

Abstract
Chronic kidney disease (CKD) is a public health concern that affects over 200 million people worldwide and is associated with a tremendous economic burden. Therefore, deciphering the mechanisms underpinning CKD is crucial to deaccelerate its progression towards end-stage renal disease. Renal tubular cells are populated with a high number of mitochondria, which produce cellular energy and modulate several important cellular processes, including generation of reactive oxygen species, calcium homeostasis, proliferation, and apoptosis. Over the past few years, increasing evidence has implicated renal mitochondrial damage in the pathogenesis of common etiologies of CKD, such as diabetes, hypertension, metabolic syndrome, chronic renal ischemia, and polycystic kidney disease. However, most compelling evidence is based on preclinical studies because renal biopsies are not routinely performed in many patients with CKD. Previous studies have shown that urinary mitochondrial DNA (mtDNA) copy numbers may serve as non-invasive biomarkers of renal mitochondrial dysfunction. Emerging data also suggest that CKD is associated with altered expression of mitochondria-related microRNAs (mitomiRs), which localize in mitochondria and regulate the expression of mtDNA and nuclear-encoded mitochondrial genes. This review summarizes relevant evidence regarding the involvement of renal mitochondrial injury and dysfunction in frequent forms of CKD. We further provide an overview of non-invasive biomarkers and potential mechanisms of renal mitochondrial damage, especially focusing on mtDNA and mitomiRs.
Keywords: Chronic kidney disease, Metabolic syndrome, Polycystic Kidney Disease, Mitochondria, microRNA, mtDNA
Introduction
Chronic kidney disease (CKD) is increasingly recognized as a major public health problem worldwide that affects more than 15% of Americans and over 200 million people across the globe1. CKD, historically termed chronic renal failure (CRF), refers to the slow and irreversible deterioration of kidney function over time, and is commonly associated with adverse renal and cardiovascular outcomes and premature death. Renal impairment in CKD may gradually progress to end-stage renal disease (ESRD), the final and permanent stage of CKD, requiring renal replacement therapy. Although many patients can access to treatment with kidney transplant or dialysis, marked differences remain in the availability of treatment for ESRD according to race, ethnicity, and socioeconomic status2.
The National Kidney Foundation defined CKD as either kidney damage or a decline in glomerular filtration rate (GFR) to less than 60 mL/min/1.73 m2 for at least 3 months3. The Kidney Disease Outcomes Quality Initiative (KDOQI) subsequently established guidelines to classify CKD as G1-G5, based on the levels of kidney function and/or evidence of renal parenchymal damage3. The etiology of CKD varies globally, but the top 2 leading causes are diabetes and hypertension, which account for up to two-thirds of the cases4. Metabolic syndrome (MetS), which refers to the co-occurrence of obesity, hypertension, insulin resistance, and hyperlipidemia, is a strong and independent risk factor for CKD associated with increased risk of ESRD5, 6. Autosomal dominant polycystic kidney disease (ADPKD) is the most common inherited cause of CKD and the fourth leading cause of renal failure in adults worldwide7. Other common primary diseases causing CKD include glomerulonephritis, chronic tubulointerstitial nephritis, other inherited kidney disorders, infections, obstructive uropathy, plasma cell dyscrasias, and neoplasms8.
The prevalence of CKD has been increasing over recent years and is projected to increase to 17% by 20304. This growing prevalence imposes a tremendous economic burden worldwide. In the US, annual costs associated with ESRD and CKD exceed $30 and $50 billion dollars, respectively, consuming more than 20% of the total annual budget9. Likewise, the burden of CKD per million of the population with diabetes in the UK is expected to rise to £11.4 billion by 202510, whereas the high costs of dialysis and kidney transplantation in Latin America warrants cost-effective forms of renal replacement therapy11.
Beyond the economic impact, CKD is independently associated with serious complications. Patients with CKD have increased cardiovascular risk manifesting as heart failure with reduced or preserved ejection fraction12, which might be present in more than 50% of patients with CKD13. Similarly, CKD might be associated with mineral bone disease, anemia, acidosis, hyperphosphatemia, hyperkalemia, and all-cause mortality14–17. A worldwide cohort study revealed that CKD resulted in 1.2 million deaths and was responsible for 1.4 million of all cardiovascular disease deaths in 201718. What is more, deaths associated with CKD are predicted to increase further over the next decade4. Therefore, deciphering the mechanisms underpinning chronic renal injury in CKD is crucial to deaccelerate its progression towards ESRD and death.
The kidney is equipped with a very high number of mitochondria19, which not only produce cellular energy, but also modulate several important cellular processes, including redox status, calcium homeostasis, proliferation, and programmed cell death20. Accumulating experimental evidence suggest that several forms of CKD are associated with important mitochondrial structural and functional abnormalities in different renal cell types21–23. However, most data were originated from preclinical studies because renal biopsies are not routinely performed in many patients with CKD, underscoring the need to identify surrogate markers of mitochondrial injury. Likewise, the primary mechanisms implicated in CKD-induced renal mitochondrial damage remain to be clarified.
Over the last couple of years, several studies in patients with several forms of CKD have shown that damaged or dying renal cells may release fragments of their mitochondrial genome into the urine, and urinary mitochondrial DNA (mtDNA) copy numbers may serve as non-invasive biomarkers of renal mitochondrial injury and dysfunction24–26. More recently, studies revealed that CKD is associated with altered expression of mitochondria-related microRNAs (mitomiRs)27, 28, a group of miRNAs that localize in mitochondria and regulate the expression of mtDNA and nuclear-encoded mitochondrial genes29. This review summarizes current knowledge of the contribution of renal mitochondrial injury and dysfunction in the pathogenesis of common forms of CKD. In particular, we discuss the role of mtDNA as non-invasive biomarkers of renal mitochondrial injury and mitomiRs as potential mediators of renal mitochondrial damage in CKD.
Renal mitochondrial damage in CKD
Mitochondrial density varies greatly among different organs and cell types. In the kidney, renal tubular cells, responsible for active transport of solutes, contain high numbers of mitochondria19, whereas these organelles occupy only a small fraction of the cytoplasmic volume of endothelial cells30. Mitochondria are characterized by two lipid bilayers, the outer membrane, freely permeable to small molecules, and the inner membrane permeable only to oxygen, carbon dioxide, and water. This double-membrane structure generates two major compartments, the intermembrane space, which houses only 5% of the mitochondrial proteome, and the matrix, which contains mtDNA and the enzymes of the tricarboxylic acid (TCA) cycle20, a series of reactions that produce NADH and FADH2 and provide metabolites for several biosynthetic processes31. To maximize energy production, numerous folds (cristae) of the inner membrane project towards the matrix, increasing its total surface area. These cristae contain the electron transport chain (ETC) system that generates ATP by shuttling electrons from NADH and FADH2 to molecular oxygen.
Although the primary function of mitochondria is the production of cellular energy by oxidative phosphorylation, these organelles are also key regulators of essential cell processes, such as proliferation, survival, and calcium homeostasis32. Mitochondria play a central role in intrinsic apoptosis. Release of cytochrome-c and second mitochondria-derived activator of caspase from mitochondria to the cytosol activates the caspase pathway, initiating apoptosis33. In addition, mitochondria represent an important cellular source of reactive oxygen species (ROS). Mitochondrial superoxide is primarily generated due to leakage of electrons at complex-I and complex-III at the electron transport chain34. However, major mitochondrial antioxidant systems, such as manganese superoxide dismutase (SOD-2), peroxiredoxin, and thioredoxin reductase may counterbalance ROS production, modulating the cellular redox state35. Importantly, mitochondrial ROS impact on multiple cellular signalling pathways, including the nuclear factor (NF)-κB, the mitogen-activated protein kinase, and the phosphoinositide-3-kinase-Akt pathways, ultimately coordinating cell homeostasis, fate, and function36.
An increasing body of evidence suggest that mitochondrial structural damage and dysfunction might be implicated in the pathogenesis of common forms of CKD, including diabetes37 and hypertension38. Exposure of mouse podocytes to high glucose results in apoptosis and NADPH superoxide generation both in vitro and in vivo39 (Figure 1). NADPH oxidase (NOX)-4-derived ROS inactivates mitochondrial respiratory chain complex I40 and promotes extracellular matrix accumulation in mesangial cells41, 42. However, the apoptotic effect of glucose is prevented by inhibitors of the mitochondrial respiratory chain and NADPH oxidases43, underscoring the role of mitochondria in glucose-induced podocyte damage. Likewise, spontaneously hypertensive rats exhibit renal mitochondrial structural abnormalities (swelling, enlargement, less defined cristae), impaired bioenergetics44, and reduced expression of uncoupling protein (UCP)-245, a mitochondrial anion carrier protein that uncouples oxygen consumption from ATP synthesis46. A high salt diet in SOD-2-deficient mice is associated with intrarenal inflammation and increased production of ROS47. Furthermore, proteomic analysis of hypertensive rat renal tubular cells identified several differentially expressed mitochondrial proteins implicated in glucose metabolism and oxygen utilization48, suggesting that renal mitochondrial damage might be implicated in the pathogenesis of hypertensive CKD.
Figure. 1. Potential mechanisms of renal mitochondrial damage in hypertensive and diabetic CKD.
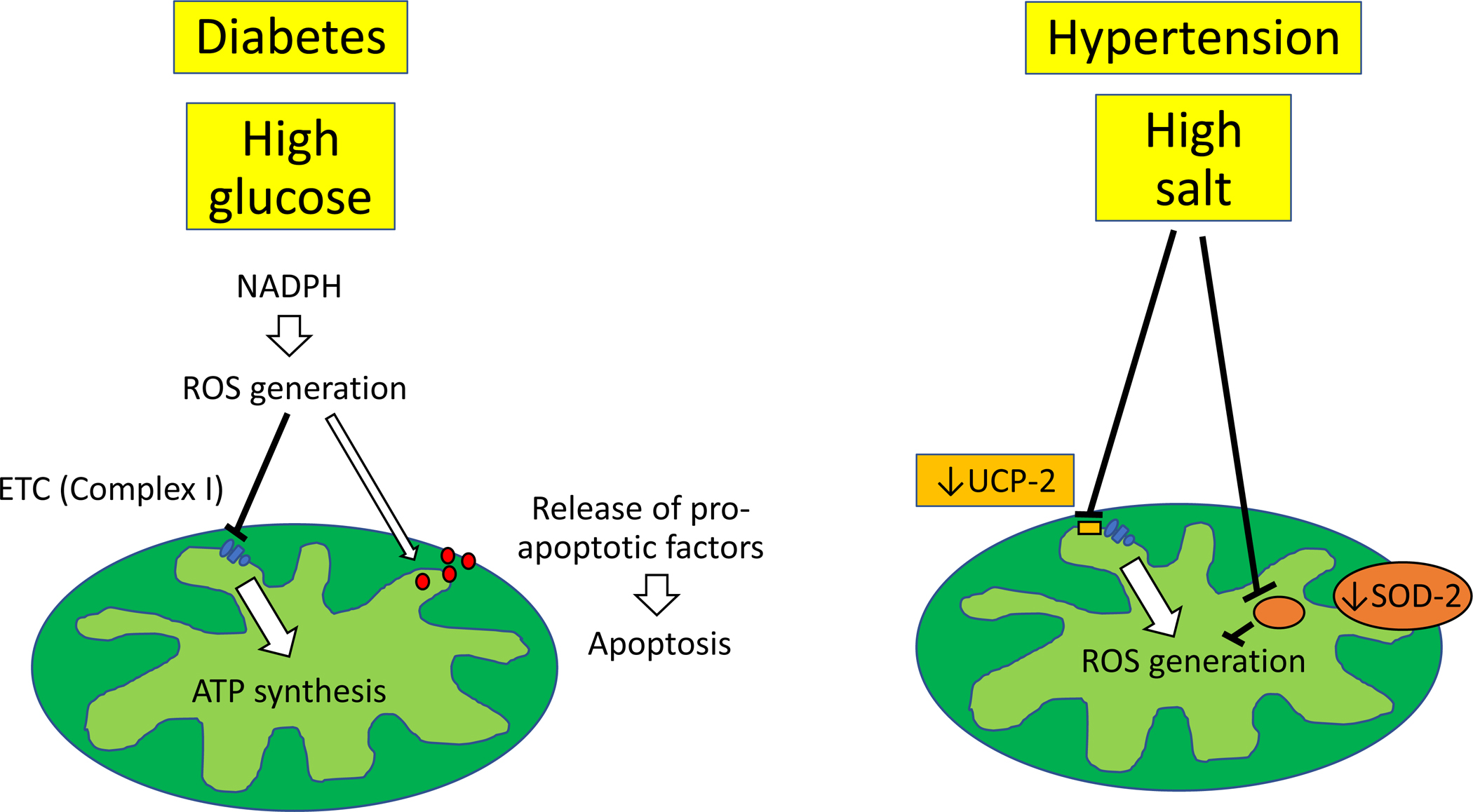
Exposure of podocytes to high glucose results in apoptosis and NADPH superoxide generation. This in turn inactivates mitochondrial respiratory chain complex I and favors release of pro-apoptotic factors, contributing to impaired bioenergetics and apoptosis. A high salt diet induces structural mitochondrial abnormalities and impaired bioenergetics, associated with reduced expression of uncoupling protein-2 and increased production of ROS.
