


Abstract
Transposons are mobile genetic elements prevalent in the genomes of most species. The distribution of transposons within a genome reflects the actions of two opposing processes: initial insertion site selection, and selective pressure from the host. By analyzing whole-genome sequencing data from transposon-activated Drosophila melanogaster, we identified 43 316 de novo and 237 germline insertions from four long-terminal-repeat (LTR) transposons, one LINE transposon (I-element), and one DNA transposon (P-element). We found that all transposon types favored insertion into promoters de novo, but otherwise displayed distinct insertion patterns. De novo and germline P-element insertions preferred replication origins, often landing in a narrow region around transcription start sites and in regions of high chromatin accessibility. De novo LTR transposon insertions preferred regions with high H3K36me3, promoters and exons of active genes; within genes, LTR insertion frequency correlated with gene expression. De novo I-element insertion density increased with distance from the centromere. Germline I-element and LTR transposon insertions were depleted in promoters and exons, suggesting strong selective pressure to remove transposons from functional elements. Transposon movement is associated with genome evolution and disease; therefore, our results can improve our understanding of genome and disease biology.
INTRODUCTION
Transposons are mobile genetic elements that can compose up to 85% of a metazoan genome (1). Transposon movement can introduce genetic variations and drive genome evolution (2–4), but insertions into genes and regulatory elements are often detrimental to the host, causing diseases like infertility and cancer (5–8). Therefore, understanding transposon movement provides valuable insights into genome function and disease biology (9).
Transposons differ in how they mobilize. DNA transposons use a ‘cut-and-paste’ mechanism: a transposase excises a progenitor copy and integrates it directly into a new target site (10,11). In contrast, retrotransposons replicate using a ‘copy-and-paste’ mechanism via an RNA intermediate made by transcribing an existing genomic copy of the transposon. The RNA is then converted to DNA in the cytoplasm by a transposon-encoded reverse transcriptase, and the resulting DNA is inserted into the genome by a mechanism that differs between subfamilies of retrotransposons. For example, long terminal repeat (LTR) transposons behave like retroviruses, using an integrase to insert the reverse-transcribed DNA (12). In contrast, long interspersed nuclear elements (LINEs) perform target-primed reverse transcription by nicking one genomic strand of the target site and exposing a 3′ hydroxyl, which then serves as the primer for reverse transcription, and the resulting DNA is directly incorporated into the target site (13).
The target sites of different transposons exhibit distinct structural and sequence characteristics (14). Deciphering insertion site preferences is challenging because most genomic transposon copies have been rendered immobile by mutations and truncations or repressed by DNA methylation, piRNAs (in germ cells), and siRNAs (in somatic cells) (15–19). The distribution of transposon copies in the genome reflects two ongoing processes: initial insertion and subsequent selection by the host. Some transposons actively produce new copies, but distinguishing new insertions from pre-existing ones is challenging (20). Sequencing of cells transfected with mobilization-competent transposons or infected with retroviruses has revealed initial insertion site preferences for a few transposons (21–28), but the preferences of many others remain unknown.
P-element (a DNA transposon), used by the Gene Disruption project to generate a public resource of Drosophila melanogaster strains with disrupted genes, is one of the best-studied fly transposons. More than ten independent screens using a variety of engineered P-elements have resulted in 18 214 fly lines with unselected insertions (29,30). Seventy percent of P-element insertions were reported to be near gene promoters, with most of them in 200–400 ‘hotspot’ loci (30). However, Spradling et al. analyzed the P-element insertions in the Gene Disruption project and showed that these insertions were most enriched in the sites bound by origin recognition complex (ORC) proteins which function as replication origins in tissue-culture cells (26); the apparent enrichment of P-element insertions in promoters occurred because a subset of promoters also functions as replication origins. Their study provided a mechanistic model for how P-element increases its copy number in the D. melanogaster genome (26). Subsequently, this enrichment in replication origins was reported for P-element insertions in D. simulans (31).
In D. melanogaster ovaries, piRNAs guide their PIWI protein partners, providing sequence-specific repression of transposons (15,32); lack of complementary maternal piRNAs can derepress a transposon in the ovaries of the progeny and cause hybrid dysgenesis. Alternatively, disrupting the piRNA pathway can activate multiple endogenous transposons and lead to sterility (33–35). Zhang and colleagues used such approaches to mobilize three families of transposons: the DNA transposon P-element, I-element in the LINE family, and four LTR transposons—HMS-Beagle and blood in the Gypsy subfamily, and 3S18 and Max-element in the BEL-Pao subfamily (36,37). They concluded that I-element and LTR transposons did not share insertion hotspots, but they did not perform a detailed analysis of insertion site preferences for these transposons.
The whole-genome sequencing data by Zhang et al. (36,37) provide a unique opportunity to study de novo insertions of transposons belonging to all three major families in vivo. Therefore, we used our recently developed TEMP2 algorithm (38) to identify 43 316 de novo and 237 germline insertions of these six transposons. We found that although the different families of transposons shared a preference for euchromatin and active promoters, in other ways insertion patterns for each family were distinct. Subsequent host selection alters these initial insertion patterns, resulting in substantially different germline patterns. We confirmed Spradling et al.’s finding that P-element preferred replication origins (26). We found that de novo insertions of LTR transposons were enriched in the promoters and exons of highly expressed genes and that the histone modifications H3K4me3 and H3K36me3 likely direct LTR transposons to their targets. For I-element, a very different target site preference emerged; more insertions were observed toward the telomeres of each chromosome.
Our study provides new insight into transposon insertion site preferences, revealing substantial differences among DNA, LINE and LTR transposons. Furthermore, our findings highlight two opposing forces: the innate preferences of transposon movement and the evolutionary pressure to maintain a functional genome for the host.
MATERIALS AND METHODS
Annotations
Centromere-adjacent heterochromatin regions, defined by H3K9me3 ChIP-seq in ovaries, were downloaded from our previous study (39), and the remaining regions were defined as euchromatin. Transposon and gene annotations were downloaded from Flybase (version 104).
Source of data
Accessions and mappability statistics for DNA-seq, RNA-seq, ChIP-seq and ATAC-seq data used in this study are listed in Supplementary Table S1.
Detection of de novo and germline transposon insertions
To detect new transposon insertion events, we used ‘TEMP2 insertion’ (version 0.1.4) with default parameters (38). TEMP2 detects insertions that are present in input samples and absent in the D. Melanogaster reference (dm6) genome. Insertions with any supporting reads were preserved. We used the insertion sites identified by TEMP2 for downstream analyses. To determine the genomic interval for transposon insertions that could not be precisely located because of insufficient split reads (reads that cover the boundary between the transposon and the surrounding genomic sequence), we applied a method that involved calculating the 95th percentile of fragment length (the length of the DNA fragments used to create the sequencing library, also known as the insert size) minus the read length of the library and 25 bp (the minimum sequence alignment length needed to accurately map a transposon to the genome). In the DNA-seq datasets analyzed in this study, the intervals were 183–409 bp and they were used directly to identify de novo insertions as described below. For the insertions in potential insertion intervals, we used the centers of the insertion intervals computed by TEMP2 as the insertion sites for enrichment analysis in various types of genomic regions, e.g. promoters, replication origins, exons, introns and intergenic regions. For these enrichment analyses, the inaccuracy of insertion site identification could be as large as half of the interval (92–205 bp).
For each transposon, we defined de novo insertions as those that were present in the offspring but not at the same locus in the corresponding parents (Figure 1A). Using BEDTools, we compared insertion sites/intervals from the offspring (F2 eggs for I-element and LTR transposons; F1 ovaries for P-element) with the insertion sites/intervals in their corresponding parents (F1 eggs) (Supplementary Figure S1; Supplementary Table S1), and only retained those insertion sites/intervals in the offspring that did not overlap any insertion site/intervals in the parent (40). For example, to identify I-element insertions, we used F1 eggs from the wk female to w1118 male cross as the parents and F2 eggs from the F1 female to sperm-less wk male cross as the offspring (Supplementary Figure S1A). Similarly, for LTR transposons, we used F1 eggs from the untreated female to Ago3&Aub-depleted male cross as the parents, and F2 eggs from the F1 female to sperm-less male cross as the offspring (Supplementary Figure S1B). For de novo P-element insertions, we considered F1 ovaries from the w1 female to Har male cross as the offspring and compared them with F1 eggs as the parents (Supplementary Figure S1C).
Figure 1.
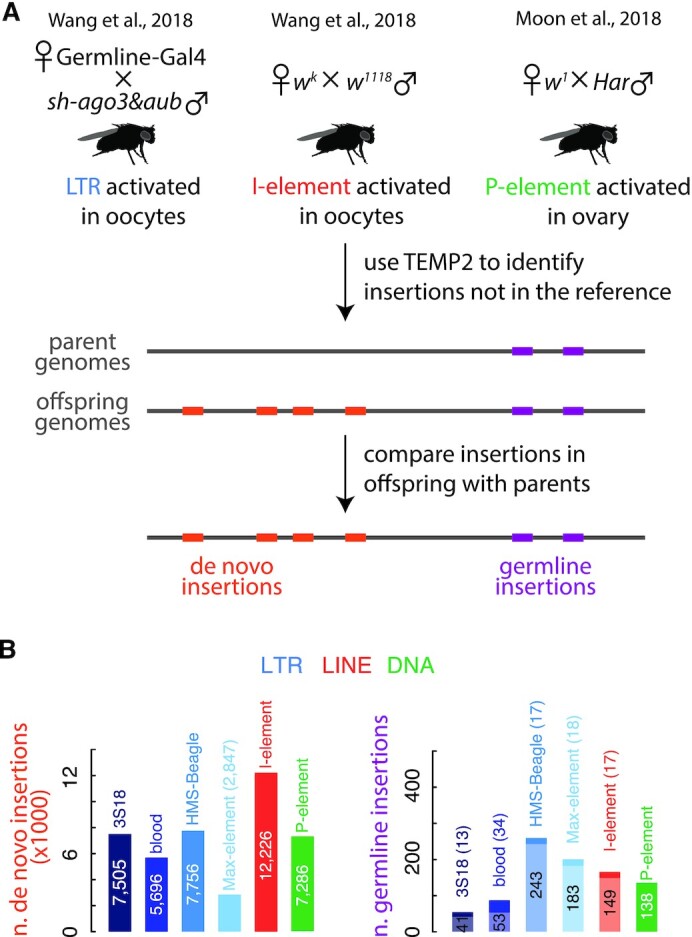
Identification of de novo and germline transposon insertions. (A) A workflow for detecting de novo and germline transposon insertions, illustrated as orange and purple boxes, respectively. First, new insertions were identified by comparison with the reference fly genome using TEMP2. Then, de novo and germline insertions were identified by comparing parent and offspring genomes. (B) Numbers of de novo and germline transposon insertions identified by our workflow. In the right panel, newly detected germline transposon insertions are marked in dark colors, and those germline insertions present in both the reference genome and the fly genome we used are marked in light colors. LTR transposons (3S18, blood, HMS-Beagle and Max-element) are colored in different shades of blue; LINE (I-element) in red; and DNA transposon (P-element) in green. This color scheme is used throughout the paper.
Germline insertions were defined as those shared by the offspring and the parents (Figure 1A). Due to the large number of reads covering germline insertions, we identified most (86.1%) of them at the resolution of target-site duplications (3–10 bp); we defined two insertions as shared when they overlapped by at least 1 bp. We supplemented our identified germline insertions with the insertions that are annotated in the fly reference genome and also present in our samples (both offspring and parents). To identify the latter, we used ‘TEMP2 absence’ (version 0.1.4) with default parameters to detect the reverse—insertions that are present in the fly reference genome but absent in our samples—and then subtracted them from the insertions annotated in the reference genome.
