

Abstract
Kaposiform lymphangiomatosis (KLA) is a life-threatening rare disease that can cause substantial morbidity, mortality, and social burdens for patients and their families. Diagnosis often occurs long after initial symptoms, and there are few centers in the world with the expertise to diagnose and care for patients with the disease. KLA is a lymphatic anomaly and significant advancements have been made in understanding its pathogenesis and etiology since its first description in 2014. This review provides multi-disciplinary, comprehensive, and state-of-the-art information on KLA patient presentation, diagnostic imaging, pathology, organ involvement, genetics, and pathogenesis. Finally, we describe current therapeutic approaches, important areas for research, and challenges faced by patients and their families. Further insights into the pathogenesis of KLA may advance our understanding of other vascular anomalies given that similar signaling pathways may be involved.
Keywords: Kaposiform lymphangiomatosis, lymphatic anomalies, rare diseases
Introduction
Kaposiform lymphangiomatosis (KLA) is a rare and devastating lymphatic anomaly that affects children and young adults. The first reported series of 20 patients by Croteau et al. found the median age of onset to be 6.5 years (range, 0 to 44 years), 51% 5-year and 34% overall survival rates, and a mean interval from diagnosis to death of 2.75 years (range, 1 to 6.5 years).1A more recent study primarily focused on KLA pathology reported an improved survival rate of 79% and a mean interval from disease onset to death of 6.7 years (range, 1.9 to 14.3 years) in 38 patients available to follow-up.2 New therapies have likely improved the prognosis, although updated epidemiological studies are needed to better quantify the prevalence of KLA and survival with available treatments.3 The diagnosis of KLA is often delayed due to variable presenting symptoms, apprehension concerning obtaining a biopsy due to coagulopathy and risk of lymphatic leakage, and lack of familiarity with this disease.4–6 Treatment remains challenging as many patients show no or only partial responses, and so there is an urgent need to identify new therapeutic targets and more effective medications.7 Mortality is typically the result of cardiorespiratory failure or coagulopathy.6
The International Society for the Study of Vascular Anomalies (ISSVA) 2018 updated classification defines KLA as a subset of generalized lymphatic anomaly (GLA). However, whether KLA is actually distinct from GLA remains an ongoing debate.8 KLA is characterized by malformed lymphatic channels accompanied by clusters of lymphatic endothelial cells (LECs) with a spindled morphology that is referred to as “kaposiform.” Lung, mediastinum, skin and subcutaneous fat, trabecular bone, and spleen are typically involved.9,10 GLA has broad diagnostic criteria defined by multifocal overgrowth of lymphatic vessels.11 Other complex lymphatic anomalies that can be confused with KLA are central conducting lymphatic anomaly (CCLA) and Gorham-Stout disease (GSD).12,13 CCLA is characterized by a failure of lymphatic drainage by central channels, including the thoracic duct, leading to pericardial and pleural effusions and peripheral lymphedema.14 KLA can be associated with central conducting issues leading to pericardial and pleural effusions, but the disease also affects soft tissues and other organs. In GSD, abnormal lymphangiogenesis in bone is predominant, leading to progressive osteolysis and loss of cortical bone. GSD lesions lack kaposiform cells and patients do not have coagulopathy. KLA must also be distinguished from kaposiform hemangioendothelioma (KHE), another rare disease characterized by kaposiform cells and coagulopathy. KHE usually presents in infancy or early childhood with an indurated purpuric skin lesion.15 In contrast to KLA, KHE is typically unifocal in the trunk or extremities and is not associated with central conducting abnormalities.
In this review, the symptoms, diagnosis, pathology, organ involvement, genetics, pathogenesis, and treatments for KLA will be discussed as well as emerging therapies and future research directions. Importantly, we also address the challenges that patients and their families experience. We discuss the role of patients and their families in advocacy, support, and the care of those living with KLA.
Clinical Presentation and Imaging
KLA patients often present with variable and complex symptoms depending on the primary location of the diseased tissues (Table 1). Lesions commonly involve the thoracic cavity, including the mediastinum, lung, and heart, and may be associated with pleural and pericardial effusions.16,17 The effusions are often hemorrhagic, even in the absence of coagulopathy, and this can be important in the diagnosis of KLA. Patients may present with fatigue, shortness of breath, cough, and fever.1,6 Skeletal involvement can lead to back, joint, soft tissue, or bone pain. Organs of the abdominal cavity, including the spleen and liver, may be involved, but hepatic dysfunction is uncommon. Pelvic disease is sometimes seen with genital swelling that may be confused with hernia diagnoses. Consumptive coagulopathy of unknown etiology can occur—it can manifest as bruising or bleeding. It is highly variable but can present with a combination of markedly elevated D-dimer, low or moderate fibrinogen, and low or moderate platelet levels.4,5,12,18,19 Unlike Kasabach-Merritt Phenomenon (KMP), which is associated with KHE and characterized by profound thrombocytopenia and hypofibrinogenemia, decreases in platelet levels are generally, but not always, more mild in cases of KLA.20 Additional investigation into the coagulopathy in KLA is much needed. Biomarkers have been identified, especially serum angiopoietin-2 (Ang-2) levels, which are elevated.21,22 Other rare symptoms include pseudotumor cerebri, which may be due to blockage of venous return in the neck by lymphangiomatous tissue, and cerebrospinal fluid-lymphatic fistula causing spontaneous intracranial hypotension.23,24 The complex and diverse nature of presenting symptoms, along with the lack of familiarity of clinicians with this rare disorder, frequently results in the misdiagnosis of KLA as pneumonia, cancer, bone marrow failure, or other vascular anomalies, sometimes leading to years of diagnostic uncertainty and delays in effective treatment.
TABLE 1.
