


Abstract
It has been known for decades that codon usage contributes to translation efficiency and hence to protein production levels. However, its role in protein synthesis is still only partly understood. This lack of understanding hampers the design of synthetic genes for efficient protein production. In this study, we generated a synonymous codon-randomized library of the complete coding sequence of red fluorescent protein. Protein production levels and the full coding sequences were determined for 1459 gene variants in Escherichia coli. Using different machine learning approaches, these data were used to reveal correlations between codon usage and protein production. Interestingly, protein production levels can be relatively accurately predicted (Pearson correlation of 0.762) by a Random Forest model that only relies on the sequence information of the first eight codons. In this region, close to the translation initiation site, mRNA secondary structure rather than Codon Adaptation Index (CAI) is the key determinant of protein production. This study clearly demonstrates the key role of codons at the start of the coding sequence. Furthermore, these results imply that commonly used CAI-based codon optimization of the full coding sequence is not a very effective strategy. One should rather focus on optimizing protein production via reducing mRNA secondary structure formation with the first few codons.
INTRODUCTION
Due to degeneracy in the genetic code, a protein with a single amino acid sequence can be encoded by an extremely large number of different coding sequences (CDS). While different synonymous codons do not alter the amino acid sequence, they are known to influence translation efficiency and in some cases even protein folding properties (1–5). However, many questions about the roles of codons and their often subtle and intertwined effects are still unanswered. Several studies have revealed the importance of the first few codons on overall protein production but there is still no full consensus on the underlying mechanisms (2,4,6–10). Also it is unclear to what extent codons further downstream the coding sequence influence protein production (1). Understanding codon usage is key to grasping one of the fundamental processes of life: the translation of mRNA into proteins. In addition, precise control over translation efficiency is highly desirable in both biotechnology and synthetic biology to make the process of protein production and cell engineering more predictable.
Since the early days of DNA sequencing it has been observed that, depending on the organism, specific codons are overrepresented (11). This led to the hypothesis that frequently occurring codons could be translated more efficiently, e.g. due to a higher abundance of corresponding tRNAs. It also led to the so-called Codon Adaptation Index (CAI) (11), which is defined as the geometric mean of the relative codon usage in a specific coding sequence (based on the average codon usage in the genome or a subset of highly expressed genes). In other words, a CDS with a high CAI primarily uses frequent codons, while a CDS with a low CAI contains more rare codons. However, the hypothesis that a high CAI is related to high protein production has been disputed in several studies in recent years (2,4,12). Especially the hallmark study by Kudla et al., in the bacterium Escherichia coli, revealed that the CAI does not seem a major determinant of high protein production. In this study, a set of 154 codon variants of the Green Fluorescent Protein (GFP) was generated. The authors could not correlate the CAI to the protein production levels. However, the predicted folding energy of the mRNA around the start codon did correlate with protein production. They hypothesised that the protein production efficiency in E. coli is mostly influenced by the availability of the ribosome binding site (RBS) for translation initiation. Relatedly, already in the 1980s it had been observed that the codon identity in the 5′ of the CDS has major effects on protein production efficiency of some recombinantly expressed genes in E. coli (7,13). Several studies have since followed up on this and suggest a key role of mRNA secondary structures (4,6,14,15) around the RBS and start codon. The codon usage at the 5′ CDS has also been hypothesised to be involved in a so-called codon ramp, as these regions generally contain more rare codons (9,16). This ramp would result in a slow initial translation elongation speed, reducing the risk of detrimental ribosomal collisions along the length of the CDS. Finally, codon usage has also been associated with mRNA stability (1,3,17). Especially in eukaryotes, slow-moving ribosomes can initiate RNA decay, thereby linking translation elongation efficiency to mRNA decay (18).
Despite a range of studies in this field, there are still many open questions about the complex role of codon usage on protein production in different organisms and which are the key determinants and if weaker determinants play a role and in what cases. Consequently, reliable models for predicting protein production based on codon usage are unavailable. Many of the algorithms used for ‘codon optimization’ are based on the CAI score or variations thereof, which in practice often fail to give optimal results (19).
To further contribute to the understanding and predictability of protein production based on codon usage, aiming to understand influences throughout the whole coding sequence, we decided to generate a large, gene-wide synonymous codon library of the gene encoding the monomeric Red Fluorescent Protein (mRFP), and express this library in E. coli. This reporter protein has rarely been used in studies that focus on codon usage, as opposed to GFP, which is predominantly used in this field. We chose a different reporter protein to examine if a different gene candidate would lead to new findings on the determinants of codon usage. To improve our fundamental understanding of the impact of codon usage, and in an attempt to improve the predictability of optimal codon usage for protein production, we decided to test different interpretable machine learning approaches. Very recently, some studies have successfully utilised machine learning methods to predict protein production efficiency based on randomised sequence libraries for non-coding gene regions, such as promoters and 5′ untranslated regions (5′ UTR) in E. coli and Saccharomyces cerevisiae (20,21), as well as for a factorially designed 5′ end of the CDS (22).
In this study, we constructed mRFP gene libraries for which the codon usage throughout almost the whole gene was fully randomised. To this end, we used an assembly approach based on type IIS restriction and ligation, which has not been used before for whole-gene codon randomization. After library assembly, high-quality curated protein production levels were measured for 1459 individual clones, and their specific coding sequences were accurately determined. We then used these pairs of CDS sequences and corresponding expression values as training data for our machine learning algorithm MEW (mRNA Expression Wizard), to establish an algorithm that can predict protein production from the CDS. Remarkably, we show that only a window covering codons 2–8 is required to accurately predict mRFP production, based on sequence information only. This further strengthens the conclusions from previous studies and demonstrates that other codons later in the CDS in this study are not important to explain protein production. This also underlines that future studies aiming to optimize protein production should focus mostly on the codon usage of the 5′ start of the coding sequence, rather than the current practice of full CDS optimization.
MATERIALS AND METHODS
mRFP codon randomization
The amino acid sequence of the monomeric Red Fluorescent Protein (mRFP) was used to generate three degenerate DNA sequences representing our libraries (CAIL, CAIM and CAIH). Libraries were designed as such that they could be assembled from DNA oligos via a Type IIS restriction enzyme-based assembly method. Each degenerate library sequence was split into blocks of roughly equal sizes (80–90 nucleotides) in such a way that each block has a unique 4-bp overhang with neighbouring blocks. Overhangs were selected from a set that is optimised for high ligation fidelity via Type IIS assembly (23). To create each required fixed overhang sequence, we attempted to fix degenerate codons in such a way that the separate blocks were roughly equal in size, and that loss of degeneracy stayed limited. For example, fixing the degenerate sequence ARAT to AAAT would result in the loss of 1 codon possibility, while fixing the degenerate sequence YGCN to CGCC would result in the loss of seven codon possibilities. 5′ and 3′ flanking sequences containing recognition sites for the Type IIS restriction enzyme BsaI-HF®v2 (NEB, R3733) were added to each DNA block, to generate unique single-stranded overhangs after digestion. The 5′ end of the first block and the 3′ end of the last block contained SapI (NEB, R0569) recognition sites instead. Each block was ordered as a DNA oligo (Ultramer® DNA Oligonucleotides, IDT) (Supplementary Table S2) and using a strand-displacing Taq polymerase (NEB, M0482), the ssDNA was converted to double-stranded DNA via PCR. PCR reactions containing the dsDNA block were cleaned and concentrated to 20 μl mQ using the DNA Clean & Concentrator™-5 kit (Zymo, D4004). 4 μl Gel Loading Dye, Purple (6×) (NEB, B7024) was added to each block and they were loaded on a 1% agarose gel and ran for 30 min at 100 V. The dsDNA blocks were excised from the gel and purified to 20 μl mQ using the Zymoclean™ Gel DNA Recovery Kit (Zymo, D4002). 5 μl of the dsDNA was used to quantify the DNA concentration with the Qubit assay (Invitrogen, Q32853) according to the manufacture's protocol.
