

Abstract
Damage to the repository of genetic information in cells has plagued life since its very beginning 3–4 billion years ago. Initially, in the absence of an ozone layer, especially damage from solar UV radiation must have been frequent, with other sources, most notably endogenous sources related to cell metabolism, gaining in importance over time. To cope with this high frequency of damage to the increasingly long DNA molecules that came to encode the growing complexity of cellular functions in cells, DNA repair evolved as one of the earliest genetic traits. Then as now, errors during the repair of DNA damage generated mutations, which provide the substrate for evolution by natural selection. With the emergence of multicellular organisms also the soma became a target of DNA damage and mutations. In somatic cells selection against the adverse effects of DNA damage is greatly diminished, especially in postmitotic cells after the age of first reproduction. Based on an abundance of evidence, DNA damage is now considered as the single most important driver of the degenerative processes that collectively cause aging. Here I will first briefly review the evidence for DNA damage as a cause of aging since the beginning of life. Then, after discussing the possible direct adverse effects of DNA damage and its cellular responses, I will provide an overview of the considerable progress that has recently been made in analyzing a major consequence of DNA damage in humans and other complex organisms: somatic mutations and the resulting genome mosaicism. Recent advances in studying somatic mutagenesis and genome mosaicism in different human and animal tissues will be discussed with a focus on the possible mechanisms through which loss of DNA sequence integrity could cause age-related functional decline and disease.
Keywords: Somatic mutation, ageing, DNA damage, transcriptional noise, pathogenic Consequences
Graphical abstract

Mutations accumulate in the somatic cells organs and tissues during the aging process. Some mutations clonally expand (orange cells), with cancer as an example.
1. Introduction
Life is based on replication and its accompanying errors. Replication assures self-perpetuation, and replication errors provide for the diversity of life when acted upon by natural selection. The first protocells that arose 3–4 billion years ago were likely based on RNA as their information carrier (Joyce and Szostak, 2018). These cells were no more than a few catalytic RNAs (ribozymes) that exhibited polymerase-like activity. These RNA replicases became shielded from the outside world by a fatty acid membrane and could make copies of each other. They were subdivided among daughter cells when the continuously added lipids to the membrane made the protocell unstable, prompting it to split in two. Errors in the replication process would most of the time lead to defective replicases, but sometimes better ones. Occasionally, erroneous replication must also have generated other ribozymes that could carry out additional biochemical functions. This error-generated diversity was driving evolution by natural selection. Errors and natural selection do not strictly need compartmentalization, but this has the major advantage that all interacting molecules stay close to one another. In a non-compartmentalized world they would diffuse away, with improved ribozymes benefiting other molecular systems, which makes selection based on individual entities more difficult. Hence, selectable traits arose very early in the history of life (de Duve, 2005).
In the proto-world of life, RNA acted as both genotype and phenotype. Soon, the phenotype became more complicated with the introduction of proteins. As demonstrated by Miller and Urey in 1952, amino acids could, under the right conditions, form spontaneously in the primordial soup (Miller, 1955). These developed into peptides and then proteins, made contact with the RNA genome and eventually gave rise to the genetic code, i.e., a correspondence between nucleotide and amino acid coding units (Alberti, 1997). The switch from RNA to DNA was mostly driven by the greater chemical stability of DNA based on the deoxyribose in its sugar-phosphate backbone. This must have been a major factor in allowing much greater lengths of nucleic acid template to be maintained without breakage and, therefore, the possibility to encode more complex information (Leu et al., 2011). That was also the time of our last universal common ancestor (LUCA) before life would split into the three domains: Archaea, Bacteria and Eukarya. LUCA could still have had an RNA genome (Glansdorff et al., 2008).
DNA and protein-based systems are now common to all life on planet Earth. However, DNA is not a stable molecule and it is difficult to see how the evolution from short RNA genomes to ever longer, more complex DNA genomes could have taken place without the early emergence of a system to repair the ubiquitous DNA damage. This explains why DNA repair is such a highly conserved system and shared by all organisms we know. It has been argued that the oldest form of DNA repair is recombinational repair, hypothesized to have arisen as a form of sexual reproduction and then coopted to remove DNA damage through exchange with a partner generating a genetically altered individual (Bernstein et al., 1987). Indeed, RecA recombinase was already present in LUCA (Aravind et al., 1999). However, in spite of the critical need for DNA repair for short-term survival, ‘evolvability’, that is, the ability to generate a certain level of mutations for evolution to work with, is equally important on the long term. Hence, depending on the niche conditions a fine balance between error-free and error-prone repair must be kept.
Interestingly, an aging process is already implicit in these early life forms. In between each protocell generation there was a time period in which the biochemical processes were carried out necessary for growth of the protocell and replication of its biomolecules, with its inevitable errors. We could call this intergenerational process aging, the progressive loss of function at the macromolecular level (Figure 1). Aging in unicellular organisms has often been considered as non-existent because of the lack of a clear distinction between the soma and the germline. But this view has now been abandoned based on careful studies of unicellular organisms, such as Escherichia coli and Schizosaccharomyces pombe. These studies indicate that E. coli (Proenca et al., 2019) and S. pombe (Minois et al., 2006) do age without necessarily resulting in mortality of a whole population of cells. In unicellular organisms competition for those least damaged can provide indefinite survival of the population despite individual degradation.
Figure 1.
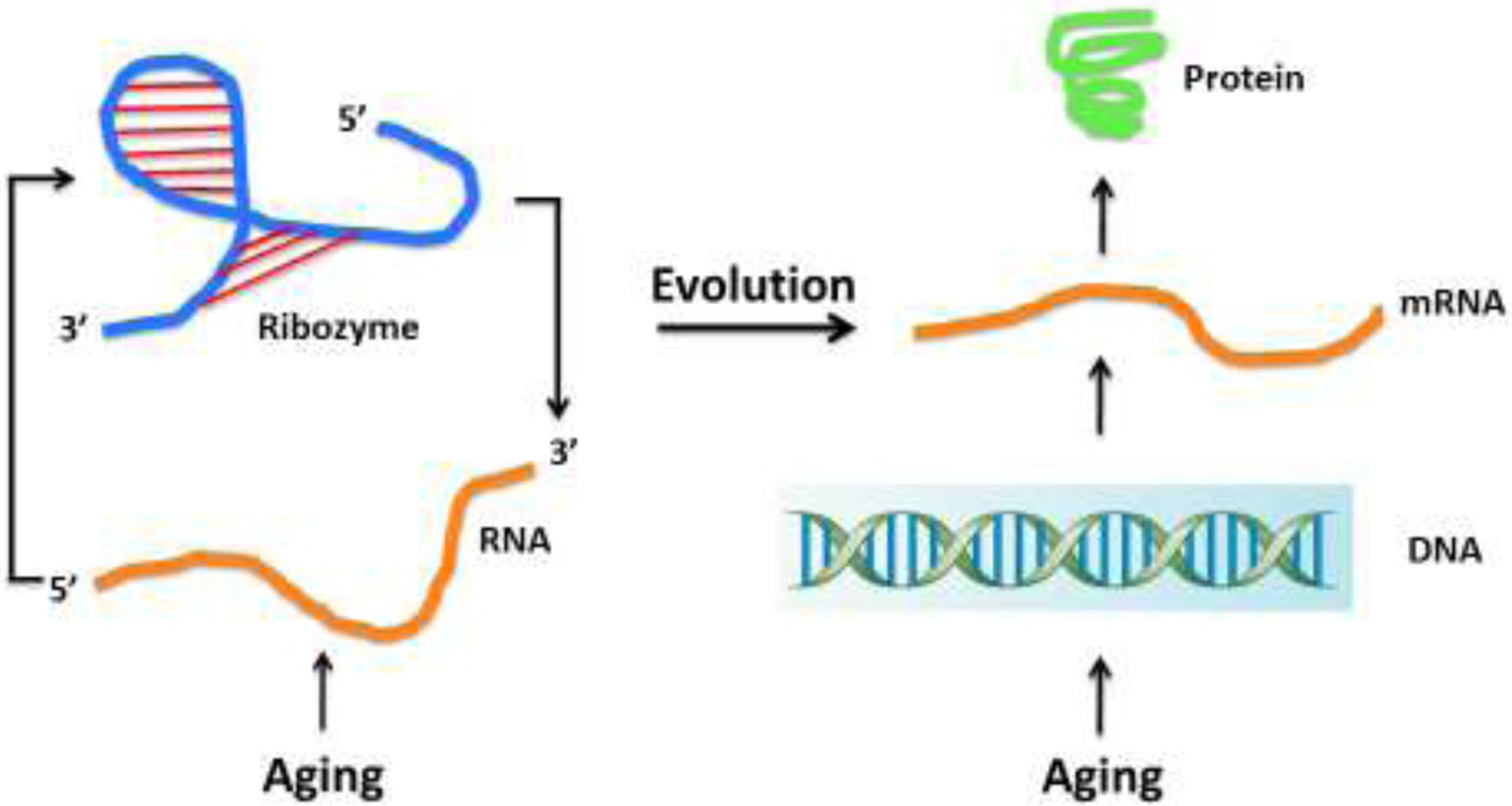
Evolution from the RNA world to modern living systems based on DNA and proteins. In both cases aging could occur as error-based mutations between two generations.
Like populations of unicellular organisms, multicellular organisms also accumulate damaged cells. However, selection in somatic tissues for least damaged cells is very limited. Hence, aging of the soma eventually results in the death of the individual organism, with some exceptions. The freshwater polyps of the genus Hydra, when asexually reproducing, can live indefinitely. This is probably for the same reason most unicellular species are immortal, i.e., the continuous renewal of somatic cells from stem cells with their ample opportunity for selecting the least damaged cells (Martinez, 1998; Sun et al., 2020).
Based on the above it is not surprising that the primary cause of aging was initially sought in random errors in DNA and/or protein. While theories of DNA and protein aging have been proposed since the 1950s, shortly after the discovery of DNA and the genetic code, definite answers remain lacking mostly due to technical limitations. Instead, the focus in aging research has moved to more accessible features such as hallmarks of aging and the discovery of interventions that appear to delay these processes. However, I would argue that in the absence of a detailed understanding of the basic mechanisms of aging, research into therapies will essentially targeting symptoms. Here I will focus on the first primary mechanism of aging that has shown itself amenable to quantitative testing: postzygotic accumulation of mutations in the genome of somatic cells. I will first describe how genome instability in humans and most other metazoa arises, how mutations accumulate in different cells and tissues of aging humans and mice, and how they could possibly contribute to the functional loss and increased disease risk at old age. Finally, I will briefly discuss possible strategies to therapeutically target the effects of somatic mutations.
2. DNA damage as a driver of aging
DNA is only sufficiently stable to serve as the carrier of genetic information in the presence of systems to maintain its integrity by repairing damage accurately. As mentioned above, RNA and DNA damage must have occurred frequently during early life conditions, which explains why DNA repair emerged as one of the most conserved genetic traits. However, under physiological conditions in “modern” organisms, DNA is also subject to extensive decay mostly from endogenous sources (Lindahl, 1993). The most common source of damage is hydrolysis. Due to extensive hydrolysis in the aqueous environment of the human body there is a high frequency of base loss, mostly depurinations, estimated at 10,000 per cell per day. Depyrimidinations also occur but at a much slower rate. Then, there is deamination of cytosine and 5-methylcytosine, various forms of alkylation damage and, most of all oxidative damage. The same aerobic conditions that led Harman to postulate his free radical hypothesis of aging (Harman, 1956), also guarantee a large influx of oxidative DNA damage, which can be several tens of thousands of lesions a day, as byproducts of oxidative respiration, metabolic processing of environmental toxins or products of macrophages and neutrophils at sites of inflammation and infections (De Bont and van Larebeke, 2004).
Under normal conditions environmental sources of DNA damage are less important than endogenous damage. The most frequently encountered environmental DNA lesions are pyrimidine dimers and other photoproducts induced by UV mostly in skin, due to sun exposure, and the large variety of lesions induced by tobacco smoke predominantly in lung. Less frequent are the DNA single- and double-strand breaks induced by radioactive radon gas that can accumulate indoors or through radiation associated with various medical diagnostic and treatment procedures. Rarer still are the guanine adducts of aflatoxins, a genotoxicant found in contaminated peanuts, and various other less frequent types of damage (Li and Sancar, 2020). While environmental damage is related to lifestyle factors, endogenous damage is unavoidable although there is evidence that lifestyle also in this case can make a difference. For example, dietary restriction extends life span in multiple animal species and was found to reduce age-related DNA damage (Vermeij et al., 2016) and mutations (Garcia et al., 2008).