Summary of features of KLA.*
| Approach | Features |
|---|---|
| Clinical presentation | Symptoms are secondary to lesion
location. General: i) Fatigue; ii) Nausea Pulmonary: i) Hemoptysis; ii) Shortness of breath; iii) Recurrent pneumonia Hematology: Bruising Bone and tissue: i) Pain; ii) Soft tissue swelling |
| Imaging | Thoracic: i) Pleural effusions; ii)
Pericardial effusion; iii) Mediastinal infiltration; iv) Thickened lung
interstitium Abdominal: Cystic splenic lesions Skeletal: Cystic, infiltrative, or fat-containing bone lesions |
| Laboratory findings | Blood levels†: i) Elevated angiopoietin-2‡; ii) Elevated D-dimer; iii) Low fibrinogen; iv) Low platelets |
| Pathology | Tissue lesions#: i) Spindled lymphatic endothelial cells; ii) Malformed lymphatic channels; iii) D2–40, LYVE-1, and PROX-1 positive immunostaining |
| Genetic testing | Somatic mutations#: i) NRAS Q61R in tissue samples (90% of patients)2,8 and in plasma and effusions56; ii) CBL mutation in CD31-selected cells from pleural chylous effusions (one patient)51 |
Abbreviation: KLA, Kaposiform lymphangiomatosis
Laboratory values may normalize with treatment.
Serum angiopoietin-2 levels can be determined with a clinical test at Cincinnati Children’s Hospital Medical Center’s Hemostasis and Thrombosis Laboratory (https://www.cincinnatichildrens.org/service/c/cancer-blood/hcp/clinical-laboratories/hemostasis-thrombosis-lab)
Can be difficult to obtain and prone to sampling errors.
Imaging is essential in the diagnosis and management of KLA and should be interpreted in the context of each patient’s clinical presentation. Initial imaging often includes thoracic radiography, which may show pulmonary parenchymal abnormalities, mediastinal widening, pleural effusions, and/or bone abnormalities (Figs. 1A–1C). However, computed tomography (CT) and magnetic resonance imaging (MRI) are the primary modalities for diagnosis and management.12,16 Chest CT often reveals pleural effusions, pericardial effusion, and extensive smooth thickening of the peribronchovascular and interlobular interstitium with mediastinal fat infiltration by poorly-defined fluid-attenuation lesions without discrete macrocysts (Figs. 2A–2B).16,25–27 CT may also reveal randomly distributed osteolytic lesions.28 MRI is particularly useful to identify KLA lesions in the thorax, abdomen, pelvis, and skeleton.29 MRI chest findings largely parallel those of CT, with interstitial thickening, mediastinal infiltration, and effusions generally appearing bright on T2-weighted images (Figs. 3A–3D). If present, splenic lesions can appear cystic. On MRI, lesions in the bone marrow may be well defined and cystic-appearing or poorly defined and demonstrate increased fat deposition.30
Figure 1.

Chest radiography of patients with kaposiform lymphangiomatosis. A) Upright posteroanterior view in a 12-year-old male shows thickening of the peribronchovascular interstitium (solid white arrows). Widening of the left paraspinal stripe inferiorly (solid black arrow) is due to mediastinal infiltration by the disease. B) Upright lateral chest view in the same patient shows thickening of the peribronchovascular interstitium (solid white arrows) with visualization of peripheral interlobular septal thickening anteriorly (dotted white arrows). C) Supine frontal chest view in a 25-year-old male shows generalized interstitial thickening with left greater than right pleural effusions (solid black arrows). There is also mediastinal widening due to infiltration by abnormal lymphatic channels/fluid (solid white arrows).
Figure 2.

Computed tomography images of patient with kaposiform lymphangiomatosis. A) Axial contrast-enhanced chest image in lung windows in an 11-year-old female shows variable degrees of interstitial thickening, visible along the bronchial walls centrally (white arrows) and secondary pulmonary lobules peripherally (black arrows). B) Coronal contrast-enhanced chest image in the same patient shows expansion of the mediastinum by poorly defined fluid attenuation tissue (solid white arrows), corresponding to infiltration by disease. Numerous lesions are also noted in the spleen (dotted white arrow).
Figure 3.

Coronal magnetic resonance imaging of patients with kaposiform lymphangiomatosis. A) Short-T1 inversion recovery (STIR) image of the body in a 5-year-old male shows large pleural effusions (solid white arrows), multiple small cystic-appearing splenic lesions (dotted white arrows), and numerous cystic-appearing lesions throughout the skeleton (purple arrows). B) STIR image in the same patient shows abnormal confluent foci of increased signal intensity throughout the right pelvis and femoral neck (solid white arrows). There is also abnormally decreased signal intensity throughout the right femoral head (yellow arrow). A more normal pattern of red marrow is seen in most of the left pelvis and femur. Note also the abnormal poorly defined lymphatic tissue in the pelvic soft tissues (dotted white arrow). C) T1 image in the same patient shows intermixed regions of increased signal intensity (yellow arrow) due to increased fat deposition as well as regions of decreased signal intensity (solid white arrow) that are more typical of abnormal lymphatics. D) STIR image in a 10-month-old male shows cystic-appearing splenic lesions (dotted white arrow). Bilateral pleural disease (purple arrows) is noted, as well as mediastinal (blue arrow) and neck (yellow arrow) lymphatic malformations.
Other imaging techniques that can be used include ultrasonography (US) and dynamic contrast-injected magnetic resonance lymphangiography (MRL).27,31 US may be used to evaluate pleural and pericardial effusions, which appear largely anechoic as they are typically serosanguinous, chylous, or chylosanguinous. MRL is an advanced technique that can be very helpful in fully delineating the extensive abnormalities, including dilation and tortuosity of the lymphatic vessels and patterns of lymph flow in KLA (Figs. 4A–4F).31,32 There are two components to performing MRL: the first is non-invasive T2 space imaging to visualize slow moving fluids and edema. The second is injection of magnetic resonance contrast solution into the lymphatics, typically via the inguinal lymph nodes, liver lymphatics, and mesenteric lymphatics, and subsequent time resolved and high-resolution static imaging to visualize the central lymphatic system. In KLA, there is a wide variation of the central lymphatics ranging from little to no perfusion abnormalities to extensive multiorgan involvement.
Figure 4.