The dsDNA blocks were mixed in an equal molar ratio to a total volume of 41 μl, with 5 μl T4 Ligase Buffer (NEB, B0202), 400 units T4 Ligase (NEB, M0202) and 60 units BsaI-HF®v2 (NEB, R3733). Assembly reaction was done overnight at 37°C for 18 h, followed by 5 min at 60°C and a holding step at 12°C. The assembly is cleaned and concentrated to 15 μl mQ using the DNA Clean & Concentrator™-5 kit (Zymo, D4004). 3 μl Gel Loading Dye, Purple (6×) (NEB, B7024) was added and the assembly mixture was loaded on a 1% agarose gel and was run for 40 min at 100 V. The full-length assembled product was excised from the gel and purified to 44 μl mQ using the Zymoclean™ Gel DNA Recovery Kit by Zymo (D4002). 10 units of SapI (NEB, R0569) were added with 5 μl CutSmart Buffer (NEB, B7204) and digested for 2 h at 37°C. The digested codon random mRFP library with single-stranded overhangs was cleaned and concentrated to 15 μl mQ using the DNA Clean & Concentrator™-5 kit by Zymo (D4004). The complete 15 μl containing the codon random mRFP library was used in a ligation reaction to generate the plasmid library.
Plasmid preparation and library generation
The pFAB3909 plasmid (24) (Addgene #47812) with a P15A origin, kanamycin resistance gene and bicistronic design element was modified to be able to accept the codon randomised mRFP library and include a constitutively expressed GFPuv gene. The relatively weak bla promoter was used to drive the mRFP expression, keeping the total protein yield relatively low to prevent overburdening of the protein production machinery and negative growth effects on production for high producing mRFP codon variants. A strong terminator was used for efficient transcription termination and to enhance mRNA stability (25). The open reading frame was replaced by SapI recognition sites to generate the sticky overhangs that accept the mRFP library and a large fragment of nonsense DNA was inserted between the SapI sites to be able to easily separate the double SapI digested plasmid from linear product based on size on a gel. A GFPuv gene, driven by the P4 promoter (24), was added to the plasmid as an internal standard for gene expression. Expression of GFPuv by this promoter is weak as to not interfere with the mRFP expression efficiency, but strong enough for detection with flow cytometry, to allow for data normalization.
About 3 μg plasmid was digested with 20 units SapI (NEB, R0569) and dephosphorylated with 3 units rSAP (NEB, M0371) with 6 μl CutSmart Buffer (NEB, B7204) in a total volume of 60 μl for 3 h at 37°C, followed by an inactivation step at 65°C for 20 min. The linear plasmid was excised from the gel and purified to 30 μl mQ using the Zymoclean™ Gel DNA Recovery Kit by Zymo (D4002). The codon random mRFP library (15 μl) was ligated into 30 ng linear plasmid with 400 units of T4 ligase (NEB, M0202) and 2 μl T4 Ligase Buffer (NEB, B0202) in a total volume of 30 μl for 18 h at 16°C. The ligation mixture was cleaned and concentrated to 10 μl mQ using the DNA Clean & Concentrator™-5 kit by Zymo (D4004). 1 μl of the codon randomised mRFP library was transformed into electrocompetent DH10B cells (20 μl competent cells, 2 mm cuvette, voltage: 2500 V, resistor: 200 Ω, capacitor 25 μF, BTX® ECM630). Cells were recovered in 1 ml NEB® 10-beta/Stable Outgrowth Medium (NEB, B9035) at 37°C for 1 h. The cells were transferred to a 50 ml tube and 9 ml LB (10 g/l Peptone (OXOID, LP0037), 10 g/l NaCl (ACROS, 207790010) and 5 g/l yeast extract (BD, 211929)) were added with 50 μg/l kanamycin (ACROS, 450810500) and incubated for 18 h at 37°C.
Expression range enrichment and selection
A Fluorescence Activated Cell Sorter (FACS) (Sony, SH800S Cell Sorter; GFPuv excitation at 488 nm, emission at 525/50 nm; mRFP excitation at 561 nm, emission at 617/30 nm) was used to sort 50 000 cells of the overnight cell culture into three groups based on protein production levels. The left and right tail of the normal distribution and a part of the middle peak were sorted to create 3 groups of low, medium and high production. The three cell groups were put on individual agar plates (10 g/l Peptone (OXOID, LP0037), 10 g/l NaCl (ACROS, 207790010), 5 g/l yeast extract (BD, 211929), 15 g/l agar (OXOID, LP0011), 50 mg/l kanamycin (ACROS, 450810500)) and grown overnight at 37°C. From these plates, individual colonies were picked and grown in 2 ml 96-well plates with 200 μl LB with kanamycin (10 g/l peptone (OXOID, LP0037), 10 g/l NaCl (ACROS, 207790010), 5 g/l yeast extract (BD, 211929), 15 g/l agar (OXOID, LP0011), 50 mg/l kanamycin (ACROS, 450810500)) for 18 h at 37°C.
Measurements and sequencing
The cell cultures were diluted 100× in PBS (8 g/l NaCl (ACROS, 207790010), 200 mg/l KCl (ACROS, 196770010), 144 mg/l Na2HPO4 (ACROS, 12499010), 240 mg/l KH2PO4 (ACROS, 447670010)). mRFP expression was measured using a flow cytometer (Thermo, Attune NxT Flow Cytometer; GFPuv excitation at 405 nm, emission at 512/25 nm; mRFP excitation at 561 nm, emission at 620/15 nm; stop option 200 000 single cells). A gate was used to exclude GFPuv outliers (±10% of the total population) aiming to reduce unrelated biological variance as GFPuv expression levels are expected to stay constant. From the overnight cultures, 1 μl of cells were used in a PCR reaction to amplify the DNA for Sanger sequencing using Q5 (NEB, M0492). The PCR reaction was sent to Macrogen Europe B.V. for sample clean-up and Sanger sequencing.
All cell cultures were also measured using a microplate reader (BioTek, Synergy Mx). 50 μl overnight cell cultures were diluted in 50 μl PBS (8 g/l NaCl (ACROS, 207790010), 200 mg/l KCl (ACROS, 196770010), 144 mg/l Na2HPO4 (ACROS, 12499010), 240 mg/l KH2PO4 (ACROS, 447670010)). The plates were incubated at room temperature for 1 h before measuring (cell density measured at 600 nm; GFPuv excitation at 395/9 nm, emission at 508/9 nm; mRFP excitation at 584/9, emission at 607/9 nm). The microplate reader fluorescent readings were normalised with the OD600 for both the GFPuv and mRFP readings.