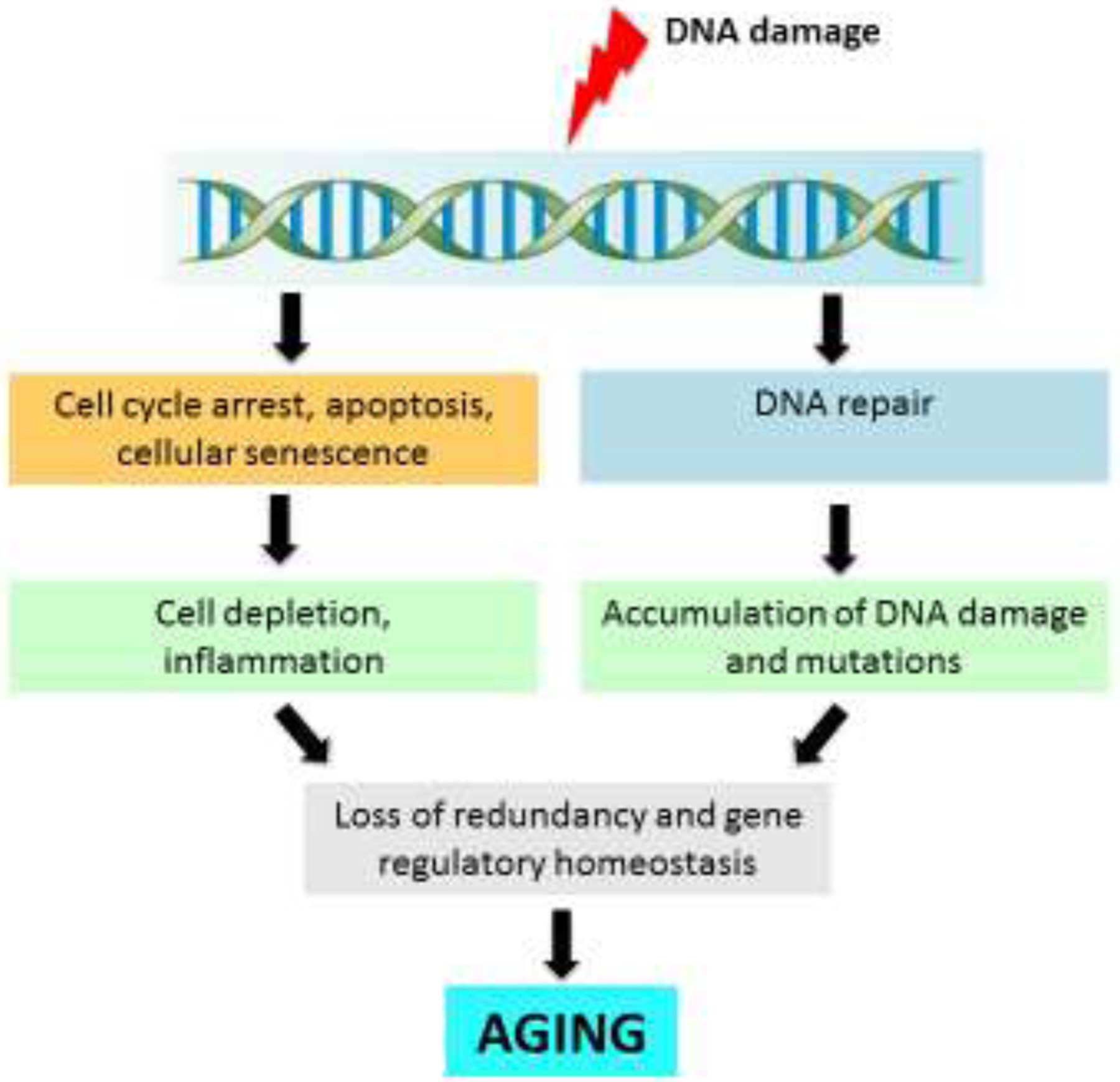
The very high numbers of DNA lesions induced in a cell each day are quickly processed by the different components of the genome maintenance system in coordination with other cellular processes, such as replication, transcription, chromatin remodeling, apoptosis and cellular senescence (Jackson and Bartek, 2009). While loss of individual DNA repair pathways is compatible with life, DNA repair overall is highly conserved and absolutely essential for cell survival. This is why DNA repair appeared so early in the evolution of life forms (above). Virtually all DNA damage as soon as it is induced in a cell is repaired, usually in minutes. This is true for the most frequent lesions, such as base loss and single-strand breaks, but not for all forms of damage. For example, DNA double-strand breaks are rare, difficult to repair and extremely toxic. There may be many unidentified, rare forms of DNA damage that may not be repaired or repaired very slowly (Schmeiser et al., 2014).
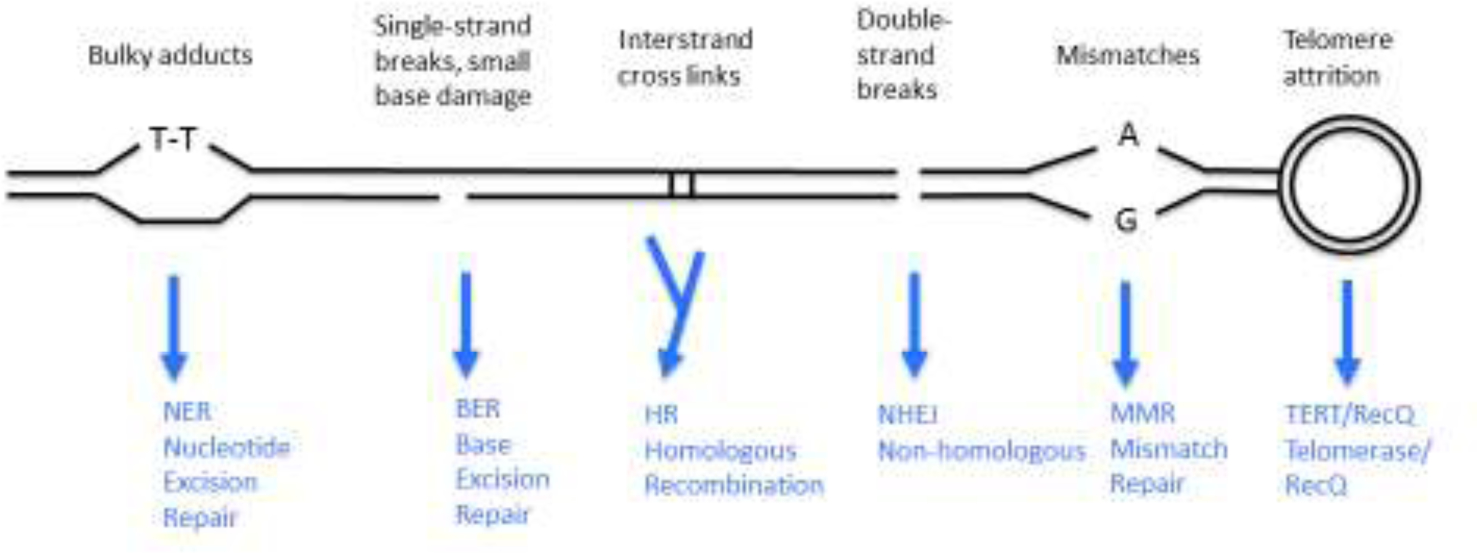
The main DNA repair pathways are schematically depicted in Figure 2 (Chatterjee and Walker, 2017). Specific DNA lesions can be excised by base excision repair or nucleotide excision repair, exchanged for a correct copy of the affected strand through homologous recombination, usually with a sister chromatid as template, breaks can be reannealed and telomere breakdown fixed or prevented by the enzyme telomerase. Small lesions can also be ignored during replication through a tolerance mechanism.
Figure 2.
DNA damage as a driver of aging.
While DNA repair has come into existence very early after the origin of life to guarantee survival (above), it has never been perfect. Then, as now, DNA repair or DNA replication errors are the ultimate source of mutations, i.e., permanent changes in the genome’s information content. Such errors are responsible for the perpetuation of life and now plague us as the cause of genetic disease, including cancer. DNA mutation rates differ greatly between species and are likely to be lower in cells from long-lived species (Vijg, 2007). However, mutation rate will never evolve to zero because of the high cost of near perfect genome maintenance (Sniegowski et al., 2000). Of course, the absence of mutations would no longer allow evolutionary adaptation. The cost of fidelity is not the only factor that determines mutation rate of a species. Sexual recombination is capable to clear deleterious mutations from the genome or combine beneficial mutations (Barton and Charlesworth, 1998).
Hence, damage to the genome has always been a critical component of life rather than just being a nuisance to our modern world. However, that does not necessarily make it a primary cause of aging. Still, there are objective reasons for DNA as the primary target of the aging process. First, there is the nature of the genome as the irreplaceable repository of all genetic information a cell needs to provide function. While other cellular components, most notably proteins, have been proposed as major targets of aging, these can theoretically all be replaced provided there is still an intact primary DNA template. Hence, damage to the primary template would be most critical. Albeit not new (Hirsch, 1978), this argument has lost nothing of its logic.
Second, there is general consensus that cancer is caused by somatic mutations (Nordling, 1953; Nowell, 1976). Cancer risk increases exponentially with age, which is best explained by an accumulation of mutations due to the continuous exposure to a wide variety of exogenous and endogenous DNA damaging agents in interaction with other factors such as changes in apoptosis rate or immune surveillance (Rozhok and DeGregori, 2019). A critical issue is whether DNA damage can also explain aging phenotypes other than cancer. This point has been argued in the past, most notably by Burnet, who considered the accumulation of somatic mutations as the most logical explanation of a range of aging-related phenotypes, from the decline in immune response to arteriosclerosis and senile dementia (Burnet, 1974). This remains a critical question, which I will further address below.
A third argument, and one that emerged fairly recently by comparison, is the series of observations that led to the realization that heritable mutations in components of genome maintenance often lead to premature aging. Human progeroid syndromes were considered as such almost a century before the discovery of DNA (Martin and Oshima, 2000; Navarro et al., 2006). It was only much later that all these syndromes proved to be based on a defect in genome maintenance. The last decade or two has seen a remarkable stream of experimentation indicating that all the genetic traits underlying progeroid diseases, such as Werner’s and Cockayne’s syndrome, are components of DNA repair, with even the lamin defect underlying Hutchinson Gilford Progeria Syndrome associated with DNA damage responses (Liu et al., 2005). Mutations targeted to these and other genome maintenance genes in mice often also result in premature aging (Hasty et al., 2003). It has been argued that such premature aging is often far from a complete phenocopy of normal aging and may not share the same mechanisms (Miller, 2004). While this is a valid argument, it overlooks the fact that only heritable mutations that affect genome maintenance have been found associated with progeroid phenotypes. Heritable mutations in other complex gene families that can be considered as longevity assurance systems, such as P450-based detoxification, autophagy, proteostasis, immune response and antioxidant defense, have never been found widely associated with multiple symptoms of aging occurring prematurely.
How could DNA damage drive aging in humans or mammals in general? There are essentially three types of adverse consequences of the steady stream of DNA lesions and their processing by the DNA damage response systems: (1) direct effects of the lesions on DNA replication or transcription; (2) cell elimination or cessation of cell replication; and (3) DNA mutations (Figure 3). These adverse consequences are not mutually exclusive. Here I will discuss the first two consequences only briefly and focus mostly on DNA somatic mutations, an area of research that has recently seen some remarkable progress.
Figure 3.
The principle DNA repair pathways active in most organisms including humans and mice.
3. Direct effects of DNA damage
DNA damage as a possible direct cause of aging was first postulated by Alexander (Alexander, 1967) and became generally accepted as a bona fide theory of aging once the very high levels of spontaneous DNA damage and the absolute requirement of advanced DNA repair were realized (Hoeijmakers, 2009; Lindahl, 1993). However, the problems with this theory became apparent when it proved to be almost impossible to quantitatively detect spontaneous DNA damage in primary cells or tissues. While many lesions are induced in DNA under physiological conditions, DNA repair keeps the steady state level very low. The methods most commonly used, i.e., gas chromatography, HPLC, proved to be fraud with error (Collins et al., 2004). The limit of detection of most of these methods is in the order of 1 lesion per 100,000 base pairs and it proved difficult to validate the frequencies of various lesions, most notably oxidative lesions. However, there are some exceptions. Using a very sensitive mass spectrometry method cyclopurines, rare substrates for nucleotide excision repair, have been found to accumulate with age in different tissues of the mouse, up to about 3 per million nucleotides (Wang et al., 2012). This underscores the very low frequency of DNA lesions accumulating with age. However, even very low numbers of DNA lesions can interfere with DNA replication by blocking replication forks. This would adversely affect mitotic activity, especially at higher levels of damage. A relatively low number of lesions can also interfere with transcription due to the very large size of primary transcripts.