Coronal T2-weighted magnetic resonance imaging (MRI) and magnetic resonance lymphangiography (MRL) of patients with kaposiform lymphangiomatosis. A) T2-weighted MRI of a 10 year-old male shows significant mediastinal thickening with pulmonary edema and bilateral supraclavicular edema. B) The corresponding MRL shows a significantly dilated and tortuous thoracic duct (yellow arrows) that terminates in the mediastinum with significant perfusion of the mediastinal and pulmonary lymphatics on MRL. C) T2-weighted MRI of a 4 year-old male shows mediastinal thickening with pulmonary edema, dermal edema, and ascites. D) The corresponding MRL shows no clear thoracic duct but several abnormal lymphatic channels coursing into the thorax leading to mediastinal and pulmonary lymphatic perfusion. E) T2-weighted MRI of a 12 year-old female shows mediastinal thickening with bilateral pleural effusions. F) The corresponding MRL shows a dilated and mildly tortuous thoracic duct (white arrows), but with limited mediastinal or pulmonary lymphatic perfusion.
Bone and Bone Marrow Involvement
Lymphatic vessels are not found in normal healthy bone; however, bone involvement in KLA is observed on imaging and in tissue specimens.33,34 Osseous lesions in KLA localize to trabecular bone and marrow, and they may significantly affect structural integrity leading to fractures, pain, and disability.35 Treatment with steroids, used in the initial treatment of KLA, can weaken bone due to glucocorticoid-induced osteoporosis.36 Bone resorption markers, such as carboxyterminal cross-linking telopeptide (CTX-1), may be elevated.18 Treatment of KLA may include antiresorptive drugs such as bisphosphonates or denosumab to mitigate the disease’s effects on skeletal health.18,37
Pathology
The pathology of KLA lesions is characterized by dysmorphic lymphatic channels that are dilated, abnormally shaped, variable in size, and lined by attenuated endothelium (Fig. 5A). In addition, there is a variable number of clusters of spindled and often hemosiderotic LECs (Figs. 5B–5D). These are usually dispersed but occasionally are focally aggregated.15 The spindled cells are immunopositive for the lymphatic markers D2–40, LYVE1, and PROX1 (Figs. 5E–5I).10,38 Spindled LECs in KLA lesions can also express mesenchymal stem cell markers (CD73, CD90, CD105, CD146) and proliferate and migrate rapidly.10 It is believed, but not proven, that spindled kaposiform LECs may contribute to malformed lymphatic vessels within lesions.39 Splenic histopathology can show focal or diffuse spongiform changes with widened cords of Billroth containing lymphocytes, red blood cells, hematopoietic precursors, histiocytes, stromal cells, and reticulin (Fig. 5J). Dilated sinusoids are lined by large, spindled, and focally redundant endothelial cells that can be hemosiderotic (Fig. 5K). Sometimes, the spongioform changes progress to overt macrocysticifcation. Small nodules, several millimeters in size, of spindled kaposiform cells are seen in some specimens.
Figure 5.
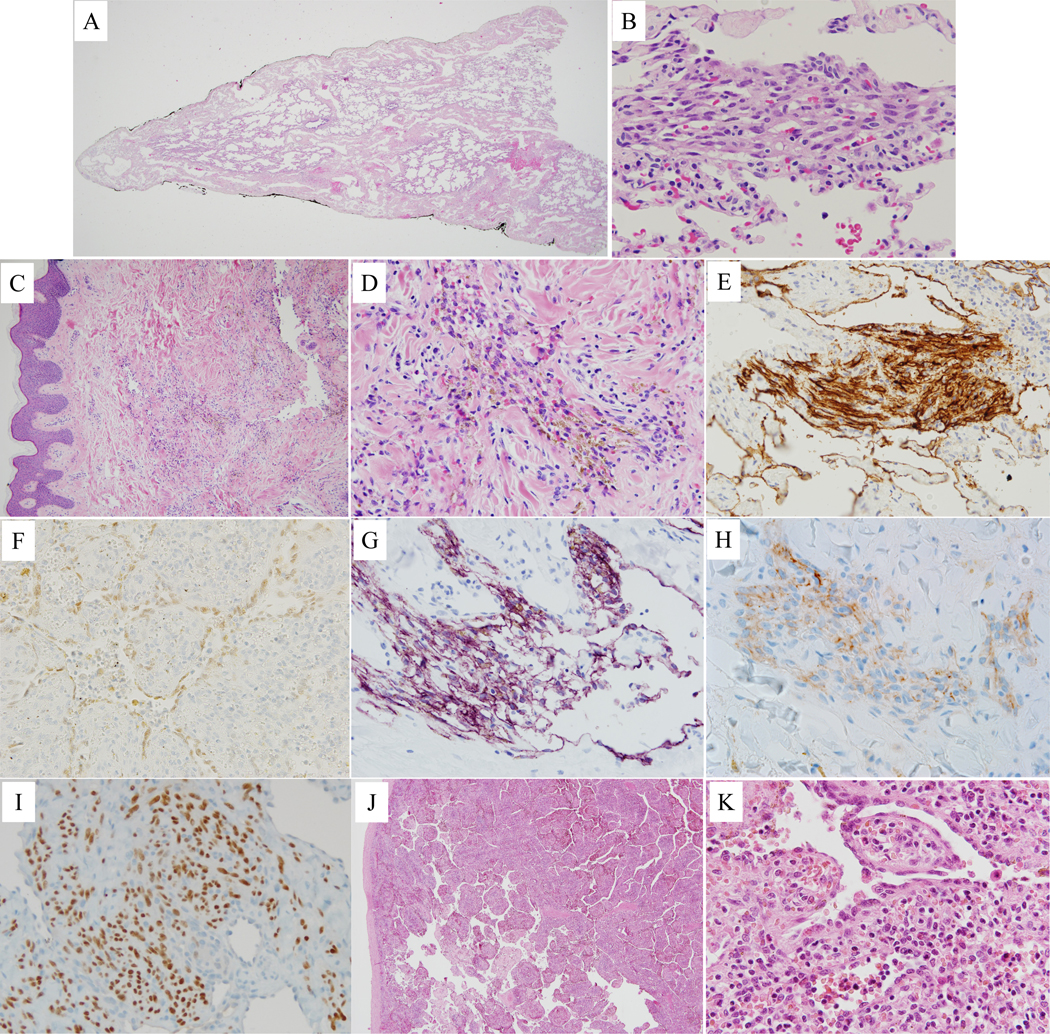
Histopathology of kaposiform lymphangiomatosis lesions. A) Lung with marked dilated lymphatic channels in pleura and interlobular septa. B) Lung with cluster of spindled cells and interspersed erythrocytes. C) Single, dilated lymphatic channel in reticular dermis and small cellular cluster in middle of the field. D) Dermal cellular cluster of hemosiderotic spindled cells with interspersed erythrocytes. E) Cytoplasmic immunopositivity for D2–40 in spindled cell cluster and adjacent dilated lymphatic vessels in lung. F) Nuclear immunopositivity for PROX1 in splenic sinusoidal lining cells. Dermal spindled cell clusters with cytoplasmic immunopositivity for D2–40 (G) and LYVE1 (H), and nuclear immunopositivity for PROX1 (I). J) Spleen with markedly expanded cords of Billroth and dilated sinusoids. K) Splenic hyperplastic sinusoidal cells, some with phagocytosed red blood cells and cytoplasmic eosinophilic globules.
