

The vertebrate body displays a segmental organization which is most conspicuous in the periodic organization of the vertebral column and peripheral nerves. This metameric organization is first implemented when somites, which contain the precursors of skeletal muscles and vertebrae, are rhythmically generated from the presomitic mesoderm (PSM). Somites then become subdivided into anterior and posterior compartments essential for vertebral formation and segmental patterning of the peripheral nervous system1–4. How this key somitic subdivision is established remains poorly understood. Here we introduce novel tridimensional culture systems of human pluripotent stem cells (PSCs), called Somitoids and Segmentoids, which recapitulate the formation of somite-like structures with antero-posterior (AP) identity. We identify a key function of the segmentation clock in converting temporal rhythmicity into the spatial regularity of anterior and posterior somitic compartments. We show that an initial salt-and-pepper expression of the segmentation gene MESP2 in the newly formed segment is transformed into compartments of anterior and posterior identity via an active cell sorting mechanism. Our work demonstrates that the major patterning modules involved in somitogenesis including the clock and wavefront, AP polarity patterning and somite epithelialization can be dissociated and operate independently in our in vitro systems. Together we define a novel framework for the symmetry breaking process initiating somite polarity patterning. Our work provides a valuable platform to decode general principles of somitogenesis and advance knowledge of human development.
Our peripheral nerves exhibit a striking periodic organization which coincides with that of vertebrae. This arrangement can be traced back to the original body segmentation resulting from somite formation. Somites, which form from the presomitic mesoderm (PSM), define the prepattern on which vertebral metamery is established1. They are repeatedly arrayed in two bilaterally symmetric columns which give rise to the skeletal muscles and axial skeleton. The PSM, which is initially mesenchymal in the posterior part of the embryo, becomes progressively epithelial as it matures. At its anterior tip, somites form rhythmically as epithelial blocks surrounding a mesenchymal core. The periodicity of somite formation involves a molecular oscillator called the segmentation clock1,5. This gene regulatory network controls the rhythmic activation of Notch, Wnt and FGF pathways which manifest as traveling waves of target gene expression in the posterior PSM. These periodic signals are interpreted at the level of the determination front, whose position is defined by posterior gradients of FGF and Wnt signaling in the PSM. This “Clock and Wavefron” mechanism eventually leads to the activation of the transcription factor MESP2 in a stripe which prefigures the future segment. MESP2 is next involved in the subdivision of forming somites into an anterior and posterior compartment6,7. This partition is critical for peripheral nervous system segmentation as the migration of neural crest cells and peripheral axons is initially restricted to the anterior somitic compartment8. It is also essential for vertebrae which form from the fusion of a posterior somite compartment with the anterior compartment of the next posterior somite3. The mechanism controlling the formation of these anterior and posterior somitic domains remains poorly understood.
Our understanding of vertebrate segmentation only relies on studies performed in model organisms such as mouse, chicken, and zebrafish embryos. Very little is known about human somitogenesis which takes place very early during pregnancy, between 3- and 4-weeks post-fertilization9. The recent development of in vitro systems recapitulating paraxial mesoderm development from pluripotent stem cells (PSCs) demonstrated a high degree of conservation of the gene regulatory networks involved in PSM patterning between mouse and human embryos10–15. Monolayers of human PSCs differentiating to a PSM fate in vitro recapitulate the posterior FGF and Wnt gradients and the oscillations of the segmentation clock with a ~5 hour period. However, a limitation of these 2D systems is that they do not allow examination of the morphogenesis of the tissues generated in vitro. In mouse, a striking recapitulation of all somitogenesis stages including epithelial somite and antero-posterior (AP) compartments formation has been achieved in 3D organoids that contain cells of all three germ layers16,17. Similar systems have begun to be introduced for human PSCs very recently suggesting that reproducing key aspects of segmentation such as the clock oscillations or somitic rosette formation is possible in vitro18,19. However, whether the mechanisms involved in somite formation and patterning described in embryos of other vertebrates are conserved in humans remains unclear.
Making somites in vitro
To study human somitogenesis, we set out to develop PSC-derived 3D culture systems. We first generated human iPSC spheroids in suspension, and then treated them with the Wnt agonist CHIR and the BMP inhibitor LDN for 48 hours to induce the PSM fate (Fig.1a). Subsequently we transferred the spheroids to a laminin coated substrate and used confocal microscopy to characterize gene expression dynamics as the spheroids spread out (Extended Data Fig.1a, Supplementary Video1). We used an iPS cell line harboring a destabilized Achilles (YFP) reporter at the HES7 locus to detect segmentation clock oscillations and a mCherry reporter at the MESP2 locus to monitor the onset of segmental determination10 (Extended Data Fig.1b). This line was engineered by introducing a t2A-mCherry construct by homologous recombination into the MESP2 locus and thus reports for MESP2 protein production10. mCherry is much more stable than MESP2 and thus it is retained for some time by cells that have previously expressed MESP2 after they stop transcribing the gene. Live imaging showed that HES7, a core component of the segmentation clock, starts to oscillate with a 4–5 hour period (Fig.1b,c, Extended Data Fig.1c) as the spheroids spread out. HES7 signals initiated from the peripheral region of the spreading organoid and propagated as concentric waves toward the center (Fig.1b, Extended Data Fig.1c, Supplementary Video2). After about 4 cycles of HES7 oscillations, between ~ 64 h and 72 h, expression of the reporter ceased and the MESP2 reporter became simultaneously expressed across spheroids (Fig.1b,c, Supplementary Video2). Thus, the onset of MESP2 immediately follows the arrest of HES7 oscillations. PAX3 is a transcription factor first expressed in the anterior PSM and epithelial somites20 soon after activation of MESP2. In the differentiating organoids of a PAX3-YFP reporter line, we observed the onset of PAX3 activation in all cells around 78 h (Fig.1c). At 90 h, numerous PAX3-positive somite-like epithelial rosettes started to emerge and they were visible under bright-field microscopy by 120 h (Fig.1d,e). These rosettes had a diameter of ~80 µm as in mouse embryos and published in vitro models17–19, and they did not scale with the overall Somitoid size (Fig.1e, Extended Data Fig.1d–f). They displayed typical somitic features such as enriched apical N-Cadherin and F-actin, a laminin-rich basal lamina, and a core region filled with mesenchymal cells (Fig.1f,g, Extended Data Fig.1g,h). Similar rosette formation was observed on gelatin-coated plates or in suspension culture (Extended Data Fig.1i,j). We performed RNAseq at 48 h, 66 h, and 120 h of the differentiation protocol, and observed the expression of signature genes associated with PSM, Determination Front, and somites respectively (Fig.1h, Extended Data Fig.1k, Supplementary Table 1). Therefore, these organoids, which we term “Somitoids”, successfully recapitulate the timely progression of gene expression from PSM to somites as well as major aspects of epithelial somite morphogenesis.
Fig.1. Characterization of the Somitoid model.
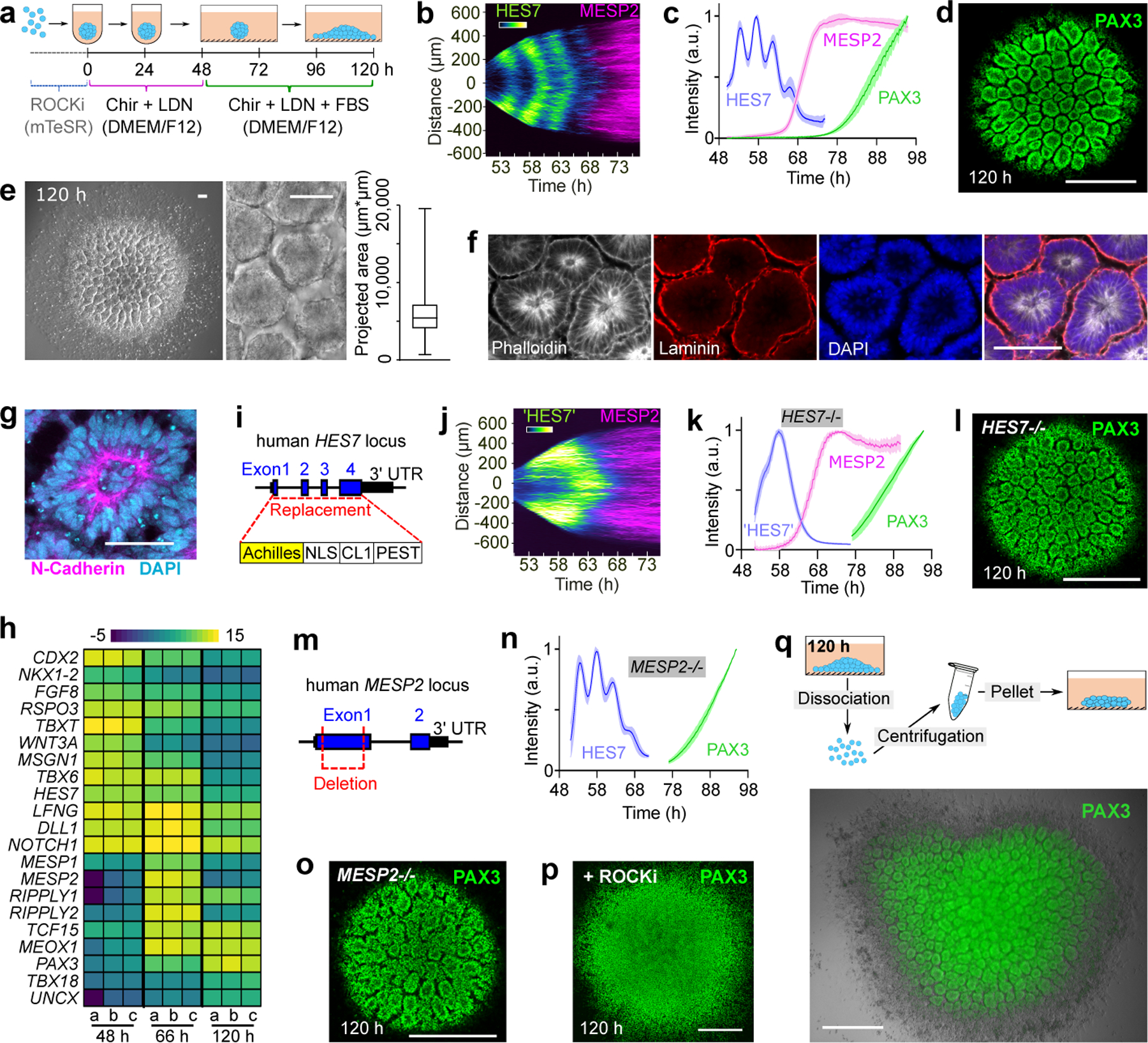
a, Protocol illustration. b, Reporter kymograph from a line scan across the center of Somitoid. c, Temporal profiles (mean±s.d.) of reporters for HES7 (n=5 Somitoids), MESP2 (n=6 Somitoids), and PAX3 (n=6 Somitoids). d, Image of PAX3 reporting Somitoid. e, Bright field images and rosette projected areas (n=1957 rosettes from 20 Somitoids). center line, median; box limits, upper and lower quartiles; whiskers, maximum and minimum. f-g, Immunostaining images of rosettes (n>10 Somitoids). h, Heat map of selected genes in 48 h, 66 h, and 120 h Somitoids (n=3 experiments; 48 Somitoids in each n), as measured by RNA sequencing. Expression levels were calculated by log2 (TPM+0.01). i, HES7 knockout strategy. j, Kymograph of pseudoHES7 and MESP2 reporters in HES7-null Somitoid. k, Temporal profiles (mean±s.d.) of reporters for pseudoHES7 (n=6 Somitoids), MESP2 (n=6 Somitoids), and PAX3 (n=9 Somitoids) in HES7-null Somitoids. l, PAX3 reporter in HES7-null Somitoid. m, MESP2 knockout strategy. n, Temporal profiles (mean±s.d.) of reporters for HES7 (n=6 Somitoids) and PAX3 (n=8 Somitoids) in MESP2-null Somitoids. o, PAX3 reporter in MESP2-null Somitoid. p, Image of PAX3-reporting Somitoid treated with 10 µM ROCKi for 48 h. q, Experiment scheme and image of re-aggregating 120 h Somitoids. PAX3 reporter and bright field images are overlayed. Scale bars 500 µm (d, l, o, p, q); 100 µm (e, f); 50 µm (g).
To explore the role of the segmentation clock in rosette formation, we replaced the coding sequence of HES7 with a destabilized Achilles (YFP) reporter to generate a null mutant (Fig.1i). The YFP signal thus represents the activity of the HES7 promoter in absence of HES7 protein, and we confirmed that the periodic dynamics was ablated (Fig.1j,k, Supplementary Video3). Yet the HES7-null explants proceeded with sequential expression of MESP2 and PAX3 followed by rosette formation as observed in controls (Fig.1j–l). We next generated a MESP2-null mutant iPS line which exhibited normal PAX3 expression and generated epithelial rosettes similar to wild type controls (Fig.1m–o). Formation of the rosettes could be blocked without altering PAX3 expression by inhibiting myosin contractility using Y-27632 (ROCKi) or Blebbistatin (Fig.1p, Extended Data Fig.1l). We also dissociated the Somitoids to single cells after rosettes appeared, and then re-aggregated cells by centrifugation prior to culture (Fig.1q). Strikingly, cells re-formed similar rosettes in the new aggregates (Fig.1q). Together, these experiments suggest that rosette formation is an acto-myosin dependent self-organizing property of cells differentiated to the somite stage and does not depend on a prior prepattern established by the clock and wavefront system. This conclusion is supported by previous experiments in vivo and in vitro where somite-like epithelial structures could be induced in the absence of normal AP patterning17,21.
Cell sorting drives somite AP patterning
We next investigated whether the epithelial rosettes exhibit an AP polarity as observed in somites. We used an iPS line harboring the MESP2 reporter (mCherry) to mark the nascent anterior compartment and a UNCX reporter (YFP) for mature posterior identity (Extended Data Fig.2a). At 120 h, we observed rosettes mostly composed of either YFP-high/mCherry-low or mCherry-high cells (Fig.2a). Some bias in the distribution of rosette types was observed with an enrichment in UNCX-positive ones in the periphery (Extended data Fig.2b). As reported in mouse embryos, UNCX trailed MESP2 expression in time (Fig.2b, Supplementary Video4). RNAseq performed on FACS-sorted YFP-high and mCherry-high cell fractions from 120 h cultures showed that they express signature genes associated with posterior (UNCX, DLL1) and anterior somite (FGFR1, TBX18) compartments (Extended Data Fig.2c, Supplementary Table 2). In mouse, Notch signaling is required for the clock oscillations and for Mesp2 expression and the establishment of AP identities7,22. Accordingly, treatment of cultures with the Notch inhibitor DAPT prematurely arrested HES7 oscillations and prevented expression of UNCX and MESP2 and rosette formation but not PAX3 expression (Extended Data Fig.2d,e). In the presence of ROCKi or Blebbistatin, no rosette formed but YFP and mCherry-positive cells still appeared and aggregated into separate clusters (Extended Data Fig.2f). HES7-null Somitoids showed similar UNCX expression and patterning as WT (Extended Data Fig.2g,h). MESP2 deletion resulted in an expansion of UNCX positive cells and formed only rosettes exhibiting a posterior identity (Extended Data Fig.2g.h), consistent with the reported role of MESP2 in inhibiting the posterior fate to promote the anterior one22. Therefore, human iPS cells differentiating to the somitic fate in this in vitro system acquire distinct AP identities. However, unlike embryos, these identities do not coexist within the same epithelial somite but are mostly found in distinct epithelial rosettes. These experiments argue that acquisition of the anterior and posterior fates operates independently of the segmentation clock and rosette morphogenesis.
Fig.2. Antero-Posterior polarity patterning in the Somitoid model.
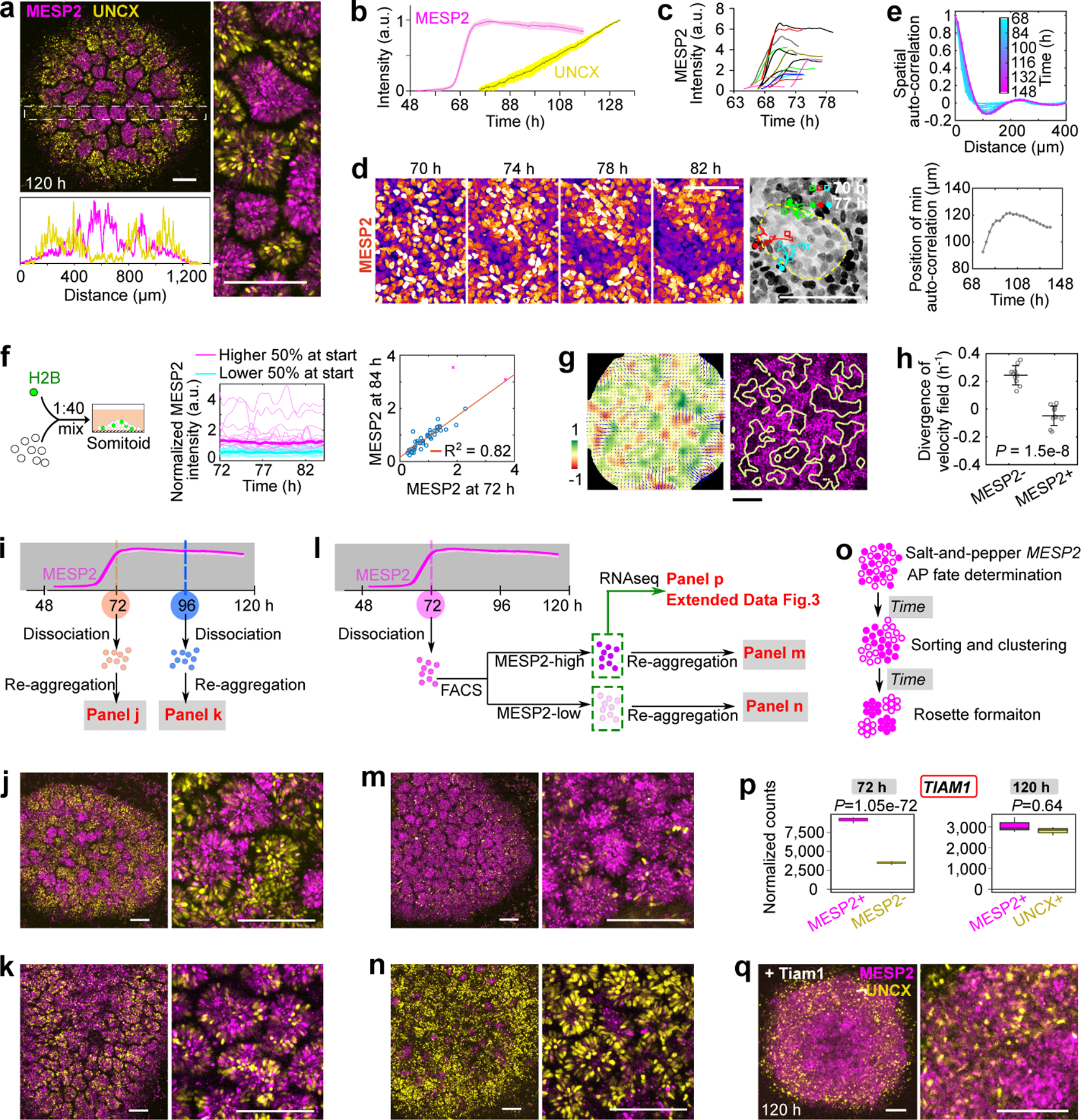
a, Images of MESP2-mCherry and UNCX-YFP reporter and fluorescence intensity profiles across the dotted-line box. b, Temporal profiles (mean±s.d.) of MESP2 (n=6 Somitoids) and UNCX (n=10 Somitoids) over entire Somitoids. c, Temporal profiles of MESP2-mCherry in single cells. d, Time lapse images of MESP2-mCherry reporter and tracks of MESP2-high cells (yellow dotted line represents a forming MESP2-low region). e, Top, spatial auto-correlation of MESP2-mCherry and UNCX-YFP signals over time. Bottom, abscissa-position of the trough of the spatial auto-correlation function over time. f, Experiment design (left), temporal MESP2-mCherry profiles in single cells (middle; n=46 cells from 1 Somitoid; thickened lines represent median), and correlation analysis of MESP2-mCherry intensities at 72 and 84 h (right). F-test (one-sided), P=3.89e-17 after 2 outliers removed (magenta cross; Methods). g, Left, velocity field (arrows) and the corresponding divergence (heatmap) of PIV analysis. Right, positive divergence regions (yellow lines), extracted from the left panel, overlayed on 84 h MESP2-mCherry image. h, Divergence of velocity field of MESP2-high and low regions (mean±s.d., n=10 regions from 2 Somitoids; unpaired two-tailed t-test). i-k, Scheme (i) and images of re-aggregated Somitoids at 72 h (j) or 96 h (k). l-n, Scheme (l) and images of re-aggregated MESP2-high (m) and low (n) cells separated at 72h. o, Model illustration. Solid circles, MESP2-high cells; Hollow circles, MESP2-low cells. p, Normalized RNA counts of TIAM1 in separated cell fractions at 72 h or 120 h, measured by RNA sequencing (n=3 experiments, 96 Somitoids in each n; DESeq2 with two-sided Wald test). MESP2-high cells are shown on the left (magenta), and MESP2-low (72 h) or UNCX+ (120 h) cells on the right (yellow). q, Reporter images of Somitoid overexpressing Tiam1 (Doxycycline since 48 h). Scale bars 150 µm (a, g, j, k, m, n, q); 100 µm (d).