Data validation
Before using the measurements in our machine learning approach, we set a few criteria that the data had to meet, in order to exclude artefacts in our dataset. First, the sequencing data should be of sufficient quality and the encoded amino acid sequence should be correct. The raw sequence data was validated by extracting the open reading frame sequence using in-house scripts. All bases in the open reading frame needed a Phred quality score >20 (a base call accuracy of at least 99%) and the translated sequence should match the mRFP amino acid sequence in order to pass. Second, any double populations or clear changes in cell morphology or culture density were excluded. Double populations were apparent in the flow cytometry data (see Supplementary Figure S2A) but were already automatically excluded due to the sequencing quality criteria as a double population will result in poor Sanger sequencing data. However, very rarely, we observed an unexplained shift in cell morphology as increased forward and side scatter values were obtained during flow cytometry (Supplementary Figure S2B). Finally, rarely, a difference was observed in fluorescence between our flow cytometry measurements and microplate reader measurements. Based on a set threshold of 25% deviation from the average relationship between the two measurement methods we excluded these deviating cell cultures (see Materials and Methods and Supplementary Figure S3). All exclusions were made prior to our analysis in an attempt to generate high-quality data to feed the machine learning algorithm. For the remaining data points, we assessed dataset-wide biases and correlations, such as assembly bias and the correlation between protein production level and GC content to ensure the dataset as a whole was appropriate for machine learning. All raw data and validated data are available in Supplementary Data S1 and S2.
Proteomics sample preparation and digestion
For 10 strains with a wide linear range of fluorescence levels, mRFP levels were also verified using proteomics. For each, 10 ml cell culture was grown overnight at 37°C. The cell pellets were resuspended in 1 ml lysis buffer containing 20 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 20 units/ml DNAse and cOmplete protease inhibitor cocktail (Roche) (pH 7.8). 400 μl of lysate was sonicated using a Qsonica Q500 sonicator (Qsonica LLC) operating at 80% amplitude with on/off interval of 2 and 8 s, respectively, then pelleted at 13 000 RPM for 15 min. Protein concentrations for supernatants (soluble fractions) were measured using the Qubit Protein Assay Kit (Invitrogen) and concentration for the pellet fraction were estimated by assuming 1.5 mg total protein in a 10 ml culture and subtracting the measured amount of soluble protein. Pellets were resuspended in 400 μl lysis buffer (insoluble fractions). Urea was added to all soluble and insoluble fraction samples to a final concentration of 8 M. 100 μl (∼12.5 μg protein) of each sample was reduced and alkylated by incubation with 8.4 μM dithiothreitol at 60°C for 1 h, followed by incubation with 19 μM iodoacetamide in the dark at room temperature for 30 min. Samples were incubated overnight at 37°C with trypsin (Sigma-Aldrich) and LysC (Wako) with enzyme:substrate ratios of 1:26 and 1:44, respectively. Samples were acidified with 10% trifluoroacetic acid to pH <3 to quench the digestion, then desalted via SPE using an Oasis PRiME HLB 96 well plate (Waters) and stored at –20°C until further analysis.
Liquid chromatography–mass spectrometric proteomics analysis
Proteomics samples were reconstituted in 10 μl 2% formic acid and analysed using an Ultimate 3000 HPLC system coupled on-line to an Orbitrap Fusion mass spectrometer (both Thermo Fisher Scientific). Trapping was performed on a 300 μm × 5 mm PepMap Neo trap cartridge (Thermo Fisher) in 100% solvent A (0.1% formic acid) at 300 nl/min for 5 min, prior to separation on a 75 μm × 500 mm column packed in-house with ReproSil-Pur 120 C18-AQ 2.4 μm resin (Dr Maisch) at 300 nl/min using a 90 min gradient as follows: 9% B (80% acetonitrile, 0.1% formic acid) for 1 min, 9–13% B for 1 min, 13–44% B for 70 min, 44–99% B for 3 min, 99% B for 4 min, 99–9% B for 1 min, 9% B for 10 min. Peptides were ionized using a 2.0 kV spray voltage. MS scans were acquired within a 375–1500 m/z range with a maximum injection time of 50 ms at a mass resolution of 60 000 and an automatic gain control (AGC) target value of 4 × 105 in the Orbitrap mass analyzer. Dynamic exclusion was set to 12 s for an exclusion window of 10 ppm with a cycle time of 1 s. MS/MS scans were performed for precursors with 2+ to 8+ charge states and intensities above 5 × 104 at a constant normalized collision energy of 30%. MS/MS scans were acquired within a 100–2300 m/z range with a maximum injection time of 22 ms at a mass resolution of 15 000 at AGC target of 5 × 104 in the Orbitrap mass analyzer.
Proteomics data analysis
Raw files were processed using MaxQuant version 2.0.3.1. Proteins and peptides were identified using a target-decoy approach with a reversed database, using the Andromeda search engine integrated into the MaxQuant environment. The database search was performed against the E. coli (strain K12) Swiss-Prot database (version October 2022) supplemented with the full protein sequence for mRFP, and against the common contaminants database integrated in MaxQuant. Default search settings were used, including methionine oxidation and protein N-term acetylation as variable modifications, enzyme specificity set to trypsin with maximum two missed cleavages, a minimum peptide length of seven amino acids, a maximum peptide mass of 4600 Da and 1% false discovery rates. Label-free quantification via MaxLFQ algorithm (26) was performed, and ‘match between runs’ was enabled. Microsoft Excel 2016 and GraphPad Prism 9 were used for further data analysis and plotting graphs. Adjusted mRFP LFQ intensities were calculated to estimate the truly insoluble mRFP abundance, as part of the pellet contained unlysed cells with soluble protein. The adjusted insoluble mRFP LFQ intensity was calculated by multiplying the LFQs of mRFP for the peptide fraction by the ratio of pellet:soluble LFQ intensities for peptide deformylase (DEF, UniProtKB P0A6K3, a highly soluble cytosolic protein) in the same fraction, and then subtracting this value from the original insoluble LFQ intensity:
 |
Building machine learning regressors
To assess if protein production levels could be predicted from sequence, we employed two different machine learning approaches: Random Forest (RF) Regressor and LASSO. We implemented RF and LASSO using the scikit-learn package (v0.23.0, ref) in python (v3.7.6), with the sklearn.ensemble.RandomForestRegressor and sklearn.linear_model.Lasso modules respectively. For RF, default settings were used, while for LASSO, various values for alpha were assessed (0.05, 0.1, 0.5, 1.0, 2.0, 5.0, 10.0, 20.0, 50.0 and 100.0 for all regressors, in addition to 500.0 and 1000.0 for regressors trained on full-length sequences; max iterations = 10 000). An alpha of 100.0 gave rise to the best predictions for full-length sequences (Supplementary Figure S4); for windows, different alphas performed better for different featurizations, but differences were minimal. Separate regressors were constructed for full-length featurised mRNA sequences and for featurised sliding windows of 10, 20, 30, or 40 bases. Prior to training, an independent test set comprising 10% of the data was set aside. Regressor accuracies were then evaluated on the remaining training data through 10-fold cross-validation, where 90% of the training data were used to predict the translation efficiency of the other 10%. This was done for each 10% of the data, such that we obtained a predicted translation efficiency, measured with flow cytometry, for each data point. From these predictions, Pearson and Spearman correlations were computed for actual flow versus predicted flow and used as measures for model accuracy. Feature importances were extracted from all ten regressors built in cross-validation, averaged, and plotted and visualized with matplotlib (v3.2.1). Finally, for regressors trained on the full-length sequences and the best-performing sequence windows (Supplementary Figures S5 and S6), new regressors were trained using all training data, and model accuracies were re-evaluated on the independent test-set. Code and regressors are made available at https://zenodo.org/record/7547381#.Y8jvJi8w1qs.