While pathologic examination can aid in the diagnosis of KLA, it is not always performed given the risk involved in obtaining a biopsy. The procedure can exacerbate coagulopathy and bleeding and can also cause lymphatic leakage with the new development or worsening of effusions, contributing to morbidity and mortality. Biopsy should be discussed by the entire healthcare team and the benefits of solidifying the diagnosis should be weighed carefully against the risks of the procedure for each patient. If a biopsy is determined to be absolutely necessary, soft tissue biopsies are preferable and rib biopsies should be avoided due to high risk for chylous leak. It is possible that the combination of advanced imaging techniques and measuring biomarkers may be sufficient to arrive at a diagnosis without the risk of obtaining a biopsy.
Genetics, Pathogenesis, and Biomarkers
Barclay et al. were the first to report a somatic activating NRAS p.Q61R mutation in the lesional tissue of 10 of 11 patients with KLA, with an allele frequency ranging from 1–28% within the lesions.8 This study suggests that the NRAS p.Q61R mutation occurs in approximately 90% of patients with KLA. In an earlier report this mutation was identified in a patient given a diagnosis of GLA but with overlapping features of KLA.40 The NRAS p.Q61R mutation activates mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) pathways.11,41 RAS mutations are implicated in many cancers and other vascular anomalies. For example, NRAS p.Q61R is present in 29% of cutaneous melanomas and 10% of pyogenic granulomas, as well as other benign and cancerous tumors.42–46 In cancers, MAPK-ERK signaling downstream of the NRAS p.Q61R mutation is known to drive cell proliferation, tumor extracellular matrix degradation, and angiogenesis.47 The PI3K-AKT-mTOR pathway is also frequently activated in numerous cancers and RASopathies and plays critical functions in cell proliferation, migration, angiogenesis, and tumorigenesis.48 Overactivation of this pathway in KLA may provide an explanation of why treatment with the mTOR inhibitor sirolimus produced a partial response in some KLA patients.49,50 Instead of an NRAS mutation, one patient with a KLA phenotype had a point mutation in the Casitas B lineage lymphoma (CBL) gene detected with an allele frequency of 4% in CD31-selected cells from pleural chylous effusions.51 CBL mutations associated with leukemia can result in activation of the Ras-MAPK pathway, and this patient had a good response to MAPK inhibition.52Currently, the genetics of KLA are uncertain but seem to relate to hyperactive Ras signaling due to somatic mutations.
Elevated Ang-2 levels have been reported in the blood of patients with KLA and KHE prior to treatment (Supplemental Fig. S1A).21,22 Ang-2 is a context-dependent signaling ligand which can act as an agonist or antagonist for the Tie2 receptor to drive blood and lymphatic vessel growth and destabilization, although its role in the pathogenesis of KLA remains uncertain.53,54 Le Cras et al. showed that serum Ang-2 levels were approximately 9-fold higher in 7 patients with KLA compared to age and sex-matched controls.21 Ozeki et al. also reported elevated serum Ang-2 (approximately 10-fold) in another cohort of 11 patients with KLA and identified additional biomarkers including increases in soluble vascular endothelial growth factor receptor 3 (sVEGFR3; 7-fold).22 Clinical testing of serum Ang-2 levels may be of use as a noninvasive method to both aid in diagnosis and monitor treatment response in KLA.18
Identification of somatic mutations via isolation and sequencing of cell free DNA from blood or other body fluids is an emerging diagnostic technique that has proven useful in identifying the genetic etiology of KLA.55 Ozeki et al. reported 5 patients with KLA from whom tissue biopsies, plasma samples, and pleural effusions were collected and analyzed for mutations.56 The NRAS p.Q61R mutation was detected in both lesions and cell free DNA extracted from plasma and pleural effusions in all the patients. Identification of somatic mutations using cell free DNA from plasma or effusions holds great promise for expediting the diagnosis, avoiding the risk of biopsy, and directing targeted treatment.
Experimental Models
The origin of the NRAS p.Q61R mutation during development and its role in KLA remain unclear. However, an experimental model of human endothelial progenitor cells (EPCs) with doxycycline-inducible expression of NRAS p.Q61R or wild-type NRAS may provide insight.57 In in vitro assays, EPCs expressing NRAS p.Q61R developed a spindled morphology with elevated proliferation and migration and increased phosphorylation of ERK1/2. In a fibrin bead in vitro angiogenesis model, NRAS p.Q61R-expressing EPCs formed enlarged vascular channels. In a xenograft model, NRAS p.Q61R-expressing EPCs developed large diameter vascular channels. Taken together, these studies indicate that NRAS p.Q61R-expressing EPCs mimic many of the features of the spindled LECs seen in KLA and therefore suggest that the NRAS p.Q61R mutation may play a key role in driving KLA pathogenesis.