Assessing the false discovery rate (FDR) for identifying de novo transposon insertions
Because the number of false-positive insertions correlates with the sequencing depth of the parent sample (e.g. F1 eggs for I-element) and negatively correlates with the sequencing depth of the offspring sample (e.g. F2 eggs for I-element), and because we want to be on the conservative side with FDR calculations, for I-element and P-element false positive insertion detection by the first strategy (Supplementary Table S2B), we downsampled the offspring dataset to the same level of genome coverage (corrected for PCR duplication bias) as the parent dataset. To determine the number of insertions for each transposon type identified in the parent sample but not in the depth-matched offspring sample (Ncontrol), we then identified I-element and LTR transposon insertions for each pair of parent-offspring comparisons. Finally, we computed FDR by dividing Ncontrol over the number of de novo insertions identified in the offspring sample but not in the parent sample (Nde novo), with the latter normalized by the ratio of the PCR-duplication-corrected depth of the parent sample (F1 eggs) over the PCR-duplication-corrected depth of the offspring sample (F2 eggs).
To calculate the FDR for detecting de novo I-element and LTR transposon insertions, we used two strategies based on the knowledge that these transposons are silenced in the F0 germline, F1 carcasses, and F1 eggs (they only become activated in the F1 germline, i.e. F2 eggs): (i) we reciprocally identified the insertions in the parents but not in the offspring and (ii) we identified the insertions in the matching F1 carcasses but not in F1 eggs (Supplementary Figure S1; Supplementary Table S2; sample information listed in Supplementary Table S1). For I-element and P-element false positive insertion detection by the first strategy (Supplementary Table S2B), we downsampled the offspring dataset to the same level of genome coverage (corrected for PCR duplication bias) as the parent dataset. To determine the number of insertions for each transposon type identified in the parent sample but not in the depth-matched offspring sample (Ncontrol), we then identified I-element and LTR transposon insertions for each pair of parent-offspring comparisons. Finally, we computed FDR by dividing Ncontrol over the number of de novo insertions identified in the offspring sample but not in the parent sample (Nde novo), with the latter normalized by the ratio of the PCR-duplication-corrected depth of the parent sample (F1 eggs) over the PCR-duplication-corrected depth of the offspring sample (F2 eggs):
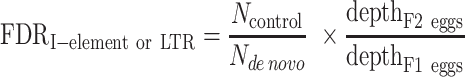 |
To compute FDR for the second strategy, we first calculated Ncontrol as the number of insertions for each transposon type identified in the F1 carcass sample but not in the depth-matched F1 egg sample, Nde novo as the number of insertions for each transposon type identified in the F2 egg sample but not in the F1 eggs sample, and computed FDR as:
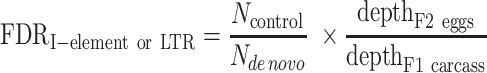 |
For P-element, we used two similar strategies to estimate the FDR of de novo insertions: (i) comparing F1 carcasses versus F1 eggs and (ii) comparing the ovaries of F1 females versus F1 ovaries. The following equations were used:
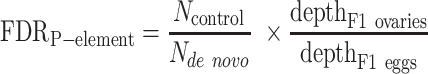 |
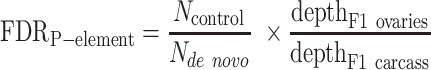 |
Calculation of PCR duplication rates for DNA-seq datasets
We first aligned each DNA-seq dataset to the D. Melanogaster reference genome (dm6) using BWA mem (41) with the parameter ‘-T 20’. We then used picard MarkDuplicates (42) to assess PCR duplication rates for each dataset.
Normalization of de novo and germline insertion numbers for genome mappability using simulated random insertions
The detection of transposon insertions using short-read whole-genome sequencing relies on finding read pairs in which one read aligns uniquely to the reference genome while the other aligns to the transposon consensus sequence. This approach is more successful in detecting transposon insertions in genomic regions with high mappability, where short reads can be uniquely aligned. We asked whether the insertions are enriched or depleted in certain types of genomic regions, including exons, introns, promoters, intergenic regions, euchromatin regions, and heterochromatin regions. Because only read pairs with one read mapping uniquely to a single location in the reference genome can be used to support transposon insertions, we needed to account for the varying levels of mappability across different genomic regions.
To quantify the mappability variation across the fly genome, we divided the genome into 100-bp nonoverlapping bins and simulated sequencing reads using the ART tool (43) by uniformly sampling the genome sequence. We then used these simulated reads to compute a mappability score for each genomic bin. Specifically, we simulated 75-bp paired-end Illumina reads (the read length in our actual sequencing data) totaling 30x genome coverage using ART (43) with parameters ‘-ss MSv3 -p -na -l 75 -f 30 -m 450 -s 10’. The simulated reads were aligned to the fly reference genome dm6 using BWA mem with parameter ‘-T 20’. If a read mapped uniquely to the loci from which it was originally obtained, the read was assigned to the corresponding 100-bp non-overlapping genomic bin. We considered a read uniquely aligned when its second-best alignment score was lower than 80% of the best alignment score (the same as the TEMP2 default). We defined the mappability scores for a genomic bin as the percentage of reads from the bin that can be uniquely aligned back to the bin. As shown in Supplementary Figure S2C, the mappability score is 0 for 12.7% of bins in the fly genome and 1 for 82.2% of the bins. However, the distributions are very different between euchromatin and heterochromatin portions of the genome, highlighting the need for normalization.
To determine whether transposon insertions are enriched or depleted in certain types of genomic regions, we simulated random insertions in each genomic bin and compared them with the actual insertions. Because most de novo insertions are supported by only one read, while germline insertions are supported by multiple reads (Supplementary Figure S2A), we used slightly different strategies to simulate random insertions for comparison with de novo and germline insertions.
For comparison with de novo insertions, we generated random insertions from each genomic bin with a probability proportional to the mappability score of the bin. This is because the likelihood of detecting a de novo insertion supported by only one read is directly correlated with the mappability of the bin in which it occurs. For example, no random insertions were generated from bins with 0% mappability, and 50% fewer random insertions were generated from bins with 50% mappability compared with bins with 100% mappability.
For comparison with germline insertions, we generated random insertions with equal probability from the mappable genomic bins, which have mappability scores greater than zero (i.e. at least one simulated read can be uniquely aligned to that bin). As expected, all 237 new germline insertions we identified are located in mappable bins. We did not scale the probability of random insertions in a bin by the bin's mappability score because most germline insertions are supported by multiple reads (Supplementary Figure S2A), while just one uniquely anchored supporting read is sufficient to detect an insertion. Another way to think about it is that for a certain probability (P) that a read can map to a genomic bin (defined by the bin's mappability score, as shown in Supplementary Figure S2C), the probability that at least one of n supporting reads can map to the bin is 1 − (1 − P)n, which approaches 1 quickly when n is large.
Assigning insertions to genomic elements
We assigned each transposon insertion to one of the following genomic elements according to their coordinates in the reference genome in decreasing priority: protein-coding gene promoters, protein-coding gene exons, protein-coding gene introns, lncRNA promoters, lncRNA exons, lncRNA introns, small noncoding RNA promoters, small noncoding RNA exons, and intergenic regions. Promoters were defined as regions ±500 bp of TSSs. Transposon insertions in protein-coding gene exons were further assigned to CDSs, 5′-UTRs, and 3′-UTRs in decreasing priority. To determine whether insertion sites were enriched in key epigenetic marks, we divided insertions into five groups: protein-coding gene promoters, protein-coding gene exons, protein-coding gene introns, promoters and gene bodies of noncoding RNAs, and intergenic regions. Furthermore, throughout our study, we used the replication origins defined by Spradling et al. using ChIP-seq data of ORC proteins in fly cell lines (26).
Assessing sequencing coverage at different genomic elements
We filtered out repetitive regions identified by RepeatMasker (44) and low-mappability regions as described above. Using only properly paired reads with BWA (41) map scores higher than 10, we then calculated the sequencing coverage for all 19 datasets we used in defining de novo insertions in promoters, exons and introns of protein-coding genes and intergenic regions and replication origins (41). In addition, we calculated the average coverages at replication origins ±1k bp and the transcription start sites (TSSs) of protein-coding genes ± 1k bp. To account for the difference in sequence depths across datasets, we further normalized the coverages by the average sequencing depth of the genomic regions, excluding repetitive and low-mappability regions.
Random simulation of promoters with de novo P-element insertions
To test whether de novo P-element insertions and EY P-element insertions tend to land in the same promoters, we first randomly drew two sets of promoters as those with de novo or EY insertions (e.g. n = 2349 for de novo and n = 3347 for EY when a promoter is defined as TSS ± 500 bp). We then compared these two sets of randomly drawn promoters with each other to calculate the numbers of shared promoters, promoters unique to each set, as well as the percentages of the promoters in each of these three groups that overlapped replication origins. We repeated this procedure 1000 times and reported the mean and standard deviations of these measures.
RNA-seq data analysis
We collected six RNA-seq datasets in fly ovaries from Moon et al. and three RNA-seq datasets in fly oocytes from Hocaoglu et al. (37,45). RNA-seq data was processed by first removing reads mapping to rRNAs using Bowtie2 (version 2.3.5.1) with default parameters (46). After filtering, reads were aligned to the dm6 genome using STAR (version 020201) with default parameters (47). Here, and throughout the study, SAMtools (version 1.10) was used to convert alignment results files from SAM to BAM format (48). HTSeq (version 0.11.1) with default parameters was then used to count uniquely mapped reads (49). For each gene, we calculated its expression level (in RPKM) by normalizing the number of uniquely mapped reads to the full length of the gene transcript. Pearson's correlation coefficients of logged gene expression were >0.96 between the three oocyte RNA-seq datasets (45) and >0.97 between the six ovary RNA-seq datasets (37) (Supplementary Table S3); therefore, we used the average gene expressions for oocytes and ovaries, respectively, for downstream analyses (Pearson's correlation coefficient between them is 0.83).
To assess the enrichment of transposon insertions near TSSs (±5 kb; Supplementary Figure S6A) or protein-coding genes (±500 bp; Supplementary Figure S6B), we divided the gene list into five groups based on expression level (measured in RPKM: <0.1, [0.1–1), [1–10), [10–100) and ≥100). Meta-analysis was then performed on each group. To analyze the enrichment of transposon insertions at each genomic element (promoters, exons, introns, CDS, 5′-UTR, 3′-UTR), protein-coding genes were divided into 20 equal groups by expression level from low to high.
ChIP-seq and ATAC-seq data analysis
To process ChIP-seq and ATAC-seq data, we mapped reads to the dm6 genome using Bowtie2 with the parameter ‘very-sensitive’. Fold enrichment of ChIP signal over input was computed from the alignment files (.bam) using MACS2 (version 2.2.7.1) bdgcmp with default parameters (50). Enrichment of ATAC signal was calculated by normalizing read density with sequencing depth. For peak calling of ATAC-seq and ChIP-seq of Pol II, H3K4me1, H3K4me2, H3K4me3, H3K9ac, H3K18ac, H3K27ac, H4K8ac, H1, H2AV, H2B and H3, we identified narrow peaks using MACS2 callpeak with default parameters. For CHIP-seq of H3K36me1, H3K36me2, H3K36me3, H3K36, H3K9me1, H3K9me2, H3K9me3, H3K27me1, H3K27me2 and H3K27me3, we used MACS2 callpeak with the parameter ‘broad’.
