

Abstract
Fibroblast growth factor 23 (FGF23) is a therapeutic target for treating hereditary and acquired hypophosphatemic disorders, such as X-linked hypophosphatemic (XLH) rickets and tumor-induced osteomalacia (TIO), respectively. FGF23-induced hypophosphatemia is mediated by signaling through a ternary complex formed by FGF23, the FGF receptor (FGFR), and α-Klotho. Currently, disorders of excess FGF23 are treated with an FGF23-blocking antibody, burosumab. Small-molecule drugs that disrupt protein/protein interactions necessary for the ternary complex formation offer an alternative to disrupting FGF23 signaling. In this study, the FGF23:α-Klotho interface was targeted to identify small-molecule protein/protein interaction inhibitors since it was computationally predicted to have a large fraction of hot spots and two druggable residues on α-Klotho. We further identified Tyr433 on the KL1 domain of α-Klotho as a promising hot spot and α-Klotho as an appropriate drug-binding target at this interface. Subsequently, we performed in silico docking of ~5.5 million compounds from the ZINC database to the interface region of α-Klotho from the ternary crystal structure. Following docking, 24 and 20 compounds were in the final list based on the lowest binding free energies to α-Klotho and the largest number of contacts with Tyr433, respectively. Five compounds were assessed experimentally by their FGF23-mediated extracellular signal-regulated kinase (ERK) activities in vitro, and two of these reduced activities significantly. Both these compounds were predicted to have favorable binding affinities to α-Klotho but not have a large number of contacts with the hot spot Tyr433. ZINC12409120 was found experimentally to disrupt FGF23:α-Klotho interaction to reduce FGF23-mediated ERK activities by 70% and have a half maximal inhibitory concentration (IC50) of 5.0 ± 0.23 μM. Molecular dynamics (MD) simulations of the ZINC12409120:α-Klotho complex starting from in silico docking poses reveal that the ligand exhibits contacts with residues on the KL1 domain, the KL1–KL2 linker, and the KL2 domain of α-Klotho simultaneously, thereby possibly disrupting the regular function of α-Klotho and impeding FGF23:α-Klotho interaction. ZINC12409120 is a candidate for lead optimization.
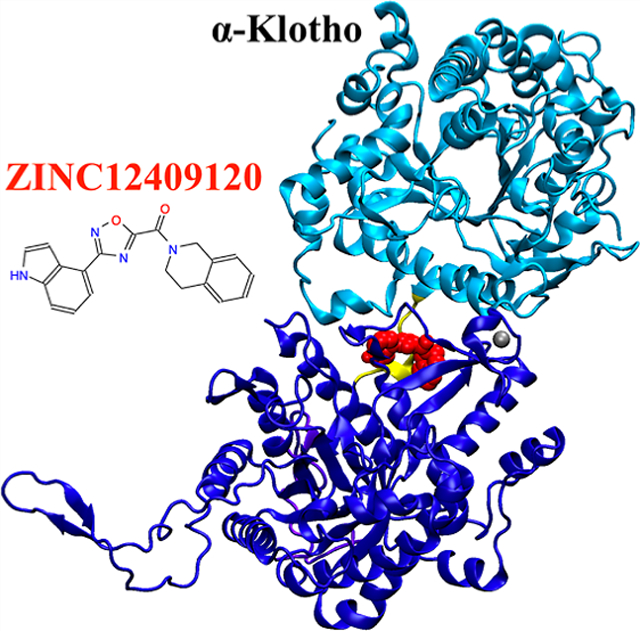
Graphical Abstract
INTRODUCTION
Fibroblast growth factor 23 (FGF23) is a bone-derived hormone that regulates phosphate and vitamin D homeostasis by forming a ternary complex with the FGF receptor (FGFR) and α-Klotho in renal proximal tubules.1,2 Excess FGF23 impairs phosphate reabsorption and vitamin D production, causing hereditary and acquired hypophosphatemic disorders, such as X-linked hypophosphatemic (XLH) rickets and tumor-induced osteomalacia (TIO), respectively.3
In the past, treatment for XLH consisted of phosphate and vitamin D supplements, which can cause excess phosphate and vitamin D and nephrocalcinosis.4,5 TIO can often be cured by resection of the FGF23-producing tumor, but the tumor removal is only successful in ~50% of the patients.6,7 Recently, the antibody burosumab has been approved by the Food and Drug Administration (FDA) to treat XLH and TIO. Burosumab binds to and blocks FGF23 activation of the FGFR:α-Klotho complex8,9 and increases serum phosphate levels in patients with XLH without reported toxicity,10 but it has a long 16 day biological half-life.11 In addition, peptides can also be used to partially block the FGF23 C-terminal tail to significantly reduce extracellular signal-regulated kinase (ERK) activity,12 one of the critical pathways for FGF23 signal transduction.13,14
Small-molecule drugs are a promising alternative to burosumab and peptides because of their potential oral availability and lower cost. We previously used high-throughput virtual screening and ensemble docking to the N-terminal domain of FGF23 [Protein Data Bank (PDB) code: 2P39]15 to identify a compound ZINC13407541 (N-[[2-(2-phenylethenyl)cyclopenten-1-yl]methylidene]hydroxylamine), which was designed to bind to FGF23 and experimentally verified to inhibit α-Klotho-dependent FGF23 signaling with a half maximal inhibitory concentration (IC50) of 0.45 ± 0.24μM.13 Further development of a series of analogues based on ZINC13407541 structure–activity relationships identified two more compounds with enhanced drug-like properties,13a [(E)-2-((E)-4-methylstyryl)benzaldehyde oxime] and 8n [(E)-2-(4-(tert-butyl)phenyl)cyclopent-1-ene-1-carbaldehyde oxime] that blocked FGF23 signaling in vitro and significantly increased serum phosphate and vitamin D concentrations in the mouse model of XLH (Hyp).16,17
Developing small-molecule drugs disrupting the FGF23:FGFR:α-Klotho complex is a promising strategy toward discovering novel inhibitors. Recently, the crystal structure of the ternary complex of FGF23:FGFR1c ectodomain:α-Klotho ectodomain 1:1:1 (PDB code: 5W21) was obtained (Figure 1),18 which shows that α-Klotho tethers the C-terminal tail of FGF23 and FGFR1c simultaneously and that α-Klotho acts as a non-enzymatic molecular scaffold for FGF23 signaling. The crystal structure provides the structural information needed for computationally exploring small molecules that disrupt the protein/protein interactions/interfaces (PPIs) to reduce FGF23 signaling.
Figure 1.
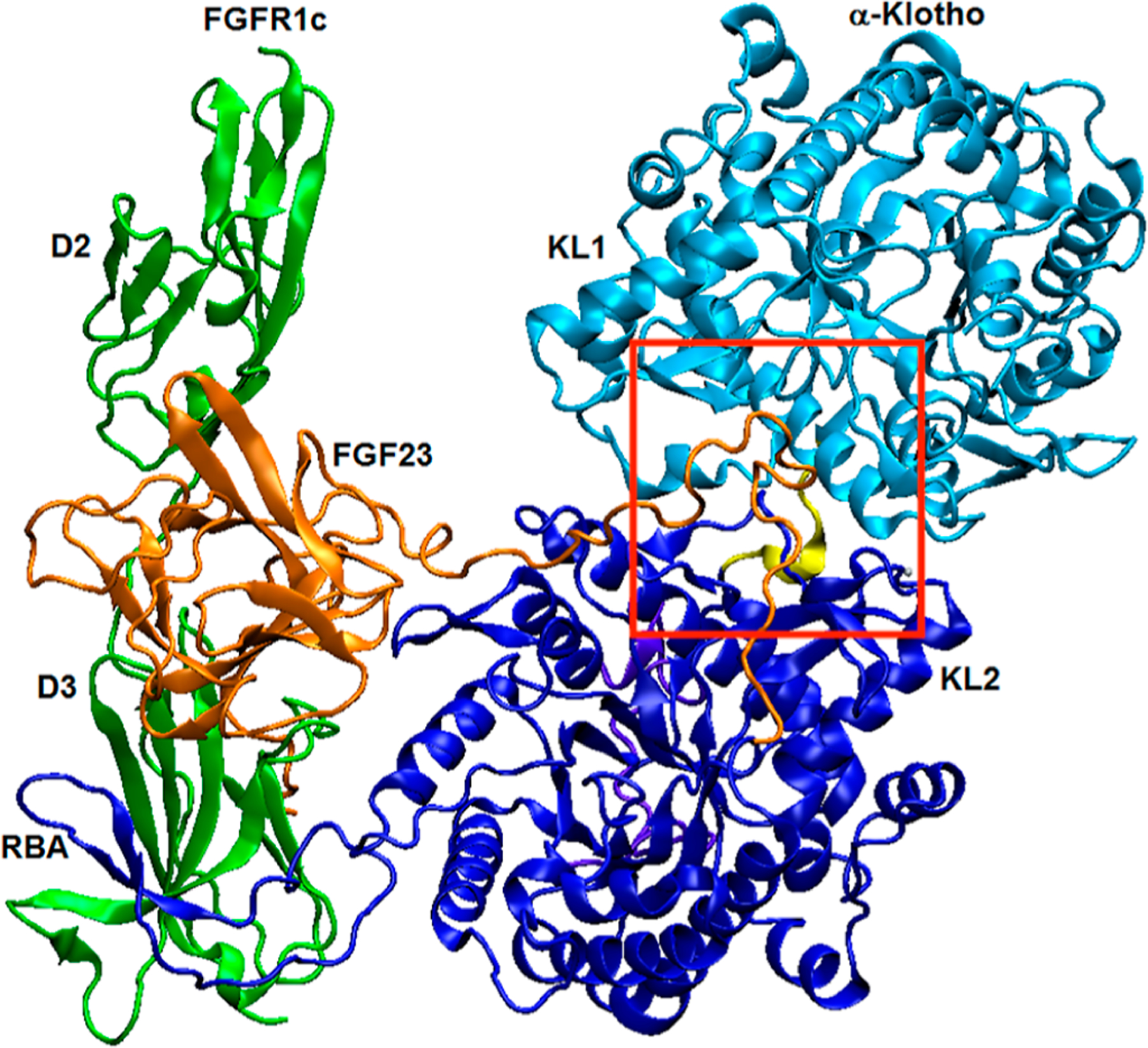
Crystal structure of the FGF23:FGFR1cecto:α-Klothoecto 1:1:1 ternary complex (PDB code: 5W21).18 The KL1 (Glu34-Phe506) and KL2 (Leu515-Ser950) domains of α-Klotho are light and dark blue, respectively, the KL1–KL2 linker (Pro507-Pro514) is yellow, the receptor-binding arm (RBA) of α-Klotho is dark blue, the C-terminal tail (Asn951-His977) of α-Klotho is purple, and the Zn atom is silver. FGF23 is orange with its C-terminal tail in the KL1–KL2 region. The D2 and D3 domains of FGFR1c are green. The red box shows the region where molecular docking to α-Klotho was performed.