Treatments
Medical treatments for KLA can involve suppression of the abnormal signaling pathways induced by the underlying somatic mutation, anti-inflammatory medication, and supportive therapies. Because these treatments are not curative, pharmacotherapy is expected to be lifelong. Most importantly, given the complexity of the disease, patients should receive care at an institution with experience. Based on a 2016 prospective clinical trial, current guidelines indicate that, in the absence of a genetic diagnosis, first-line therapy for KLA is treatment with sirolimus.3,7 Adams et al. designed the trial after observing disease regression in patients with complicated vascular anomalies who, after failing to respond to other treatments, responded to sirolimus. The study showed that age-based dosing of sirolimus with a goal trough of 10–15 ng/mL is safe and efficacious for KLA, with patients having variable but often significant improvement within 6 months of starting treatment. However, doses should be individualized so that patients receive the lowest possible dose for therapeutic effect to minimize drug toxicities. Le Cras et al. also showed that treatment with sirolimus decreases serum Ang-2 levels (Supplemental Fig. S1B).21 However, it remains unclear whether decreases in Ang-2 contribute to the response to therapy in patients with KLA.
Supportive therapies must be individualized to each patient depending on their symptoms and disease severity, although rigorous studies of these therapies for KLA are lacking. If coagulopathy is present, cryoprecipitate can be used to maintain adequate fibrinogen levels. Platelets can be given sparingly for active bleeding or surgery. Pleural or pericardial effusions can be addressed if present and causing significant symptoms. Chest tubes and pericardial windows have been reported to provide acute improvement in cases of severe organ compromise, although sustained improvement after these interventions is rare.1 Ultimately, medical management is usually required for long-term stability. Any intervention must be approached with caution after multi-disciplinary evaluation of the numerous risks and potential benefits and discussion with patients and their families. Corticosteroids and/or vincristine can also be added for exacerbations to control coagulopathy and symptoms in patients with KLA.1,7,9,18,58 In the presence of bony disease, bisphosphonates, most commonly zoledronic acid, can be used in combination with sirolimus. In one case, this multimodal approach also improved coagulopathy and reduced blood Ang-2 levels, and the patient had an excellent outcome.18 Similarly, combination sirolimus and bisphosphonates have been used for treatment of GSD suggesting that these drugs may have a synergistic effect in this condition as well.59,60 The bisphosphonate zoledronic acid may have additional inhibitory effects on Ras-ERK signaling, unlike other medications in its class.61,62
Unfortunately, first-line therapy is not always effective in treating KLA, and many patients ultimately experience disease progression. In patients who do not respond to first-line therapies, or in patients that have a known Ras-MAPK pathway mutation, MAPK inhibition is beginning to be used. However, there have not been any clinical trials in these patients to support this approach.7 Of the MAPK inhibitors, trametinib (Mekinist) has been used most frequently, although there can be significant side effects including rash, acneiform dermatitis, diarrhea, fatigue, hypertension, and ventricular dysfunction.39,51,63 Clinical trials are underway with additional MAPK inhibitors, including solumetinib and cobimetinib, for other MAPK-pathway disorders.61 Continued evaluation of genomic mutations is necessary as other mutations may be implicated in the KLA phenotype.
Patient Perspective, Experience, & Support
As with many rare diseases, support for patients with KLA is complicated, stressful, and anxiety-provoking for all involved. From the very beginning, when symptoms are first identified, obtaining a definitive diagnosis of KLA is challenging and burdensome. Patients with rare diseases often face misdiagnoses and diagnostic delays, and they sometimes receive inappropriate treatments that may cause complications and even contribute to death.64 Furthermore, the time during which patients with KLA are undiagnosed or misdiagnosed delays treatment and allows the disease to progress, potentially leading to irreversible complications and diminished efficacy of treatments.
Approaches to patient support can be modeled from guidelines already established in pediatric oncology care.65 Multidisciplinary healthcare teams should establish a physician-patient partnership that includes the patient’s caregivers, and care must be individualized (Supplemental Fig. S2).66 Furthermore, providers should offer family-centered education on KLA. This can help empower a patient’s family to better understand the disease so that the family unit feels less isolated with the diagnosis. Secondly, it can encourage families to pursue a team-approach to caring for the patient and establish a patient-centered partnership between patients, families, and providers. Another useful resource for patients and their families are vascular anomaly patient advocacy groups that help patients and families to empathize as they share their unique struggles, insights, and perspectives.67 Advocacy groups also facilitate exchange of information about specialized care centers and new therapies. Current patient advocacy groups for patients with complex lymphatic anomalies include: i) Lymphangiomatosis and Gorhams Disease Alliance (LDGA, https://lgdalliance.org/) and ii) Lymphatic Education & Research Network (LE&RN, https://lymphaticnetwork.org/). For research advocacy there is the Consortium of iNvestigators of Vascular AnomalieS (CaNVAS, https://www.research.chop.edu/canvas). Finally, providers can offer patients the opportunity to engage with ongoing research by encouraging participation in registries such as The Lymphatic Anomalies Registry at Boston Children’s Hospital (https://www.childrenshospital.org/programs/lymphatic-anomalies-registry) and natural history studies to advance understanding of the disease and help improve treatment.
Future Directions
There are many important questions left to answer about the underlying disease processes in KLA. While the NRAS p.Q61R activating mutation has been identified in several studies, and a CBL mutation was identified in one patient with KLA, it is important to determine whether there are other mutations that can cause a KLA phenotype. Given that the somatic mutations found in KLA and other vascular anomalies also play a role in cancer, there is the potential for repurposing emerging cancer treatments as new therapies for KLA. The role of Ang-2 and other pro-angiogenic biomarkers in KLA pathogenesis is important to determine as these could also be novel therapeutic targets in addition to being diagnostic tools. The lymphatic conducting problems that often contribute to the morbidity and mortality of KLA via pleural and pericardial effusions are also poorly understood. MRL clearly shows abnormalities of the central conducting system, such as a tortuous thoracic duct, but the cause of this phenotype of the disease is unclear. It is uncertain whether the endothelial cells lining the thoracic duct are abnormal and/or express the same somatic mutations as KLA lesions.
Regarding the etiopathogenesis of KLA, further research is needed to link the findings of somatic mutations and elevated biomarkers to the phenotype of abnormal spindled endothelial cells seen on histology and the atypical tangles of lymphatics found on imaging. Researchers must fully clarify the derangements in mutant LEC intracellular signaling and their complex interactions with local tissues and the immune system. Single cell techniques such as proteomics and RNA sequencing analysis can reveal the complex transcriptional, translational, and signaling effects of mutations. These novel techniques have the potential to uncover new pathways that refine the taxonomy, improve molecular diagnosis, and identify new targets for therapy. Furthermore, the knowledge gained by a better understanding of normal and abnormal endothelial cell development may find broader applicability in cardiology and oncology.