Similarly, in MetS, nutrient surplus supplies excessive amounts of electrons to the respiratory chain, favoring superoxide formation and mitochondrial dysfunction49 (Figure 2). MetS is also associated with increased systemic markers of lipid oxidation, such as oxidized low-density lipoprotein (Ox-LDL), which trigger mitochondrial superoxide production and promote SOD-2 protein degradation, contributing to cell apoptosis50. Studies in mouse models of MetS have shown that renal alterations in energy metabolism and lower tissue ATP levels are associated with decrease kidney mitochondrial density and increase oxidative stress21. High fat diet in mice induces mitochondrial structural damage and apoptosis in different renal cell types, including tubular cells, podocytes, and endothelial cells51. Intracellular lipid accumulation not only favors oxidative stress, but also uncouples oxidative phosphorylation, inhibiting ATP production52. In line with this, we found that renal mitochondrial structural damage and dysfunction in swine MetS are associated with renal lipid peroxidation and oxidative stress, reflected in increased expression of Ox-LDL53. This particle obtained from circulating LDL can damage the inner mitochondrial membrane phospholipid cardiolipin, which plays a central role in preserving mitochondrial structure and function54. Indeed, in obese mouse51 and pigs53 restoration of cardiolipin preserved mitochondrial bioenergetics and attenuated renal damage, supporting an important role for renal mitochondrial injury and dysfunction in experimental MetS.
Figure 2. Mechanisms of renal mitochondrial damage in obesity and MetS.
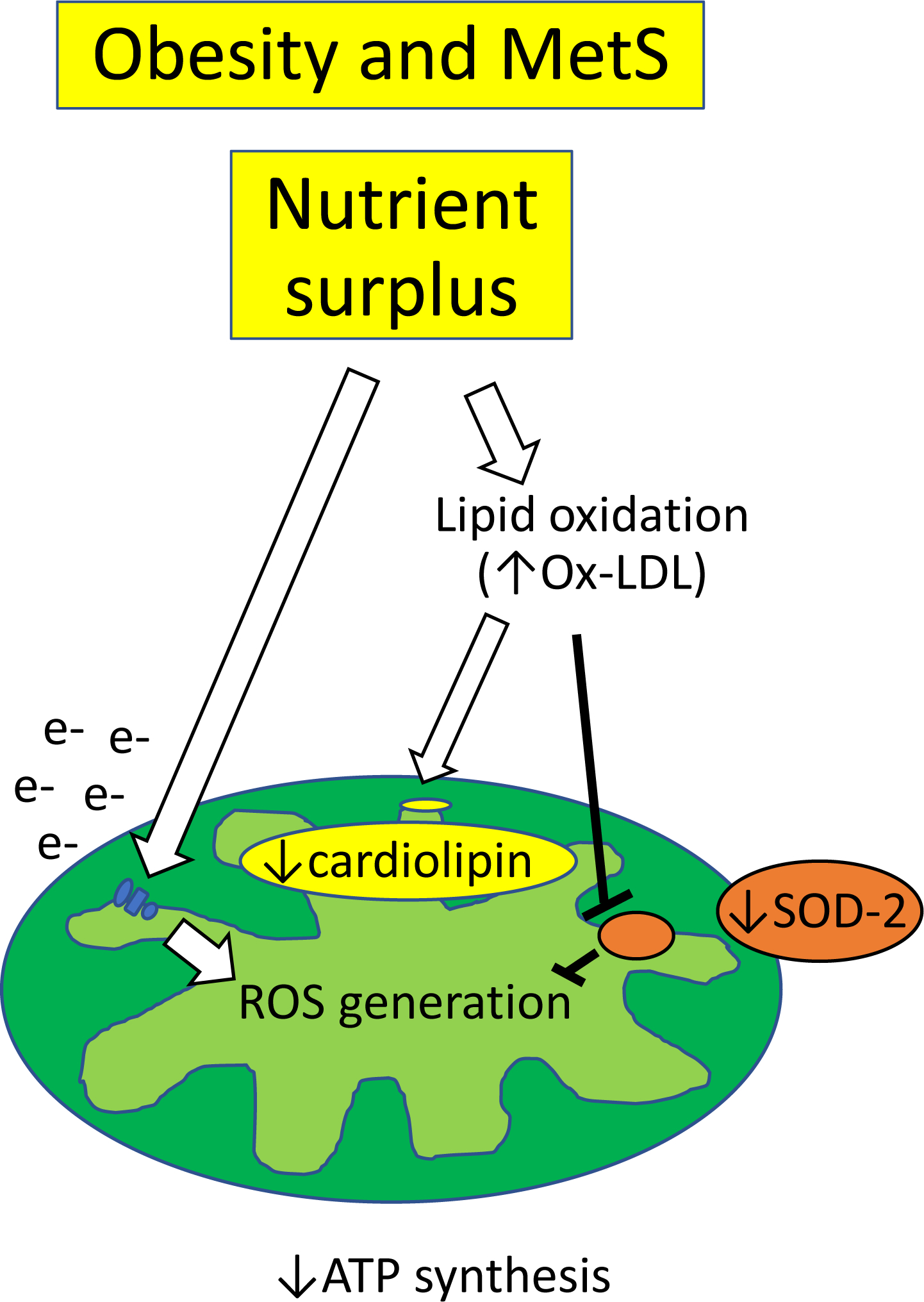
Nutrient surplus supplies excessive amounts of electrons to the respiratory chain, favoring superoxide formation. Increased lipid oxidation, such as oxidized low-density lipoprotein (Ox-LDL), triggers mitochondrial superoxide production by promoting SOD-2 protein degradation. Ox-LDL can also damage cardiolipin, leading to excessive ROS formation and impaired bioenergetics.
Renal mitochondrial abnormalities have been also described in murine models of PKD, as well as in human PKD tubular epithelial cells55–57. Comparative proteomic analysis of human cystic kidney tissue and histological and functional studies in mouse and human PKD cells as well as animal models of PKD suggest that mitochondrial injury and dysfunction may contribute to the development and progression of the disease58–61. Polycystin-1 deficiency is associated with changes in the expression of mitochondria-endoplasmic reticulum associated membranes and impaired mitochondrial calcium uptake62. Renal expression of peroxisome proliferator-activated receptor γ coactivator (PGC-1α), the master regulator of mitochondrial biogenesis was decreased in both mouse and rat models of ADPKD and correlated inversely with the levels of oxidative stress63 (Figure 3). In agreement, we found that PCK rats have increased NOX-4-induced oxidative stress and mitochondrial abnormalities predominantly in cyst-lining tubular cells and renal endothelial cells, which correlate with endothelial dysfunction and worsening of renal disease23. Expression of the mitochondrial SOD-2 is downregulated in cpk mice (64 and patients with ADPKD65, creating a vicious cycle of excessive ROS generation and impaired antioxidant defenses that aggravates mitochondrial damage. Treatment with mitochondria-specific antioxidants reduce intracellular superoxide levels and ameliorate cyst epithelial cell proliferation stress63, linking renal mitochondrial injury and dysfunction to disease progression in experimental ADPKD. Yet, additional studies are needed to define the exact role of these organelles in energy metabolism in this prevalent form of CKD.
Figure 3. Mitochondria-mediated mechanisms of disease progression in ADPKD.
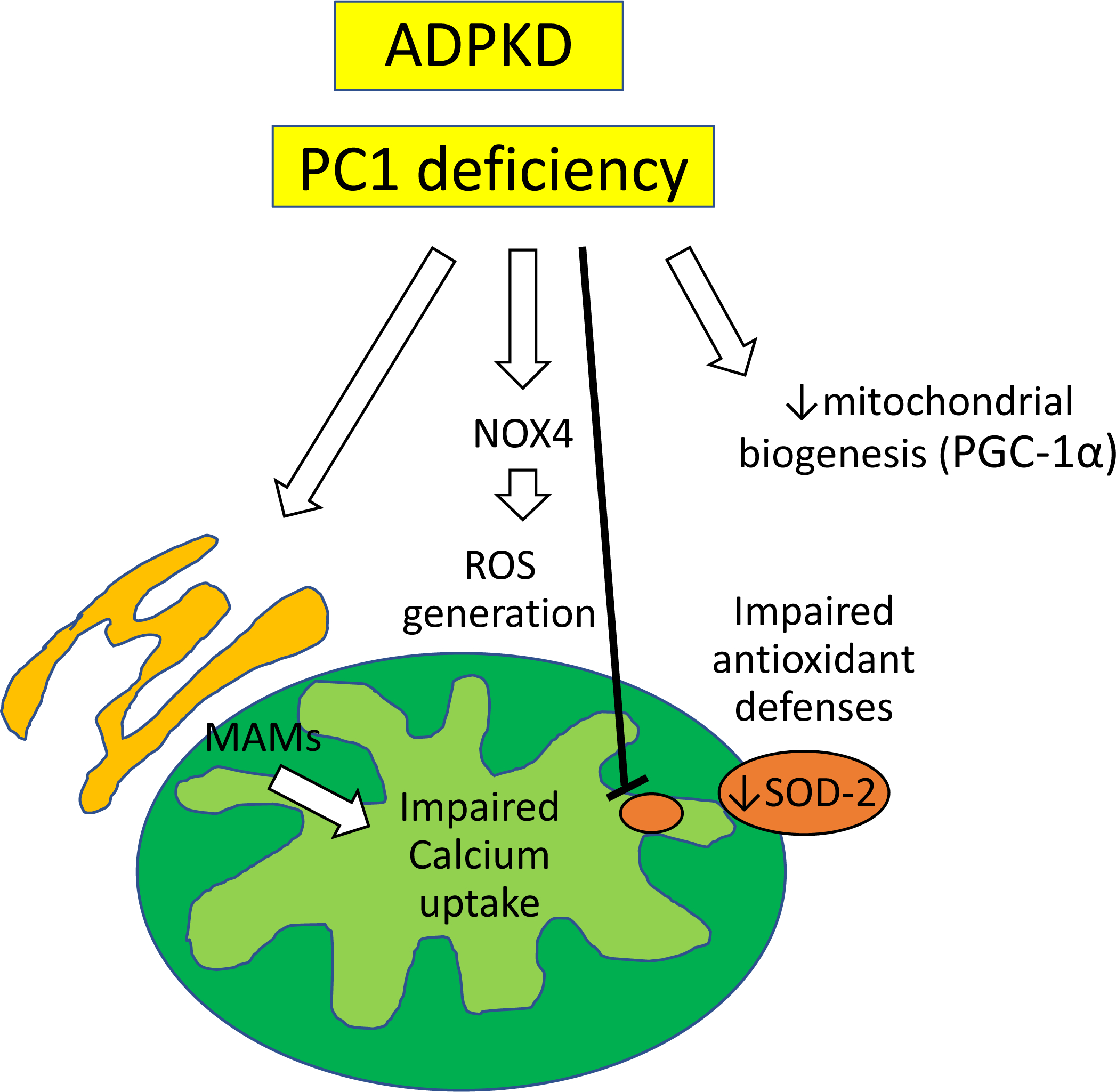
In ADPKD, polycystin-1 deficiency is associated with changes in the expression of mitochondria-associated endoplasmic reticulum membranes (MAMs) and impaired mitochondrial calcium uptake. Peroxisome proliferator-activated receptor γ coactivator (PGC-1α) expression, the main regulator of mitochondrial biogenesis is also suppressed. Expression of NOX-4 increases, whereas expression of SOD-2 decreases, creating a vicious cycle of excessive ROS generation and impaired antioxidant defenses that aggravates mitochondrial damage.
