

Abstract
Background
Cardiovascular disease medications such as aspirin (ASP), statins like atorvastatin (ATR), and blood pressure-lowering drugs including ACE inhibitors like ramipril (RAM) have been included in the World Health Organization (WHO) Essential Medicines List (EML) for many years. Therefore, there is a strong demand to develop a simple, rapid, and sensitive analytical method that can detect and quantitate the ternary mixture of these analytes in pharmaceutical preparations in a short run time. Lately, the analytical community focused on eliminating or reducing hazardous chemicals and solvents usage.
Results
A green, fast, selective, and cost-effective micellar HPLC method was established and validated for the concurrent determination of ternary combination of ASP, ATR, and RAM in the pure form and pharmaceutical preparations. Resolution of the three drugs was achieved by using a monolithic column and a micellar mobile phase consists of 0.3% triethylamine (TEA) in 90: 10 an aqueous solution of 0.12 M sodium dodecyl sulfate (SDS): n-propanol, (v/v). The pH was adjusted to 2.5 using orthophosphoric acid and a flow rate of 1.5 mL/min. was applied. To ensure method reproducibility, Valsartan (VAL) was utilized as an internal standard (IS). The UV detection of the studied drugs was performed at 210 nm. Good linearity for the three drugs was obtained over the concentration ranges of 1.0-200.0 mg/mL, 0.5-200.0 mg/mL, and 5.0-100.0 mg/mL with correlation coefficients of 0.9998,0.9999 and 0.9999 for ASP, ATR, and RAM respectively. The method sensitivity was revealed by the relatively small values of limits of detection (LOD) (0.19, 0.13 and 0.30 mg/mL) and limits of quantitation (LOQ) (0.63, 0.44 and 0.99 mg/mL) for ASP, ATR, and RAM, respectively. The retention times of ASP, ATR and RAM were 1.50, 2.3 and 4.3 min., respectively.
Conclusions
The suggested technique was employed for the analysis of the three drugs in their prepared tablets maintaining the recommended pharmaceutical ratio without any interference from excipients. The method was further extended to content uniformity testing of RAM. The results were validated according to international council for harmonisation (ICH) guidelines.
Keywords: Micellar HPLC, Aspirin, Atorvastatin, Ramipril, Laboratory prepared tablet, Content uniformity testing
Introduction
Aspirin (ASP): (Fig. 1a) is chemically named 2-Acetyloxybenzoic acid [1]. It has been used mainly in the relief of pain, inflammation and in some fever cases. ASP is used in cardiovascular disorders due to its ability to inhibit platelet aggregation [2].
Fig. 1.

The structural formulae of the studied drugs: (a) Aspirin (b) Atorvastatin (c) Ramipril
Atorvastatin (ATR); (Fig. 1b) is chemically named R-R*,R*-2-(4-Fluorophenyl)-b,d-dihydroxy-5-(1-methylethyl)-3-phenyl-4-(phenylamino)carbonyl-1 H-pyrrole-1-heptanoic acid [1]. It is used in the treatment of hyperlipidemia, as it regulates the lipid by its action on plasma lipids and reduces LDL cholesterol. Atorvastatin can be effective as adjunctive therapy in patients with homozygous familial hypercholesterolemia who have some LDL-receptor function. It was also used for primary and secondary prophylaxis of cardiovascular incidents in patients with numerous risk factors [2].
Ramipril; (Fig. 1c) is chemically (2 S,3aS,6aS)-1-(2 S)-2-(2 S)-1-Ethoxy-1-oxo-4-phenylbutan-2-yl-amino-propanoyl-3,3a,4,5,6,6a-hexahydro-2 H-cyclopenta[b]pyrrole-2-carboxylicacid [1]. Ramipril is an Angiotensin-converting enzyme (ACE) inhibitors and is used to treat hypertension, heart failure, and after myocardial infarction. It is also used to decrease the risk of cardiovascular effects in patients with certain risk factors [2].
All the three drugs were official in British [3] and United States [4] pharmacopoeia. Cardiovascular disease medications such as aspirin, statins, and blood pressure-lowering drugs including ACE inhibitors have been included in the World Health Organization (WHO) Essential Medicines List (EML) for many years. The first year of listing for aspirin was 1990, for an ACE inhibitor was 2003 and for a statin, 2007. This application seeks to add a fixed-dose combination (FDCs) of these three types of medication “a polypill” to the EML for the prevention of recurrent events in individuals with prior heart disease or stroke.