RESULTS AND DISCUSSION
Type IIS assembly method allows for generation of codon randomized libraries at different CAI levels
To randomize synonymous codon usage throughout the whole mRFP CDS, we developed a randomization-assembly method based on Type IIS restriction and ligation. We used this approach to generate three codon randomized mRFP libraries with either fully randomized codon usage or with a focus on more frequent or rare codons. The first CAI library (‘medium’, CAIM) is fully randomized and uses an equal distribution of all synonymous codons for each amino acid. Only for the 6-codon amino acids arginine, leucine and serine, as well as the stop codons, the full codon space could not be covered due to the sequence limitations in degenerate oligos. For each of the three 6-codon amino acids four of the six possible codons were included, while the stop codon was kept constant at TGA. Theoretically, the maximum number of CDSs coding for the mRFP protein is 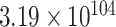 . By limiting the aforementioned amino acids, the stop codon and a few fixed codons needed for assembly purposes the library still contains at most
. By limiting the aforementioned amino acids, the stop codon and a few fixed codons needed for assembly purposes the library still contains at most 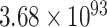 variants. Obviously, this is an astronomically large number and generated libraries and experimental efforts can only cover a very small fraction of this diversity.
variants. Obviously, this is an astronomically large number and generated libraries and experimental efforts can only cover a very small fraction of this diversity.
The randomized design approach results in a uniform codon bias distribution across the gene, with an overall medium CAI of 0.67 (Supplementary Figure S1). To see if codon randomization with an overrepresentation of frequent or rare codons affects translation differently, we generated two additional libraries. By restricting the allowed relative adaptiveness (the usage ratio of a codon to that of the most abundant synonymous codon), we generated libraries that use more frequent or rare codons. The library limited to rare codons used only synonymous codons with a relative adaptiveness <0.60, or the lowest relative adaptiveness in the case the synonymous codons are used in a close to equal ratio. This rare codon library (CAIL) had an overall CAI of 0.41 (Supplementary Figure S1). The library using frequent codons (CAIH) used only synonymous codons with a relative adaptiveness >0.50, resulting in a library with an overall CAI of 0.83 (Supplementary Figure S1). The sequence spaces for each of the libraries are reported in Supplementary Table S1.
To create the three libraries, we divided the complete degenerate CDS into eight blocks of ∼85 bases, which can be assembled with unique overhangs between each adjacent part. In order to generate complementary four base pair overhangs between the parts, some codons with multiple synonymous options needed to be fixed to a single codon. The eight DNA parts were ordered as single-stranded oligos with additional Type IIS restriction sites flanking the blocks. The oligos were converted to double-stranded DNA using PCR and consequently assembled using Type IIS restriction and ligation (Figure 1A, B). Only a small fraction of the total DNA parts assembled into the full product of 707 bp (Figure 1B, indicated with the ‘mRFP’ label). Seven intermediate products were observed that did not further assemble into the full gene. The assembly limitations could for example originate from synthesis errors in the initial oligos, preventing Type IIS restriction or resulting in incorrect overhangs. The band corresponding to the fully assembled product was purified and ligated into an expression vector (Figure 1C).
Figure 1.

Codon randomized library generation and analysis. (A) PCR is used to generate dsDNA from oligos and an electrophoresis gel yields the eight dsDNA blocks used to build codon randomized mRFP. (B) The assembly reaction and the electrophoresis gel result of the assembly. The complete assembly of all eight blocks is indicated with the mRFP tag. The seven bands below are intermediate products. (C) The expression vector used to express the codon random mRFP. (D) FACS enrichment for a wide expression range within the library is used to obtain a higher representation of the high and low expressing codon variants. (E) Flow cytometry analysis of cultures and Sanger sequencing data are QA passed and used in machine learning models.
The expression vector contains a native, relatively weak, beta-lactamase promoter (Pbla). A weak promoter poses less burden on transcriptional and/or translational processes, reducing the risk of reaching an upper limit in the protein production process and thus preserving the full expression range as determined by the coding sequences. For the 5′ UTR, a medium-strength bicistronic design (BCD) was chosen based on the work of Mutalik et al. (27). This BCD element (BCD5) was previously reported to reduce the influence of mRNA secondary structures on expression. Including this element allows us to study the more nuanced features associated with codon usage and should reduce strong effects on translation caused by mRNA structure formation between the coding sequence and the constant 5′ UTR.
Expression of libraries in E. Coli results in a wide expression range and allows for high-quality data collection
The library containing codon randomized mRFP was transformed into E. coli DH10B. A single transformation of the libraries in E. coli yielded between 150 000 and 320 000 colonies. After 18-h cultivation on liquid, roughly 70% of the cells gave a detectable level of red fluorescence (measured using flow cytometry, Figure 2). The remaining 30%, for which no or very little fluorescence was measured, was later confirmed via sequencing to mainly comprise constructs that had a frameshift in the ORF. This is not unexpected, as some blocks are likely missing one or multiple nucleotides since the coupling efficiency of oligos is not 100%. These errors eventually lead to frameshifts and thus protein truncations or mutations.
Figure 2.
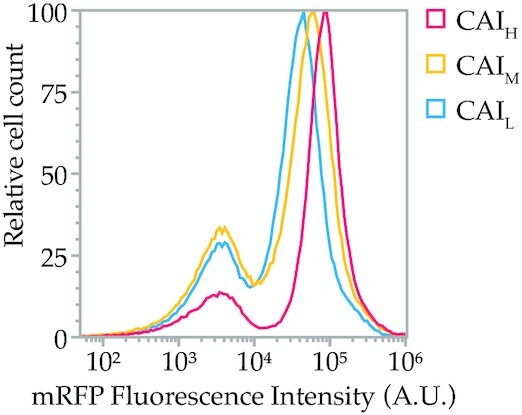
Normalized flow cytometry overlay of the mRFP fluorescence intensity from the CAIL, CAIM and CAIH libraries. The left peak is part of the population showing no fluorescence, mainly due to assembly errors in the CDS. The right peak shows the mRFP expression of each library. The average expression of the CAIL, CAIM and CAIH increases with average library CAI, but high expressing variants are found in all libraries (right tail). The ratio between the left and right peaks is a measure of library fidelity, as the left peak consists of autofluorescence of non-expressing or non-functional variants.
The flow cytometric evaluation of the three library populations showed that the average expression of the CAIL, CAIM and CAIH libraries increases with average library CAI (Figure 2). This suggests that an overall higher CAI leads to higher expressing constructs on average. However, for all three libraries, expression could be observed at the highest end of the expression spectrum, suggesting that a high CAI is not the leading factor for high protein production.
To obtain high-quality expression and sequence data for the downstream machine learning analyses, we decided to study expression levels and related sequences of individual clones. We favoured this method over previously used FlowSeq methods, which perform sequencing analysis on large mixes of clones obtained in certain bins during fluorescence activated cell sorting (FACS). FlowSeq typically employs short-read sequencing (Illumina), which will not cover the full CDS length in a single read and due to the whole-gene codon degeneracy in this study, it would be difficult to assemble reads into contigs. Alternative long-read single-molecule methods (e.g. PacBio) would offer a solution, but it was questionable whether a sufficiently high coverage could be achieved to reach meaningful conclusions. FlowSeq has another limitation, as the fluorescence level detected from cells with the same genotype already can cover a relatively wide range (28). This increases the likelihood that individual cells are binned incorrectly and that the resulting dataset is too noisy to be analysed meaningfully through statistical analyses and machine learning. This can potentially be solved by sequencing with a very high coverage; however, this is hard to achieve for high-quality long reads as mentioned above. We chose to select a limited number of individual clones for which mean expression values can be accurately determined, as well as their full gene sequences using Sanger sequencing.
