targeting YY1 was previously described (14). A non-targeting LG and siRNA were used as negative controls. LGs were lipofected using the RNAi Max transfection reagent (Invitrogen) according to the manufacturer recommendations. LGs experiments were performed at a final concentration of 50 nM (30 nM siRNAs) using a reverse transfection protocol. All samples were collected 48h post-lipofection either in TRIzol for RNA extraction or Laemmli for western blot analysis. The LGs sequences are listed below:
Control (AACACGTCTATACGC) mLG1 (GGACGCCGCCATTTTA), mLG2 (GCACATCTTTAGAAGC), mLG3 (CTCTTCTTAATGACAA); hLG1 (CGTCAGTAGAAGTTAG), hLG2 (TCGTCAGTAGAAGTTA), hLG3 (TTCGTCAGTAGAAGTT).
Total RNA extraction and RT-qPCR
Total RNAs were collected using TRIzol (Thermo Fisher Scientific) and extracted following the manufacturer's instruction. RNA Samples were treated using the DNA free Kit (Thermo Fisher Scientific) following the manufacturer recommendations. RNAs were reverse transcribed for 30 min at 50°C using the Superscript IV kit (Thermo Fisher Scientific). cDNAs were diluted 1:5 in water and transcripts expression level was assessed by real-time quantitative PCR (RT-qPCR) using the Power SYBR Green Master Mix (Thermo Fisher Scientific). All samples were run in duplicate on a ViiA-7 real-time thermal cycler (Applied Biosystems). Transcripts RNA levels were normalized against a reference gene following the 2-ΔCt method. Unless stated, the Rplp0 gene was used as a reference mouse samples and GAPDH for human samples. All the RT-qPCR primers used in this study are listed in the Supplementary Table S1.
Western blot
Total proteins were extracted with Laemmli lysis buffer (4% SDS; 20% glycerol; 10% 2-mercaptoethanol; 0.004% bromophenol blue; 0.125M Tris–HCl) and sonicated on a Bioruptor Sonication System (Diagenode, UCD-200). After 5 min denaturation at 95°C, the samples were loaded into a 4–12% gradient polyacrylamide gel (Invitrogen) for SDS-PAGE electrophoresis and transferred onto Invitrolon PVDF membranes (Invitrogen). The membranes were blocked for 1h with 5% milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween 20) and incubated overnight at 4°C with antibodies targeting YY1 (1:500, mouse sc-7341, H-10, Santa Cruz Biotechnology) or VINCULIN (1:2000, mouse V9131, Sigma Aldrich) proteins. Proteins of interest were detected using a Peroxidase-conjugated antibody (Goat anti-mouse, 1:10 000, Sigma Aldrich) with the Pierce ECL Western blotting substrate (Thermo Scientific).
Nascent RNA pulldown
Nascent RNAs were purified using the Click-iT Nascent RNA capture kit (Invitrogen). Briefly, cells were incubated for 1 h at 37°C with DMEM supplemented with 0.5 mM final of ethynyl uridine and total RNAs were extracted using TRIzol reagent. 2 μg of total RNAs were used for the biotinylation reaction using 0.5 mM of biotin azide. 1 μg of biotinylated RNAs were used for the pulldown assay using Dynabeads MyOne Streptavidin T1 magnetic beads. Reverse transcription was performed using the Superscript VILO cDNA Synthesis Kit (Invitrogen) for 1 h at 42°C. cDNAs were diluted 1:2 before RT-qPCR to quantify nascent gene expression. For each sample, a condition without EU was processed in parallel (EU–) and a 10% input (biotinylated RNA before IP) was used to assess enrichment of EU-labelled transcripts after pulldown, in both EU+ and EU– conditions. The values presented in the figures represents Δ(–Ct) values levels normalized to the nascent level of the H2A gene.
RNA-FISH
Cells preparation: Primed hESCs were grown on coverslips. pMEFs and IMR90 were centrifuged onto Superfrost Plus slides (VWR) using the Cytospin 3 Cytocentrifuge (Shandon). The cells were fixed for 10 min in a 3% paraformaldehyde solution (Electron Microscopy Science) and permeabilized for 5–10 min in ice-cold CSK buffer (10 mM PIPES; 300 mM sucrose; 100 mM NaCl; 3 mM MgCl2; pH 6.8) supplemented with 0.5% Triton X-100 (Sigma-Aldrich), 2 mM EGTA (Sigma-Aldrich) and 2 mM VRC (New England Biolabs).
Probes preparation: RNA-FISH probes were obtained after Nick translation of fosmids/BAC constructs purified using the Large Construct kit (Qiagen): 1 μg of purified DNA was labelled for 3 h at 15°C with fluorescent dUTPs (SpectrumOrange and SpectrumGreen from Abott Molecular and Cy5-UTPs from GE HealthCare Life Science). The templates used in this study are listed below:
mouse Xist (p510 from (23)), mouse Jpx (WI1-1581E8, BACPAC), human ATRX (RP11-42M11, BACPAC), human XIST (10 kb Exon 5–6 gift from Dr C. Brown, University of British Columbia.), human JPX (WI2-1796L6, BACPAC), human FTX (RP11-570B23, BACPAC), human POLA1 (RP11-11104L9, BACPAC), human XACT (RP11-35D3, BACPAC), mouse Xist intron 1 (Oligo-FISH probes were a gift from E. Heard Lab.), human XIST intron 1/2 (Oligo-FISH probes (Stellaris) are listed in Supplementary Table S2).
Hybridization: 100 ng of probes were supplemented with 1μg of Cot-I DNA (Invitrogen) and/or 3μg of Sheared Salmon Sperm DNA (Invitrogen). After precipitation, the probes were resuspended in deionized formamide (Sigma Aldrich), denatured for 7 min at 75°C and further incubated for 15 min at 37°C if Cot-I DNA was used. Probes were mixed with an equal volume of 2× Hybridization Buffer (4× SSC, 20% dextran sulfate, 2 mg/ml BSA, 2 mM VRC). Coverslips were dehydrated in 80–100% ethanol washes and incubated with the hybridization mix at 37°C overnight in a humid chamber. Next, the coverslips were washed for 4 min at 42°C three times with 50% formaldehyde/2× SSC (pH 7.2) and three times with 2× SSC. The coverslips were mounted in Vectashield plus DAPI (Vector Laboratories). RNA-FISH experiments have been performed at least twice and the percentage displayed were computed from pooled values of biological replicates.
Immunofluorescence coupled to RNA-FISH
Immunofluorescence coupled to RNA-FISH was performed as described previously (24). The antibodies used for IF H3K27me3 (Upstate, Cat#07-449), Alexa Fluor 568 nm anti-rabbit antibody (Thermo Fisher Scientific, Cat#A10042).
Microscopy and image analysis
All fluorescent microscopy images were taken on a fluorescence DMI-6000 inverted microscope with a motorized stage (Leica), equipped with a CCD Camera HQ2 (Roper Scientifics) and an HCX PL APO 100X oil objective (numerical aperture, 1.4, Leica) using the Metamorph software (version 7.04, Roper Scientifics). Depending on the cell line, 30–60 optical z-sections were collected at 0.2, 0.25 or 0.3 μm steps, at different wavelengths depending on the signal (DAPI [360 nm, 470 nm], FITC [470 nm, 525 nm], Cy3 [550 nm, 570 nm], Texas Red [596 nm, 612 nm] and Cy5 [647 nm, 668 nm]). Stacks were processed using ImageJ 1.48, and are represented as a 2D ‘maximum projection’ throughout the manuscript. The volume of XIST RNA clouds was assessed on stacks using the plugin 3D object counter from ImageJ.
Cellular fractionation
Cellular fractionation was performed on at least 5 million cells to allow precise estimation of the cells (V) and nuclei (V’) volumes. Fresh pellets of cells were resuspended in 3 volumes (V) of hypotonic buffer (20 mM HEPES pH 7; 10 mM KCl; 0.15 mM EDTA; 0.15 mM EGTA; 0.15 mM spermidine; 0.15 mM spermine). Lysis was performed by adding NP-40 (1% final, IGEPAL CA-630) and was stopped with the addition of 0.9 V of SR buffer (50 mM HEPES pH 7; 0.25 mM EDTA; 10 mM KCl; 70% sucrose; 0.15 mM spermidine; 0.15 mM spermine). The cytosolic fraction was separated from the nuclei by 5 min centrifugation at 4°C, 2000g and collected in TRIzol. The pellet of nuclei was washed in 3 V of nuclei wash buffer (10 mM HEPES pH 8; 0.1 mM EDTA; 100 mM NaCl; 25% glycerol; 0.15 mM spermidine; 0.15 mM spermine) to remove cytoplasmic contaminations. The volume of the nuclei pellet was estimated (V′) and the nuclei were resuspended in one V′ of sucrose buffer (20 mM Tris pH 7.65; 60 mM NaCl; 15 mM KCl; 0.34 M sucrose; 0.15 mM spermidine; 0.15 mM spermine). The nuclei were incubated for 30 min at 4°C with 0.29V′ of high salt buffer (900 mM NaCl; 20 mM Tris pH 7.65; 25% glycerol; 1.5 mM MgCl2; 0.2 mM EDTA) to empty the nuclei of their soluble content. After 30 min centrifugation at 4°C/10 000g, the supernatant and the pellet were collected separately in TRIzol, representing respectively the soluble and non-soluble nuclear fractions. After RT-qPCR, the absolute abundance of the transcripts (Δ-Ct) was normalized to the RNA quantity present in each fraction, from which we computed the abundance of the transcript in a given fraction.