How MESP2 expression resolves from its initial wide segmental domain which marks the future somite to an anterior half-somite stripe defining the future anterior somite compartment1,2 is not understood. To see if our Somitoid system could help shed light on this process, we analyzed the dynamics of MESP2 expression during AP patterning in vitro using the MESP2 reporter line. The temporal profile of the reporter suggests a rapid activation of MESP2 from ~64 h to 72 h (Fig. 2b). This phase is followed by a stabilization of the reporter expression in a salt-and-pepper pattern, spanning a 10-fold range of intensities (Fig.2c, Extended Data Fig.2i). Given the long lifetime of mCherry protein, this suggests that no new MESP2 is expressed in the plateau phase of the reporter profile. These observations contrast with the established notion of an initial uniform MESP2 expression in all cells of the future segmental domain1,2. Time lapse movies showed that, after 72 h, cells progressively segregated together according to their MESP2 expression levels defined by mCherry intensity. This led to the gradual formation of MESP2-high and MESP2-low clusters which eventually formed independent rosettes (Fig.2d, Extended Data Fig.2j,k, Supplementary Video5). We tracked individual cells and observed that MESP2-high cells move out of prospective regions of MESP2-low clusters (Extended Data Fig.2l, Supplementary Video6). To characterize this process, we measured the spatial auto-correlation of the mCherry (MESP2 levels at 72 h) and emerging YFP (UNCX) signals (Fig.2e, Extended Data Fig.2m,n). Before ~80 h, the auto-correlation functions were merely decreasing, suggesting the absence of a periodic spatial pattern (Fig.2e). At ~80 h, a trough formed at 90 microns and then quickly increased to 120 microns, suggesting a rapid formation of cell clusters. After ~84 h, the spatial auto-correlation function retained a damped oscillator-like shape, as typical for periodic patterns23. The onset of the periodic pattern that precedes rosette formation also corresponds to a slowing down of cell motility as evidenced by a decrease in mean squared displacement (Extended Data Fig.2o).
To verify that cell sorting underlies this process, we monitored mCherry dynamics in individual cells. To allow efficient tracking of the MESP2-expressing cells, we generated Somitoids from H2B-GFP/MESP2-mCherry reporter cells combined with unlabeled MESP2-mCherry cells at a 1:40 ratio. We performed single cell tracking of the H2B-GFP labeled cells and recorded MESP2 intensity in the same cells. Indeed, we found that the mCherry intensity in individual cells remained largely unchanged from 72 h to 84 h (Fig.2f, Extended Data Fig.2p) with very few outliers (9 out of 98). These results suggest that although fate switching may occur, the vast majority of cells kept their MESP2 levels. At 84 h, the surrounding mCherry intensities of the tracked MESP2-high cells were higher than for MESP2-low cells (Extended Data Fig.2q). Cells starting in a wrong region – MESP2-high cells in the region of a future MESP2-low cluster or vice versa – maintained constant MESP2 intensity but showed larger displacement (Extended Data Fig.2r,s), indicating cellular rearrangements. We further performed a quantitative analysis of the cellular movements during the patterning phase using Particle Image Velocimetry (PIV) to reconstruct the entire velocity field of MESP2-high cells (Fig.2g, Extended Data Fig.2t). To characterize the global pattern of cell movements, we measured the divergence of the velocity field which describes whether regions in a continuum are expanding or contracting. We expected MESP2-high cells to flow out of regions that become MESP2-low clusters (positive divergence) and flow in regions that become MESP2-high. This prediction was confirmed with high statistical confidence (Fig.2h). Taken together, from 72 h to 120 h, the salt-and-pepper mCherry distribution became sorted into mCherry-high and low clusters and then mCherry-high and low rosettes without further MESP2 expression (Extended Data Fig.2u).
To test the role of cell sorting, we dissociated and re-aggregated Somitoids at 72 h when cells are still mesenchymal, and at 96 h when epithelial rosettes start emerging (Fig.2i). Rosettes mostly formed with either high mCherry or high YFP-expressing cells appeared in re-aggregates from 72 h (Fig.2j), while homogeneous rosettes containing mixed YFP- and mCherry-positive cells were formed in re-aggregates from 96 h (Fig.2k). Thus, cell sorting before epithelialization plays an important role in AP patterning of Somitoids. To investigate when AP fates in individual cells are determined in this process, we separated the MESP2-high or MESP2-low fractions from cultures dissociated at 72 h and re-aggregated them separately (Fig.2l). At 120 h, similar rosette morphogenesis was observed in both types of aggregates with MESP2-low re-aggregates expressing significantly higher levels of UNCX than MESP2-high re-aggregates (Fig.2m,n, Extended Data Fig.2v). This suggests that AP cell fates are largely determined before cell sorting and rosette formation. Altogether, our experiments show that an initial heterogeneity of MESP2 expression levels is translated into defined compartments of anterior and posterior identity via an active cell sorting mechanism (Fig.2o).
To gain insights into the cell sorting mechanism, we performed RNAseq on FACS-sorted MESP2-high and MESP2-low cell fractions dissociated at 72 h (Fig.2l). Among the differentially expressed genes (Supplementary Table 3), we identified several cell surface proteins including PCDH8 and EPHA4, which are known to play a role in somitogenesis24–28 (Extended Data Fig.3a–d). We also found a range of genes encoding cytoskeleton regulators such as TIAM1, which acts as a prominent regulator of the small GTPase RAC1 also implicated in somite formation29 (Fig.2p, Extended Data Fig.3e). These differential expression patterns were transient and disappeared after cell epithelialization (Extended Data Fig.3a,c,e). We engineered a Doxycycline inducible expression system30 to overexpress the catalytic domain of mouse Tiam1 in the triple reporter cell line monitoring the expression dynamics of HES7 (destabilized YFP), MESP2 (mCherry) and UNCX (YFP). Forced activation of Tiam1 in entire Somitoids abolished MESP2 and UNCX periodic patterning as shown by spatial auto-correlation analysis, while HES7 oscillations are maintained and the UNCX-positive population is slightly increased (Fig.2q, Extended Data Fig.3f–h). This suggests that different expression levels of TIAM1 in the anterior and posterior somitic compartments are required for sorting to proceed. As TIAM1 regulates the small GTPase RAC1 that plays a central role in regulating the cytoskeleton, this suggests that the differential expression of TIAM1 downstream of MESP2 differentially alters the mechanical properties of the cells allowing the sorting of MESP2-high and low cells. Rosette formation was also inhibited in these experiments in agreement with the proposed role of RAC1 in epithelial somite formation29.
Segmentoids recapitulate PSM development
To further test whether this cell sorting mechanism explains AP polarization of somites, we next set out to establish an in vitro model reproducing the spatial features of somitogenesis, including sequential formation and patterning of somites. We treated iPSCs with CHIR and LDN for 24 hours, and then dissociated the cultures to single cells to generate spheroids using low adhesion wells (Fig.3a). We then embedded these spheroids into low-percentage Matrigel (10%) at 48 h and cultured them in N2B27 media. By 96 h, initially symmetric spheroids become elongated and develop into rod-shaped tissues exhibiting somite-like rosettes at one extremity (Fig.3b,c). Time lapse movies showed that these rosettes form sequentially starting from one end (which we define as anterior) while the other unsegmented end (the posterior end) kept extending (Extended Data Fig.4a,b, Supplementary Video7). Segmentoids formed close to 20 rosettes, which displayed similar size and shape as those of Somitoids (Extended Data Fig.4c,d). Varying concentrations of Matrigel showed that 1% was sufficient for rosette formation but not for elongation. 5–10% Matrigel promoted efficient tissue elongation with a significant number of organoids presenting more than one axis (Extended Data Fig.4e–g). We termed these structures “Segmentoids”. Live imaging of a differentiating PAX3-YFP reporter line showed that PAX3 expression initiated from the anterior end and propagated towards the posterior growing end accompanying rosette formation, indicating sequential maturation of the Segmentoids (Extended Data Fig.4h). TBXT/SOX2-positive cells were scattered in the spheroids at 48 h (Extended Data Fig.5). At 72 h, TBXT/SOX2-positive cells congregated at the posterior end of the elongating Segmentoids, where they remained up to 96 h (Fig.3d, Extended Data Fig.5). At 120 h, we could barely detect TBXT while SOX2-only positive cells located at the posterior tip of the tissue. These data suggest that the posterior growing end of the Segmentoids resembles the tail bud end of embryos which contains the SOX2/TBXT positive Neuro-Mesodermal Progenitors (NMPs)31.
Fig.3. Characterization of the Segmentoid model.
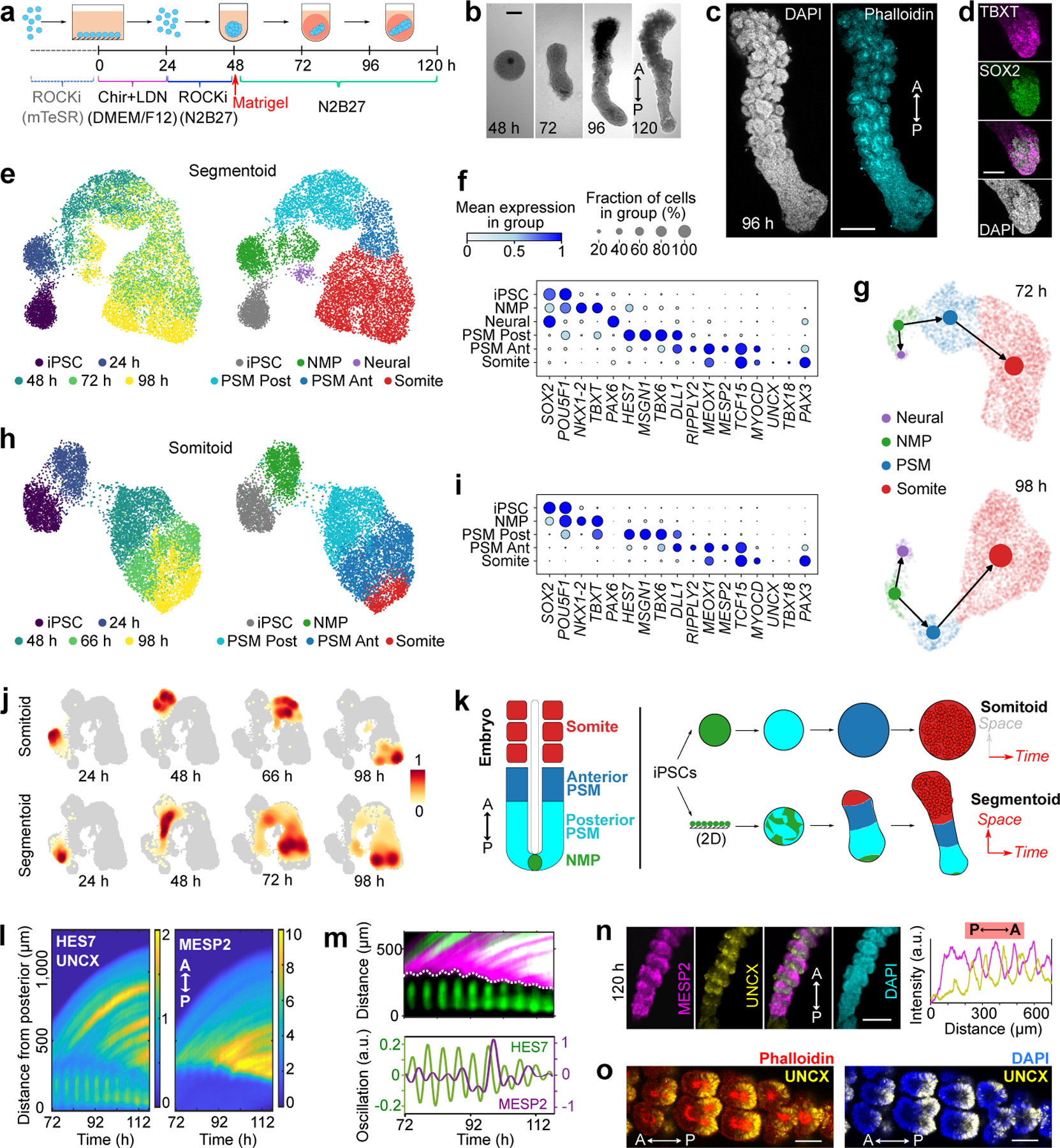
a, Protocol illustration. b, Developmental sequence of a Segmentoid. A, anterior; P, posterior. c, Segmentoid at 96 h (n>10 Segmentoids). d, Posterior tip of 96 h Segmentoid immunostained with TBXT and SOX2 (n>10 Segmentoids). e, UMAP embedding (10,861 cells) colored with timepoints (left) and cell types (right) identified with Leiden clustering. iPSC, 1,491 cells; 24 h, 1,066 cells; 48 h, 1,577 cells from 76 Segmentoids; 72 h, 3,539 cells from 64 Segmentoids; 98 h, 3,188 cells from 32 Segmentoids. f, Dot plot showing expression of selected cell type specific genes in Segmentoids’ clusters. Mean expression of each cluster is scaled per gene. g, PAGA graphs with velocity-directed edges in 72 h (top) and 98 h (bottom) Segmentoids. h, UMAP embedding (8,690 cells) colored with timepoints (left) and cell types (right) following Leiden clustering. iPSC, 1,491 cells; 24 h, 1,265 cells from 96 Somitoids; 48 h, 2,335 cells from 96 Somitoids; 66 h, 2,246 cells from 80 Somitoids; 98 h, 1,353 cells from 48 Somitoids. i, Dot plot showing expression of selected cell type specific genes in Somitoids’ clusters. j, Heatmap of cell density (scaled per timepoint) in UMAP of cells from merged datasets of Somitoids and Segmentoids (19,551 cells). k, Illustration of in vitro models. Each cell type is represented by the same color. l, Kymographs of HES7-Achilles (posterior part in the kymograph), UNCX-YFP, and MESP2-mCherry reporters. Each time point is aligned to the posterior tip. m, Top, kymograph showing expression of the HES7/UNCX (green) and MESP2 (magenta) reporters. Dotted line highlights the start of MESP2 expression. Bottom, HES7 and MESP2 oscillations (Methods). n, Reporter images (left) and intensity profiles (right) along Segmentoid at 120 h (n>10 Segmentoids). o, Images of UNCX-YFP merged with Phalloidin or DAPI (n>10 Segmentoids). Scale bars 200 µm (b, c, n); 100 µm (d, o).
We next used single-cell RNA sequencing (scRNAseq) to characterize the identity and developmental trajectory of Segmentoid cells. Using the 10X Chromium v3.1 platform, we sequenced a total of ~10,000 cells including iPSCs and Segmentoids at 24 h, 48 h, 72 h, and 98 h. When all time points were merged and analyzed together, cells on the UMAP spontaneously organized into a developmental trajectory reflecting the progression of somitogenesis (Fig.3e,f). Cells were clustered using the Leiden algorithm and the identity of clusters was defined based on differentially expressed genes. The clusters included iPSCs, NMPs (expressing SOX2, TBXT, and NKX1.2), Posterior PSM (expressing MSGN1, TBX6, and HES7), Anterior PSM (expressing TCF15 and MESP2), and Somite (expressing PAX3, UNCX, and TBX18). A small Neural cluster (expressing SOX2 and PAX6) was also observed. NMP and PSM populations gradually decreased with time while the somite population increased (Extended Data Fig.6a). Velocity combined with PAGA analysis confirmed that both Neural and Mesodermal cells arise from the NMP progenitors (Fig.3g, Extended Data Fig.6b,c). Altogether, we have established a 3D system in which differentiating human iPSC recapitulates the spatiotemporal progression of somitogenesis.
We also performed a similar scRNAseq analysis of the Somitoids. We sequenced a total of ~10,000 cells at 24 h, 48 h, 66 h and 98 h, and observed clusters similar to those of Segmentoids except for the neural cluster which was absent (Fig.3h,i). In contrast to Segmentoids, cells from a defined time point could be ascribed to a single cluster (Fig.3h), indicating synchronized differentiation across the entire culture. We created a merged dataset containing all cells from the two systems. Cells from the two datasets with the same identity merged into one single cluster (Extended Data Fig.7a,b), indicating that the cell types generated in the two systems are similar. We trained a machine learning classifier on the clusters identified in a scRNAseq dataset of the posterior region of E9.5 mouse embryos10. This classifier accurately identified the equivalent clusters of human Segmentoids and Somitoids (Extended Data Fig.7c–e). Training the classifier on the human Segmentoids or Somitoids clusters also demonstrated the similarity between the corresponding clusters (Extended Data Fig.7f–h). Using density plots we showed that Somitoid time points are clearly defined by a homogenous cell identity (Fig.3j). In contrast, the Segmentoid time points contain multiple differentiation stages (Fig.3j), recapitulating the progression of differentiation observed during somitogenesis in embryos. We extracted the somite population from the merged dataset and investigated the onset of the maturing anterior and posterior identities focusing on the expression of TBX18 and UNCX (Extended Data Fig.7i). We found that the expression of the two genes occurred at the somite stage and was mutually exclusive (0/76 in Somitoids and 9/757 in Segmentoids were double positive cells). Yet, these cells did not segregate into distinct clusters suggesting that they share a similar transcriptome at these stages despite their different AP identities. This probably reflects the fact that the somite cluster cells are quite immature by 98 h and still at the equivalent of the forming somite stage. This is supported by the lack of expression of differentiation markers such as MYF5 or PAX1 in the datasets. The larger number of cells expressing UNCX and TBX18 in the Segmentoids suggests that cell maturation proceeds faster than for Somitoids. Somitoids and Segmentoids dynamically activate HOX genes up to the HOX9 group by 98 h suggesting a thoracic identity of the cells (Extended Data Fig.7j,k). In the Segmentoids, HOX genes were first activated in the NMPs and their activation showed a collinear pattern for HOXA and HOXD but not for HOXB or HOXC (Extended data Fig.7j, l). These analyses demonstrate that both systems can recapitulate somitogenesis in vitro with Somitoids showing synchronized cell differentiation while Segmentoids exhibit a spatially organized progressive maturation similar to that of the embryonic tissue (Fig.3k).
We next investigated somite formation and patterning in Segmentoids using the HES7/MESP2/UNCX triple-reporter line. We observed periodic waves of HES7 traveling along the posterior PSM and arresting before MESP2 expression starts (Fig.3l,m, Supplementary Video8). The domain of MESP2 expression progressed posteriorly in a staggered manner by one segment increments, closely in sync with HES7 oscillations (Fig.3m). Time auto-correlation analysis shows oscillations with a period of 4.6±0.1 h for HES7 and 5.4±0.5 h for MESP2 (Extended Data Fig.8a,b). Thus, the coupling between the segmentation clock and MESP2 induction observed in mouse embryos32 is recapitulated in Segmentoids. At 120 h, alternating stripes of mCherry (MESP2) and YFP (UNCX) were observed (Fig.3n,o, Extended Data Fig.8c). From posterior to anterior, mCherry first appeared as a broad stripe followed by narrower bands with complementary YFP bands emerging (Fig.3n), recapitulating the expression patterns observed in mouse in vivo. The most anterior region of Segmentoids was often composed of grape-like rosettes that did not show clear AP identities (Extended data Fig. 8c). In segments consisting of one mCherry and one YFP stripe, one rosette predominantly formed along the Anterior-Posterior axis (Extended Data Fig.8d), while 1–4 rosettes could be generated along the Medial-Lateral axis (Extended Data Fig.8e). This suggests that antero-posterior organization of the rosettes is geometrically constrained by the forming segment along the AP axis.