The combination used for blood pressure and cholesterol lowering and antiplatelet treatments has been recommended as potential strategy to decrease the global burden of Atherosclerotic Cardiovascular Disease given its potential for better adherence, lower costs and improved operational logistics especially in health systems in low-resource settings [5]. Due to importance of this combination the “21st Expert Committee on the Selection and use of essential medicines” recommended a fixed-dose combination of atorvastatin, ramipril and aspirin [6].
The literature of the methods reported for the simultaneous assay of that ternary combination declares that a few number of studies have been performed including HPLC [7–10], HPTLC [11] and UV spectrophotometric [12] methods, but all the reported chromatographic methods utilized mobile phases containing large quantities of expensive and hazardous organic solvents. As far as we know, no micellar HPLC methods published for the concurrent determination of this combination either in pure synthetic mixture or in tablets. According to the above details, it was desirable to develop an isocratic, simple and green HPLC method to determine these drugs in their combined formulations in short run time.
The green analytical chemistry concept aims at saving environment. It focus on replacing the required harmful chemicals used in analytical techniques with safer substitutes, using limited energy and reducing the waste, without influencing the analytical performance [13]. The consumption of organic solvents in large amounts in liquid chromatography makes it sometimes harmful technique [14]. Replacement of the common harmful mobile phases that by greener alternatives, such as water [15], ethanol or isopropanol [16]. The usage of micellar liquid chromatographic method also is a solution as it is easy to handle, time and cost saving and neither damaging the ecosystem nor the analyst. Unlike the available HPLC techniques which depend mainly on consumption of large quantities of harmful organic solvents [17] in the mobile phase. Decreasing the usage of toxic solvents and reagents is a crucial goal in terms of environment conservation and human health [18, 19]. Assessment of the proposed method was achieved through its validation according to ICH regulations. It was successfully applied to analysis of the three drugs in pharmaceutical dosage forms and content uniformity testing. In addition, the green character of the suggested method was evaluated with the three assessment tools.
Experimental
Equipment
Merck Hitachi LaChrom HPLC system model L-7100 with a Rheodyne injection valve having a 20 µL loop.
Millipore filter membranes (SIBATA) were used for filtration of the mobile phase.
Merck solvent L-7612 degasser was used for degassing of the mobile phase.
Consort P-901 pH-meter was used for pH measurements.
Materials and reagents
All used solvents were HPLC grade.
Aspirin, 100.05 g%, Atorvastatin, 99.78 g%, and Ramipril, 100.09 g%, were provided by El Arabeya Company for pharmaceutical & Chemical Industries (ADCO), Cairo, Egypt, delta pharma, Cairo, Egypt and Sanofi-Aventis, Pakistan respectively.
Valsartan, 100.23 g%, (IS), was provided by Memphis Co. for pharm.
Riedel-de Häen (Seelze, Germany) was source of Triethylamine (TEA), Sodium dodecyl sulfate (SDS) and orthophosphoric acid 85%.
Methanol, n-propanol and acetonitrile were from Sigma-Aldrich (St. Louis, MO, USA).
Rivo® 320 mg ASP/tablet, Atorstat® 10 mg ATR/tablet and Tritace® 1.25 mg RAM/tablet tablets, were bought from the local pharmacy in Egypt. They were manufactured under license of El Arabeya Co. for pharmaceutical and chemical Industries (ADCO), Cairo, Egypt, delta pharma, Cairo, Egypt and Sanofi-Aventis, Pakistan, respectively.
Chromatographic conditions
A mobile phase consisting of 90%: 10%, v/v of aqueous solution of 0.12 M SDS: n-propanol was used. To each 100 mL of the mobile phase 0.3 mL of TEA was added, finally pH was adjusted to 2.5 using orthophosphoric acid. The stationary phase was C8 monolithic column. Detection was performed at 210 nm using flow rate of 1.5 mL/min. at room temperature. Valsartan was selected as the internal standard of choice.
Standard solutions and synthetic mixtures
Methanolic stock standard solutions containing 500 µg/mL of ASP, ATR, RAM and VAL (IS) were prepared in four flasks by dissolving 50 mg of ASP, ATR, RAM and VAL separately in 100 mL methanol. Working standard solutions were obtained by subsequent dilution of the stock solutions with the mobile phase. ASP is freshly prepared every day while other solutions remain stable for 7 days when kept in the refrigerator.