Mitochondrial structural and functional alterations may also play a pivotal role in chronic renal ischemia. The clipped kidneys of the Goldblatt’s 2 kidney 1 clip (2K1C) rat animal model of RVD is characterized by impaired mitochondrial biogenesis and increased mitophagy66 (Figure 4), a form of macroautophagy that selectively degrades damaged mitochondria67. Importantly, RVD-induced renal mitochondrial damage was associated with renal oxidative stress, fibrosis, and necrotic death, which were prevented by inhibition of the pro-death protein BCL2 Interacting Protein 3. Surgically induced RVD in swine is also associated with post-stenotic kidney mitochondrial structural damage and impaired biogenesis, associated with oxidative stress, microvascular loss, fibrosis, and renal dysfunction68. Interestingly, restoration of mitochondrial cardiolipin preserved mitochondrial damage and improved renal function, implicating mitochondrial injury in renal deterioration in chronic experimental RVD. The mechanisms by which renal ischemia and hypertension contribute to renal mitochondrial damage are multifactorial. Activation of the NADPH oxidase complex in response to mechanical stretch favors ROS generation and alter mitochondrial structure69. Extracellular matrix turnover may also compromise the integrity of mitochondrial membranes and mtDNA, increasing mitochondrial permeability70. Furthermore, activation of angiotensin-II receptors in the inner mitochondrial membrane may compromise mitochondrial respiration and membrane potential, promoting local ROS generation71, 72.
Figure 4. Mitochondrial structural and functional alterations in chronic renal ischemia.
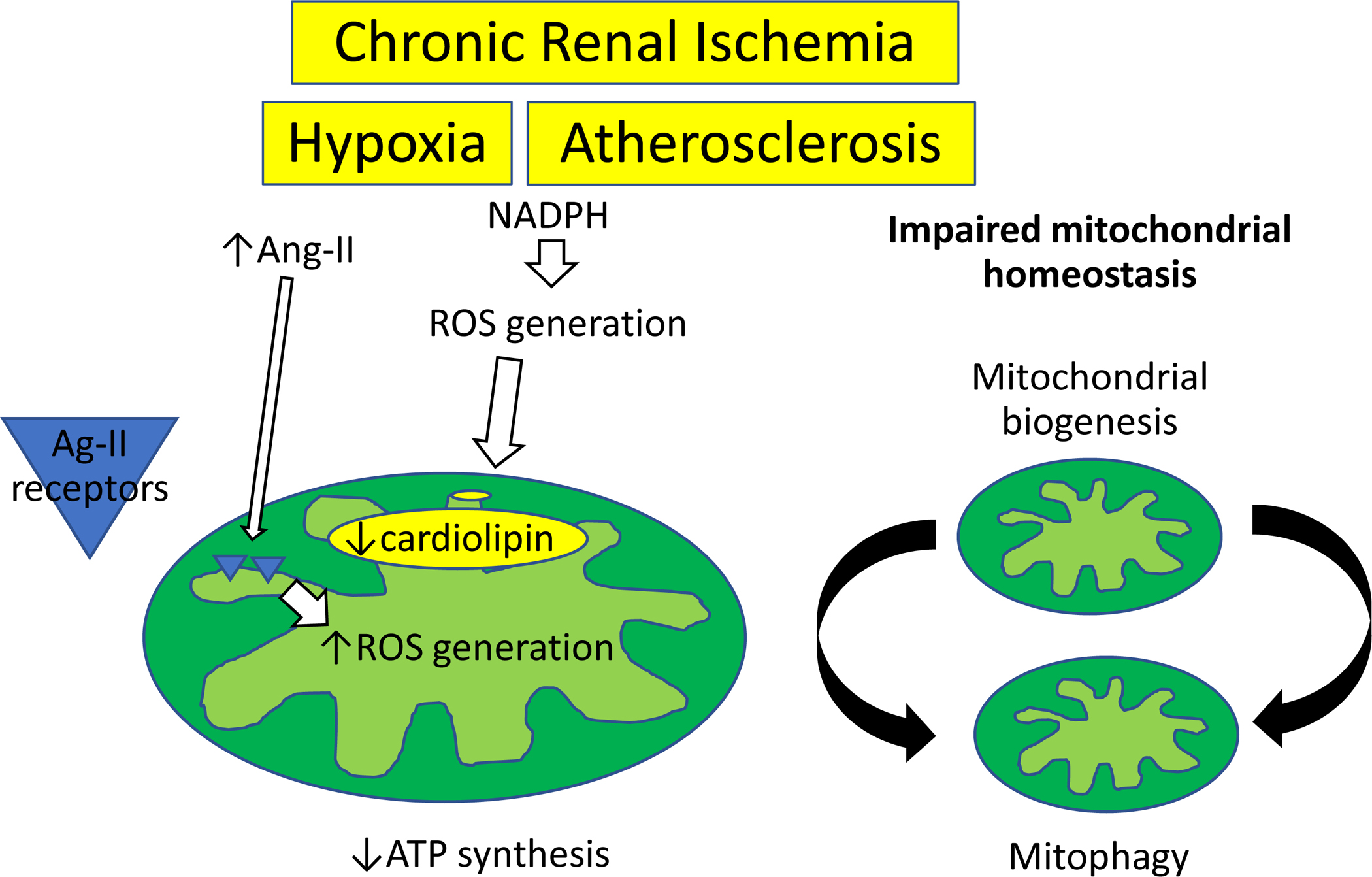
Experimental RVD alters renal mitochondrial biogenesis and mitophagy and decreases cardiolipin content. Activation of the NADPH oxidase complex in response to mechanical stretch favors ROS generation, whereas activation of angiotensin (Ang)-II receptors in the inner mitochondrial membrane may compromise mitochondrial respiration and promote local ROS generation.
Importantly, the coexistence of MetS and CKD may synergistically aggravate mitochondrial structural damage and dysfunction. We have recently shown in swine that the concurrence of MetS and RVD amplify renal tubular mitochondrial damage (Figure 5) and impair energy production in the poststenotic kidney, leading to greater renal fibrosis22. Notably, both MetS and CKD can also induce mitochondrial damage in endogenous repair cells, such as mesenchymal stem cells28, 73 and scattered tubular-like cells (STCs)27, 74, a dedifferentiated phenotype that can be adopted by surviving tubular epithelial cells to repair neighboring injured renal tubular cells75. Taken together, these studies implicate renal mitochondrial damage in the pathogenesis of CKD, and position mitochondria as a potential therapeutic target. Nevertheless, additional studies are needed to establish a cause-effect relationship and test the safety and efficacy of mitoprotective drugs in patients with CKD.
Figure. 5. Experimental MetS and CKD induce renal tubular cell mitochondrial injury.
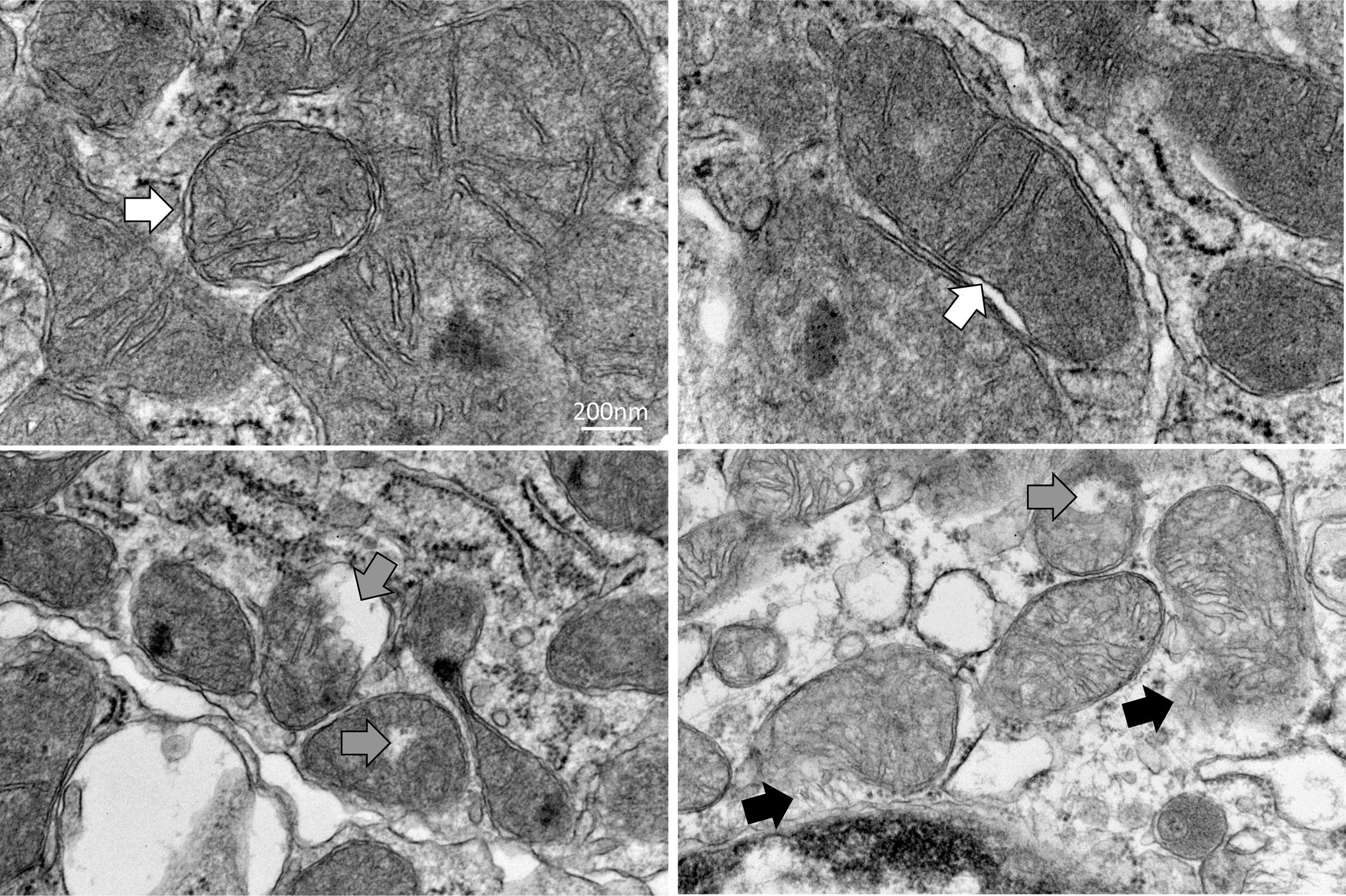
Representative transmission electron microscopic images of renal tubular cell mitochondrial from MetS+CKD pigs showing mitochondrial swelling and decreased cristae membranes (white arrows). Disruption of the outer mitochondrial membrane favors entry of water to the organelle (grey arrows) and release of matrix content to the cytosol (black arrows).