In silico screening of virtual compound libraries is a promising way to identify effective PPI inhibitors for subsequent experimental validation.19,20 For example, pharmacophore screening followed by molecular docking has led to PPI inhibitors with micromolar binding affinity.21 A critical step for the success of this approach is to select appropriate binding sites to dock the compounds (ligands) to. Promising ligand-binding sites in this scenario would be “hot spot” residues. These residues confer a disproportionate amount of the binding energy at the PPI and may be druggable by small molecules.22 Hot spots can be predicted computationally,23–31 and here, we used the KFC2a method,30 which is based on a machine learning predictive model and recognizes structural features of PPIs.
In this study, we used the ternary crystal structure (PDB code: 5W21)18 to computationally identify the FGF23:α-Klotho interface as one with the largest fraction of hot spots and druggable ligand-binding sites on α-Klotho. We further identified Tyr433 on α-Klotho as a promising hot spot and α-Klotho as an appropriate drug-binding target at this interface.~5.5 million lead-like compounds were then docked computationally to an isolated α-Klotho crystal structure near its interface with FGF23 (see the red box in Figure 1). We identified 24 compounds with the highest protein–ligand binding affinities to α-Klotho and 20 compounds with the largest number of contacts with Tyr433. 5 of the 44 compounds were selected for in vitro assays based on their predicted ligand efficiency and vendor availability. We found that ZINC12409120 {3,4-dihydro-1H-isoquinolin-2-yl-[3-(1H-indol-4-yl)-1,2,4-oxadiazol-5-yl]methanone} disrupted FGF23:α-Klotho interaction to reduce FGF23-mediated ERK activities by 70%, and it had an IC 50 of 5.0 ± 0.23 μM. Molecular-dynamics (MD) simulations of the ZINC12409120:α-Klotho complex starting from in silico docking poses show that ZINC12409120 interacts with residues on the KL1 domain, the KL1–KL2 linker, and the KL2 domain of α-Klotho simultaneously to potentially disrupt the function of α-Klotho and deter FGF23:α-Klotho interaction. More compounds will be tested, and more functional assays of ZINC12409120 will be conducted in future studies.
METHODS
Hot Spot and Surface Pocket Analyses.
We used the crystal structure of the FGF23:FGFR1cecto:α-Klothoecto 1:1:1 ternary complex (PDB code: 5W21).18 The N-acetyl glucosamine residues on α-Klotho were not included since they are not at the KL1:KL2:FGF23 interface,18 whose structural flexibility is in the interest of this study. The four missing residues (Glu957-Glu960) on the C-terminal tail of α-Klotho were patched using alignment followed by an automodel class without refinement in MODELLER,32 and the missing atoms within each residue on the complex were then built using VMD.33 Protonation states were determined using the pdb2gmx command in GROMACS, where they are assigned using the pKa of isolated amino acids and pH = 7.34,35 The complex for docking was prepared using MGLTools36 (https://ccsb.scripps.edu/mgltools) to remove its non-polar hydrogen atoms. This structure was uploaded to the KFC Server30,31 (https://mitchell-web.ornl.gov/KFC_Server) and FTMap Server29 (https://ftmap.bu.edu) for predicting hot spots using the KFC2a method30 and druggable sites at the PPI, respectively. In addition, the CASTp method37 (http://sts.bioe.uic.edu/castp) was used to obtain solvent-accessible surface area (SASA) and residue information of the surface pockets using a default probe radius of 1.4 Å. A brief description of these three online servers is in the following paragraph.
The KFC2a method30,31 calculates each PPI residue’s eight features, which are mainly related to its SASA and local plasticity, and compares them with those of experimentally determined hot spots and outputs hot spot confidence scores. In the independent test set of the KFC2a method, 82% of experimentally validated hot spots have positive confidence scores, and 70% of non-hot spots have negative ones. Therefore, positive confidence scores suggest prospective hot spots. The FTMap Server29 distributes 16 small organic probe molecules, varying in size, shape, and polarity, on the protein surface, finds the most energetically favorable positions for each probe type, and then clusters the probes. The residue with the largest number of probe clusters is considered as the main druggable site. CASTp37 uses the alpha shape method38 to identify topographic features (e.g., area and volume) of proteins.
Molecular Docking to α-Klotho.
Docking was performed on the α-Klotho structures extracted from the ternary complex18 and from snapshots selected from MD simulations as described below. The latter approach, known as ensemble docking, takes into account the thermal fluctuations of the binding-site atoms.39
MD Simulations for Ensemble Docking.
α-Klotho was centered in a dodecahedron box with a 15 Å padding between α-Klotho and the box. The box was then solvated with water, and eight Na+ ions were also included to neutralize the system. To ensure our findings do not depend on one initial configuration, five independent MD simulations (i.e., instances) with different initial velocity distributions were performed using the CHARMM36 force field parameters40,41 for α-Klotho, the TIP3P model42 for water, and the GROMACS simulation code.34,35
For each of the five simulations, energy minimization was performed, followed by equilibration in the NVT and then the NPT ensembles with a time step of 2 fs at 310 K and 1 bar for 1 ns, with non-hydrogen atoms on the α-Klotho restrained using a force constant of 1000 kJ/mol/nm2 in each dimension. Subsequently, each of the five simulations was continued for production in the NPT ensemble for 200 ns, with only the Zn atom on the α-Klotho restrained using the above-mentioned force constant to ensure its coordination state. The instantaneous energies and configurations were saved every 10 ps, and the last 100 ns were used for clustering. The details of the simulation settings and validation of data in the last 100 ns are included in the Supporting Information.
Root-mean-square deviation (rmsd)-based clustering of α-Klotho was performed using the GROMOS43 method on all non-hydrogen atoms of nine residues at the KL1:KL2:FGF23 interface (i.e., interface residues) identified in the ternary crystal structure18 (Table 1). The GROMOS method counts the number of neighbor structures using the specified cutoff, takes the structure with the largest number of neighbors and all its neighbors as a cluster, eliminates these structures from the pool of clusters, and then repeats for remaining structures in the pool.43 The cutoff of rmsd from the crystal structure18 was varied between 1.5 and 2.0 Å so as to identify the optimal rmsd cutoff, such that the total number of clusters is ~40, and the number of 1-frame clusters is minimal.44 The rmsd cutoff chosen here was 1.52 Å which resulted in 38 clusters, only one of which contained 1 frame. The 10 central structures, those with the smallest average rmsd from all other structures, each from the 10 most populated clusters #1–10 representing 98.4% of MD frames, were selected as an ensemble of α-Klotho, named MD clusters #1–10 (Table S1), whose Zn atom has the same coordination state as that in the crystal structure18 (Table S2).
Table 1.
15 Residues Defining the KL1:KL2:FGF23 Interface in the Ternary Crystal Structure18 and Additional Two Residues in Bold Were Predicted as Druggable Sites Using FTMap.29a
| protein | residue | number | hot spot | confidence score | |
|---|---|---|---|---|---|
| α-KlothoKL1 domain | Lys | 429 | yes | 1.05 | 1 |
| Tyr | 432 | No | −1.21 | 1,2 | |
| Tyr | 433 | Yes | 1.43 | 1 | |
| Lys | 435 | N/A | N/A | 2 | |
| Phe | 437 | No | −0.30 | 1 | |
| Ser | 471 | N/A | N/A | N/A | |
| α-KlothoKL2 domain | Lys | 823 | Yes | 0.26 | 1 |
| Met | 833 | Yes | 1.38 | 1 | |
| Thr | 834 | Yes | 1.37 | 1 | |
| Ile | 836 | Yes | 0.20 | 1 | |
| Gln | 844 | No | −0.74 | 1 | |
| FGF23C-terminal tail | Asp | 188 | Yes | 0.42 | |
| Pro | 189 | Yes | 1.31 | ||
| Leu | 190 | Yes | 1.32 | ||
| Asn | 191 | No | −1.19 | ||
| Val | 192 | Yes | 1.94 | ||
| Leu | 193 | Yes | 1.96 |
The hot-spot identification and confidence scores were evaluated by the KFC2a method.30 Positive confidence scores suggest prospective hot spots, and 11 out of 15 interface residues are predicted hot spots. The rightmost column shows the surface pocket(s) (See Figure 2 for visualization), the residues on α-Klotho belong to using CASTp,37 and 10 out of 11 residues are in the pockets. N/A: not available.
Preparation of Receptors and Ligands Prior to Docking.
The interface residues in each MD cluster were aligned with those in the crystal structure18 using a least-squares fit. The files in the PDB format of the aligned structures are included in the “alpha-Klotho.zip” as the Supporting Information. For docking to the crystal structure18 and each of the 10 MD clusters, a cubic box of 30 × 30 × 30 Å was centered at the geometric center of Tyr432 (Table S3), a potentially druggable site (Table 1), to include the 6 computationally predicted hot spots at the KL1:KL2:FGF23 interface (Tables 1 and S4), and the entire box was used for searching ligand poses.
5,450,731 compounds in the lead-like45 and clean (without aldehydes and thiols) subsets of the ZINC database46 (http://zinc12.docking.org/subsets/clean-leads) were used since we aimed at finding lead molecules in in vitro assays and keeping them safe for clinical trials in mice and humans in future studies, and these compounds with different protonation states but the same ZINC ID were included. Million-compound docking has been previously shown to discover protein inhibitors with submicro to nanomolar binding affinity.47,48 The ligands for docking were prepared using MGLTools36 (https://ccsb.scripps.edu/mgltools) where non-polar hydrogen atoms were removed, and Gasteiger partial atomic charges were not added to ligands. The atomic charges built in the ligands’ original.mol2 files from the ZINC database46 were used.
Three-Phase Docking.
In the first phase, VinaMPI,49 a modified version of AutoDock Vina50 optimized for performing calculations on supercomputers, was used to dock the 5,450,731 compounds to the α-Klotho crystal structure18 with an exhaustiveness of 10. For each compound, nine binding poses were generated, each with estimated protein–ligand free energy of binding (ΔG). We note that AutoDock Vina has been shown to be one of the best docking protocols in identifying the native ligand binding pose.51,52
In the second phase, those 820,835 compounds whose ΔG was within 3 kcal/mol from the ΔG of the best-ranked ligand were re-docked to the same region of the α-Klotho crystal structure18 with a larger exhaustiveness of 20 to potentially enhance binding pose prediction. 3 kcal/mol was used as it is the uncertainty of ΔG in AutoDock Vina.50 Subsequently, 1063 compounds were chosen for further evaluation: 476 compounds were selected based on the lowest ΔG (≤ −10.4 kcal/mol), and the other 587 compounds were selected because one of their binding poses had the largest number of contacts with Tyr433 (≥ 17), a critical hot spot residue at the KL1:KL2:FGF23 interface of α-Klotho18 (Table 1). A contact here is defined as non-hydrogen interatomic distance between the protein and ligand ≤4.0 Å.