Assessing insertions and epigenetic marks across protein-coding genes
We separated exons into three categories: first, intermediate and last exon. Exons of single-exon genes were classified as both first and last exons. Using the same classification, we separated introns into first, intermediate, and last introns. To produce an overview of the profile of transposon insertions and epigenetic marks, we aggregated the enrichment in the following order: first exon, first intron, intermediate exon, intermediate intron, intermediate exon, last intron and last exon. We separated each exon and intron into 20 equally sized bins. We then calculated the enrichment of transposon insertions over random insertions, enrichment of ChIP-seq signal over input, and normalized ATAC-seq signal in each exon and intron bin. Exons and introns were normalized by size using the formula,
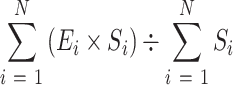 |
where E denotes enrichment or signal of insertions or epigenetic marks, S bin size and N total number of exons or introns. Finally, enrichment or signal was aggregated for each bin of each type of exon or intron. Size normalization enables longer exons and introns to contribute more to the aggregated signal than short exons and introns.
Investigating possible truncations of new I-element and P-element insertions
We wanted to determine if de novo and new germline insertions were truncated. To do this, we analyzed the distribution of the locations of insertion-supporting reads relative to the transposon's consensus sequence. To minimize the impact of chimeric reads, TEMP2 usually removes singleton supporting reads that map to the center of the consensus sequence when analyzing de novo insertions (38); However, we did not perform this step while analyzing transposon truncation. Instead, to determine the frequency of transposon truncation we used all singleton insertion-supporting reads to compute the distribution of their positions relative to the consensus sequence. For germline insertions, which are typically supported by multiple reads, TEMP2 does not filter out supporting reads that map to the transposon center, and these reads were included in our analysis of transposon truncation by default.
RESULTS
Identification of de novo transposon insertion sites
Two forces shape the transposon profiles of metazoan genomes: initial insertion and post-insertion selection (14,51–52). Therefore, to determine initial insertion site preferences, we must locate de novo insertions before they are subject to selection pressures. Here, we investigate transposition site preferences using data from two published studies in which Zhang and colleagues experimentally activated transposons in D. melanogaster ovaries using hybrid dysgenic crosses (36,37). Wang et al. used two methods to activate transposons. First, they created a hybrid dysgenic cross using two fly strains: w1118 males, which had I-element transposons and I-element-targeting piRNAs, and wk females, which did not have either. Without maternal I-element-targeting piRNAs, I-element became active in the progeny gonads, causing infertility. Second, they depleted the core piRNA pathway proteins Ago3 and Aub, leading to uncontrolled transposition in the oocytes. Similarly, Moon et al. mobilized P-elements by mating P-element-lacking w1 females with P-element-containing Har males, causing infertility in their progeny (F1).
We re-analyzed the whole-genome resequencing data from these transposon reactivated flies using our recently developed transposition detection software TEMP2 and classified transposon insertions into de novo and germline insertions by comparing offspring with their corresponding parents (Figure 1A; Supplementary Figure S1; see Materials and Methods) (38). We focused on highly activated transposons with >1000 de novo insertions, altogether identifying 43 316 de novo insertions in oocytes or ovaries for three transposon families: 12 226 insertions for I-element, which belongs to the LINE family, 7286 insertions of P-element, which is a DNA transposon; and for the LTR family, we identified 7756 HMS-Beagle, 7505 3S18, 5696 blood and 2847 Max-element insertions (Figure 1B, left panel; Supplementary Table S4). In addition to the de novo insertions, we detected 237 new (i.e. not in the reference D. melanogaster genome) insertions shared between the progeny and a parent sample (Figure 1B, right panel; Supplementary Table S4). Of these germline insertions, 138 were P-elements, and 99 were from the other five transposons. Only wild flies possess P-element, and the reference genome is from a laboratory strain; therefore, the reference genome lacks P-element insertions but contains insertions for the other transposons. For insertions supported by many sequencing reads (36,37), we combined the newly identified inherited insertions and the reference-genome-annotated insertions supported by our sequencing reads (Methods), collectively calling them germline insertions. Such germline insertions should be under selective pressure from the host (Figure 1B, right panel; Supplementary Table S4).
De novo insertions were present in only a few cells; therefore, 79.7% were singleton insertions, supported by a single paired-end read (Supplementary Figure S2A). Consequently, the median frequency of the identified de novo insertions was 0.042. The 20.3% de novo insertions supported by multiple reads could be caused by mosaicism (insertions at an early developmental stage) or PCR duplicates in the sequencing libraries. High PCR duplication rates (22–57%) in the F2 egg libraries (Supplementary Figure S2B) suggest that many of their multi-read de novo LTR and I-element insertions were introduced by PCR. In contrast, the ovary libraries showed <10% PCR duplication rates (Supplementary Figure S2B), indicating the existence of de novo P-element insertions at early developmental stages, likely because P-element is an active transposon.
In contrast, the median frequencies of the newly identified germline insertions were 0.48 for 3S18, 0.37 for blood, 0.55 for HMS-Beagle, 0.39 for Max-element, 0.28 for I-element and 0.33 for P-element. Only four of the 237 were singleton insertions, revealing a significantly broader distribution for germline insertion frequencies in oocytes and ovaries (Supplementary Figure S2A). There are several reasons that the frequency of a germline insertion might be lower than 0.5 in F1 ovaries. One reason is that some germline insertions are absent in some sequenced individuals. Among the 138 germline P-element insertions in the Har strain, 29 have frequencies <0.9, and 17 have frequencies <0.5. The frequencies of all P-element insertions are halved in the dysgenic F1 (w1 × Har) ovaries (Supplementary Figure S3B), as previously shown in a similar data set (38). The mean frequency for monomorphic P-element insertions in Har ovaries was 0.38 in the dysgenic F1 ovaries, still lower than the expected value of 0.5. A similar pattern is observed for the germline insertions of other transposons, but the mean frequency for monomorphic insertions of non-P-element transposons in Har ovaries is 0.45 in the dysgenic F1 ovaries, close to the expected value of 0.5 (Supplementary Figure S3C). The lower-than-expected insertion frequency of P-element in the dysgenic F1 ovaries may be because P-element is derepressed in these ovaries. P-element is a DNA transposon, and it moves through a cut-and-paste mechanism. A germline P-element can excise itself from the genome of a fly and reinsert itself into another location in the genome, resulting in a de novo insertion. This process could lead to a decrease in the frequency of the germline P-element. To test this hypothesis, we analyzed the data on the non-dysgenic, reciprocal-cross F1 (Har × w1) ovaries, where P-element remains repressed. Indeed, the mean frequency of monomorphic P-element insertions in Har ovaries is 0.44 in the non-dysgenic F1 ovaries, which is close to the expected value of 0.5 (Supplementary Figure S3D). Another reason is that newly inserted P-elements may contain large internal truncations, which can hinder the detection of some insertion-supporting reads and lead to a decrease in the computed insertion frequencies. Finally, some germline insertions, particularly paternally inherited P-element or I-element insertions, may be under selection, which can cause their frequencies to be lower than 0.5 in F1 ovaries.
Because most de novo insertions are supported by a single paired-end read, either chimeric reads in the progeny library or false-negative detection in the parent library could lead to false positives (38,53). To investigate the FDR of our pipeline for identifying de novo insertions, we took advantage of the fact that LTR transposons and I-element are inactive in the parents (F1 eggs) and the soma (F1 carcasses). Therefore, we used our pipeline to identify insertions present in parent or soma samples but not in the progeny sample—false positives—and calculated the FDR for each transposon (Supplementary Figure S3A; Materials and Methods). After normalization by sequencing depth, we found fewer than seven false-positive LTR or I-element insertions among all the comparisons, corresponding to FDRs <6.31 × 10−4 (Supplementary Table S2). Despite the repression by P-element-targeting piRNAs, P-element is active in the germline of all flies that it has invaded, including in F1 eggs and carcasses (presumably from the paternal (Har) germline), albeit at a much lower level than in F1 ovaries, which also lack maternal P-element-targeting piRNAs. We identified 34 P-element insertions in the F1 egg sample that were absent in the F1 ovary sample and 135 non-overlapping insertions in the F1 carcass sample that were absent in the F1 ovary sample (333 and 361 insertions, respectively, after normalizing by sequencing depth, to be compared with the 7286 de novo P-element insertions in F1 ovaries and not in F1 eggs), corresponding to FDR estimates of 0.045 and 0.050, respectively (Supplementary Table S2). However, some of these 34 and 135 false-positive P-element insertions may be bona fide de novo insertions; thus, the true FDR of our pipeline for detecting de novo P-element insertions is lower than 0.05. Further supporting our approach of FDR estimation, we detected similar numbers of insertions in the parent or the soma sample but not in the progeny sample to those in the progeny sample but not in the parent sample for other transposons (Supplementary Figure S3A, gray dots fall on the diagonal),. In conclusion, with an FDR of less than 0.05, we are confident in most of the 43 316 de novo transposon insertions identified by our pipeline.
In summary, we identified 43 316 de novo insertions at an FDR of <0.05 and 237 new germline insertions for six transposons from three transposon families. The de novo insertions occur at low frequencies and have few supporting reads, suggesting that they are not subject to post-insertion selection.
De novo P-element insertions are enriched in replication origins
To examine the insertion preferences for all six transposons at the gene level, we divided the D. melanogaster genome into promoters (TSS ± 500 bp), exons, introns, and intergenic regions and measured the percentage of each transposon's insertions in each type of regions. For comparison, we simulated 10 000 random insertions across the genome repeatedly 100 times and assigned them to genic or intergenic regions (Materials and Methods). Insertions around long-noncoding RNAs (lncRNAs) or small noncoding RNAs (ncRNAs) were uncommon. All six transposons exhibited lower enrichment in ncRNA genes than in protein-coding genes (Figures 2A and 3D). One explanation for this result is that lncRNAs and small ncRNAs are expressed at substantially lower levels than protein-coding genes in D. melanogaster ovaries (median expression = 0.006, 0.02 and 2.97 RPKM for lncRNAs, small ncRNAs, and protein-coding genes, respectively; Wilcoxon rank-sum test P-values < 2.2 × 10−16) and oocytes (median expression = 0.05, 0 and 8.12 RPKM for lncRNAs, small ncRNAs, and protein-coding genes, respectively; P-values < 2.2 × 10−16). Therefore, we focused on protein-coding genes in subsequent analyses.
Figure 2.

De novo insertions prefer while germline insertions disfavor promoters and exons. (A) Fold enrichment of de novo and germline transposon insertions in promoters, exons, and introns of protein-coding genes and intergenic regions. The fold enrichment in each genomic element has been normalized by their mappability, i.e. the percentage of mappable nucleotides. For all transposons, insertions in each genomic element differ significantly from random insertions (multiple testing corrected t-test q-values < 0.001). (B) Average fold enrichment of the observed over the expected number of insertions in the ±5 kb window (100 bps per bin) centered on the TSSs of protein-coding genes. (C) Numbers of promoters (TSS ± 500 bp) with de novo P-element insertions and EY P-element insertions, and the percentage of these promoters overlap with replication origins. Promoters with only de novo P-element insertions are colored in green, promoters with only EY P-element insertions are in purple, and promoters with both types of P-element insertions are in blue. We randomly sampled two sets of active promoters (>1 RPKM) as negative controls, and the numbers and percentages for them are shown in black as mean ± standard deviation. (D) Number of transposons inserted into the sense strand or antisense strand of protein-coding genes. De novo insertions and germline insertions are shown in the left and right panels, respectively. The numbers of transposon insertions in the CDSs, UTRs and introns of the protein-coding genes are labeled.
Figure 3.