Synthetic mixture solutions were obtained by mixing certain volumes of ASP, ATR and RAM stock solutions in 100 mL volumetric flasks and diluting to the volume with mobile phase keeping the pharmaceutical ratio of 10:4:1 for ASP, ATR and RAM, respectively [6].
Calibration graphs
Different concentrations from ASP, ATR and RAM within the range of 1-200.0, 0.5–200.0 and 5-100 µg/mL, respectively were transferred into three sets of 10-mL volumetric flasks. Volume of VAL was added to obtain a final concentration of 10 µg/mL. The flasks were completed with the mobile phase to the mark and mixed well. 20 µL of each concentration was injected (three times) under the optimum chromatographic parameters. The average peak area ratios (Drug/I.S.) were plotted against the final concentrations of each drug in µg/mL to obtain corresponding calibration curves then the corresponding regression equations were obtained.
Assay of ASP, ATR and RAM in synthetic mixtures
Different aliquots of the stock solutions were moved to a set of 10 mL volumetric flasks achieving the ratio of 10:4:1 for ASP, ATR and RAM, respectively. The flasks were then completed with the mobile phase. The previous steps were repeated, and the percent recoveries were then calculated from the corresponding regression equations.
Pharmaceutical application
Analysis of the studied drugs in tablets
Ten tablets of Rivo 320 mg, Atorstat 10 mg and tritac 1.25 mg were weighed accurately and ground to fine powder individually. A weight equivalent to 12.5, 5 and 1.25 mg of Aspirin, Atorvastatin and Ramipril, respectively were dissolved in about 80 mL of methanol in a 100 mL measuring flask with 30 min. sonication. The flask was completed with the same solvent to the mark and filtered into another 100 mL volumetric flask. The final working concentrations were 125 µg/mL aspirin, 50 µg/mL of atorvastatin and 12.5 µg/mL of ramipril. Different volumes of the filtrate were analyzed with the previous mentioned procedures and the nominal contents were computed from the plotted calibration curves.
Application of the proposed method to content uniformity testing for Tritace® 1.25 mg RAM/tablet
The suggested technique was capable of testing the content uniformity of RAM in Tritace 1.25 mg RAM tablets. Content uniformity test is needed for the tablets containing ≤ 25 mg active ingredient. Owing to the small content of RAM in tablet (1.25 mg), this test can be applied for RAM uniformity testing. The test steps were followed according to the USP procedure [4] and percentage recoveries were computed by from the regression equation of RAM.
Results and discussion
The simultaneous separation of ASP, ATR and RAM ternary mixture was carried out using miceller HPLC-UV analysis. After parameters investigation, good separation was resulted as illustrated in Fig. 2. Well-defined peaks for a synthetic mixture of the three studied drugs using the optimized micellar HPLC method. The retention times for ASP, ATR, VAL and RAM were 1.5, 2.3, 3.2 and 4.3 min., respectively, so it was found that the total runtime did not exceed 5 min. The suggested technique has superiority over other published methods. Less amount of green organic modifier was needed in the proposed method, which complies with green analysis. Shorter elution time and higher sensitivity were obtained in the current study. It is also convenient for content uniformity testing due to its ability to measure the peak area with sufficient accuracy.
Fig. 2.

Typical chromatogram of 60.0 µg/mL ASP, 60 µg/mL ATR, 60 µg/mL RAM and 10.0 µg/mL VAL (I.S) using the optimum chromatographic conditions.
Optimization of the chromatographic parameters
Various experimental trials were made to obtain sharp symmetrical peaks as summarized in the following subsections:
Column choice
Three columns were tried including: Shim-pack VP-ODS column C18 (250 mm x 4.6 mm i.d., 5 μm particle size), Onyx monolithic C18 100 mm x 4.6 mm i.d., 2 μm pore size highly porous column and Onyx monolithic C8 100 mm x 4.6 mm i.d., 2 μm pore size highly porous column. The study revealed that the last column showed symmetrical sharp peaks with the highest resolution, so it was the column of choice.
Wavelength selection
Three wavelengths were tried 210,220 and 230; a wavelength of 210 nm was the most suitable one showing the highest sensitivity because the RAM was a weak UV-absorbing compound (Fig. 3).
Fig. 3.