Urinary mtDNA – non-invasive biomarkers of renal injury, mitochondria dysfunction, or decreased mitochondria content
Mitochondrial DNA (mtDNA) is a very small circular (16,500 base pairs) double-stranded DNA caged within the double mitochondrial membrane. It contains very few introns and only 37 genes encoding for 13 proteins, 22 transfer-RNA, and 2 ribosomal-RNA, which are critically important to sustain energy production and modulate several mitochondrial functions. Indeed, mtDNA damage has been linked to impaired cellular bioenergetics, proliferation, and increased apoptosis76. Unlike nuclear DNA, mtDNA is inherited maternally, and is located near the primary sites of generation of ROS, which can induce oxidative damage such as DNA strand breaks, base modification or removal, and cross linking77. Fragments of mtDNA may escape from the matrix to the cytosol and then outside the cell, and may be ultimately released into the systemic circulation78. In renal cells, disruption of mitochondrial integrity may result in release of mtDNA genes into the urine, which may serve as surrogate markers of renal mitochondrial injury and dysfunction. On the other hand, a decrease in urinary or circulating mtDNA copy number, which is proportional to the cell mtDNA content, has been associated with mitochondrial dysfunction in several diseases and could serve as a surrogate marker of mitochondrial integrity and function79–82.
Previous studies in patients with acute kidney injury (AKI) have shown that elevated urinary mtDNA copy numbers correlate with mitochondrial dysfunction and renal injury. Urinary mtDNA levels are significantly elevated in patients with severe sepsis-induced AKI, and positively correlated with plasma creatinine, urinary neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule (KIM)-1, and inversely with the estimated GFR (eGFR)83. Likewise, urinary mtDNA copy number is associated with progression of renal dysfunction in patients with AKI after cardiac surgery84, could identify newly developed AKI, and predict renal replacement therapy or hospital mortality in surgical intensive care unit patients85.
Similarly, studies in patients with different forms of CKD suggest that urinary copies of mtDNA genes, such as cytochrome-c oxidase-3 (COX3) and NADH dehydrogenase subunit-1 (ND1) may serve as novel markers of mitochondrial stress and damage (Table 1). We have previously shown that urinary mtDNA copy number is elevated in patients with essential hypertension and renovascular RVD and correlate with markers of renal injury (NGAL and KIM-1) and dysfunction (serum creatinine and eGFR)86. Treatment with renal revascularization leads to an acute rise in urinary mtDNA levels in RVD patients, likely reflecting renal ischemia-reperfusion injury-induced mitochondrial damage25. Importantly, urinary mtDNA levels vary as a function of serum creatinine and eGFR 3 months after medical therapy and renal revascularization, implicating mitochondrial injury in kidney damage in human hypertensive CKD.
Table 1.
Studies reporting changes in urinary mtDNA levels in patients with different etiologies of CKD
| Condition | Main Findings | References |
|---|---|---|
|
| ||
| Essential hypertension / RVD | • Elevated urinary copies of COX3 and ND1 • mtDNA correlates with markers of renal injury and dysfunction |
• Eirin A, et al86 |
| RVD revascularization | • Revascularization leads to an acute rise in urinary mtDNA levels • mtDNA levels correlate with renal function 3 months after therapy |
• Eirin A, et al25 |
| IgA nephropathy / Minor glomerular abnormalities | • Elevated urinary copies of COX3 and ND1 • mtDNA correlates positively with changes in proteinuria, but inversely with changes in eGFR |
• Yu BC, et al87, 88 |
| Diabetic nephropathy | • Urinary supernatant mtDNA level correlates inversely with eGFR and positively with interstitial fibrosis | • Wei, et al89 |
| Obesity / Bariatric surgery | • Elevated urinary copies of COX3 and ND1 • Bariatric surgery reduces urinary mtDNA copy numbers 6 months later |
• Seo M, et al24 |
| ANCA-associated Vasculitis | • Increase urinary and plasma mtDNA levels that correlate with the severity of kidney injury | • Wu SJ, et al91 |
| Obesity / Hypertension | • Elevated urinary copies of COX3 and ND1 • mtDNA correlates with renal hyperfiltration |
• Eirin A, et al26 |
| Diabetic nephropathy | • Decreased mtDNA levels in urine exosomes | • Sharma K, et al92 |
| Non-diabetic CKD | • Urinary mtDNA levels correlate with baseline renal function, proteinuria, and the severity of glomerulosclerosis and tubulointerstitial fibrosis | • Wei PZ, et al93 |
mtDNA: mitochondrial DNA; CKD: Chronic kidney disease; RVD: renovascular disease; COX3: cytochrome-c oxidase-3, ND1: NADH dehydrogenase subunit-1; eGFR: estimated glomerular filtration rate; ANCA: anti-neutrophil cytoplasmic antibody
Urinary levels of ND1 and COX3 are also elevated in patients with IgA nephropathy87 and minor glomerular abnormalities88, correlate positively with changes in proteinuria, but inversely with changes in eGFR. Glomerular hyperfiltration in obese African American essential hypertensive patients is also associated with elevated urinary levels of COX3 and ND1, suggesting that mitochondrial injury may aggravate renal damage and contribute to hypertension-related morbidity and mortality rates in this population26. In agreement, studies in patients with biopsy-proven diabetic nephropathy observed that urinary supernatant mtDNA levels correlate inversely with eGFR and positively with interstitial fibrosis89. Urinary copy numbers of ND1 and COX3 genes are also higher in patients with obesity compared to healthy volunteers and further increased in those with coexisting type-2 diabetes24. Interestingly, bariatric surgery reduced urinary mtDNA copy numbers 6 months later, underscoring the potential of this intervention to ameliorate renal mitochondrial damage in obesity and diabetes.
In individuals with glomerular diseases, mtDNA is highly filtered by the kidney. Thus, increased systemic mtDNA levels are often associated with higher urinary levels. Circulating mitochondrial DNA is extremely high in untreated patients with anti-neutrophil cytoplasmic antibody-associated vasculitis90, and their urinary levels increase with the severity of kidney injury and neutrophil infiltration in pathology91. In contrast, we found discrepancies between circulating and urinary mtDNA levels in hypertensive patients. Although urinary levels of COX3 and ND1 were elevated in essential hypertensive and RVD patients, their plasma levels were comparable to healthy volunteers and remained unaltered 3 months after medical therapy or revascularization, suggesting primarily renal production25, 86. Similarly, urinary (but not plasma) mtDNA levels were further elevated in obese African American hypertensive patients26. Yet, serum COX-3 copy numbers have been reported to be higher in obese patients with or without diabetes compared to controls24.
Contrarily, the urine exosomes of patients with diabetic nephropathy demonstrate decreased mtDNA levels, associated with reduced mitochondrial metabolites and low renal mitochondrial protein compared to healthy controls92. Along the same lines, impaired renal mitochondrial biogenesis is associated with reduced mtDNA copy number in the kidney tissue of mice and rats with PKD63. Nevertheless, studies suggest that the role of urinary mtDNA level may be limited in certain etiologies of CKD. A prospective study of 102 non-diabetic CKD patients followed for 48 months found that urinary mtDNA levels have no significant association with the rate of renal function decline, but correlate with baseline renal function, proteinuria, and the severity of glomerulosclerosis and tubulointerstitial fibrosis93. Although higher mtDNA copy number in peripheral blood has been associated with the lower prevalence of microalbuminuria79, studies in patients with diabetic nephropathy reported a significant inverse correlation between urinary supernatant and intra-renal mtDNA levels, implying that intra-renal mitochondrial loss may result in increased urinary mtDNA levels89.
Most studies assessed mtDNA levels by quantitative real-time PCR (qPCR), but genotyping microarray probe intensities and DNA sequencing read counts, such as whole genome and whole exome sequence may be more accurate and reliable methods for detecting mtDNA levels94. Analysis of somatic mtDNA mutations may also assist in developing early detection and monitoring strategies for patients with renal urothelial cell carcinoma95. Collectively, these observations suggest that although urinary mtDNA levels might serve as surrogate markers of permanent renal damage, they should be interpreted with caution in several etiologies of CKD. No doubt future large prospective cohort studies are needed to explore the exact role of urinary and plasma mtDNA in renal mitochondrial damage in the development and progression of CKD.
Mechanisms of renal mitochondrial injury - mitomiRs
Notwithstanding the evidence implicating mitochondrial injury in the pathogenesis of CKD, the precise mechanisms responsible for CKD-induced renal mitochondrial damage are incompletely understood. Deciphering these mechanisms is fundamental to develop novel therapies to preserve renal mitochondrial morphology and function and decrease the progression towards ESRD.
Several studies in humans and animal models have postulated miRNAs as master regulators of renal gene expression implicated in the pathogenesis of AKI and CKD96, 97. These small (19–23 nucleotides), endogenous, single-stranded noncoding RNAs regulate expression of protein-coding genes by repressing mRNA translation or promoting its degradation98. There are currently over 2,000 human miRNAs known. MiRNA biosynthesis encompases several enzymatic steps in both the nucleus and cytoplasm99, 100 (Figure 6). MiRNA genes are transcribed in the nucleus by the RNA polymerase II (Pol II) which produces primary miRNAs (pri-miRNAs), which are then modified by the RNAse III class enzyme, Drosha, to form precursor-miRNAs (pre-miRNAs)101. Pre-miRNAs are then exported from the nucleus to the cytoplasm by exportin 5 (EXP5) and subsequently processed to produce mature miRNAs102. Although most mature miRNAs are present in the cytosol, studies have revealed the presence of few miRNAs, known as ‘mitomiRs’, in the mitochondrion29, 103, 104.
Figure. 6. mitomiR biosynthesis.