The preference of transposon insertions correlates with gene activity. (A) Meta analysis quantifying the average enrichment of transposon insertions in the ±500 bp window surrounding protein-coding genes. We divided the entire region into 160 bins, with ten bins each for the upstream and downstream 500-bp regions and 20 bins each for the first exon, the first intron, the first intermediate exon, the last intermediate intron, the last intermediate exon, the last intron and the last exon. The first exon, first intron, middle exon, last intron and last exon in our schematic are drawn in proportion to their average sizes in the fly genome. (B, C) Protein-coding genes were divided into 20 equal-sized sets by expression level in ovaries (for comparison with P-element insertions) and oocytes (for comparison with LTR transposon and I-element insertions). Enrichment of transposon insertions in the promoters or exons in each of the 20 quantiles is shown in ascending order of transcription level. Each quantile contains 697 genes; their average expression levels in ovaries and oocytes are provided in RPKM (reads per kilobase pair per million aligned reads). (D) Barplots depicting the percentage of de novo and germline transposon insertions in the CDS, 5′-UTR and 3′-UTR of protein-coding genes; the promoter, exon and intron of lncRNAs; and the promoter of small ncRNAs. Error bars indicate the standard deviations of random insertions. The percentages of observed insertions were compared with random insertions in each type of genomic element. All observed insertions differ significantly from random insertions in each type of genomic element (multiple testing corrected t-test q-values < 0.05).
De novo insertion of all six transposons primarily targeted promoters of protein-coding genes, but the degree and enrichment patterns differed among the transposons. Most (60.8%) de novo P-element insertions were in promoters, with a 4.73-fold enrichment over random insertions; germline insertions exhibited a similar level of enrichment (Figure 2A and Supplementary Figure S5C). De novo P-element insertions clustered tightly around TSSs (Figures 2B and 3A). Spradling et al. analyzed the EY collection of 18 213 independent P-element insertions in the Gene Disruption project (30). They showed high enrichment of these insertions in the sites bound by ORC proteins, and these sites function as replication origins in cultured cells (26). Although they observed enrichment of these insertions in promoters (defined by them as TSS ± 100 bp), they argued that many promoters functioned as replication origins. The 7286 de novo and 138 germline P-element insertions we identified were highly enriched in promoters (also defined as TSS ± 100 bp), albeit not as enriched as the EY P-element insertions. Using wider promoter definitions (TSS ± 500 bp or TSS ± 1000 bp), P-element insertions in promoters approached that of EY P-element insertions (Supplementary Figure S4A). The EY P-element insertions were identified using inverse PCR followed by direct sequencing of the flanking sequences (29), while our de novo insertions were identified using short-read DNA-seq data, which did not contain sufficient split read-pairs to identify breakpoints accurately. This difference likely explains the lower enrichment of our de novo insertions in the narrowly-defined promoters. Thus, we used TSS ± 500 bp as the default definition for promoters. For both EY and our de novo insertions, the more insertions that fell into a promoter, the more likely the promoter was to overlap a replication origin (Supplementary Figure S4B). Both EY and de novo insertions strongly preferred the promoters of genes expressed in ovaries; however, as was reported for EY P-elements (26), for genes with expression levels above the 50th percentile, we did not observe a significant correlation between gene expression level and the number of de novo insertions in a promoter (Pearson's correlation coefficient = 0.32; P-value = 0.37; Figure 3B; Supplementary Figure S4C).
We next asked whether our de novo P-element insertions and EY P-element insertions tended to land in an overlapping set of promoters (TSS ± 500 bp). Using randomly drawn promoters from expressed genes (expression level ≥1 RPKM in ovaries) as controls, we found that promoters with de novo insertions were twice more likely than random to also have EY insertions (Figure 2C). Specifically, 1527 promoters contained both de novo and EY insertions, whereas random simulation yielded only 1122 ± 20 shared promoters (t-test P-value < 2.2 × 10–16). Accordingly, significantly fewer promoters contained only de novo or only EY insertions than random simulation (t-test P-values < 2.2 × 10−16). Promoters with both de novo and EY insertions were more likely to overlap replication origins (76%) than randomly chosen, expressed promoters with EY insertions (27 ± 1.3%; t-test P-value < 2.2 × 10−16). Furthermore, even though promoters with EY insertions but no de novo insertions and promoters with de novo insertions but no EY insertions were less likely to overlap replication origins than promoters with both de novo and EY insertions (53% and 49% versus 76%; chi-squared test P-values ≤ 1.1 × 10–10), they were significantly more likely to overlap replication origins than randomly selected, expressed promoters (27% for random; P-values < 2.2 × 10–16). These results held for the narrow promoter definition (TSS ± 100 bp; Supplementary Figure S4D). The 138 germline P-element insertions also exhibited significant co-localization with EY P-element insertions in promoters, and germline-insertion-containing promoters exhibited high enrichment in replication origins (Supplementary Figure S4E). Furthermore, de novo P-element insertions showed significant enrichment at replication origins genomewide (Supplementary Figure S4F).
The similarity and extensive overlap between the P-element insertions identified by our pipeline and the EY collection confirm the previous finding that P-element insertions target replication origins (26). These results also validate our approach to identifying new transposon insertions, including challenging-to-detect de novo insertions.
De novo LTR transposon insertions favor active promoters and expressed exons
Like P-element, LTR transposons preferentially integrated into promoters, although their enrichment (1.94–2.28-fold higher than random insertions) was much lower than P-element enrichment (4.73-fold; Figures 2A and 3A; t-test P-values < 2.2 × 10−16). I-element insertions also showed a modest enrichment in promoters (1.74-fold higher than random insertions; Figure 2A; t-test P-values < 2.2 × 10−16). Although the coverage of DNA-seq reads depends on nucleotide composition of a genomic region and could be affected by the replication timing of the cells used for preparing the DNA-seq library (54–57), we found no coverage bias at replication origins or promoters and only small biases at exons (1.11-fold), introns (1.05-fold), and intergenic regions (0.87-fold; Supplementary Figure S5A; Materials and Methods). There is also little variation in coverage around replication origins and TSSs (Supplementary Figure S5B). Thus, the 1.74–2.28-fold enrichment of LTR transposon and I-element insertions in promoters was not an artifact introduced by biased sequencing read coverage. In contrast to de novo P-element insertions, which were clustered tightly around TSS (both upstream and downstream), de novo insertions of I-element and LTR transposons frequently localized to proximal promoters (∼150 bp upstream TSS) (Figures 2B and 3A). The differences in promoter enrichment and preferred position relative to TSSs between I-element and LTR transposons, and P-element, suggest that, unlike P-element, I-element and LTR transposons do not use an ORC-dependent insertion mechanism.
All four LTR transposons exhibited similar gene-level insertion preferences, often targeting promoters and exons of protein-coding genes for de novo insertions. Exons accounted for 46.8–53.5% of de novo LTR insertions, 2.15–2.46-fold higher than random insertions at 21.8% (t-test P-value < 2.2 × 10−16; Figures 2A, 3A, and Supplementary Figure S5C). Two of the LTR transposons, 3S18 and Max-element, which both belong to the BEL-Pao subfamily, also showed enrichments in their de novo insertions at the end of genes, which might suggest interactions between the integrase of BEL-Pao family transposons, RNA polymerase II, and the poly-adenylation machinery (Figure 3A). Compared with random insertions (Figure 2A and Supplementary Figure S5C), the percentage of de novo insertions in exons was significantly lower for P-element (6.0%; t-test P-value < 2.2 × 10−16) and I-element (15.1%; t-test P-value < 2.2 × 10−16). Exons include protein-coding sequences (CDSs) and untranslated regions (UTRs), and de novo LTR insertions exhibited enrichment in both (2.17–2.52-fold higher in CDS, 1.34–1.82-fold higher in 5′-UTR and 1.83–3.39-fold higher in 3′-UTR; Figure 3D). In contrast, de novo LTR insertions were depleted in introns and intergenic regions—2.10–2.53-fold lower in introns (t-test P-value < 2.2 × 10−16) and 3.58–5.04-fold lower in intergenic regions (t-test P-value < 2.2 × 10−16). Finally, de novo P-element insertions displayed similar depletion in introns and intergenic regions, whereas I-element insertion levels in these regions were comparable to random insertions (Figures 2A, 3A, and Supplementary Figure S5C). It is important to note that these are relative levels of enrichment or depletion for transposon insertions in each type of genomic region. This is because the various types of genomic regions are interdependent, and their percentages sum to one, representing the entire genome. Therefore, an enrichment in one region type may result in a corresponding depletion in another region type.
The preference of LTR transposons for inserting into promoters and exons suggested that gene transcription might influence their insertion site selection. To examine this possibility, we divided 13 950 protein-coding genes into 20 equal-sized sets by their expression levels in oocytes; since LTR transposons and I-element become active in oocytes (36), we assessed the enrichment of de novo LTR transposon and I-element insertions in each gene set (Supplementary Table S5; see Methods). Because P-element is generally active in ovaries (37), we similarly assessed the enrichment of de novo P-element insertions using gene expression in ovaries. For genes with expression levels above the 50th percentile (the 3 RPKM bin for ovaries and the 8 RPKM bin for oocytes), we observed a strong correlation between gene expression level and enrichment of de novo LTR insertions in promoter and exons (Pearson's correlation coefficient = 0.78–0.99; P-values = 7.2 × 10−3–2.9 × 10−8; Figure 3B, C). In contrast, as described above, de novo P-element insertions strongly favored active promoters and were enriched in promoters where the expression level exceeded a threshold (above the 0.32 RPKM bin for ovaries). However, above the 3 RPKM bin, de novo P-element insertions did not correlate with gene expression (Pearson's correlation coefficient = 0.32; P-value = 0.37; Figure 3B; see also Supplementary Figure S4C). The density of replication origins in promoters follows the same trend as the enrichment of P-element insertions—it is much higher in active promoters than inactive promoters but is not highly correlated with the strength of the promoters (Supplementary Figure S5D). The two quantities are highly correlated (Pearson's correlation coefficient = 0.92), lending further support to the idea that the preference of P-element insertions for the promoters of high-expression genes is the result of the ORCs. In promoters, de novo I-element insertions increased moderately with expression level (Figure 3B). However, in exons we observed no correlation between either P-element or I-element insertions and gene expression. Dividing genes into five bins with an equal range of log-expression yielded the same conclusions (Supplementary Figures S6A, B). Compared with random insertions, de novo P-elements preferred promoters, even in the low expression group (0.1–1 RPKM), exhibiting a five-fold enrichment around TSSs; LTR transposon insertions were rare at such promoters (Supplementary Figure S6A). The enrichment of de novo I-element insertions in the most highly expressed promoters (six times the random level) was much lower than the enrichment of P-element and LTR transposons in such promoters (∼15 fold). These results suggest that LTR transposons are highly dependent on gene expression for insertion. Finally, we asked whether LTR and other transposons preferred inserting into the sense (i.e. coding) or antisense strand of genes, as LINEs reportedly show such a preference (I-element is a LINE) (20,58). In protein-coding genes, 3S18 and blood showed slight but opposite strand preferences but no overall strand bias, nor did I-element and P-element (Figure 2D), suggesting that for the six transposons we investigated, the process of de novo insertion is not directly coupled to transcription.
In summary, LTR transposons prefer to integrate into the promoters and exons of transcribed protein-coding genes, and their de novo insertion rates are strongly correlated with gene expression without showing strand bias. I-element and LTR transposons prefer active proximal promoters (∼150 bp upstream TSS), while P-element insertions occur predominantly around TSSs (both upstream and downstream), including those of genes expressed at low levels. I-element is less discriminating about the non-promoter genomic regions into which it integrates.