Zero-order UV spectrophotometric spectra of 20 µg/mL ASP, ATR, RAM and ternary mixture in methanol
Mobile phase composition
The partition coefficient and logP were reported to be -1.1 and pka = 3.5 for ASP, for ATR logP = 6.36 and pka = 4.46 and for RAM logP = 3.32 and pka is 3.1, 5.6 [1]. Large difference between the hydrophobicities of the analytes made micellar mobile phase is appropriate for their separation. To improve the separation efficiency, TEA and short chain alcohol were added [20]. By changing the organic modifier type and concentration, SDS concentration, and pH we reached the optimum conditions as summarized in Table 1.
Table 1.
Effect of experimental parameters on the number of theoretical plates
| Parameter | Number of theoretical plates (N) | Resolution, RS | Selectivity, α | |||||
|---|---|---|---|---|---|---|---|---|
| ASP | ATR | RAM | ASP/ATR | ATR/RAM | ASP/ATR | ATR/RAM | ||
| % Concentration n- propanol (v/v) | ||||||||
| 6 | 770 | 1271 | 746 | 2.9 | 4.2 | 3.5 | 2.78 | |
| 8 | 510 | 931 | 1005 | 2.7 | 4.7 | 3.7 | 2.9 | |
| 10 | 504 | 971 | 1317 | 2.7 | 4.6 | 3.1 | 2.7 | |
| 12 | 465 | 892 | 1336 | 1.9 | 4.42 | 3.6 | 3.4 | |
| 14 | 423 | 743 | 1402 | 3.5 | 3.6 | 16 | 2.7 | |
| 15 | 567 | 1112 | 363 | 5.1 | 2.7 | 18 | 2.2 | |
| Concentration of SDS, M | ||||||||
| 0.05 | 467 | 1045 | 395 | 4.6 | 8 | 3 | 3.2 | |
| 0.08 | 970 | 1766 | 1172 | 4.36 | 7.2 | 3.4 | 3.2 | |
| 0.1 | 659 | 1229 | 1491 | 3 | 5 | 3.3 | 3.3 | |
| 0.12 | 515 | 869 | 1485 | 2 | 4.44 | 4 | 3.5 | |
| 0.15 | 480 | 859 | 1158 | 1.7 | 3.6 | 3.75 | 3.3 | |
| 0.18 | 414 | 658 | 1143 | 1.5 | 2.2 | 6 | 3.5 | |
| pH of the medium | ||||||||
| 2.5 | 515 | 971 | 1485 | 3.2 | 5 | 3 | 2.66 | |
| 3 | 651 | 606 | 1173 | 2.2 | 5.87 | 3.75 | 4.1 | |
| 4 | 308 | 1207 | 207 | 0.32 | 6.35 | 1.17 | 26.76 | |
| 5 | Overlapped | 343 | ||||||
| 6 | Overlapped peaks | |||||||
| Flow rate, mL/min. | ||||||||
| 0.8 | 417 | 1094 | forked | 2.6 | 3.36 | 4.42 | 2.64 | |
| 1 | 469 | 987 | 1380 | 2.38 | 4.4 | 4.16 | 3.12 | |
| 1.2 | 458 | 974 | 1376 | 2.8 | 4.16 | 4.24 | 3.76 | |
| 1.5 | 515 | 871 | 1485 | 2.54 | 4.2 | 4 | 3.37 | |
| 2 | 473 | 964 | 1210 | 2.26 | 4.34 | 5.5 | 3.36 | |
Type of organic modifier
The effect of different organic modifiers, including methanol, n-butanol and n-propanol, was studied in order to achieve the best separation of the studied drugs. N-propanol gave well-resolved sharp peaks within a reasonable short analysis time (less than 4.5 min.) so it was the organic modifier of choice.
Organic modifier concentration
Eluents containing 6 to 15% v/v of n-propanol were tried. The optimum chromatographic performance was attained using 10% v/v of n-propanol as it yields the maximum NTP with good Rs. Lower concentrations result in broadening of RAM peak, so decreasing the sensitivity. While higher concentrations lead to overlapping of ASP peak with solvent one. Regarding resolution and number of theoretical plates of the drugs, the optimum n-propanol concentration was selected to be 10% v/v.