Conclusions
KLA is a complex lymphatic anomaly with a poor prognosis. Diagnosis is difficult, and current treatments are often only partially effective and can cause severe side effects. Further research is urgently needed to improve our understanding of the disease process, reduce time to diagnosis, and develop more effective treatments for KLA. Not only will this have important impacts on outcomes for patients with KLA, but it holds additional implications for related diseases given the overlap in signaling pathways, disease pathogenesis, and therapeutics.
Supplementary Material
Supplemental Figure S1. A) Serum angiopoietin-2 (Ang-2) levels prior to treatment are elevated in kaposiform lymphangiomatosis (KLA) and kaposiform hemangioendothelioma patients compared to age and sex-matched controls. Serum Ang-2 levels were not elevated in patients with generalized lymphatic anomaly compared to controls. Standard error of the mean and p-value comparisons from analysis of variance and Tukey’s multiple comparison test are shown. ns = not significant. B) Serum Ang-2 levels in patients with KLA decrease after treatment with sirolimus. Dotted lines show maximum (max) and mean serum Ang-2 levels in control group.
Supplemental Figure S2. Disciplines that must work together in the care of a patient with kaposiform lymphangiomatosis.
Funding/Support:
NHLBI R01 HL156866
Abbreviations
- KLA
Kaposiform lymphangiomatosis
- DIC
Disseminated intravascular coagulation
- GLA
Generalized lymphatic anomaly
- CCLA
Central conducting lymphatic anomaly
- GSD
Gorham-Stout disease
- ISSVA
International Society for the Study of Vascular Anomalies
- LEC
Lymphatic endothelial cell
- KHE
Kaposiform hemangioendothelioma
- KMP
Kasabach-Merritt Phenomenon
- Ang-2
Angiopoietin-2
- CT
Computed tomography
- MRI
Magnetic resonance imaging
- US
Ultrasonography
- MRL
Magnetic resonance lymphangiogram
- CTX-1
Carboxyterminal cross-linking telopeptide
- MAPK
Mitogen-activated protein kinase
- PI3K
Phosphoinositide 3-kinase
- CBL
Casitas B lineage lymphoma
- sVEGFR3
Soluble vascular endothelial growth factor receptor 3
- EPC
Endothelial progenitor cell
Footnotes
Contributors Statement Page
CGM conceptualized and designed the review, drafted the initial manuscript, and critically reviewed and revised the manuscript.
DMA conceptualized and designed the review, provided critical clinical expertise for KLA presentation and treatment, drafted the initial manuscript, and critically reviewed and revised the manuscript.
KES conceptualized and designed the review, provided personal experiences about patient perspectives and support, drafted the initial manuscript, and critically reviewed and revised the manuscript.
TDLC conceptualized and designed the review, provided expertise in experimental models and scientific approaches, drafted the initial manuscript, and critically reviewed and revised the manuscript.
AMH, and JLC provided critical clinical expertise for KLA presentation and treatment and critically reviewed and revised the manuscript.
ACM and CLS provided expertise in diagnostic techniques and imaging and critically reviewed and revised the manuscript.
HPWK provided pathology images and expertise in pathology and critically reviewed and revised the manuscript.
All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Conflict of interest disclosure: DMA is a consultant and steering committee member for Novartis and a consultant for Alderis and Nobias. AMH provides clinical research support for Merck (ongoing), Novartis (ongoing), Venthera (within the past year), and Palvella (within the past year); is a steering committee member for Novartis; and is a consultant for Novartis, Ideaya, and Aytu. ACM is an author/consultant/editor for Elsevier, Inc for royalties and fees.
References
- 1.Croteau SE, Kozakewich HP, Perez-Atayde AR, et al. Kaposiform lymphangiomatosis: a distinct aggressive lymphatic anomaly. J Pediatr. 2014;164(2):383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Atayde AR, Debelenko L, Al-Ibraheemi A, et al. Kaposiform Lymphangiomatosis: Pathologic Aspects in 43 Patients. Am J Surg Pathol. 2022;46(7):963–976. [DOI] [PubMed] [Google Scholar]
- 3.Adams DM, Trenor CC, Hammill AM, et al. Efficacy and Safety of Sirolimus in the Treatment of Complicated Vascular Anomalies. Pediatrics. 2016;137(2):e20153257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandes VM, Fargo JH, Saini S, et al. Kaposiform lymphangiomatosis: unifying features of a heterogeneous disorder. Pediatr Blood Cancer. 2015;62(5):901–904. [DOI] [PubMed] [Google Scholar]
- 5.Wang Z, Li K, Yao W, et al. Successful treatment of kaposiform lymphangiomatosis with sirolimus. Pediatric blood & cancer. 2015;62(7):1291–1293. [DOI] [PubMed] [Google Scholar]
- 6.Bundy JJ, Ootaki Y, McLean TW, et al. Thoracic duct embolization in kaposiform lymphangiomatosis. J Vasc Surg Venous Lymphat Disord. 2020;8(5):864–868. [DOI] [PubMed] [Google Scholar]
- 7.Adams DM, Ricci KW. Vascular Anomalies: Diagnosis of Complicated Anomalies and New Medical Treatment Options. Hematol Oncol Clin North Am. 2019;33(3):455–470. [DOI] [PubMed] [Google Scholar]
- 8.Barclay SF, Inman KW, Luks VL, et al. A somatic activating NRAS variant associated with kaposiform lymphangiomatosis. Genet Med. 2019;21(7):1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams DM, Brandão LR, Peterman CM, et al. Vascular anomaly cases for the pediatric hematologist oncologists-An interdisciplinary review. Pediatr Blood Cancer. 2018;65(1). [DOI] [PubMed] [Google Scholar]
- 10.Glaser K, Dickie P, Dickie BH. Proliferative Cells From Kaposiform Lymphangiomatosis Lesions Resemble Mesenchyme Stem Cell-like Pericytes Defective in Vessel Formation. Journal of pediatric hematology/oncology. 2018;40(8):e495–e504. [DOI] [PubMed] [Google Scholar]