Probably the most functional way of testing for increased DNA damage with age is through the tendency of lesions to inhibit DNA template-dependent synthesis, such as in PCR or transcription. Inhibition of long distance PCR has been used as a method for estimating DNA damage levels, using different doses of UV light for calibration. Using this method PCR inhibition in liver of old mice as compared to liver of young mice has been interpreted as a slight increase in spontaneous DNA damage. Such inhibition was observed in liver but not in brain (Maslov et al., 2013). Interestingly, a decline of expression of long relative to short genes consistent with stochastic transcription blocking lesion accumulation has also been reported for aging rat liver and aging human hippocampus. This transcription stress was found to be increased in mice harboring DNA repair defects and showing multiple symptoms of premature aging (Vermeij et al., 2016). Subjecting these mice to dietary restriction, a condition that increases life span in different species including the mouse, was demonstrated to reduce transcription stress and increase life span of these DNA repair mutant mice almost 3-fold (Vermeij et al., 2016). In addition, prematurely aging mice due to a DNA repair defect were found to display a time-dependent decline of nascent RNA production, which also suggests the accumulation of transcription-blocking DNA damage in the genome during (premature) aging (Milanese et al., 2019). These results suggest that DNA damage accumulates over the lifetime, which could be a primary causal factor in the aging process through its effects on transcription.
4. Cellular consequences of DNA damage
In addition to direct effects of DNA damage on transcription, also the cellular responses to DNA damage could contribute to aging. For example, as part of the DNA damage response, when threatened to be overwhelmed by DNA damage, cells can undergo apoptosis or cellular senescence (Ciccia and Elledge, 2010; d’Adda di Fagagna, 2008). When occurring frequently enough such responses could lead to atrophy and a loss of mitotic capacity. The latter could adversely affect the regenerative capacity of stem cells. In addition, cellular senescence, i.e., permanent cessation of cell division capacity, could not only affect cellular redundancy but may also exert other deleterious effects, most notably inflammation. Increased inflammation, or inflammaging, a term coined by Franceschi (Franceschi et al., 2018), is a hallmark of aging.
Both apoptosis and cellular senescence are hallmarks of aging. However, these processes are rare events in vivo, even at very old age (Hinkal et al., 2009) and there is no evidence that we age and die because we run out of cells. But even very few senescent cells could cause inflammation through the senescence-associated secretory phenotype (SASP). First described by Campisi (Coppe et al., 2008), senescent cells secrete high levels of inflammatory cytokines, immune modulators, growth factors, and proteases. Systemic chronic inflammation can lead to age-related diseases, such as cardiovascular disease, cancer, diabetes, chronic kidney disease, osteoarthritis, fatty liver disease and autoimmune and neurodegenerative disorders. Senescent cells are drivers of a large number of these diseases (Campisi et al., 2019).
Hence, while apoptosis in response to DNA damage is unlikely to play a major causal role in aging, accumulated senescent cells even in small numbers could contribute to many age-related diseases. Whether DNA damage-induced cellular senescence is a major driver of aging is unknown. In this respect it would be important to better define cellular senescence (Gorgoulis et al., 2019). A lack of suitable markers makes it very difficult to conclusively argue that a cell is senescent, i.e., driven into a permanently quiescent state as an end point of the DNA damage response, or merely a dysfunctional, worn-out cell.
5. Somatic DNA mutations and genome mosaicism
As explained earlier, mutations result from the inherent infidelity of DNA replication and repair of a damaged template. In contrast to DNA damage itself, mutations are irreversible because the original template for making a new copy is lost. Mutations in somatic cells of multicellular organisms are therefore bound to accumulate since there is no evolutionary advantage to drive mutation rates to zero, not even in somatic cells. Indeed, maximization of organismal functions, including maintenance of the integrity of the somatic genome, for extended periods of time after first reproduction does not have a selective advantage (Charlesworth, 2000; Williams, 1957). As discussed above, this would also be energetically too costly (Sniegowski et al., 2000).
Shortly after the structure of DNA had been first described (Watson and Crick, 1953), somatic mutation was implicated as a fundamental cause of aging. Based on observations that ionizing radiation, known to cause mutations (then defined as changes in genetic traits based on phenotypes), appeared to accelerate age-related pathology and shorten life span, two physicists Gioacchino Failla (Failla, 1958) and Leo Szilard (Szilard, 1959) hypothesized that aging is due to the accumulation of mutations in the genetic material of somatic cells. At that time the relationship between DNA damage, DNA repair and mutations was not understood. The hypothesis was rejected based on the perceived extremely low frequency of mutations (Maynard Smith, 1959). At that time the only way to estimate mutation rates was by deducing germ line mutation rates from heritable changes in proteins. For humans this was found to be about one in 10−8 mutations per basepair per generation (Drake et al., 1998). This is not very far from what more recently has been directly measured by counting the number of de novo mutations in offspring as compared to parents after whole genome sequencing (Kong et al., 2012).
When samples from pedigrees are available germ line mutation rates can be measured for large numbers of people and tested for a correlation with life span. Recently, age-adjusted mutation rates were determined in 61 women and 61 men from the Utah CEPH (Centre d’Etude du Polymorphisme Humain) families, with higher mutation rates significantly associated with higher all-cause mortality in both sexes and a shorter reproductive life span in women (Cawthon et al., 2020).
Mutation rates in germ and somatic cells are not necessarily the same. Direct comparisons of somatic and germline mutation rates in humans and mice showed that the former are 1–2 orders of magnitude higher than the latter in both species (Milholland et al., 2017). Because of their obvious importance, germ cells have developed specialized quality control systems that monitor DNA damage (the source of mutations) and remove cells unable to remove damage, such as DNA double-strand breaks (Gebel et al., 2020; Helle, 2012; Vanneste et al., 2009).
An elegant, early experiment conducted to test the hypothesis that aging is caused by somatic mutations was based on the idea that haploid organisms should age faster than their diploid equivalents, which would always have a backup copy of each gene. Such haploid-diploid pairs of species were available in the form of the parasitic wasp, Habrobracon. Survival of haploid and diploid variants of Habrobracon after X irradiation was compared and the results showed that the diploid variant was indeed more resistant (Clark and Rubin, 1961). However, without irradiation no difference in life span as a function of ploidy was found. From this result it was concluded that normal aging could not be caused by mutations. However, as also argued by others (Morley, 1982), this result is far from conclusive since irradiation is toxic due to DNA damage, not mutations, a difference not realized at the time. Mutations on the other hand can be co-dominant and have also effects when recessive. Indeed, the idea that a mutation is harmless as long as the sister allele is still intact has proved to be too simplistic. For example, de novo mutations may give rise to haploinsufficiency, which can have a major impact on gene expression and gene regulation (Morrill and Amon, 2019).
With the success of generating animals through somatic cell nuclear transfer (SCNT) another argument against somatic mutation as a cause of aging has arisen. SCNT appears to work by resetting the aging clock to zero in the somatic nuclei inserted into the enucleated oocyte. This should not be possible when mutation loads in somatic nuclei are high enough to cause adverse effects, or so goes the argument. In fact, this is a variant on a much older argument: if somatic mutations would ever rise to toxic levels in somatic cells they should do so also in germ cells. So why aren’t we all extinct? In both cases the explanation is the same. There is no clock resetting; instead, selection is responsible for what seems to be rejuvenation. First of all, adverse effects of reproduction among older individuals, especially males, have been observed and have been associated with increased germline mutations (Crow, 2000; Gavrilov et al., 1997). Indeed, while exact data are lacking, at least 50% of all conceptuses perish early on (Jarvis, 2016), with chromosomal abnormalities accounting for approximately 50% of clinically recognizable spontaneous abortions (Daniely et al., 1998). This is of course an underestimate because not all spontaneous abortions are detected. Interestingly, even successful fertilizations are subject to mutations, with virtually all blastomeres carrying multiple mutations, i.e., chromosomal anomalies and copy number variations (Vanneste et al., 2009). In such cases a successful outcome is probably facilitated by the elimination of mutant cells through apoptosis (Vanneste et al., 2009). Even then, all offspring carries de novo mutations sometimes resulting in disease.
SCNT has not been studied as exhaustively, but also in this case the procedure involves ample opportunity for selection. Indeed, efficiency of SCNT is no more than about 1% per reconstructed oocyte and the nuclei are generally obtained from mitotically active cells, such as fibroblasts or epithelial cells, from fetal or young adults. Hence, a sufficiently high number of cells with low mutation loads should be present to give rise to a new organism at low frequencies. As expected, cells from fetuses and newborn animals appeared to be most efficient in nuclear transfer, but cells from adult animals frequently do give rise to healthy animals and there are rare reports that even cells from aged animals can be successful (Tian et al., 2003). Nevertheless, selection against heavily mutated cells could easily account for successful SCNT, even from aged tissue. Hence, the term rejuvenation to indicate epigenetic reprogramming is misplaced. Cells cannot be truly rejuvenated for the simple reason that mutations once incurred cannot be removed except by selecting against such cells.
5.1. Quantitative analysis of somatic mutations
The first question that needs to be addressed in considering mutations as a cause of aging is whether they can be demonstrated to accumulate with age in different organs and tissues. Approaches to test the stochastic accumulation of DNA mutations has proved to be a major challenge. When measuring age-related changes in bulk tissue samples it is often not realized that such studies target age-related adaptive processes rather than primary causes. Any age-related change that similarly affects all or most cells in a tissue is generally adaptive, i.e., a response to other age-related changes or changes in the environment. There is general consensus that aging is non-adaptive and not based on a genetic program acting to provide some benefit to the group or species (Kirkwood, 2005; Vijg and Kennedy, 2016). Hence, the primary causes of aging must be sought in stochastic processes that occur above and beyond Darwinian selection. Only the timing of such processes would be subject to selection, for example, through longevity assurance systems (Sacher, 1982).
As a consequence of the inconvenient truth that aging is likely to be primarily caused by stochastic processes, the elucidation of the mechanisms underlying such processes is difficult. This is illustrated by the fact that the two most prominent theories of aging have never been successfully tested. The protein error catastrophe theory proposed by Orgel in the 1960s (Orgel, 1963) is still a likely primary mechanism of aging but has never been put to the test (Martin, 2012). Protein errors are random and different from cell to cell. By measuring bulk tissues or cell populations the true error loads in individual cells is camouflaged by the average. Even if thousands of protein errors accumulate in each cell and they are all different you will detect nothing at bulk level. However, such a situation is very serious and cells with such a load of protein errors will certainly suffer fitness loss. The solution is to take a single cell approach and comprehensively analyze the proteome in many single cells. Only very recently enough progress has been made to analyze a few thousand proteins in single cells, a tiny fraction of the proteome (Slavov, 2020).
The problem is similar in detecting mutations in the somatic genome. However, the genome is much simpler than the proteome and lends itself very well for high-throughput analysis due to the possibility of amplifying nucleic acids based on their simple structure as a code of 4 different bases organized in complementary pairs in a double helix. But even in this case it is only very recent that so much progress has been made that most somatic mutations can now be quantitatively analyzed in multiple tissues during in vivo aging of mammals, including humans. Below I will briefly review progress in this area, including the pivotal role of next-generation sequencing in analyzing somatic mutations, the frequency of the different types of mutations found to accumulate with age, and their possible functional consequences in the context of other causal contributors to the aging process.
5.2. Evidence for somatic mutation accumulation with age
Before the 1980s the only method available to detect somatic mutations was cytogenetics. Early work established how large chromosomal abnormalities could be identified in metaphase plates of a dividing cell population by Giemsa staining. This led to the realization that while in peripheral blood lymphocytes (PBLs) of young people about 1% of cells harbor at least one chromosomal abnormality, in cells from old people that number is close to 5% (Jacobs et al., 1964). This was later amply confirmed using more advanced fluorescence in situ hybridization (FISH) methods (Ramsey et al., 1995).