To allow the selected clones to cover a wide range of low, medium and high expressing constructs, and exclude non-expressing (e.g. frameshifted) constructs, three expression-level groups were preselected for each CAI library using FACS (Figure 1D). After sorting, we picked colonies from each group. These clones were all inoculated in liquid culture (for a total of 480, 1440 and 480 individual cultures for CAIL, CAIM and CAIH respectively). The fluorescence of the cell cultures was measured using both flow cytometry and a microplate reader. mRFP expression was normalized through comparison with the constant constitutively expressed GFP (Figure 1C). The mRFP coding sequence (and untranslated regions) was amplified using colony PCR and amplicons were analysed by Sanger sequencing. Next, the data were evaluated to exclude low-quality sequencing reads, amino acid mutations, mixed populations (Supplementary Figure S2A), and rare deviations in cell morphology (notable increase in FSC and SSC as observed in the flow cytometer, Supplementary Figure S2B) or culture density (deviation between measurements by flow cytometry and microplate reader, Supplementary Figure S3). After this exclusion, 1459 sequences which showed a high sequence diversity for each of the variable bases in the CDS (Supplementary Data S3) were selected for further analysis. The exclusion criteria are further described in the Materials and Methods – Data validation section. This yielded 1459 high-quality data points that we could use in our machine learning approach (Figure 1E).
LC–MS/MS-based proteomics demonstrates fluorescence intensities correlate well with mRFP levels
It was previously reported that codon changes can also influence the folding properties, which can result in different protein functionalities or misfolding for some proteins (29,30). Misfolded or differently folded mRFP could in theory influence fluorescence levels. So far, this potential influence of codon variation on fluorescence levels has been typically ignored in studies using fluorescent proteins (mostly GFP) as reporter protein. In this study, we verified that the measured fluorescence levels correlate to the mRFP protein levels by cross-checking abundances with quantitative LC–MS/MS-based proteomics. We selected 10 mRFP gene variants that cover the complete observed fluorescence range in our libraries. For strains harbouring these variants, the soluble and insoluble protein fractions were quantified from the mass spectrometry data. This analysis revealed that fluorescence levels correlated well with the determined abundances (Pearson correlation 0.901) and that only a minor part of mRFP ends up in the insoluble fraction (Figure 3). Hence, we conclude that the fluorescence levels are an effective approximation of mRFP protein abundances in the cell and that fluorescence intensity data can be used for machine learning.
Figure 3.
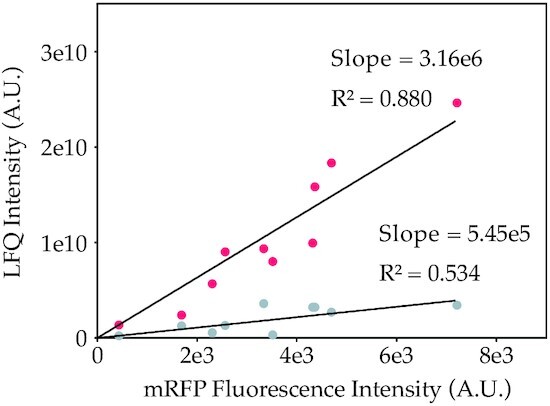
Relation between LC–MS/MS based quantification of mRPF and fluorescent intensity for the soluble (red) and insoluble (gray) protein fractions of 10 mRFP codon variants. LC–MS/MS based quantification of mRFP was performed to determine relative, label-free quantities (LFQ) of both soluble and insoluble mRPF. Insoluble LFQ intensities are adjusted for soluble mRFP ending up in the pellet fraction (in non-lysed cells) by correcting these values based on LFQ intensities for a highly soluble E. coli protein in the pellet fraction.
Different machine learning approaches can predict mRPF production levels
To identify the determinants of protein production levels and to assess if the protein production levels could be predicted from gene sequence, we employed two different machine learning approaches: Random Forest Regressor (RFR) and LASSO (Least Absolute Shrinkage and Selection Operator). For this purpose, we developed MEW: the mRNA Expression Wizard, which can train and test a variety of machine learning models to predict the protein production level from mRNA sequence, using different types of featurizations. These featurizations include methods that focus on the base pair composition of the coding sequence, to observe the effects of factors like translation elongation efficiency, and featurizations that reflect the probability that a base is paired in the context of an mRNA secondary structure.
Our rationale to use both LASSO and RFR is that due to their stepwise decision making, RFRs can model non-linear interdependencies between bases, while LASSO is better suited to straight-forward linear regression and feature selection. Importantly, for each regressor we extracted the feature importances as this could help to identify determinants of translation efficiency. We trained separate regressors for both full-length featurized mRNA sequences and for sliding windows of varying sizes along the entirety of the mRNA, to assess if certain windows are more predictive of translation efficiency than others.
As performance of machine learning algorithms depends greatly on their input data, featurizing our mRFP data in a way that captures most information was key. We used three featurization methods: one based on one-hot encoding of base identity, another based on predicted base pairing probabilities for each individual base via calculating the mRNA secondary structure probabilities or the whole transcript using ViennaRNA (31), and a third featurization method which combines these two. One-hot encoding could also capture base pairing probability. However, we decided to include a specific, predicted mRNA secondary structure featurization as this can to some extent ‘isolate’ this feature from all other features linked to specific bases and codons. We will call the three types of featurizations one-hot, BPP (base pairing probability), and one-hot + BPP respectively.
We trained and validated the RFR and LASSO regressors with our flow cytometry data and Sanger sequencing data, using 10-fold cross-validation, yielding a value of predicted mRFP production level for each data point. Dependent on the method used (LASSO or RFR) and the featurization method, the prediction accuracy somewhat varies. However, all methods can predict protein levels reasonably well, with Pearson correlation coefficients ranging from 0.546 up to 0.776 (Figure 4, Supplementary Table S3).
Figure 4.

Actual expression data vs predicted expression using various machine learning algorithms and featurizations. Blue, yellow and red points indicate data points from the leave-out test sets for the CAIL, CAIM and CAIH libraries respectively. (A) Actual expression data vs predicted expression using LASSO. (B) Actual expression data vs predicted expression using the Random Forest Regressor (RFR). Regressor accuracies were evaluated through 10-fold cross-validation.
The predictive strength of one-hot encoding featurization is stronger than that of BPP featurization. This is not unexpected, as BPP featurization assumes that mRNA secondary structures are the only cause of expression variance, and base pairing featurization is done based on calculated mRNA secondary structure probabilities (ViennaRNA), which likely cannot perfectly predict exact base pairing. Still, BPP featurization yields reasonable performance, which suggests that mRNA structures are a key factor in predicting protein production levels. One-hot encoding captures all information in the sequence and expectedly gives substantially better performance. Combining one-hot encoding with BPP featurization does not substantially improve predictions, in line with our expectation that all information on base pairing probability should already be captured by one-hot encoding. No large differences in performance between the two regression methods LASSO and RFR are observed. When only BPP is used as feature, RFR performs slightly better than LASSO; when one-hot or one-hot + BPP are provided, LASSO slightly outperforms RFR.
Bases surrounding the start codon and the RBS are most predictive of translation efficiency
Next, we assessed which features, and by extension which bases, are most predictive for translation efficiency. We did this by extracting the coefficients for LASSO and the feature importances for RFR and plotting them against sequence position (Figure 5). For the BPP featurization, information can be obtained for every nucleotide, including constant nucleotides in the CDS and the constant UTRs, as they are still involved in the overall secondary structure formation predictions. For the one-hot featurization however, the information is only limited to changing nucleotides and can therefore be plotted per codon as only every third base varies across gene variants.
Figure 5.

Feature importances for various machine learning algorithms and featurizations. LASSO feature importances are coefficients: a positive coefficient indicates a positive correlation between a base and translation efficiency, a negative coefficient indicates a negative correlation. In RFR, feature importances are always positive and therefore not indicative of the directionality of the correlation. (A) Feature importances for algorithms using BPP featurization. (B) Feature importances for algorithms using one-hot encoding. Since only every third one-hot encoded base of the coding sequence varies, only every third base of the coding sequence was plotted. (C) Feature importances for algorithms using BPP + one-hot featurization. BCD = bicistronic design 5′ untranslated region; RBS = ribosome binding site; CDS = coding sequence; TER = terminator.