Chromatin immunoprecipitation
ChIP experiments were performed as described previously (25). Cells were crosslinked in 1% formaldehyde (Cliniscience) for 10 min and quenched with 0.125 mM glycine for 5 min. Nuclei were extracted after 30 min incubation in Swelling Buffer (5 mM PIPES pH 8.0; 85 mM KCl; 0.5% NP-40). Samples were then sonicated in TSE150 buffer (0.1% SDS; 1% Triton; 2 mM EDTA; 20 mM Tris–HCl pH8; 150 mM NaCl) using a Bioruptor Sonication System (Diagenode, UCD-200). 1–2 μg of antibody were incubated overnight with 5–20 μg of chromatin and protein A Magnetic Beads (Thermo Scientific). The following mix was then washed in TSE150, TSE500 (20 mM Tris–HCl pH8; 2mM EDTA; 0.1% SDS; 1% Triton X-100; 500 mM NaCl), Washing buffer (10 mM Tris–HCl pH 8; 1 mM EDTA; 250 mM LiCl; 0.5% NP-40; 0.5% Na-deoxycholate), twice in TE (10 mM Tris–HCl pH 8; 1 mM EDTA) and eluted in TE/1% SDS. After reverse-crosslink (overnight, 65°C), the samples were purified using a phenol–chloroform extraction, resuspended in water and further analyzed by qPCR in duplicates on both IP and input DNA. All values were processed following the 2-ΔCt method and normalized to the input. The primers used for qPCR are available in Supplementary Table S3. The antibodies used in this study: CTCF (Millipore, Cat#07-729, Lot: 2452497), YY1 (Abcam, Cat#ab109237, Lot: GR188694-6), H3K9me3 (Diagenode, Cat#pAb-193–050, Lot: A1671-001P), RNA Pol2 (Active Motif, Cat#91151), RNA Pol2 CTD Ser5P (Active Motif, Cat#91119).
Lentivectors production
Lentiviral particles were produced by transient transfection of HEK293T cells using the calcium-phosphate transfection method. The lentiviral constructs of interest were co-transfected with pMD2.G (Addgene #12259) and psPAX2 (Addgene #12260) plasmids (kindly provided by Didier Trono). After 48 h, the culture media was collected, and lentiviral particles were concentrated by ultracentrifugation. For each construct, we assessed the lentiviral titer by infection of HEK293T with serial dilution (1:3) of the lentivirus into DMEM and FACS analysis.
CRISPR inhibition
The CRISPR inhibitor system (26) was used to inhibit JPX and FTX transcription in primed H9 hESCs. DNA oligonucleotides corresponding to the sgRNAs sequences were obtained with the online software CCTop (https://crispr.cos.uni-heidelberg.de/index.html). Oligonucleotide pairs were annealed to generate short double-stranded DNA fragments with overhangs compatible with ligation into the BsmbI-digested plasmid pLKO5.sgRNA.EFS.tGFP (Addgene #57823). Production of stable cell lines. H9 cells were infected with the dCas9-mCherry-KRAB construct (3) and sorted by FACS (INFLUX 500-BD BioSciences). A second lentiviral infection was performed with the constructs containing the sgRNAs (∼80–85% GFP positive).
Targeting sequences are: Sg-A: GCAATCACTGCGTCCTTACG, Sg-B: GACGCCTTGCAACCCCCGTA, Sg-C: GATCGCGTGGCCTGAGTCGG, sgFTX1: AAACTAGGGAAGTTGAATCG, sgFTX2: ACACTGCGGCGATTCTGGAG.
CRISPR/cas9-mediated deletion of the JPX promoter region
JPX promoter was deleted in primed H9 hESCs using the CRISPR-Cas9 system. To proceed, plasmid constructs harboring both the sgRNA sequence and the Cas9 fused to a reporter gene were used to allow subsequent selection of transfected cells by FACS. sgRNAs downstream of JPX TSS were cloned into a Cas9-GFP construct while upstream guides were cloned into a Cas9-mCherry construct. Therefore, double GFP+/mCherry + positive cells represent the fraction of cells simultaneously transfected with the two sgRNAs, where the probability for a direct deletion event was increased.
Guides design and cloning. DNA oligonucleotides corresponding to the sgRNAs sequences were obtained with the online software Zifit (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx). Oligonucleotide pairs were annealed to generate short double-stranded DNA fragments with overhangs compatible with the ligation into the BbsI-digested plasmid (pSpCas9(BB)-2A-GFP, Addgene #48138, Feng Zhang Lab). We also replaced the GFP by a mCherry reporter to produce a pSpCas9(BB)-2A-mCherry plasmid using the NEBuilder HiFi DNA Assembly Cloning Kit (New England Biolabs). The sequences of the guides are: hJU1 (GGGGCATGAAGCTTGCACCG), hJD1-(ATGCCATCACTATACATAGT), hJU3 (GGAAGCATTATTCGAGAAAT), hJD3 (AAATCTAAGTGATCCATATA).
One million of hESCs were transfected with 5 μg of plasmid DNA for each guide, using the 4D-Nucleofector system (Lonza) as recommended by the manufacturer. 48h post nucleofection, cells were sorted by FACs (INFLUX 500 BD BioSciences) and double positive GFP+ / mCherry + cells were plated onto Laminin-521 coated plates (Stemcell technologies) at low density in mTeSR™1 supplemented with 1× CloneR™ (Stemcell technologies). Individual colonies were picked and screened by PCR for deletions and inversions events using the following primers:
WT allele (GGTCCAGGACGTGGAATTTA, TCGTCAATGCAATTTCAAACA); deletion (AGGAAAAGTGGGTTTCCACA, GGGTGACAAGAGCAAGACTTC); inversion (GGGGAAATGTGAGTGAGTGG, GGGGTGCATGTTTAGTTGGT.
For each clone, the number of X-chromosomes was validated by qPCR on genomic DNA using:
Outside deletion (ATTTCTACCTTGTACCTAGCACAG, AGATTACATTCAAATCGGAGAGG)
Inside deletion (TGTGGGGGTCTCGTAGAAAA, TGCTTCACCGGTAAGGAAAA).
Capture HiC
Tilling capture probes targeting a 3 Mb locus centered around the human XIST gene (hg38, chrX:72413462–75413462) were designed and ordered through the Agilent Sure Design platform. Probes coordinates can be found in Supplementary Table 1. In situ Capture HiC was performed as previously published (27) with the following modifications. Cells were crosslinked with 2% formaldehyde. In short, 107 cells were lysed on ice in 10 mM Tris–HCl pH8.0, 10 mM NaCl, 0,2% NP40, 1× complete protease inhibitor cocktail (Roche). De-crosslinking was performed in 100 μl 0,5% SDS at 37°C for 1h under agitation (1400 rpm). Nuclei permeabilization was done by adding 50 μl of 10% Triton X-100 and 290μl H2O and incubated for 15 min at 37°C and swirling at 1400 rpm. To prepare for digestion, 50 μl of 10× DpnII buffer was added and 50μl were taken as an undigested control. At mid-day, a first 10 μl of DpnII (NEB, 50 000 U/ml) was added and incubated for 4 h at 37°C, swirling at 1400 rpm. Another 10 μl was added and incubation was carried on overnight. A final 10μl of DpnII was added the following morning and left for 4 h. DpnII was inactivated by heating at 65°C for 20 min and 50 μl were taken as a digested-unligated control. Ligation was carried by adding 800 μl of ligation cocktail containing 240U T4 ligase (Thermo Scientific 30 U/μl), 1× T4 ligase buffer, and incubated overnight at 16°C swirling at 1000 rpm. For DNA purification, samples and controls were incubated with 150 μg of Proteinase K (Eurobio) for 4 h at 65°C. Then, DNA was precipitated, resuspended and incubated with RNAseA (Thermo Scientific). DNA quantities in sample and controls were determined using the Qbit broad range kit (Thermo Scientific) and 100 ng of each were loaded on an agarose gel as quality control of the digestion and ligation steps. The purified samples were sent to the EMBL Gene Core facility in Heidelberg, Germany, for ligation products capture, library preparation and sequencing.
Capture HiC data analysis
Sequencing reads from both technical replicates were processed using the HiC-pro pipeline (28), including, reads alignment on the human reference genome (hg38, mapping quality > 23) and valid interaction pairs detection. Raw contact matrices were built by binning in genomic windows of 2, 4, 6, 8 and 10 kb, using the allValidPairs2cooler.sh utility. Correlation analysis between the raw contact matrices of both replicates was done using the hicCorrelate package from the HiCExplorer toolbox (29). The 4 kb binning was retained based on the high correlation score (R² = 0.67) and the contact matrices of both replicates were merged. The raw contact interaction frequencies of the captured region were normalized using the Iterative Correction approach (30). Insulation score was computed on the normalized contact matrices using the hicFindTads package with –minDepth 12000 –maxDepth 48 000 –step 8000 (29). Viewpoints data were extracted using the chicViewpoint package with default parameters. Chromatin loops were called on ICE normalized contact matrix using the mustache software with default parameters (31), non-significant loop interactions (FDR P-value < 0,05) were filtered before downstream analysis.
Resources for genomic data
We downloaded data generated by the ENCODE Project Consortium corresponding to CTCF, RAD21, SMC3, CEPBP, H3K27Ac and H3K4me3 ChIP-seq performed in IMR90 cells; K562 chromatin state hidden Markov model (ChromHMM) and H3K27me3 ChIP-seq. H9 RNA-seq (32) were obtained from the GEO repository under the accession numbers GSM978784, GSE62562 and H9 CTCF ChIP-seq was retrieved from the ENCODE portal. mESC CTCF ChIP-Seq data were downloaded from the GEO repository under the accession numbers GSE90994 (33). We obtained sequence conservation of the human XIC from the UCSC Genome Browser (34) corresponding to the 100 vertebrates Base-wise Conservation by PhyloP.