Somatic mutations lead to tissue genome mosaicism, which is more extensive when a mutation occurs earlier and the cell that contains the mutation expands into a clone. The earliest and best documented example of somatic mosaicism is Y chromosomal loss from bone marrow cells (Pierre and Hoagland, 1972). Such loss is very frequent, has also been observed in peripheral blood lymlphocytes and can affect more than 20% of all cells in almost half of all men at 70-years of age (Thompson et al., 2019). Advanced SNP arrays and next-generation sequencing has now demonstrated that genome mosaicism is not limited to the Y chromosome but is widespread in human tissues. Most studies have been done in blood in which the phenomenon is known as clonal hematopoiesis. The detection of such mutations is dependent on the clonal expansion of a somatic mutation either because of a fitness advantage or stochastic neutral drift. If the resulting clone comprises a sufficiently large fraction of the DNA sample it can be detected, for example, by deep sequencing (Jaiswal et al., 2014; Laurie et al., 2012; Martincorena et al., 2018). Most human tissues have now been shown to contain clones of expanded somatic mutations significantly increasing with age (Garcia-Nieto et al., 2019; Yizhak et al., 2019).
Interestingly, the level of genome mosaicism, such as LOY or clonal hematopoiesis, has been associated with risk for all-cause mortality and common age-related diseases, such as cancer, Alzheimer disease and atherosclerosis (Jaiswal et al., 2014; Thompson et al., 2019). In certain cases the nature of the mutation may increase susceptibility to a disease. For example, somatic mutations in the gene encoding the epigenetic modifier enzyme TET2 promote expansion of the mutant cell in blood. This was found associated with increased IL-1β production, which is essential for accelerated atherosclerosis (Fuster et al., 2017). This could explain why clonal hematopoiesis has been found to correlate with an increased risk of atherosclerotic cardiovascular disease. However, it seems unlikely that all or even most somatic mutations have such direct effects. Indeed, this would be difficult to imagine for loss of a Y chromosome, which has only few functional genes. Instead, it seems much more likely that extensive genome mosaicism is a surrogate marker for genome instability in general, providing evidence for a causal relationship between somatic mutation rate and aging.
The vast majority of somatic mutations will not expand or expand to very small clones that cannot be detected by direct analysis. As explained above increased somatic mutation loads in cells may be undetectable by direct analysis, but could still adversely affect cell fitness and therefore fitness of the organism. To get access to such low-abundant somatic mutations we (Gossen et al., 1989) and later others too (Kohler et al., 1991; Leach et al., 1996) developed mouse models harboring chromosomally integrated reporter genes that can be rescued into E. coli and selected for mutations that had inactivated the reporter genes in vivo. These mouse models were used to study mutation frequencies in different organs and tissues and the results showed that in virtually all tissues mutations accumulate with age (Dollé et al., 1997; Dollé et al., 2002; Dollé et al., 2000; Ono et al., 2000; Stuart et al., 2000). Importantly, these studies showed that mutation frequencies and spectra differ between tissues. One of these mouse models allowed to determine the frequency of genome structural variations (SVs) based on extrapolation from breaks in the reporter gene to the genome overall. Based on this crude estimate SVs were found to vary from about 5 per cellular genome in mouse brain at old age to almost 40 in the aged mouse heart (Dolle and Vijg, 2002). Point mutations were higher but still greatly underestimated because most base substitutions do not inactivate the reporter gene and go undetected. Genome structural variations always inactivate the reporter and their impact is much greater because these events can involve thousands to over a million bases (see also below).
While endogenous reporters such as the hypoxanthine phosphoribosyltransferase 1 gene (HPRT), have been used in studying somatic mutation in humans (Martin et al., 1996; Trainor et al., 1984) and also showed an age-related increase, such assays can only be used in human or mouse primary cells that can be propagated in culture. Only transgenic reporters in the mouse allow easy access to different organs and tissues. However, since reporter genes do not allow analyzing somatic mutations in humans and it can also not be assumed that mutation frequencies at reporter gene loci are fully representative for the genome overall, methods based on direct, genome wide sequencing were recently developed. In view of the stochastic nature of somatic mutation accumulation such methods must be based on single cell analysis.
As a surrogate for true single-cell analysis, which remains a challenge, primary human cells have been cloned and mutations identified after whole genome sequencing of the DNA from these clones, which should be representative for the original single cell. The results indicated an age-related increase in single-nucleotide variants (SNVs) in liver, colon, muscle satellite cells and kidney from about 600 mutations per cell at young age to several thousands in samples derived from older individuals (Blokzijl et al., 2016; Franco et al., 2019; Franco et al., 2018). These numbers are considerably higher than the number of de novo mutations in germlines, which are about 60 per genome per generation (Kong et al., 2012). Indeed, as mentioned earlier, somatic mutation rate is 1–2 orders of magnitude higher than germline mutation rate (Milholland et al., 2017).
Sequencing clones rather than single cells obviously limits studies to cells that can be cloned in culture. This restricts analysis mostly to stem cells, which have been demonstrated to contain significantly less de novo mutations than differentiated cells (see further below and (Cervantes et al., 2002; Thompson et al., 2020). Hence, mutation frequencies obtained from stem cell clones should be considered underestimates of the true frequency of mutations as they occur in the differentiated cells that provide function to different organs and tissues. A problem in directly analyzing differentiated cells for mutations is the need for whole genome amplification which is fraught with error. Also in this area major advances have recently been made. We and others developed reliable computational methods to correct for amplification errors in detecting SNVs in single cells based on genetic linkage with germline heterozygous single nucleotide polymorphisms (hSNPs)(Bohrson et al., 2019; Dong et al., 2017). It should be realized that all single-cell approaches require appropriate controls because once analyzed the cell is destroyed and the results cannot be replicated. Such controls can include sequencing kindred cells from clones derived from the same cell population and sequenced without amplification, analyzing germ line variants in each single cell to test sensitivity, and various positive controls, such as cell populations treated with known mutagens (Dong et al., 2017; Gundry et al., 2012).
Thus far, information on mutation frequencies and age is available for human B lymphocytes, neuronal cells and liver hepatocytes (Brazhnik et al., 2020; Lodato et al., 2018; Zhang et al., 2019). In all three cell types mutations were found to increase with age albeit at different rates. The highest mutation frequencies, i.e., of about 5,000 SNVs per cell in 70–80 year old individuals, were found in liver. This may simply be a consequence of increased tolerance of liver for mutation, due to the high level of polyploidy in this organ, a layer of additional redundancy likely to provide protection against random mutations and cancer (Zhang et al., 2018). However, liver stem cells, obtained by growing organoids from hepatocyte cell populations from middle aged individuals, had mutation frequencies of only about 1000 mutations per cell as compared to at least twice that number in the differentiated hepatocytes from the same individuals (Brazhnik et al., 2020). A comparison between clones, sequenced without amplification and whole genome amplified single cells derived from these clones showed no difference, confirming the validity of the single cell data. These results suggest that while all cells, including stem cells, undergo an age-related accumulation of mutations, cells further in the hierarchy of the differentiation process accumulate more mutations. This could be explained simply by replication errors during the differentiation process but may also reflect increased repair accuracy in stem cells.
In most of these experiments also small insertions and deletions (INDELs) were analyzed. They are generally much more deleterious than base substitutions and were found to occur at much lower frequency, i.e., about 100 per cell in young tissues to about 300 per cell in old subjects. Much less is known about larger insertions and deletions and genome structural variations (SVs) in general. Some of these events have been analyzed in single cells, but not in relation to aging. For example, copy number variation (CNV) has been analyzed in single human neurons directly taken from the in vivo situation (Cai et al., 2014; McConnell et al., 2013). The results generally show about one large CNV (>1Mb) on average in most cells analyzed. However, many submegabase CNVs have been found in single neurons in the developing mouse brain (Rohrback et al., 2018).
Also other SVs, such as deletions larger than 50 bp and complex genome structural variation has been observed in organoid clones from human liver, colon and small intestine, with about 50% of clones harboring one SV (Blokzijl et al., 2016). Because of these small numbers no quantitative information in relation to aging is as yet known, except for the very large chromosomal events that can be analyzed by cytogenetics (above). It is here where next-generation sequencing meets cytogenetics. Note that also in the mouse at reporter gene loci genome structural variations have been detected, from about 5 per cell in brain to almost 40 in heart (above).
The detection of SVs after whole genome sequencing is generally difficult and has a sensitivity not higher than about 20% (Kosugi et al., 2019). Detection of SVs is generally based on finding abnormal positions of short, paired-end sequencing reads when aligning to the reference genome. In this way it is possible to map the two breakpoints of the SV, indicating deletions, inversions or translocations (Medvedev et al., 2009). The problem with detecting SVs in this way is the high probability of mapping errors especially with larger events. To some extent this problem is addressed by long-read sequencing, but the generally high error rate of these approaches poses new challenges (Mahmoud et al., 2019). SV detection in single cells is much more difficult than in DNA from bulk tissue because in multiple displacement amplification (MDA), the method of choice for mutation analysis in single cells, artificial chimeric sequences are often generated because nascent DNA strands can be displaced from their original template and prime a second template resulting in artificial SV calls (Lasken and Stockwell, 2007).
True quantitative analysis still remains to be done for SVs, an important class of mutations that has been proposed as the mutation type most likely to have major adverse effects at old age (Vijg, 2002). Nevertheless, improvements are made continuously and it seems highly likely that within a decade we will be able to build a reliable quantitative inventory of the various types of somatic mutations in aging humans and model organisms using single-cell or single nucleus approaches. Based on such comprehensive somatic mutation maps for different tissues in aging humans we should be able to address the question to what extent somatic DNA mutations cause aging.
5.3. Can accumulating mutations affect cell fitness?
A major question in the biology of aging is how stochastic changes can lead to the aging phenotypes that are repeatedly observed in humans or animals. While there is great individual variation in the timing of the major aging phenotypes, including diseases, such as cancer, cardiovascular disease and diabetes, they all appear as very similar from individual to individual. Indeed, there is even great similarity in aging phenotypes across species (Vijg and Campisi, 2008). How can a stochastic accumulation of somatic mutations cause all or most of these phenotypes?
Elsewhere we proposed three mechanisms as to how somatic mutations can cause disease and aging (Vijg and Dong, 2020). The first two are based on clonal expansion of mutations either as a consequence of drift or of a fitness advantage of the mutation. It now seems likely that through this mechanism most if not all genetic diseases, i.e., diseases caused by a germline mutation, have a somatic mutational variant (Erickson, 2010; Li, 2013). A few examples are neurofibromatosis, paroxysmal nocturnal hemoglobinuria, Alport syndrome, autism spectrum disorders, certain autoimmune diseases and even Alzheimer’s disease (Erickson, 2010; Poduri et al., 2013). Of note, sometimes disease symptoms occur with only a small fraction of cells in the target tissue harboring the mutation and there are forms of somatic genetic disease, apart from cancer, that do not even occur as germline mutations because it would not be compatible with life when present in all somatic cells (Li, 2013).
An important mechanism in the expansion of a somatic mutation, long recognized as the cause of cancer, is somatic evolution, i.e., the clonal expansion of a mutation that provides a cell with a growth advantage, ultimately leading to a full-blown tumor (Fearon and Vogelstein, 1990; Nowell, 1976). There is now ample evidence that clonal expansion of mutations is frequently observed in aged tissues (above) and there is increasing evidence that this can causally contribute not only to cancer but also to non-neoplastic, age-related diseases, including COPD (Anderson and Bozinovski, 2003), cardiovascular disease (Fuster et al., 2017; Sano et al., 2018), type 2 diabetes (Bonnefond et al., 2013) and inflammatory bowel disease (Nanki et al., 2020; Olafsson et al., 2020). Hence, both clonal expansion by genetic drift and somatic evolution can explain age-related disease phenotypes. Here I will focus on the third mechanism that does not require clonal enrichment but can cause cell-autonomous and cell type-specific functional decline.