Overall, we found that independent of the algorithms used, the most predictive bases were always close to the start of the CDS, including the 5–10 bases before the start codon for BPP featurizations and the first 25 bases following the start codon for all featurizations. In comparison, the remaining codons play a minimal role in predicting translation efficiency. This strongly suggests that mRNA secondary structure or other factors around the start of the coding sequence play a dominant role in determining translation efficiency. This is in agreement with previous studies that found the same effect for GFP and attributed this mostly to the necessity for an accessible RBS for translation initiation (2,4). However, in this study we used a BCD design in the 5′ UTR, which should in theory improve RBS accessibility, independent of sequence context. The BCD design encodes an RBS1 which initiates the translation of a short leader peptide. This RBS1 should lead to efficient ribosome recruitment for the translation of mRFP, as the ribosomes translating the leader peptide can unfold the mRNA structures around the RBS2. The latter RBS is the translation initiation site for mRFP. Due to the translating ribosomes from the leader peptide, this ‘unfolded’ mRNA region should be better available for translation initiation and mRNA structures should have less influence on translation initiation. Still, we observe that the first codons and their base pairing probabilities explain a large part of the protein production levels. This could be explained by the fact that the BCD design may be unable to completely resolve inhibitory secondary structure effects, or other factors at the start of the coding sequence may still play a dominant role.
Interestingly, the LASSO regressor using BPP for featurization assigns positive coefficients to the bases immediately succeeding the RBS2 in the BCD region (Figure 5A, first panel). A positive coefficient indicates that involvement in mRNA secondary structures at this position is positively correlated with gene expression levels. In contrast, the bases in the RBS2 itself and the 5′ of the coding region are overwhelmingly assigned negative coefficients, which supports the hypothesis that minimal secondary structure for bases involved in the RBS is beneficial for high protein production levels. In concordance, the presence of A or T bases in the 5′ of the coding region, particularly ‘A’, is strongly positively correlated with protein production levels, while the presence of G or C bases tends to be negatively correlated with protein production levels in this region (Figure 5B, first panel). As A–T base pairs, and their A–U equivalents in mRNA, only form two hydrogen bonds versus three in G–C base pairs, the resulting secondary structures are weaker, and as a result, the RBS may be more accessible.
A sequence window covering first eight codons can predict protein production
To further substantiate our finding that bases surrounding the start codon dictate translation efficiency, we trained regressors on sliding windows of 10, 20, 30 or 40 bases to visualize which regions of the mRNA were most predictive of translation efficiency. For each sliding window, we performed a 10-fold cross-validation and plotted the correlation between actual expression data and the predicted expression data as a function of the position of the sliding window (Figure 6). Clear peaks of increased predictive power can be observed around the start codon, which corroborates our earlier finding that this region primarily dictates translation efficiency. This is especially apparent in models trained with one-hot encoded features and BPP + one-hot encoded features. Specifically, the 20 nucleotides surrounding base 15 (bases 6–25) lead to high prediction accuracy (Figure 6B, C, window size 20; Supplementary Figures S5 and S6). This window covers codons 2–8 and logically does not cover the start codon or the first two nucleotides of the second codon, as these are constant in our design and thus cannot have any predictive power in one-hot featurization.
Figure 6.

Predictive regions of translation efficiency in mRFP mRNA. The x-axis represents the central base of a sliding window of indicated lengths, the y-axis the correlation between actual expression data and the expression as predicted by a machine learning algorithm trained on solely that sliding window. (A) Predictive regions found by algorithms trained with BPP featurization. (B) Predictive regions found by algorithms trained with one-hot encoding. As the one-hot encoded features for the UTRs are constant and thus contain no predictive information, windows that only contain residues in the UTR were omitted. (C) Predictive regions found by algorithms trained with BPP + one-hot featurization.
It should be noted that the remainder of the CDS, while a lot less predictive than its 5′ region, still holds some predictive power (Pearson correlation ∼ 0.4). We ascribe this to the inclusion of the CAIH and CAIL libraries in our dataset. These datasets use completely different sets of codons, meaning that the machine learning approaches can infer from small sequence windows the identity of the library from which the sequence originated. Since data points from the CAIH library on average display higher expression levels than data points from the CAIL library (Figure 2), we attribute the non-zero Pearson correlations observed for windows downstream of the 5′ end of the CDS mostly to the algorithm's ability to detect the library of origin of CAIH and CAIL data points. Inspection of scatter plots for windows in these regions confirmed this (Supplementary Figure S7).
We also observed some ‘dips’ in predictability performance in the sliding window analysis. One such dip can be seen for small window sizes in the 3′ UTR with the BPP featurization and LASSO regressor (Figure 6A, C). This region is very invariable both in terms of sequence and secondary structure: since the terminator almost always forms a strong secondary structure, the bases directly before it are less likely to be involved in secondary structures. As a result, the BPP features representing this region hold practically no information. The effect is exacerbated for small windows, as they are less likely to capture predictive residues upstream or downstream of an information-devoid region. In contrast, the secondary structure of the terminator itself does appear to be slightly informative. A perhaps unlikely but possible explanation could be that certain codon sequences interfere with the terminator stem formation and thereby influence mRNA stability. However, it is important to keep in mind that correlations between actual and predicted expression data for regressors trained on this region are still extremely low. Therefore, while the 3′ UTR region holds some information, it is not likely to be very influential.
A second dip is located around base 165 and 166 for regressors using one-hot-encoded featurizations (Figure 6B). This information valley is caused by an unusually constant region in the mRFP gene, particularly in the CAIH library, due to the low codon variability of local amino acids and a fixed boundary region of two assembly blocks. This is an artefact of our method, and hence not a biologically relevant observation. This dip is not observed for featurization methods that also include base pairing probabilities, as base pairing interactions of constant regions with other bases can still be informative.
To better understand which sequence elements in the 20-base window surrounding base 15 affect translation efficiency, we plotted feature importances for each regressor trained on this window (Supplementary Figure S8). From this, we inferred that especially at position 15 (codon 5), low probabilities of involvement in mRNA secondary structure are predictive of high expression. This is in line with the current consensus that minimal mRNA secondary structure surrounding the 5′ end of the coding region is conducive to efficient translation. In the case of mRFP, this low base pairing probability seems to be primarily achieved by placing an ‘A’ at position 15 (Supplementary Figure S8B, C).
Of all our regressors, six random forest regressors outperformed the rest (Supplementary Figures S5 and S6). As these six regressors were comparable in performance, we selected the model that used the fewest features, and retrained the model using our full training set. We then plotted actual expression data against predicted expression for each data point in our leave-out test set. This revealed a very strong correlation (Pearson r = 0.762) for all three libraries (Figure 7), which demonstrates that mRFP protein production can be correlated extremely well to sequence by just looking at bases 6–25 of the entire coding sequence.
Figure 7.