The clock controls cell sorting timing
To investigate the role of the segmentation clock in somite polarity patterning21, we examined HES7-null Segmentoids. The YFP signal, reporting activity of the HES7 promoter in absence of HES7 protein (pseudoHES7), was confined to the posterior tip of the elongating tissue. It progressively shrank in a non-oscillatory pattern as the end grew (Fig.4a–c, Extended Data Fig.8f, Supplementary Video9). The onset of MESP2 expression was still coordinated with the down-regulation of pseudoHES7. In contrast to its staggered progression in WT Segmentoids, the MESP2 expression domain moved continuously towards the posterior end in the HES7-null mutants (Fig.4a,b). At 120 h, no alternating stripes of mCherry (MESP2) or YFP (UNCX) could be observed in the mutant (Extended Data Fig.8g). Cells of posterior and anterior identity formed randomly distributed clusters as well as intermixed regions (Extended Data Fig.8g,h, Supplementary video9), consistent with the segmental polarity defects reported in HES7-null mouse embryos33. To confirm this apparent disorganization in HES7-null Segmentoids, we used the nematic order parameter34 of the MESP2/UNCX signal as a measure of anisotropy; we found that during differentiation the nematic order parameter was lower in HES7-null Segmentoids than in controls (Extended Data Fig. 8i). Thus, the segmentation clock is not required for the expression of AP identity genes in individual cells, but its output conferring rhythmicity to MESP2 induction and segment determination appears to play an important role in the spatial organization of stripes of anterior and posterior identity in the forming somites (Extended Data Fig.8j).
Fig.4. Formation of anterior and posterior somite compartments in Segmentoids.
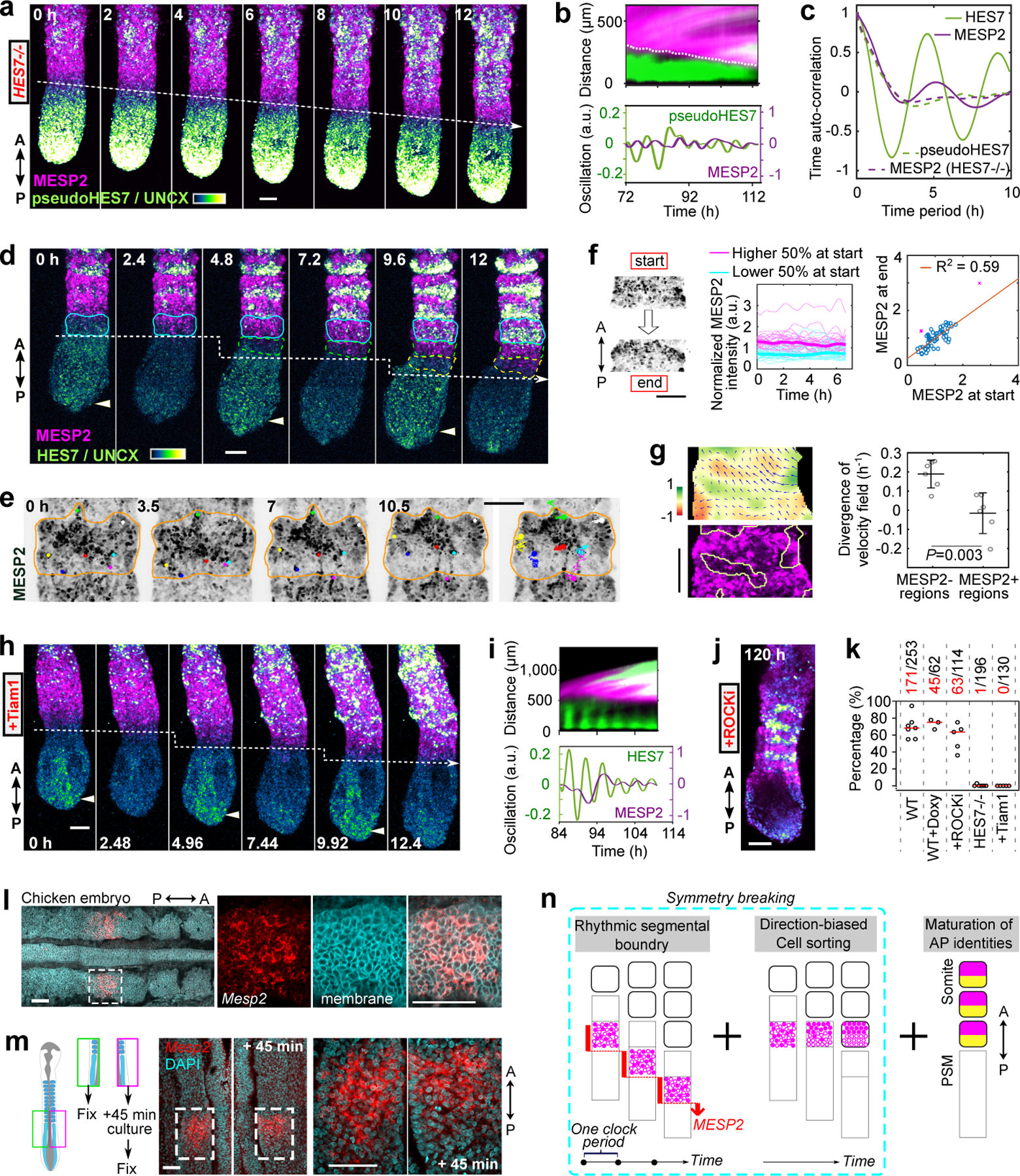
a, Time lapse reporter images of HES7-null Segmentoid. b, Merged kymographs (top) and pseudoHES7-Achilles and MESP2-mCherry oscillations (bottom; Methods). c, Average time auto-correlation of reporter oscillations in WT (n=7 Segmentoids) and HES7-null (n=6 Segmentoids). d, Time lapse reporter images of WT Segmentoid. White arrowheads indicate peaks of HES7 oscillation. e, Time lapse MESP2-mCherry images and tracks of MESP2-high cells. Dots of same color represent the same cell. f, Images showing “start” and “end” of tracking (left), temporal MESP2-mCherry profiles in single cells (middle; n=63 cells from 7 segments of 3 Segmentoids; thickened lines represent median), and correlation analysis of MESP2-mCherry intensities at start and end (right). F-test (one-sided), P = 7.47e-13 after 3 outliers removed (magenta cross; Methods). g, Left, Velocity field with the corresponding divergence of PIV analysis (top) and positive divergence regions overlayed on MESP2-mCherry image (bottom). Right, divergence of velocity field of MESP2-high and low regions (mean±s.d., n=6 segments of 3 Segmentoids; unpaired two-tailed t-test). h, Time lapse reporter images after Tiam1 overexpression. Doxycycline (Doxy) was added 24 h before. i, Merged kymographs (top) and reporter oscillations (bottom) after Tiam1 overexpression (Doxy since 72 h). j, Image of reporters after ROCKi treatment for 72 h. k, Percentages with proper patterning (Methods). Each data represents an independent experiment (WT, n=7; Doxy, n=3; ROCKi, n=5; HES7-/-, n=6; Tiam1, n=5); red bars, median. Total numbers scored and positive numbers (red) are shown. One-way ANOVA Tukey, compared with WT, P=0.98 (Doxy), 0.31 (ROCKi), 5.03e-10 (HES7-/-), 1.14e-9 (Tiam1). l, Images of chicken embryo stained with MESP2 HCR probe and membrane dye. m, Experiment scheme and images of half-embryo pairs stained with MESP2 HCR probe and DAPI. n, Model illustration. Scale bars 100 µm (a, d, e, f, g, h, j); 50 µm (l, m).
To explore whether the cell sorting mechanism observed in Somitoids could be involved in the generation of alternate AP stripes, we analyzed the formation of individual segments in Segmentoids. We first observed a segment-wide transient expression of MESP2 lasting for ~1 clock period followed by UNCX expression in MESP2-low cells during the next clock period (Fig.4d, Extended Data Fig.9a, Supplementary Video10). As with Somitoids, the initial induction of MESP2 resulted in cells displaying a broad distribution of expression levels throughout the newly specified segment (Extended Data Fig.9b). Cells showing high MESP2 expression levels gradually congregated in the anterior compartment while the overall mCherry intensity in the segment stayed constant (Extended Data Fig.9c,d). In time lapse movies of the developing Segmentoids, we observed that MESP2-high cells starting in the posterior part of the forming segment predominantly migrated anteriorly toward the prospective MESP2-high compartment, or posteriorly toward the MESP2-high region of the next segment (Fig.4e, Extended Data Fig.9e,f, Supplementary Video11). Next, individual cells were tracked in Segmentoids in which H2B-GFP/MESP2-mCherry reporter cells were spiked in MESP2-mCherry reporter cells at a 1:40 ratio (Fig.4f). MESP2 reporter intensities of individual cells stayed constant in the time window of pattern emergence with very few outliers (4 out of 63). We further performed Particle Image Velocimetry (PIV) on MESP2 signal to quantitatively analyze the cellular movements in forming segments. As predicted by the cell sorting model, prospective MESP2-low regions showed positive divergence representing MESP2-high cells moving out (Fig.4g, Extended Data Fig.9g). Furthermore, overexpressing Tiam1 abolished AP patterning, further supporting its role in the segregation of the AP compartments (Fig.4h,i, Extended Data Fig.9h,i, Supplementary Video12). HES7 and MESP2 oscillations were observed, although the discrete MESP2 progression appeared less robust (Fig.4i, Extended Data Fig.9h). Tiam1 overexpression also prevented rosette formation. In contrast, Segmentoids treated with ROCKi formed mCherry/YFP stripes but did not form rosettes (Fig.4j,k, Extended Data Fig.9i). Altogether, this suggests that cell sorting leads to the formation of the stripes of the anterior and posterior somitic compartments, rather than the differential regulation of MESP2 expression classically postulated.
To test whether such a heterogeneous MESP2 expression is observed in vivo, we used the quantitative in situ Hybridization Chain Reaction (HCR), to examine the onset of MESP2 expression in the anterior PSM of chicken and mouse embryos. Indeed, we observed a clear salt-and-pepper pattern of MESP2 expression among cells of the future segmental domain (Fig.4l, Extended Data Fig.10a,b). To examine how the MESP2 band evolves in time, we split chicken embryos into halves with one half fixed immediately while the other was subjected to in vitro culture before fixation35 (Fig.4m). Both halves were then hybridized with the MESP2 HCR probe. Our analysis shows that in 45 min (half a clock period) more cells with high MESP2 expression were located in the anterior portion of the segmental domain resulting in decreased MESP2 signal heterogeneity (Fig.4m, Extended Data Fig.10c–f), consistent with a cell sorting mediated mechanism. Together, these suggest that the sorting mechanism that we uncovered in vitro is likely operating in vivo.
In summary, we established two iPSC-derived 3D models recapitulating human somitogenesis, Somitoids and Segmentoids (Fig.3k). In contrast to gastruloids or Trunk-Like Structures17,36,37 which harbor cell lineages derived from the three germ layers, our two models contain almost exclusively paraxial mesoderm. Somitoids recapitulate the temporal sequence of somitogenesis, with all cells undergoing differentiation and morphogenesis in a synchronous manner. This system can provide unlimited amounts of cells precisely synchronized in their differentiation. It will allow deconstructing these patterning processes to understand how they integrate in the complex morphogenetic program of somitogenesis. On the other hand, Segmentoids recapitulate the spatio-temporal features of somitogenesis, including gene expression dynamics, tissue elongation, sequential somite morphogenesis, and polarity patterning. They therefore provide an excellent proxy to study human somitogenesis at an unprecedented level of detail. The developmental strategy of the Somitoids is very reminiscent of the synchronized development of the fly embryo, while the Segmentoids evokes the sequential development of vertebrates.
Together our work suggests a novel framework (Fig.4n) explaining how somite AP polarity is coordinated with segmental determination. We propose that the segmentation clock contributes to AP patterning by defining regular boundaries of maturation which organize cell sorting. In each cycle, the segmentation clock defines a stripe of Notch activation where the salt and pepper onset of MESP2 expression is synchronized among all cells, possibly involving a mechanism such as lateral inhibition; Since the cell sorting ability is transient, this synchronization restricts the sorting within the stripe. Within each stripe, the same cell sorting process rearranges MESP2-high and -low cells into an anterior and a posterior domain respectively in response to an AP cue that remains to be characterized. As this process repeats in time, the modular compartmentalization in each segment eventually constitutes the spatially regular, alternative stripes defining somite polarity along the body axis. In conclusion, our work exemplifies how the resolution offered by PSC-derived in vitro systems can be used to answer long-standing developmental biology questions.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized, and investigators were not blinded to allocation during experiments and outcome assessment.
Human iPS cell culture
Human stem cell work was approved by Partners Human Research Committee (Protocol Number 2017P000438/PHS). We complied with all relevant ethical regulations. NCRM1 iPS cells (RUCDR, Rutgers University) were used throughout this study. Written informed consent from the donor was obtained by Rutgers University at the time of sample collection. Cells were maintained in Matrigel-coated plates (Corning 35277) in mTeSR1 medium (StemCell Technologies 05851) and passed every four days. Briefly, cultures of 90% confluency were dissociated in Accutase (Corning 25058CI) and 500,000 cells were seeded into one well of a 6-well plate with mTeSR1 and 10μM Y-27362 dihydrochloride (Tocris Bioscience1254). Fresh mTeSR1 medium was supplemented daily in the following days. All cell lines were maintained no longer than 15 passages and were regularly tested for mycoplasma contamination.
Generation of reporter cell lines
The CRISPR-Cas9 system for genome editing38 was used to generate all reporter lines. The following reporter lines were generated from previous studies10: Line#1, double reporter line of HES7-T2A-Achilles-NLS-CL1-PEST and MESP2-T2A-H2B-mCherry; Line#2, Double reporter line of pCAG-H2B-mCherry and HES7-T2A-Achilles-NLS-CL1-PEST; Line#3, reporter line of PAX3-T2A-YFP-NLS. To make the triple reporter line reporting HES7/MESP2/UNCX (Line#4), a single-guide RNA targeting the 5′ end of UNCX (CGTCCATCATCTCGCGCGGG GGG) was designed using the online Tool (http://chopchop.cbu.uib.no/) and cloned into the pGuide-it-tdTomato vector (Takara 632604). We also generated a repair plasmid consisting of 1-kb 3′ and 5′ homology arms flanking a YFP variant Achilles, a nuclear localization signal (NLS), and a self-cleaving T2A peptide sequence in a pUC19 vector backbone by In-Fusion cloning (Takara Bio 638909). Both the pGuideit-tdTomato and repair plasmids were delivered to Line#1 iPS cells by nucleofection (Lonza VPH-5022) using a NEPA 21 electroporator. 24 hours after nucleofection, cells were sorted by TdTomato expression using an S3 cell sorter (Biorad) and seeded at low density (500 cells per 35mm dish) in Matrigel coated plates in mTeSR1 with 10μM Y-27362 and CloneR (Stemcell Technologies 05888). Single cells were expanded clonally, and individual colonies were screened by PCR for targeted homozygous insertion of Achilles-NLS-T2A immediately after the start codon of UNCX. Only one clone out of 70 colonies showed a positive insertion by PCR, with Achilles-NLS-T2A inserted into only one allele of UNCX locus. Both the tagged and untagged allele were sequenced to ensure that no undesired mutations had been introduced by the genome-editing process.
An identical approach was used to generate HES7-null and MESP2-null cell lines. For HES7-null, two single-guide RNA plasmids were made to target sequences near both the start codon of HES7 locus (CCTATTCTCAGCTCGATCCC GGG) and the stop codon (CGCCGCCTCACAGACAAGAC GGG). The repair plasmid consisted of 1-kb 3′ and 5′ homology arms flanking Achilles-NLS and two destabilization domains CL1 and PEST (Achilles-NLS-CL1-PEST) in a pUC19 vector backbone. These three plasmids were delivered to Line#1 iPS cells to generate homozygous Line#5 (HES7-null and MESP2-reporting), Line#3 to generate homozygous Line#6 (HES7-null and PAX3-reporting), and Line#4 to generate homozygous Line#7 (HES7-null and MESP2/UNCX-reporting). For MESP2-null, two single-guide RNA plasmids were made to target sequences in exon1 with 662bp between PAM sites (GTCCCTTGGGACGAATACGG GGG and GCACGAACCCGACGAATCGG AGG). These two plasmids with no repair plasmids were delivered to Line#3 iPS cells to generate homozygous Line#8 (MESP2-null and PAX3-reporting) and Line#4 to generate homozygous Line#9 (MESP2-null and HES7/UNCX-reporting).
To insert the constitutively expressed pCAG-H2B-mCherry in the safe harbor AAVS1 locus of Line#3 or pCAG-H2B-GFP to Line#1, we used a previously described approach39. In brief, we cloned the H2B-mCherry or H2B-GFP sequence into the AAVS1-pCAG vector (Addgene 80490) and co-transfected it along with the pXAT2 vector (Addgene 80494) into corresponding iPS cells. One day after nucleofection, we selected positive clones by supplementing mTeSR1 with puromycin (0.5 μg/ml, Sigma-Aldrich P7255) for a total of 4 days. We generated Line#10 (H2B-mCherry and PAX3-T2A-YFP-NLS) and Line#11 (H2B-GFP and MESP2-T2A-H2B-mCherry) confirmed by PCR and fluorescence expression. To generate Line#12 (pTRE3G-CFP-Tiam1-CAAX and HES7/MESP2/UNCX triple reporter), we cloned the murine Tiam1 (Addgene 85221) with CFP tagged at the N-terminus and CAAX sequence at the C-terminus into the AAVS1-pTRE3G vector30 (Addgene 52343), followed by similar transfection and selection.
Somitoid protocol
(Day -1) Mature iPS cell cultures (80% confluency) were dissociated in Accutase and 3,000–4,000 cells were seeded into one well of 96-well U-bottom non-treated plates (Greiner bio-one 650185 or Falcon 351177) with 100 μl mTeSR1 medium and 10 μM Y-27362. The plate was centrifuged at 300 rcf for 2 min and the cell pellet can be seen at the bottom of the well. (Day 0) All medium was carefully removed under a dissection microscope and replaced with 150 μl DICL medium: DMEM/F12 GlutaMAX (Gibco10565042) supplemented with 1% insulin-transferrin-selenium (ITS; Gibco 41400045), 3 μM CHIR99021 (Tocris 4423) and 0.5 μM LDN193189 (Stemgent 04–0074). (Day 1) 130 μl medium was removed and 150 μl fresh DICL medium was added to each well. (Day 2) The spheroids were transferred to a flat surface coated with Laminin-521 (Stemcell Technologies 77003) or gelatin (Stemcell Technologies 07903) and cultured in somite-inducing medium (SIM): DMEM/F12 supplemented with 1% ITS, 1.5 μM CHIR99021, 0.5 μM LDN193189, and 5% Fetal Bovine Serum (FBS; Sigma-Aldrich EmbryoMax ES-009-B). Note the concentration change of CHIR99021. Laminin-521 was preferred for glass or ibidi polymer vessels since gelatin coating on these surfaces often lead to insufficient spreading. No further medium change was needed.