- 11.Ozeki M, Fukao T. Generalized Lymphatic Anomaly and Gorham-Stout Disease: Overview and Recent Insights. Adv Wound Care (New Rochelle). 2019;8(6):230–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iacobas I, Adams DM, Pimpalwar S, et al. Multidisciplinary guidelines for initial evaluation of complicated lymphatic anomalies-expert opinion consensus. Pediatr Blood Cancer. 2020;67(1):e28036. [DOI] [PubMed] [Google Scholar]
- 13.ISSVA Classification of Vascular Anomalies 2018. Paper presented at: International Society for the Study of Vascular Anomalies; Accessed 2022 May 4. [Google Scholar]
- 14.McCormick A, Rosenberg S, Tier K, et al. A Case of a Central Conducting Lymphatic Anomaly Responsive to Sirolimus. Pediatrics. 2016;137(1). [DOI] [PubMed] [Google Scholar]
- 15.Ji Y, Chen S, Peng S, et al. Kaposiform lymphangiomatosis and kaposiform hemangioendothelioma: similarities and differences. Orphanet J Rare Dis. 2019;14(1):165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ozeki M, Fujino A, Matsuoka K, et al. Clinical Features and Prognosis of Generalized Lymphatic Anomaly, Kaposiform Lymphangiomatosis, and Gorham-Stout Disease. Pediatr Blood Cancer. 2016;63(5):832–838. [DOI] [PubMed] [Google Scholar]
- 17.Chen G, Graf NS, Pang T. Isolated abdominal kaposiform lymphangiomatosis: a novel presentation of a rare entity. Pathology. 2021;53(2):279–282. [DOI] [PubMed] [Google Scholar]
- 18.Crane J, Manfredo J, Boscolo E, et al. Kaposiform lymphangiomatosis treated with multimodal therapy improves coagulopathy and reduces blood angiopoietin-2 levels. Pediatr Blood Cancer. 2020;67(9):e28529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Itkin M, McCormack FX. Nonmalignant Adult Thoracic Lymphatic Disorders. Clin Chest Med. 2016;37(3):409–420. [DOI] [PubMed] [Google Scholar]
- 20.Kelly M. Kasabach-Merritt Phenomenon. Pediatric Clinics of North America. 2010;57(5):1085–1089. [DOI] [PubMed] [Google Scholar]
- 21.Le Cras TD, Mobberley-Schuman PS, Broering M, et al. Angiopoietins as serum biomarkers for lymphatic anomalies. Angiogenesis. 2017;20(1):163–173. [DOI] [PubMed] [Google Scholar]
- 22.Ozeki M, Nozawa A, Kawamoto N, et al. Potential biomarkers of kaposiform lymphangiomatosis. Pediatr Blood Cancer. 2019;66(9):e27878. [DOI] [PubMed] [Google Scholar]
- 23.Barceló-López C, López-Guerrero AL, García-López A, et al. Pseudotumor cerebri in kaposiform lymphangiomatosis: a case report and pathogenetic hypothesis. Childs Nerv Syst. 2018;34(8):1609–1611. [DOI] [PubMed] [Google Scholar]
- 24.Soderlund KA, Mamlouk MD, Shah VN, et al. Cerebrospinal fluid-lymphatic fistula causing spontaneous intracranial hypotension in a child with kaposiform lymphangiomatosis. Pediatr Radiol. 2021;51(11):2093–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kato H, Ozeki M, Fukao T, et al. Chest imaging in generalized lymphatic anomaly and kaposiform lymphangiomatosis. Pediatr Int. 2018;60(7):667–668. [DOI] [PubMed] [Google Scholar]
- 26.Ricci KW, Iacobas I. How we approach the diagnosis and management of complex lymphatic anomalies. Pediatr Blood Cancer. 2021:e28985. [DOI] [PubMed] [Google Scholar]
- 27.Goyal P, Alomari AI, Kozakewich HP, et al. Imaging features of kaposiform lymphangiomatosis. Pediatr Radiol. 2016;46(9):1282–1290. [DOI] [PubMed] [Google Scholar]
- 28.Trenor CC 3rd, Chaudry G. Complex lymphatic anomalies. Seminars in pediatric surgery. 2014;23(4):186–190. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura F, Kato H, Ozeki M, et al. CT and MRI Findings of Focal Splenic Lesions and Ascites in Generalized Lymphatic Anomaly, Kaposiform Lymphangiomatosis, and Gorham-Stout Disease. J Clin Imaging Sci. 2021;11:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato H, Ozeki M, Fukao T, et al. MR imaging findings of vertebral involvement in Gorham-Stout disease, generalized lymphatic anomaly, and kaposiform lymphangiomatosis. Jpn J Radiol. 2017;35(10):606–612. [DOI] [PubMed] [Google Scholar]
- 31.Itkin M. Magnetic Resonance Lymphangiography and Lymphatic Embolization in the Treatment of Pulmonary Complication of Lymphatic Malformation. Seminars in interventional radiology. 2017;34(3):294–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chavhan GB, Lam CZ, Greer MC, et al. Magnetic Resonance Lymphangiography. Radiol Clin North Am. 2020;58(4):693–706. [DOI] [PubMed] [Google Scholar]
- 33.Allen-Rhoades W, Al-Ibraheemi A, Kohorst M, et al. Cellular Variant of Kaposiform Lymphangiomatosis: A Report of Three Cases, Expanding the Morphologic and Molecular Genetic Spectrum of this Rare Entity. Hum Pathol. 2022. [DOI] [PubMed] [Google Scholar]
- 34.Edwards JR, Williams K, Kindblom LG, et al. Lymphatics and bone. Hum Pathol. 2008;39(1):49–55. [DOI] [PubMed] [Google Scholar]
- 35.Solorzano E, Alejo AL, Ball HC, et al. Osteopathy in Complex Lymphatic Anomalies. Int J Mol Sci. 2022;23(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Briot K, Roux C. Glucocorticoid-induced osteoporosis. RMD open. 2015;1(1):e000014-e000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Anthony MD, Swilling A, Jiwani ZM, et al. Multidisciplinary Multiagent Treatment of Complex Lymphatic Anomalies with Severe Bone Disease: A Single-Site Experience. Lymphatic Research and Biology. 2022;20(2):118–124. [DOI] [PubMed] [Google Scholar]
- 38.Safi F, Gupta A, Adams D, et al. Kaposiform lymphangiomatosis, a newly characterized vascular anomaly presenting with hemoptysis in an adult woman. Annals of the American Thoracic Society. 2014;11(1):92–95. [DOI] [PubMed] [Google Scholar]