Germline P-element insertions are enriched in promoters but depleted in exons
To examine the effects of post-insertion selection, we compared de novo and germline insertions for the six transposons (Figure 1B, right panel). In promoters, there was little difference in enrichment between de novo and germline P-element insertions (Figure 2A and Supplementary Figure S4A; also compare Figure 2C with Supplementary Figure S4E). This similarity suggests that de novo P-element insertions into replication origins that function as promoters are not under stringent natural selection. Supporting this notion, genes with germline P-element insertions in their promoters were not depleted of genes identified as cell essential using a CRISPR screen (59).
Although both de novo and germline P-element insertions were depleted in exons, introns, and intergenic regions, germline insertions were far more depleted in exons (15.00-fold depletion) than other genomic regions (t-test P-value < 2.2 × 10−16, Figure 2A). Moreover, germline P-element favors insertion upstream of TSSs rather than downstream of TSSs (83 versus 38 insertions in the 500 bp TSS-upstream versus 500 bp TSS-downstream windows; Supplementary Figure S6C). Together, these suggest that evolutionary pressure acts against germline P-element insertions in exons.
Germline LTR transposon and I-element insertions are depleted in promoters and exons
In sharp contrast to the enrichment of de novo LTR insertions in promoters and exons of protein-coding genes, germline LTR insertions were 5.57–6.92-fold depleted in promoters and 2.70–43.72-fold depleted in exons compared with random insertions (Figure 2A; t-test P-values < 2.2 × 10−16). While de novo LTR insertions were depleted in introns and intergenic regions, the frequency of germline LTR insertions in introns was the same as that of random insertions. Germline LTR insertion levels in intergenic regions were 1.70–2.66-fold higher than random (Figure 2A; t-test P-value < 2.2 × 10−16).
De novo I-element insertions were moderately enriched in promoters, slightly depleted in exons, and at random levels in introns and intergenic regions. In comparison, germline I-element insertions were 7.08-fold depleted in promoters (t-test P-value < 2.2 × 10−16), 12.03-fold depleted in exons (t-test P-value < 2.2 × 10−16), at a random level in introns, and 2.36-fold enriched in intergenic regions (Figure 2A; t-test P-value < 2.2 × 10−16). These results indicate that although I-element prefers to insert into promoters, and LTR transposons prefer to insert into promoters and exons, these insertions are detrimental to the host and strongly selected against.
Finally, we examined the preferences of germline insertions in components of protein-coding genes. Unlike de novo insertions, which showed no preference for the sense/antisense strand or exon/UTR/intron (Figure 2D, left panel), the germline insertions of all six transposons showed a strong bias against the protein-coding portion of the sense strand (CDS)—almost no sense strand insertions were in CDS (zero for 3S18, two blood, one HMS-Beagle, zero Max-element, one I-element, and one P-element), compared with 270–1631 de novo insertions in the sense strand of CDS (Figure 2D, right versus left panel). Furthermore, there were only a few germline insertions in UTRs (Supplementary Figure S5E). While there were fewer sense than antisense germline insertions in introns for some of the transposons, the differences were not statistically significant after multiple-testing correction (Supplementary Figure S5E). In conclusion, transposon insertions in both CDS and UTRs are under strong negative selection.
De novo P-element insertions are enriched in high chromatin accessibility regions, which likely contain additional replication origins
Chromatin state, histone modifications and transcription factor binding are correlated with transposon insertion site selection (25,27,60–61); therefore, we assessed the effects of different epigenetic marks on de novo insertion site preference by examining chromatin accessibility (ATAC-seq), binding of RNA polymerase II (ChIP-seq), and histone modifications in fly ovaries (ChIP-seq; see Materials and Methods; Supplementary Table S1) (62–67). Figure 4A shows the insertion frequency in the peaks of each epigenetic mark assessed in this study for each transposon. I-element transposition was not highly correlated with any epigenetic mark—none of the marks accounted for >25% of I-element insertions in their peaks. In contrast, de novo P-element insertions strongly correlated with chromatin accessibility—74.3% of its de novo insertions were in ATAC-seq peaks (6.34-fold higher than random; t-test P-value < 2.2 × 10−16). De novo P-element insertions were also highly enriched in Pol II (14.27-fold higher than random; t-test P-value < 2.2 × 10−16), H3K4me3 (4.25-fold higher than random; t-test P-value < 2.2 × 10−16), and H3K27ac peaks (4.13-fold higher than random; t-test P-value < 2.2 × 10−16). As a reciprocal analysis, we examined the enrichment of the signals of epigenetic marks in the ±5 kb windows centered on the de novo insertions of each transposon. Again, we observed strong enrichment for ATAC, Pol II and H3K27ac signals sharply centered on de novo P-element insertions (Figure 4B).
Figure 4.

Chromatin accessibility defines insertion sites of P-elements. (A) Percentage of insertions in the peaks of ATAC-seq and the ChIP-seq data of Pol II, H3K4me1–3, H3K9ac, H3K18ac, H3K27ac, H4K8ac, H3K36me1–3, H3K9me1–3, H3K27me1–3, H1, H2AV, H2B and H3 identified in the respective samples. Observed insertions are illustrated as colored dots, and random insertions as gray bars. Error bars represent the standard deviation of random insertions. (B) Normalized signal of ATAC-seq and fold enrichment over input of Pol II, H3K4me3, H3K27ac, H3K36me3 and H3K9me3 ChIP-seq data are shown for the ±5 kb window centered on transposon insertions. (C) Normalized Pol II ChIP-seq and ATAC-seq signals are shown for the ±5 kb window centered on transposon insertions stratified into five groups: in the promoters of protein-coding genes, in the exons or introns of protein-coding genes, in intergenic regions, and in the promoters or bodies of noncoding genes. (D) The percentages of de novo P-element, germline P-element, and EY P-element insertions localized to ATAC and replication origins, representing open chromatic and origins of replication, respectively.
To investigate which genomic regions contributed the most to the enrichments, we refined our analysis by dividing epigenetic peaks into five gene-level categories: promoters, exons, introns, intergenic regions, and noncoding genes (Materials and Methods). The enrichment of de novo P-element insertions in ATAC and H3K27ac peaks was the strongest in promoters and remained strong outside promoters, e.g. 4.20–6.30-fold over random in introns and 6.25–7.42-fold over random in intergenic regions (t-test P-values < 2.2 × 10−16; Supplementary Figure S7). Furthermore, we observed enrichment of de novo P-element insertions in the H3K27ac peaks of intronic and intergenic regions (Supplementary Figure S7); H3K27ac is a histone mark characteristic of TSS-distal enhancers (68). The reciprocal analysis confirmed the enrichment of ATAC, H3K27ac and H3K4me1 signals around the de novo P-element insertions in introns and intergenic regions, while the signals of Pol II and the promoter mark H3K4me3 were mainly restricted to promoter insertions (Figure 4C and Supplementary Figure S8). We further quantified the average epigenetic signals ±500 bp around de novo transposon insertions and observed statistical enrichment for ATAC, Pol II and H3K27ac signals at de novo P-element insertions in promoters and ATAC and H3K27ac signals such insertions outside promoters (Supplementary Figure S9).
The enrichment in Pol II and H3K4me3 peaks reflects the known enrichment of P-element insertions at replication origin-overlapping promoters. As replication origins are characterized by open chromatin, we hypothesized that the enrichment of P-element insertions in ATAC peaks would reveal additional replication origins in fly ovaries not captured in the set of replication origins curated by Spradling et al. using ORC ChIP-seq data in cell lines (26) (ORC ChIP-seq data are not available for fly ovaries). Indeed, 46.7% of de novo P-element insertions fell inside ATAC peaks that did not overlap replication origins, and these ATAC-only peaks were as enriched in de novo P-element insertions as replication origins that did not overlap ATAC peaks. Germline and EY P-element insertions (26) were similar in this regard (Figure 4D and Supplementary Figure S4G).
In summary, chromatin accessibility and the epigenetic marks associated with open chromatin regions are the most enriched epigenetic features of de novo P-element insertions. Nearly half of P-element insertions fall in ATAC peaks that did not overlap previously annotated replication origins (26), and many of these ATAC peaks are likely used as replication origins in fly ovaries.
De novo LTR transposon insertions are most enriched in regions with high H3K36me3 signals
H3K4me3 and H3K36me3 (and the related H3K36me2) were two epigenetic marks that showed the highest signals in the genome regions with LTR transposon insertions (Figure 4A). These results are consistent with the above-described enrichment of de novo LTR transposon insertions in the promoters and exons of expressed genes (Figure 3A, C). Nucleosomes at the promoters of actively transcribed genes are highly enriched in H3K4me3, while H3K36me3 is enriched in nucleosomes toward the end of actively transcribed genes (69,70). Like the H3K36me3 signal, there is also an increasing trend of de novo LTR transposon insertions from the beginning to the end of a gene (Supplementary Figure S7F). Furthermore, H3K36me3 and other histone marks are known to be at higher levels in exons than in introns (71,72), and indeed we observe the elevated H3K36me3 levels at exons (Supplementary Figure S6D), which further underscores the consistency between the enrichments of de novo LTR transposon insertions in exons and H3K36me3 peaks. Roughly half of the de novo LTR insertions (46.1–56.3%) were localized to H3K36me3 peaks, 3.00–3.66-fold higher than random (t-test P-value < 2.2 × 10−16), and aggregation plots centered on the breakpoints of all four LTR transposons displayed high H3K36me3 signals (Figure 4A, B).
Only 17.5–22.4% of LTR insertions were outside promoters and exons, and most of these insertions were in introns (70.1–75.2%), with the remaining insertions in intergenic regions (21.6–25.8%) and noncoding RNAs (3.1–5.4%). Despite the overall low H3K36me3 signal in introns (Supplementary Figure S6D), there was a significant enrichment of the H3K36me3 signal around the de novo insertions of LTR transposons in introns (Figure 5A), further suggesting that high H3K36me3 may directly recruit the integration of LTR transposons.
Figure 5.

LTR transposon insertions are enriched in promoters and H3K36me3 ChIP-seq peaks, while P-element insertions are enriched in ATAC-seq peaks. (A) Normalized H3K36me3 ChIP-seq signal in the ±5 kb window centered on transposon insertions stratified into five groups: in the promoters of protein-coding genes, in the exons or introns of protein-coding genes, in intergenic regions, and in the promoters or bodies of noncoding genes. (B, C) Genome browser views of a 28-kb and a 52-kb region in D. melanogaster chromosome 2R and X. RNA-seq signal, gene annotation, transposon insertions, replication origins, ATAC-seq signal, H3K36me3 ChIP-seq signal, Pol II ChIP-seq signal and H3K4me4 signals are displayed from top to bottom.
Figure 5B illustrates the de novo transposon insertions near Vha16-1, which codes for the vacuolar H+ ATPase 16kD subunit 1, a membrane channel protein highly expressed in oocytes (89.4 RPKM). High H3K36me3 marks the last two exons of this gene, and 11 de novo LTR transposon insertions fall in this region; 14 more de novo LTR transposon insertions nearby loosely colocalize with H3K36me3 and H3K4me3 peaks. In contrast, de novo P-element insertions were more segregated, forming three clumps, two with two high ATAC regions containing replication origin-overlapping peaks. The larger high-ATAC region covers the long second intron of Vha16-1 is crowded with de novo P-element insertions. EY P-element insertions cluster near de novo P-element insertions, corroborating the inherent selectivity of P-element insertions.
Another example is shown in Figure 5C at the locus of nej, which encodes the transcriptional co-activator CBP, an acetyltransferase whose targets include histone 3 on lysines 18 and 27 and histone 4 on lysine 8. nej is expressed at a high level (86.1 RPKM) in oocytes. It has several long exons and short introns, corresponding to rises and falls of the H3K36me3 signal; its promoter lacks H3K36me3 but shows high H3K4me3. There are two dozen or so de novo insertions from all four LTR transposons throughout nej. In contrast, de novo and EY P-element insertions are tightly clustered around ATAC peaks, most of which overlap replication origins. In both examples, de novo I-element insertions are scattered loosely throughout, with a slight preference for promoters as described above.