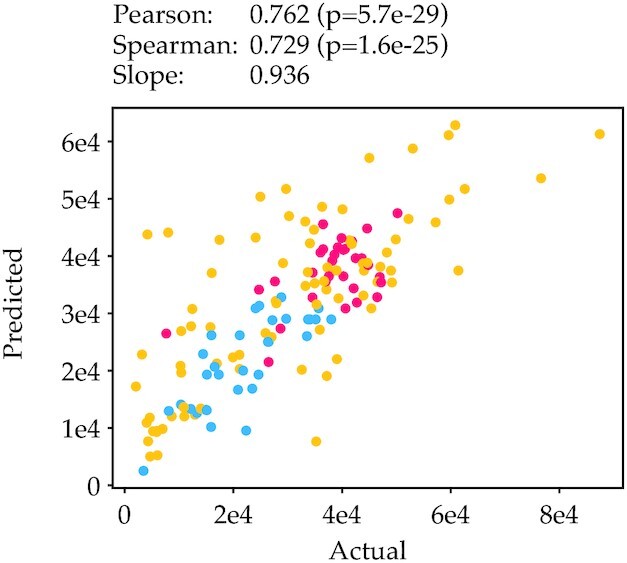
Actual vs predicted expression for one of our six best performing regressors. The RFR regressor on one-hot featurization and a window size of 20 located at base 15 showed a high correlation between actual and predicted expression (Pearson correlation 0.762).
The bases 6–25 correspond to codons 2–8. The nucleotides in the third position of these codons that contribute to high expression are mostly A, as well as T in codon 2 (as shown earlier by the feature importances, Figure 5B). An important question in the field is if the benefit of these codons at the start of the coding sequence is related to mRNA secondary structures and/or the efficient translation of these codons. To approximate which codons can be translated efficiently, commonly indices such as the CAI and tRNA adaptation index (tAI) (32) are used. Interestingly, the beneficial A-ending codons in most cases have a lower CAI and tAI index than other synonymous codons (Supplementary Table S4, Supplementary Figures S9 and S10), but still are highly important for high expression. This indicates that there is an advantage of these codons, likely unrelated to translation elongation efficiency. The most plausible explanation of their mechanistic roles is the lower secondary structure propensity of A/T-ending codons due to general weaker A-T/U binding. This is further supported by the second codon: the T-ending variant is also related to high expression but is the ‘least efficient’ codon, based on CAI and tAI. However, for the eighth and last codon of the important codon window, there is a reverse situation where a C is favoured over an A for high expression. The A-ending codon has a very low tAI and CAI value, lower than any available codon in the first 8 codons. For this codon, the relevance of ‘optimal translation’ seems to be dominant over the influence of secondary structure binding propensity of C/G-ending codons. Since this codon within our codon window is furthest away from the translation initiation region it is less likely to form detrimental RBS-obscuring secondary structures.
Because translation initiation is the major rate-limiting factor, the effects of codon usage throughout the gene seem less apparent. This is also exemplified by our finding that the highest expressing variants originate from our CAIM library. This library contains more codon variance than the CAIH library (Supplementary Table S1), which is particularly important for the 5′ of the CDS.
Altogether, our results show that while the CAI has some influence on gene expression (Figure 2), the majority of the translation regulation arises from codon usage in the 5′ of the CDS. This matches the conclusion made by Kudla et al., which proposed the secondary structures predicted a window of base -4 to +37 (spanning codon 1–13) as a key determinant for GFP production. In our analysis a slightly smaller window of codon 2–8 is sufficient to predict protein production. The same 8-codon window was also found in another systematic effort parallel to our study, using an alternative non-fluorescent reporter system (Bxb1 recombinase). This suggests that the relevance of this window may be a general phenomenon, at least for gene expression in E. coli (33).
Our study also suggests that the 5′ UTR may influence expression based on the observed BPP feature importance for the bases in that region. However this region was kept constant in this work on purpose. The parallel study by Höllerer et al. (this issue) randomized bases –25 to –1 in the 5′ UTR alongside codons 2–16 of their reporter protein Bxb1. Indeed, this study confirmed a large contribution to expression variance from both the 5′ UTR (50%) and the CDS (20%) (33).
Due to the partial black box nature of machine learning, design rules for the 5′ CDS are not fully apparent. However, our analysis suggests that secondary structure is likely a key determinant of translation efficiency for these codons. It is clear that if high protein production is desired the focus should be on optimizing the start of the coding sequence and the 5′ UTR in E. coli, and possible in other bacterial hosts. Typically, codon optimization algorithms and approaches optimize for parameters such as CAI over the full CDS but ignore the 5′ UTR sequence, and therefore may introduce detrimental secondary structures. We suggest a shift in these approaches to specifically tackle optimization of the first ∼8 codons. Some previous studies have proposed randomization approaches in the 5′ UTR and/or first codons (13,34), but these approaches are currently hardly applied. Alternatively, existing or new in silico design tools considering secondary structures in the 5′ UTR—CDS start region (such as RBS Calculator (35)) could be considered to improve gene expression. This systematic study provides a clear rationale for adopting these methods, rather than commonly used whole-gene codon optimization algorithms, to improve protein production.
DATA AVAILABILITY
MEW: the mRNA Expression Wizard is available at https://zenodo.org/record/7547381#.Y8jvJi8w1qs.
Supplementary Material
ACKNOWLEDGEMENTS
We acknowledge Markus Jeschek and Sjoerd Creutzburg for fruitful discussions and feedback on this work, as well as Rob Joosten and Christian Sudfeld for experimental support for flow cytometry and FACS operation.
Contributor Information
Thijs Nieuwkoop, Laboratory of Microbiology, Wageningen University, Wageningen, Stippeneng 4, 6708 WE, The Netherlands.
Barbara R Terlouw, Bioinformatics Group, Wageningen University, Wageningen, Droevendaalsesteeg 1, 6708 PB, The Netherlands.
Katherine G Stevens, Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, University of Utrecht, Padualaan 8, 3584 CH Utrecht, The Netherlands; Netherlands Proteomics Center, Padualaan 8, 3584 CH Utrecht, The Netherlands.
Richard A Scheltema, Biomolecular Mass Spectrometry and Proteomics, Bijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, University of Utrecht, Padualaan 8, 3584 CH Utrecht, The Netherlands; Netherlands Proteomics Center, Padualaan 8, 3584 CH Utrecht, The Netherlands.
Dick de Ridder, Bioinformatics Group, Wageningen University, Wageningen, Droevendaalsesteeg 1, 6708 PB, The Netherlands.
John van der Oost, Laboratory of Microbiology, Wageningen University, Wageningen, Stippeneng 4, 6708 WE, The Netherlands.
Nico J Claassens, Laboratory of Microbiology, Wageningen University, Wageningen, Stippeneng 4, 6708 WE, The Netherlands.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Nederlandse Organisatie voor Wetenschappelijk Onderzoek [024.003.019, SPI 93–537 to J.v.d.O., VI.Veni.192.156 to N.J.C.]; Wageningen University via the Fellowship program Data Science/Artificial Intelligence (to B.T.). Funding for open access charge: Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO).
Conflict of interest statement. T.N., J.v.d.O. and N.C. have filed a patent regarding the gene assembly approach for codon randomisation. J.v.d.O. is scientific advisor of NTrans Technologies, Hudson Biotechnology and Scope Biosciences.
This paper is linked to: doi:10.1093/nar/gkad040.