- 39.Chowers G, Abebe-Campino G, Golan H, et al. Treatment of severe Kaposiform lymphangiomatosis positive for NRAS mutation by MEK inhibition. Pediatr Res. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Manevitz-Mendelson E, Leichner GS, Barel O, et al. Somatic NRAS mutation in patient with generalized lymphatic anomaly. Angiogenesis. 2018;21(2):287–298. [DOI] [PubMed] [Google Scholar]
- 41.Boscolo E, Pastura P, Glaser K, et al. Signaling pathways and inhibitors of cells from patients with kaposiform lymphangiomatosis. Pediatr Blood Cancer. 2019;66(8):e27790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129(7):1287–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degirmenci U, Wang M, Hu J. Targeting Aberrant RAS/RAF/MEK/ERK Signaling for Cancer Therapy. Cells. 2020;9(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Muñoz-Couselo E, Adelantado EZ, Ortiz C, et al. NRAS-mutant melanoma: current challenges and future prospect. Onco Targets Ther. 2017;10:3941–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Queisser A, Boon LM, Vikkula M. Etiology and Genetics of Congenital Vascular Lesions. Otolaryngol Clin North Am. 2018;51(1):41–53. [DOI] [PubMed] [Google Scholar]
- 46.Lim YH, Douglas SR, Ko CJ, et al. Somatic Activating RAS Mutations Cause Vascular Tumors Including Pyogenic Granuloma. J Invest Dermatol. 2015;135(6):1698–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo YJ, Pan WW, Liu SB, et al. ERK/MAPK signalling pathway and tumorigenesis (Review). Exp Ther Med. 2020;19(3):1997–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Castellano E, Downward J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes & cancer. 2011;2(3):261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nature reviews Drug discovery. 2014;13(2):140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karar J, Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front Mol Neurosci. 2011;4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster JB, Li D, March ME, et al. Kaposiform lymphangiomatosis effectively treated with MEK inhibition. EMBO Mol Med. 2020;12(10):e12324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loh ML, Sakai DS, Flotho C, et al. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood. 2009;114(9):1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yancopoulos GD, Davis S, Gale NW, et al. Vascular-specific growth factors and blood vessel formation. Nature. 2000;407(6801):242–248. [DOI] [PubMed] [Google Scholar]
- 54.Akwii RG, Sajib MS, Zahra FT, et al. Role of Angiopoietin-2 in Vascular Physiology and Pathophysiology. Cells. 2019;8(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chemi F, Pearce SP, Clipson A, et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat Cancer. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozeki M, Aoki Y, Nozawa A, et al. Detection of NRAS mutation in cell-free DNA biological fluids from patients with kaposiform lymphangiomatosis. Orphanet J Rare Dis. 2019;14(1):215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boscolo E, Pastura P, Schrenk S, et al. NRASQ61R mutation in human endothelial cells causes vascular malformations. Angiogenesis. 2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou J, Yang K, Chen S, et al. Sirolimus in the treatment of kaposiform lymphangiomatosis. Orphanet J Rare Dis. 2021;16(1):260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anthony MD, Swilling A, Jiwani ZM, et al. Multidisciplinary Multiagent Treatment of Complex Lymphatic Anomalies with Severe Bone Disease: A Single-Site Experience. Lymphat Res Biol. 2021. [DOI] [PubMed] [Google Scholar]
- 60.Angelini A, Mosele N, Pagliarini E, et al. Current concepts from diagnosis to management in Gorham-Stout disease: a systematic narrative review of about 350 cases. EFORT open reviews. 2022;7(1):35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Braicu C, Buse M, Busuioc C, et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel). 2019;11(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu J, Pan Q, Ju W. Ras inhibition by zoledronic acid effectively sensitizes cervical cancer to chemotherapy. Anti-cancer drugs. 2019;30(8):821–827. [DOI] [PubMed] [Google Scholar]
- 63.Thota R, Johnson DB, Sosman JA. Trametinib in the treatment of melanoma. Expert Opin Biol Ther. 2015;15(5):735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schieppati A, Henter J-I, Daina E, et al. Why rare diseases are an important medical and social issue. The Lancet. 2008;371(9629):2039–2041. [DOI] [PubMed] [Google Scholar]
- 65.Landier W, Ahern J, Barakat LP, et al. Patient/Family Education for Newly Diagnosed Pediatric Oncology Patients. J Pediatr Oncol Nurs. 2016;33(6):422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pomey M-P, Ghadiri DP, Karazivan P, et al. Patients as partners: a qualitative study of patients’ engagement in their health care. PloS one. 2015;10(4):e0122499-e0122499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Delisle VC, Gumuchian ST, Rice DB, et al. Perceived Benefits and Factors that Influence the Ability to Establish and Maintain Patient Support Groups in Rare Diseases: A Scoping Review. Patient. 2017;10(3):283–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1. A) Serum angiopoietin-2 (Ang-2) levels prior to treatment are elevated in kaposiform lymphangiomatosis (KLA) and kaposiform hemangioendothelioma patients compared to age and sex-matched controls. Serum Ang-2 levels were not elevated in patients with generalized lymphatic anomaly compared to controls. Standard error of the mean and p-value comparisons from analysis of variance and Tukey’s multiple comparison test are shown. ns = not significant. B) Serum Ang-2 levels in patients with KLA decrease after treatment with sirolimus. Dotted lines show maximum (max) and mean serum Ang-2 levels in control group.
Supplemental Figure S2. Disciplines that must work together in the care of a patient with kaposiform lymphangiomatosis.