I-Element prefers to insert in chromosomal locations far away from centromeres
To investigate large-scale insertion site preferences, we assessed the distribution of de novo transposon insertions across D. melanogaster chromosomes by dividing each chromosome into 500-kb bins. Detecting transpositions using typical short-read sequencing data depends highly on mappability; therefore, we normalized the number of insertions in each chromosome bin accordingly (Materials and Methods). D. melanogaster females possess three major chromosomes: 2, 3 and X (73). These chromosomes contain two types of chromatin—lightly packed euchromatin and densely packed heterochromatin. Heterochromatin concentrates around the centromeres of chromosomes 2 and 3 and at the 3′ end of chromosome X (74). For all six transposons, de novo insertions were more frequent in euchromatin than heterochromatin, with 1.47–2.37-fold more insertions per Mb (Figure 6A). On the other hand, germline insertions were enriched in heterochromatin, likely reflecting selection against insertions that cause adverse developmental or reproductive outcomes (Supplementary Figure S10A).
Figure 6.

The number of de novo I-element insertions is correlated with the distance to the centromere. (A) Normalized number of insertions of three transposon types (colored dots) per Mb in euchromatin (black) and heterochromatin (gray) regions of each chromosome arm. The number of insertions is normalized by mappability. Wilcoxon signed-rank tests were performed between euchromatin and heterochromatin, and their P-values are provided. (B) Average C + G percentage in the ±1 kb window (with a bin size 10 bp) centered on the breakpoints of de novo insertions for each transposon. (C) Normalized number of de novo insertions in 500-kb non-overlapping windows for HMS-Beagle, I-element and P-element. The percentage of C + G in the same sliding windows is shown in the bottom panel. For each chromosome arm, Pearson correlation coefficients (r) between the number of insertions in a window and the window's distance to the centromere are provided. Heterochromatin regions are shaded gray.
I-element belongs to the LINE family, whose propagation requires target DNA as the reverse transcription primer (13); accordingly, I-elements preferred integration into AT-rich sites that can base-pair with the poly-A tails of I-element transcripts (Figure 6B and Supplementary Figure S10B). Conversely, LTR transposons and P-elements preferred GC-rich insertion sites, with LTR transposons favoring broader GC-rich regions (Figure 6B and Supplementary Figure S10B). Despite their preference for AT-rich sites, the broader regions into which I-elements integrated exhibited slightly higher C + G% than random (Supplementary Figure S10B).
Although LTR transposon and P-element preferred insertion into euchromatin, they integrated into peritelomeric and pericentromeric regions at comparable frequencies (0.94–1.33 folds of random) (Figure 6C and Supplementary Figure S10C). In contrast, the density of I-element insertions correlated significantly with distance from the centromere; the correlation coefficient ranged from 0.63 (P-value = 8.7 × 10−7) for the right arm of chromosome 2 to 0.87 (P-value = 9.0 × 10−16) for the left arm of chromosome 2 (Figure 6C). We found 2,074 de novo I-element insertions in the 2-Mb bins around telomeres; this is 2.5-fold more than the number of I-elements (n = 817) inserted into the 2-Mb euchromatic regions around centromeres. C + G% is higher in bins near telomeres than in bins around centromeres (Figure 6C), but the C + G% in a bin is less correlated with I-element insertion density than is the bin's genomic distance from the centromere (Figure 6C). Because I-element only shows a slight preference for broadly high C + G% regions (Supplementary Figure S10B), other factors may account for their preferential insertions in telomeric regions.
LINE elements are sometimes truncated at the 5′ end, and 20 copies of the I-element (out of a total of 158 copies) in the reference fly genome are truncated at the 5′ end, starting mostly between 2100 and 3900 nucleotides in the consensus sequence. However, reads that map to the I-element consensus sequence show an even coverage (Supplementary Figure S11A), and almost all insertion-supporting reads map to the 5′ or 3′ end of the I-element consensus (Supplementary Figure S11B; see Methods), indicating that de novo and new germline I-element insertions detected in the dysgenic oocytes are not 5′ or 3′ truncated. In comparison, some internal truncations are supported by reads that map to the P-element consensus sequence (Supplementary Figure S11C). However, the details of such internal truncations cannot be studied using the Illumina sequencing data due to the short read length (Supplementary Figure S11D, with the regions reachable by the sequencing reads shaded at the two ends).
In summary, all six transposons prefer insertion into euchromatin and disfavor heterochromatin. LTR transposon and P-element insertions show no other specific preferences at the chromosome scale. However, I-element insertions are highly correlated with the distance from the centromere, but the mechanism needs further investigation. Locally, I-elements prefer AT-rich sites, while P-elements and LTR transposons favor GC-rich sites. Our data also suggested de novo and new germline I-element insertions are not 5′ or 3′ truncated in the dysgenic oocytes, while some P-element insertions have internal truncation.
DISCUSSION
Transposons are a major force driving genome evolution, but they can interrupt genes and cause genome instability (33,75–77). Different transposons use different transposition mechanisms, resulting in diverse target site preferences (25–27,78). The transposon profiles recorded in metazoan genomes reflect the outcomes of initial insertion, natural selection, and genetic drift. Uncoupling these processes and deciphering insertion site preferences for each transposon is difficult. In this study, we analyzed Zhang and colleagues’ data on derepressed, genome-encoded transposons in piRNA-deficient fly oocytes (LTR transposons and I-element) and fly ovaries (P-element), identifying both de novo and germline insertions in vivo. We observed very different preferences for the three families of transposons (Figure 7): LTR transposons tend to insert into the core promoters and exons of expressed protein-coding genes, and have a general preference for high-H3K36me3 regions; P-element prefers to insert into replication origins, many of which have open chromatin and a subset of which are promoters; while I-element has a weak preference for core promoters, does not show a strong tendency to insert near genes, and shows a local preference for high-AT sites and a chromosome-level tendency toward telomeres.
Figure 7.
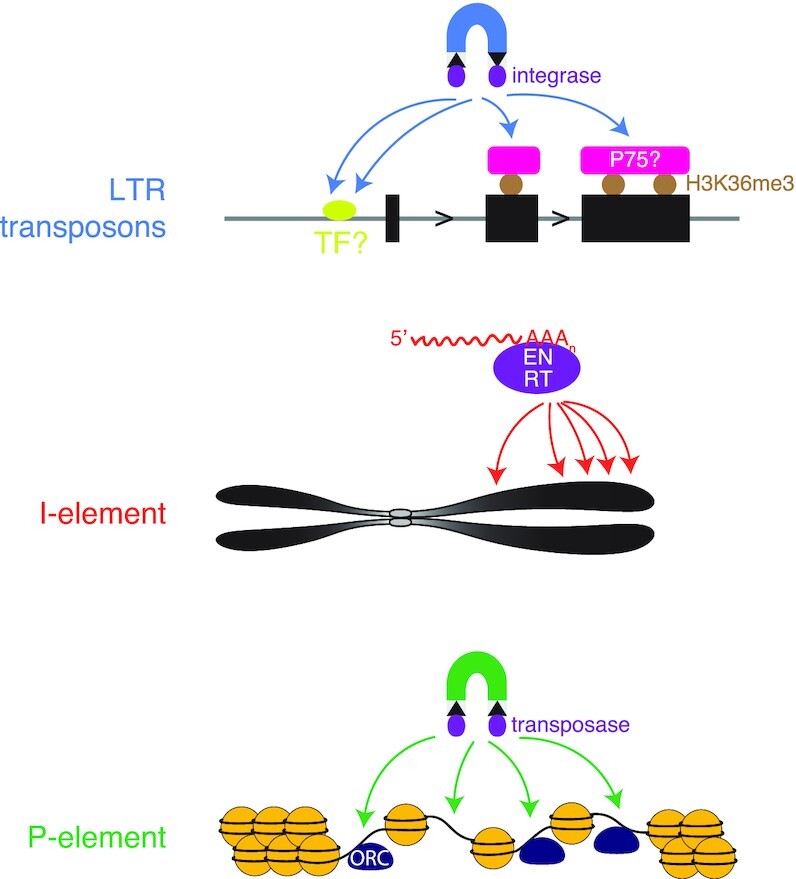
Different genome insertion preferences for three types of transposable elements. A diagram depicting the preference of insertion site selection for LTR transposons, I-element and P-element in D. melanogaster. LTR transposon insertions prefer active proximal promoters and H3K36me3 marked gene bodies, possibly due to the tethering by transcription factors and PWWM-domain-containing proteins like dP75. I-element tends to reside close to centromeres. P-element integration is likely driven by replication origins with open chromatin.
Gene interruption using single insertions of an engineered DNA transposon has served as a powerful tool for studying gene function. The Drosophila Gene Disruption Project used three engineered DNA transposons—P-element, piggyBac and Minos—to generate mutant fly lines for most protein-coding genes (29,30). Detailed analyses of the insertion sites of these transposons revealed a lack of insertions for all three transposons in Polycomb-regulated regions and a strong preference for P-element insertions in promoters and replication origins (26,30). Using engineered human LINE1 expression plasmids, studies in cell lines revealed that the LINE1 endonuclease predominantly cleaves the DNA replication templates for lagging strand synthesis, facilitating subsequent LINE1 insertion (20,58). Similar work on human cell lines infected with HIV and HIV-based vectors revealed insertion hotspots, possibly connected to super-enhancers and located near nuclear speckles (61,79–80).
The preference of transposon insertions in promoters has been extensively studied. One study showed insertions of Mu element in maize and P-element in D. melanogaster are specifically associated with the transcriptional initiation of highly expressed genes by Pol II (81). Similarly, other studies showed Ty1 and Ty3 specifically insert into Pol III promoters in S. cerevisiae (82) while Tf1 favors Pol II promoters in S. pombe (83), and, in particular, Tf1 insertions impact the adaptation of gene expression to environmental stress (84). Our results expand on these previous studies, providing an analysis of six transposons in three major families (DNA, LTR and LINE) and adding valuable knowledge about transposon target site preferences. Our results are notable as they derive from data of endogenous transposons in live animals under hybrid dysgenic or piRNA pathway disruption conditions that could occur in the wild.
In whole-genome short-read sequencing data, de novo insertions are supported by very few sequencing reads; most de novo insertions are supported by a single read pair. Therefore, accurate identification of de novo insertions must overcome noise in the sequencing data, notably chimeric reads from library construction (38). Furthermore, due to the low read coverage, we have limited precision in mapping the exact location of breakpoints of de novo insertions, which prevents us from identifying sequence motifs near breakpoints. However, the high concordance between our results for de novo P-element insertions in hybrid dysgenic fly ovaries and the results of Spradling et al. using the EY collection, a collection consisting of tens of thousands of fly lines harboring engineered P-elements, supports our approach of using TEMP2 (38) to identify de novo insertions in whole-genome short-read sequencing data. In addition, the concordance indicates that the target site specificity of P-element resides in its sequence and implies that, as concluded previously, P-element hijacks the host replication machinery for transposition (26).