While somatic mutations accumulate across the genome randomly, only those that end up in regions relevant for the specific function of that cell type can have adverse effects. In differentiated cells most of the genome is non-functional with only a fraction of the about 1% protein-coding sequences relevant for that cell. However, the part of the genome that regulates these active genes is substantially larger and estimated at approximately 10% of the genome in a typical differentiated cell (Kellis et al., 2014). Function in such cells is not provided by individual genes but through gene regulatory networks (GRNs). GRNs consist not only of genes but also of enhancers, promoters and other sequences that affect gene transcript levels. Naturally, SVs have a much higher impact on the GRNs that control cell functions than SNVs. Indeed, large deletions can wipe out multiple genes and regulatory sequences. As mentioned earlier, haploinsufficiency can have dominant effects, for example, when the gene product interacts with a ligand that requires the correct relative amounts of its interactive partner. Hence, recessive mutations, and all de novo somatic mutations are recessive, can still have a major impact on gene expression and gene regulation (Morrill and Amon, 2019).
Hence, mutation accumulation can only functionally impact a cell when occurring in its functional genome. Random mutations that beneficially affect the organism are extremely rare. Hence, apart from the mutations that have no effect at all, somatic mutagenesis can only lead to a gradual loss of function of the tissue or organ. Therefore, it is not surprising that stochastic mutation accumulation has similar effects in a particular tissue or organ from individual to individual and to some extent even across species. There are of course cell-to-cell differences and the age-related mutation burden will vary between individuals and between tissues within individuals, but the effect is always the same. Therefore, somatic mutation accumulation as a stochastic process could reasonably explain age-related functional decline and disease patterns that are similar across individuals. However, what is the evidence that somatic mutations do affect cell function?
It has been demonstrated in human B lymphocytes that SNVs accumulate with age but do so much more rapidly in the functional part of the genome than the genome overall (Zhang et al., 2019). This suggests selection against cells harboring high mutation loads, possibly because of an adverse effect on fitness. Indeed, after whole genome sequencing of single human muscle satellite cell clones of the leg muscle from healthy individuals of different ages, mutation load was found to correlate with functional defects in satellite cells from aged individuals (Franco et al., 2018).
One possible effect of mutation accumulation in GRNs is an increase in transcriptional noise. Depending on where random mutations are incurred, they could slightly up- or down-regulate gene expression levels. This is known as transcriptional noise, i.e., different levels of mRNA among otherwise identical cells. While a certain level of transcriptional noise is inherent to gene expression, an increase with age has now been demonstrated in different cell types in mice and humans (Angelidis et al., 2019; Bahar et al., 2006; Enge et al., 2017).
Transcriptional noise levels can be measured by quantifying individual gene transcript levels across cells in single-cell RNA-seq. However, this is not very sensitive because as mentioned above, genes do not operate in a vacuum but are connected in GRNs. Recently, a new measure of transcriptional noise has been developed called global coordination level (GCL), which is based on the average multivariate dependency between expression levels of random subsets of genes in single-cell RNA-seq data sets. GCL values higher than zero reflect gene-to-gene regulatory interactions. Using this new measure it was indeed found that GCL consistently declines with age in different organisms and cell types (Levy et al., 2020). Moreover, increased mutations in tissues at old age appeared to correlate with reduced GCL values. Analyzing a data set on human pancreatic cells in which both mRNA levels and somatic mutations were assessed in the same cells (Enge et al., 2017) those cells with the highest mutation loads generally also had the lowest GCL values (Levy et al., 2020). While a relationship of DNA mutations with transcriptional noise has been suggested before (Vijg et al., 2005), this is the strongest evidence to date for a cause and effect relationship between somatic mutations, transcriptional noise and aging.
How could an increase in transcriptional noise causally contribute to aging? One example is cell fate drift. Enge et al demonstrated an age-associated increase in both somatic mutations and transcriptional noise in human pancreas cells (Enge et al., 2017). They found that islet endocrine cells from older donors displayed not only increased levels of transcriptional noise but also an increase in non-cell-type-specific hormone expression in endocrine cells. Such “fate drift” would naturally have physiological consequences in terms of decreased fitness and organ function at old age if happening on a large enough scale.
Increased transcriptional noise could also explain defects in cell signaling that have been observed with age. Indeed, even slight up- or down-regulation of genes active in stress or immune responses are likely to significantly reduce efficiency of such adaptive processes. This could for example explain the increased susceptibility to infections in elderly and their reduced response to vaccines (Lord, 2013) or the well-documented age-related impairment of temperature regulation (Blatteis, 2012).
Finally, transcriptional noise could lead to a stoichiometric imbalance in the assembly of multiprotein complexes. Macromolecular complexes are responsible for most of the essential mechanisms in cells and transcriptional noise could, therefore, contribute to the observed protein aggregation with age (Santra et al., 2019).
Based on the above, it is conceivable that somatic mutation accumulation during aging in vivo leads to increased transcriptional noise, which in turn could explain the functional decline observed in most cell types during aging. Together with the increased disease risk found associated with the clonal development of mutations, this would make mutation accumulation a most likely primary cause of aging. Like mutations, also epimutations have been suggested as a cause of transcriptional noise (Enge et al., 2017). Epigenetic change has been proposed as a major cause of aging almost since its discovery (Holliday, 1991). The main reason it is so often mentioned as a possible cause of aging is the relatively simply way epigenetic changes can be demonstrated, either as changes in histone modification or changes in patterns of DNA methylation. However, the exact same reasoning used earlier in this review also applies here, i.e., any changes readily observed in bulk tissue are adaptive changes and unlikely to be primary causes.
Of course, epimutations can also occur stochastically. DNA methylation maintenance during replication and repair is not error-free (Fu et al., 2010; Laird et al., 2004) and extensive cell-to-cell variation in DNA methylomes has been demonstrated (Gravina et al., 2016). In principle, therefore, random changes in the epigenome could be a more likely cause of transcriptional noise than mutations. However, while this possibility cannot be excluded for now, it is in conflict with the rare observation of epimutations as causal factors in disease, most notably cancer, and then often only indirectly through genetic mutations in epigenetic modifiers that affect chromatin (Zoghbi and Beaudet, 2016). It is possible that epimutations only have an effect when occurring as blocks of change rather than individual epimutations. That is unlikely to happen at the single cell level but readily explains how methylation changes can underlie adaptive changes, such as memory formation in the brain (Heyward and Sweatt, 2015).
6. Conclusions and future prospects
Aging can be defined as the progressive decline in fitness that brings life to a close. The process provides a definite limit to life, in the sense that improved environmental and medical conditions are ultimately unable to change the maximum life span of our species once optimal conditions have been achieved (Dong et al., 2016). However, life span varies considerably among species, of which there are several with greater maximum life span than humans (Ma and Gladyshev, 2017). The genetic basis of these differences is likely to be extremely complex and not well understood, but there is evidence that mutation rates are higher in short lived species, such as rodents, than in long-lived primates (Britten, 1986). This may be a result of superior DNA repair in long-lived species for which there is also evidence (Hart and Setlow, 1974; Ma et al., 2016; MacRae et al., 2015).
According to the general consensus, aging is ultimately a consequence of the decline in efficacy of natural selection with age (Kirkwood, 2005; Medawar, 1952; Williams, 1957). In stark contrast, the process has thus far escaped an explanation in terms of proximal causation. As mentioned earlier in this paper, age-related functional decline and increased disease risk is now usually discussed in terms of hallmarks of aging, i.e., age-related phenotypes that are often conserved among species (Lopez-Otin et al., 2013). But most hallmarks of aging are either adaptive responses to more fundamental causes of aging or late adverse effects of otherwise beneficial processes. Various stress responses are an example of the former while inflammation as an adverse side effect of the immune response illustrates the latter category. Hence, most hallmarks of aging are evolutionary adaptations for optimal functioning over time in a continuously changing internal and external environment.
Adaptive changes of aging are by definition not the primary causes of the process, which have to be sought in stochastic processes of wear and tear that can be delayed but ultimately not prevented. As also discussed elsewhere, the most likely mechanistic driver of aging appears to be DNA damage (Schumacher, 2021). In this present review I discuss the evidence that one of the multiple consequences of DNA damage, i.e., somatic mutations, are a universal, primary cause of aging. This remains untested and other consequences of DNA damage, such as persistent lesions and cellular responses to DNA damage, may eventually prove to be more important. However, one conclusion from this review is that major technological advances have now made it possible to quantitatively analyze somatic mutations directly in tissues of humans and experimental animals as a function of age. This should soon allow the somatic mutation theory of aging to be tested in sufficient depth to begin drawing definite conclusions as to its validity.
A second conclusion we can now draw is that there can be no longer any doubt that the frequency of somatic mutations increases with age in various tissues and organs of humans to levels that could potentially lead to disease and fitness loss. This is not the case for DNA damage per se, which remains very difficult to quantitatively analyze. By contrast, for mutations there are now many independent observations that most if not all human tissues show an age-related increase of somatic DNA mutations, often in the form of mutant clones carrying expanded mutations (schematically depicted in Figure 4). These clonal expansions should now no longer be considered as only relevant for cancer, but for many age-related diseases as well (Lawson et al., 2020). Somatic mutation frequency in tissues of the elderly appears to be highest in differentiated somatic cells, less in stem cells (Brazhnik et al., 2020; Cervantes et al., 2002; Thompson et al., 2020) and lowest in germ cells (Milholland et al., 2017). While there is indirect evidence that high mutation frequencies reduce fitness of individual cells (Franco et al., 2018; Zhang et al., 2019), we still don’t know its mechanistic basis. As discussed, through their penetration in DNA sequence components (promoters, enhancers) of gene regulatory networks, accumulating mutations could lead to transcriptional noise. In principle, this can now be directly tested through high-throughput, single-cell sequencing at DNA and RNA level in the same single cells (Hou et al., 2016; Li et al., 2015).
Figure 4.
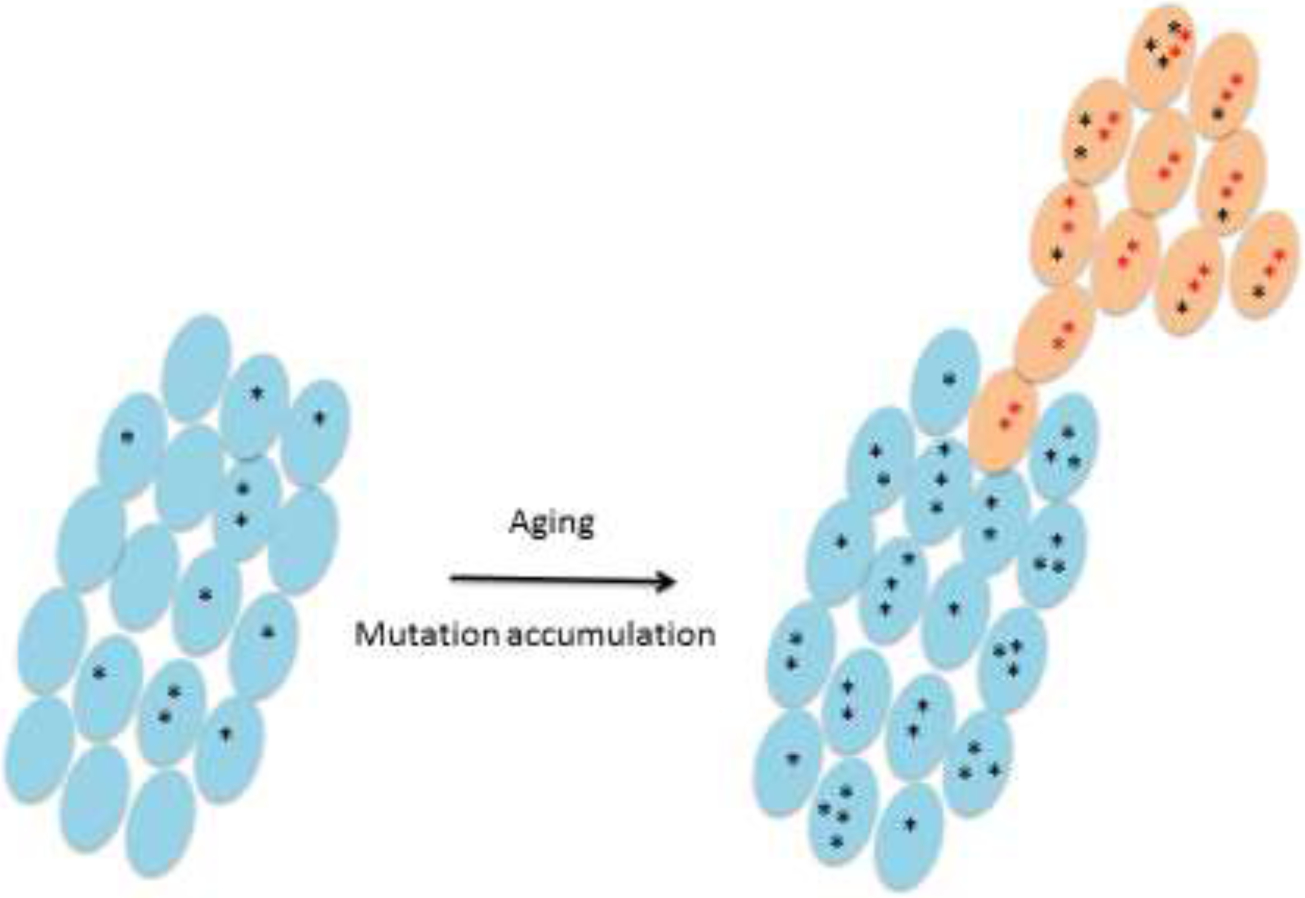
Schematic depiction of mutation accumulation in a tissue, including expansion into clones. De novo mutations are in black. Mutations can be clonally amplified due to genetic drift or a survival advantage. This is indicated by the two red asterisks in the orange cells. Additional mutations (in black) can subsequently accumulate but all cells will retain the two original mutations in the ancestor cell. The best example of such somatic evolution, but by no means the only one, is the generation of tumors.