SDS concentration
Different concentrations of SDS were tried using mobile phases containing SDS in the range 0.05 to 0.18 M, while keeping the concentration of n-propanol and pH constant. The retention times of RAM increase and sensitivity decrease significantly upon decreasing the concentration of SDS. This indicated that the best chromatographic performance was using 0.12 M SDS regarding resolution and number of theoretical plates of the studied drugs. Concentrations less than 0.08 M SDS result in significant increasing in the retention time and decreasing in sensitivity of RAM, while concentrations higher than 0.12 M SDS result in decreasing the resolution of the peaks.
pH
The effect of varying mobile phase pH on the chromatographic performance of the developed method was studied using different mobile phases with pH ranging from of 2 to 6 (using H3PO4 or TEA), while keeping the concentration of n-propanol and SDS constant. It was found that the sensitivity and the resolution of three drugs decrease upon decreasing the pH below 2.5. At pH above 3, significant decrease in the sensitivity and increase in retention time of both ATR and RAM. pH 2.5 was found to be the most appropriate pH.
Flow rate
The flow rate was changed from 0.8 to 2 mL/min. and a flow rate 1.5 ml/min. was found to be optimum for good separation in a reasonable time.
The nature of internal standard
Both the internal standard and drug are analyzed simultaneously to compensate for any possible variation during the entire process of sample preparation and analysis. Different internal standards including valsartan, clopidogrel, rivaroxaban and amlodipine were investigated. Valsartan (VAL) was the internal standard of choice showing the highest resolution from the peaks of the three drugs.
The final optimized conditions were as follow: an aqueous solution containing 0.12 M SDS, 10% (v/v) n-propanol and 0.3% (v/v) TEA, adjusted to pH 2.5 using orthophosphoric acid. The mobile phase was pumped at a flow rate of 1.5 mL/min. and UV detector was set at 210 nm.
Method validation
Validation of the proposed method was accomplished as per ICH recommendations [21] as follows:
Linearity, range, limit of quantitation (LOQ) and limit of detection (LOD)
Linearity was studied for ASP, ATR and RAM by plotting relationship between peak area ratio [drug/I.S.] and each drug concentrations (C) in µg/mL. Table 2 presents the equations of the regression lines, linearity ranges and the analytical data of the calibration curves. Statistical analysis [22] of the data are abridged in Table 2.
Table 2.
Analytical performance data for the determination of the ASP, ATR and RAM by the proposed Micellar HPLC method
| Parameter | ASP | ATR | RAM |
|---|---|---|---|
| Range (µg/mL) | 0.1–200 | 0.5–200 | 5.0-100 |
| Intercept (a) | 0.0077 | 0.0557 | -0.1199 |
| Slope (b) | 0.0337 | 0.0498 | 0.0734 |
| Correlation coefficient (r) | 0.9998 | 0.9999 | 0.9999 |
| S.D. of residuals (Sy/x) | 0.0027 | 0.00335 | 0.00148 |
| S.D. of intercept (Sa) | 0.00005 | 0.00005 | 0.00007 |
| S.D. of slope (Sb) | 0.00001 | 0.00002 | 0.00002 |
| Regression equation |
*P = 0.0337 C + 0.0077 |
*P = 0.0498 C + 0.0557 |
*P = 0.0734 C − 0.1199 |
| Percentage relative standard deviation, % RSD | 1.315 | 1.027 | 1.078 |
| Percentage relative error, % Error | 0.412 | 0.309 | 0.407 |
| Limit of detection, LOD (µg/mL) | 0.19 | 0.13 | 0.30 |
| Limit of quantitation, LOQ (µg/mL) | 0.63 | 0.44 | 0.99 |
*P is the peak area ratio, C is the concentration of the drug in µg/mL and r is the correlation coefficient
The limit of quantitation (LOQ) and the limit of detection (LOD) were calculated practically according to ICH recommendations [21]. Limit of detection (LOD) and limit of quantification (LOQ) results are given in Table 2.
Accuracy
The accuracy of the proposed method was evaluated through comparing the results of assay of the proposed method with those obtained using the comparison method [9]. The comparison method was a RP-HPLC using C-18 column, a mobile phase composed of methanol and acetate buffer in the ratio (70:30 v/v) at pH to 3.1.
When comparing the proposed approach with a comparison method, student’s t-test and variance ratio F-test [22] revealed no-significant difference as shown in Table 3.
Table 3.