All heatmap from in situ Hi-C datasets (35–38) represents raw observed matrix visualized at a 5 kb resolution and were visualized using the Juicebox suit (39). Domains and loops coordinates were obtained from the corresponding studies.
For ChiA-PET datasets, long-range chromatin interactions and signals tracks were obtained from: (i) the ENCODE project (https://www.encodeproject.org) for POLR2A (ENCSR000BZY, ENCSR035PVZ) and CTCF (ENCSR000CAC, ENCSR278IZK) (40); (ii) the GEO repository for SMC1 ChIA-PET (GSE69643) (41). SMC1 ChIA-PET was processed using Juicer Tools for heatmap visualizations.
Resources for single-cell RNA-seq
RPKM (reads per kilobase millions) tables of single-cell RNAseq datasets performed on human embryos (42) were obtained from a previous analysis (24). Briefly, RPKM values were computed following a gene-based model and counts falling on regions overlapping two genes were discarded. Unless stated, we used log2 (RPKM + 0.001) as expression levels for representation and for computation of Pearson's correlation scores. For lineage assignments, we used the metadata from (43). All graphical plots were obtained using R (version 3.0.2) with the ggplot2 package (version 1.0.1).
Statistical information
Throughout the manuscript, RT-qPCR bar plots are presented as the mean value with error bars corresponding to standard deviation. The exact number of biological replicates are indicated by the value ‘n’. Statistical tests used to compute statistical significance are specified in figures legend.
Code availability
The codes used in this study are available from the corresponding author on request.
RESULTS
Identification of candidate regulators of XIST during early human embryonic development
To screen for human XIC-linked genes that contribute to the upregulation of XIST in vivo, we investigated their kinetics of expression during early embryogenesis using published single-cell RNA-seq datasets obtained from human pre-implantation embryos (Figure 1A) (42,44). XIST expression initiates between the four- and eight-cell stages (Supplementary Figure S1A), corresponding to embryonic day 4 (E4, Figure 1B), and increases hereafter, more predominantly in females than in males. While most of XIC-linked genes including FTX remained lowly expressed throughout pre-implantation development, RLIM and JPX showed the highest levels at early embryonic days, but displayed different expression trajectories (Figure 1C). The expression of the protein-coding gene RLIM was the highest at E3 in both male and female embryos and rapidly decreased in the following days, prior to XIST induction (Supplementary Figure S1B). This pattern likely reflects strong maternal inheritance of RLIM transcripts, which is consistent with previous observations made in the mouse (45).
Figure 1.

Identification of candidate regulators of XIST during early human embryonic development. (A) Single cell RNA-seq data from E3 to E7 pre-implantation embryos (42) were used to probe for XIST regulators. Also shown is the timing of XIST induction along with observed XCI dynamics. (B) XIST expression was upregulated in male and female embryos from E4. RPKM: Reads Per Kilobase Million (C) Analysis of single cell expression of XIC-linked genes from E3 to E6 revealed that JPX induction precedes that of XIST. (D) JPX was expressed with comparable kinetics in male and female embryos. (E) Combined analysis of JPX and XIST expression in single cells showed that the proportion of cells expressing JPX alone decreased during development, while the percentage of cells co-expressing the two genes increased (Chi-square test). n.s., not significant, ***P < 0.001; ****P < 0.0001.
In contrast, low levels of JPX could be detected at the 2–4 cells stage, followed by a major burst of expression at the 8-cell stage (Supplementary Figure S1A) or E4 (Figure 1D), coinciding with XIST initial induction. JPX was broadly expressed from the E4 stage onwards, independently from the sex of the embryos. JPX levels were almost double in females compared to male embryos, suggesting transcription from the two active X-chromosomes (Figure 1D). Using an RPKM threshold to define XIST and JPX expressing cells in female embryos (Supplementary Figure S1C), we found that the majority of the cells were expressing either JPX only or JPX and XIST concomitantly, with very few XIST-only expressing cells (Figure 1E). This pattern suggests that JPX activation precedes XIST induction and that the two genes become eventually co-expressed in a vast proportion of cells as development progresses. Altogether, these results point toward JPX, but not FTX, as a candidate regulator for the initial induction of XIST expression during pre-implantation development.
JPX mature RNA is dispensable for XIST expression in human
The JPX LRG derived from the pseudogenization of the protein-coding gene USPL after the divergence of eutherians and marsupials, and evolved concomitantly to XIST (46,47), although the two genes display distinct evolutionary trajectories. While XIST presents strong signs of positive selection in both intronic and exonic regions, the JPX LRG evolved through a quasi-neutral selection, as illustrated by a conservation score close to zero along the entire locus (Figure 2A). This strong sequence turnover is essentially due to species-specific integration of transposable elements in this region (48,49), resulting in poor multiple alignment of the homologous region of five eutherian species. As observed for numerous LRGs (5,8,47), signs of purifying selection on the JPX gene are concentrated toward the promoter region, including the first exon that contains two highly conserved regions of ∼20 nucleotides embedded within the mouse and human transcripts.
Figure 2.

JPX RNA is dispensable for XIST expression in human. (A) JPX genomic sequence is weakly conserved, as illustrated by the conservation score in 100 vertebrates and alignment of the human genomic region to five other mammalian species. Red shade highlights the JPX locus. (B) JPX escapes XCI in female fetal fibroblasts and primed H9 hESCs, as assessed by JPX/XIST double RNA-FISH. (C) Scheme of hLGs lipofection strategy in primed H9 hESCs and of JPX RNA isoforms (red line: LG-targeted exon). (D) JPX RNA levels were reduced following hLGs transfection, RT-qPCR, n = 3. (E) XIST RNA levels were unaffected following JPX RNA depletion in primed H9 hESC, RT-qPCR, n = 3. (F-G) JPX KD did not affect the number of cells expressing XIST (chi-square test) nor the volume of XIST RNA territory, RNA-FISH, Mann-Whitney test. Scale bars are 5 μm. Unless stated otherwise, unpaired two-tailed t-test were performed and error bars represent standard deviation; n.s., not significant; *P < 0.05, **P < 0.01 and ***P < 0.001. Number of counted cells is in brackets.
As the human and mouse genes bear limited sequence identity (48), we examined several features of JPX in human, such as its expression pattern and inactivation status in multiple human cell lines. We found that JPX expression is not restricted to pre-implantation development but is ubiquitous across a wide range of human tissues (Supplementary Figure S2A). Similarly to the pre-XCI state, JPX transcripts levels appeared consistently higher in females compared to males, suggesting expression from both active and inactive X and, thus, escape from XCI. To confirm this hypothesis, we performed RNA fluorescence in situ hybridization (RNA-FISH) to detect simultaneously sites of JPX active transcription and of XIST RNA accumulation. In both pluripotent and differentiated cellular contexts, every cell displayed two pinpoints of JPX transcription and one was associated in cis to a XIST RNA cloud in about ∼80 to 85% of cells (Figure 2B), confirming the strong tendency of JPX to escape XCI.
We next analyzed the function of JPX transcripts as they were described as the major functional component in the mouse (50). We used an LNA-GapmeR (LGs)-mediated knockdown (KD) strategy (Figure 2C) that has been previously used for the functional analysis of several lncRNAs (3,51–53). To address JPX function in an embryonic context, we carried out this analysis in primed female hESCs, with a percentage of XIST-expressing cells >90% (54). Robust depletion of JPX mature transcripts could be achieved in both H9 and WIBR2 lines (Figure 2D and Supplementary Figure S2B) using three distinct hLGs targeting different sequences within JPX second exon. In both hESC lines, XIST RNA levels and accumulation within the nuclei remained unaffected by JPX KD, as monitored by RT-qPCR (Figure 2E and Supplementary Figure S2B) and RNA-FISH (Figure 2F, G and Supplementary Figure S2C). Similar results were obtained in differentiated cells such as fetal fibroblasts (Supplementary Figure S2D, E). As previous studies reported that oligonucleotides containing LNA bases could result in transcriptional inhibition (55), we performed JPX RNA-FISH to monitor JPX nascent transcription. No difference could be seen between KD and control conditions in H9 cells, suggesting that JPX-targeting LGs are not affecting JPX ongoing transcription (Supplementary Figure S2F). These results suggest that JPX mature RNA is dispensable for XIST expression once XCI has been established.
XIST promoter anchors interactions within the XIC
Considering that JPX is the closest gene 5’ to XIST (56), we wondered whether the JPX locus could be part of the cis-regulatory landscape of XIST. Using capture HiC (cHiC), we surveyed at high resolution (bin size of 4kb) the three-dimensional chromatin organization of a 3 Mb region spanning the XIC in primed hESC H9. This revealed a partitioning of the human XIC into 3 topological associating domains (TADs) (16,57) with noticeable differences compared to the mouse syntenic region (Figure 3A). Notably, the boundaries of the XIST-associated TAD are located in the vicinity of XIST 5’ promoter region and downstream the SLC16A2 gene in human, whereas in the mouse, Xist-associated TAD is delimited by a genomic region encompassing the 3’ end of the Xist gene and the upstream region of the Rlim gene.
Figure 3.