Hence, more than half a century after it was first proposed the plausibility of somatic mutations as a primary mechanism of aging has been demonstrated experimentally. Nevertheless, testing for a causal role of somatic mutations in aging remains difficult. The most straightforward way would be to experimentally elevate the natural level of somatic mutations across tissues and then test for an effect on aging rate and life span. Unfortunately, as of yet there are no approaches to accomplish this. Alternatively, it is now possible to correlate functional measures of cells in aged organisms with mutation burden in the cells. An example of such an approach is the correlation between growth rate of cell clones and their mutation burden as mentioned above (Franco et al., 2018). However, probably the best possible approach in providing a quantitative link between cause and effect would be mathematical modeling based on the multitude of quantitative data that are now emerging. This approach has been pioneered by Kirkwood and Proctor (Kirkwood and Proctor, 2003) even in the absence of our current rich data sets.
It is important to note that DNA mutation cannot be the sole causal factor in aging. Evolutionary logic immediately rules out any single cause of aging. During evolution there must have been many genetic variants with deleterious effects late in life that for this reason escaped natural selection, which declines with age (Kirkwood, 2005). Indeed, many of these variants were positively selected because of an early beneficial effect, for example, variants in lipoprotein genes, immune response genes or growth factor genes. However, while causally contributing to aging, the processes associated with these genetic variants are not primary causes of the process.
A major question for the future is if somatic mutation as a major causal mechanism in aging can be therapeutically targeted. In this respect several options could be entertained. For example, the clonally amplified postzygotic mutations in tissues can be identified and further analyzed for phenotypic differences from the normal tissues. Once identified these differences could serve as targets for developing drugs to eliminate such cells. Likewise, cells that suffer loss of function due to the accumulation of mutations in gene regulatory networks could also be eliminated. Cells could be eliminated by driving them into apoptosis or through cell competition. While still far away, such options would be the first approaches to intervene in a basic molecular cause of aging rather than in one or more of its symptoms.
Somatic mutations as a cause of aging is in keeping with Muller’s ratchet, i.e., the inevitable accumulation of deleterious mutations in asexually propagated cell populations (Muller, 1964). Indeed, the ratchet was proposed to explain the advantage of sexual reproduction and has been experimentally verified in Salmonella typhymurium (Andersson and Hughes, 1996). As we have seen, sexual reproduction through recombinational repair to promote survival from deleterious DNA mutations may have begun with the first replicators. But mutation is of course also the driver of evolution and underlies the great diversity of life forms. It would indeed be ironic if the same process that has given us life would also drive its demise.
Highlights.
There is abundant evidence for DNA damage as a cause of aging.
DNA damage can act as causal factor in aging through direct effects on transcription, by signaling apoptosis or cellular senescence and through somatic DNA mutations.
Somatic mutations can now be quantitatively analyzed by single-cell whole genome sequencing.
Somatic mutations accumulate with age in virtually all human tissues and organs.
Somatic mutations can cause cell functional loss and age-related disease by clonal expansion or through interference in gene regulatory networks.
Somatic mutations may lead to age-related loss of function through transcriptional noise.
Acknowledgements
My work is supported by NIH grants AG017242, AG047200, AG056278, AG038072, ES029519, HL145560, the Glenn Foundation for Medical Research and Shanghai Jiaotong University School of Medicine. I thank Dr. Joris Pothof for critically reading the manuscript, Drs. Claudia Gravekamp and Alex Maslov for making the figures and two anonymous reviewers for multiple constructive comments and suggestions to improve the manuscript.
Table 1. List of Abbreviations
- CEPH
Centre d’Etude du Polymorphism Humain
- CNV
Copy Number Variation
- COPD
Chronic Obstructive Pulmonary Disease
- FISH
Fluorescent In Situ Hybridizating
- GRN
Gene Regulatory Network
- GCL
Global Coordination Level
- HPLC
High-Performance Liquid Chromatography
- HPRT
Hypoxanthine PhosphoRibosylTransferase
- INDELs
Insertions or deletions
- LOY
Loss of chromosome Y
- LUCA
Last Universal Common Ancestor
- PBL
Peripheral Blood Lymphocyte
- PCR
Polymerase Chain Reaction
- SASP
Senescence-Associated Secretory Phenotype
- SCNT
Somatic Cell Nuclear Transfer
- SNP
Single-Nucleotide Polymorphism
- SNV
Single-Nucleotide Variant
- SV
Genome Structural Variation
- UV
UltraViolet
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberti S, 1997. The origin of the genetic code and protein synthesis. J Mol Evol 45, 352–358. [DOI] [PubMed] [Google Scholar]
- Alexander P, 1967. The role of DNA lesions in the processes leading to aging in mice. Symposia of the Society for Experimental Biology 21, 29–50. [PubMed] [Google Scholar]
- Anderson GP, Bozinovski S, 2003. Acquired somatic mutations in the molecular pathogenesis of COPD. Trends Pharmacol Sci 24, 71–76. [DOI] [PubMed] [Google Scholar]
- Andersson DI, Hughes D, 1996. Muller’s ratchet decreases fitness of a DNA-based microbe. Proc Natl Acad Sci U S A 93, 906–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelidis I, Simon LM, Fernandez IE, Strunz M, Mayr CH, Greiffo FR, Tsitsiridis G, Ansari M, Graf E, Strom TM, Nagendran M, Desai T, Eickelberg O, Mann M, Theis FJ, Schiller HB, 2019. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nature communications 10, 963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Walker DR, Koonin EV, 1999. Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res 27, 1223–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dollé MET, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J, 2006. Increased cell-to-cell variation in gene expression in aging mouse heart. Nature 441, 1011–1014. [DOI] [PubMed] [Google Scholar]
- Barton NH, Charlesworth B, 1998. Why sex and recombination? Science 281, 1986–1990. [PubMed] [Google Scholar]
- Bernstein H, Hopf FA, Michod RE, 1987. The molecular basis of the evolution of sex. Adv Genet 24, 323–370. [DOI] [PubMed] [Google Scholar]
- Blatteis CM, 2012. Age-dependent changes in temperature regulation - a mini review. Gerontology 58, 289–295. [DOI] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, Nijman IJ, Martincorena I, Mokry M, Wiegerinck CL, Middendorp S, Sato T, Schwank G, Nieuwenhuis EE, Verstegen MM, van der Laan LJ, de Jonge J, JN IJ, Vries RG, van de Wetering M, Stratton MR, Clevers H, Cuppen E, van Boxtel R, 2016. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohrson CL, Barton AR, Lodato MA, Rodin RE, Luquette LJ, Viswanadham VV, Gulhan DC, Cortes-Ciriano I, Sherman MA, Kwon M, Coulter ME, Galor A, Walsh CA, Park PJ, 2019. Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nat Genet 51, 749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefond A, Skrobek B, Lobbens S, Eury E, Thuillier D, Cauchi S, Lantieri O, Balkau B, Riboli E, Marre M, Charpentier G, Yengo L, Froguel P, 2013. Association between large detectable clonal mosaicism and type 2 diabetes with vascular complications. Nat Genet 45, 1040–1043. [DOI] [PubMed] [Google Scholar]
- Brazhnik K, Sun S, Alani O, Kinkhabwala M, Wolkoff AW, Maslov AY, Dong X, Vijg J, 2020. Single-cell analysis reveals different age-related somatic mutation profiles between stem and differentiated cells in human liver. Sci Adv 6, eaax2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ, 1986. Rates of DNA sequence evolution differ between taxonomic groups. Science 231, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Burnet M, 1974. Intrinsic mutagenesis: A genetic approach to ageing. Wiley, New York. [Google Scholar]
- Cai X, Evrony GD, Lehmann HS, Elhosary PC, Mehta BK, Poduri A, Walsh CA, 2014. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell reports 8, 1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Kapahi P, Lithgow GJ, Melov S, Newman JC, Verdin E, 2019. From discoveries in ageing research to therapeutics for healthy ageing. Nature 571, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawthon RM, Meeks HD, Sasani TA, Smith KR, Kerber RA, O’Brien E, Baird L, Dixon MM, Peiffer AP, Leppert MF, Quinlan AR, Jorde LB, 2020. Germline mutation rates in young adults predict longevity and reproductive lifespan. Scientific reports 10, 10001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervantes RB, Stringer JR, Shao C, Tischfield JA, Stambrook PJ, 2002. Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc Natl Acad Sci U S A 99, 3586–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, 2000. Fisher, Medawar, Hamilton and the evolution of aging. Genetics 156, 927–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, Walker GC, 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 58, 235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Elledge SJ, 2010. The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AM, Rubin MA, 1961. The modification by X-irradiation of the life span of haploids and diploids of the wasp, Habrobracon SP. Radiat. Res 15, 244–253. [PubMed] [Google Scholar]
- Collins AR, Cadet J, Moller L, Poulsen HE, Vina J, 2004. Are we sure we know how to measure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys 423, 57–65. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J, 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF, 2000. The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 1, 40–47. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, 2008. Living on a break: cellular senescence as a DNA-damage response. Nat Rev Cancer 8, 512–522. [DOI] [PubMed] [Google Scholar]
- Daniely M, Aviram-Goldring A, Barkai G, Goldman B, 1998. Detection of chromosomal aberration in fetuses arising from recurrent spontaneous abortion by comparative genomic hybridization. Hum Reprod 13, 805–809. [DOI] [PubMed] [Google Scholar]
- De Bont R, van Larebeke N, 2004. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis 19, 169–185. [DOI] [PubMed] [Google Scholar]
- de Duve C, 2005. The onset of selection. Nature 433, 581–582. [DOI] [PubMed] [Google Scholar]
- Dollé ME, Giese H, Hopkins CL, Martus HJ, Hausdorff JM, Vijg J, 1997. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet 17, 431–434. [DOI] [PubMed] [Google Scholar]
- Dollé ME, Snyder WK, Dunson DB, Vijg J, 2002. Mutational fingerprints of aging. Nucleic Acids Res 30, 545–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé ME, Snyder WK, Gossen JA, Lohman PH, Vijg J, 2000. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci U S A 97, 8403–8408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolle ME, Vijg J, 2002. Genome dynamics in aging mice. Genome research 12, 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong X, Milholland B, Vijg J, 2016. Evidence for a limit to human lifespan. Nature 538, 257–259. [DOI] [PubMed] [Google Scholar]
- Dong X, Zhang L, Milholland B, Lee M, Maslov AY, Wang T, Vijg J, 2017. Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat Methods 14, 491–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drake JW, Charlesworth B, Charlesworth D, Crow J, 1998. Rates of Spontaneous Mutation. Genetics 148, 1667–1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enge M, Arda HE, Mignardi M, Beausang J, Bottino R, Kim SK, Quake SR, 2017. Single-Cell Analysis of Human Pancreas Reveals Transcriptional Signatures of Aging and Somatic Mutation Patterns. Cell 171, 321–330 e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson RP, 2010. Somatic gene mutation and human disease other than cancer: an update. Mutat Res 705, 96–106. [DOI] [PubMed] [Google Scholar]