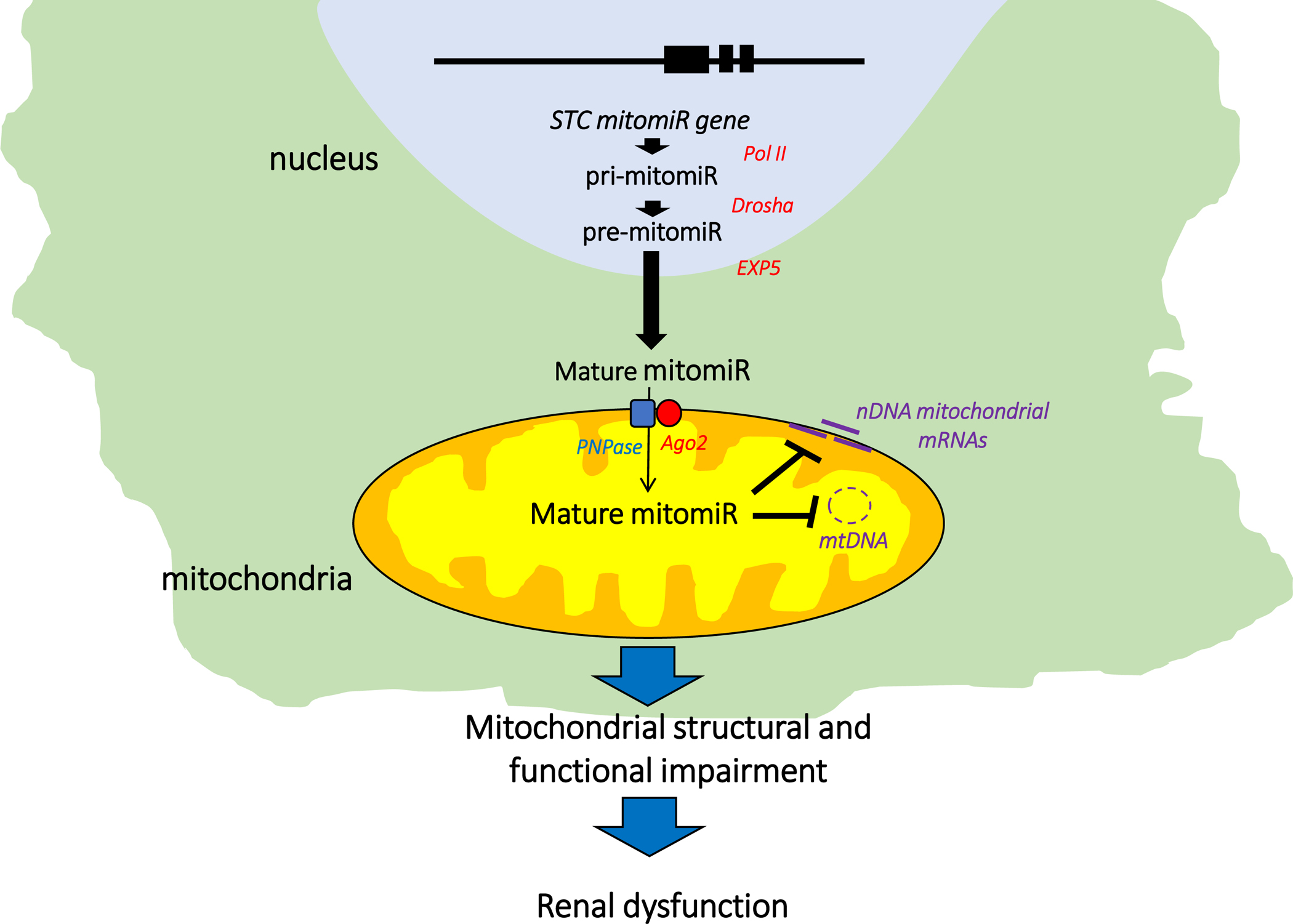
mitomiRs are transcribed in the nucleus by the RNA polymerase II (Pol II) which produces primary miRNAs (pri-mitomiRs), which are then modified by the RNAse III class enzyme, Drosha, to form precursor-miRNAs (pre-mitomiRs). Pre-mitomiRs are exported to the cytoplasm by exportin 5 (EXP5) and subsequently processed to produce mature mitomiRs, which will be subsequently imported into the organelle by the polynucleotide phosphorylase (PNPase) and RNA-induced silencing complex protein Argonaute 2 (Ago2) to target mtDNA genes or induce post-transcriptional repression of nuclear-encoded mitochondrial genes on the mitochondrial surface.
mitomiRs can either bind to and repress mRNA expression in the cytoplasm or being imported into mitochondria to target mtDNA genes or induce post-transcriptional repression of nuclear-encoded mitochondrial genes on the mitochondrial surface105. The later are imported by the polynucleotide phosphorylase (PNPase) situated in the inner mitochondrial membrane and intermembrane space106. PNPase interacts with the RNA-induced silencing complex protein Argonaute 2 (Ago2) to shuttle mitomiRs from the cytosol into the mitochondrion. Therefore, expression and localization of PNPase and Ago2 modulate mitochondrial translocation of mitomiRs, ultimately regulating mitochondrial gene expression and function.
Recent studies suggest that regulation of mitochondrial function by mitomiRs could contribute to renal disease progression in CKD (Table 2). Studies in mice with unilateral ureteral obstruction have shown that the mitomiR miR-30e, which targets UCP2, has an important role in mediating transforming growth factor (TGF)-β1-induced epithelial-mesenchymal transition and kidney fibrosis107. Importantly, this mitomiR can also target the translocase of inner mitochondrial membrane-22 (TIMM22) and the mitochondrial inner membrane organizing system-1 (MINOS1), which maintains crista junctions, inner membrane architecture, and formation of contact sites to the outer membrane. Other studies have shown that upregulation of miR-21 in tubular epithelial cells in response to TGF-β promotes progression of renal fibrosis in established obstructive nephropathy108. This mitomiR modulates mitochondrial-mediated apoptosis by altering the BAX/BCL2 ratio and mitochondrial fission, which in turn alter mitochondrial membrane potential, cytochrome-c release, and caspase activity109.
Table 2.
mitomiRs commonly implicated in CKD
| mitomiR | Disease/Model | Main mitochondrial target genes | References |
|---|---|---|---|
|
| |||
| miR-30e | • Unilateral ureteral obstruction | • UCP2, UCP3, MINOS1, GOT2, TIMM22 | • Jiang L, et al107 |
| miR-21 | • Obstructive nephropathy | • BAX, BCL2, MRPL49, MRPL45, CMC1 | • Zhong X, et al108 |
| miR-27a | • Diabetic nephropathy | • ATPAF1, TIMM10, CLPP, ATP5G3, SLC25A25, TOMM40L, GPD2, MICU3, MRPS14, SLC25A16 | • Hou X, et al110 |
| miR-29 | • Diabetic nephropathy | • MCL1, CPS1, GPAM, GRPEL2, ATP5G3, CLPX, DIABLO, SLC25A22, TSFM, MIEF1, SLC25A29 | • Du B, et al111 |
| miR-17 | • ADPKD | • PPARA, GPD2, TIMM8B, CLUH, MALSU1 | • Hajarnis S, et al60 |
| miR-335, miR-34a | • Aging | • SOD2, TXNRD2, PTPMT1, TACO1, ATP5S, MRPL3, MRPL52 | • Bai Xy, et al117 |
| miR-15a, miR-181a, miR-196a, miR-296-3p | • RVD | • ND2, ND4, ND4L, ND5, ATP6, GLS2, TCAIM, SLC25A22, SLC25A37, UCP2, AIFM1, MINOS1, MTFR1L, CLUH, TOMM20, MALSU1, SLC25A37, SLC25A4, GOT2, GPD2, SLC25A24, SLC25A25, TSFM, MCUR1, CMPK2, CLUH, TFAM, IARS2, GRPEL2, MARS2, MIEF1, SLC25A22, MRRF | • Farahani RA, et al27 |
| miR-15a, miR-137, miR-181c, miR-196a miR-296-5p | • MetS | • SLC25A5, ATPAF1, MPC1, GPD2, AIFM2, GPAM, GLS2, TCAIM, SLC25A22, SLC25A37, UCP2, AIFM1, MINOS1, MTFR1L, CLUH, TOMM20, MALSU1, GRPEL2, MARS2, MIEF1, SLC25A22, MRRF, TSFM, SLC25A23 | • Farahani RA, et al28, Aghajani Nargesi A, et al120 |
| miR-196a | • FSGS | • MIEF1, GRPEL2, MARS2, MIEF1, SLC25A22, MRRF | • Zhang W, et al121 |
| miR-15a | • Membranous nephropathy | • TFAM, GLS2, TCAIM, SLC25A22, SLC25A37, UCP2, AIFM1, MINOS1, MTFR1L, CLUH, TOMM20, MALSU1 | • Chen W, et al122 |
ADPKD: Autosomal dominant polycystic kidney disease; RVD: Renovascular disease; MetS: Metabolic syndrome; FSGS: Focal segmental glomeruslosclerosis.
Similarly, miR-27a promotes renal tubulointerstitial fibrosis via suppressing peroxisome proliferator-activated receptor pathway in streptozotocin-induced diabetic rats110. In addition, miR-29, which targets the mitochondrial Apoptosis Regulator, BCL2 Family Member (MCL1), modulates the production of collagen IV in proximal tubular cells exposed to high glucose111. MCL1 is a critical modulator of the mitochondrial fusion and fission machinery112. Therefore, miR-29 may represent a mechanism for regulating mitochondrial dynamics and intracellular matrix components in proximal tubular cells during the progression of diabetic nephropathy.
The mitomiR miR-17 is induced in kidney cysts of mouse and human ADPKD, and its genetic deletion inhibits cyst proliferation and disease progression in several ADPKD mouse models irrespective of the mutated gene (Pkd1 or Pkd2)60. These observations have important clinical implications as miR-17 inhibition also suppresses proliferation and growth of primary ADPKD cysts cultures derived from human donors. Mechanistic studies revealed that the deleterious effect of mitomiR miR-17 was in part mediated by inhibition of mitochondrial fatty acid oxidation and oxidative phosphorylation through direct repression of peroxisome proliferator-activated receptor-(PPAR)-α, one of the top downregulated genes in human ADPKD113. Therefore, miRNA-based approaches that specifically target this mitomiR may provide hope for therapies to attenuate disease progression in ADPKD.
Intriguing insights into the contribution of mitomiRs to renal mitochondrial damage may be gleaned from studies assessing cellular senescence, a state of stable and irreversible cell cycle arrest that plays an important role in the pathogenesis of different forms of renal damage, including CKD114. Senescent cells contain dysfunctional mitochondria, which play a major role in the promotion of the senescence-associated secretory phenotype115. Recent studies suggest that mitomiRs may influence the energetic, oxidative, and Inflammatory status of senescent cells by translocating to mitochondria and targeting numerous mRNAs encoding for proteins implicated in vital mitochondrial functions116. Studies in rats have shown that miR-335 and miR-34a contribute to renal aging by inhibiting intracellular pathways such as those involving the mitochondrial antioxidative enzymes SOD-2 and thioredoxin reductase-2117. Our group has recently shown that experimental RVD may induce senescence in endogenous STCs and impair their in vivo reparative capacity118. Moreover, we found that swine RVD-STCs exhibited increased expression of the mitomiRs miR-15a, miR-181a, miR-196a, and miR-296-3p, which targeted and reduced the expression of several mtDNA genes implicated in oxidative phosphorylation, such as the complex I genes ND2, ND4, ND4L, and ND5, and the complex V gene ATP627.
Similar findings can be noted in pig MSCs exposed a high fat and high fructose diet for 16 weeks, a model that develops many features of clinical MetS119. We found that expression of several mitomiRs (miR-15a, miR-137, and miR-181c) was higher in MetS-MSCs compared to Lean-MSCs, which modulate expression of genes encoding for mitochondrial proteins primarily implicated in energy pathways and mitochondrial dynamics28. Interestingly mitochondrial fusion and ATP production were impaired in MetS-MSCs compared to their lean counterparts, suggesting that MetS-induced post-transcriptional regulation of mitochondrial genes might have accounted for mitochondrial damage in MSCs. Likewise, increased expression of the mitomiRs miR-196a and miR-296-5p in MetS-MSCs interfered with mitochondrial protein import and impaired mitochondrial homeostasis and energy production, highlighting the important role of mitomiRs in mitochondrial damage in this endogenous repair mechanism120.
mitomiRs have been also implicated in the pathogenesis of glomerulopathies, including focal segmental glomerulosclerosis (FSGS) and membranous nephropathy. For example, miR-196a, which targets the mitochondrial fission protein mitochondrial elongation factor-1 (MIEF1), is elevated in the urine of patients with FSGS121, whereas miR-15a, which targets the transcription factor A mitochondrial (TFAM) that promotes mitochondrial DNA replication and repair, is upregulated in peripheral blood lymphocyte cells from patients with membranous nephropathy122. Collectively, these observations suggest that dysregulation of mitomiRs capable of compromising critical functions of mitochondria might be implicated in the pathogenesis of several forms of CKD. However, further mechanistic studies using miRNA mimics and antagomiRs are needed to establish a cause-effect relationship between mitomiRs and renal mitochondrial damage in CKD.