In the third phase, these 1063 compounds were docked to the same region of the α-Klotho crystal structure18 and to the 10 MD clusters (i.e., ensemble docking, which considers protein flexibility and has proven to be useful in early drug discovery39) with an exhaustiveness of 30 to possibly further refine binding poses. 44 compounds were in the final list: 24 compounds were selected based on the lowest ΔG (≤ −10.9 kcal/mol) from the 476-compound set, and the other 20 compounds were selected based on the largest number of contacts with Tyr433 (≥ 22) from the 587-compound set. The SASA for each of the 44 compounds in the ZINC database46 was obtained using the SASA command in GROMACS.34,35 We note that 44 out of 1063 (4%) compounds in the ensemble docking are in the final list, and this percentage is similar to that (5%) in another recent study.53 For each of the 24 and 20 compounds, the binding pose with the lowest ΔG from Vina and that with the largest number of contacts with Tyr433, respectively, were rescored using KDEEP,54 and the ligand efficiency was also derived by dividing ΔG (KDEEP) by the number of non-hydrogen atoms in a ligand to provide a useful metric for lead selection.55KDEEP is a protein–ligand absolute binding affinity predictor based on deep convolutional neural networks (CNNs),54 and its training set is the refined set of the PDBbind database (v.2016), which contains 4057 diverse rather than a handful of families of protein–ligand complexes experimentally determined with reported binding affinities and deposited in the PDB.56 SwissADME57 and https://www.cbligand.org/PAINS58 were used to evaluate drug-likeness and to identify Pan Assay Interference compounds (PAINS) of these 44 compounds, respectively. The files in the PDB format for these binding poses and the simplified molecular-input line-entry system (SMILES) in the CSV format for each of the 44 compounds are included in “Compounds.zip” as the Supporting Information. In addition, an average ΔG from Vina over all 11 α-Klotho structures, the crystal structure,18 and the 10 MD clusters from the ensemble docking was given to each of the 1063 compounds. The 50 compounds with the lowest average ΔG (≤ −9.45 kcal/mol) are discussed in the Results and Discussion section.
In Silico Ligand:α-Klotho Interaction Analysis.
Two sets of MD simulations of the solvated ZINC12409120:α-Klotho complex, with starting structures obtained from the docking to MD cluster #8 and to the crystal structure,18 were performed.The force field parameters of ZINC12409120 which did not exist in latest CHARMM3640,41 were obtained using the CHARMM General Force Field (CGenFF)59,60 (https://cgenff.umaryland.edu). The resulting MD trajectories were analyzed by calculating 1) contacts between non-hydrogen atoms of specific residues and ZINC12409120 using a maximum contact distance of 4.0 Å and 2) hydrogen bonds between all atoms of specific residues and ZINC12409120 using a donor–acceptor cutoff radius of 4.0 Å.
The contacts of ZINC12409120 and ZINC05326903 with α-Klotho in the ensemble docking were analyzed using LigPlot+.61 For non-bonded contact calculation, the minimum and maximum contact distances were set at 2.0 and 4.0 Å, respectively. For hydrogen-bond calculation, the maximum hydrogen-acceptor and maximum donor–acceptor distances were set at 3.0 and 4.0 Å, respectively.
In Vitro Functional Assays.
The criteria used to select compounds for experimental validation were (1) no PAINS;58 (2) consensus ranking, obtained by combining the ligand efficiency η55 with the ΔG (Vina) in Table 2 or the number of contacts with Tyr433 in Table 3; and (3) in stock availability from the vendor [AKos Consulting and Solutions Deutschland GmbH (Lörrach, Germany)]. All the ΔG values in Tables 2 and 3 are within the uncertainty, so we selected compounds by also considering the KDEEP54 ligand efficiency,55 which is a useful metric for lead selection. Unavailability at the time of purchase is the reason some compounds that are ranked high in Tables 2 and 3 were not selected for experimental validation.
Table 2.
21 Unique Compounds and Their SASA in the Database46 Ranked by the Lowest Estimated Free Energies of Binding (ΔG) to α-Klotho in the Ensemble Docking Where Receptor Conformations and ΔG Are Shown in the Fourth and Fifth Columns, Respectively.a
| ligand # | ZINC ID | SASA [Å2] | receptor | ΔG (Vina) [kcal/mol] | ΔG(KDEEP) [kcal/mol] | η (KDEEP) [kcal/mol] |
|---|---|---|---|---|---|---|
| 1a | 12409120 | 575 | cluster #8 | −11.2 | −8.31(−0.42) | −0.32 |
| 1b | 12409120 | 575 | crystal | −11.1 | −8.29(−0.36) | −0.32 |
| 2 | 70700439 | 545 | crystal | −11.1 | −8.20(−0.40) | −0.32 |
| 3 | 65044553 | 560 | crystal | −11.0 | −9.06(−0.35) | −0.35 |
| 4 | 19373000 | 572 | crystal | −11.0 | −8.66(−0.38) | −0.33 |
| 5 | 38747189 | 598 | crystal | −11.0 | −8.55(−0.44) | −0.33 |
| 6a | 98150430 | 543 | crystal | − 11.0 | −8.48(−0.38) | −0.33 |
| 7 | 33020020 | 540 | crystal | −11.0 | −8.37(−0.39) | −0.32 |
| 8 | 89914674 | 535 | crystal | −11.0 | −8.27(−0.29) | −0.32 |
| 9 | 89915984 | 555 | crystal | −11.0 | −8.25(−0.47) | −0.32 |
| 10 | 05326903 | 535 | crystal | −11.0 | −8.12(−0.30) | −0.32 |
| 11 | 17322022 | 587 | cluster #8 | −11.0 | −7.82(−0.43) | −0.30 |
| 12 | 03908210 | 565 | cluster #2 | −11.0 | −7.72(−0.42) | −0.30 |
| 13 | 88338906 | 556 | crystal | −11.0 | −7.69(−0.52) | −0.30 |
| 6b | 98150430 | 540 | crystal | −10.9 | −8.52(−0.41) | −0.33 |
| 14 | 65236018 | 595 | crystal | −10.9 | −8.46(−0.40) | −0.33 |
| 15 | 96129233 | 586 | crystal | −10.9 | −8.43(−0.56) | −0.32 |
| 16 | 36391530 | 561 | crystal | −10.9 | −8.39(−0.26) | −0.32 |
| 17 | 69840438 | 580 | crystal | −10.9 | −8.27(−0.52) | −0.32 |
| 18 | 05089013 | 540 | crystal | −10.9 | −8.04(−0.39) | −0.32 |
| 19 | 92506891 | 537 | crystal | −10.9 | −8.02(−0.39) | −0.32 |
| 20a | 65514802 | 498 | crystal | −10.9 | −8.00(−0.42) | −0.31 |
| 20b | 65514802 | 503 | crystal | −10.9 | −8.00(−0.40) | −0.31 |
| 20c | 65514802 | 583 | crystal | −10.9 | −8.00(−0.39) | −0.31 |
| 21 | 89364390 | 530 | crystal | −10.9 | −7.79(−0.30) | −0.31 |
| Average | N/A | 556 | N/A | −11.0 | −8.23 (−0.40) | −0.32 |
ΔG rescored by KDEEP54 with standard deviation in parentheses and their corresponding ligand efficiencies (η)55 are shown in the 6th and 7th columns, respectively. The 3 unique compounds in bold were selected for in vitro assays based on their η and vendor availability. The compound in ligand #4 row was identified as pan assay interference compounds (PAINS),58 and the explanation is included in “PAINS-ZINC19373000.pdf” as the Supporting Informationa. Ligand #1b is shown for discussion purposes, and ligands #6a-b and #20a-c each with the same ZINC ID have different protonation states in the database.46 N/A: not applicable.
Table 3.
19 Unique Compounds and Their SASA in the Database46 Ranked by the Largest Number of Non-Hydrogen Atomic Contacts with the Hot Spot Tyr433 on α-Klotho in the Ensemble Docking Where the Receptor Conformations, Number of Contacts with Tyr433, and ΔG Are Shown in the Fourth, Fifth, and Sixth Columns, Respectively.a
| ligand # | ZINC ID | SASA [Å2] | receptor | Tyr433 contact # | ΔG(Vina) [kcal/mol] | ΔG(KDEEP) [kcal/mol] | η (KDEEP)[kcal/mol] |
|---|---|---|---|---|---|---|---|
| 22 | 69770999 | 541 | crystal | 26 | −8.2 | −8.07(−0.47) | −0.32 |
| 23a | 72289860 | 553 | crystal | 24 | −8.3 | −8.25(−0.33) | −0.34 |
| 24 | 74046068 | 583 | crystal | 23 | −8.8 | −8.37(−0.44) | −0.32 |
| 25 | 31607018 | 529 | crystal | 23 | −8.8 | −8.37(−0.43) | −0.35 |
| 26 | 92169003 | 521 | crystal | 23 | −8.7 | −7.87(−0.32) | −0.33 |
| 27 | 72329523 | 557 | crystal | 23 | −8.4 | −8.22(−0.32) | −0.33 |
| 28 | 72329507 | 552 | crystal | 23 | −8.3 | −8.33(−0.31) | −0.33 |
| 29 | 89510830 | 569 | crystal | 23 | −8.3 | −8.01(−0.43) | −0.32 |
| 30 | 24208749 | 477 | crystal | 23 | −8.2 | −8.03(−0.44) | −0.33 |
| 31 | 27986924 | 524 | crystal | 23 | −8.2 | −7.67(−0.43) | −0.33 |
| 32 | 04626551 | 547 | crystal | 22 | −8.9 | −7.86(−0.53) | −0.33 |
| 33 | 29506278 | 543 | crystal | 22 | −8.6 | −8.24(−0.35) | −0.33 |
| 34 | 58903720 | 585 | crystal | 22 | −8.3 | −8.32(−0.41) | −0.33 |
| 35 | 38767403 | 505 | crystal | 22 | −8.3 | −7.45(−0.52) | −0.32 |
| 23b | 72289860 | 542 | crystal | 22 | −8.2 | −8.40(−0.39) | −0.35 |
| 36 | 21836253 | 542 | crystal | 22 | −8.2 | −8.11(−0.44) | −0.32 |
| 37 | 21914858 | 560 | crystal | 22 | −8.1 | −8.16(−0.49) | −0.33 |
| 38 | 56338067 | 534 | crystal | 22 | −8.1 | −7.97(−0.42) | −0.33 |
| 39 | 12812109 | 589 | crystal | 22 | −8.1 | −7.95(−0.44) | −0.32 |
| 40 | 80057881 | 566 | crystal | 22 | −8.1 | −7.80(−0.47) | −0.33 |
| Average | N/A | 546 | N/A | 23 | −8.4 | −8.07 (−0.42) | −0.33 |
ΔG rescored by KDEEP54 with standard deviation in parentheses and their corresponding ligand efficiencies (η)55 are shown in the seventh and eighth columns, respectively. The two unique compounds in bold were selected for in vitro assays based on their η and vendor availability. Ligands #23a-b with the same ZINC ID have different protonation states in the database.46 N/A: not applicable.