One advantage of our approach is that we compared de novo and germline insertions of the same set of transposons in fly oocytes (LTR transposons and I-element) and fly ovaries (P-element). This approach allowed us to make inferences about both initial insertion site preferences and post-insertion selection pressures. All six transposons prefer to insert into euchromatic regions, especially promoters (additionally in exons for LTR transposons); however, such insertions are depleted in the germline, indicating that most of them are harmful to the host and that individuals harboring these mutations are quickly eliminated from the population. Strikingly, all four LTR transposons prefer to insert into exons, but exonic insertions, particularly those overlapping CDS, have almost all been removed in the germline. The only insertion preference that does not seem to be under strong selection is the enrichment of P-element insertions in promoters; it is unclear why P-element insertions in promoters do not seem to impact the gene function. Overall, the contrast between de novo insertions and germline insertions indicates that untangling the impact of evolution from the inherent target site preference requires studying both types of insertions.
Our analysis revealed that LTR transposons prefer to insert into core promoters and high-H3K36me3 regions. Core promoters tend to have low H3K36me3 signals, even for highly expressed genes; thus, the preference for promoters is distinct from the preference for high-H3K36me3 regions. Because they use similar mechanisms to replicate, one might expect parallels between LTR transposons and retroviruses such as ASV, HTLV-1, MLV and HIV. Like the four fly LTR transposons, MLV also prefers promoters; however, ASV and HTLV-1 do not specifically target active genes and promoters in the human genome (85–88). HIV is known to prefer human genes, but unlike fly LTR transposons, does not favor exons over introns. This difference between fly LTR transposons and a human retrovirus could reflect the tendency toward much longer introns in humans than in flies. What might explain the preference of LTR transposons for active genes? The ubiquitously expressed, evolutionarily conserved transcriptional co-activator lens epithelium-derived growth factor (LEDGF; also known as LEDGF/p75) contains a conserved amino-terminal PWWP domain that binds to H3K36me3 (89) and a carboxy-terminal domain that binds to the integrase of HIV and directs its integration into actively transcribed human genes (90). The structurally conserved D. melanogaster ortholog of LEDGF/p75 has been shown to be involved in the transcriptional regulation of genes in the oogenesis and piRNA production pathways (91). Although there is currently no data supporting the interaction between the fly LEDGF/p75 and the integrases of LTR transposons (personal communication, Kun Dou), it is possible that LTR transposons and HIV use a similar mechanism for transposition.
Among the transposons that we studied, I-element is unique, and the explanation for its chromosome-scale integration preferences is unknown. I-element has a modest preference for active promoters and strongly prefers chromosomal regions more distant from centromeres. In contrast, the human LINE1, which belongs to the same class of transposons, integrates throughout the human genome with no preference for active genes (58). In addition, human LINE1 inserts more frequently into the leading strand of DNA replication. When replication fork direction data for D. melanogaster become available, it will be interesting to determine if this is also true for I-element. Like LTR transposons, I-element tends to insert into the promoter region 100–200 bp upstream TSSs in a manner correlated with expression level. This pattern suggests that both I-element and LTR transposons are associated with the transcriptional machinery, although LTR transposons exhibit a stronger association.
In summary, we performed detailed analyses on the insertion preferences of three types of transposons in D. melanogaster. Different transposon types display distinct patterns of insertion, which provide a window into their different mechanisms of transposition. Dramatic differences between de novo and germline insertions reflect the dynamic arms race between transposons and their hosts. Our results advance several testable hypotheses and questions, the answers to which will further our understanding of the molecular mechanisms of I-element and LTR transposons.
DATA AVAILABILITY
No new data were generated or analysed in support of this research. The public data used in this article are available in NCBI Bioproject database at https://www.ncbi.nlm.nih.gov/bioproject, and can be accessed with PRJNA382678 and PRJNA454868.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the members of Weng laboratories for their critical comments. We thank Dr Edith Pfister for critically editing the manuscript and making helpful suggestions on the writing. We thank Dr Kun Dou for discussing with us possible interactions between the fly LEDGF/p75 and the integrases of LTR transposons.
Author contributions: T.Y., J.C. and Z.W. conceived the project. T.Y. and J.C. performed the computational analyses with the help of B.X. and Z.H.; T.Y., J.C., and Z.W. wrote the manuscript. All authors proofread and approved the manuscript.
Contributor Information
Jichuan Cao, The School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Tianxiong Yu, Program in Bioinformatics and Integrative Biology, University of Massachusetts Chan Medical School, Worcester, MA, USA.
Bo Xu, The School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Zhongren Hu, The School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
Xiao-ou Zhang, The School of Life Sciences and Technology, Tongji University, Shanghai 200092, China.
William E Theurkauf, Program in Molecular Medicine, University of Massachusetts Chan Medical School, Worcester, MA, USA.
Zhiping Weng, Program in Bioinformatics and Integrative Biology, University of Massachusetts Chan Medical School, Worcester, MA, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [HD049116, in part]. Funding for open access charge: NIH [HD049116].
Conflict of interest statement. Z. Weng is a co-founder of Rgenta Therapeutics and she serves on its scientific advisory board.
REFERENCES
- 1. Huang C.R.L., Burns K.H., Boeke J.D.. Active transposition in genomes. Annu. Rev. Genet. 2012; 46:651–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Britten R.J. Transposable element insertions have strongly affected human evolution. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:19945–19948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hedges D.J., Belancio V.P.. Restless genomes humans as a model organism for understanding host-retrotransposable element dynamics. Adv. Genet. 2011; 73:219–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bennetzen J.L. Transposable element contributions to plant gene and genome evolution. Plant Mol. Biol. 2000; 42:251–269. [PubMed] [Google Scholar]
- 5. Belancio V.P., Hedges D.J., Deininger P.. Mammalian non-LTR retrotransposons: for better or worse, in sickness and in health. Genome Res. 2008; 18:343–358. [DOI] [PubMed] [Google Scholar]
- 6. Iskow R.C., McCabe M.T., Mills R.E., Torene S., Pittard W.S., Neuwald A.F., Van Meir E.G., Vertino P.M., Devine S.E.. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010; 141:1253–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shukla R., Upton K.R., Muñoz-Lopez M., Gerhardt D.J., Fisher M.E., Nguyen T., Brennan P.M., Baillie J.K., Collino A., Ghisletti S.et al.. Endogenous retrotransposition activates oncogenic pathways in hepatocellular carcinoma. Cell. 2013; 153:101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Solyom S., Kazazian H.H. Jr. Mobile elements in the human genome: implications for disease. Genome Med. 2012; 4:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bourque G., Burns K.H., Gehring M., Gorbunova V., Seluanov A., Hammell M., Imbeault M., Izsvák Z., Levin H.L., Macfarlan T.S.et al.. Ten things you should know about transposable elements. Genome Biol. 2018; 19:199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greenblatt I.M., Alexander Brink R.. Transpositions of modulator in maize into divided and undivided chromosome segments. Nature. 1963; 197:412–413. [Google Scholar]
- 11. Rubin G.M., Kidwell M.G., Bingham P.M.. The molecular basis of P-M hybrid dysgenesis: the nature of induced mutations. Cell. 1982; 29:987–994. [DOI] [PubMed] [Google Scholar]
- 12. Brown P.O., Bowerman B., Varmus H.E., Michael Bishop J.. Correct integration of retroviral DNA in vitro. Cell. 1987; 49:347–356. [DOI] [PubMed] [Google Scholar]
- 13. Luan D.D., Korman M.H., Jakubczak J.L., Eickbush T.H.. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993; 72:595–605. [DOI] [PubMed] [Google Scholar]
- 14. Sultana T., Zamborlini A., Cristofari G., Lesage P.. Integration site selection by retroviruses and transposable elements in eukaryotes. Nat. Rev. Genet. 2017; 18:292–308. [DOI] [PubMed] [Google Scholar]
- 15. Brennecke J., Malone C.D., Aravin A.A., Sachidanandam R., Stark A., Hannon G.J.. An epigenetic role for maternally inherited piRNAs in transposon silencing. Science. 2008; 322:1387–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Saito K., Nishida K.M., Mori T., Kawamura Y., Miyoshi K., Nagami T., Siomi H., Siomi M.C.. Specific association of Piwi with rasiRNAs derived from retrotransposon and heterochromatic regions in the Drosophila genome. Genes Dev. 2006; 20:2214–2222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nishida K.M., Saito K., Mori T., Kawamura Y., Nagami-Okada T., Inagaki S., Siomi H., Siomi M.C.. Gene silencing mechanisms mediated by Aubergine piRNA complexes in Drosophila male gonad. RNA. 2007; 13:1911–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ghildiyal M., Seitz H., Horwich M.D., Li C., Du T., Lee S., Xu J., Kittler E.L.W., Zapp M.L., Weng Z.et al.. Endogenous siRNAs derived from transposons and mRNAs in Drosophila somatic cells. Science. 2008; 320:1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Slotkin R.K., Keith Slotkin R., Martienssen R.. Transposable elements and the epigenetic regulation of the genome. Nat. Rev. Genet. 2007; 8:272–285. [DOI] [PubMed] [Google Scholar]
- 20. Sultana T., van Essen D., Siol O., Bailly-Bechet M., Philippe C., Zine El Aabidine A., Pioger L., Nigumann P., Saccani S., Andrau J.-C.et al.. The landscape of L1 retrotransposons in the human genome is shaped by pre-insertion sequence biases and post-insertion selection. Mol. Cell. 2019; 74:555–570. [DOI] [PubMed] [Google Scholar]
- 21. Hickey A., Esnault C., Majumdar A., Chatterjee A.G., Iben J.R., McQueen P.G., Yang A.X., Mizuguchi T., Grewal S.I.S., Levin H.L.. Single-nucleotide-specific targeting of the Tf1 retrotransposon promoted by the DNA-binding protein Sap1 of Schizosaccharomyces pombe. Genetics. 2015; 201:905–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Philippe C., Vargas-Landin D.B., Doucet A.J., van Essen D., Vera-Otarola J., Kuciak M., Corbin A., Nigumann P., Cristofari G.. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife. 2016; 5:e13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maldarelli F., Wu X., Su L., Simonetti F.R., Shao W., Hill S., Spindler J., Ferris A.L., Mellors J.W., Kearney M.F.et al.. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014; 345:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gangadharan S., Mularoni L., Fain-Thornton J., Wheelan S.J., Craig N.L.. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21966–21972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bridier-Nahmias A., Tchalikian-Cosson A., Baller J.A., Menouni R., Fayol H., Flores A., Saïb A., Werner M., Voytas D.F., Lesage P.. Retrotransposons. An RNA polymerase III subunit determines sites of retrotransposon integration. Science. 2015; 348:585–588. [DOI] [PubMed] [Google Scholar]
- 26. Spradling A.C., Bellen H.J., Hoskins R.A.. Drosophila P elements preferentially transpose to replication origins. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:15948–15953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. LaFave M.C., Varshney G.K., Gildea D.E., Wolfsberg T.G., Baxevanis A.D., Burgess S.M.. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res. 2014; 42:4257–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao G.-C., Jay Rehm E., Rubin G.M.. Insertion site preferences of the P transposable element in Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:3347–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bellen H.J., Levis R.W., Liao G., He Y., Carlson J.W., Tsang G., Evans-Holm M., Hiesinger P.R., Schulze K.L., Rubin G.M.et al.. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004; 167:761–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bellen H.J., Levis R.W., He Y., Carlson J.W., Evans-Holm M., Bae E., Kim J., Metaxakis A., Savakis C., Schulze K.L.et al.. The Drosophila gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011; 188:731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kofler R., Hill T., Nolte V., Betancourt A.J., Schlötterer C.. The recent invasion of natural Drosophila simulanspopulations by the P-element. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:6659–6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Brennecke J., Aravin A.A., Stark A., Dus M., Kellis M., Sachidanandam R., Hannon G.J.. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007; 128:1089–1103. [DOI] [PubMed] [Google Scholar]