XIST promoter anchors interactions within the XIC. (A) Capture HiC of the syntenic XIC locus in human (top) and mouse (bottom) ESC (37) with a 4kb binning window. CTCF ChIP-Seq track are displayed for each species (human : H9 ENCODE, mouse : mESC. Arrowheads position the TAD borders. (B) CTCF binding sites coincides with strong interactions between XIST and JPX (cHi-C, H9). Viewpoint anchors are displayed above the corresponding CTCF ChIP-Seq peak and corresponding interaction frequency tracks are plotted below. (C, D) XIST and JPX loci interact in IMR90 (female) and in HCT116 (male) cells (Hi-C) through CTCF-mediated loops (ChIA-PET from K562 and HCT116 cells). RNA-PolII-mediated loops between XIST promoter and JPX Ht1 locus are readily detected in IMR90 but not in HCT116 cells. Called loops are highlighted by colored arches.
The high resolution of our data reveals the complex 3D architecture of the human XIC, where the XIST promoter appeared to be a local connection hub, with multiple binding sites of the structural protein CTCF (Supplementary Figure S3A). Indeed, using publicly available ChIP-seq datasets for CTCF in H9 cells, we identified 3 binding sites in the vicinity of the XIST promoter region (Supplementary Figure S3B). The most intense CTCF ChIP-seq signal (XIST-CTCF1) is located 2.9 kb upstream of XIST transcriptional start site (TSS), while XIST-CTCF2 and -CTCF3 are located within XIST first exon, at respectively 2.2 kb and 4.6 kb of the annotated TSS.
To determine which regions of the XIC these XIST-CTCF sites were the most frequently interacting with, we extracted the interaction frequencies of each individual site using our cHiC data. This viewpoint analysis revealed that these XIST-CTCF sites show the highest interaction frequencies with two sites located within the JPX (Ht1) and FTX (Ht2) LRGs (Figure 3B and Supplementary Figure S3A), although they also interact with other CTCF binding sites delineating the XIC chromatin organization. Moreover, reciprocal viewpoints analysis using Ht1 and Ht2 as anchors confirmed their preferential interactions with XIST-CTCF sites (Figure 3B). This analysis suggests that XIST distal regulatory elements are hosted within the body of the JPX and FTX genes and brought in the XIST promoter-proximal region through CTCF mediated interactions, consistent with the Promoter-Enhancer contact model (58–60).
We also verified these observations in human female differentiated cell lines, and found that all anchors were co-occupied by CTCF and the cohesin complex, but were not enriched in chromatin marks or proteins associated with active enhancers (Supplementary Figure S3C). Inspection of the XIST chromatin organization in female differentiated cells (41,61,62) revealed that CTCF–CTCF loops formed an insulated chromatin neighborhood, hosting preferential contacts between the XIST promoter region and the JPX and FTX genes (Figure 3C). Interestingly, we found that only XIST/JPX, but not XIST/FTX, long-range interactions were associated with RNA polymerase II (RNAPII), suggesting that this loop is transcriptionally competent. This hypothesis is supported by the fact that no such RNAPII-mediated loops can be detected in male fibroblasts where XIST is not expressed (Figure 3D), although the overall organization of the XIST-associated TAD on the sole active X (Xa) chromosome is similar to that of females (Figure 3C, D) (40). Altogether, this suggests a female-specific transcriptional association of XIST and JPX that likely occurs on the Xi. This hypothesis is further supported by the high number of cells (>80%) co-transcribing XIST and JPX from the Xi as assessed by RNA-FISH in several cell lines (Figure 2B).
JPX transcription is required for proper XIST expression
As CTCF-mediated loops are thought to favor promoter-enhancer interactions to maximize interaction frequencies between genes and their regulatory elements (37,41,60,63), we hypothesized that XIST/JPX long-range interactions could provide a structural framework for JPX transcription to regulate XIST expression.
To test this hypothesis, we used a CRISPR inhibition strategy (CRISPRi) (26) in female H9 hESCs, where three guide RNAs were used independently to recruit a catalytically inactive Cas9 (dCas9) fused to a KRAB co-repressor to the JPX 5’ region, in order to prevent its transcription (Figure 4A). This system efficiently triggered the local deposition of the H3K9me3 repressive mark on a restricted region surrounding JPX TSS (Figure 4B). As a result, RNAPII recruitment to JPX TSS was compromised (Figure 4C) and transcription at the locus was severely impaired; JPX remained transcribed in less than ∼15% of the cells as assessed by RNA-FISH (Figure 4D) and JPX RNA levels were reduced by 90% (Figure 4E). RNAPII occupancy was also decreased at the Ht1 but not Ht2 (Figure 4F), indicating that this strategy efficiently reduced RNAPII processing along the 70 kb of the JPX gene. In these conditions, XIST steady-state RNA levels were significantly reduced (Figure 4G), as were the percentage of cells with XIST accumulation (Figure 4H) and focal enrichment of the H3K27me3 repressive mark (Figure 4I). In addition, the cells that retained XIST expression displayed smaller XIST RNA cloud compared to the control condition (Figure 4H), indicating that most cells in the population were affected by JPX inhibition. These results were reproduced in another primed hESCs line (WIBR2, Supplementary Figure S4A, B).
Figure 4.

JPX transcription is required for proper XIST expression. (A) Scheme of CRISPRi strategy to inhibit JPX transcription in primed H9 hESCs. (B) A strong and local enrichment of H3K9me3 was observed at JPX promoter upon CRISPRi. Results are presented as fold enrichment of H3K9me3 in cells expressing compared to cells not expressing the sgA, ChIP-qPCR, n = 4. (C) JPX CRISPRi in primed H9 cells prevented RNAPII recruitment at JPX promoter (ChIP-qPCR, n = 3). (D) The number of cells expressing JPX was strongly reduced in CRISPRi conditions, with both JPX alleles being efficiently silenced one week after lentiviral infection with the guides. Left: Representative images. Right: scoring of JPX RNA-FISH signals (Chi-square test). (E) JPX CRISPRi in primed H9 cells resulted in a strong decrease of JPX RNA levels, RT-qPCR, n = 4. (F) Inhibition of JPX transcription reduced RNAPII availability at the Ht1, ChIP-qPCR, n = 3. (G) XIST steady state RNA levels were reduced upon inhibition of JPX transcription (RT-qPCR, n = 4). (H) JPX CRISPRi resulted in a decreased number of cells expressing XIST (chi-square test) and on the volume of XIST RNA cloud (Mann-Whitney test) by RNA-FISH. (I) JPX CRISPRi in primed H9 cells led to the simultaneous loss of XIST RNA clouds (RNA-FISH) and H3K27me3 foci (IF); right panel represents the fraction of double positive cells for XIST and H3K27me3 cells, Fischer's exact test. Error bars represent standard deviation; n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired two-tailed t-tests to the empty condition unless stated otherwise. Number of counted cells is in brackets.
The loss of XIST expression upon JPX inhibition was not linked to a perturbation of the binding of YY1, a known regulator of XIST expression in human cells (14), on XIST promoter (Supplementary Figure S4C), suggesting that JPX acts through YY1-independent mechanisms. We also verified that reduced XIST expression did not result from ectopic deposition of H3K9me3 at XIST promoter due to the CRISPRi strategy (Supplementary Figure S4D).
As long-range interactions associated with RNAPII could also be detected between FTX gene body and XIST promoter in female human cells notably via the Ht2 region (Figure 3C), we tested whether FTX contributes to XIST transcriptional regulation in human, as we previously showed in the mouse (3). CRISPRi-mediated repression of FTX had little to no effect on XIST expression (Supplementary Figure S4E). In addition, FTX does not escape XCI, in contrast to mouse post-XCI cells (pMEFs), and is therefore never expressed in cis with XIST (Supplementary Figure S4F, G). Altogether, these results clearly demonstrate that transcription of JPX, but not FTX, is required to sustain XIST expression. It is important to note that we cannot exclude a role for nascent JPX transcripts as these are also impacted by the CRISPRi strategy.
JPX controls XIST in cis
To further explore the contribution of JPX transcription to XIST regulation, we generated deletion of the JPX promoter region using the CRISPR-Cas9 technology in H9 hESCs. Based on JPX promoter features and its expression in H9 cells, we designed guides RNAs to delete a ∼7 kb region encompassing the three first exons of JPX (Figure 5A, B, Supplementary Figure S5A). We used a strategy where hESCs co-transfected with two sgRNAs, each targeting a region upstream and downstream of JPX TSS, could be selected based on the expression of fluorescent genes, GFP and mCherry, respectively. Therefore, FACS-sorting of double positive cells maximized the probability of obtaining clones with a direct deletion of JPX promoter (64). This approach allowed us to interrogate, in an allele-specific manner, the contribution of JPX transcription and/or JPX DNA elements to XIST regulation and to tease apart cis- from trans-effects of the LRG.
Figure 5.