Assay results for determination of ASP, ATR and RAM drugs in pure forms by the proposed micellar HPLC method
| Compound | Proposed method | Comparison method [9] | |
|---|---|---|---|
| Amount taken, (µg/mL) | % Found | % Found | |
| ASP | 1 | 100.20 | 99.37 |
| 40 | 100.59 | 100.83 | |
| 100 | 99.94 | 99.69 | |
| 150 | 101.91 | ||
| 200 | 98.85 | ||
| Mean ± SD | 100.31 ± 1.11 | 99.69 ± 0.77 | |
| t-test | 0.45 (2.45) | ||
| F- test | 2.09 (19.25) | ||
| ATR | 0.5 | 99.00 | 99.05 |
| 40 | 100.63 | 101.34 | |
| 100 | 99.74 | 99.49 | |
| 150 | 101.05 | ||
| 200 | 99.55 | ||
| Mean ± SD | 99.99 ± 0.83 | 99.96 ± 1.22 | |
| t-test | 0.05 (2.45) | ||
| F-test | 2.13 (6.95) | ||
| RAM | 5 | 100.86 | 101.03 |
| 20 | 99.53 | 98.73 | |
| 60 | 99.43 | 100.46 | |
| 80 | 98.94 | ||
| 100 | 100.72 | ||
| Mean ± SD | 99.91 ± 0.85 | 100.07 ± 1.21 | |
| t-test | 0.25 (2.45) | ||
| F-test | 2.00 (6.95) | ||
Precision
Repeatability and intermediate precision of the proposed method were tested. The repeatability was examined by testing three different concentration of each drug three replicates in the same day and in three consecutive days for intermediate precision. The studied concentrations were: 80.0,100.0,120.0 µg/mL for ASP & ATR and 40.0,050.0,and 60.0 µg/mL for RAM. The results indicate acceptable repeatability and intermediate precision of the proposed method as shown in Table 4.
Table 4.
Precision data for the determination of ASP, ATR and RAM by the proposed Micellar HPLC method
| Am. taken, µg/mL | ASP concentration | ATR concentration | RAM concentration | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 80.0 | 100.0 | 120.0 | 80.0 | 100.0 | 120.0 | 40.0 | 50.0 | 60.0 | ||
| Intraday | % Found | 99.14 | 99.26 | 99.45 | 100.46 | 100.45 | 99.85 | 100.46 | 99.75 | 98.85 |
| 99.62 | 98.83 | 99.12 | 100.73 | 99.75 | 100.37 | 101.32 | 99.93 | 98.77 | ||
| 99.35 | 98.46 | 98.20 | 101.52 | 99.24 | 100.48 | 101.22 | 99.57 | 99.18 | ||
| Mean | 99.37 | 98.85 | 98.92 | 100.90 | 99.81 | 100.23 | 101 | 99.75 | 98.93 | |
| ±SD | 0.24 | 0.40 | 0.65 | 0.55 | 0.61 | 0.34 | 0.47 | 0.18 | 0.22 | |
| %RSD | 0.24 | 0.40 | 0.66 | 0.55 | 0.61 | 0.34 | 0.47 | 0.18 | 0.22 | |
| %Error | 0.14 | 0.23 | 0.38 | 0.32 | 0.35 | 0.19 | 0.27 | 0.10 | 0.13 | |
| Interday | % Found | 100.13 | 99.36 | 99.82 | 99.66 | 100.78 | 100.46 | 99.68 | 100.58 | 99.87 |
| 99.78 | 99.03 | 98.96 | 100.04 | 101.26 | 99.82 | 101.24 | 101.17 | 101.00 | ||
| 99.94 | 99.58 | 99.91 | 100.11 | 101.04 | 101.03 | 100.52 | 100.34 | 100.03 | ||
| Mean | 99.95 | 99.32 | 99.56 | 99.94 | 101.03 | 100.44 | 100.48 | 100.70 | 100.30 | |
| ±SD | 0.18 | 0.28 | 0.52 | 0.24 | 0.24 | 0.61 | 0.78 | 0.43 | 0.61 | |
| %RSD | 0.18 | 0.28 | 0.53 | 0.24 | 0.24 | 0.60 | 0.78 | 0.42 | 0.61 | |
| %Error | 0.10 | 0.16 | 0.30 | 0.14 | 0.14 | 0.35 | 0.45 | 0.25 | 0.35 | |
Robustness
The robustness was evaluated, and some parameters showed an acceptable range for variation including n-propanol concentration (10.0 ± 0.5% v/v), SDS concentration (0.12 M ± 0.005) and pH of the mobile phase (2.5 ± 0.2). As shown in Table 5, these slight changes did not affect the peak area ratios or the retention times of the three analytes.
Table 5.