- Failla G, 1958. The aging process and carcinogenesis. Annals of the New York Academy of Science 71, 1124–1135. [DOI] [PubMed] [Google Scholar]
- Fearon ER, Vogelstein B, 1990. A genetic model for colorectal tumorigenesis. Cell 61, 759–767. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, Santoro A, 2018. Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Franco I, Helgadottir HT, Moggio A, Larsson M, Vrtacnik P, Johansson A, Norgren N, Lundin P, Mas-Ponte D, Nordstrom J, Lundgren T, Stenvinkel P, Wennberg L, Supek F, Eriksson M, 2019. Whole genome DNA sequencing provides an atlas of somatic mutagenesis in healthy human cells and identifies a tumor-prone cell type. Genome Biol 20, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco I, Johansson A, Olsson K, Vrtacnik P, Lundin P, Helgadottir HT, Larsson M, Revechon G, Bosia C, Pagnani A, Provero P, Gustafsson T, Fischer H, Eriksson M, 2018. Somatic mutagenesis in satellite cells associates with human skeletal muscle aging. Nature communications 9, 800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu AQ, Genereux DP, Stoger R, Laird CD, Stephens M, 2010. Statistical Inference of Transmission Fidelity of DNA Methylation Patterns over Somatic Cell Divisions in Mammals. The annals of applied statistics 4, 871–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K 2017. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 355, 842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AM, Busuttil RA, Calder RB, Dolle ME, Diaz V, McMahan CA, Bartke A, Nelson J, Reddick R, Vijg J, 2008. Effect of Ames dwarfism and caloric restriction on spontaneous DNA mutation frequency in different mouse tissues. Mech Ageing Dev 129, 528–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Nieto PE, Morrison AJ, Fraser HB, 2019. The somatic mutation landscape of the human body. Genome Biol 20, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilov LA, Gavrilova NS, Kroutko VN, Evdokushkina GN, Semyonova VG, Gavrilova AL, Lapshin EV, Evdokushkina NN, Kushnareva YE, 1997. Mutation load and human longevity. Mutat Res 377, 61–62. [DOI] [PubMed] [Google Scholar]
- Gebel J, Tuppi M, Sanger N, Schumacher B, Dotsch V, 2020. DNA Damaged Induced Cell Death in Oocytes. Molecules 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glansdorff N, Xu Y, Labedan B, 2008. The last universal common ancestor: emergence, constitution and genetic legacy of an elusive forerunner. Biol Direct 3, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, Jurk D, Maier AB, Narita M, Niedernhofer L, Passos JF, Robbins PD, Schmitt CA, Sedivy J, Vougas K, von Zglinicki T, Zhou D, Serrano M, Demaria M, 2019. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827. [DOI] [PubMed] [Google Scholar]
- Gossen JA, de Leeuw WJ, Tan CH, Zwarthoff EC, Berends F, Lohman PH, Knook DL, Vijg J, 1989. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci U S A 86, 7971–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina S, Dong X, Yu B, Vijg J, 2016. Single-cell genome-wide bisulfite sequencing uncovers extensive heterogeneity in the mouse liver methylome. Genome Biol 17, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundry M, Li W, Maqbool SB, Vijg J, 2012. Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res 40, 2032–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D, 1956. Aging: A theory based on free radical and radiation chemistry. Journal of Gerontology 2, 298–300. [DOI] [PubMed] [Google Scholar]
- Hart RW, Setlow RB, 1974. Correlation between deoxyribonucleic acid excision-repair and life-span in a number of mammalian species. Proc Natl Acad Sci U S A 71, 2169–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J, 2003. Aging and genome maintenance: lessons from the mouse? Science 299, 1355–1359. [DOI] [PubMed] [Google Scholar]
- Helle F, 2012. Germ cell DNA-repair systems-possible tools in cancer research? Cancer Gene Ther 19, 299–302. [DOI] [PubMed] [Google Scholar]
- Heyward FD, Sweatt JD, 2015. DNA Methylation in Memory Formation: Emerging Insights. Neuroscientist 21, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkal GW, Gatza CE, Parikh N, Donehower LA, 2009. Altered senescence, apoptosis, and DNA damage response in a mutant p53 model of accelerated aging. Mech Ageing Dev 130, 262–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch GP, 1978. Somatic mutations and aging, in: Schneider EL (Ed.), The Genetics of Aging. Plenum, New York, pp. 91–127. [Google Scholar]
- Hoeijmakers JH, 2009. DNA damage, aging, and cancer. N Engl J Med 361, 1475–1485. [DOI] [PubMed] [Google Scholar]
- Holliday R, 1991. Mutations and epimutations in mammalian cells. Mutat Res 250, 351–363. [DOI] [PubMed] [Google Scholar]
- Hou Y, Guo H, Cao C, Li X, Hu B, Zhu P, Wu X, Wen L, Tang F, Huang Y, Peng J, 2016. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs PA, Brunton M, Brown WM, 1964. Cytogenetic Studies in Leucocytes on the General Population: Subjects of Ages 65 Years and More. Annals of human genetics 27, 353–365. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan BF, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, Ebert BL, 2014. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvis GE, 2016. Early embryo mortality in natural human reproduction: What the data say. F1000Res 5, 2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce GF, Szostak JW, 2018. Protocells and RNA Self-Replication. Cold Spring Harbor perspectives in biology 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, Dunham I, Elnitski LL, Farnham PJ, Feingold EA, Gerstein M, Giddings MC, Gilbert DM, Gingeras TR, Green ED, Guigo R, Hubbard T, Kent J, Lieb JD, Myers RM, Pazin MJ, Ren B, Stamatoyannopoulos JA, Weng Z, White KP, Hardison RC, 2014. Defining functional DNA elements in the human genome. Proceedings of the National Academy of Sciences of the United States of America 111, 6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB, 2005. Understanding the odd science of aging. Cell 120, 437–447. [DOI] [PubMed] [Google Scholar]
- Kirkwood TB, Proctor CJ, 2003. Somatic mutations and ageing in silico. Mech Ageing Dev 124, 85–92. [DOI] [PubMed] [Google Scholar]
- Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Sorge JA, Putman DL, Short JM, 1991. Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America 88, 7958–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G, Gudjonsson SA, Sigurdsson A, Jonasdottir A, Wong WS, Sigurdsson G, Walters GB, Steinberg S, Helgason H, Thorleifsson G, Gudbjartsson DF, Helgason A, Magnusson OT, Thorsteinsdottir U, Stefansson K, 2012. Rate of de novo mutations and the importance of father’s age to disease risk. Nature 488, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosugi S, Momozawa Y, Liu X, Terao C, Kubo M, Kamatani Y, 2019. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol 20, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird CD, Pleasant ND, Clark AD, Sneeden JL, Hassan KM, Manley NC, Vary JC Jr., Morgan T, Hansen RS, Stoger R, 2004. Hairpin-bisulfite PCR: assessing epigenetic methylation patterns on complementary strands of individual DNA molecules. Proc Natl Acad Sci U S A 101, 204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasken RS, Stockwell TB, 2007. Mechanism of chimera formation during the Multiple Displacement Amplification reaction. BMC Biotechnol 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie CC, Laurie CA, Rice K, Doheny KF, Zelnick LR, McHugh CP, Ling H, Hetrick KN, Pugh EW, Amos C, Wei Q, Wang LE, Lee JE, Barnes KC, Hansel NN, Mathias R, Daley D, Beaty TH, Scott AF, Ruczinski I, Scharpf RB, Bierut LJ, Hartz SM, Landi MT, Freedman ND, Goldin LR, Ginsburg D, Li J, Desch KC, Strom SS, Blot WJ, Signorello LB, Ingles SA, Chanock SJ, Berndt SI, Le Marchand L, Henderson BE, Monroe KR, Heit JA, de Andrade M, Armasu SM, Regnier C, Lowe WL, Hayes MG, Marazita ML, Feingold E, Murray JC, Melbye M, Feenstra B, Kang JH, Wiggs JL, Jarvik GP, McDavid AN, Seshan VE, Mirel DB, Crenshaw A, Sharopova N, Wise A, Shen J, Crosslin DR, Levine DM, Zheng X, Udren JI, Bennett S, Nelson SC, Gogarten SM, Conomos MP, Heagerty P, Manolio T, Pasquale LR, Haiman CA, Caporaso N, Weir BS, 2012. Detectable clonal mosaicism from birth to old age and its relationship to cancer. Nat Genet 44, 642–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson ARJ, Abascal F, Coorens THH, Hooks Y, O’Neill L, Latimer C, Raine K, Sanders MA, Warren AY, Mahbubani KTA, Bareham B, Butler TM, Harvey LMR, Cagan A, Menzies A, Moore L, Colquhoun AJ, Turner W, Thomas B, Gnanapragasam V, Williams N, Rassl DM, Vohringer H, Zumalave S, Nangalia J, Tubio JMC, Gerstung M, Saeb-Parsy K, Stratton MR, Campbell PJ, Mitchell TJ, Martincorena I, 2020. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 370, 75–82. [DOI] [PubMed] [Google Scholar]
- Leach EG, Gunther EJ, Yeasky TM, Gibson LH, Yang-Feng TL, Glazer PM, 1996. Frequent spontaneous deletions at a shuttle vector locus in transgenic mice. Mutagenesis 11, 49–56. [DOI] [PubMed] [Google Scholar]
- Leu K, Obermayer B, Rajamani S, Gerland U, Chen IA, 2011. The prebiotic evolutionary advantage of transferring genetic information from RNA to DNA. Nucleic Acids Res 39, 8135–8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy O, Amit G, Vaknin D, Snir T, Efroni S, Castaldi P, Liu YY, Cohen HY, Bashan A, 2020. Age-related loss of gene-to-gene transcriptional coordination among single cells. Nat Metab 2, 1305–1315. [DOI] [PubMed] [Google Scholar]
- Li C, Williams SM, 2013. Human Somatic Variation: It’s Not Just for Cancer Anymore. Current Genetic Medicine Reports 1, 212–218. [Google Scholar]
- Li W, Calder RB, Mar JC, Vijg J, 2015. Single-cell transcriptogenomics reveals transcriptional exclusion of ENU-mutated alleles. Mutat Res 772, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Sancar A, 2020. Methodologies for detecting environmentally induced DNA damage and repair. Environ Mol Mutagen 61, 664–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl T, 1993. Instability and decay of the primary structure of DNA. Nature 362, 709–715. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, Huang JD, Li KM, Chau PY, Chen DJ, Pei D, Pendas AM, Cadinanos J, Lopez-Otin C, Tse HF, Hutchison C, Chen J, Cao Y, Cheah KS, Tryggvason K, Zhou Z, 2005. Genomic instability in laminopathy-based premature aging. Nat Med 11, 780–785. [DOI] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava CN, Yang P, Chittenden TW, Hatem NE, Ryu SC, Woodworth MB, Park PJ, Walsh CA, 2018. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G, 2013. The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord JM, 2013. The effect of ageing of the immune system on vaccination responses. Hum Vaccin Immunother 9, 1364–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Gladyshev VN, 2017. Molecular signatures of longevity: Insights from cross-species comparative studies. Semin Cell Dev Biol 70, 190–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Upneja A, Galecki A, Tsai YM, Burant CF, Raskind S, Zhang Q, Zhang ZD, Seluanov A, Gorbunova V, Clish CB, Miller RA, Gladyshev VN, 2016. Cell culture-based profiling across mammals reveals DNA repair and metabolism as determinants of species longevity. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacRae SL, Croken MM, Calder RB, Aliper A, Milholland B, White RR, Zhavoronkov A, Gladyshev VN, Seluanov A, Gorbunova V, Zhang ZD, Vijg J, 2015. DNA repair in species with extreme lifespan differences. Aging 7, 1171–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud M, Gobet N, Cruz-Davalos DI, Mounier N, Dessimoz C, Sedlazeck FJ, 2019. Structural variant calling: the long and the short of it. Genome Biol 20, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, 2012. Stochastic modulations of the pace and patterns of ageing: impacts on quasi-stochastic distributions of multiple geriatric pathologies. Mech Ageing Dev 133, 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GM, Ogburn CE, Colgin LM, Gown AM, Edland SD, Monnat RJ Jr., 1996. Somatic mutations are frequent and increase with age in human kidney epithelial cells. Hum Mol Genet 5, 215–221. [DOI] [PubMed] [Google Scholar]