Segmentoid protocol
(Day -1) Mature iPS cell cultures (100% confluency) were dissociated in Accutase and 200,000–240,000 cells were seeded into one Matrigel-coated well of a 6-well plate with 2 ml mTeSR1 medium and 10 μM Y-27362. The seeding number was important for further differentiation, with a lower range working best. (Day 0) All medium was replaced directly with 2 ml DICL medium: DMEM/F12 supplemented with 1% ITS, 3 μM CHIR99021, and 0.5 μM LDN193189. (Day 1) Cells were dissociated with Accutase and 6,000–8,000 cells were seeded into one well of the 96-well cell-repellent U-bottom plate (Greiner bio-one, 650970) with 150 μl N2B27 medium and 10 μM Y-27362. The plate was centrifuged at 300 rcf for 2 min and a cell pellet can be seen at the bottom of the well. N2B27 medium was prepared from 1:1 mix of DMEM/F12 and Neurobasal medium (Gibco 21103049), supplemented with 1% N2 Supplement-B (Stemcell Technologies 07156), 2% B-27 Supplement (Gibco 17504044), and 0.1% 2-Mercaptoethanol (Gibco 21985023). (Day 2) A single spheroid was formed in each well. All medium was removed under a dissection microscope and 30 μl ice-cold 10% Matrigel (Corning CB-40234A; diluted with cold N2B27) was added to each well. The plate was then incubated at 37°C for one hour. After the Matrigel solidified, 150 μl N2B27 medium was carefully added to each well. No further medium change was needed.
Dissociation and Re-aggregation of Somitoids
About 20 Somitoids were cultured in one well of a 6-well plate coated with gelatin. Somitoids at 72 h or 96 h were dissociated with Accutase and single cells were resuspended in SIM (DMEM/F12 with 1% ITS, 1.5 μM CHIR99021, 0.5 μM LDN193189, and 5% FBS) with 10 μM Y-27362. The cell resuspension was split into five 1.5 ml Eppendorf tubes and centrifuged at 400 rcf for 4 min. Under a dissection microscope, each pellet was loosened from the bottom of the Eppendorf tube with gentle pipetting and transferred to a new gelatin-coated plate containing SIM medium with 10 μM Y-27362. After 24 h, the medium was changed to just SIM and no further media change was performed. After another 24 h, imaging was performed on each re-aggregate. For the experiment in Fig.1l–n, about 60 Somitoids were dissociated in each independent experiment. MESP2-high (top 10% mCherry histogram) and MESP2-low (bottom 10% mCherry histogram) cells were collected separately via flow cytometry and counted. About 50,000 cells were re-aggregated and cultured in SIM medium with 10 μM Y-27362 followed by just SIM after 24 h.
RNA sequencing and analysis
Line#4 was used for all RNA sequencing experiments. In each experiment of Fig.1h, Somitoids-48h (n=48) were pooled together and subjected to RNA extraction using the NucleoSpin RNA XS kit (Macherey-Nagel 740902.50). Similar RNA extraction was performed on Somitoids-66h (n=48) or Somitoid-120h (n=48) growing on two gelatin-coated wells of a 6-well plate. For each experiment in Extended Data Fig.2c, Somitoids-120h (n=96) were dissociated with Accutase. Cells with top 10% mCherry fluorescence and top 10% YFP fluorescence were sorted by flow cytometry and collected separately and subjected to RNA extraction with the RNeasy micro kit (Qiagen 74004). For each experiment in Fig.2p and Extended Data Fig.3a–e, Somitoids-72h (n=96) were dissociated with Accutase. Cells with top 10% mCherry fluorescence and bottom 10% mCherry fluorescence were collected separately and subjected to RNA extraction. Three independent experiments were performed in all cases.
For RNA sequencing, libraries were prepared with Genewiz using the polyA selection method and were sequenced using 150bp-paired end sequencing on an Illumina HiSeq platform. The sequencing quality was assessed by FastQC (version 0.11.9). The sequence reads were then mapped to the human transcriptome by Salmon (version 1.4.0) and quantified using the quasi-mapping method. To assess the quality and origin of the mapped reads, a further quality control step was performed by aligning the reads to the human genome (GRCh38) using STAR (version 2.7.6a) and by subsequently subjecting the resulting alignment data files (BAM files) to further analysis using Qualimap (version 2.2.2a). The transcript per million (TPM) mapped reads count of each transcript calculated by Salmon was thereafter used for differential gene expression analysis, which was performed using the DESeq2 package. After regularized log (rlog) transformation, the top 500 most variable genes evaluated by DESeq2 were used to perform principal component analysis (PCA). To generate the heatmap in Fig.1h, gene-level TPM values were log2 transformed after adding an offset of 0.01 to each value. To compare gene expression between cells with top 10% mCherry fluorescence and those with top 10% YFP or bottom 10% mCherry fluorescence, the Wald test was performed with multiple test correction of p-values using the Benjamini-Hochberg method.
Flow cytometry analysis
To determine the fraction of 120 h Somitoid cells that express YFP-UNCX, cultures from Line#4, Line#7, and Line#9 were dissociated in Accutase and analyzed by flow cytometry using an S3 cell sorter (Biorad). Cells from Somitoids formed from parental NCRM1 cells, which do not express the fluorescent protein, were used as a negative control for gating purposes. Results were presented as the percentage of YFP+ cells in the sorted fraction with debris and doublets excluded.
Immunostaining
Somitoids growing on Laminin-521 or gelatin coated plates were fixed in a 4% paraformaldehyde solution (PFA; Electron Microscopy Sciences 15710) overnight at 4°C, then washed 3 times with phosphate buffered saline (PBS). Typically, samples were washed 3 times for 3 min each in Tris buffered saline (TBS) with 0.1% Tween (TBST), permeabilized in PBS with 1% Triton for 10min, and blocked for 1 h at room temperature in TBS with 0.1% Triton and 3% FBS. Primary antibodies were diluted in blocking solution and incubated overnight at 4 °C with gentle rocking. Following 3 TBST washes and a short 10-min block, cultures were incubated with Alexa-Fluor-conjugated secondary antibodies (1:500), Phalloidin (1:200) and Hoechst33342 (1:1000) overnight at 4°C with gentle rocking. Three final TBST washes and a PBS rinse were performed, and cells were imaged in PBS. To clear samples for the z-stack acquisition in Extended Data Fig.1g,h, cultures were mounted with a 3D cell culture clearing reagent from the 3D cell culture clearing kit (Abcam ab243299). Segmentoids were fixed in the same cell-repellent wells. Briefly, medium and Matrigel were removed with a pipette as much as possible, then 150 μl 4% PFA was added before overnight incubation at 4°C. Following similar washes and permeabilization, Segmentoids were transferred to a well of a 24-well plate and proceeded with further immunostaining. Imaging was performed in PBS. Information of the primary antibodies is as follows: Laminin (Sigma-Aldrich L9393, 1:200), N-Cadherin (abcam ab76057; 1:350), TBXT (R&D AF2085; 1:350), SOX2 (Millipore AB5603; 1:500).
Microscopy
Wide-field fluorescence imaging was performed on an EVOS cell imaging system, and confocal imaging was performed on a Zeiss LSM 780 point-scanning confocal inverted microscope fitted with a large temperature incubation chamber and a CO2 module. To acquire time-lapse imaging of Somitoids, the ibidi 24-well plate with polymer coverslip bottom (ibidi 82426) coated with Laminin-521 was used. Glass coverslip did not support stable spreading of Somitoids. To promote HES7 wave propagation, 5µM EHT1864 (Tocris 3872) was added to all media. To acquire time-lapse imaging of Segmentoids, the culture was carefully transferred in a droplet to the center of a non-coated well of the 24-well glass-bottom plate (Cellvis P24–1.5H-N) with a P1000 pipette. The medium was removed as much as possible before a droplet of 30 µl ice-cold 10% Matrigel was added to immerse the Segmentoid. After 1-hour incubation at 37°C, N2B27 medium was added to the well without disturbing the solidified Matrigel. A 20× Plan Apo (N.A. 0.8) objective was used for most time-lapse fluorescence imaging, with an Argon laser at 514 nm to excite the YFP fluorophore and a DPSS 561 laser at 561 nm to excite mCherry.
Image analysis
To generate the kymographs in Fig.1b,j and Extended Data Fig.3f, a line scan crossing the center of the Somitoid was selected and the Multi Kymograph analysis in ImageJ was used. To measure the temporal intensity profiles of reporter gene expression, a box covering the center region of the Somitoid was selected in ImageJ and the average fluorescence intensity was measured for each time point. The single cell MESP2 intensity profiles in Fig.2c were generated by manually tracking single cells with a 6 min interval and the plots were smoothed over 10 timepoints. The Mean Squared Displacement (MSD) in Extended Data Fig.2o was measured using 6 min time-lapse movies of Somitoids comprising 1:40 mix of H2B labeled (Line#11) and unlabeled cells (Line#1). Nuclei were segmented and tracked using the Particle Tracking pipeline in Arivis Vision4D 3.5. Movies were divided into 3-hour time subsets and MSD was calculated for each subset using the software’s MSD function. A total of n= 3,422 tracks from n=2 30-hour long movies were used to calculate the MSD.
Morphometric analysis
To measure the size and shape of individual rosettes and overall Somitoids, all structures were outlined manually using a stylus pen for touch screens and characterized in ImageJ. 1,957 rosettes from 20 Somitoids-120h were measured. Similarly, 345 rosettes from 20 Segmentoids were outlined and measured. To measure the lengths, the longest axis of each Segmentoid were marked with a stylus pen and measured in ImageJ. Number of organoids measured in each experiment: 16, 33, 24 (No Matrigel); 12, 12, 13 (1% Matrigel); 32, 29, 42 (5% Matrigel); 34, 58, 30, 41, 33 (10% Matrigel). Segmentoids showing at least three UNCX expression stripes were defined as organoids with proper polarity patterning (Fig.4k). Number of Segmentoids scored in each experiment: 40, 31, 27, 58, 41, 36, 20 (wt); 20, 18, 24 (wt+Doxy); 16, 22, 30, 25, 21 (wt+ROCKi); 44, 23, 35, 48, 21, 25 (HES7-null); 22, 35, 19, 24, 30 (+Tiam1).
Analysis of gene expression reporters in each segment
To plot the reporter expression profiles for individual segments of Segmentoids in Extended Data Fig.9a, HES7 oscillations were first defined from trough-to-trough by measuring HES7 reporter intensity in the elongating region for every slice over the entire movie. For each segment of the segmentoids, a measurement region of interest (ROI) was drawn manually in imageJ based on the UNCX boundary of the previous segment and the posterior HES7 signal. The ROI was then fixed in position and the intensity of the MESP2 and UNCX reporter within that ROI was measured every 10 slices (every 1 hour) for 3 full oscillations. Measurement for each segment begins at the trough of HES7 oscillation immediately preceding the onset of MESP2 reporter expression. 3 full HES7 oscillations was defined as the endpoint as this was when a sharp UNCX boundary was observed for that segment, indicating the mature somite. Measurements were taken from 2 separate videos, each providing 3 segments for a total of 6 segments measured. Reporter intensities of each segment are internally normalized as 0% to 100% peak fluorescence, where the lowest and highest measured expression observed for each segment over the 3 oscillations is set as the limit, and the values in between are scaled linearly to this new range. Measurements across segments were aligned at time 0 using the trough of the initial HES7 oscillation. No alignment was needed or performed beyond this as the HES7 oscillations were very similar across segments and videos, enabling the data to be directly overlayed from different segments or videos. All graphs above were generated with Graphpad Prism 9.
Spatial auto-correlation analysis
The formation of MESP2 and UNCX clusters in Somitoids was investigated using the radial part of the two-point spatial auto-correlation function. This function typically has a “damped oscillator”-like shape in the case of alternating clusters of opposite polarity. The position of the first trough is related to the characteristic distance between two clusters of opposite polarity23.
First, we segmented Somitoids in the first frame of each time-series using the MESP2-mCherry signal and applying gaussian filtering, thresholding and morphological operations; a 50 µm erosion was applied to remove the external rim of Somitoids. These Somitoid masks were used for all time-points to avoid spurious effects due to change in Somitoid boundary. To construct a “polarity” image, we integrated the information of MESP2-mCherry (anterior) and UNCX-YFP (posterior) signals. To this end, we normalized MESP2-mCherry and UNCX-YFP images to their mean intensity in the Somitoid mask region and subtracted the normalized MESP2-mCherry signal from the normalized UNCX-YFP signal. Hereafter, comparable results were obtained when using MESP2-mCherry alone or the polarity MESP2-UNCX image; similar results were also obtained using UNCX-YFP alone, although for later time-points, as UNCX is expressed later than MESP2.
Image resolution was reduced to 1./5.5 px µm−1 by bilinear down-sampling. Then, images were smoothed using a gaussian filter (SD: 25 µm, filter size: 50 µm). To eliminate long-range intensity variations (detrending), images were divided by their “background”, obtained by long-range gaussian filtering (SD and filter size: 280 µm). Two-point auto-correlation was calculated using the MATLAB function xcorr2, after subtracting the image mean, and normalizing to the image variance (calculations were restricted to the Somitoid mask region). The auto-correlation function was expressed in polar coordinates and averaged over polar angles, thus obtaining the radial part of the auto-correlation function. The radial auto-correlation function was averaged across time using a 3 h-span moving average. The first putative trough of the averaged radial auto-correlation function was identified using a smoothing spline and the MATLAB function findpeaks (minProminence: 0.01). To distinguish troughs from noise, a putative trough was accepted when its auto-correlation value was lower than -0.02 and lower than minus the standard deviation of the averaged radial auto-correlation function for distances 50 µm to 300 µm larger that the x-coordinate (distance) of the putative trough itself. A similar procedure was applied to calculate the two-point radial auto-correlation function of Segmentoid images; in this case, individual Segments were manually selected.
Segmentoid kymograph generation
Segmentoid data were 2D max z-projections organized in time-series (typical pixel size 1.38 µm). For each Segmentoid, we first generated a mask. Thus, we max projected the time-series along its time coordinate and added MESP2-mCherry and HES7-Achilles/UNCX-YFP signals, to obtain a 2D image of the Segmentoid path; the Segmentoid mask was generated from the Segmentoid path using gaussian filtering, thresholding and morphological operations. Then, the orientation angle of the Segmentoid mask was calculated. This angle was used to rotate the original time-stack and the Segmentoid mask to become parallel to the x-axis; the posterior of the Segmentoid (manually assigned) was set to be on its right side.
A midline profile of the Segmentoid was calculated as the mean y-coordinate of the Segmentoid mask for each x-coordinate, followed by smoothing. A midline mask was generated using the midline profile and dilating it by 75 pixels, thus generating a stripe running across the Segmentoid. Signals were averaged at each time-point along the y-coordinate in the midline mask region, thus obtaining a signal x-profile. The position of the posterior end of the Segmentoid at each time-point was detected from the HES7-Achilles/UNCX-YFP or H2B-mCherry x-profiles; this position was chosen as the x-coordinate in which the x-profile decreased past a certain threshold (either manually selected or calculated as 1.2 times the x-profile averaged 100 to 200 pixels past its maximum). At each-time point, intensity profiles were aligned to bring the posterior end of the Segmentoid at the same x-coordinate (10 pixels away from the right end of the image). Aligned intensity x-profiles were assembled in a kymograph.
Signal time-profile averaging, detrending and period calculation
Signal time-profiles were calculated from aligned kymographs at a select position; this position was 150 µm away from the posterior end of the Segmentoid for HES7-Achilles, while for MESP2-mCherry it was taken as the coordinate in which its x-profile in the first timeframe decreased to half its maximum. For detrending, a trend was calculated by smoothening time-profiles with a moving average (9 h span) and subtracted from time-profiles. Time two-point auto-correlation functions were calculated from detrended time-profiles, after subtracting their mean, and normalizing to their standard deviation. Oscillation periods were calculated as the first peak (time lag>0) of time auto-correlation functions (a smoothing spline was used). For visualization purpose, signal time-profiles were smoothed using a 1.5 h-span moving average and a Savitzky-Golay filter (order=3, frame = 2*round(1.8/dt)+1, where dt is 1/frame-rate in h−1).
Segmentoid nematic order parameter
To characterize the elongated shape of Segments, we used a method based on the Fourier Transform of fluorescence intensity images that does not rely on segmentation34. For each Segmentoid, their time-stacks were rotated and shifted along the x-coordinate to bring the Segmentoid parallel to the x-axis and aligned to their posterior end, as described above.
We continued by generating a mask for the MESP2-UNCX portion of the Segmentoid at each time-point, as follows. To determine the location of the transition point from the MESP2-UNCX to the HES7 posterior regions, we generated a mask based on the MESP2-mCherry signal alone, using gaussian filtering, thresholding and morphological operations. The transition point was determined from the MESP2-mCherry average x-profile over the MESP2 mask region. Then, we generated a Segmentoid mask at each time-point, by summing MESP2-mCherry and HES7-Achilles/UNCX-YFP aligned images normalized to their respective average intensities at each time point, followed by gaussian filtering, thresholding and morphological operations. MESP2-UNCX time-series were generated by subtracting the normalized HES7-Achilles/UNCX-YFP aligned images from MESP2-mCherry aligned images, both normalized to their mean intensity in the Segmentoid mask region. Finally, to crop out the HES7 posterior region, the MESP2-UNCX mask was set to zero in the region anterior to the transition point calculated above.
The nematic order parameter was calculated in square interrogation windows (124 µm-side) moving along the midline of the MESP2-UNCX mask (14 µm step, 750 to 250 µm anterior to the posterior end of the Segmentoid) and then averaged. The nematic order parameter was not calculated when less than 90% of the moving window was contained within the MESP2-UNCX mask. The Fourier power spectrum G was calculated in each interrogation window and expressed in polar coordinates, where k is the circular wave-number polar radius and the polar angle (here, with being the wavelength). We calculated a nematic tensor with
where is a cut-off used to eliminate long wavelengths. indicates alignment to the x-axis, to the y-axis, to the y=x line and to the y=-x line. To consider all possible alignments, we used the positive eigenvalue of the nematic tensor Q, i.e., as nematic order parameter.
Single cell MESP2 intensity quantification
For single cell tracking and MESP2 reporter quantification, we used chimeric Somitoids and Segmentoids from 1:40 mix of Line#11 (H2B-GFP and MESP2-T2A-H2B-mCherry) and Line#4, in which the nuclei were sparsely labeled with H2B-GFP to facilitate segmentation. For Somitoids, single nuclei that were labeled by both reporters were segmented and tracked with Ilastik 1.3.3 software40, using the Pixel Classification – Tracking with Learning pipeline. For Segmentoids, sparsely labeled nuclei were also segmented in Ilastik, but the tracking was done using a custom developed particle-tracking software41. The segmentation-tracking outputs were subsequently manually curated to remove apparently faulty cell tracks.
To correct for the decrease in overall intensity in z, a calibration curve was obtained for each time point of the movie using the H2B-GFP channel as reference. Specifically, the calibration curve was calculated by taking the average pixel intensity value in each z-plane for all pixels selected by the nuclei-segmentation mask. The MESP2 average pixel intensity for each segmented nucleus is then normalized by the corresponding value in the calibration curve.
To account for any batch effect between Segmentoids or Somitoids, another normalization has been performed. For Somitoid data, each MESP2 intensity value was further normalized by the mean MESP2 intensity of all cells belonging to the same Somitoid at the corresponding time point. For Segmentoids, different segments from the same Segmentoid develop at different time, hence the normalization factor was calculated based on the cells belonging to the same segment. This normalization allows for merging MESP2 intensity data from different Segmentoids or Somitoids, while adjusting for the global intensity decrease caused by decay of fluorescence reporter. We have measured the cell cycle duration in 2D cultures of iPS-derived human PSM which is around 20 hours. Thus, there is an important time difference between the sorting phase and the cell cycle suggesting that dilution of mCherry due to cell division is unlikely to strongly affect the mCherry intensity profile during the sorting phase. Nevertheless, cell division together with photobleaching, likely accounts for the general slow downward trend of MESP2 plateau phase.
For plotting, MESP2 intensity at the start was calculated as median intensity of the first five frames, and likewise, MESP2 intensity at the end was calculated as median intensity of the last five frames. In the correlation plot for MESP2 intensities at the start and the end, outliers were detected using Mahalanobis distances with a threshold of 8 (unitless). To facilitate visualization, each curve in the MESP2 intensity – time profile was smoothened via a Savitzky-Golay filter (order = 5, frame = 21).