Summary and future perspectives
There is considerable evidence suggesting that dysfunctional mitochondria may be implicated in the development and progression of renal damage in common causes of CKD. While the effect of mitochondrial dysfunction in the pathogenesis of CKD has been studied extensively, most compelling evidence were generated in preclinical studies as renal biopsies are not routinely performed in many patients with CKD. Although great progress was made in understanding the role of urinary mtDNA as non-invasive markers of renal mitochondrial damage, contradicting findings emphasize the need of additional studies to elucidate the biological mechanisms and exact role of mtDNA in the pathogenesis of CKD. miRNA localizing in mitochondria (mitomiRs) may contribute to CKD-induced mitochondrial damage by post-transcriptional regulation of mtDNA and nuclear-encoded gene expression related to mitochondrial functions. It is therefore hopeful that continued studies on the precise mechanism of mitomiRs in mitochondrial injury will advance our understanding of their role in renal damage and contribute to develop novel therapeutic strategies for patients with chronic renal injury.
Acknowledgments
This work was supported by the NIH grants: DK129240, DK104273, DK118391, DK128017, GM104357, HL095638, the PKD Foundation (PKDF243G20a), and Regenerative Medicine Minnesota (RMM 091620 DS 004).
Footnotes
Conflict of Interest
None.
References
- 1.Saran R, Robinson B, Abbott KC, Agodoa LY, Albertus P, Ayanian J, Balkrishnan R, Bragg-Gresham J, Cao J, Chen JL, Cope E, Dharmarajan S, Dietrich X, Eckard A, Eggers PW, Gaber C, Gillen D, Gipson D, Gu H, Hailpern SM, Hall YN, Han Y, He K, Hebert H, Helmuth M, Herman W, Heung M, Hutton D, Jacobsen SJ, Ji N, Jin Y, Kalantar-Zadeh K, Kapke A, Katz R, Kovesdy CP, Kurtz V, Lavalee D, Li Y, Lu Y, McCullough K, Molnar MZ, Montez-Rath M, Morgenstern H, Mu Q, Mukhopadhyay P, Nallamothu B, Nguyen DV, Norris KC, O’Hare AM, Obi Y, Pearson J, Pisoni R, Plattner B, Port FK, Potukuchi P, Rao P, Ratkowiak K, Ravel V, Ray D, Rhee CM, Schaubel DE, Selewski DT, Shaw S, Shi J, Shieu M, Sim JJ, Song P, Soohoo M, Steffick D, Streja E, Tamura MK, Tentori F, Tilea A, Tong L, Turf M, Wang D, Wang M, Woodside K, Wyncott A, Xin X, Zang W, Zepel L, Zhang S, Zho H, Hirth RA and Shahinian V. US Renal Data System 2016 Annual Data Report: Epidemiology of Kidney Disease in the United States. Am J Kidney Dis. 2017;69:A7–A8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall YN. Racial and ethnic disparities in end stage renal disease: access failure. Clin J Am Soc Nephrol. 2012;7:196–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levey AS, Coresh J, Balk E, Kausz AT, Levin A, Steffes MW, Hogg RJ, Perrone RD, Lau J, Eknoyan G and National Kidney F. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med. 2003;139:137–47. [DOI] [PubMed] [Google Scholar]
- 4.Hoerger TJ, Simpson SA, Yarnoff BO, Pavkov ME, Rios Burrows N, Saydah SH, Williams DE and Zhuo X. The future burden of CKD in the United States: a simulation model for the CDC CKD Initiative. Am J Kidney Dis. 2015;65:403–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Navaneethan SD, Schold JD, Kirwan JP, Arrigain S, Jolly SE, Poggio ED, Beddhu S and Nally JV Jr. Metabolic syndrome, ESRD, and death in CKD. Clin J Am Soc Nephrol. 2013;8:945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng HT, Huang JW, Chiang CK, Yen CJ, Hung KY and Wu KD. Metabolic syndrome and insulin resistance as risk factors for development of chronic kidney disease and rapid decline in renal function in elderly. J Clin Endocrinol Metab. 2012;97:1268–76. [DOI] [PubMed] [Google Scholar]
- 7.Torres VE, Harris PC and Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. [DOI] [PubMed] [Google Scholar]
- 8.Webster AC, Nagler EV, Morton RL and Masson P. Chronic Kidney Disease. Lancet. 2017;389:1238–1252. [DOI] [PubMed] [Google Scholar]
- 9.Honeycutt AA, Segel JE, Zhuo X, Hoerger TJ, Imai K and Williams D. Medical costs of CKD in the Medicare population. J Am Soc Nephrol. 2013;24:1478–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nguyen NTQ, Cockwell P, Maxwell AP, Griffin M, O’Brien T and O’Neill C. Chronic kidney disease, health-related quality of life and their associated economic burden among a nationally representative sample of community dwelling adults in England. PLoS One. 2018;13:e0207960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez-Bedat M, Rosa-Diez G, Pecoits-Filho R, Ferreiro A, Garcia-Garcia G, Cusumano A, Fernandez-Cean J, Noboa O and Douthat W. Burden of disease: prevalence and incidence of ESRD in Latin America. Clin Nephrol. 2015;83:3–6. [DOI] [PubMed] [Google Scholar]
- 12.Escoli R, Carvalho MJ, Cabrita A and Rodrigues A. Diastolic Dysfunction, an Underestimated New Challenge in Dialysis. Ther Apher Dial. 2019;23:108–117. [DOI] [PubMed] [Google Scholar]
- 13.Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C, van Heerebeek L, Hillege HL, Lam CS, Navis G and Voors AA. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail. 2016;18:588–98. [DOI] [PubMed] [Google Scholar]
- 14.Hallan SI, Matsushita K, Sang Y, Mahmoodi BK, Black C, Ishani A, Kleefstra N, Naimark D, Roderick P, Tonelli M, Wetzels JF, Astor BC, Gansevoort RT, Levin A, Wen CP, Coresh J and Chronic Kidney Disease Prognosis C. Age and association of kidney measures with mortality and end-stage renal disease. JAMA. 2012;308:2349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gansevoort RT, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J and Chronic Kidney Disease Prognosis C. Lower estimated GFR and higher albuminuria are associated with adverse kidney outcomes. A collaborative meta-analysis of general and high-risk population cohorts. Kidney Int. 2011;80:93–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chronic Kidney Disease Prognosis C, Matsushita K, van der Velde M, Astor BC, Woodward M, Levey AS, de Jong PE, Coresh J and Gansevoort RT. Association of estimated glomerular filtration rate and albuminuria with all-cause and cardiovascular mortality in general population cohorts: a collaborative meta-analysis. Lancet. 2010;375:2073–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, Jong PE, Coresh J, Chronic Kidney Disease Prognosis C, Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, de Jong PE, Coresh J, El-Nahas M, Eckardt KU, Kasiske BL, Wright J, Appel L, Greene T, Levin A, Djurdjev O, Wheeler DC, Landray MJ, Townend JN, Emberson J, Clark LE, Macleod A, Marks A, Ali T, Fluck N, Prescott G, Smith DH, Weinstein JR, Johnson ES, Thorp ML, Wetzels JF, Blankestijn PJ, van Zuilen AD, Menon V, Sarnak M, Beck G, Kronenberg F, Kollerits B, Froissart M, Stengel B, Metzger M, Remuzzi G, Ruggenenti P, Perna A, Heerspink HJ, Brenner B, de Zeeuw D, Rossing P, Parving HH, Auguste P, Veldhuis K, Wang Y, Camarata L, Thomas B and Manley T. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int. 2011;79:1331–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carney EF. The impact of chronic kidney disease on global health. Nat Rev Nephrol. 2020;16:251. [DOI] [PubMed] [Google Scholar]
- 19.Pfaller W and Rittinger M. Quantitative morphology of the rat kidney. Int J Biochem. 1980;12:17–22. [DOI] [PubMed] [Google Scholar]
- 20.McFarland R, Taylor RW and Turnbull DM. Mitochondrial disease--its impact, etiology, and pathology. Curr Top Dev Biol. 2007;77:113–55. [DOI] [PubMed] [Google Scholar]
- 21.Andres-Hernando A, Lanaspa MA, Kuwabara M, Orlicky DJ, Cicerchi C, Bales E, Garcia GE, Roncal-Jimenez CA, Sato Y and Johnson RJ. Obesity causes renal mitochondrial dysfunction and energy imbalance and accelerates chronic kidney disease in mice. Am J Physiol Renal Physiol. 2019;317:F941–F948. [DOI] [PubMed] [Google Scholar]
- 22.Nargesi AA, Zhang L, Tang H, Jordan KL, Saadiq IM, Textor SC, Lerman LO and Eirin A. Coexisting renal artery stenosis and metabolic syndrome magnifies mitochondrial damage, aggravating poststenotic kidney injury in pigs. J Hypertens. 2019;37:2061–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahveci AS, Barnatan TT, Kahveci A, Adrian AE, Arroyo J, Eirin A, Harris PC, Lerman A, Lerman LO, Torres VE and Irazabal MV. Oxidative Stress and Mitochondrial Abnormalities Contribute to Decreased Endothelial Nitric Oxide Synthase Expression and Renal Disease Progression in Early Experimental Polycystic Kidney Disease. Int J Mol Sci. 2020;21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Seo M, Kim H, Noh H, Jeon JS, Byun DW, Kim SH, Kim HJ, Suh K, Park HK and Kwon SH. Effect of bariatric surgery on circulating and urinary mitochondrial DNA copy numbers in obesity with or without diabetes. BMJ Open Diabetes Res Care. 2020;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eirin A, Herrmann SM, Saad A, Abumoawad A, Tang H, Lerman A, Textor SC and Lerman LO. Urinary mitochondrial DNA copy number identifies renal mitochondrial injury in renovascular hypertensive patients undergoing renal revascularization: A Pilot Study. Acta Physiol (Oxf). 2019;226:e13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eirin A, Saad A, Woollard JR, Juncos LA, Calhoun DA, Tang H, Lerman A, Textor SC and Lerman LO. Glomerular Hyperfiltration in Obese African American Hypertensive Patients Is Associated With Elevated Urinary Mitochondrial-DNA Copy Number. Am J Hypertens. 2017;30:1112–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farahani RA, Zhu XY, Tang H, Jordan KL, Lerman LO and Eirin A. Renal ischemia alters expression of mitochondria-related genes and impairs mitochondrial structure and function in swine scattered tubular-like cells. Am J Physiol Renal Physiol. 2020;319:F19–F28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farahani RA, Farah MC, Zhu XY, Tang H, Saadiq IM, Lerman LO and Eirin A. Metabolic Syndrome Impairs 3D Mitochondrial Structure, Dynamics, and Function in Swine Mesenchymal Stem Cells. Stem Cell Rev Rep. 2020. [DOI] [PubMed] [Google Scholar]
- 29.Duarte FV, Palmeira CM and Rolo AP. The Emerging Role of MitomiRs in the Pathophysiology of Human Disease. Adv Exp Med Biol. 2015;888:123–54. [DOI] [PubMed] [Google Scholar]