Two sets of assays were conducted to test the effects of these five compounds identified computationally on (1) FGF23:FGFR1c:α-Klotho-mediated ERK activation13,14 and (2) epidermal growth factor (EGF):epidermal growth factor receptor (EGFR)-mediated ERK activation.62 First, human embryonic kidney (HEK) 293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10 wt % of fetal bovine serum (FBS) and 1 wt % of penicillin and streptomycin (P/S). Since HEK 293T cells expressed FGFR1c but not transcripts encoding α-Klotho or the EGFR,13 they were transiently transfected with the full-length human α-Klotho or EGFR along with the ERK luciferase reporter system and Renilla luciferase-null as the internal control plasmid. Transfection was performed by electroporation using Lonza Cell Line Nucleofector Kit R (Walkersville, MD), and further treatment as described below was done 36 h after transfection. For measuring FGF23-mediated ERK reporter activities, the α-Klotho-transfected cells were treated with (1) thevehicle only as the control; (2) FGF23 only at 1 μM; (3) each of the five compounds at 10 μM in the presence of FGF23 at 1 μM; and (4) ZINC12409120 in a range of 10–9–10–4 M in the presence or absence of FGF23 at 1 μM to obtain the half maximal inhibitory concentration (IC50). For measuring EGF-mediated ERK reporter activities, the EGFR-transfected cells were treated with (1) the vehicle only as the control; (2) EGF only at 20 ng/mL; and (3) each of the five compounds at 10 μM or erlotinib at 1 μM in the presence of EGF at 20 ng/mL. Erlotinib [N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine] was purchased from Sigma-Aldrich (St. Louis, MO). After 5 h, the cells were lysed, and luciferase activities were measured using a BioTek Synergy H4 Hybrid Multi-Mode Microplate Reader (Winooski, VT) and Promega Dual-Luciferase Reporter Assay System (Madison, WI).13 Three independent experiments were run for each scenario to obtain sufficient statistics. Statistical significance between two groups was evaluated by unpaired the 2-tailed t-test and that between multiple groups was evaluated by one-way analysis of variance (ANOVA) with the Newman–Keuls multiple comparison test. These calculations were performed using GraphPad Prism 5.0 (San Diego, CA). The IC50 of ZINC12409120 was obtained graphically from concentration–effect curves using GraphPad Prism 5.0 (San Diego, CA). The half-lives of ZINC12409120 and ZINC05326903 were derived from their volume of distribution and clearance,63 as predicted by pkCSM64 (http://biosig.unimelb.edu.au/pkcsm/). More details are described in our previous work.13
RESULTS AND DISCUSSION
Hot Spot and Surface Pocket Analyses.
The KL1:KL2:FGF23 interface is perhaps the most promising target for small-molecule drug discovery among the five interfaces identified in the ternary crystal structure,18 as it has predicted druggable sites, and 73% of the interface residues are predicted hot spots (Table 4). We note that 75% of the KL2:FGF23 interface residues are predicted hot spots, but all of them are on the FGF23 C-terminal tail (Table S6), which is coil-like and thus unlikely to be a competent drug-binding region.
Table 4.
| interface | druggable site | hot spot % | details in table |
|---|---|---|---|
| KL1:KL2:FGF23 | yes | 73% | 1 |
| D3:FGF23 | yes | 44% | S5 |
| KL2:FGF23 | no | 75% | S6 |
| D2:FGF23 | no | 64% | S7 |
| RBA:D3 | no | 50% | S8 |
Tyr432 and Lys435 on the KL1 domain are the two most druggable sites close to the KL1:KL2:FGF23 interface, and Tyr433 on the KL1 domain is the most promising predicted hot spot on α-Klotho as it has the highest confidence score (Table 1). Tyr433 was recently suggested in the ternary crystal structure as a key residue on α-Klotho that tethers FGF23,18 which agrees with our hot-spot prediction. In addition to Tyr433, the other six residues (i.e., Met833 and Thr834 on the KL2 domain and Pro189, Leu190, Val192, and Leu193 on the FGF23 C-terminal tail) also have relatively high confidence scores. A recent experimental study shows that Pro189 and Leu190 on FGF23 are critical for binding to the KL1 domain.65 However, out of these seven hot spots, only Tyr433 on a KL1 α7-helix (Ala428-Leu447) and Met833 on a KL2 β-strand (Val830-Met833) (Figure 3) may offer prospective binding sites for the subsequent docking campaign. The other five hot spots may not be good drug-binding targets since they are located on a coil. Most reported PPI inhibitors bind strongly to a PPI with well-defined secondary structures, which exists in the unbound protein but becomes buried when the PPI complex is formed.66 Table 1 also shows that at the KL1:KL2:FGF23 interface, eight out of nine α-Klotho residues are in the surface pocket (Pocket 1). This suggests that interface residues on α-Klotho would be appropriate targets for small molecules to disrupt FGF23:α-Klotho interactions.
Figure 3.
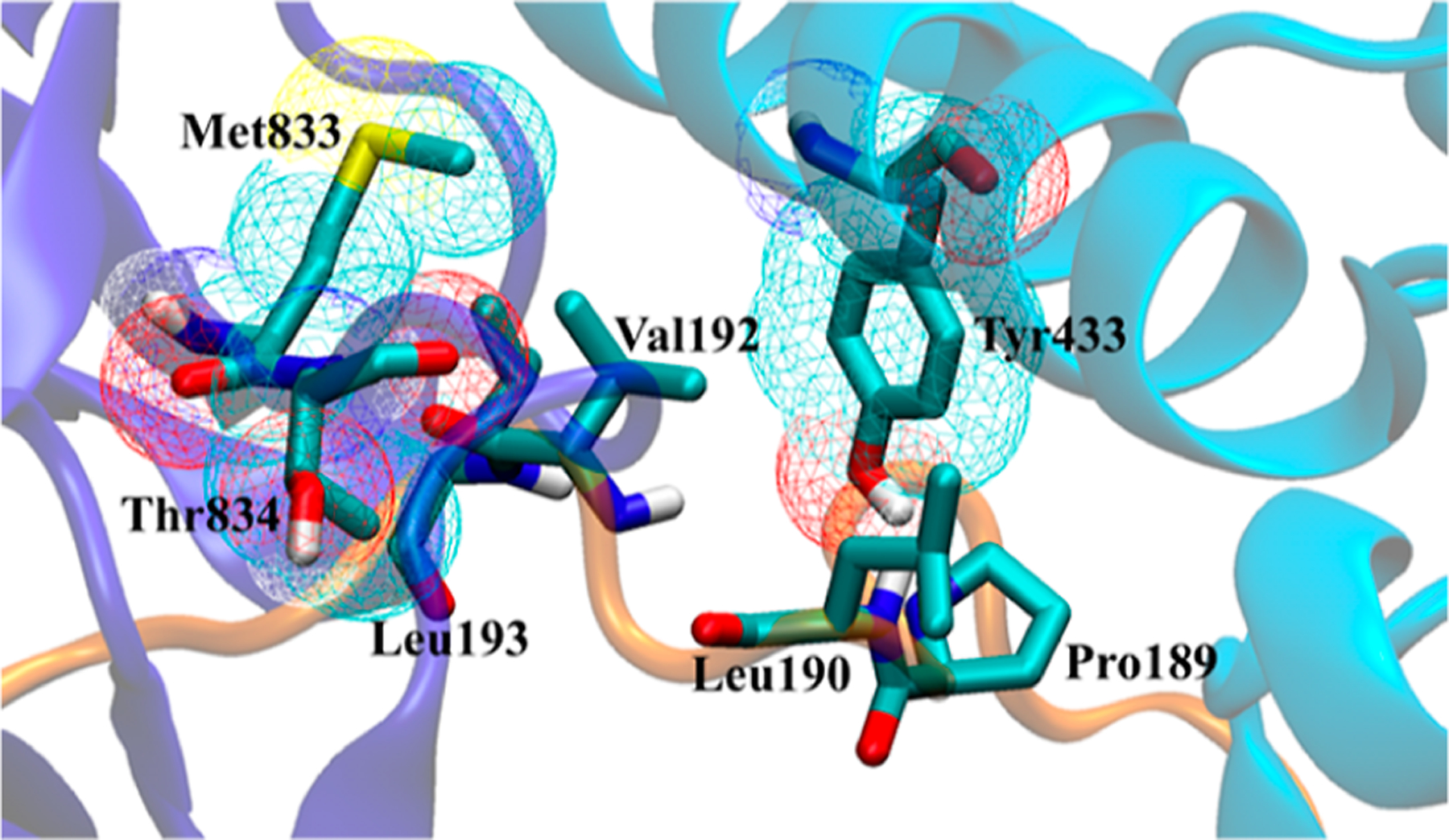
Visualization of the seven calculated hot spots with relatively high confidence scores in Table 1 at the KL1:KL2:FGF23 (light blue:dark blue:orange) interface of the ternary crystal structure.18 The solvent-accessible surface of the three hot spots on α-Klotho is shown in the mesh.
Molecular Docking to α-Klotho.
To select compounds for experimental validation, we applied two approaches. The first approach is to identify compounds with the most favorable binding energies predicted with Vina (Table 2). ZINC12409120 ranks first in ΔG (Vina) and in silico binds to α-Klotho cluster #8 and the crystal structure18 with nearly the same ΔG. 18 of the 21 unique compounds bind to Pocket 2 in the α-Klotho crystal structure,18 and only 3 bind to α-Klotho clusters obtained from the MD simulations (Table 2 and Figure 4). The reason is that the size of these ligands with an average SASA of 556 Å2 (Table 2) fits better into that of Pocket 2 in the crystal structure18 (814=Å2, see Figure 2). However, Pocket 2 collapses or shrinks in the MD clusters (Figure S4), preventing ligands from binding to Pocket 2. Furthermore, K 54 DEEP was used to rescore ΔG for these 25 protein–ligand complexes generated by Vina. The ligand efficiency (η) for each compound was also obtained. The lower the value of η, the better the potency of a ligand.55 All 21 unique compounds are drug-like,57 and only ZINC19373000 is PAINS.58 Three compounds (ZINC12409120, ZINC05326903, and ZINC36391530) were chosen based on their η and vendor availability for in vitro assays to test their efficacies.
Figure 4.