Single cell MESP2 surrounding intensity quantification in Somitoids
The MESP2 surrounding intensity aims to describe local structure in the Somitoid. The “surrounding intensity map” for a Somitoid was calculated by applying a gaussian filter (σ = 35 µm) to max z-projection of the MESP2 channel. The MESP2 surrounding intensity value for each cell was then assigned to the intensity value on the smoothened map at a location specified by the x, y-coordinates of the cell’s centroid. Cells at the most outer region of the movie canvas were removed since no clear Somitoid structure was detected in this region, where outer region was defined as 10% of the canvas width from each of the four edges. For plotting, cells were grouped into higher and lower 50% separately for each Somitoid to account for possible batch effect. To facilitate visualization, each curve in the surrounding intensity – time profile was smoothened via a Savitzky-Golay filter (order = 5, frame = 21).
Comparing displacement and MESP2 intensity between cells at the “correct” and “wrong” starting positions in Somitoids
The “correct” and “wrong” grouping aims to compare cells which begin with the “correct” or "wrong" MESP2 levels for their eventual surroundings. In this analysis, the “correct” group is defined as the cells with high “MESP2 intensity at the start” and high “eventual surroundings”, combined with the cells with low “MESP2 intensity at the start” and low “eventual surroundings”. By contrast, the “wrong” group is defined as the cells with high “MESP2 intensity at the start” and low “eventual surroundings”, combined with the cells with low “MESP2 intensity at the start” and high “eventual surroundings”. More specifically, the “eventual surrounding” of a cell was calculated as the value of the last frame “surrounding intensity map” at a location specified by the x, y-coordinates of the cell’s centroid at the first frame. The “MESP2 intensity at the start” was calculated in the same way as described in the “Single cell MESP2 intensity quantification” section. Moreover, “high” and “low” are defined as whether a cell belongs to the higher or lower 50% among all the cells using the corresponding measurements. Cells were grouped separately for each Somitoid to account for possible batch effect. Lastly, the displacement of a cell in each frame was calculated relative to the position of the same cell in the first frame.
Particle Image Velocimetry (PIV)
In the sorting process, MESP2-high (MESP2+) cells migrate away from the prospective MESP2-low (MESP2-) clusters. Therefore, we reasoned that their velocity field would have positive divergence in this prospective MESP2-low regions. To calculate the velocity field, we registered MESP2 images using the ImageJ plugin “Linear Stack Alignment with SIFT”42 and then performed PIV (PIVlab43–45). For PIV, we used interrogation areas of 40, 20 and 10 pixels (pixel size is 0.692 µm) with 50% overlap. In the case of Somitoids, we manually picked a time-window starting when MESP2 signal appears and ending when Somitoids are patterned, but rosettes have not formed yet. In the case of Segmentoids, segments form at different times and therefore we considered them separately. For each segment, we manually picked a time-window starting when the entire segment region expresses the MESP2 reporter to the end of the patterning process before the segment contracts and gets rounder. Velocities were averaged over this time-window. We tested various choices of the measurement time-window and we found similar results.
In each movie, we calculated a mask corresponding to the Somitoid/Segmentoid using thresholding and morphological operations of MESP2 signal. We restricted the velocity field to this mask. The velocity field was smoothed using a moving average filter (size 35 µm) and then divergence was calculated. To compare the divergence in MESP2+ and MESP2- regions, we selected regions at the end of the patterning process (MESP2 movies). Then, we averaged the divergence in MESP2+ and MESP2- regions. Statistics was performed using Student’s t test. For visualization purposes, the divergence was smoothed using a gaussian filter of 35 µm size and 17.5 µm standard deviation. The boundary of the regions with positive divergence was calculated using a threshold of 0.15 h−1.
Finally, we asked whether the divergence measurement might have been biased by the presence of MESP2- cells that have low, yet non-null, MESP2 expression. To test this potential effect, we subtracted the typical intensity of MESP2- cells from each Segmentoid video (calculated as the 25th percentile of the distribution of MESP2 in each cell during the first hour of the patterning process), thus removing MESP2- cells. We repeated the divergence analysis in these movies and obtained similar results.
Single-cell RNA sequencing
Line#4 was used for all single-cell RNA experiments. Different dissociation methods were used for various time points. iPSC culture was dissociated with Accutase. (Somitoid) Somitoids-24h (n=96) or Somitoids-48h (n=96) were collected into a 15 ml conical tube and incubated at 37°C with 1ml Accutase for 10min. At the end of incubation, gentle pipetting was performed for further dissociation. Somitoids-66h (n=80) growing on 4 gelatin-coated wells of a 6-well plate were incubated with 0.125% Trypsin-EDTA (Gibco 25200056; diluted with PBS) for 20 min and further dissociated with gentle pipetting after incubation. Somitoid-98h (n=48) growing on 3 gelatin-coated wells were incubated with 0.25% Trypsin-EDTA for 20 min and further dissociated with gentle pipetting. (Segmentoid) 3 wells of Segmentoid-24h were dissociated with Accutase. Segmentoids-48h (n=76) were pooled and incubated with Accutase for 10 min before gentle pipetting. Segmentoids-72h (n=64) or Segmentoids-98h (n=32) were collected into a 15 ml conical tube with as little Matrigel or medium as possible. To dissolve Matrigel, 1ml pre-chilled Cell Recovery Solution (Corning 47743–696) was added. After incubation at 4°C for 10 min, the solution was gently mixed with pipetting and removed without breaking Segmentoids. 1ml fresh Cell Recovery Solution was added before another 10 min incubation at 4°C. After washed with PBS, Segmentoids-72h was incubated with 0.125% Trypsin-EDTA for 20 min at 37°C and Segmentoids-98h with 0.25% Trypsin-EDTA followed by gentle pipetting for thorough dissociation. After dissociation, all timepoints were blocked with 4-fold volume of DMEM/F12, centrifuged at 300 rcf for 4 min, resuspended with 1 ml room-temperature (RT) PBS with 0.04% Bovine Serum Albumin (BSA; Sigma-Aldrich 9048–46-8), filtered with 30 μm filters (CellTrics 04–0042-2316), and collected into 1.5 ml DNA LoBind tubes (Eppendorf 022–43-102–1).
Multiplexing was performed using the 10x Genomics 3’ CellPlex Kit (PN-1000261). Briefly, each sample was centrifuged at RT at 300 rcf for 4 min and the supernatant was removed. Each sample was then resuspended with 100 μl CellPlex oligo and incubated at RT for 5 min. 1 ml ice-cold PBS with 1% BSA was added and centrifuged at 4°C. After supernatant removal, three additional washes at 4°C using 1 ml PBS with 1% BSA were performed. Each sample was then resuspended in PBS with 1% BSA, counted, and kept on ice. Three multiplexed pools were generated. Pool #1 contains an equal cell number of iPSC (oligo301), Somitoid-24h (oligo302), Segmentoid-24h (oligo303), and Segmentoid-48h (oligo304); Pool #2 includes an equal cell number of Somitoid-48h (oligo301, 302), Somitoid-66h (oligo303, 304), and Somitoid-98h (oligo305, 306); Pool #3 includes an equal cell number of Segmentoid-72h (oligo307, 308) and Segmentoid-98h (oligo309, 310).
The Chromium Next GEM Single Cell 3ʹ Kit v3.1 was used to encapsulate single cells. To target 10,000 cell recovery for each pool, 16,500 cells from each pooled sample were loaded to one individual lane of the Chromium chip. The encapsulation and library preparation were performed using 10x Genomics protocols. All libraries were pooled and sequenced on an S1 full flow cell of the NovaSeq 6000 V1.5.
Analysis of Single-cell RNA sequencing data
Transcriptome libraries were demultiplexed and aligned to the Homo sapiens (human) genome assembly GRCh38 (hg38) using cell ranger 6.1.1 with the following parameters: expect-cells 3,000, include-introns FALSE, min-assignment-confidence 0.9. Raw count matrices were converted to anndata objects using Scanpy46 1.8.1 and custom python code. Cells with counts below 4,000, above 65,000 and number of genes below 3,000 and above 8,000 were filtered. Cells with more than 10% of mitochondrial gene expression were also filtered. Raw data were normalized, log-transformed and scaled. Cell cycle genes were regressed linearly, 1,500 highly variables genes were identified, PCA with 50 components was performed along with bbknn47 batch correction before final UMAP embedding. Leiden clustering and ranking of all genes per cluster (wilcoxon test p-value corrected by benjamin-hochberg) allowed us to annotate the cell populations present in the datasets. Matplotlib48 and seaborn were used to plot the number of cells in every cluster. Dot plots were made with a list of signature genes with a gene-based standard scale and plotted using a custom matplotlib color palette on processed data. Density embeddings are made with identified clusters on individual datasets before the merge on the merge embedding. Somite subcluster was done by isolation of the merge somite cluster and re-processing of the data starting from the raws. Velocity was calculated on BAM files using velocyto: spliced, unspliced and ambiguous matrices were merged to the cellranger raw count matrices outputs for the individual Segmentoid timepoints processing to keep the same processing on all the datasets. Scvelo49 was used to process the velocities and differential kinetics was performed to consider the multiple dynamics of the multiple lineages inside the same time point (neural and mesodermal). Then, PAGA-graph was done with velocity-directed edges. CellRank50 was then used to plot smoothed gene expression toward the somite as the terminal population and we used Generalized Perron Cluster Cluster Analysis (GPCCA) as an estimator to predict cell fates using the transitions derived from a mix of the velocity kernel (90%) and the connectivity kernel (10%) that compute transition probabilities based on similarities among cells.
RNA fluorescence in situ hybridization
Fertilized chicken eggs were obtained from Charles River Laboratories. Eggs were incubated at 37°C in a humidified incubator, and the embryos were staged according to Hamburger and Hamilton51. Chicken embryos at Hamburger-Hamilton stage (HH) 10–11 (10–13 somites) were used for RNA fluorescence in situ hybridization (RNA FISH) via hybridization chain reaction (HCR)52. Embryos were collected with ring-shaped filter paper and immediately fixed with 4% formaldehyde-2 mM EDTA-PBS solution. Custom HCR probe set for Gallus gallus MESP2 was designed by Molecular Instruments, Inc. upon request based on the published mRNA sequence on NCBI (NCBI Reference Sequence: XM_003641832.4; probe set size=14). HCR amplifier (B1-Alexa Fluor 594) and buffers were also purchased from Molecular Instruments, Inc. HCR was performed following the standard protocol provided by Molecular Instruments. The CellBrite Red Cytoplasmic Membrane dye (Biotium 30023; 1:1,000) and Hoechst 33342 (Invitrogen H3570; 1:1,000) were added to the final wash of HCR RNA-FISH protocol. Stained embryos were rinsed with PBS, mounted on MatTek glass bottom dish with Fluoromount-G (SouthernBiotech 0100–01), and imaged from the ventral side (Fig.4l) with Zeiss LSM 780 confocal microscope.
E8.5 mouse embryos were generated through timed mating between C57Bl/6J adult mice purchased from Jackson laboratories. Mice were kept under a light cycle from 7am-7pm, a temperature of 20–23°C, and a humidity level of 35%-65%. Embryos were dissected in PBS and fixed overnight at 4oC in RNase-free 4% paraformaldehyde (PFA) in PBS. Embryos were transferred to 100% methanol (MeOH) where they were stored at -20oC until processing. Custom HCR probe set for Mus musculus Mesp2 was designed by Molecular Instruments, Inc. upon request based on the full-length published mRNA sequence from UCSC genome browser (Reference Sequence: NM_008589; probe set size = 20). The samples were processed for HCR according to the E9.5 mouse embryo whole-mount HCR protocol on the Molecular Instruments website. All steps were performed on an orbital shaker with gentle shaking sufficient to agitate the solution without damaging the embryos. Embryos were mounted in 1% low-melt agarose (RPI A20070–25.0) in water on glass-bottom dishes (MatTek P35G-1.5–20-C) and imaged at 10X and 60X on an FV1000 confocal microscope. All mice were handled according to local regulations, consistent with national and international guidelines. We complied with all relevant ethical regulations. The study protocol was approved by Brigham and Women’s Hospital IACUC/CCM (protocol number 2016N000478). Sample sizes were not estimated, nor were randomization or blinding performed.
Half-embryo in vitro culture
Fertilized transgenic chicken eggs, which express cytoplasmic GFP, were obtained from Dr. Susan Chapman’s laboratory at Clemson University53. Half-embryo in vitro culture experiment was performed as previously described35 with some modifications. GFP-chick embryos between 10–19 somite stage were bilaterally divided with the custom-made microsurgical scalpel. Right sides were fixed immediately with 4% formaldehyde-2 mM EDTA-PBS solution. Left sides were cultured in vitro on the membrane filter (Millipore HATF01300) floating on the Dulbecco’s Modified Eagle Medium (Gibco 11965–092) supplemented with 10% fetal bovine serum (R&D Systems S11150) and 1% Pen/Strep (Gibco 15140122). After 45 min culture at 37°C, the left sides were also fixed. Next, HCR RNA FISH for Gallus gallus MESP2 was performed as described above. The stained left-right pairs were mounted on the glass bottom dish (MatTek Corporation) with Fluoromount-G (SouthernBiotech 0100–01) and imaged from the dorsal side with Zeiss LSM 880 confocal microscope at the NeuroTechnology Studio (Brigham and Women’s Hospital).
Statistics and reproducibility
Details of statistical analyses are indicated in the figure legends. More than five independent experiments were repeated for data shown in Fig.1d,o,p,q, Fig.2a,d,q, Fig.4a,d,h,j, Extended Data Fig.1a,c,l, Extended Data Fig.2e,f,j,k,l, Extended Data Fig.4h, and Extended Data Fig.9b,c. Three independent experiments were repeated for Fig.2j,k,m,n. More than five embryos were repeated for Fig.4l and three embryos for Extended Data Fig.10a,b. Fig.4m and Extended Data Fig.10c represent three out of ten embryos that captured the peak of the broad band phase of MESP2 expression at Time 0. Step-by-step protocols of Somitoid and Segmentoid are available on Protocol Exchange54.
Extended Data
Extended Data Fig.1. Characterization of the Somitoid model.
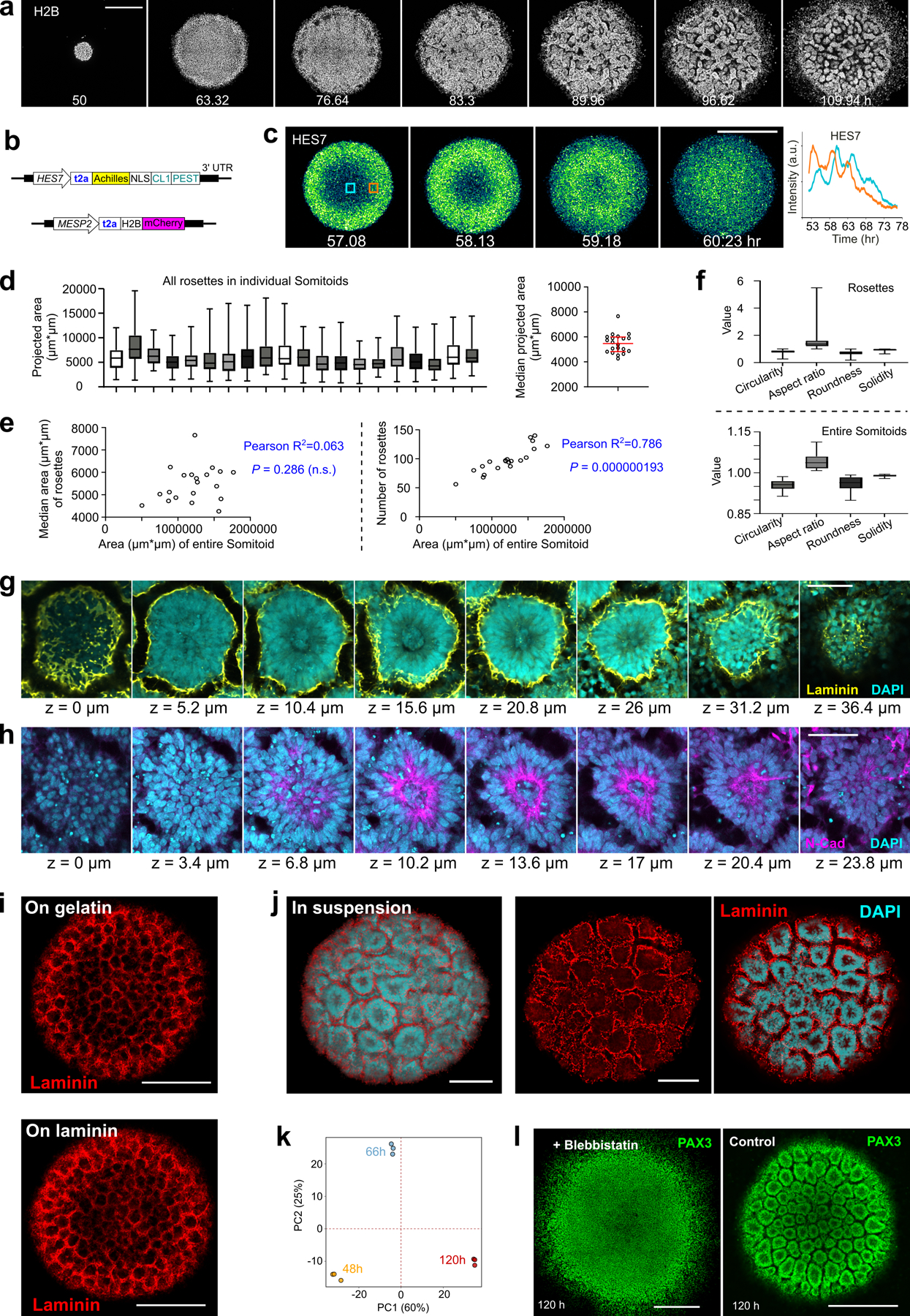
a, Time lapse confocal images of H2B-mCherry in a spreading Somitoid. b, Illustration of the design of the HES7/MESP2 double-reporter cell line. c, Left, time-lapse confocal images of HES7 wave; Right, temporal profiles of HES7 reporter in two different regions indicated by the blue and orange boxes. d, Left, box plots of projected areas of all rosettes in individual Somitoids. Right, plot of median rosette area of each Somitoid (n=20 Somitoids). Red bars indicate median with interquartile range. e, Correlation analysis (n=20 Somitoids; two-sided) between the entire Somitoid area and median rosette area (left); between the entire Somitoid area and total rosette number (right). f, Shape descriptors of individual rosettes (top) and entire Somitoids (bottom). n=1,957 rosettes from 20 Somitoids. g, h, Confocal slices from the bottom (z=0 µm) to the top of a rosette in 120 h Somitoid stained with Laminin (g) and N-Cadherin (h) (n=2 Somitoids). i, Representative images of a Somitoid cultured on gelatin (n=5 Somitoids) or laminin (n=5 Somitoids) coated surface, stained with Laminin. j, 3D reconstruction image of a Somitoid cultured in suspension (left; n=2 Somitoids) and a confocal section (right), stained with Laminin. k, Principal components analysis using the same RNA sequencing datasets shown in Fig. 1h. l, Confocal images of 120 h PAX3-reporting Somitoids treated with 5 µM Blebbistatin (left) and control (right). In box-and-whiskers plots, the middle hinge corresponds to median, the lower and upper hinges correspond to the first and third quartiles, respectively, and the lower and upper whiskers correspond to the minimum and maximum, respectively. Scale bars represent 500 µm (a, c, i, l), 50 µm (g, h) and 100 µm (j).
Extended Data Fig.2. Antero-Posterior patterning in Somitoids.