REFERENCES
- 1. Nieuwkoop T., Finger-Bou M., van der Oost J., Claassens N.J.. The ongoing quest to crack the genetic code for protein production. Mol. Cell. 2020; 80:193–209. [DOI] [PubMed] [Google Scholar]
- 2. Kudla G., Murray A.W., Tollervey D., Plotkin J.B.. Coding-sequence determinants of gene expression in Escherichia coli. Science. 2009; 324:255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Boël G., Letso R., Neely H., Price W.N., Wong K.-H., Su M., Luff J.D., Valecha M., Everett J.K., Acton T.B.et al.. Codon influence on protein expression in E.coli correlates with mRNA levels. Nature. 2016; 529:358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cambray G., Guimaraes J.C., Arkin A.P.. Evaluation of 244,000 synthetic sequences reveals design principles to optimize translation in Escherichia coli. Nat. Biotechnol. 2018; 36:1005–1015. [DOI] [PubMed] [Google Scholar]
- 5. Harigaya Y., Parker R.. The link between adjacent codon pairs and mRNA stability. BMC Genomics [Electronic Resource]. 2017; 18:364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Goodman D.B., Church G.M., Kosuri S.. Causes and effects of N-terminal codon bias in bacterial genes. Science. 2013; 342:475–479. [DOI] [PubMed] [Google Scholar]
- 7. Looman A.C., Bodlaender J., Comstock L.J., Eaton D., Jhurani P., de Boer H.A., van Knippenberg P.H.. Influence of the codon following the AUG initiation codon on the expression of a modified lacZ gene in Escherichia coli. EMBO J. 1987; 6:2489–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stenström C.M., Jin H., Major L.L., Tate W.P., Isaksson L.A.. Codon bias at the 3′-side of the initiation codon is correlated with translation initiation efficiency in Escherichia coli. Gene. 2001; 263:273–284. [DOI] [PubMed] [Google Scholar]
- 9. Tuller T., Zur H.. Multiple roles of the coding sequence 5′ end in gene expression regulation. Nucleic Acids Res. 2015; 43:13–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quax T.E.F., Wolf Y.I., Koehorst J.J., Wurtzel O., van der Oost R., Ran W., Blombach F., Makarova K.S., Brouns S.J.J., Forster A.C.et al.. Differential translation tunes uneven production of operon-encoded proteins. Cell Rep. 2013; 4:938–944. [DOI] [PubMed] [Google Scholar]
- 11. Sharp P.M., Li W.-H.. The codon adaptation index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987; 15:1281–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Welch M., Govindarajan S., Ness J.E., Villalobos A., Gurney A., Minshull J., Gustafsson C.. Design parameters to control synthetic gene expression in Escherichia coli. PLoS One. 2009; 4:e7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Isacchi A., Sarmientos P., Lorenzetti R., Soria M.. Mature apolipoprotein AI and its precursor proApoAI: influence of the sequence at the 5’ end of the gene on the efficiency of expression in Escherichia coli. Gene. 1989; 81:129–137. [DOI] [PubMed] [Google Scholar]
- 14. Kelsic E.D., Chung H., Cohen N., Park J., Wang H.H., Kishony R.. RNA structural determinants of optimal codons revealed by MAGE-Seq. Cell Syst. 2016; 3:563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frumkin I., Schirman D., Rotman A., Li F., Zahavi L., Mordret E., Asraf O., Wu S., Levy S.F., Pilpel Y.. Gene architectures that minimize cost of gene expression. Mol. Cell. 2017; 65:142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tuller T., Carmi A., Vestsigian K., Navon S., Dorfan Y., Zaborske J., Pan T., Dahan O., Furman I., Pilpel Y.. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344–354. [DOI] [PubMed] [Google Scholar]
- 17. Hanson G., Coller J.. Translation and Protein Quality Control: codon optimality, bias and usage in translation and mRNA decay. Nat. Rev. Mol. Cell Biol. 2018; 19:20–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Radhakrishnan A., Chen Y.-H., Martin S., Alhusaini N., Green R., Coller J.. The DEAD-box protein Dhh1p couples mRNA decay and translation by monitoring codon optimality. Cell. 2016; 167:122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Parret A.H., Besir H., Meijers R.. Critical reflections on synthetic gene design for recombinant protein expression. Curr. Opin. Struct. Biol. 2016; 38:155–162. [DOI] [PubMed] [Google Scholar]
- 20. Höllerer S., Papaxanthos L., Gumpinger A.C., Fischer K., Beisel C., Borgwardt K., Benenson Y., Jeschek M.. Large-scale DNA-based phenotypic recording and deep learning enable highly accurate sequence-function mapping. Nat. Commun. 2020; 11:3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vaishnav E.D., de Boer C.G., Molinet J., Yassour M., Fan L., Adiconis X., Thompson D.A., Levin J.Z., Cubillos F.A., Regev A.. The evolution, evolvability and engineering of gene regulatory DNA. Nature. 2022; 603:455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nikolados E.-M., Wongprommoon A., Mac Aodha O., Cambray G., Oyarzún D.A.. Accuracy and data efficiency in deep learning models of protein expression. Nat. Commun. 2022; 13:7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Potapov V., Ong J.L., Kucera R.B., Langhorst B.W., Bilotti K., Pryor J.M., Cantor E.J., Canton B., Knight T.F., Evans T.C.et al.. Comprehensive profiling of four base overhang ligation fidelity by T4 DNA ligase and application to DNA assembly. ACS Synth. Biol. 2018; 7:2665–2674. [DOI] [PubMed] [Google Scholar]
- 24. Mutalik V.K., Guimaraes J.C., Cambray G., Lam C., Christoffersen M.J., Mai Q.-A., Tran A.B., Paull M., Keasling J.D., Arkin A.P.et al.. Precise and reliable gene expression via standard transcription and translation initiation elements. Nat. Methods. 2013; 10:354–360. [DOI] [PubMed] [Google Scholar]
- 25. Cetnar D.P., Salis H.M.. Systematic quantification of sequence and structural determinants controlling mRNA stability in bacterial operons. ACS Synth. Biol. 2021; 10:318–332. [DOI] [PubMed] [Google Scholar]
- 26. Cox J., Mann M.. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008; 26:1367–1372. [DOI] [PubMed] [Google Scholar]
- 20. Nieuwkoop T., Claassens N.J., van der Oost J.. Improved protein production and codon optimization analyses in Escherichia coli by bicistronic design. Microb. Biotechnol. 2019; 12:173–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peterman N., Levine E.. Sort-seq under the hood: implications of design choices on large-scale characterization of sequence-function relations. BMC Genomics (Electronic Resource]. 2016; 17:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kimchi-Sarfaty C., Oh J.M., Kim I.-W., Sauna Z.E., Calcagno A.M., Ambudkar S.V., Gottesman M.M.. A ‘silent’ polymorphism in the MDR 1 gene changes substrate specificity. Science. 2007; 315:525–528. [DOI] [PubMed] [Google Scholar]
- 30. Zhou M., Guo J., Cha J., Chae M., Chen S., Barral J.M., Sachs M.S., Liu Y.. Non-optimal codon usage affects expression, structure and function of clock protein FRQ. Nature. 2013; 495:111–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lorenz R., Bernhart S.H., Höner zu Siederdissen C., Tafer H., Flamm C., Stadler P.F., Hofacker I.L.. ViennaRNA package 2.0. Algorith. Mol. Biol. 2011; 6:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sabi R., Volvovitch Daniel R., Tuller T.. stAIcalc: tRNA adaptation index calculator based on species-specific weights. Bioinformatics. 2016; 33:589–591. [DOI] [PubMed] [Google Scholar]
- 33. Jeschek M., Höllerer S.. Ultradeep characterisation of translational sequence determinants refutes rare-codon hypothesis and unveils quadruplet base pairing of initiator tRNA and Transcript. Nucleic Acids Res. 2023; 10.1093/nar/gkad040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mirzadeh K., Martínez V., Toddo S., Guntur S., Herrgård M.J., Elofsson A., Nørholm M.H.H., Daley D.O.. Enhanced protein production in Escherichia coli by optimization of cloning scars at the vector–coding sequence junction. ACS Synth. Biol. 2015; 4:959–965. [DOI] [PubMed] [Google Scholar]
- 35. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MEW: the mRNA Expression Wizard is available at https://zenodo.org/record/7547381#.Y8jvJi8w1qs.