Robustness of the proposed method using ASP (100.0 µ/mL), ATR (100.0) and RAM (50.0 µ/mL)
| Parameter | % Found, % | ||
|---|---|---|---|
| ASP | ATR | RAM | |
| pH | |||
| 2.3 | 98.40 | 101.14 | 99.96 |
| 2.5 | 98.22 | 100.11 | 99.64 |
| 2.7 | 99.12 | 99.15 | 100.14 |
| Mean | 98.58 | 100.13 | 99.91 |
| ± S.D. | 0.48 | 0.99 | 0.25 |
| % RSD | 0.48 | 0.99 | 0.25 |
| % Error | 0.28 | 0.57 | 0.15 |
| Concentration of n-Propanol (%v/v) | |||
| 9.5 | 99.48 | 100.23 | 101.76 |
| 10 | 99.45 | 99.67 | 100.44 |
| 10.5 | 100.34 | 100.78 | 101.06 |
| Mean | 99.76 | 100.23 | 101.09 |
| ± S.D. | 0.51 | 0.56 | 0.66 |
| % RSD | 0.51 | 0.55 | 0.65 |
| % Error | 0.29 | 0.32 | 0.38 |
| SDS concentration, M | |||
| 0.115 | 99.75 | 100.45 | 99.47 |
| 0.12 | 98.65 | 99.67 | 100.34 |
| 0.125 | 99.88 | 99.96 | 100.12 |
| Mean | 99.43 | 100.03 | 99.98 |
| ± S.D. | 0.68 | 0.39 | 0.45 |
| % RSD | 0.68 | 0.39 | 0.45 |
| % Error | 0.39 | 0.23 | 0.26 |
Selectivity and specificity
The suggested technique was able to determine ASP, ATR or RAM in presence of each other with high accuracy without any interference.
The proposed method specificity was tested by observing any interference resulted from the used dosage forms excipients such as: microcrystalline cellulose, magnesium stearate, citric acid anhydrous, colloidal silicon dioxide, butylated hydroxyanisol, talc, croscarmellose sodium, lactose monohydrate, maize starch, hypromellose, pregelatinised, sodium stearyl fumarate. All these compounds did not show any interference with the results of the suggested technique as shown in Table 6.
Table 6.
Assay results for the determination of the ASP, ATR and RAM in prepared tablet
| Compound | Proposed method | Comparison method [9] | |
|---|---|---|---|
| Amount taken, (µg/mL) | % Found, % | % Found, % | |
| ASP | 62.5 | 98.95 | 100.52 |
| 125 | 101.02 | 99.45 | |
| 187.5 | 99.77 | 100.18 | |
| Mean ± SD | 99.91 ± 1.31 | 100.05 ± 0.547 | |
| t-test | 0.20 (2.78) | ||
| F- test | 3.64 (19.00) | ||
| ATR | 25 | 99.16 | 98.54 |
| 50 | 99.91 | 101.18 | |
| 75 | 99.67 | 99.63 | |
| Mean ± SD | 99.58 ± 0.383 | 99.78 ± 1.33 | |
| t-test | 0.26 (2.68) | ||
| F-test | 11.99 (19.00) | ||
| RAM | 6.25 | 99.44 | 100.67 |
| 12.5 | 101.22 | 99.42 | |
| 18.75 | 100.11 | 100.18 | |
| Mean ± SD | 100.26 ± 0.90 | 100.09 ± 0.63 | |
| t-test | 0.26 (2.78) | ||
| F-test | 2.04 (19.00) | ||
System suitability
The resolution and reproducibility of the produced peaks was checked to make sure if the selected chromatographic conditions are appropriate for the analysis or not. By comparing to the reference official values, acceptable parameters were resulted as shown in Table 7.
Table 7.
Parameters of system suitability for the proposed HPLC method for the determination of VAL and AML
| Parameter | ASP/ATR | ATR/IS | IS/RAM | Reference value |
|---|---|---|---|---|
| Retention time, min | 1.5 | 2.3 | 4.3 | |
| Capacity factor, K’ | 0.36 | 1.09 | 2.9 | 0–10 |
| Symmetry factor | 0.718 | 0.829 | 1.05 | ~ 1 |
| Resolution, RS | 2.67 | 2.4 | 2. 2 | RS > 2 |
| Selectivity, α | 3.0 | 1.75 | 1.52 | α > 1 |
| Theoretical plates, N | 400 | 690.93 | 603.75 |
Greenness assessment
Green analytical chemistry plays a major role in preserving and protecting the environment. The analytical Eco-Scale [23] is a green metric that is used to assess the greenness of an analytical method. It has unique advantages compared to other metrics. It is easy to calculate its score that makes comparison of analytical procedures very easy [24]. It includes varied aspects of environmental impact in its assessment procedure. It is calculated by allotting penalty points to any factor in the analytical procedure. The analytical Eco-Scale score is calculated by subtracting penalty points from the basis of 100 points. A score of 100 represents an ideal green analysis. The higher the value is, the greener the method will be [25–27].