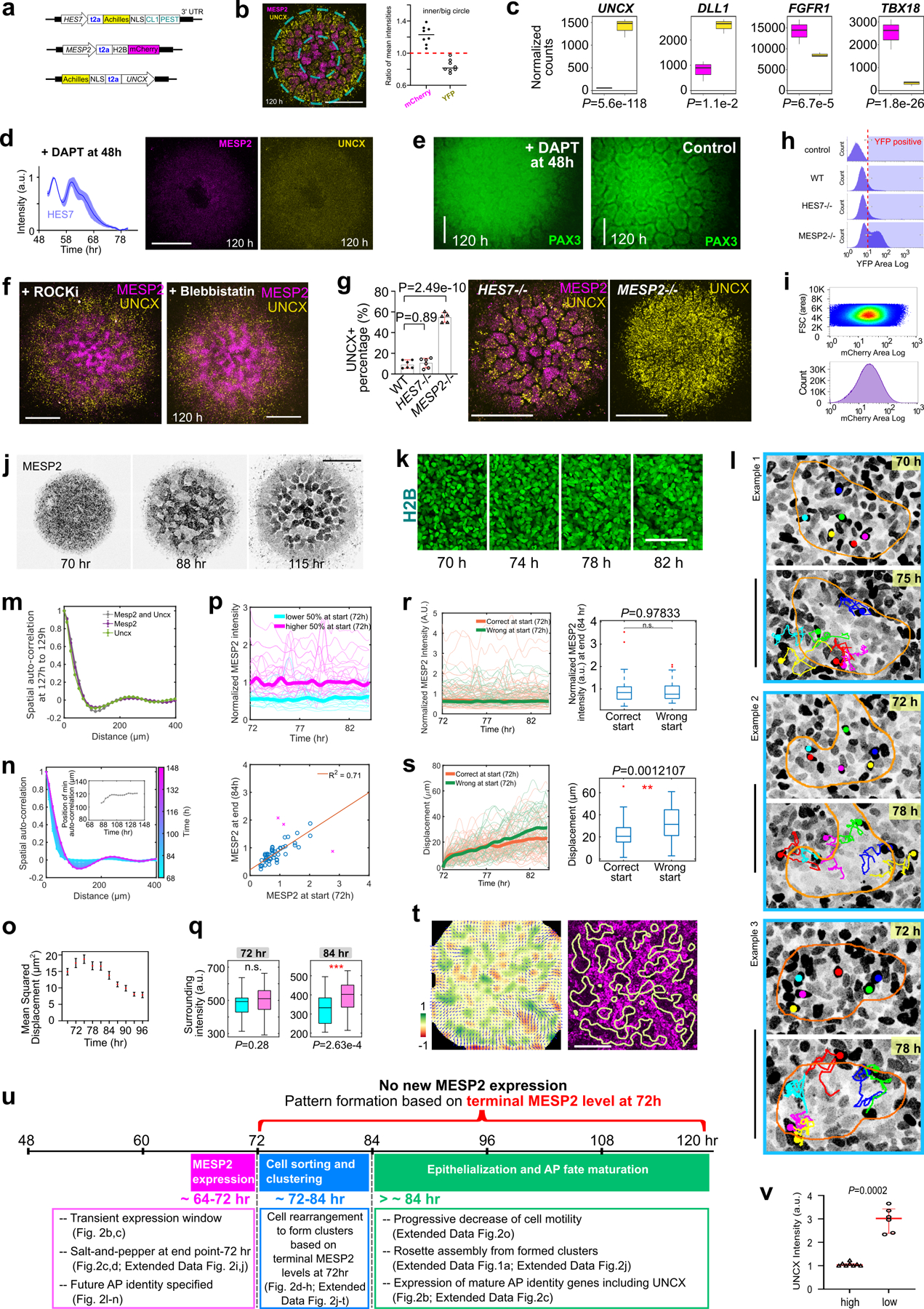
a, Illustration of the design of the HES7/MESP2/UNCX triple-reporter cell line. b, Ratio of mean mCherry or YFP intensities in the center circle vs in the big circle (n=8 Somitoids and the bars indicate median). c, Normalized RNA counts of selected polarity genes in cell fractions separated by flow cytometry, as measured by RNA sequencing (n=3 independent experiments, 96 Somitoids in each n). Cells with top 10% mCherry fluorescence are shown on the left (magenta) and top 10% YFP fluorescence on the right (yellow). All four genes were identified as differentially expressed genes by DESeq2 using the Wald test (two-sided). d, Temporal plot of HES7 reporter (mean±s.d., n=3 Somitoids) and images of an UNCX and MESP2 reporting Somitoid treated with 50 µM DAPT added at 48 h. e, Wide-field images of PAX3-reporting Somitoids treated with 50 µM DAPT (left) since 48 h and control (right). f, Maximum-z-projection confocal images of UNCX and MESP2 reporting Somitoids treated with 10 µM ROCKi (left) or 5 µM Blebbistatin (right) since 48 h. g, Left, percentage of UNCX-positive cells characterized by flow cytometry in 120 h WT (n=6 experiments), HES7-null (n=6 experiments), and MESP2-null (n=5 experiments) Somitoids; Data are represented as mean±s.d., one-way ANOVA, compared with WT, P = 0.89 (HES7-null); 2.49e-10 (MESP2-null). Right, images of MESP2 and UNCX reporters in HES7-null Somitoids, and UNCX reporter in MESP2-null Somitoids. h, Histograms of flow cytometry analysis of UNCX-YFP in 120 h Somitoids (control, WT, HES7-null, and MESP2-null cell lines) with debris and doublets removed. Control is the parental NCRM1 cell line. Fractions on the right side of the red dotted line in the histograms are defined as YFP-positive. i, Scattered plot (top) and histogram (bottom) of flow cytometry analysis on MESP2-mCherry Somitoids at 72 h with debris and doublets removed. j, Time-lapse images of MESP2 reporter in a Somitoid. k, Time-lapse maximum-z-projection confocal images of H2B-GFP in the same region of a Somitoid as in Fig.2d. l, Cell tracks of MESP2-high cells overlayed on images of MESP2 reporter. The orange outlines represent the forming MESP2-low regions. m, Spatial auto-correlation (sole MESP2 signal, sole UNCX signal or them combined together) once rosettes are formed (representative example from n=3 Somitoids). n, Additional example of spatial auto-correlation analysis and abscissa-position of the auto-correlation trough (inset) of MESP2/UNCX double reporting Somitoid over time. o, Temporal plot (mean±95%CI) of mean squared displacement (n=3,422 tracks from 2 Somitoids). p, Additional example of normalized temporal profiles of MESP2 reporter in individual cells (top; n=52 cells from one Somitoid), and correlation analysis of MESP2 intensities at 72 h and 84 h (bottom; F-test, one-sided, P = 4.67e-14 after removing 3 outliers identified by calculating Mahalanobis distance, as explained in Methods, marked by magenta cross). Temporal profiles are colored based on relative MESP2 intensity among tracked cells at 72 h, with higher 50% in magenta and lower 50% in cyan throughout the time window. q, Surrounding MESP2 intensity (Methods) of tracked cells at 72 h and 84 h (n=98 cells from two Somitoids; unpaired two-tailed t-test). Cells at both time points are grouped based on relative MESP2 intensity at 72 h, with lower 50% on the left (cyan) and higher 50% on the right (magenta). r, Left, temporal profile of MESP2 intensity in cells starting in a correct (orange) or wrong (green) region (Method). Right, end-time-point MESP2 intensity of cells with correct or wrong start. n=98 cells from two Somitoids; unpaired two-tailed t-test. s, Left, temporal profile of displacement in cells starting in a correct (orange) or wrong (green) region (Method). Right, end-time-point displacement of cells with correct or wrong start. n=98 cells from two Somitoids; unpaired two-tailed t-test. t, Velocity field (arrows) and the corresponding divergence (heatmap) of Particle Image Velocimetry analysis (left) on an additional Somitoid and regions of positive divergence overlayed on the MESP2 reporter image (right; yellow outlines). u, Summary of MESP2 expression and pattern formation processes in the timeline of the Somitoid differentiation. v, Quantification of UNCX reporter in MESP2-high (n=8 re-aggregates from 3 experiments) and MESP2-low (n=6 re-aggregates from 3 experiments) re-aggregates in Fig.2l–n, paired two-sided t-test. In all box and whisker plots, the center indicates the median, the upper bound indicates 75th percentile, and the lower bound indicates 25th percentile. The maxima and minima of the whiskers represent the most extreme non-outlier data points. The outliers are defined as data points greater than the upper bound or smaller than the lower bound by more than 1.5 times the interquartile range. Scale bars represent 500 µm (b, d, f, g, j), 200 µm (e, t), and 100 µm (k, l).
Extended Data Fig.3. Differential gene expression during cell sorting and perturbations.
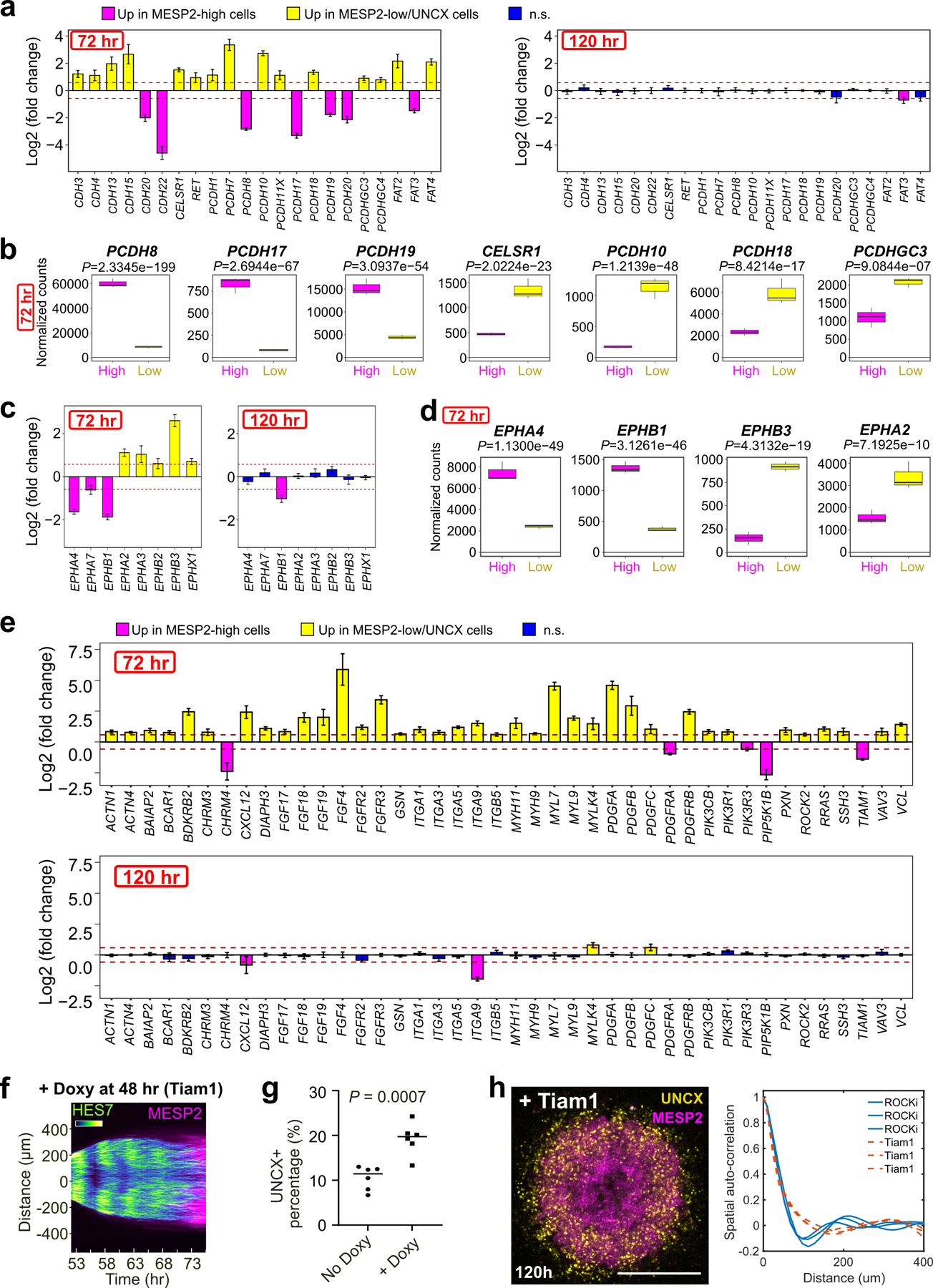
a, Expression fold change plots of selected adhesion proteins between MESP2-low vs MESP2-high cells at 72 h, or between MESP2-high vs UNCX cells at 120 h (n=3 independent experiments for each time point, with 96 Somitoids in each n). The genes plotted are differentially expressed cadherin and protocadherin encoding genes from the comparison between MESP2-low and MESP2-high cells at 72 h. The dashed (dark red) lines represent log2 fold change values of -0.58 and 0.58. The error bar represents the estimated standard error for the log fold change from the model (DESeq2) which is represented as the center of the bar. Genes with fold changes greater than 1.5 (above or below the dash line) and padj < 0.05 (estimated by Deseq2 using two-sided Wald test) are considered to be differentially expressed and colored in either yellow (upregulated in MESP2 low cells) or magenta (upregulated in MESP2 high cells). Genes in blue color from the comparison between MESP2-high and UNCX cells at 120 h are non-differentially expressed genes. The exact P values for each gene are shown in Supplementary Table 3. b, Normalized RNA counts of selected genes encoding adhesion proteins in MESP2-high and MESP2-low cell fractions at 72 h (n=3 independent experiments for each time point, with 96 Somitoids in each n; DESeq2 with two-sided Wald test). Cells with top 10% mCherry fluorescence are shown on the left (magenta) and top 10% YFP fluorescence on the right (yellow). c, Expression fold change plots of selected Ephrin protein encoding genes between MESP2-low vs MESP2-high cells at 72 h, or between MESP2-high vs UNCX cells at 120 h (n=3 independent experiments for each time point, with 96 Somitoids in each n). The error bar represents the estimated standard error for the log fold change from the model (DESeq2) which is represented as the center of the bar. Genes with fold changes greater than 1.5 (above or below the dash line) and padj < 0.05 (estimated by Deseq2 using two-sided Wald test) are considered to be differentially expressed and colored in either yellow (upregulated in MESP2 low cells) or magenta (upregulated in MESP2 high cells). Genes in blue color from the comparison between MESP2-high and UNCX cells at 120 h are non-differentially expressed genes. The exact P values for each gene are shown in Supplementary Table 3. d, Normalized RNA counts of selected genes encoding Ephrin proteins in MESP2-high and MESP2-low cell fractions at 72 h (n=3 independent experiments for each time point, with 96 Somitoids in each n; DESeq2 with two-sided Wald test). e, Expression fold change plots of selected cytoskeleton regulating proteins between MESP2-low vs MESP2-high cells at 72 h, or between MESP2-high vs UNCX cells at 120 h (n=3 independent experiments for each time point, with 96 Somitoids in each n). The error bar represents the estimated standard error for the log fold change from the model (DESeq2) which is represented as the center of the bar. Genes with fold changes greater than 1.5 (above or below the dash line) and padj < 0.05 (estimated by Deseq2 using two-sided Wald test) are considered to be differentially expressed and colored in either yellow (upregulated in MESP2 low cells) or magenta (upregulated in MESP2 high cells). Genes in blue color from the comparison between MESP2-high and UNCX cells at 120 h are non-differentially expressed genes. The exact P values for each gene are shown in Supplementary Table 3. After differential gene expression analysis using Deseq2, differentially expressed genes from the MESP2-high vs MESP2-low comparison (72 h) were used to do KEGG functional analysis. The 42 genes plotted represent those that appear in the KEGG pathway “hsa04810” (Regulation of actin cytoskeleton). f, Kymograph of HES7 and MESP2 reporters obtained from a line scan across the center of a Somitoid overexpressing Tiam1 induced by Doxycycline addition at 48 h. g, percentage of UNCX-positive cells characterized by flow cytometry in 120 h control and Somitoids overexpressing Tiam1 induced by Doxycycline addition at 48 h. Bars represent median. Unpaired two-tailed t-test n=6 replica from 2 independent experiments, with 12–18 Somitoids in each replica. h, Left, maximum-z-projection confocal image of a MESP2/UNCX-reporting Somitoid at 120 h, overexpressing Tiam1 induced by Doxycycline addition at 48 h. Right, spatial auto-correlation analysis of MESP2 and UNCX signals (n=3 Somitoids for each condition). All scale bars represent 500 µm.
Extended Data Fig.4. Characterization of the Segmentoid model.
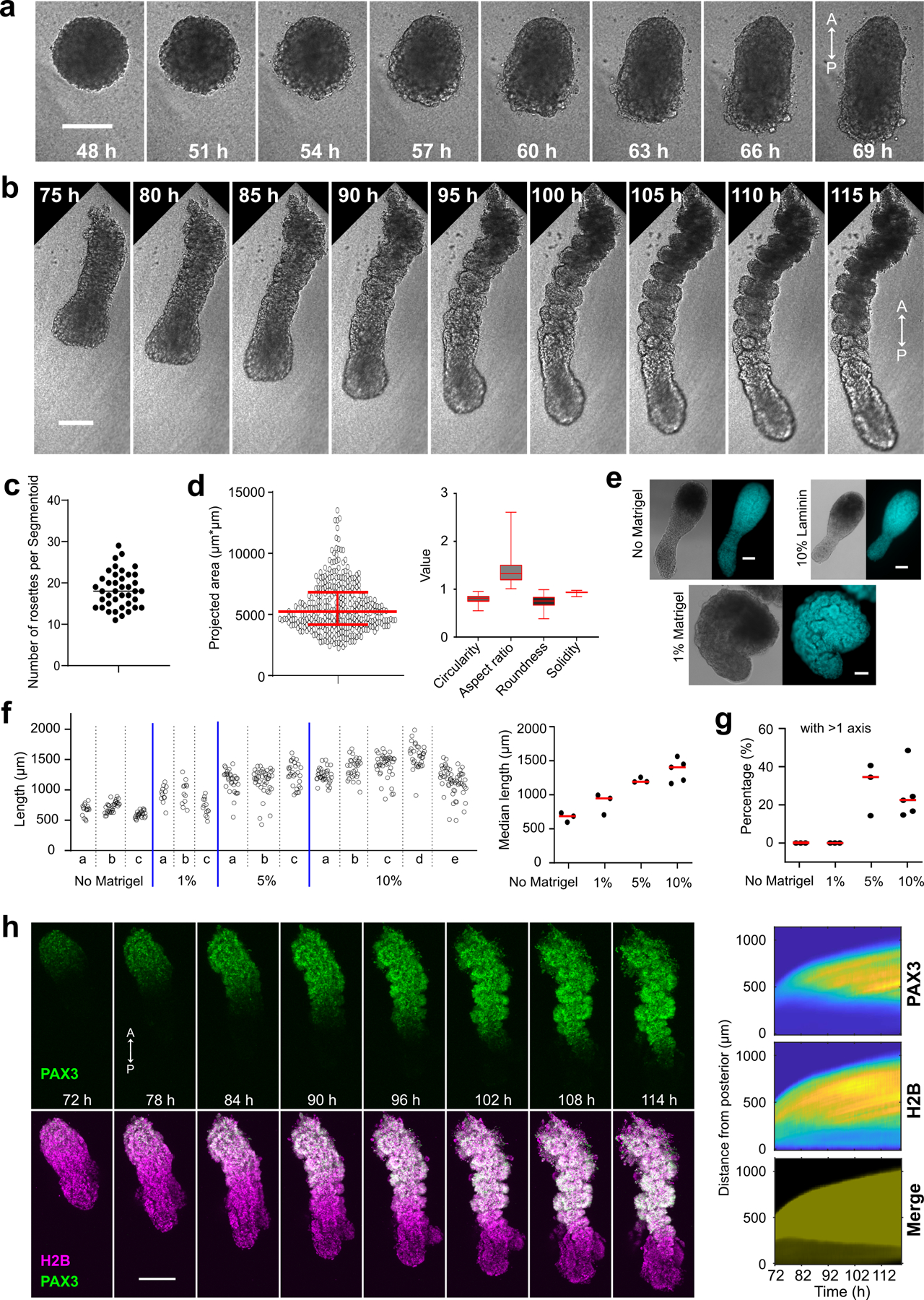
a, b, Time-lapse bright field images of the Segmentoid model. A, anterior; P, posterior. c, Number of rosettes in each Segmentoid (n=40 Segmentoids) with the bar representing median. d, Left, projected areas of rosettes in Segmentoids (n=345 rosettes from 20 Segmentoids). Red bars indicate median with interquartile range. Right, shape descriptors of rosettes in Segmentoids (n=345 rosettes from 20 Segmentoids). The middle hinge corresponds to median, the lower and upper hinges correspond to the first and third quartiles, respectively, and the lower and upper whiskers correspond to the minimum and maximum, respectively. e, Representative bright-field and DAPI images of organoids without Matrigel, with 10% Laminin supplemented, and embedded in 1% Matrigel (n>10 Segmentoids for each condition). f, Length of organoids in suspension (n=3 experiments) or embedded in Matrigel (1%, 5%, n=3 experiments; 10%, n=5 experiments). Individual structure lengths in each experiment are plotted on the left. The median lengths of each experiment are plotted on the right with red bars indicating median, ordinary one-way ANOVA, P=0.26 (1%), 0.0023 (5%), 0.00014 (10%) compared with No Matrigel condition. g, Percentage of structures with more than 1 axis in different conditions with red bars indicating median. Ordinary one-way ANOVA, P>0.999 (1%), =0.0154 (5%), 0.0205 (10%) compared with No Matrigel condition. h, Left, time-lapse maximum-z-projection confocal images of PAX3-YFP reporter (top) and PAX3-YFP merged with H2B-mCherry (bottom) in a Segmentoid. Right, kymographs of PAX3 reporter (top), H2B (middle), and merged channels (bottom) in the same Segmentoid. Segmentoids are aligned to the posterior tip at each time point. Scale bars represent 200 µm (a, b, h) and 100 µm (e).