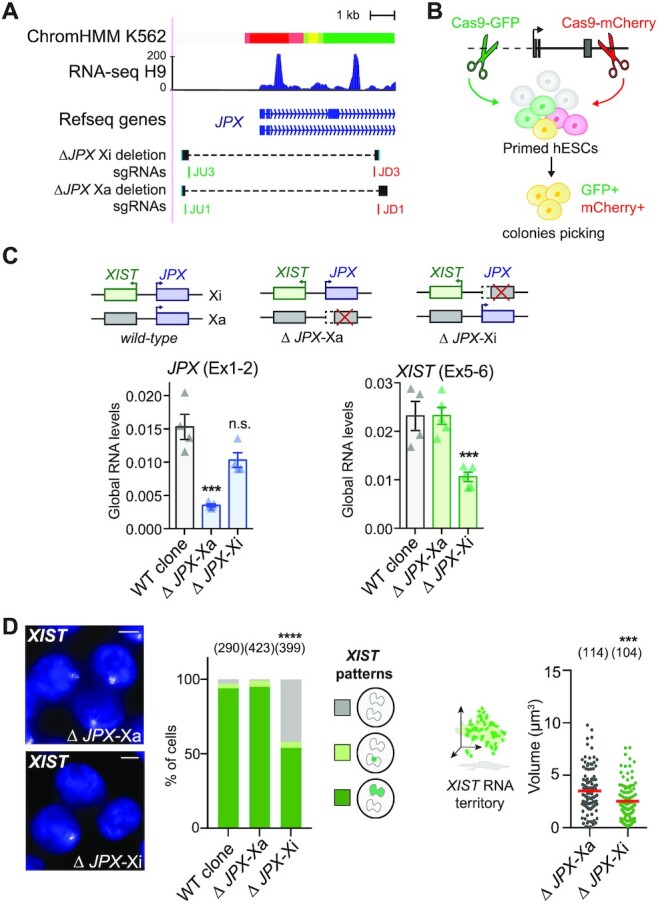
JPX controls XIST in cis. (A) Features of the JPX 5’ region. Top: chromatin state from chromHMM, with active promoter highlighted in red; below, RNA-seq in H9 and Ref-seq genes. The bottom tracks show guide RNA positions (in green, sgRNAs coupled to a Cas9-GFP; in red sgRNAs coupled to a Cas9-mCherry) and graphical depiction of the deleted region to the reference genome in the two clones. (B) Scheme of the strategy to produce genomic deletion using the CRISPR-Cas9 system coupled to FACs-sorting in hESCs. (C) ΔJPX-Xa cells displayed a ∼80% reduction of JPX RNA levels compared to WT, while a moderate decrease was observed in ΔJPX-Xi cells (∼20%), suggesting an asymmetric expression of JPX from the two X chromosomes. However, reduction of XIST RNA levels are observed only when JPX promoter was deleted in cis (ΔJPX-Xi), not in trans (ΔJPX-Xa). (RT-qPCR, n = 4). (D) In ΔJPX-Xi cells, both the number of cells expressing XIST (Chi-square test) and the volume of XIST RNA cloud (Mann-Whitney test) were reduced by RNA-FISH. Error bars represent standard deviation; n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired two-tailed t-tests to the empty condition unless stated otherwise. Number of counted cells is in brackets.
To determine on which of the two X-chromosomes (Xa or Xi) JPX was deleted, we performed simultaneous JPX and ATRX RNA-FISH. Using this strategy, we selected two clones carrying heterozygous deletions of JPX promoter region for further investigation, in which the deletion occurred either on the active X-chromosome (ΔJPX-Xa) or on the inactive X (ΔJPX-Xi) (Supplementary Figure S5B). Of note, the two clones displayed different JPX spliced RNA levels depending on the deleted allele, with the expression level in ΔJPX-Xa clone reaching only ∼15% of that of the WT clone, and ∼80% in the ΔJPX-Xi clone (Figure 5C). This is in agreement with JPX being predominantly expressed from the Xa in WT cells, as found for most genes that escape XCI (65).
Remarkably, we found that XIST expression was perturbed exclusively in the ΔJPX-Xi clone; XIST RNA levels were reduced by half compared to the WT and ΔJPX-Xa cells (Figure 5C), and only ∼55% of the cells displayed XIST RNA accumulation (Figure 5D) and H3K27me3 foci (Supplementary Figure S5C). Moreover, XIST RNA territory in the remaining XIST-positive cells were significantly smaller in the ΔJPX-Xi clone (Figure 5D), indicating that XIST expression was also impacted in those cells. Altogether these results show that transcription originating from the JPX promoter region is required in cis to sustain XIST expression in human post-XCI cells. Moreover, the fact that JPX RNA levels were the least perturbed in the ΔJPX-Xi clone confirms that JPX mature RNA is not required for XIST expression in human.
Jpx RNA regulates XIST expression in mouse post-XCI cells
Previous studies have shown that the deletion of a single allele of the Jpx gene or shRNA-mediated knockdown of Jpx is sufficient to prevent Xist upregulation (50,66,67), with Jpx acting through RNA-based mechanisms, both in cis and in trans. Considering the results we obtained in human cells, we decided to revisit the function of Jpx RNA in the mouse, by matching both our experimental approaches and the cellular models. We performed LGs-mediated KD experiments in both primary mouse embryonic fibroblasts (mEFs) and murine epiblast-derived stem cells (mEpiSCs). mEpiSCs share similarities with primed hESCs in terms of transcriptional signatures, signaling pathways and XCI status (19,68), while primary mEFs parallel the primary fibroblasts of fetal origin used in this study. Both cell types express Jpx and display an Xi coated by Xist.
First, we investigated mouse Jpx RNA function in primary mouse embryonic fibroblasts (mEFs) derived from 13.5 days post-coitum mouse embryos, using three distinct LNA Gapmers targeting different exons (Figure 6A). This approach was efficient in depleting Jpx RNA (Figure 6B), and the strongest effect was obtained with the mLG1. The three LGs induced a decrease in spliced Xist RNA levels (Figure 6C) that correlated with the extent of Jpx RNA depletion (Pearson correlation = 0.96, P-val. = 0.035), suggesting a dose-dependent effect of Jpx RNA. RNA-FISH analyses revealed that most of the cells were affected by Jpx KD, as both the percentage of Xist positive cells and the volume of the remaining Xist RNA clouds were reduced (Figure 6D, E). As previously, we verified that the observed effects of Jpx mLGs were not due to Jpx transcriptional inhibition through quantification of Jpx nascent transcripts both by RNA-FISH and after ethynyl uridine (EU) incorporation followed by EU pull-down (Figure 6F, G). We could thus conclude that the observed Xist downregulation can be attributed to the depletion of Jpx mature transcripts only, and not to alterations of Jpx ongoing transcription.
Figure 6.

Jpx RNA regulates XIST expression in mouse post-XCI cells. (A) Schematic representation of mLGs lipofection in primary MEFs; mLGs-targeted regions (red lines) are indicated on Jpx RNA isoforms. (B, C) mLGs-transfected pMEFs showed reduced Jpx and Xist RNA levels, RT-qPCR; n = 4. (D, E) Jpx KD reduced the number of Xist expressing cells (chi-square test) and the volume of Xist RNA cloud (Mann–Whitney test) by RNA-FISH. Red bars: median. (F) Jpx KD did not affect the number of cells displaying foci of nascent Jpx transcription, as shown by RNA-FISH 48h after mLGs lipofection in pMEFs. Left panel: representative images. Right panel: quantification of Jpx positive cells (blue fill). Number of counted cells is in brackets. (G) Jpx KD did not affect nascent Jpx transcription, as observed by nascent RNA pulldown of EU-labelled nascent transcripts, RT-qPCR, n = 2. (H) In EpiSCs, Jpx KD led to a decrease in Xist RNA levels, RT-qPCR, n = 3. (I) Tsix was not re-expressed in EpiSC transfected with Jpx-targeting mLGs. (J) Log2 expression fold change for a selection of markers (19,68) in EpiSCs transfected with control or Jpx-targeting mLGs, normalized to expression in mESC, RT-qPCR, n = 3. Error bars represent standard deviation; n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired two-tailed t-tests to the control LG unless stated otherwise.
Subcellular fractionation revealed that mature Jpx RNA is equally distributed between the cytoplasm and the nucleus (Supplementary Figure S6A). In order to determine which fraction of Jpx intervenes in Xist regulation, we compared the effect of Jpx siRNA vs mLG1 (both targeting Jpx exon1) on Xist accumulation (Supplementary Figure S6B). Both strategies led to strong reduction of spliced Jpx RNA levels; however, only the LNA-mediated depletion affected mature Xist levels (Supplementary Figure S6C, D). Considering that the siRNA is mainly targeting the cytoplasmic RNA fraction while the LNA gapmer is effective on nuclear transcripts, this suggest that only the nuclear fraction of Jpx has regulatory relevance for Xist RNA accumulation.
Depletion of Jpx RNA in pluripotent mEpiSCs similarly led to a decrease in Xist RNA levels (Figure 6H). This effect is likely to be direct as no ectopic expression of known mouse Xist negative regulators, such as its antisense Tsix (Figure 4I) or the pluripotency factors Rex1 and Klf4, was detected (Figure 6J) (25). These data demonstrate the contribution of mouse Jpx RNA to the maintenance of Xist expression in post-XCI cells and confirm the role of Jpx as a potent regulator of Xist.
Mechanisms of XIST regulation by JPX have diversified during evolution
To further decipher the mechanisms underlying the different molecular function of JPX, we investigated which step of Xist/XIST biogenesis is under the control of the Jpx/JPX LRG in mouse and human.
Since previous work reported that Jpx RNA could activate Xist by evicting the CTCF protein from its TSS (69), we investigated CTCF binding profile across the Xist promoter upon Jpx KD in primary mEFs. Binding of CTCF to a position ∼1 kb upstream of Xist TSS was significantly increased upon Jpx KD (Figure 7A), an effect we also observed on the imprinting control region of H19, but not on other control positions (Supplementary Figure S7A). We probed the impact of this change of CTCF binding on Xist transcription by measuring Xist premature transcripts. While decrease in Xist premature transcript levels could be observed, it was not statistically significant (Figure 7B). The conclusion that transcription of Xist remains largely unaffected by the depletion of Jpx RNA was confirmed using two orthogonal approaches, nascent RNAs pulldown (Figure 7C) and detection of ongoing transcription at the single-cell level by RNA-FISH with stranded oligo-probes detecting Xist first intron (Figure 7D). No change in Xist transcription was detected upon Jpx KD, while Xist nascent RNA levels were strongly impacted by Yy1 KD, a known regulator of Xist transcription (14) (Figure 7C, D and Supplementary Figure S7B–F). Altogether, these data demonstrate that Jpx RNA is required for proper Xist RNA accumulation but acts downstream of Xist transcription.