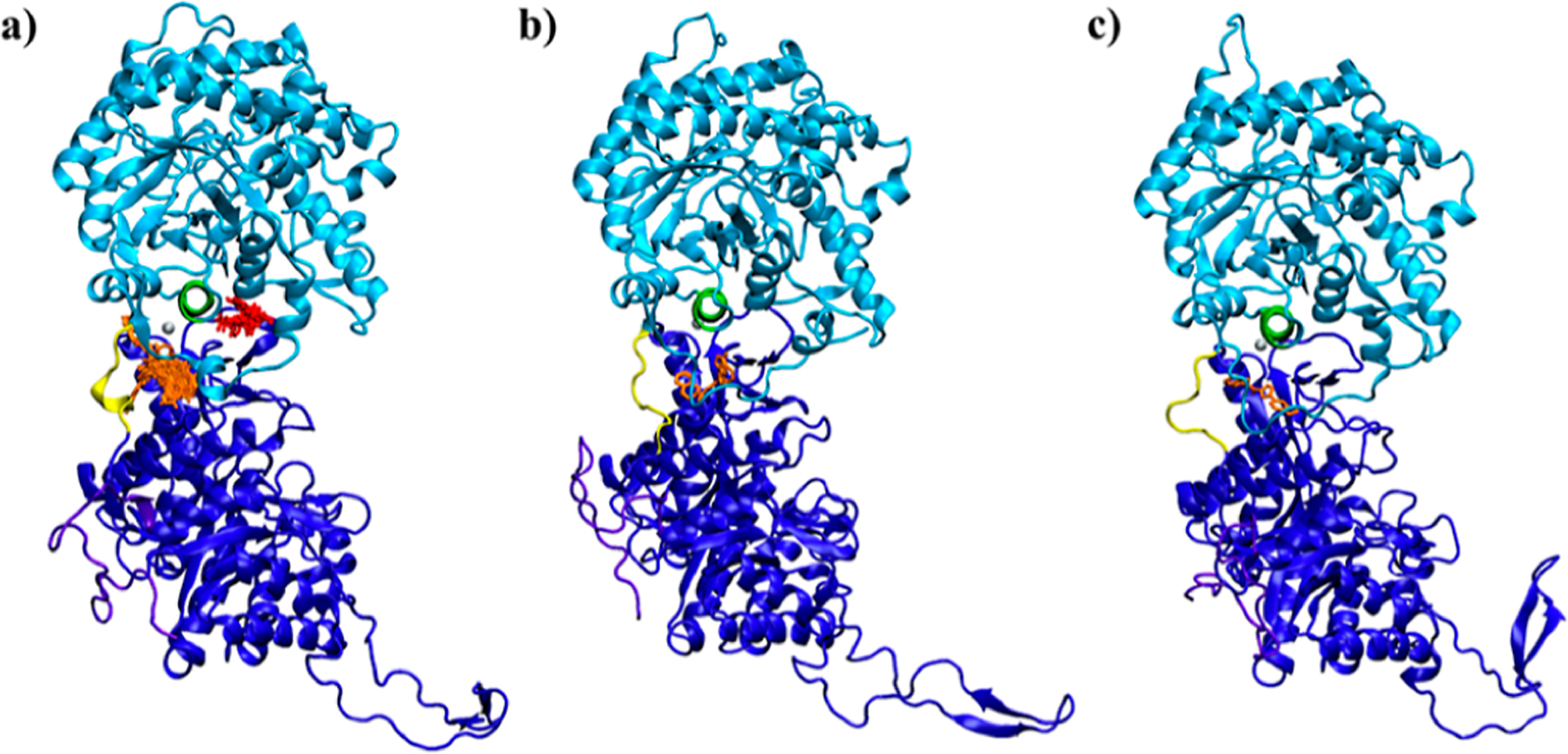
Docking poses of ligands listed in Tables 2 (orange) and 3 (red) on α-Klotho (a) crystal structure,18 (b) cluster #8, and (c) cluster #2. The KL1 α7-helix (Ala428-Leu447) is green for locating ligands. The KL1 domain, KL2 domain, and C-terminal tail of α-Klotho are light blue, dark blue, and purple, respectively. The KL1–KL2 linker is yellow, and the Zn atom is silver. The residues in surface pockets with which these ligands have contacts are listed in Table S10.
Figure 2.
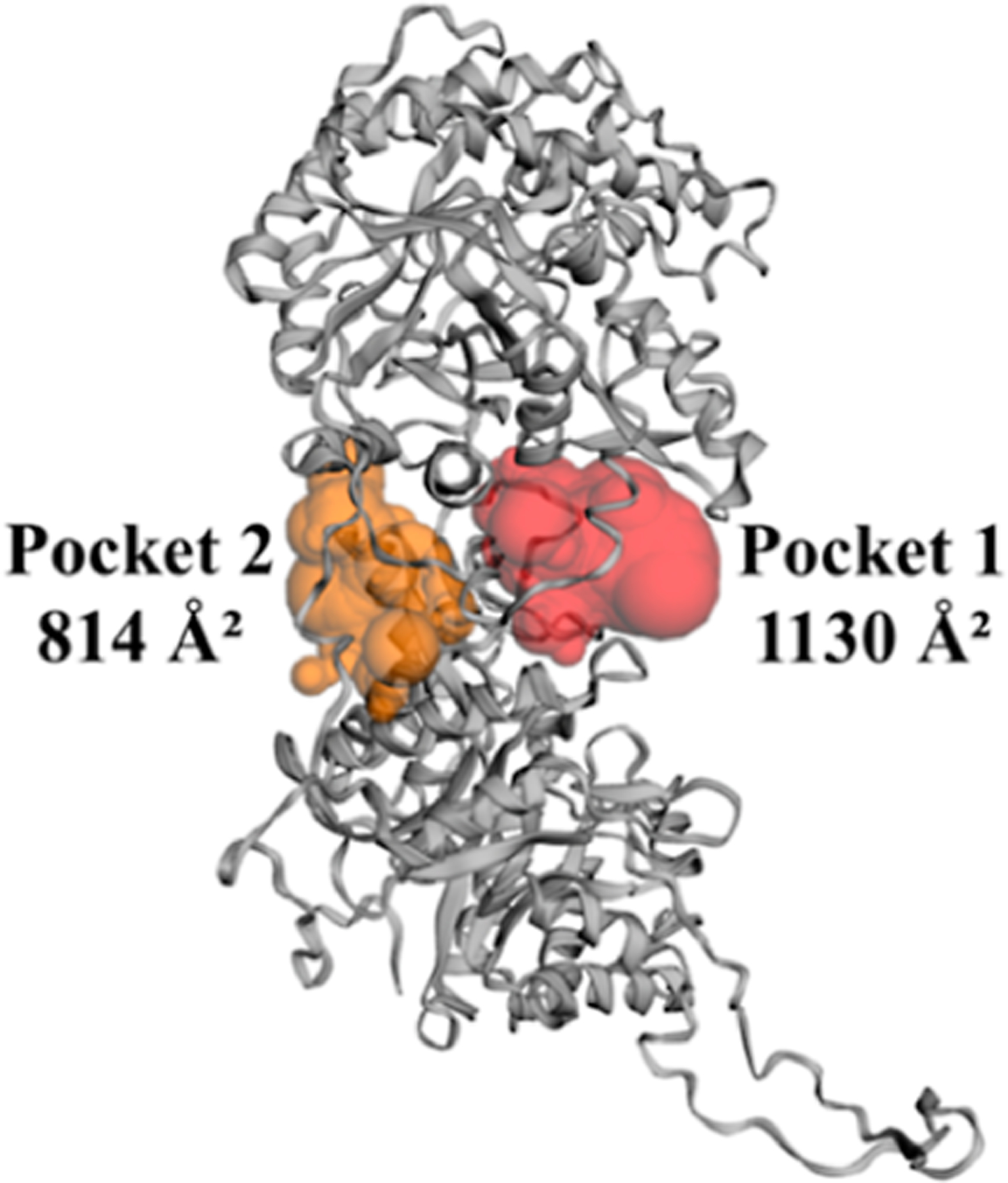
Two surface pockets closest to the KL1:KL2:FGF23 interface with their SASA in Å2 calculated using CASTp37 for the crystal structure18 of α-Klotho. The residues participating in these pockets are listed in Table S9.
The second approach to identifying candidates for experimental validation involves 19 unique compounds that primarily contact the hot spot residue Tyr433 (Table 3), but they have less favorable ΔG on average than those in Table 2. All of these compounds bind to Pocket 1 in the α-Klotho crystal structure (Figure 4a) because the SASA of this pocket is the smallest in the crystal structure (1130 Å2, see Figure 2) than that in the MD clusters (Figure S4). We note that Tyr433 is not found in Pocket 2 of the crystal structure (Table S9). All 19 unique compounds are drug-like,57 and none is PAINS.58 Two compounds (ZINC31607018 and ZINC72289860) were chosen based on their η and vendor availability for in vitro assays to test their efficacies.
Furthermore, there is weak linear correlation (Pearson’s correlation coefficient I54 = 0.28) between ΔG (Vina) and ΔG (KDEEP) in Tables 2 and 3 (Figure S5). The results suggest that there is significant room to improve KDEEP.54
In addition to the above-mentioned two approaches to selecting compounds from the ensemble docking, 50 compounds with the lowest average ΔG over all α-Klotho structures, the crystal structure,18 and the 10 MD clusters from the ensemble docking are listed in Table S12. These 50 compounds also appear in the 476-compound set in the first approach, and ZINC12409120 remains in the first place as it is in Table 2. Furthermore, Table S12 and Table 2 have seven unique compounds in common, which are of interest for in vitro assays in future work.
In Vitro Functional Assays.
The ERK is one of the critical pathways for FGF23 signal transduction and can be activated by formation of the FGF23:α-Klotho complex in the presence of FGFR1c.13,14 Reduced ERK activities are thus consistent with partial disruption or inhibition of the FGF23:α-Klotho complex by small molecules. In addition, ERK activities can also be induced by EGF/EGFR interaction.62 Therefore, the effects of the five compounds on FGF23- and EGF-mediated ERK reporter activities were measured. Out of the five compounds we tested, ZINC12409120 and ZINC05326903 reduce FGF23-mediated ERK reporter activities by 70 and 31% on average, respectively, if FGF23 alone and the control are considered as full and null activities, respectively (Figure 5a). We note that reduction of ERK activities by each of ZINC12409120 and ZINC05326903 is statistically significant (Figure 5a). In addition, the calculated half-lives of ZINC12409120 and ZINC05326903 using pkCSM 63,64 are 8.4 and 7.7 h, respectively. A half-life in this range would generally require twice-daily dosing. The shorter half-lives as compared with the 16 day half-life of burosumab11 may allow dose titration of the compounds to achieve their optimal efficacy and safety profiles. Furthermore, ZINC12409120 has an IC50 of 5.0 ± 0.23 μM and a smooth dose–response curve (Figure 5b). None of the five compounds exhibits EGFR tyrosine kinase inhibition and disrupts EGF/EGFR interaction and EGF-mediated ERK activation (Figure 5c). In contrast, erlotinib, a small-molecule EGFR tyrosine kinase inhibitor,67 completely abolishes EGF-mediated ERK reporter activities (Figure 5c). Comparing Figure 5a with 5c indicates that ZINC12409120 specifically disrupts FGF23:α-Klotho interaction to reduce ERK reporter activities.Optimization of this lead compound could potentially result in FGF23:α-Klotho interaction inhibitors with sub-micromolar to nanomolar binding affinities to α-Klotho.
Figure 5.