Extended Data Fig.5. Expression of TBXT and SOX2 in Segmentoids.
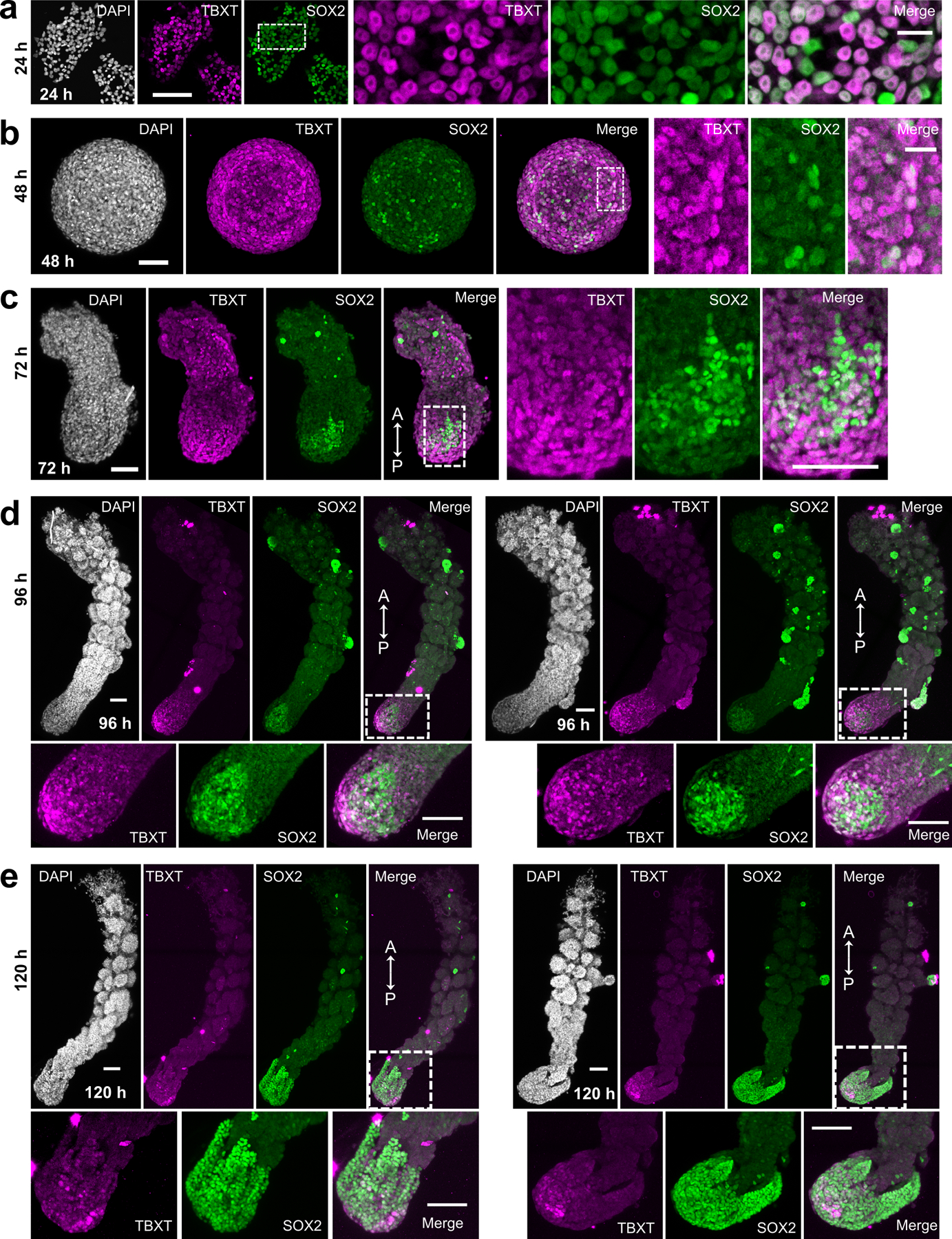
Confocal images of immunostaining of TBXT and SOX2 at 24 h (a), 48 h (b), 72 h (c), 96 h (d), and 120 h (e) of the Segmentoid model. Representative maximum-z-projection images are shown from b-e. 48 h, n=3 Segmentoids; 72 h, n=11 Segmentoids; 96 h, n=17 Segmentoids; 120 h, n=21 Segmentoids, with 7 Segmentoids still showing apparent TBXT and SOX2 double positive pole. A, anterior; P, posterior. Scale bars (a, b) represent 100 µm and 20 µm in corresponding enlarged views; Scale bars (c, d, e) represent 100 µm.
Extended Data Fig.6. Single-cell RNAseq of the Segmentoid model.
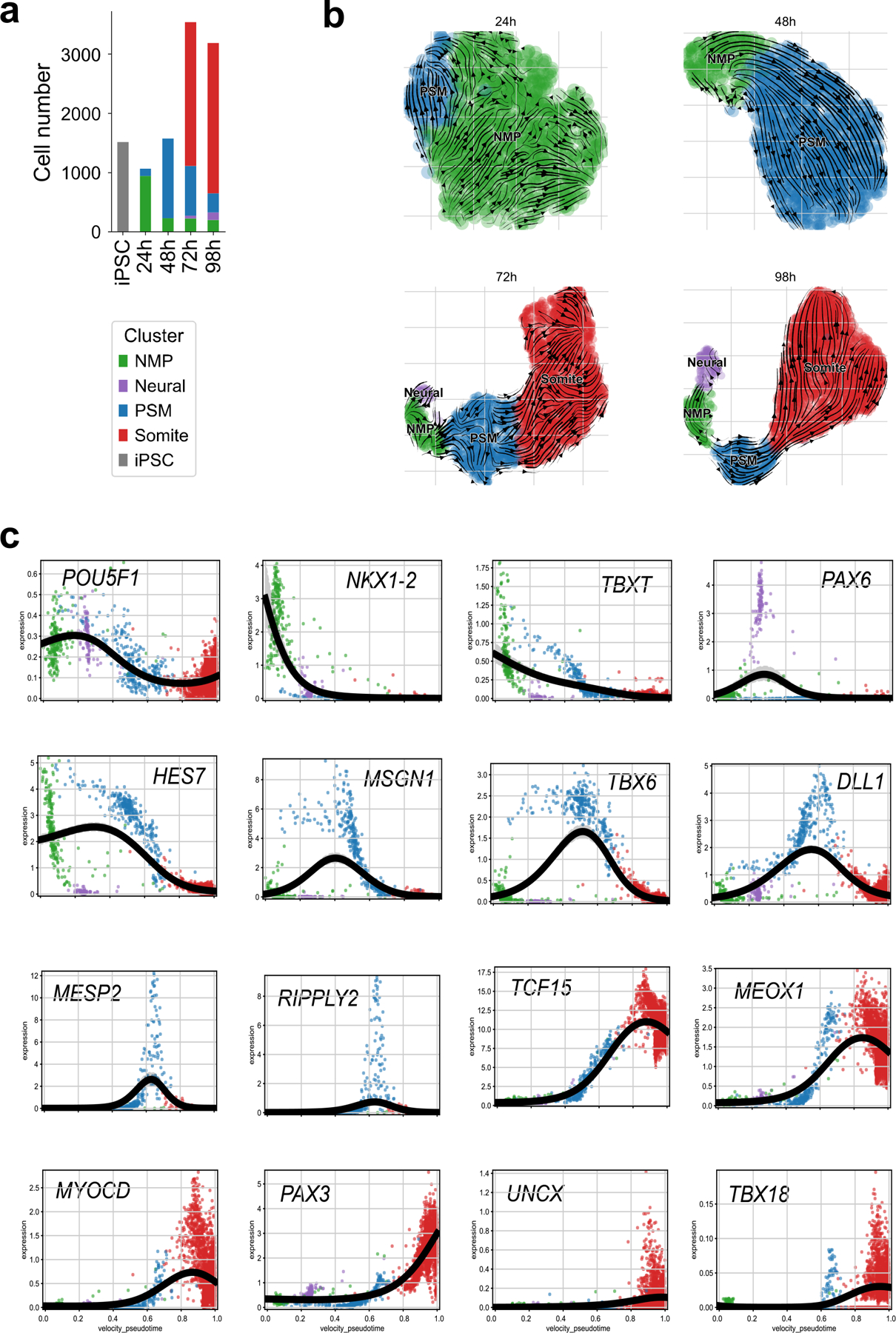
a, Proportion of cell types identified with Leiden clustering at different timepoints of the Segmentoid model. b, Stream plots of velocities on the UMAP after correction for differential kinetics recapitulating trajectory of cell types at various timepoints. c, Signature gene expression trends (Log2/Normalized) toward somite as the specific terminal population.
Extended Data Fig.7. Single-cell RNAseq of the Segmentoid and the Somitoid model.
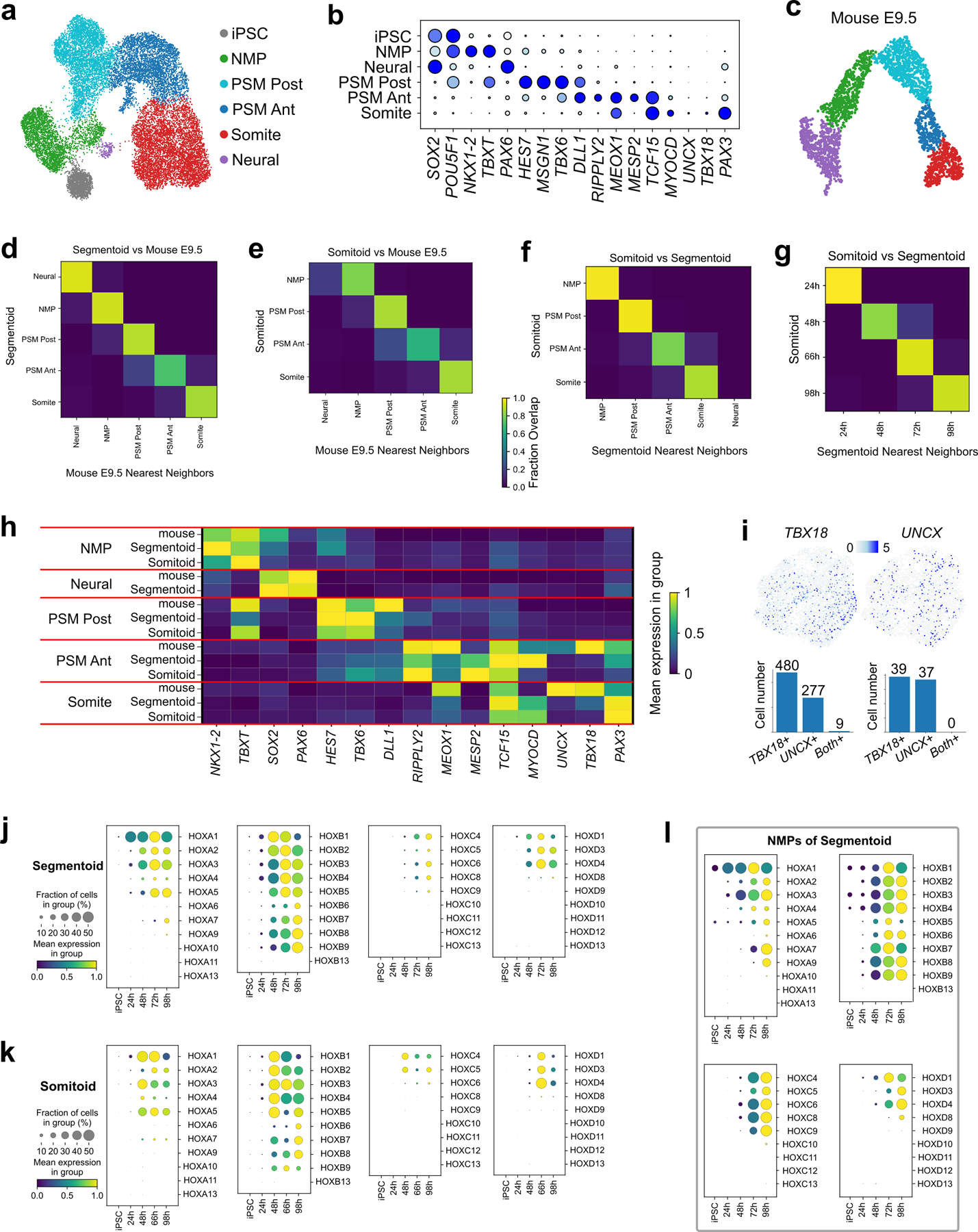
a, UMAP embedding of cells merged from both models (19,551 cells) colored with cell types identified with Leiden clustering. b, Dot plot of selected genes in cell type clusters from both models. c, Machine-learning classification of a previous data set of E9.5 mouse embryo. d, e, Classifier analysis on cell types comparing the in vitro models with mouse E9.5. A k-NN classifier trained on clusters of mouse clusters was used to predict identities of the human in vitro models. f, g, Classifier analysis on cell types (f) and time points (g) comparing Somitoids with Segmentoids. h, Mean expression heatmap of selected genes in the three datasets. i, Top, somite sub-cluster highlighting cells expressing TBX18 (left) and UNCX (right); Bottom, number of cells expressing TBX18, UNCX, or both in Segmentoids (left) and Somitoids (right). j, k, l, Dot plots of HOX-family genes expression at various timepoints of the Segmentoid model (j), the Somitoid model (k), and the NMP cells of the Segmentoid (l). The mean expression of each cluster is scaled per gene.
Extended Data Fig.8. Antero-Posterior patterning and the segmentation clock.
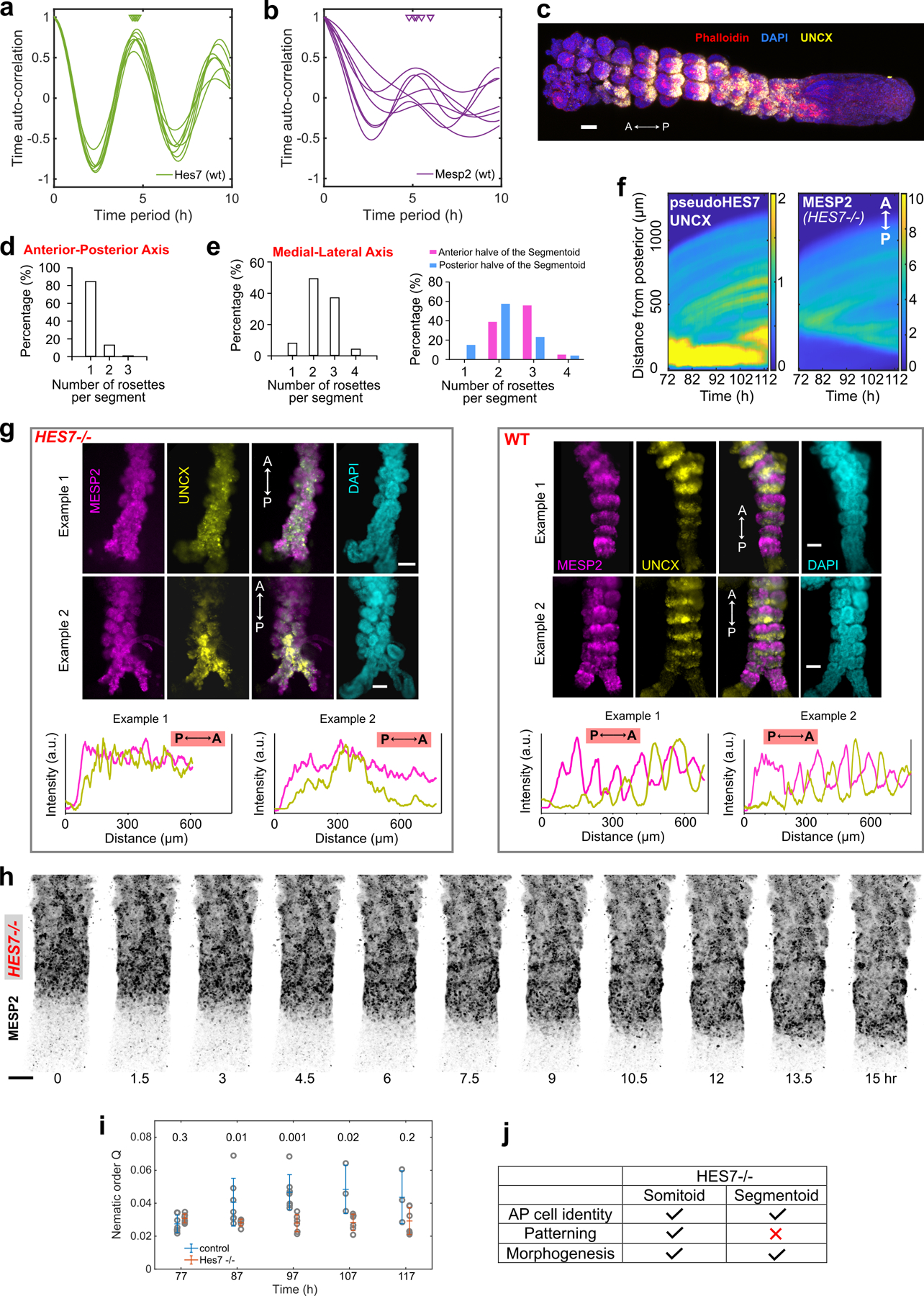
a, b, Time auto-correlation of HES7 (a) and MESP2 (b) reporter oscillations in individual WT Segmentoids. Triangles indicate auto-correlation peaks, which in turn indicate oscillation period. c, Merged maximum-z-projection confocal image of a Segmentoid with UNCX reporter, DAPI, and Phalloidin staining (n>10 Segmentoids). d, Distribution of rosette numbers in each segment along the anterior-posterior axis. A segment is defined as the posterior boundary of the UNCX stripe to that of the next posterior UNCX stripe. The maximal number of rosettes along the AP axis observed was used to represent the entire segment (n=24 Segmentoids). e, Distribution of rosette numbers in each segment along the medial-lateral axis (left; n=25 Segmentoids). The data is re-grouped based on relative AP location in the Segmentoids (right). f, Kymographs of reporters for pseudoHES7, UNCX, and MESP2 in the same HES7-null Segmentoid. g, Wide-field images and graphs of reporter intensities from posterior (P) to anterior (A) end along 120 h HES7-null (left) and WT (right) Segmentoids (n>10 Segmentoids for each condition). h, Time-lapse, maximum-z-projection confocal images of MESP2 reporter in a HES7-null Segmentoid. i, Average nematic order of MESP2/UNCX signals in WT and HES7-null Segmentoids as a function of time (mean±s.d; n=7 WT Segmentoids and n=6 HES7-null Segmentoids). Statistics was performed with a Wilcoxon rank-sum test (two-sided) and P-value is shown. j, Summary of HES7-null phenotypes in Somitoid and Segmentoid. All scale bars represent 100 µm.
Extended Data Fig.9. Antero-Posterior patterning and cell sorting.