Figure 7.

Mechanisms of XIST regulation by JPX have diversified during evolution. (A) In the mouse, CTCF binding to Xist promoter region was increased upon Jpx KD using mLGs in pMEF, ChIP-qPCR, n = 3. (B) KD of Jpx RNA did not impact significantly on Xist premature transcript levels, intronic RT-qPCR, n = 3. (C) Xist transcription was affected by Yy1 KD, but not by Jpx KD, in pMEFs when quantified after pulldown of EU-labelled nascent transcripts, RT-qPCR, n = 3. (D) Jpx KD did not affect Xist ongoing transcription (intron 1 stranded oligo-FISH probes) but only Xist accumulation (p510 probe) (Fisher's exact test). (E) In human, JPX CRISPRi in primed H9 hESCs resulted in reduction of XIST premature transcripts levels, intronic RT-qPCR, n = 3. (F) JPX CRISPRi in primed H9 hESCs did not affect CTCF enrichment at XIST promoter nor at interaction hotspots Ht1 and Ht2, ChIP-qPCR, n = 3. (G) Inhibition of JPX transcription in primed hESCs prevented RNAPII (CTD-phospho-Serine5) recruitment at XIST promoter, ChIP-qPCR, n = 4. (H) The number of cells with XIST RNA accumulation (G1A probe) and transcription (intronic stranded oligo-FISH probes) was reduced following JPX CRISPRi in primed hESCs. Error bars represent standard deviation; n.s., not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Unpaired two-tailed t-tests to control condition unless stated otherwise.
In striking contrast, in human, intronic and exonic XIST RNA levels were strongly decreased following inhibition of JPX transcription (Figures 7E and 4G, respectively), while CTCF binding across the XIST 5’ region was unaltered (Figure 7F). This was accompanied by reduction of phospho-Ser5 RNAPII recruitment at XIST promoter (Figure 7G). Severe impairment of XIST ongoing transcription following JPX transcriptional inhibition was also evident at the single-cell level, when stranded oligo-FISH probes were used to detect human XIST intronic regions (Figure 7H). This altogether suggests a transcriptional crosstalk between JPX and XIST in human, where ongoing transcription across the JPX locus favors the recruitment of the transcription machinery at XIST, possibly through local 3D interaction and chromosomal looping. Our results therefore demonstrate that not only the functional entity of the Jpx/JPX LRG differs between human and mouse, but also the mechanism through which these orthologues regulate Xist/XIST.
DISCUSSION
Various modes of LRG emergence in eukaryotic genomes have been proposed, from pseudogenization of protein coding genes to cooption of ‘junk’ transcript in gene regulatory networks (70). How their function is conserved in the wake of genome evolution accompanying speciation has not currently been explored.
Here, by interrogating whether and how orthologous LRGs-based regulatory networks operate in different species, we provide evidence for a marked divergence in the contribution of XIC-linked LRGs in regulating XIST in human compared to mouse. Through the unbiased analysis of XIC-linked genes expression dynamics, we identified JPX as the best candidate for promoting XIST activation in human. Functional investigation in various human cellular contexts further demonstrated a major role for JPX in XIST expression. JPX acts in cis to facilitate XIST transcription, through mechanisms that involve transcription across the JPX locus, or nascent, but not spliced JPX RNA. The report of a female patient carrying an heterozygous deletion encompassing the JPX-SLC16A2 loci, in which XCI is skewed towards the intact chromosome (71) further supports our conclusion. This deletion includes FTX, but the data reported here argue against a significant contribution of FTX to XIST regulation. These findings contrast with the established role of mouse Jpx and Ftx, where both Ftx transcription and Jpx RNA are required to promote Xist transcriptional up-regulation at the onset of XCI (72).
To exclude a context-dependent effect, we revisited the function of mouse Jpx in post-XCI cellular systems that match those used to address human JPX function. The current model of mouse Jpx function at the onset of XCI is somewhat controversial, with some experimental settings suggesting Jpx acts in cis (73), while others supporting a role in trans for mature Jpx RNA in regulating the initiation Xist transcription, through the eviction of CTCF from Xist 5’ region (69). Our results in mouse post-XCI cells indicate that Jpx mature RNA is required to maintain Xist expression. We, however, could not link this regulation to a CTCF-based transcriptional control. Indeed, the nuclear fraction of Jpx RNA seems to act on Xist RNA metabolism (accumulation) downstream of the transcription step. Whether different mechanisms of Xist regulation by Jpx operate at the various stages of the XCI process (initiation versus maintenance) remains to be determined. Nevertheless, although we cannot exclude that transcription across Jpx also favors Xist transcription in the mouse, our study clearly demonstrates that JPX/Jpx operates through different regulatory mechanisms in mouse and human.
Addressing the functional conservation of LRGs is a challenge given their fast-evolutionary rate. Overexpression of human JPX RNA in trans was shown to complement heterozygous deletion of mouse Jpx during the establishment of XCI, suggesting that the human RNA might be functional in an ectopic context (67). Such rescue experiments using orthologous LRGs, as opposed to our strategy to tackle the role of mouse and human Jpx/JPX in the respective species, reveal the effect of the environment on LRG mode of action but do not interrogate LRGs’ function in their endogenous contexts. Disentangling the contribution of mature RNAs from that of the act of transcription is also challenging as many strategies, such as insertion of polyA signal, blocks transcription and the synthesis of mature, full length RNA. Intrinsic RNA-encoded regulatory activity can be indirectly probed through various experimental settings such as RNA overexpression in trans, RNA-pull-down or CRISPR-display based strategy, each with pros and cons (74). Nevertheless, the absence of XIST deregulation upon JPX mature RNA depletion in all human cellular contexts that we tested, together with the fact that deleting JPX impacts on XIST only in cis, strongly argues against a major role of JPX mature RNA during human XCI.
It is interesting to note that mouse Ftx and human JPX genes appears to act through similar mechanisms, involving the act of transcription and not the mature RNA. That they also share several features might provide clues as to the evolutionary processes underlying the mechanistic plasticity of LRGs that we have uncovered: Ftx is located 141 kb upstream of Xist in the mouse, which is comparable to the distance bridging XIST promoter to the interaction hotspot Ht1 (∼163 kb) within the human JPX; Ftx escapes XCI and interacts through CTCF-mediated loops with Xist (3). The mechanistic diversification we observed between mouse and human Jpx/JPX might therefore be a consequence of the changes within the chromatin neighborhood encompassing the XIC. It is within this preferentially interacting chromatin neighborhood, which has been reshaped between mouse and human, that constraints on the JPX locus might have favored diversification of JPX mode of action on XIST. One compelling hypothesis from this model is that XIST transcriptional cis-regulators in eutherian species could have been co-opted based on features such as linear distance from XIST promoter and local 3D organization. Importantly, this scenario is reminiscent of what has been observed for enhancer evolution (75), and is thus likely not restricted to JPX evolution but may apply to other orthologous LRGs.
It remains to be seen whether our finding that LRG orthologues may act differently in various species is relevant for processes other than XCI. Diversification of LRGs across evolution could confer molecular plasticity to developmental processes, thus contributing to species adaptability and fitness: their higher turnover offers a plausible mechanism for generating phenotypic diversity in the control of gene expression across evolution. Whether mechanistic or functional, this plasticity appears to be an essential parameter to take into consideration in the context of animal modelling of human diseases involving LRGs. Our study paves the way for systematic experimental investigations of LRGs functional conservation with the aim to provide a definitive understanding of underlying rules.
DATA AVAILABILITY
Capture HiC have been deposited in GEO under accession number GSE200317 (https://www.ncbi.nlm.nih.gov/geo/). All other data generated or analyzed during this study are included in the published article (and its supplementary information).
Supplementary Material
ACKNOWLEDGEMENTS
We thank lab members for critical evaluation of the work leading to this publication. We also thank Antonin Morillon, Pierre-Antoine Defossez, Claire Francastel, Jonathan Weitzman and Céline Morey for critical reading of the manuscript. We thank the Epigenomic, the Microscopy, the Vectorology and the BIBS Platforms, all hosted in UMR7216 Epigenetic and Cell Fate, for technical advices and access to instruments. We thank Laura Villacorta from EMBL GeneCore for NGS and Nicolas Servant for the help in the analysis of Capture Hi-C data. We acknowledge the ImagoSeine core facility of the Institut Jacques Monod, member of the France BioImaging (ANR-10-INBS-04) and the support of the Region Île-de-France (E539).
Notes
Present address: Olga Rosspopoff. Global Health Institute, School of Life Sciences, EPFL, Lausanne, Switzerland.
Contributor Information
Olga Rosspopoff, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
Emmanuel Cazottes, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
Christophe Huret, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
Agnese Loda, Directors' research, European Molecular Biology Laboratory (EMBL), Heidelberg, Germany.
Amanda J Collier, Epigenetics Programme, The Babraham Institute, Cambridge CB22 3AT, UK; Wellcome Trust – Medical Research Council Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 1QR, UK.
Miguel Casanova, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
Peter J Rugg-Gunn, Epigenetics Programme, The Babraham Institute, Cambridge CB22 3AT, UK; Wellcome Trust – Medical Research Council Cambridge Stem Cell Institute, University of Cambridge, Cambridge CB2 1QR, UK.
Edith Heard, Directors' research, European Molecular Biology Laboratory (EMBL), Heidelberg, Germany; Collège de France, Paris, France.