- 30.Barth E, Stammler G, Speiser B and Schaper J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol. 1992;24:669–81. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Reyes I and Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW 2nd, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ, American Heart Association Council on Basic Cardiovascular Sciences CoCC, Council on Functional G and Translational B. Mitochondrial Function, Biology, and Role in Disease: A Scientific Statement From the American Heart Association. Circ Res. 2016;118:1960–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang C and Youle RJ. The role of mitochondria in apoptosis*. Annu Rev Genet. 2009;43:95–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Handy DE and Loscalzo J. Redox regulation of mitochondrial function. Antioxid Redox Signal. 2012;16:1323–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ramzan F, D’Souza RF, Durainayagam BR, Milan AM, Markworth JF, Miranda-Soberanis V, Sequeira IR, Roy NC, Poppitt SD, Mitchell CJ and Cameron-Smith D. Circulatory miRNA biomarkers of metabolic syndrome. Acta Diabetol. 2020;57:203–214. [DOI] [PubMed] [Google Scholar]
- 36.Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y and Dong W. ROS and ROS-Mediated Cellular Signaling. Oxid Med Cell Longev. 2016;2016:4350965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang PN, Zhou MQ, Guo J, Zheng HJ, Tang J, Zhang C, Liu YN, Liu WJ and Wang YX. Mitochondrial Dysfunction and Diabetic Nephropathy: Nontraditional Therapeutic Opportunities. J Diabetes Res. 2021;2021:1010268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eirin A, Lerman A and Lerman LO. Mitochondria: a pathogenic paradigm in hypertensive renal disease. Hypertension. 2015;65:264–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K and Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58:1201–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koziel R, Pircher H, Kratochwil M, Lener B, Hermann M, Dencher NA and Jansen-Durr P. Mitochondrial respiratory chain complex I is inactivated by NADPH oxidase Nox4. Biochem J. 2013;452:231–9. [DOI] [PubMed] [Google Scholar]
- 41.Papadimitriou A, Peixoto EB, Silva KC, Lopes de Faria JM and Lopes de Faria JB. Inactivation of AMPK mediates high phosphate-induced extracellular matrix accumulation via NOX4/TGFss-1 signaling in human mesangial cells. Cell Physiol Biochem. 2014;34:1260–72. [DOI] [PubMed] [Google Scholar]
- 42.Eid AA, Lee DY, Roman LJ, Khazim K and Gorin Y. Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent endothelial nitric oxide synthase uncoupling and matrix protein expression. Mol Cell Biol. 2013;33:3439–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Susztak K, Raff AC, Schiffer M and Bottinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55:225–33. [PubMed] [Google Scholar]
- 44.Domondon M, Polina I, Nikiforova AB, Sultanova RF, Kruger C, Vasileva VY, Fomin MV, Beeson GC, Nieminen AL, Smythe N, Maldonado EN, Stadler K and Ilatovskaya DV. Renal Glomerular Mitochondria Function in Salt-Sensitive Hypertension. Front Physiol. 2019;10:1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de Cavanagh EM, Toblli JE, Ferder L, Piotrkowski B, Stella I and Inserra F. Renal mitochondrial dysfunction in spontaneously hypertensive rats is attenuated by losartan but not by amlodipine. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1616–25. [DOI] [PubMed] [Google Scholar]
- 46.Tian XY, Ma S, Tse G, Wong WT and Huang Y. Uncoupling Protein 2 in Cardiovascular Health and Disease. Front Physiol. 2018;9:1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jin K and Vaziri ND. Salt-sensitive hypertension in mitochondrial superoxide dismutase deficiency is associated with intra-renal oxidative stress and inflammation. Clin Exp Nephrol. 2014;18:445–52. [DOI] [PubMed] [Google Scholar]
- 48.Zheleznova NN, Yang C, Ryan RP, Halligan BD, Liang M, Greene AS and Cowley AW Jr. Mitochondrial proteomic analysis reveals deficiencies in oxygen utilization in medullary thick ascending limb of Henle in the Dahl salt-sensitive rat. Physiol Genomics. 2012;44:829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonnard C, Durand A, Peyrol S, Chanseaume E, Chauvin MA, Morio B, Vidal H and Rieusset J. Mitochondrial dysfunction results from oxidative stress in the skeletal muscle of diet-induced insulin-resistant mice. J Clin Invest. 2008;118:789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Takabe W, Li R, Ai L, Yu F, Berliner JA and Hsiai TK. Oxidized low-density lipoprotein-activated c-Jun NH2-terminal kinase regulates manganese superoxide dismutase ubiquitination: implication for mitochondrial redox status and apoptosis. Arterioscler Thromb Vasc Biol. 2010;30:436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szeto HH, Liu S, Soong Y, Alam N, Prusky GT and Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int. 2016;90:997–1011. [DOI] [PubMed] [Google Scholar]
- 52.Wojtczak L and Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta. 1993;1183:41–57. [DOI] [PubMed] [Google Scholar]
- 53.Eirin A, Woollard JR, Ferguson CM, Jordan KL, Tang H, Textor SC, Lerman A and Lerman LO. The metabolic syndrome induces early changes in the swine renal medullary mitochondria. Transl Res. 2017;184:45–56 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171:2029–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilson PD, Schrier RW, Breckon RD and Gabow PA. A new method for studying human polycystic kidney disease epithelia in culture. Kidney Int. 1986;30:371–8. [DOI] [PubMed] [Google Scholar]
- 56.Thomson RB, Mentone S, Kim R, Earle K, Delpire E, Somlo S and Aronson PS. Histopathological analysis of renal cystic epithelia in the Pkd2WS25/- mouse model of ADPKD. Am J Physiol Renal Physiol. 2003;285:F870–80. [DOI] [PubMed] [Google Scholar]
- 57.Mahajan N, Kaur T, Puri V, Singla SK, Jha V and Puri S. Calcium ameliorates renal cyst growth in metanephric organ culture: a morphological study. J Environ Pathol Toxicol Oncol. 2012;31:285–93. [DOI] [PubMed] [Google Scholar]
- 58.Liu Y, Dai B, Mei C, Zhang Y, Xiong X and Sandford R. Identification of phosphoproteins in kidney tissues from patients with autosomal dominant polycystic kidney disease. Proteomics Clin Appl. 2008;2:1153–66. [DOI] [PubMed] [Google Scholar]
- 59.Li QW, Lu XY, You Y, Sun H, Liu XY, Ai JZ, Tan RZ, Chen TL, Chen MZ, Wang HL, Wei YQ and Zhou Q. Comparative proteomic analysis suggests that mitochondria are involved in autosomal recessive polycystic kidney disease. Proteomics. 2012;12:2556–70. [DOI] [PubMed] [Google Scholar]
- 60.Hajarnis S, Lakhia R, Yheskel M, Williams D, Sorourian M, Liu X, Aboudehen K, Zhang S, Kersjes K, Galasso R, Li J, Kaimal V, Lockton S, Davis S, Flaten A, Johnson JA, Holland WL, Kusminski CM, Scherer PE, Harris PC, Trudel M, Wallace DP, Igarashi P, Lee EC, Androsavich JR and Patel V. microRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat Commun. 2017;8:14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin CC, Kurashige M, Liu Y, Terabayashi T, Ishimoto Y, Wang T, Choudhary V, Hobbs R, Liu LK, Lee PH, Outeda P, Zhou F, Restifo NP, Watnick T, Kawano H, Horie S, Prinz W, Xu H, Menezes LF and Germino GG. A cleavage product of Polycystin-1 is a mitochondrial matrix protein that affects mitochondria morphology and function when heterologously expressed. Sci Rep. 2018;8:2743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Padovano V, Kuo IY, Stavola LK, Aerni HR, Flaherty BJ, Chapin HC, Ma M, Somlo S, Boletta A, Ehrlich BE, Rinehart J and Caplan MJ. The polycystins are modulated by cellular oxygen-sensing pathways and regulate mitochondrial function. Mol Biol Cell. 2017;28:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ishimoto Y, Inagi R, Yoshihara D, Kugita M, Nagao S, Shimizu A, Takeda N, Wake M, Honda K, Zhou J and Nangaku M. Mitochondrial Abnormality Facilitates Cyst Formation in Autosomal Dominant Polycystic Kidney Disease. Mol Cell Biol. 2017;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maser RL, Vassmer D, Magenheimer BS and Calvet JP. Oxidant stress and reduced antioxidant enzyme protection in polycystic kidney disease. J Am Soc Nephrol. 2002;13:991–999. [DOI] [PubMed] [Google Scholar]
- 65.Menon V, Rudym D, Chandra P, Miskulin D, Perrone R and Sarnak M. Inflammation, oxidative stress, and insulin resistance in polycystic kidney disease. Clin J Am Soc Nephrol. 2011;6:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fedorova LV, Sodhi K, Gatto-Weis C, Puri N, Hinds TD Jr., Shapiro JI and Malhotra D. Peroxisome proliferator-activated receptor delta agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS One. 2013;8:e64436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dengjel J and Abeliovich H. Musical chairs during mitophagy. Autophagy. 2014;10:706–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eirin A, Ebrahimi B, Zhang X, Zhu XY, Woollard JR, He Q, Textor SC, Lerman A and Lerman LO. Mitochondrial protection restores renal function in swine atherosclerotic renovascular disease. Cardiovasc Res. 2014;103:461–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Grote K, Flach I, Luchtefeld M, Akin E, Holland SM, Drexler H and Schieffer B. Mechanical stretch enhances mRNA expression and proenzyme release of matrix metalloproteinase-2 (MMP-2) via NAD(P)H oxidase-derived reactive oxygen species. Circ Res. 2003;92:e80–6. [DOI] [PubMed] [Google Scholar]
- 70.Odenbach J, Wang X, Cooper S, Chow FL, Oka T, Lopaschuk G, Kassiri Z and Fernandez-Patron C. MMP-2 mediates angiotensin II-induced hypertension under the transcriptional control of MMP-7 and TACE. Hypertension. 2011;57:123–30. [DOI] [PubMed] [Google Scholar]
- 71.Abadir PM, Foster DB, Crow M, Cooke CA, Rucker JJ, Jain A, Smith BJ, Burks TN, Cohn RD, Fedarko NS, Carey RM, O’Rourke B and Walston JD. Identification and characterization of a functional mitochondrial angiotensin system. Proc Natl Acad Sci U S A. 2011;108:14849–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Doughan AK, Harrison DG and Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. 2008;102:488–96. [DOI] [PubMed] [Google Scholar]
- 73.Farahani RA, Zhu XY, Tang H, Jordan KL, Lerman A, Lerman LO and Eirin A. Metabolic Syndrome Alters the Cargo of Mitochondria-Related microRNAs in Swine Mesenchymal Stem Cell-Derived Extracellular Vesicles, Impairing Their Capacity to Repair the Stenotic Kidney. Stem Cells Int. 2020;2020:8845635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nargesi AA, Zhu XY, Conley SM, Woollard JR, Saadiq IM, Lerman LO and Eirin A. Renovascular disease induces mitochondrial damage in swine scattered tubular cells. Am J Physiol Renal Physiol. 2019;317:F1142–F1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Smeets B, Boor P, Dijkman H, Sharma SV, Jirak P, Mooren F, Berger K, Bornemann J, Gelman IH, Floege J, van der Vlag J, Wetzels JF and Moeller MJ. Proximal tubular cells contain a phenotypically distinct, scattered cell population involved in tubular regeneration. J Pathol. 2013;229:645–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Greaves LC, Reeve AK, Taylor RW and Turnbull DM. Mitochondrial DNA and disease. J Pathol. 2012;226:274–86. [DOI] [PubMed] [Google Scholar]