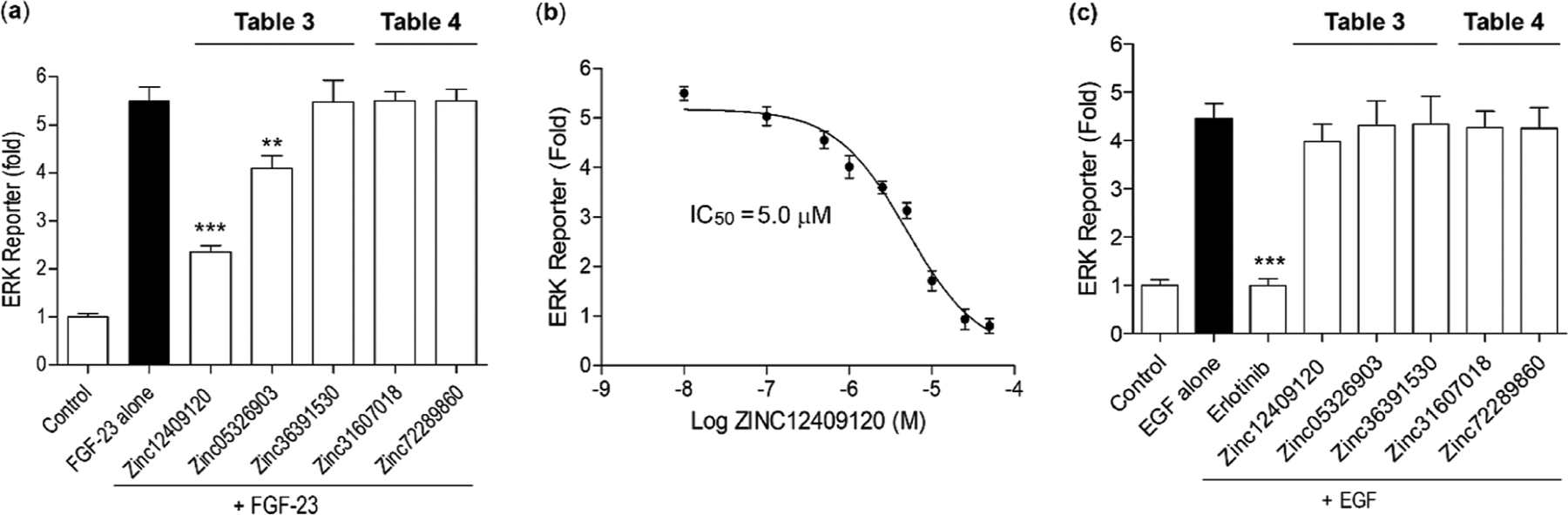
In vitro functional assays of the five compounds selected from Tables 2 and 3. (a) Effects of these compounds on FGF23-mediated ERK reporter activities in transiently α-Klotho-transfected HEK 293T cells. (b) Dose–response curve of ZINC12409120 on FGF23-mediated ERK reporter activities. (c) Effects of these compounds and erlotinib, a small-molecule EGFR tyrosine kinase inhibitor,67 on EGF-mediated ERK reporter activities in transiently EGFR-transfected HEK 293T cells. Each bar shows average ±standard deviation among three independent experiments. *** (p-value ≤ 0.001) and ** (p-value ≤ 0.01) indicate statistically significant difference from FGF23 alone or EGF alone.
In Silico ZINC12409120:α-Klotho Interaction Analysis.
To provide guidance on lead optimization of ZINC12409120 in future studies, it is useful to assess the stability of ZINC12409120:α-Klotho contacts by performing MD simulations starting from the two docking poses on cluster #8 and the crystal structure18 (i.e., Ligand 1a and 1b in Table 2).ZINC12409120 in the MD simulations interacts with the KL1-linker-KL2 region of α-Klotho: Gly55 and Leu56 on the KL1 α-helix, Asn512 on the KL1–KL2 linker, Thr837, Trp838, and Leu839 on the KL2 turn, Pro849 and Trp850 on the KL2 α6-helix, and Tyr889 on the KL2 α7-helix (Table 5 and Figure 6). Among these residues, Trp850 and Tyr889 are the most probable sites for non-hydrogen atomic contact and hydrogen bonding, respectively. In addition, ZINC05326903 also binds to the KL1-linker-KL2 region of the α-Klotho crystal structure18 (Figure S6). The consensus interaction sites of these two compounds are Asn512, Trp838, Pro849, and Trp850, suggesting that targeting at least these four residues with small-molecule drugs may disrupt FGF23:α-Klotho interaction and reduce ERK activities. Our findings also indicate that a ligand binding to residues on the KL1 domain, the KL1–KL2 linker, and the KL2 domain simultaneously may further hinder FGF23:α-Klotho interaction from forming a complex, thereby reducing ERK activities observed in the in vitro assays. Figure 6 visualizes the contacts listed in Table 5 and shows that the two docking poses are similar, and Trp838, Pro849, Trp850, and Tyr889 are their consensus binding sites.
Table 5.
Average Non-Hydrogen Atomic Contacts and Hydrogen Bonds in Parentheses (If Any) with Stand Errors of ZINC12409120 with α-Klotho Residues over Five Runs of 200 ns MD Simulations Starting from the Two Ligand-Bound Conformations (Cluster #8 and Crystal18) in the Ensemble Docking.a
| residue number | initial α-Klotho structure | |
|---|---|---|
| cluster #8 | crystal | |
| Gly55 | 5 ± 1 | 2 ± 0 |
| Leu56 | 6 ± 0 | 2 ± 1 |
| Phe57 | 0 ± 0 | 8 ± 2 |
| Lys435 | 0 ± 0 | 1 ± 0 |
| Glu511 | 0 ± 0 | 0 ± 0 |
| Asn512 | 2 ± 0 | 1 ± 0 |
| Thr837 | 2 ± 0 | 1 ± 0 |
| Trp838 | 2 ± 0 | 4 ± 0 (1 ± 0) |
| Leu839 | 6 ± 0 (1 ± 0) | 1 ± 0 |
| Val847 | 3 ± 1 | 0 ± 0 |
| Pro849 | 2 ± 0 | 2 ± 0 |
| Trp850 | 8 ± 0 | 24 ± 0 (1 ± 0) |
| Tyr889 | 5 ± 0 (1 ± 0) | 3 ± 0 (1 ± 0) |
| Asn893 | 0 ± 0 | 1 ± 0 |
The Numbers of Contacts and Hydrogen Bonds Are Rounded to the Nearest Integer, and Only Residues Contacting ZINC12409120 in Either of the Docking Poses (i.e., Ligand 1a and 1b in Table 2) Are Listed.
Figure 6.
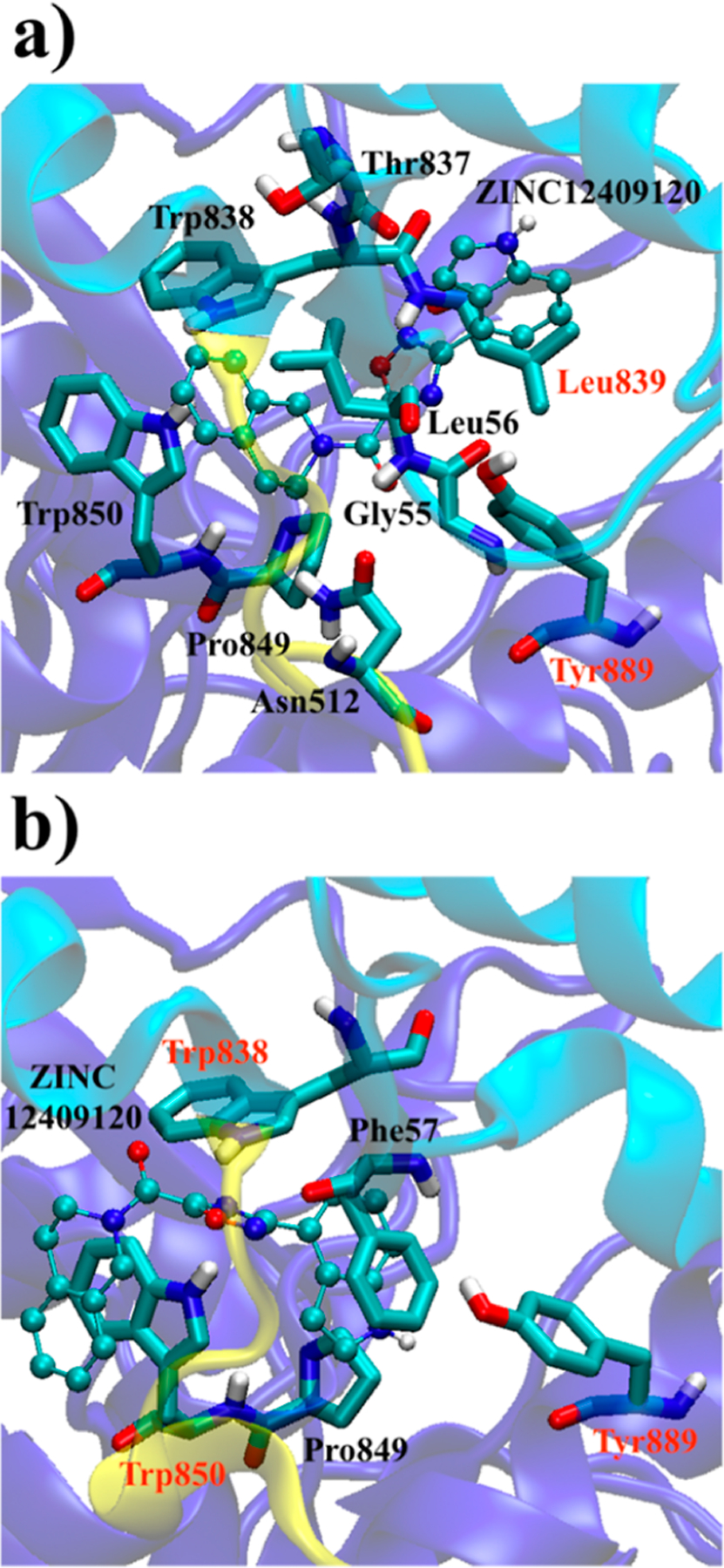
Two binding poses of ZINC12409120 in the KL1-linker-KL2 (light blue-yellow-dark blue) region of α-Klotho (a) cluster #8 and (b) crystal structure18 in the ensemble docking. Only residues contacting the ligand in the docking conformation and MD simulations (average non-hydrogen atomic contacts >1) are shown. The residues in red form hydrogen bonds with the ligand in MD simulations.
In summary, future studies may focus on how to design small molecules with a better geometric fit in between residues on the KL1 domain, the KL1–KL2 linker, and the KL2 domain (especially on Trp850 and Tyr889) of α-Klotho.
CONCLUSIONS
Of the five protein–protein interfaces in the crystal structure of the FGF23:FGFR1c:α-Klotho ternary complex (PDB code: 5W21), that between FGF23 and α-Klotho was computationally identified as the one with the largest fraction (73%) of hot spots and with two druggable residues on α-Klotho. Our hot spot analysis further identified Tyr433 on the KL1 domain of α-Klotho as a promising binding site for in silico docking, and our surface pocket analysis identified α-Klotho as an appropriate drug-binding target at this interface. Armed with this information, we screened computationally ~5.5 million lead-like compounds and identified 476 compounds with the highest predicted protein–ligand binding affinities to α-Klotho and the other 587 compounds whose binding poses had the largest number of contacts with Tyr433. We re-docked these 1063 compounds to the crystal structure and 10 clusters of α-Klotho obtained from MD simulations to potentially refine binding free energies and poses. We rescored the binding free energies of the 24 best docking-scored poses from the 476-compound set and those of the 20 poses with the largest number of contacts with Tyr433 from the 587-compound set. 5 out of 44 compounds were selected for in vitro assays based on their predicted ligand efficiency and vendor availability. Two of the five compounds significantly reduce FGF23-mediated ERK activities. These two compounds were identified based on estimated Vina binding affinities and not on contacts with the hot spot Tyr433. The most potent one (ZINC12409120) disrupts FGF23:α-Klotho interaction to reduce the ERK activities by 70% and has an IC50 of 5.0 ± 0.23 μM. MD simulations starting from the two in silico binding poses of ZINC12409120 on α-Klotho from docking reveal that ZINC12409120 is likely to be in contact with the KL1 domain, the KL1–KL2 linker, and the KL2 domain simultaneously to modulate the function of α-Klotho. This may disrupt FGF23:α-Klotho interaction to reduce the ERK activities.