Jean-François Ouimette, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
Claire Rougeulle, Université Paris Cité, CNRS, Epigenetics and Cell Fate, F-75013 Paris, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme [101020423]; European Commission Network of Excellence EpiGeneSys [HEALTH-F4-2010-257082 to C.R., P.J.R.-G.]; Agence Nationale pour la Recherche [ANR-14-CE10-0017 to C.R.]; Ligue Nationale contre le Cancer (to C.R.); LabEx ‘Who Am I?’ [ANR-11-LABX-0071]; Université de Paris IdEx [ANR-18-IDEX-0001] funded by the French Government through its ‘Investments for the Future’ program; O.R. and E.C. were supported by fellowships from the French Ministry of Education and Research and O.R. also from the French Medical Research Foundation (FRM); LabEx ‘Who Am I?’; P.J.R.-G. and A.J.C. are supported by the Biotechnology and Biological Sciences Research Council [BB/M022285/1 and BB/P013406/1]; Medical Research Council [MR/J003808/1]; European Research Council Advanced Investigator award [XPRESS — AdG671027]; A.L. is supported by a European Union Marie Skłodowska-Curie Actions Individual Fellowship [IF-838408]. Funding for open access charge: Agence Nationale pour la Recherche.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cho S.W., Xu J., Sun R., Mumbach M.R., Carter A.C., Chen Y.G., Yost K.E., Kim J., He J., Nevins S.A.et al.. Promoter of lncRNA gene PVT1 is a tumor-suppressor DNA boundary element. Cell. 2018; 173:1398–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Engreitz J.M., Haines J.E., Perez E.M., Munson G., Chen J., Kane M., McDonel P.E., Guttman M., Lander E.S.. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature. 2016; 539:452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Furlan G., Gutierrez Hernandez N., Huret C., Galupa R., van Bemmel J.G., Romito A., Heard E., Morey C., Rougeulle C.. The Ftx noncoding locus controls X chromosome inactivation independently of its RNA products. Mol. Cell. 2018; 70:462–472. [DOI] [PubMed] [Google Scholar]
- 4. Paralkar V.R., Taborda C.C., Huang P., Yao Y., Kossenkov A.V., Prasad R., Luan J., Davies J.O.J., Hughes J.R., Hardison R.C.et al.. Unlinking an lncRNA from Its associated cis element. Mol. Cell. 2016; 62:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hezroni H., Koppstein D., Schwartz M.G., Avrutin A., Bartel D.P., Ulitsky I.. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Rep. 2015; 11:1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Necsulea A., Soumillon M., Warnefors M., Liechti A., Daish T., Zeller U., Baker J.C., Grützner F., Kaessmann H.. The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature. 2014; 505:635–640. [DOI] [PubMed] [Google Scholar]
- 7. Ulitsky I., Shkumatava A., Jan C.H., Sive H., Bartel D.P.. Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 2011; 147:1537–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Washietl S., Kellis M., Garber M.. Evolutionary dynamics and tissue specificity of human long noncoding RNAs in six mammals. Genome Res. 2014; 24:616–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Romito A., Rougeulle C.. Origin and evolution of the long non-coding genes in the X-inactivation center. Biochimie. 2011; 93:1935–1942. [DOI] [PubMed] [Google Scholar]
- 10. Furlan G., Rougeulle C.. Function and evolution of the long noncoding RNA circuitry orchestrating X-chromosome inactivation in mammals. Wiley Interdiscip. Rev. RNA. 2016; 7:702–722. [DOI] [PubMed] [Google Scholar]
- 11. Mutzel V., Okamoto I., Dunkel I., Saitou M., Giorgetti L., Heard E., Schulz E.G.. A symmetric toggle switch explains the onset of random X inactivation in different mammals. Nat. Struct. Mol. Biol. 2019; 26:350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sousa E.J., Stuart H.T., Bates L.E., Ghorbani M., Nichols J., Dietmann S., Silva J.C.R.. Exit from naive pluripotency induces a transient X chromosome inactivation-like state in males. Cell Stem Cell. 2018; 22:919–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vallot C., Ouimette J.-F., Rougeulle C.. Establishment of X chromosome inactivation and epigenomic features of the inactive X depend on cellular contexts. Bioessays. 2016; 38:869–880. [DOI] [PubMed] [Google Scholar]
- 14. Makhlouf M., Ouimette J.-F., Oldfield A., Navarro P., Neuillet D., Rougeulle C.. A prominent and conserved role for YY1 in Xist transcriptional activation. Nat. Commun. 2014; 5:4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ouimette J.-F., Rougeulle C.. How many non-coding RNAs does it take to compensate male/female genetic imbalance?. Adv. Exp. Med. Biol. 2016; 886:33–49. [DOI] [PubMed] [Google Scholar]
- 16. Nora E.P., Lajoie B.R., Schulz E.G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N.L., Meisig J., Sedat J.et al.. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsai C.-L., Rowntree R.K., Cohen D.E., Lee J.T.. Higher order chromatin structure at the X-inactivation center via looping DNA. Dev. Biol. 2008; 319:416–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duret L., Chureau C., Samain S., Weissenbach J., Avner P.. The Xist RNA gene evolved in eutherians by pseudogenization of a protein-coding gene. Science. 2006; 312:1653–1655. [DOI] [PubMed] [Google Scholar]
- 19. Brons I.G.M., Smithers L.E., Trotter M.W.B., Rugg-Gunn P., Sun B., Chuva de Sousa Lopes S.M., Howlett S.K., Clarkson A., Ahrlund-Richter L., Pedersen R.A.et al.. Derivation of pluripotent epiblast stem cells from mammalian embryos. Nature. 2007; 448:191–195. [DOI] [PubMed] [Google Scholar]
- 20. Thomson J.A., Itskovitz-Eldor J., Shapiro S.S., Waknitz M.A., Swiergiel J.J., Marshall V.S., Jones J.M.. Embryonic stem cell lines derived from human blastocysts. Science. 1998; 282:1145–1147. [DOI] [PubMed] [Google Scholar]
- 21. Lengner C.J., Gimelbrant A.A., Erwin J.A., Cheng A.W., Guenther M.G., Welstead G.G., Alagappan R., Frampton G.M., Xu P., Muffat J.et al.. Derivation of pre-X inactivation human embryonic stem cells under physiological oxygen concentrations. Cell. 2010; 141:872–883. [DOI] [PubMed] [Google Scholar]
- 22. Beers J., Gulbranson D.R., George N., Siniscalchi L.I., Jones J., Thomson J.A., Chen G.. Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat. Protoc. 2012; 7:2029–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Debrand E., Heard E., Avner P.. Cloning and localization of the murine Xpct gene: evidence for complex rearrangements during the evolution of the region around the Xist gene. Genomics. 1998; 48:296–303. [DOI] [PubMed] [Google Scholar]
- 24. Vallot C., Patrat C., Collier A.J., Huret C., Casanova M., Liyakat Ali T.M., Tosolini M., Frydman N., Heard E., Rugg-Gunn P.J.et al.. XACT noncoding RNA competes with XIST in the control of X chromosome activity during human early development. Cell Stem Cell. 2017; 20:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Navarro P., Oldfield A., Legoupi J., Festuccia N., Dubois A., Attia M., Schoorlemmer J., Rougeulle C., Chambers I., Avner P.. Molecular coupling of Tsix regulation and pluripotency. Nature. 2010; 468:457–460. [DOI] [PubMed] [Google Scholar]
- 26. Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A.et al.. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013; 154:442–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davies J.O.J., Telenius J.M., McGowan S.J., Roberts N.A., Taylor S., Higgs D.R., Hughes J.R.. Multiplexed analysis of chromosome conformation at vastly improved sensitivity. Nat. Methods. 2016; 13:74–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Servant N., Varoquaux N., Lajoie B.R., Viara E., Chen C.-J., Vert J.-P., Heard E., Dekker J., Barillot E.. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 2015; 16:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wolff J., Rabbani L., Gilsbach R., Richard G., Manke T., Backofen R., Grüning B.A.. Galaxy HiCExplorer 3: a web server for reproducible Hi-C, capture Hi-C and single-cell Hi-C data analysis, quality control and visualization. Nucleic Acids Res. 2020; 48:W177–W184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Imakaev M., Fudenberg G., McCord R.P., Naumova N., Goloborodko A., Lajoie B.R., Dekker J., Mirny L.A.. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods. 2012; 9:999–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roayaei Ardakany A., Gezer H.T., Lonardi S., Ay F.. Mustache: multi-scale detection of chromatin loops from Hi-C and Micro-C maps using scale-space representation. Genome Biol. 2020; 21:256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vallot C., Huret C., Lesecque Y., Resch A., Oudrhiri N., Bennaceur-Griscelli A., Duret L., Rougeulle C.. XACT, a long noncoding transcript coating the active X chromosome in human pluripotent cells. Nat. Genet. 2013; 45:239–241. [DOI] [PubMed] [Google Scholar]