- 77.Gomez-Cabrera MC, Sanchis-Gomar F, Garcia-Valles R, Pareja-Galeano H, Gambini J, Borras C and Vina J. Mitochondria as sources and targets of damage in cellular aging. Clin Chem Lab Med. 2012;50:1287–95. [DOI] [PubMed] [Google Scholar]
- 78.Wenceslau CF, McCarthy CG, Szasz T, Spitler K, Goulopoulou S, Webb RC and Working Group on DiCD. Mitochondrial damage-associated molecular patterns and vascular function. Eur Heart J. 2014;35:1172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lee JE, Park H, Ju YS, Kwak M, Kim JI, Oh HY and Seo JS. Higher mitochondrial DNA copy number is associated with lower prevalence of microalbuminuria. Exp Mol Med. 2009;41:253–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shen J, Platek M, Mahasneh A, Ambrosone CB and Zhao H. Mitochondrial copy number and risk of breast cancer: a pilot study. Mitochondrion. 2010;10:62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xing J, Chen M, Wood CG, Lin J, Spitz MR, Ma J, Amos CI, Shields PG, Benowitz NL, Gu J, de Andrade M, Swan GE and Wu X. Mitochondrial DNA content: its genetic heritability and association with renal cell carcinoma. J Natl Cancer Inst. 2008;100:1104–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chien MC, Huang WT, Wang PW, Liou CW, Lin TK, Hsieh CJ and Weng SW. Role of mitochondrial DNA variants and copy number in diabetic atherogenesis. Genet Mol Res. 2012;11:3339–48. [DOI] [PubMed] [Google Scholar]
- 83.Hu Q, Ren J, Ren H, Wu J, Wu X, Liu S, Wang G, Gu G, Guo K and Li J. Urinary Mitochondrial DNA Identifies Renal Dysfunction and Mitochondrial Damage in Sepsis-Induced Acute Kidney Injury. Oxid Med Cell Longev. 2018;2018:8074936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Whitaker RM, Stallons LJ, Kneff JE, Alge JL, Harmon JL, Rahn JJ, Arthur JM, Beeson CC, Chan SL and Schnellmann RG. Urinary mitochondrial DNA is a biomarker of mitochondrial disruption and renal dysfunction in acute kidney injury. Kidney Int. 2015;88:1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hu Q, Ren J, Wu J, Li G, Wu X, Liu S, Wang G, Gu G, Ren H, Hong Z and Li J. Urinary Mitochondrial DNA Levels Identify Acute Kidney Injury in Surgical Critical Illness Patients. Shock. 2017;48:11–17. [DOI] [PubMed] [Google Scholar]
- 86.Eirin A, Saad A, Tang H, Herrmann SM, Woollard JR, Lerman A, Textor SC and Lerman LO. Urinary Mitochondrial DNA Copy Number Identifies Chronic Renal Injury in Hypertensive Patients. Hypertension. 2016;68:401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yu BC, Cho NJ, Park S, Kim H, Choi SJ, Kim JK, Hwang SD, Gil HW, Lee EY, Jeon JS, Noh H, Han DC, Kim YH, Jin SY, Park MY and Kwon SH. IgA nephropathy is associated with elevated urinary mitochondrial DNA copy numbers. Sci Rep. 2019;9:16068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu BC, Cho NJ, Park S, Kim H, Gil HW, Lee EY, Kwon SH, Jeon JS, Noh H, Han DC, Moon A, Park SJ, Kim JK, Hwang SD, Choi SJ and Park MY. Minor Glomerular Abnormalities are Associated with Deterioration of Long-Term Kidney Function and Mitochondrial Injury. J Clin Med. 2019;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wei PZ, Kwan BC, Chow KM, Cheng PM, Luk CC, Li PK and Szeto CC. Urinary mitochondrial DNA level is an indicator of intra-renal mitochondrial depletion and renal scarring in diabetic nephropathy. Nephrol Dial Transplant. 2018;33:784–788. [DOI] [PubMed] [Google Scholar]
- 90.Surmiak MP, Hubalewska-Mazgaj M, Wawrzycka-Adamczyk K, Szczeklik W, Musial J and Sanak M. Circulating mitochondrial DNA in serum of patients with granulomatosis with polyangiitis. Clin Exp Immunol. 2015;181:150–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu SJ, Yang X, Xu PC, Chen T, Gao S, Hu SY, Wei L and Yan TK. Urinary mitochondrial DNA is a useful biomarker for assessing kidney injury of antineutrophil cytoplasmic antibody -associated vasculitis. Clin Chim Acta. 2020;502:263–268. [DOI] [PubMed] [Google Scholar]
- 92.Sharma K, Karl B, Mathew AV, Gangoiti JA, Wassel CL, Saito R, Pu M, Sharma S, You YH, Wang L, Diamond-Stanic M, Lindenmeyer MT, Forsblom C, Wu W, Ix JH, Ideker T, Kopp JB, Nigam SK, Cohen CD, Groop PH, Barshop BA, Natarajan L, Nyhan WL and Naviaux RK. Metabolomics reveals signature of mitochondrial dysfunction in diabetic kidney disease. J Am Soc Nephrol. 2013;24:1901–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wei PZ, Kwan BC, Chow KM, Cheng PM, Luk CC, Lai KB, Li PK and Szeto CC. Urinary mitochondrial DNA level in non-diabetic chronic kidney diseases. Clin Chim Acta. 2018;484:36–39. [DOI] [PubMed] [Google Scholar]
- 94.Longchamps RJ, Castellani CA, Yang SY, Newcomb CE, Sumpter JA, Lane J, Grove ML, Guallar E, Pankratz N, Taylor KD, Rotter JI, Boerwinkle E and Arking DE. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS One. 2020;15:e0228166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Dasgupta S, Shao C, Keane TE, Duberow DP, Mathies RA, Fisher PB, Kiemeney LA and Sidransky D. Detection of mitochondrial deoxyribonucleic acid alterations in urine from urothelial cell carcinoma patients. Int J Cancer. 2012;131:158–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ichii O and Horino T. MicroRNAs associated with the development of kidney diseases in humans and animals. J Toxicol Pathol. 2018;31:23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Iranzad R, Motavalli R, Ghassabi A, Pourakbari R, Etemadi J and Yousefi M. Roles of microRNAs in renal disorders related to primary podocyte dysfunction. Life Sci. 2021;277:119463. [DOI] [PubMed] [Google Scholar]
- 98.Tetreault N and De Guire V. miRNAs: their discovery, biogenesis and mechanism of action. Clin Biochem. 2013;46:842–5. [DOI] [PubMed] [Google Scholar]
- 99.Ha M and Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15:509–24. [DOI] [PubMed] [Google Scholar]
- 100.Lin S and Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer. 2015;15:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. [DOI] [PubMed] [Google Scholar]
- 102.O’Brien J, Hayder H, Zayed Y and Peng C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front Endocrinol (Lausanne). 2018;9:402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paramasivam A and Vijayashree Priyadharsini J. MitomiRs: new emerging microRNAs in mitochondrial dysfunction and cardiovascular disease. Hypertens Res. 2020;43:851–853. [DOI] [PubMed] [Google Scholar]
- 104.Rippo MR, Olivieri F, Monsurro V, Prattichizzo F, Albertini MC and Procopio AD. MitomiRs in human inflamm-aging: a hypothesis involving miR-181a, miR-34a and miR-146a. Exp Gerontol. 2014;56:154–63. [DOI] [PubMed] [Google Scholar]
- 105.Song R, Hu XQ and Zhang L. Mitochondrial MiRNA in Cardiovascular Function and Disease. Cells. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shepherd DL, Hathaway QA, Pinti MV, Nichols CE, Durr AJ, Sreekumar S, Hughes KM, Stine SM, Martinez I and Hollander JM. Exploring the mitochondrial microRNA import pathway through Polynucleotide Phosphorylase (PNPase). J Mol Cell Cardiol. 2017;110:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang L, Qiu W, Zhou Y, Wen P, Fang L, Cao H, Zen K, He W, Zhang C, Dai C and Yang J. A microRNA-30e/mitochondrial uncoupling protein 2 axis mediates TGF-beta1-induced tubular epithelial cell extracellular matrix production and kidney fibrosis. Kidney Int. 2013;84:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhong X, Chung AC, Chen HY, Meng XM and Lan HY. Smad3-mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22:1668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wu H, Wang J, Ma H, Xiao Z and Dong X. MicroRNA-21 inhibits mitochondria-mediated apoptosis in keloid. Oncotarget. 2017;8:92914–92925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hou X, Tian J, Geng J, Li X, Tang X, Zhang J and Bai X. MicroRNA-27a promotes renal tubulointerstitial fibrosis via suppressing PPARgamma pathway in diabetic nephropathy. Oncotarget. 2016;7:47760–47776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Du B, Ma LM, Huang MB, Zhou H, Huang HL, Shao P, Chen YQ and Qu LH. High glucose down-regulates miR-29a to increase collagen IV production in HK-2 cells. FEBS Lett. 2010;584:811–6. [DOI] [PubMed] [Google Scholar]
- 112.Morciano G, Giorgi C, Balestra D, Marchi S, Perrone D, Pinotti M and Pinton P. Mcl-1 involvement in mitochondrial dynamics is associated with apoptotic cell death. Mol Biol Cell. 2016;27:20–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND and Pei Y. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18:2328–43. [DOI] [PubMed] [Google Scholar]
- 114.Li Y and Lerman LO. Cellular Senescence: A New Player in Kidney Injury. Hypertension. 2020;76:1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly YM, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, von Zglinicki T, Korolchuk VI and Passos JF. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J. 2016;35:724–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Giuliani A, Prattichizzo F, Micolucci L, Ceriello A, Procopio AD and Rippo MR. Mitochondrial (Dys) Function in Inflammaging: Do MitomiRs Influence the Energetic, Oxidative, and Inflammatory Status of Senescent Cells? Mediators Inflamm. 2017;2017:2309034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bai XY, Ma Y, Ding R, Fu B, Shi S and Chen XM. miR-335 and miR-34a Promote renal senescence by suppressing mitochondrial antioxidative enzymes. J Am Soc Nephrol. 2011;22:1252–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chen XJ, Kim SR, Jiang K, Ferguson CM, Tang H, Zhu XY, Lerman A, Eirin A and Lerman LO. Renovascular Disease Induces Senescence in Renal Scattered Tubular-Like Cells and Impairs Their Reparative Potency. Hypertension. 2021;77:507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pawar AS, Zhu XY, Eirin A, Tang H, Jordan KL, Woollard JR, Lerman A and Lerman LO. Adipose tissue remodeling in a novel domestic porcine model of diet-induced obesity. Obesity (Silver Spring). 2015;23:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aghajani Nargesi A, Zhu XY, Hickson LJ, Conley SM, van Wijnen AJ, Lerman LO and Eirin A. Metabolic Syndrome Modulates Protein Import into the Mitochondria of Porcine Mesenchymal Stem Cells. Stem Cell Rev Rep. 2019;15:427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang W, Zhang C, Chen H, Li L, Tu Y, Liu C, Shi S, Zen K and Liu Z. Evaluation of microRNAs miR-196a, miR-30a-5P, and miR-490 as biomarkers of disease activity among patients with FSGS. Clin J Am Soc Nephrol. 2014;9:1545–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chen W, Lin X, Huang J, Tan K, Chen Y, Peng W, Li W and Dai Y. Integrated profiling of microRNA expression in membranous nephropathy using high-throughput sequencing technology. Int J Mol Med. 2014;33:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]