In this study, two out of five compounds tested were found to have functional activity. This illustrates that the computationally relatively inexpensive screening of millions of compounds to a region with druggable sites and a large fraction of predicted hot spots may allow small-molecule PPI inhibitors to be identified experimentally even when only a very small number of compounds are tested. In future studies, ZINC12409120 analogues could be evaluated for binding to α-Klotho and efficacy in inhibiting FGF23:α-Klotho interaction. Furthermore, the MD clusters of α-Klotho may provide functional insights from structural biology and be useful in a variety of ensemble docking tasks. In addition, we may perform mutagenesis of α-Klotho, test other functions of α-Klotho in the presence and absence of these 44 compounds, and test compounds in a bioluminescence resonance energy transfer assay for target engagement.
DATA AND SOFTWARE AVAILABILITY
The atomic coordinates in the PDB format of all 11 α-Klotho structures and the 44 compounds in the final list from the ensemble docking are included in the Supporting Information. The SMILES in the CSV format for each of the 44 compounds is included in the Supporting Information. The explanation of the identified PAINS (ZINC19373000) is included in the Supporting Information. The atomic coordinates of the α-Klotho crystal structure (PDB code: 5W21) can be downloaded from https://www.rcsb.org/structure/5w21. The lead-like and clean subsets of the ZINC database can be downloaded from http://zinc12.docking.org/subsets/clean-leads. The CHARMM36 force field parameters can be downloaded from http://mackerell.umaryland.edu/charmm_ff.shtml. The following software or program was used: CHARMM General Force Field (CGenFF) (https://cgenff.umaryland.edu), MODELLER (https://salilab.org/modeller/), GROMACS (https://www.gromacs.org/), MGLTools (https://ccsb.scripps.edu/mgltools), KFC Server (https://mitchell-web.ornl.gov/KFC_Server), FTMap Server (https://ftmap.bu.edu), CASTp (http://sts.bioe.uic.edu/castp), AutoDock Vina (https://vina.scripps.edu/), K DEEP (https://playmolecule.com/Kdeep/), SwissADME (http://www.swissadme.ch/), https://www.cbligand.org/PAINS, LigPlot+ (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/), VMD (https://www.ks.uiuc.edu/Research/vmd/), GraphPad Prism (https://www.graphpad.com/), and pkCSM (http://biosig.unimelb.edu.au/pkcsm/).
Supplementary Material
ACKNOWLEDGMENTS
S.H.L. and L.P. were funded by the Laboratory Directed Research and Development Program of Oak Ridge National Laboratory. Z.X. and L.D.Q. were supported by grant R01-DK121132 of the National Institutes of Health. This research used resources of the Compute and Data Environment for Science (CADES) at the Oak Ridge National Laboratory, which is supported by the Office of Science of the United States Department of Energy under Contract no. DE-AC05-00OR22725. This manuscript has been authored by UT-Battelle, LLC, under Contract no. DE-AC05-00OR22725 with the U.S. Department of Energy. The United States Government retains, and the publisher, by accepting the article for publication, acknowledges that the United States Government retains a nonexclusive, paid-up, irrevocable, and worldwide license to publish or reproduce the published form of this manuscript, or allow others to do so, for United States Government purposes. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan (https://www.energy.gov/downloads/doe-public-access-plan).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.2c00633.
MD simulations of α-Klotho; clustering of α-Klotho; coordination of the Zn atom on all α-Klotho structures in the ensemble docking; docking to α-Klotho; hot spot analysis on the FGF23:FGFR1c:α-Klotho ternary complex; surface pocket analysis on α-Klotho in the ternary complex; contact analysis on the 45 compounds; surface pocket analysis on α-Klotho MD clusters; correlation between ΔG (Vina) and ΔG (KDEEP); top 50 compounds ranked by ΔG (Vina) over all α-Klotho structures in the ensemble docking; and in silico ZINC05326903:α-Klotho interaction analysis (PDF)
alpha-Klotho (ZIP)
Compounds (ZIP)
PAINS-ZINC19373000 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.2c00633
The authors declare no competing financial interest.
Contributor Information
Shih-Hsien Liu, UT/ORNL Center for Molecular Biophysics, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States; Department of Biochemistry and Cellular and Molecular Biology, University of Tennessee, Knoxville, Tennessee 37996, United States;.
Zhousheng Xiao, Department of Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Sambit K. Mishra, Biosciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States;
Julie C. Mitchell, Biosciences Division, Oak Ridge NationalLaboratory, Oak Ridge, Tennessee 37831, United States;
Jeremy C. Smith, UT/ORNL Center for Molecular Biophysics, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States; Department of Biochemistry and Cellular and Molecular Biology, University of Tennessee, Knoxville, Tennessee 37996, United States;
L. Darryl Quarles, Department of Medicine, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee 38163, United States.
Loukas Petridis, UT/ORNL Center for Molecular Biophysics, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, United States; Department of Biochemistry and Cellular and Molecular Biology, University of Tennessee, Knoxville, Tennessee 37996, United States;.
REFERENCES
- (1).Quarles LD “Dem bones” are made for more than walking. Nat. Med 2011, 17, 428–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Quarles LD Evidence for a Bone-Kidney Axis Regulating Phosphate Homeostasis. J. Clin. Invest 2003, 112, 642–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Weber TJ; Liu S; Indridason OS; Quarles LD Serum FGF23 Levels in Normal and Disordered Phosphorus Homeostasis. J. Bone Miner. Res 2003, 18, 1227–1234. [DOI] [PubMed] [Google Scholar]
- (4).Sancho PA Complications of Phosphate and Vitamin D Treatment in X-Linked Hypophosphataemia. Adv. Ther 2020, 37, 105–112. [DOI] [PubMed] [Google Scholar]
- (5).Colares Neto G; Ide Yamauchi FI; Hueb Baroni RH; de Andrade Bianchi M; Cavalanti Gomes AC; Chammas MC; Matsunaga Martin RM Nephrocalcinosis and Nephrolithiasis in X-Linked Hypophosphatemic Rickets: Diagnostic Imaging and Risk Factors. J. Endocr. Soc 2019, 3, 1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Florenzano P; Gafni RI; Collins MT Tumor-Induced Osteomalacia. Bone Rep. 2017, 7, 90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fukumoto S Diagnostic Modalities for FGF23-Producing Tumors in Patients with Tumor-Induced Osteomalacia. Endocrinol. Metab 2014, 29, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Carpenter TO; Whyte MP; Imel EA; Boot AM; Högler W; Linglart A; Padidela R; van’t Hoff W; Mao M; Chen C-Y; Skrinar A; Kakkis E; San Martin JS; Portale AA Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med 2018, 378, 1987–1998. [DOI] [PubMed] [Google Scholar]
- (9).Lamb YN Burosumab: First Global Approval. Drugs 2018, 78, 707–714. [DOI] [PubMed] [Google Scholar]
- (10).Carpenter TO; Imel EA; Ruppe MD; Weber TJ; Klausner MA; Wooddell MM; Kawakami T; Ito T; Zhang X; Humphrey J; Insogna KL; Peacock M Randomized Trial of the Anti-FGF23 Antibody KRN23 in X-Linked Hypophosphatemia. J. Clin. Invest 2014, 124, 1587–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zhang X; Imel EA; Ruppe MD; Weber TJ; Klausner MA; Ito T; Vergeire M; Humphrey J; Glorieux FH; Portale AA; Insogna K; Carpenter TO; Peacock M Pharmacokinetics and Pharmacodynamics of a Human Monoclonal Anti-FGF23 Antibody (KRN23) in the First Multiple Ascending-Dose Trial Treating Adults With X-Linked Hypophosphatemia. J. Clin. Pharmacol 2016, 56, 176–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Agrawal A; Ni P; Agoro R; White KE; DiMarchi RD Identification of a Second Klotho Interaction Site in the C terminus of FGF23. Cell Rep. 2021, 34, 108665. [DOI] [PubMed] [Google Scholar]
- (13).Xiao Z; Riccardi D; Velazquez HA; Chin AL; Yates CR; Carrick JD; Smith JC; Baudry J; Quarles LD A Computationally Identified Compound Antagonizes Excess FGF-23 Signaling in Renal Tubules and a Mouse Model of Hypophosphatemia. Sci. Signal 2016, 9, ra113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Urakawa I; Yamazaki Y; Shimada T; Iijima K; Hasegawa H; Okawa K; Fujita T; Fukumoto S; Yamashita T Klotho Converts Canonical FGF Receptor into a Specific Receptor for FGF23. Nature 2006, 444, 770–774. [DOI] [PubMed] [Google Scholar]
- (15).Goetz R; Beenken A; Ibrahimi OA; Kalinina J; Olsen SK; Eliseenkova AV; Xu CF; Neubert TA; Zhang F; Linhardt RJ; Yu X; White KE; Inagaki T; Kliewer SA; Yamamoto M; Kurosu H; Ogawa Y; Kuro-o M; Lanske B; Razzaque MS; Mohammadi M Molecular Insights into the Klotho-Dependent, Endocrine Mode of Action of Fibroblast Growth Factor 19 Subfamily Members. Mol. Cell. Biol 2007, 27, 3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Xiao Z; Liu J; Liu S-H; Petridis L; Cai C; Cao L; Wang G; Chin AL; Cleveland JW; Ikedionwu MO; Carrick JD; Smith JC; Quarles LD Novel Small Molecule Fibroblast Growth Factor 23 Inhibitors Increase Serum Phosphate and Improve Skeletal Abnormalities in Hyp Mice. Mol. Pharmacol 2022, 101, 408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Downs RP; Xiao Z; Ikedionwu MO; Cleveland JW; Lin Chin AL; Cafferty AE; Darryl Quarles LD; Carrick JD Design and Development of FGF-23 Antagonists: Definition of the Pharmacophore and Initial Structure-Activity Relationships Probed by Synthetic Analogues. Bioorg. Med. Chem 2021, 29, 115877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chen G; Liu Y; Goetz R; Fu L; Jayaraman S; Hu M-C; Moe OW; Liang G; Li X; Mohammadi M α-Klotho is a Non-Enzymatic Molecular Scaffold for FGF23 Hormone Signaling. Nature 2018, 553, 461–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Damm-Ganamet KL; Arora N; Becart S; Edwards JP; Lebsack AD; McAllister HM; Nelen MI; Rao NL; Westover L; Wiener JJM; Mirzadegan T Accelerating Lead Identification by High Throughput Virtual Screening: Prospective Case Studies from the Pharmaceutical Industry. J. Chem. Inf. Model 2019, 59, 2046–2062. [DOI] [PubMed] [Google Scholar]
- (20).Wingert BM; Camacho CJ Improving Small Molecule Virtual Screening Strategies for the Next Generation of Therapeutics. Curr. Opin. Chem. Biol 2018, 44, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Dawidowski M; Emmanouilidis L; Kalel VC; Tripsianes K; Schorpp K; Hadian K; Kaiser M; Mäser P; Kolonko M; Tanghe S; Rodriguez A; Schliebs W; Erdmann R; Sattler M; Popowicz GM Inhibitors of PEX14 Disrupt Protein Import into Glycosomes and Kill Trypanosoma Parasites. Science 2017, 355, 1416–1420. [DOI] [PubMed] [Google Scholar]