- 33. Hansen A.S., Pustova I., Cattoglio C., Tjian R., Darzacq X.. CTCF and cohesin regulate chromatin loop stability with distinct dynamics. Elife. 2017; 6:e25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kent W.J., Sugnet C.W., Furey T.S., Roskin K.M., Pringle T.H., Zahler A.M., Haussler D.. The human genome browser at UCSC. Genome Res. 2002; 12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bonev B., Mendelson Cohen N., Szabo Q., Fritsch L., Papadopoulos G.L., Lubling Y., Xu X., Lv X., Hugnot J.-P., Tanay A.et al.. Multiscale 3D genome rewiring during mouse neural development. Cell. 2017; 171:557–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dekker J., Belmont A.S., Guttman M., Leshyk V.O., Lis J.T., Lomvardas S., Mirny L.A., O’Shea C.C., Park P.J., Ren B.et al.. The 4D nucleome project. Nature. 2017; 549:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hsieh T.-H.S., Cattoglio C., Slobodyanyuk E., Hansen A.S., Rando O.J., Tjian R., Darzacq X.. Resolving the 3D landscape of transcription-linked mammalian chromatin folding. Mol. Cell. 2020; 78:539–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rao S.S.P., Huntley M.H., Durand N.C., Stamenova E.K., Bochkov I.D., Robinson J.T., Sanborn A.L., Machol I., Omer A.D., Lander E.S.et al.. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Durand N.C., Robinson J.T., Shamim M.S., Machol I., Mesirov J.P., Lander E.S., Aiden E.L.. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 2016; 3:99–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li G., Ruan X., Auerbach R.K., Sandhu K.S., Zheng M., Wang P., Poh H.M., Goh Y., Lim J., Zhang J.et al.. Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell. 2012; 148:84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ji X., Dadon D.B., Powell B.E., Fan Z.P., Borges-Rivera D., Shachar S., Weintraub A.S., Hnisz D., Pegoraro G., Lee T.I.et al.. 3D chromosome regulatory landscape of human pluripotent cells. Cell Stem Cell. 2016; 18:262–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Petropoulos S., Edsgärd D., Reinius B., Deng Q., Panula S.P., Codeluppi S., Plaza Reyes A., Linnarsson S., Sandberg R., Lanner F.. Single-cell RNA-Seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell. 2016; 165:1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stirparo G.G., Boroviak T., Guo G., Nichols J., Smith A., Bertone P.. Integrated analysis of single-cell embryo data yields a unified transcriptome signature for the human pre-implantation epiblast. Development. 2018; 145:dev158501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yan L., Yang M., Guo H., Yang L., Wu J., Li R., Liu P., Lian Y., Zheng X., Yan J.et al.. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat. Struct. Mol. Biol. 2013; 20:1131–1139. [DOI] [PubMed] [Google Scholar]
- 45. Shin J., Bossenz M., Chung Y., Ma H., Byron M., Taniguchi-Ishigaki N., Zhu X., Jiao B., Hall L.L., Green M.R.et al.. Maternal Rnf12/RLIM is required for imprinted X-chromosome inactivation in mice. Nature. 2010; 467:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Elisaphenko E.A., Kolesnikov N.N., Shevchenko A.I., Rogozin I.B., Nesterova T.B., Brockdorff N., Zakian S.M.. A dual origin of the Xist gene from a protein-coding gene and a set of transposable elements. PLoS One. 2008; 3:e2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hezroni H., Ben-Tov Perry R., Meir Z., Housman G., Lubelsky Y., Ulitsky I.. A subset of conserved mammalian long non-coding RNAs are fossils of ancestral protein-coding genes. Genome Biol. 2017; 18:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chureau C., Prissette M., Bourdet A., Barbe V., Cattolico L., Jones L., Eggen A., Avner P., Duret L.. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res. 2002; 12:894–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kolesnikov N.N., Elisafenko E.A.. Comparative organization and the origin of noncoding regulatory RNA genes from X-chromosome inactivation center of human and mouse. Genetika. 2010; 46:1386–1391. [PubMed] [Google Scholar]
- 50. Tian D., Sun S., Lee J.T.. The long noncoding RNA, Jpx, is a molecular switch for X chromosome inactivation. Cell. 2010; 143:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Leucci E., Vendramin R., Spinazzi M., Laurette P., Fiers M., Wouters J., Radaelli E., Eyckerman S., Leonelli C., Vanderheyden K.et al.. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016; 531:518–522. [DOI] [PubMed] [Google Scholar]
- 52. Luo S., Lu J.Y., Liu L., Yin Y., Chen C., Han X., Wu B., Xu R., Liu W., Yan P.et al.. Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell. 2016; 18:637–652. [DOI] [PubMed] [Google Scholar]
- 53. Tripathi V., Ellis J.D., Shen Z., Song D.Y., Pan Q., Watt A.T., Freier S.M., Bennett C.F., Sharma A., Bubulya P.A.et al.. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell. 2010; 39:925–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vallot C., Ouimette J.-F., Makhlouf M., Féraud O., Pontis J., Côme J., Martinat C., Bennaceur-Griscelli A., Lalande M., Rougeulle C.. Erosion of X chromosome inactivation in human pluripotent cells initiates with XACT coating and depends on a specific heterochromatin landscape. Cell Stem Cell. 2015; 16:533–546. [DOI] [PubMed] [Google Scholar]
- 55. Beane R.L., Ram R., Gabillet S., Arar K., Monia B.P., Corey D.R.. Inhibiting gene expression with locked nucleic acids (LNAs) that target chromosomal DNA. Biochemistry. 2007; 46:7572–7580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Johnston C.M., Newall A.E.T., Brockdorff N., Nesterova T.B.. Enox, a novel gene that maps 10 kb upstream of Xist and partially escapes X inactivation. Genomics. 2002; 80:236–244. [DOI] [PubMed] [Google Scholar]
- 57. Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J.S., Ren B.. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fulco C.P., Munschauer M., Anyoha R., Munson G., Grossman S.R., Perez E.M., Kane M., Cleary B., Lander E.S., Engreitz J.M.. Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science. 2016; 354:769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Fulco C.P., Nasser J., Jones T.R., Munson G., Bergman D.T., Subramanian V., Grossman S.R., Anyoha R., Doughty B.R., Patwardhan T.A.et al.. Activity-by-contact model of enhancer-promoter regulation from thousands of CRISPR perturbations. Nat. Genet. 2019; 51:1664–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kubo N., Ishii H., Xiong X., Bianco S., Meitinger F., Hu R., Hocker J.D., Conte M., Gorkin D., Yu M.et al.. Promoter-proximal CTCF binding promotes distal enhancer-dependent gene activation. Nat. Struct. Mol. Biol. 2021; 28:152–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dowen J.M., Fan Z.P., Hnisz D., Ren G., Abraham B.J., Zhang L.N., Weintraub A.S., Schujiers J., Lee T.I., Zhao K.et al.. Control of cell identity genes occurs in insulated neighborhoods in mammalian chromosomes. Cell. 2014; 159:374–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Hnisz D., Day D.S., Young R.A.. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell. 2016; 167:1188–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun F., Chronis C., Kronenberg M., Chen X.-F., Su T., Lay F.D., Plath K., Kurdistani S.K., Carey M.F.. Promoter-enhancer communication occurs primarily within insulated neighborhoods. Mol. Cell. 2019; 73:250–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Casanova M., Moscatelli M., Chauvière L.É., Huret C., Samson J., Liyakat Ali T.M., Rosspopoff O., Rougeulle C.. A primate-specific retroviral enhancer wires the XACT lncRNA into the core pluripotency network in humans. Nat. Commun. 2019; 10:5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Carrel L., Cottle A.A., Goglin K.C., Willard H.F.. A first-generation X-inactivation profile of the human X chromosome. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:14440–14444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Carmona S., Lin B., Chou T., Arroyo K., Sun S.. LncRNA Jpx induces Xist expression in mice using both trans and cis mechanisms. PLoS Genet. 2018; 14:e1007378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Karner H., Webb C.-H., Carmona S., Liu Y., Lin B., Erhard M., Chan D., Baldi P., Spitale R.C., Sun S.. Functional conservation of LncRNA JPX despite sequence and structural divergence. J. Mol. Biol. 2020; 432:283–300. [DOI] [PubMed] [Google Scholar]
- 68. Tesar P.J., Chenoweth J.G., Brook F.A., Davies T.J., Evans E.P., Mack D.L., Gardner R.L., McKay R.D.G.. New cell lines from mouse epiblast share defining features with human embryonic stem cells. Nature. 2007; 448:196–199. [DOI] [PubMed] [Google Scholar]
- 69. Sun S., Del Rosario B.C., Szanto A., Ogawa Y., Jeon Y., Lee J.T.. Jpx RNA activates Xist by evicting CTCF. Cell. 2013; 153:1537–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Palazzo A.F., Koonin E.V.. Functional long non-coding RNAs evolve from junk transcripts. Cell. 2020; 183:1151–1161. [DOI] [PubMed] [Google Scholar]
- 71. Quesada-Espinosa J.F., Garzón-Lorenzo L., Lezana-Rosales J.M., Gómez-Rodríguez M.J., Sánchez-Calvin M.T., Palma-Milla C., Gómez-Manjón I., Hidalgo-Mayoral I., Pérez de la Fuente R., Arteche-López A.et al.. First female with Allan-Herndon-Dudley syndrome and partial deletion of X-inactivation center. Neurogenetics. 2021; 22:343–346. [DOI] [PubMed] [Google Scholar]
- 72. Loda A., Collombet S., Heard E.. Gene regulation in time and space during X-chromosome inactivation. Nat. Rev. Mol. Cell Biol. 2022; 4:231–249. [DOI] [PubMed] [Google Scholar]
- 73. Barakat T.S., Loos F., van Staveren S., Myronova E., Ghazvini M., Grootegoed J.A., Gribnau J.. The trans-activator RNF12 and cis-acting elements effectuate X chromosome inactivation independent of X-pairing. Mol. Cell. 2014; 53:965–978. [DOI] [PubMed] [Google Scholar]
- 74. Gil N., Ulitsky I.. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020; 21:102–117. [DOI] [PubMed] [Google Scholar]
- 75. Villar D., Berthelot C., Aldridge S., Rayner T.F., Lukk M., Pignatelli M., Park T.J., Deaville R., Erichsen J.T., Jasinska A.J.et al.. Enhancer evolution across 20 mammalian species. Cell. 2015; 160:554–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Capture HiC have been deposited in GEO under accession number GSE200317 (https://www.ncbi.nlm.nih.gov/geo/). All other data generated or analyzed during this study are included in the published article (and its supplementary information).