- Martin GM, Oshima J, 2000. Lessons from human progeroid syndromes. Nature 408, 263–266. [DOI] [PubMed] [Google Scholar]
- Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, Cagan A, Murai K, Mahbubani K, Stratton MR, Fitzgerald RC, Handford PA, Campbell PJ, Saeb-Parsy K, Jones PH, 2018. Somatic mutant clones colonize the human esophagus with age. Science 362, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez DE, 1998. Mortality patterns suggest lack of senescence in hydra. Exp Gerontol 33, 217–225. [DOI] [PubMed] [Google Scholar]
- Maslov AY, Ganapathi S, Westerhof M, Quispe W, White RR, Van Houten B, Reiling E, Dolle ME, van Steeg H, Hasty P, Hoeijmakers JH, Vijg J, 2013. DNA damage in normally and prematurely aged mice. Aging Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J, 1959. A theory of ageing. Nature 184, 956–957. [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, Gage FH, 2013. Mosaic copy number variation in human neurons. Science 342, 632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medawar PB, 1952. An Unsolved Problem in Biology. H.K. Lewis, London. [Google Scholar]
- Medvedev P, Stanciu M, Brudno M, 2009. Computational methods for discovering structural variation with next-generation sequencing. Nature methods 6, S13–20. [DOI] [PubMed] [Google Scholar]
- Milanese C, Bombardieri CR, Sepe S, Barnhoorn S, Payan-Gomez C, Caruso D, Audano M, Pedretti S, Vermeij WP, Brandt RMC, Gyenis A, Wamelink MM, de Wit AS, Janssens RC, Leen R, van Kuilenburg ABP, Mitro N, Hoeijmakers JHJ, Mastroberardino PG, 2019. DNA damage and transcription stress cause ATP-mediated redesign of metabolism and potentiation of anti-oxidant buffering. Nature communications 10, 4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milholland B, Dong X, Zhang L, Hao X, Suh Y, Vijg J, 2017. Differences between germline and somatic mutation rates in humans and mice. Nature communications 8, 15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RA, 2004. ‘Accelerated aging’: a primrose path to insight? Aging Cell 3, 47–51. [DOI] [PubMed] [Google Scholar]
- Miller SL, 1955. Production of Some Organic Compounds under Possible Primitive Earth Conditions. J. Am. Chem. Soc 77, 2351–2361. [Google Scholar]
- Minois N, Frajnt M, Dolling M, Lagona F, Schmid M, Kuchenhoff H, Gampe J, Vaupel JW, 2006. Symmetrically dividing cells of the fission yeast schizosaccharomyces pombe do age. Biogerontology 7, 261–267. [DOI] [PubMed] [Google Scholar]
- Morley AA, 1982. Is ageing the result of dominant and co-dominant mutations? J Theor Biol 98, 469–474. [DOI] [PubMed] [Google Scholar]
- Morrill SA, Amon A, 2019. Why haploinsufficiency persists. Proc Natl Acad Sci U S A 116, 11866–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ, 1964. The relation of recombination to mutational advance. Mutation Research 1, 2–9. [DOI] [PubMed] [Google Scholar]
- Nanki K, Fujii M, Shimokawa M, Matano M, Nishikori S, Date S, Takano A, Toshimitsu K, Ohta Y, Takahashi S, Sugimoto S, Ishimaru K, Kawasaki K, Nagai Y, Ishii R, Yoshida K, Sasaki N, Hibi T, Ishihara S, Kanai T, Sato T, 2020. Somatic inflammatory gene mutations in human ulcerative colitis epithelium. Nature 577, 254–259. [DOI] [PubMed] [Google Scholar]
- Navarro CL, Cau P, Levy N, 2006. Molecular bases of progeroid syndromes. Hum Mol Genet 15 Spec No 2, R151–161. [DOI] [PubMed] [Google Scholar]
- Nordling CO, 1953. A new theory on cancer-inducing mechanism. Br J Cancer 7, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell PC, 1976. The clonal evolution of tumor cell populations. Science 194, 23–28. [DOI] [PubMed] [Google Scholar]
- Olafsson S, McIntyre RE, Coorens T, Butler T, Jung H, Robinson PS, Lee-Six H, Sanders MA, Arestang K, Dawson C, Tripathi M, Strongili K, Hooks Y, Stratton MR, Parkes M, Martincorena I, Raine T, Campbell PJ, Anderson CA, 2020. Somatic Evolution in Non-neoplastic IBD-Affected Colon. Cell 182, 672–684 e611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono T, Ikehata H, Nakamura S, Saito Y, Hosoi Y, Takai Y, Yamada S, Onodera J, Yamamoto K, 2000. Age-associated increase of spontaneous mutant frequency and molecular nature of mutation in newborn and old lacZ-transgenic mouse. Mutat Res 447, 165–177. [DOI] [PubMed] [Google Scholar]
- Orgel LE, 1963. The maintenance of the accuracy of protein synthesis and its relevance to ageing. Proc Natl Acad Sci U S A 49, 517–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierre RV, Hoagland HC, 1972. Age-associated aneuploidy: loss of Y chromosome from human bone marrow cells with aging. Cancer 30, 889–894. [DOI] [PubMed] [Google Scholar]
- Poduri A, Evrony GD, Cai X, Walsh CA, 2013. Somatic mutation, genomic variation, and neurological disease. Science 341, 1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenca AM, Rang CU, Qiu A, Shi C, Chao L, 2019. Cell aging preserves cellular immortality in the presence of lethal levels of damage. PLoS Biol 17, e3000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsey MJ, Moore DH 2nd, Briner JF, Lee DA, Olsen L, Senft JR, Tucker JD, 1995. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutat Res 338, 95–106. [DOI] [PubMed] [Google Scholar]
- Rohrback S, April C, Kaper F, Rivera RR, Liu CS, Siddoway B, Chun J, 2018. Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc Natl Acad Sci U S A 115, 10804–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhok A, DeGregori J, 2019. A generalized theory of age-dependent carcinogenesis. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacher GA, 1982. Evolutionary theory in gerontology. Perspect Biol Med 25, 339–353. [DOI] [PubMed] [Google Scholar]
- Sano S, Oshima K, Wang Y, MacLauchlan S, Katanasaka Y, Sano M, Zuriaga MA, Yoshiyama M, Goukassian D, Cooper MA, Fuster JJ, Walsh K, 2018. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. J Am Coll Cardiol 71, 875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santra M, Dill KA, de Graff AMR, 2019. Proteostasis collapse is a driver of cell aging and death. Proc Natl Acad Sci U S A 116, 22173–22178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeiser HH, Nortier JL, Singh R, Gamboa da Costa G, Sennesael J, Cassuto-Viguier E, Ambrosetti D, Rorive S, Pozdzik A, Phillips DH, Stiborova M, Arlt VM, 2014. Exceptionally long-term persistence of DNA adducts formed by carcinogenic aristolochic acid I in renal tissue from patients with aristolochic acid nephropathy. International journal of cancer. Journal international du cancer 135, 502–507. [DOI] [PubMed] [Google Scholar]
- Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ, 2021. The central role of DNA damage in the ageing process. Nature In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavov N, 2020. Single-cell protein analysis by mass spectrometry. Curr Opin Chem Biol 60, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sniegowski PD, Gerrish PJ, Johnson T, Shaver A, 2000. The evolution of mutation rates: separating causes from consequences. Bioessays 22, 1057–1066. [DOI] [PubMed] [Google Scholar]
- Stuart GR, Oda Y, de Boer JG, Glickman BW, 2000. Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice. Genetics 154, 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, White RR, Fischer KE, Zhang Z, Austad SN, Vijg J, 2020. Inducible aging in Hydra oligactis implicates sexual reproduction, loss of stem cells, and genome maintenance as major pathways. Geroscience 42, 1119–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilard L, 1959. On the nature of the aging process. Proc Natl Acad Sci U S A 45, 30–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson DJ, Genovese G, Halvardson J, Ulirsch JC, Wright DJ, Terao C, Davidsson OB, Day FR, Sulem P, Jiang Y, Danielsson M, Davies H, Dennis J, Dunlop MG, Easton DF, Fisher VA, Zink F, Houlston RS, Ingelsson M, Kar S, Kerrison ND, Kinnersley B, Kristjansson RP, Law PJ, Li R, Loveday C, Mattisson J, McCarroll SA, Murakami Y, Murray A, Olszewski P, Rychlicka-Buniowska E, Scott RA, Thorsteinsdottir U, Tomlinson I, Moghadam BT, Turnbull C, Wareham NJ, Gudbjartsson DF, International Lung Cancer, C., Breast Cancer Association, C., Consortium of Investigators of Modifiers of, B., Endometrial Cancer Association, C., Ovarian Cancer Association, C., Prostate Cancer Association Group to Investigate Cancer Associated Alterations in the Genome, C., Kidney Cancer, G.M.-A.P., e, Q.C., Biobank-based Integrative Omics Study, C., andMe Research, T., Kamatani Y, Hoffmann ER, Jackson SP, Stefansson K, Auton A, Ong KK, Machiela MJ, Loh PR, Dumanski JP, Chanock SJ, Forsberg LA, Perry JRB, 2019. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 575, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson O, von Meyenn F, Hewitt Z, Alexander J, Wood A, Weightman R, Gregory S, Krueger F, Andrews S, Barbaric I, Gokhale PJ, Moore HD, Reik W, Milo M, Nik-Zainal S, Yusa K, Andrews PW, 2020. Low rates of mutation in clinical grade human pluripotent stem cells under different culture conditions. Nature communications 11, 1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian XC, Kubota C, Enright B, Yang X, 2003. Cloning animals by somatic cell nuclear transfer--biological factors. Reprod Biol Endocrinol 1, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trainor KJ, Wigmore DJ, Chrysostomou A, Dempsey JL, Seshadri R, Morley AA, 1984. Mutation frequency in human lymphocytes increases with age. Mech Ageing Dev 27, 83–86. [DOI] [PubMed] [Google Scholar]
- Vanneste E, Voet T, Le Caignec C, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, Fryns JP, Verbeke G, D’Hooghe T, Moreau Y, Vermeesch JR, 2009. Chromosome instability is common in human cleavage-stage embryos. Nat Med 15, 577–583. [DOI] [PubMed] [Google Scholar]
- Vermeij WP, Dolle ME, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, Wu H, Roks AJ, Botter SM, van der Eerden BC, Youssef SA, Kuiper RV, Nagarajah B, van Oostrom CT, Brandt RM, Barnhoorn S, Imholz S, Pennings JL, de Bruin A, Gyenis A, Pothof J, Vijg J, van Steeg H, Hoeijmakers JH, 2016. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537, 427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, 2007. Aging of the Genome. Oxford, New York. [Google Scholar]
- Vijg J, Busuttil RA, Bahar R, Dolle ME, 2005. Aging and genome maintenance. Ann N Y Acad Sci 1055, 35–47. [DOI] [PubMed] [Google Scholar]
- Vijg J, Campisi J, 2008. Puzzles, promises and a cure for ageing. Nature 454, 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Dollé ME, 2002. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev 123, 907–915. [DOI] [PubMed] [Google Scholar]
- Vijg J, Dong X, 2020. Pathogenic Mechanisms of Somatic Mutation and Genome Mosaicism in Aging. Cell 182, 12–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J, Kennedy BK, 2016. The Essence of Aging. Gerontology 62, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Clauson CL, Robbins PD, Niedernhofer LJ, Wang Y, 2012. The oxidative DNA lesions 8,5’-cyclopurines accumulate with aging in a tissue-specific manner. Aging Cell 11, 714–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J, Crick F, 1953. Molecular structure of nucleic acids. Nature 171, 737–738. [DOI] [PubMed] [Google Scholar]
- Williams GC, 1957. Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411. [Google Scholar]
- Yizhak K, Aguet F, Kim J, Hess JM, Kubler K, Grimsby J, Frazer R, Zhang H, Haradhvala NJ, Rosebrock D, Livitz D, Li X, Arich-Landkof E, Shoresh N, Stewart C, Segre AV, Branton PA, Polak P, Ardlie KG, Getz G, 2019. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Dong X, Lee M, Maslov AY, Wang T, Vijg J, 2019. Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc Natl Acad Sci U S A 116, 9014–9019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Zhou K, Luo X, Li L, Tu HC, Sehgal A, Nguyen LH, Zhang Y, Gopal P, Tarlow BD, Siegwart DJ, Zhu H, 2018. The Polyploid State Plays a Tumor-Suppressive Role in the Liver. Developmental cell 47, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Beaudet AL, 2016. Epigenetics and Human Disease. Cold Spring Harb Perspect Biol 8, a019497. [DOI] [PMC free article] [PubMed] [Google Scholar]