- (22).Macalino SJY; Basith S; Clavio NAB; Chang H; Kang S; Choi S Evolution of In Silico Strategies for Protein-Protein Interaction Drug Discovery. Molecules 2018, 23, 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang H; Liu C; Deng L Enhanced Prediction of Hot Spots at Protein-Protein Interfaces Using Extreme Gradient Boosting. Sci. Rep 2018, 8, 14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Xia J; Yue Z; Di Y; Zhu X; Zheng C-H Predicting Hot Spots in Protein Interfaces Based on Protrusion Index, Pseudo Hydrophobicity and Electron-Ion Interaction Pseudopotential Features. Oncotarget 2016, 7, 18065–18075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Deng L; Guan J; Wei X; Yi Y; Zhang QC; Zhou S Boosting Prediction Performance of Protein-Protein Interaction Hot Spots by Using Structural Neighborhood Properties. J. Comput. Biol 2013, 20, 878–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Xia J-F; Zhao X-M; Song J; Huang D-S APIS: Accurate Prediction of Hot Spots in Protein Interfaces by Combining Protrusion Index with Solvent Accessibility. BMC Bioinf. 2010, 11, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cho K.-i.; Kim D; Lee D A Feature-Based Approach to Modeling Protein-Protein Interaction Hot Spots. Nucleic Acids Res. 2009, 37, 2672–2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kortemme T; Baker D A Simple Physical Model for Binding Energy Hot Spots in Protein-Protein Complexes. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 14116–14121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Kozakov D; Grove LE; Hall DR; Bohnuud T; Mottarella SE; Luo L; Xia B; Beglov D; Vajda S The FTMap Family of Web Servers for Determining and Characterizing Ligand-Binding Hot Spots of Proteins. Nat. Protoc 2015, 10, 733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhu X; Mitchell JC KFC2: A Knowledge-Based Hot Spot Prediction Method Based on Interface Solvation, Atomic Density, and Plasticity Features. Proteins 2011, 79, 2671–2683. [DOI] [PubMed] [Google Scholar]
- (31).Darnell SJ; Page D; Mitchell JC An Automated Decision-Tree Approach to Predicting Protein Interaction Hot Spots. Proteins 2007, 68, 813–823. [DOI] [PubMed] [Google Scholar]
- (32).Webb B; Sali A Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinf 2016, 54, 5.6.1–5.6.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (34).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (35).Páll S; Abraham MJ; Kutzner C; Hess B; Lindahl E Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Lect. Notes Comput. Sci 2015, 8759, 3–27. [Google Scholar]
- (36).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDockTools4 Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tian W; Chen C; Lei X; Zhao J; Liang J CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Edelsbrunner H; Mücke EP Three-Dimensional Alpha Shapes. ACM Trans. Graph 1994, 13, 43–72. [Google Scholar]
- (39).Amaro RE; Baudry J; Chodera J; Demir O; McCammon JA; Miao Y; Smith JC Ensemble Docking in Drug Discovery. Biophys.J 2018, 114, 2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Best RB; Zhu X; Shim J; Lopes PEM; Mittal J; Feig M; MacKerell AD Jr. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput 2012, 8, 3257–3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926–935. [Google Scholar]
- (43).Daura X; Gademann K; Jaun B; Seebach D; van Gunsteren WF; Mark AE Peptide Folding: When Simulation Meets Experiment. Angew. Chem., Int. Ed 1999, 38, 236–240. [Google Scholar]
- (44).Ellingson SR; Miao Y; Baudry J; Smith JC Multi-Conformer Ensemble Docking to Difficult Protein Targets. J. Phys. Chem. B 2015, 119, 1026–1034. [DOI] [PubMed] [Google Scholar]
- (45).Teague SJ; Davis AM; Leeson PD; Oprea T The Design of Leadlike Combinatorial Libraries. Angew. Chem., Int. Ed 1999, 38, 3743–3748. [DOI] [PubMed] [Google Scholar]
- (46).Irwin JJ; Sterling T; Mysinger MM; Bolstad ES; Coleman RG ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model 2012, 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Wang S; Wacker D; Levit A; Che T; Betz RM; McCorvy JD; Venkatakrishnan AJ; Huang X-P; Dror RO; Shoichet BK; Roth BL D4 Dopamine Receptor High-Resolution Structures Enable the Discovery of Selective Agonists. Science 2017, 358, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; Tolmachev AA; Shoichet BK; Roth BL; Irwin JJ Ultra-Large Library Docking for Discovering New Chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ellingson SR; Smith JC; Baudry J VinaMPI: Facilitating Multiple Receptor High-Throughput Virtual Docking on High-Performance Computers. J. Comput. Chem 2013, 34, 2212–2221. [DOI] [PubMed] [Google Scholar]
- (50).Trott O; Olson AJ AutoDock Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31, 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Su M; Yang Q; Du Y; Feng G; Liu Z; Li Y; Wang R Comparative Assessment of Scoring Functions: The CASF-2016 Update. J. Chem. Inf. Model 2019, 59, 895–913. [DOI] [PubMed] [Google Scholar]
- (52).Gaillard T Evaluation of AutoDock and AutoDock Vina on the CASF-2013 Benchmark. J. Chem. Inf. Model 2018, 58, 1697–1706. [DOI] [PubMed] [Google Scholar]
- (53).Acharya A; Agarwal R; Baker MB; Baudry J; Bhowmik D; Boehm S; Byler KG; Chen SY-C; Coates L; Cooper CJ; Demerdash O; Daidone I; Eblen JD; Ellingson S; Forli S; Glaser J; Gumbart JC; Gunnels J; Hernandez O; Irle S; Kneller DW; Kovalevsky A; Larkin J; Lawrence TJ; LeGrand S; Liu S-H; Mitchell JC; Park G; Parks JM; Pavlova A; Petridis L; Poole D; Pouchard L; Ramanathan A; Rogers DM; Santos-Martins D; Scheinberg A; Sedova A; Shen Y; Smith JC; Smith MD; Soto C; Tsaris A; Thavappiragasam M; Tillack AF; Vermaas JV; Vuong VQ; Yin J; Yoo S; Zahran M; Zanetti-Polzi L Supercomputer-Based Ensemble Docking Drug Discovery Pipeline with Application to Covid-19. J. Chem. Inf. Model 2020, 60, 5832–5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Jiménez J;Škalič M; Martínez-Rosell G; De Fabritiis G KDEEP: Protein-Ligand Absolute Binding Affinity Prediction via 3DConvolutional Neural Networks. J. Chem. Inf. Model 2018, 58, 287–296. [DOI] [PubMed] [Google Scholar]
- (55).Hopkins AL; Groom CR; Alex A Ligand Efficiency: A Useful Metric for Lead Selection. Drug DiscovToday 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- (56).Li Y; Liu Z; Li J; Han L; Liu J; Zhao Z; Wang R Comparative Assessment of Scoring Functions on an Updated Benchmark: 1. Compilation of the Test Set. J. Chem. Inf. Model 2014, 54, 1700–1716. [DOI] [PubMed] [Google Scholar]
- (57).Daina A; Michielin O; Zoete V SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep 2017, 7, 42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Baell JB; Holloway GA New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem 2010, 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- (59).Vanommeslaeghe K; Hatcher E; Acharya C; Kundu S; Zhong S; Shim J; Darian E; Guvench O; Lopes P; Vorobyov I; Mackerell AD Jr. CHARMM General Force Field: A Force Field for Drug-Like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem 2010, 31, 671–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Yu W; He X; Vanommeslaeghe K; MacKerell AD Jr. Extension of the CHARMM General Force Field to Sulfonyl-Containing Compounds and Its Utility in Biomolecular Simulations. J. Comput. Chem 2012, 33, 2451–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Laskowski RA; Swindells MB LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [DOI] [PubMed] [Google Scholar]
- (62).Pierce KL; Tohgo A; Ahn S; Field ME; Luttrell LM; Lefkowitz RJ Epidermal Growth Factor (EGF) Receptor-Dependent ERK Activation by G Protein-Coupled Receptors. J. Biol. Chem 2001, 276, 23155–23160. [DOI] [PubMed] [Google Scholar]
- (63).Mansoor A; Mahabadi N Volume of Distribution. StatPearls; StatPearls Publishing, 2022. https://www.ncbi.nlm.nih.gov/books/NBK545280/. [PubMed] [Google Scholar]
- (64).Pires DEV; Blundell TL; Ascher DB pkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem 2015, 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Suzuki Y; Kuzina E; An SJ; Tome F; Mohanty J; Li W; Lee S; Liu Y; Lax I; Schlessinger J FGF23 Contains Two Distinct High-Affinity Binding Sites Enabling Bivalent Interactions with α-Klotho. Proc. Natl. Acad. Sci. U.S.A 2020, 117, 31800–31807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Ran X; Gestwicki JE Inhibitors of Protein-Protein Interactions (PPIs): An Analysis of Scaffold Choices and Buried Surface Area. Curr. Opin. Chem. Biol 2018, 44, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Chaft JE; Oxnard GR; Sima CS; Kris MG; Miller VA; Riely GJ Disease Flare after Tyrosine Kinase Inhibitor Discontinuation in Patients with EGFR-Mutant Lung Cancer and Acquired Resistance to Erlotinib or Gefitinib: Implications for Clinical Trial Design. Clin. Cancer Res 2011, 17, 6298–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates in the PDB format of all 11 α-Klotho structures and the 44 compounds in the final list from the ensemble docking are included in the Supporting Information. The SMILES in the CSV format for each of the 44 compounds is included in the Supporting Information. The explanation of the identified PAINS (ZINC19373000) is included in the Supporting Information. The atomic coordinates of the α-Klotho crystal structure (PDB code: 5W21) can be downloaded from https://www.rcsb.org/structure/5w21. The lead-like and clean subsets of the ZINC database can be downloaded from http://zinc12.docking.org/subsets/clean-leads. The CHARMM36 force field parameters can be downloaded from http://mackerell.umaryland.edu/charmm_ff.shtml. The following software or program was used: CHARMM General Force Field (CGenFF) (https://cgenff.umaryland.edu), MODELLER (https://salilab.org/modeller/), GROMACS (https://www.gromacs.org/), MGLTools (https://ccsb.scripps.edu/mgltools), KFC Server (https://mitchell-web.ornl.gov/KFC_Server), FTMap Server (https://ftmap.bu.edu), CASTp (http://sts.bioe.uic.edu/castp), AutoDock Vina (https://vina.scripps.edu/), K DEEP (https://playmolecule.com/Kdeep/), SwissADME (http://www.swissadme.ch/), https://www.cbligand.org/PAINS, LigPlot+ (https://www.ebi.ac.uk/thornton-srv/software/LigPlus/), VMD (https://www.ks.uiuc.edu/Research/vmd/), GraphPad Prism (https://www.graphpad.com/), and pkCSM (http://biosig.unimelb.edu.au/pkcsm/).