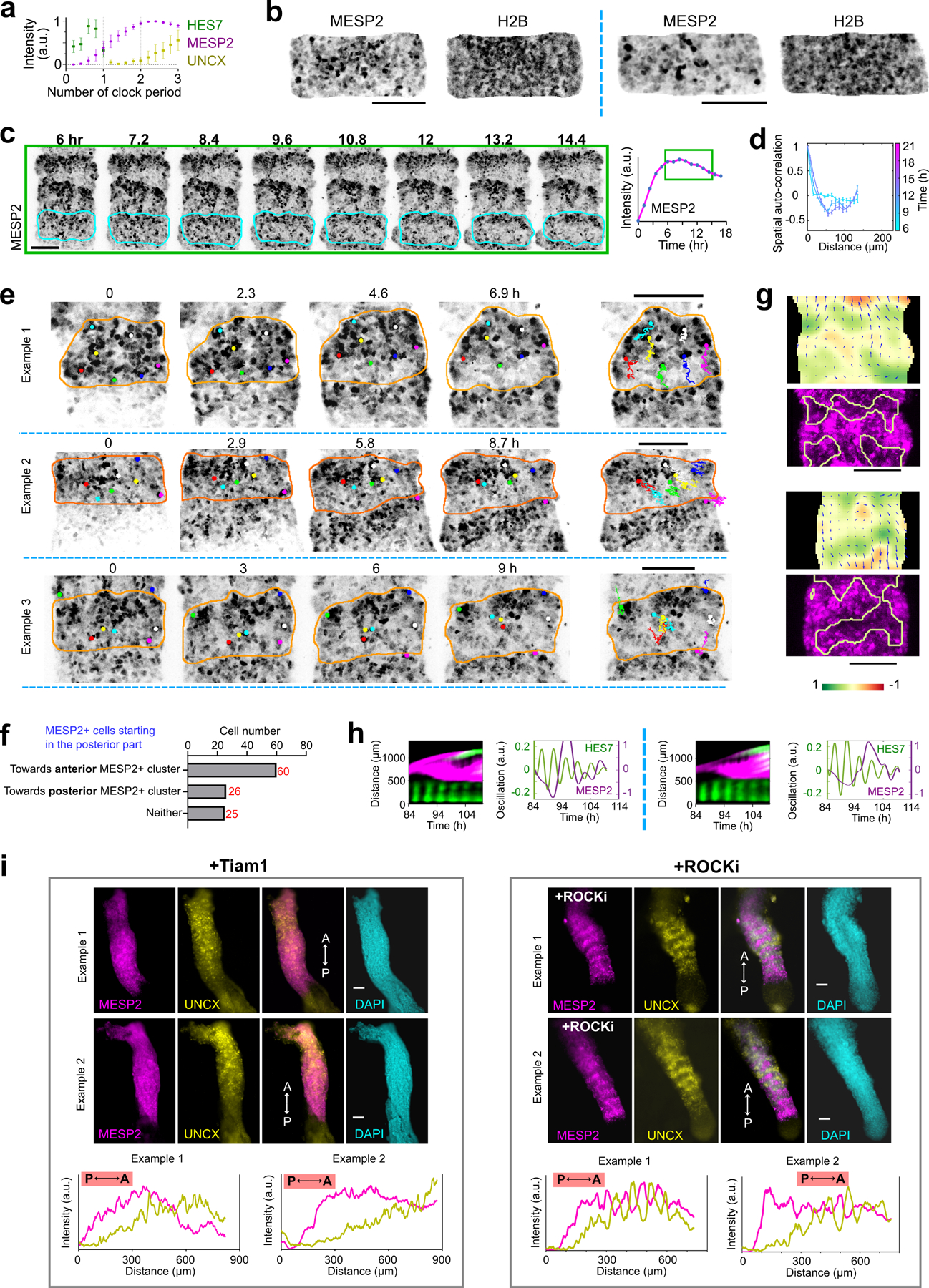
a, Reporter dynamics in forming segments aligned according to phases of HES7 oscillation (n=6 segments in 2 Segmentoids). Data are represented as mean±s.d. b, Representative images of MESP2 reporter and H2B-GFP in a segment at the salt and petter stage. c, Left, Time-lapse, maximum-z-projection confocal images of MESP2 reporter in the same Segmentoid in Fig.4d. Right, temporal profile of MESP2 intensity in the forming segment outlined in cyan. Green solid-line boxes indicate the corresponding time points. d, Representative example of spatial auto-correlation analysis (mean±s.e.m) of MESP2 and UNCX reporters as a function of time in a developing segment (n=6 segments from 2 Segmentoids). e, Cell tracking examples of MESP2-high cells. Dots of the same color represent the same cell and the orange outlines indicate the forming segment. f, Movement classification of tracked MESP2-high cells starting in the posterior part of the segment (n=111 cells from 10 segments in 5 Segmentoids). g, Additional examples of velocity field (arrows) and the corresponding divergence (heatmap) of Particle Image Velocimetry analysis and regions of positive divergence overlayed on the MESP2 reporter image. h, Additional examples of merged kymographs of HES7/UNCX (green) and MESP2 (magenta) in a Segmentoid overexpressing Tiam1 induced by Doxycycline addition at 72 h, as well as HES7 and MESP2 oscillations. i, Wide-field images and graphs of reporter intensities from posterior (P) to anterior (A) end along 120 h Segmentoids, overexpressing Tiam1 induced by Doxycycline addition at 72 h (left; n>10 Segmentoids) or treated with 10 µM ROCKi (right; n>10 Segmentoids). All scale bars represent 100 µm.
Extended Data Fig.10. Embryos stained with Mesp2 HCR probe.
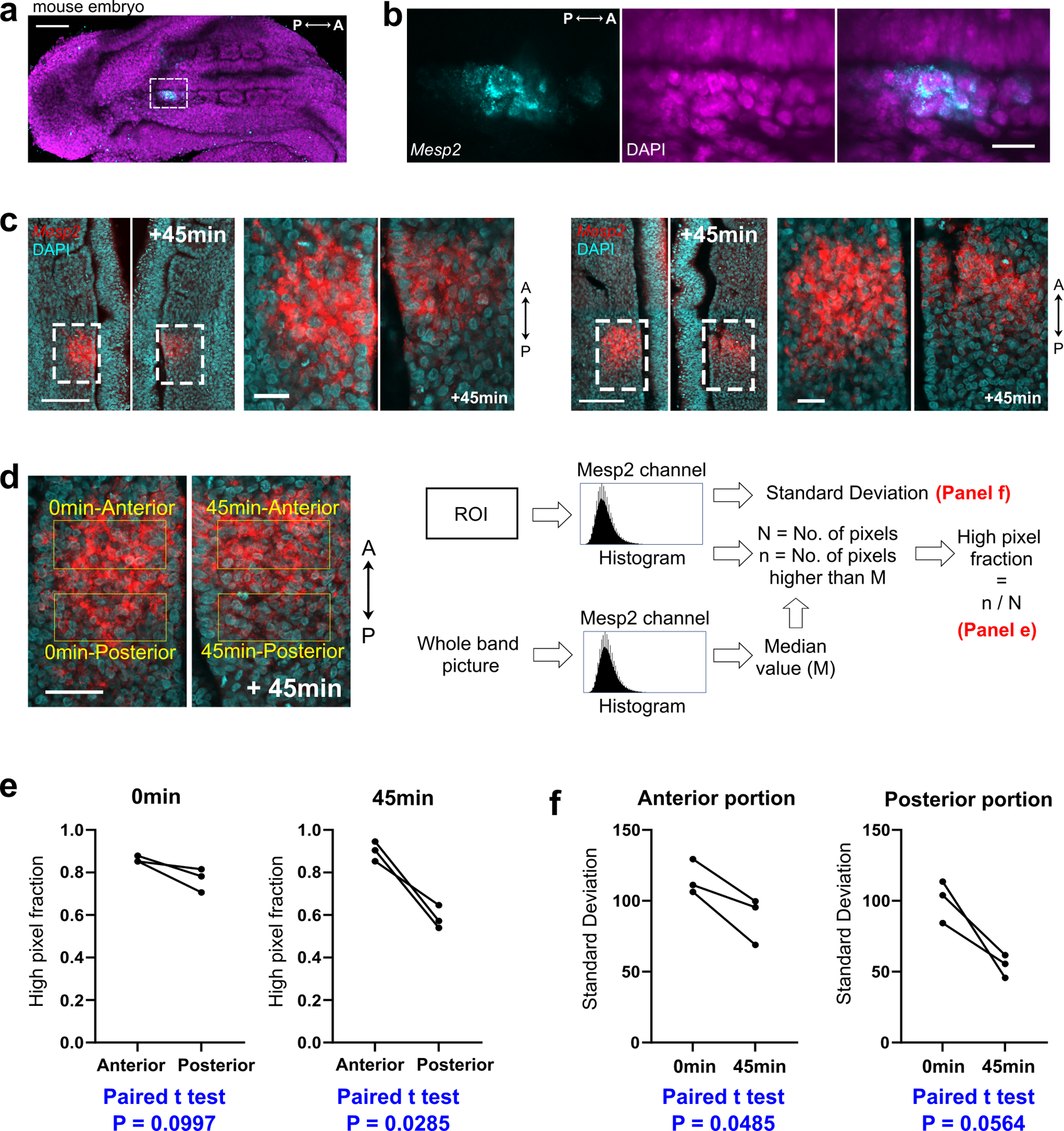
a, Merged maximum-z-projection confocal image of a mouse embryo stained with Mesp2 HCR probe (cyan) and DAPI (magenta). b, Enlarged view of the region indicated by the dotted-line box in a. c, Additional half-embryo pairs stained with MESP2 HCR probe (red) and DAPI (cyan). d, Schemes for quantification. e, High pixel fractions of the anterior and posterior portions of the MESP2 bands at 0 and 45 min. Paired t-test, two-sided. f, Standard Deviations of pixel values in the anterior/posterior portions of the MESP2 bands at 0 and 45 min. Paired t-test, two-sided. Three out of 10 embryos at Time 0 captured the peak of the broad band phase of MESP2 expression, with the criteria that the expression domain roughly occupied the whole segment and MESP2 total intensity was not significantly increased at 45 min. Scale bars represent 100 µm (a, d) and 20 µm (b); 100 µm and 20 µm in corresponding enlarged views (c).
Supplementary Material
Acknowledgements
We thank Sudhir Gopal Tattikota from N. Perrimon lab for help with scRNA sequencing experiments. We thank the Biopolymers Facility at Harvard Medical School for providing 10X Genomics Chromium Controller instrument access and sequencing consultation. We thank the NeuroTechnology Studio at Brigham and Women’s Hospital for providing microscope access and consultation on data acquisition and data analysis. We thank the Harvard Neurobiology Imaging Facility for access to the FV1000 confocal microscope (NINDS P30 Core Center grant NS072030). We thank S. Megason for critical reading of the manuscript. Research in the Pourquié lab was funded by a grant from the National Institute of Health (5R01HD085121). Y.D. is supported by Fondation pour la Recherche Médicale (FRM) PLP2020100012456.
Footnotes
Competing interests
The authors declare the following competing interests: O.P. is scientific founder of Anagenesis Biotechnologies. All other authors declare no competing interests.
Code availability
Codes for scRNA analyses can be accessed at: https://github.com/PourquieLab/Miao_Djeffal_2022.git.
Codes for quantitative image analyses are at https://github.com/desimonea/MiaoSomitogenesis2022.
Data availability
Single-cell RNA sequencing data has been deposited to NCBI Gene Expression Omnibus (GEO): GSE195467 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195467) . Bulk RNA sequencing data has been deposited to GEO: GSE220634 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220634). The Homo sapiens (human) genome assembly (GRCh38) is from: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/. The mouse embryo scRNAseq data is from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114186. The RNA sequencing data of iPSCs is from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164874. Source data are provided with this paper. All materials used in this study including stem cell lines are available by request from the corresponding author.
References
- 1.Hubaud A & Pourquié O Signalling dynamics in vertebrate segmentation. Nat. Rev. Mol. Cell Biol 15, 709–721 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Saga Y The mechanism of somite formation in mice. Curr. Opin. Genet. Dev 22, 331–338 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Fleming A, Kishida MG, Kimmel CB & Keynes RJ Building the backbone: the development and evolution of vertebral patterning . Development 142, 1733–1744 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Kuan C-YK, Tannahill D, Cook GMW & Keynes RJ Somite polarity and segmental patterning of the peripheral nervous system. Mech. Dev 121, 1055–1068 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Oates AC, Morelli LG & Ares S Patterning embryos with oscillations: structure, function and dynamics of the vertebrate segmentation clock. Development 139, 625–639 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Saga Y, Hata N, Koseki H & Taketo MM Mesp2: a novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev 11, 1827–1839 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Morimoto M, Takahashi Y, Endo M & Saga Y The Mesp2 transcription factor establishes segmental borders by suppressing Notch activity . Nature 435, 354–359 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Keynes RJ & Stern CD Segmentation in the vertebrate nervous system . Nature 310, 786–789 (1984). [DOI] [PubMed] [Google Scholar]
- 9.Schoenwolf GC, Bleyl SB, Brauer PR & Francis-West PH Larsen’s Human Embryology E-Book (Elsevier Health Sciences, 2020). [Google Scholar]
- 10.Diaz-Cuadros M et al. In vitro characterization of the human segmentation clock. Nature (2020) doi: 10.1038/s41586-019-1885-9. [DOI] [PMC free article] [PubMed]
- 11.Matsuda M et al. Recapitulating the human segmentation clock with pluripotent stem cells. Nature 580, 124–129 (2020). [DOI] [PubMed] [Google Scholar]
- 12.Chu L-F et al. An In Vitro Human Segmentation Clock Model Derived from Embryonic Stem Cells. Cell Rep 28, 2247–2255.e5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chal J et al. Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol 33, 962–969 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Matsumiya M, Tomita T, Yoshioka-Kobayashi K, Isomura A & Kageyama R ES cell-derived presomitic mesoderm-like tissues for analysis of synchronized oscillations in the segmentation clock. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chal J et al. Recapitulating early development of mouse musculoskeletal precursors of the paraxial mesoderm in vitro. Development 145, (2018). [DOI] [PubMed] [Google Scholar]
- 16.van den Brink SC et al. Single-cell and spatial transcriptomics reveal somitogenesis in gastruloids. Nature (2020) doi: 10.1038/s41586-020-2024-3. [DOI] [PubMed]
- 17.Veenvliet JV et al. Mouse embryonic stem cells self-organize into trunk-like structures with neural tube and somites. Science 370, (2020). [DOI] [PubMed] [Google Scholar]
- 18.Budjan C et al. Paraxial mesoderm organoids model development of human somites. Elife 11, e68925 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sanaki-Matsumiya M et al. Periodic formation of epithelial somites from human pluripotent stem cells. Nat. Commun 13, 1–14 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buckingham M & Relaix F The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell Dev. Biol 23, 645–673 (2007). [DOI] [PubMed] [Google Scholar]
- 21.Dias AS, de Almeida I, Belmonte JM, Glazier JA & Stern CD Somites Without a Clock. Science (2014). [DOI] [PMC free article] [PubMed]
- 22.Takahashi Y et al. Mesp2 initiates somite segmentation through the Notch signalling pathway. Nat. Genet 25, 390–396 (2000). [DOI] [PubMed] [Google Scholar]
- 23.Serini G et al. Modeling the early stages of vascular network assembly. EMBO J 22, 1771–1779 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhee J, Takahashi Y, Saga Y, Wilson-Rawls J & Rawls A The protocadherin papc is involved in the organization of the epithelium along the segmental border during mouse somitogenesis. Dev. Biol 254, 248–261 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Chal J, Guillot C & Pourquié O PAPC couples the segmentation clock to somite morphogenesis by regulating N-cadherin-dependent adhesion. Development 144, 664–676 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durbin L et al. Anteroposterior patterning is required within segments for somite boundary formation in developing zebrafish. Development 127, 1703–1713 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Nakajima Y, Morimoto M, Takahashi Y, Koseki H & Saga Y Identification of Epha4 enhancer required for segmental expression and the regulation by Mesp2. Development 133, 2517–2525 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Watanabe T, Sato Y, Saito D, Tadokoro R & Takahashi Y EphrinB2 coordinates the formation of a morphological boundary and cell epithelialization during somite segmentation. Proc. Natl. Acad. Sci. U. S. A 106, 7467–7472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakaya Y, Kuroda S, Katagiri YT, Kaibuchi K & Takahashi Y Mesenchymal-epithelial transition during somitic segmentation is regulated by differential roles of Cdc42 and Rac1. Dev. Cell 7, 425–438 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Qian K et al. A simple and efficient system for regulating gene expression in human pluripotent stem cells and derivatives. Stem Cells 32, 1230–1238 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gouti M et al. A Gene Regulatory Network Balances Neural and Mesoderm Specification during Vertebrate Trunk Development. Dev. Cell 41, 243–261.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niwa Y et al. Different types of oscillations in Notch and Fgf signaling regulate the spatiotemporal periodicity of somitogenesis. Genes Dev 25, 1115–1120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bessho Y et al. Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev 15, 2642–2647 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reymann A-C, Staniscia F, Erzberger A, Salbreux G & Grill SW Cortical flow aligns actin filaments to form a furrow. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palmeirim I, Henrique D, Ish-Horowicz D & Pourquié O Avian hairy gene expression identifies a molecular clock linked to vertebrate segmentation and somitogenesis. Cell 91, 639–648 (1997). [DOI] [PubMed] [Google Scholar]
- 36.Beccari L et al. Multi-axial self-organization properties of mouse embryonic stem cells into gastruloids. Nature 562, 272–276 (2018). [DOI] [PubMed] [Google Scholar]
- 37.Moris N et al. An in vitro model of early anteroposterior organization during human development. Nature (2020) doi: 10.1038/s41586-020-2383-9. [DOI] [PubMed]
- 38.Ran FA et al. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oceguera-Yanez F et al. Engineering the AAVS1 locus for consistent and scalable transgene expression in human iPSCs and their differentiated derivatives. Methods 101, 43–55 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Berg S et al. ilastik: interactive machine learning for (bio)image analysis. Nature Methods vol. 16 1226–1232 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Koechlein CS et al. High-resolution imaging and computational analysis of haematopoietic cell dynamics in vivo. Nat. Commun 7, 12169 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lowe DG Distinctive Image Features from Scale-Invariant Keypoints. Int. J. Comput. Vis 60, 91–110 (2004). [Google Scholar]
- 43.Thielicke W & Sonntag R Particle Image Velocimetry for MATLAB: Accuracy and enhanced algorithms in PIVlab. J. At. Mol. Phys doi: 10.5334/jors.334/print. [DOI]
- 44.Thielicke W & Stamhuis E PIVlab--towards user-friendly, affordable and accurate digital particle image velocimetry in MATLAB. Journal of open research software 2, (2014). [Google Scholar]
- 45.Thielicke W The flapping flight of birds. Diss. University of Groningen (2014).
- 46.Wolf FA, Angerer P & Theis FJ SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polański K et al. BBKNN: fast batch alignment of single cell transcriptomes. Bioinformatics 36, 964–965 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunter. Matplotlib: A 2D Graphics Environment 9, 90–95 (2007). [Google Scholar]
- 49.Bergen V, Lange M, Peidli S, Wolf FA & Theis FJ Generalizing RNA velocity to transient cell states through dynamical modeling. Nat. Biotechnol 38, 1408–1414 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Lange M et al. CellRank for directed single-cell fate mapping doi: 10.21203/rs.3.rs-94819/v1. [DOI] [PMC free article] [PubMed]
- 51.Hamburger & Hamilton. A series of normal stages in the development of the chick embryo. Dev. Dyn [DOI] [PubMed]
- 52.Choi HMT et al. Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chapman SC et al. Ubiquitous GFP expression in transgenic chickens using a lentiviral vector. Development 132, 935–940 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Miao Y & Pourquié O Reconstructing human somitogenesis with Somitoid and Segmentoid. Protocol Exchange
- 55.Tanoury ZA et al. Prednisolone rescues Duchenne muscular dystrophy phenotypes in human pluripotent stem cell–derived skeletal muscle in vitro. Proceedings of the National Academy of Sciences 118, e2022960118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Single-cell RNA sequencing data has been deposited to NCBI Gene Expression Omnibus (GEO): GSE195467 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE195467) . Bulk RNA sequencing data has been deposited to GEO: GSE220634 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE220634). The Homo sapiens (human) genome assembly (GRCh38) is from: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.26/. The mouse embryo scRNAseq data is from: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE114186. The RNA sequencing data of iPSCs is from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164874. Source data are provided with this paper. All materials used in this study including stem cell lines are available by request from the corresponding author.