- 33. Khurana J.S., Wang J., Xu J., Koppetsch B.S., Thomson T.C., Nowosielska A., Li C., Zamore P.D., Weng Z., Theurkauf W.E.. Adaptation to P element transposon invasion in Drosophila melanogaster. Cell. 2011; 147:1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aravin A.A., Sachidanandam R., Girard A., Fejes-Toth K., Hannon G.J.. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007; 316:744–747. [DOI] [PubMed] [Google Scholar]
- 35. Klattenhoff C., Xi H., Li C., Lee S., Xu J., Khurana J.S., Zhang F., Schultz N., Koppetsch B.S., Nowosielska A.et al.. The Drosophila HP1 homolog Rhino is required for transposon silencing and piRNA production by dual-strand clusters. Cell. 2009; 138:1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang L., Dou K., Moon S., Tan F.J., Zhang Z.Z. Hijacking oogenesis enables massive propagation of LINE and retroviral transposons. Cell. 2018; 174:1082–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moon S., Cassani M., Lin Y.A., Wang L., Dou K., Zhang Z.Z. A robust transposon-endogenizing response from germline stem cells. Dev. Cell. 2018; 47:660–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu T., Huang X., Dou S., Tang X., Luo S., Theurkauf W.E., Lu J., Weng Z.. A benchmark and an algorithm for detecting germline transposon insertions and measuring de novo transposon insertion frequencies. Nucleic Acids Res. 2021; 49:e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang G., Yu T., Parhad S.S., Ho S., Weng Z., Theurkauf W.E.. piRNA-independent transposon silencing by the Drosophila THO complex. Dev. Cell. 2021; 56:2623–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quinlan A.R., Hall I.M.. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li H., Durbin R.. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Broad Institute Broad Institute Picard Toolkit. https://broadinstitute.github.io/picard/. [Google Scholar]
- 43. Huang W., Li L., Myers J.R., Marth G.T.. ART: a next-generation sequencing read simulator. Bioinformatics. 2012; 28:593–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smit A.F.A., Hubley R., Green P.. RepeatMasker Open-4.0.RepeatMasker. http://www.repeatmasker.org/RepeatMasker/. [Google Scholar]
- 45. Hocaoglu H., Wang L., Yang M., Yue S., Sieber M.. Heritable shifts in redox metabolites during mitochondrial quiescence reprogramme progeny metabolism. Nat. Metab. 2021; 3:1259–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Langmead B., Salzberg S.L.. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R.. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. 1000 Genome Project Data Processing Subgroup Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Anders S., Pyl P.T., Huber W.. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W.et al.. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008; 9:R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wagstaff B.J., Hedges D.J., Derbes R.S., Campos Sanchez R., Chiaromonte F., Makova K.D., Roy-Engel A.M.. Rescuing Alu: recovery of new inserts shows LINE-1 preserves Alu activity through A-tail expansion. PLoS Genet. 2012; 8:e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Quadrana L., Silveira A.B., Mayhew G.F., LeBlanc C., Martienssen R.A., Jeddeloh J.A., Colot V.. The Arabidopsis thaliana mobilome and its impact at the species level. Elife. 2016; 5:e15716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Treiber C.D., Waddell S.. Resolving the prevalence of somatic transposition in. Elife. 2017; 6:e28297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sims D., Sudbery I., Ilott N.E., Heger A., Ponting C.P.. Sequencing depth and coverage: key considerations in genomic analyses. Nat. Rev. Genet. 2014; 15:121–132. [DOI] [PubMed] [Google Scholar]
- 55. Nakamura K., Oshima T., Morimoto T., Ikeda S., Yoshikawa H., Shiwa Y., Ishikawa S., Linak M.C., Hirai A., Takahashi H.et al.. Sequence-specific error profile of Illumina sequencers. Nucleic Acids Res. 2011; 39:e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Benjamini Y., Speed T.P.. Summarizing and correcting the GC content bias in high-throughput sequencing. Nucleic Acids Res. 2012; 40:e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hansen R.S., Thomas S., Sandstrom R., Canfield T.K., Thurman R.E., Weaver M., Dorschner M.O., Gartler S.M., Stamatoyannopoulos J.A.. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Flasch D.A., Macia Á., Sánchez L., Ljungman M., Heras S.R., García-Pérez J.L., Wilson T.E., Moran J.V.. Genome-wide de novo L1 retrotransposition connects endonuclease activity with replication. Cell. 2019; 177:837–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Viswanatha R., Li Z., Hu Y., Perrimon N.. Pooled genome-wide CRISPR screening for basal and context-specific fitness gene essentiality in Drosophila cells. Elife. 2018; 7:e36333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Singh P.K., Plumb M.R., Ferris A.L., Iben J.R., Wu X., Fadel H.J., Luke B.T., Esnault C., Poeschla E.M., Hughes S.H.et al.. LEDGF/p75 interacts with mRNA splicing factors and targets HIV-1 integration to highly spliced genes. Genes Dev. 2015; 29:2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lucic B., Chen H.-C., Kuzman M., Zorita E., Wegner J., Minneker V., Wang W., Fronza R., Laufs S., Schmidt M.et al.. Spatially clustered loci with multiple enhancers are frequent targets of HIV-1 integration. Nat. Commun. 2019; 10:4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Meers M.P., Henriques T., Lavender C.A., McKay D.J., Strahl B.D., Duronio R.J., Adelman K., Matera A.G.. Histone gene replacement reveals a post-transcriptional role for H3K36 in maintaining metazoan transcriptome fidelity. Elife. 2017; 6:e23249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Meers M.P., Adelman K., Duronio R.J., Strahl B.D., McKay D.J., Matera A.G.. Transcription start site profiling uncovers divergent transcription and enhancer-associated RNAs in Drosophila melanogaster. BMC Genomics. 2018; 19:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Le Thomas A., Rogers A.K., Webster A., Marinov G.K., Liao S.E., Perkins E.M., Hur J.K., Aravin A.A., Tóth K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013; 27:390–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sokolova M., Moore H.M., Prajapati B., Dopie J., Meriläinen L., Honkanen M., Matos R.C., Poukkula M., Hietakangas V., Vartiainen M.K.. Nuclear actin is required for transcription during Drosophila oogenesis. Iscience. 2018; 9:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kharchenko P.V., Alekseyenko A.A., Schwartz Y.B., Minoda A., Riddle N.C., Ernst J., Sabo P.J., Larschan E., Gorchakov A.A., Gu T.et al.. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011; 471:480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Huynh K., Smith B.R., Macdonald S.J., Long A.D.. Genetic Variation in Chromatin State Across Multiple Tissues in Drosophila melanogaster. 2022; bioRxiv doi:27 September 2022, preprint: not peer reviewed 10.1101/2022.09.26.509449. [DOI] [PMC free article] [PubMed]
- 68. Creyghton M.P., Cheng A.W., Welstead G.G., Kooistra T., Carey B.W., Steine E.J., Hanna J., Lodato M.A., Frampton G.M., Sharp P.A.et al.. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wagner E.J., Carpenter P.B.. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012; 13:115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hyun K., Jeon J., Park K., Kim J.. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017; 49:e324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kolasinska-Zwierz P., Down T., Latorre I., Liu T., Liu X.S., Ahringer J.. Differential chromatin marking of introns and expressed exons by H3K36me3. Nat. Genet. 2009; 41:376–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Tilgner H., Nikolaou C., Althammer S., Sammeth M., Beato M., Valcárcel J., Guigó R.. Nucleosome positioning as a determinant of exon recognition. Mapp. Anal. Chromatin State Dyn. Nine Human Cell Types. 2009; 16:996–1001. [DOI] [PubMed] [Google Scholar]
- 73. Adams M.D., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F.et al.. The genome sequence of Drosophila melanogaster. Science. 2000; 287:2185–2195. [DOI] [PubMed] [Google Scholar]
- 74. Hannah A. Localization and function of heterochromatin in Drosophila melanogaster. Adv. Genet. 1951; 4:87–125. [DOI] [PubMed] [Google Scholar]
- 75. Rahman R., Chirn G.-W., Kanodia A., Sytnikova Y.A., Brembs B., Bergman C.M., Lau N.C.. Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Res. 2015; 43:10655–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kofler R., Nolte V., Schlötterer C.. Tempo and mode of transposable element activity in Drosophila. PLos Genet. 2015; 11:e1005406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Maksakova I.A., Romanish M.T., Gagnier L., Dunn C.A., van de Lagemaat L.N., Mager D.L.. Retroviral elements and their hosts: insertional mutagenesis in the mouse germ line. PLos Genet. 2006; 2:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Ciuffi A., Llano M., Poeschla E., Hoffmann C., Leipzig J., Shinn P., Ecker J.R., Bushman F.. A role for LEDGF/p75 in targeting HIV DNA integration. Nat. Med. 2005; 11:1287–1289. [DOI] [PubMed] [Google Scholar]
- 79. Schröder A.R.W., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F.. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002; 110:521–529. [DOI] [PubMed] [Google Scholar]
- 80. Bedwell G.J., Jang S., Li W., Singh P.K., Engelman A.N.. rigrag: high-resolution mapping of genic targeting preferences during HIV-1 integration in vitro and in vivo. Nucleic Acids Res. 2021; 49:7330–7346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang X., Zhao M., McCarty D.R., Lisch D. Transposable elements employ distinct integration strategies with respect to transcriptional landscapes in eukaryotic genomes. Nucleic Acids Res. 2020; 48:6685–6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lesage P., Todeschini A.L.. Happy together: the life and times of Ty retrotransposons and their hosts. Cytogenet. Genome Res. 2005; 110:70–90. [DOI] [PubMed] [Google Scholar]
- 83. Behrens R., Hayles J., Nurse P.. Fission yeast retrotransposon Tf1 integration is targeted to 5′ ends of open reading frames. Nucleic Acids Res. 2000; 28:4709–4716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Esnault C., Lee M., Ham C., Levin H.L.. Transposable element insertions in fission yeast drive adaptation to environmental stress. Genome Res. 2019; 29:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Narezkina A., Taganov K.D., Litwin S., Stoyanova R., Hayashi J., Seeger C., Skalka A.M., Katz R.A.. Genome-wide analyses of avian sarcoma virus integration sites. J. Virol. 2004; 78:11656–11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. LaFave M.C., Varshney G.K., Gildea D.E., Wolfsberg T.G., Baxevanis A.D., Burgess S.M.. MLV integration site selection is driven by strong enhancers and active promoters. Nucleic Acids Res. 2014; 42:4257–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Meekings K.N., Leipzig J., Bushman F.D., Taylor G.P., Bangham C.R.M.. HTLV-1 integration into transcriptionally active genomic regions is associated with proviral expression and with HAM/TSP. PLoS Pathog. 2008; 4:e1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Derse D., Crise B., Li Y., Princler G., Lum N., Stewart C., McGrath C.F., Hughes S.H., Munroe D.J., Wu X.. Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses. J. Virol. 2007; 81:6731–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Eidahl J.O., Crowe B.L., North J.A., McKee C.J., Shkriabai N., Feng L., Plumb M., Graham R.L., Gorelick R.J., Hess S.et al.. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res. 2013; 41:3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Llano M., Saenz D.T., Meehan A., Wongthida P., Peretz M., Walker W.H., Teo W., Poeschla E.M.. An essential role for LEDGF/p75 in HIV integration. Science. 2006; 314:461–464. [DOI] [PubMed] [Google Scholar]
- 91. Dou K., Liu Y., Zhang Y., Wang C., Huang Y., Zhang Z.Z.. Drosophila P75 safeguards oogenesis by preventing H3K9me2 spreading. J. Genet. Genomics. 2020; 47:187–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
No new data were generated or analysed in support of this research. The public data used in this article are available in NCBI Bioproject database at https://www.ncbi.nlm.nih.gov/bioproject, and can be accessed with PRJNA382678 and PRJNA454868.