The comparison between the analytical Eco-Scale values of the proposed approach and reported methods [7–9] is carried out and results are shown in Table 8. It was found that the proposed method is greener than the reported methods giving a score of 72.
Table 8.
The penalty points of the proposed methods according to the analytical Eco-Scale
| Reagents/ Instruments | Penalty points | ||||||
|---|---|---|---|---|---|---|---|
| Reagent | Number of pictograms | Word sign | Reagent, penalty points | Proposed method | Reported method [7] | Reported method [8] | Reported method [9] |
| Methanol | 3 | Danger | 6 | 6 × 1 = 6 | 6 × 2 = 12 | 6 × 2 = 12 | |
| Acetonitrile | 2 | Danger | 4 | 4 × 2 = 8 | 4 × 2 = 8 | ||
| n- propanol | 2 | Danger | 4 | 2 × 4 = 8 | |||
| Glacial acetic acid | 2 | Danger | 4 | 2 × 4 = 8 | |||
| TEA | 3 | 3 | 3 × 1 = 3 | ||||
| o-phosphoric acid | 2 | 2 | 2 × 1 = 2 | 2 × 2 = 4 | 2 × 2 = 4 | 2 × 1 = 2 | |
| Sodium acetate | 1 | Irritant | 2 | 2 × 2 = 4 | |||
| HPLC | |||||||
| Occupational hazard, Analytical process hermitization | 0 | 0 | 0 | 0 | |||
|
Waste Flow rate x run time |
1–10 = 3 > 10 = 5 |
3 | 5 | 5 | 5 | ||
| Total penalty points | 16 | 23 | 29 | 31 | |||
| Analytical Eco- Scale total score | 84 | 77 | 71 | 69 | |||
The method greenness was further assessed using complex GAPI [28] and AGREE [29] tools. Figure 4 showed GAPI pentagram and AGREE result. The AGREE score was found to be 0.76 indicating the greenness of the proposed method.
Fig. 4.

Complex GAPI pentagram and AGREE result of the proposed method.
Applications
Analysis of studied drugs in synthetic mixtures
The developed technique was successfully used to the concurrent determination of ASP, ATR and RAM in laboratory-prepared mixtures regarding their recommended pharmaceutical ratio, 10:4:1 respectively (Fig. 2). The statistical assessment of the results confirms no significant difference with those of the comparison method [9]. Good recoveries were obtained and abridged in Table 9.
Table 9.
Assay results for the determination of the ASP, ATR and RAM drugs in synthetic mixtures in ratios of 10:4:1 (w/w) by the proposed HPLC method
| Compound | Proposed method | Comparison method [9] | |
|---|---|---|---|
| Amount taken, (µg/mL) | % Found, % | % Found, % | |
| ASP | 100 | 99.14 | 101.02 |
| 140 | 101.10 | 100.45 | |
| 200 | 99.71 | 100.58 | |
| Mean ± SD | 99.98 ± 1.01 | 100.35 ± 0.73 | |
| t-test | 1.15 (2.78) | ||
| F- test | 11.39 (19.00) | ||
| ATR | 40 | 99.33 | 99.67 |
| 56 | 100.84 | 100.28 | |
| 80 | 99.97 | 99.78 | |
| Mean ± SD | 100.05 ± 0.76 | 99.91 ± 0.33 | |
| t-test | 0.29 (2.78) | ||
| F-test | 5.43 (19.00) | ||
| RAM | 10 | 101.57 | 100.03 |
| 14 | 98.59 | 98.56 | |
| 20 | 100.31 | 101.46 | |
| Mean ± SD | 100.16 ± 1.50 | 100.017 ± 1.45 | |
| t-test | 0.12 (2.78) | ||
| F-test | 1.06 (19.00) | ||
Dosage forms
The three drugs were investigated in their laboratory prepared tablets using the proposed method as shown in Fig. 5. The results of the assay agreed with those obtained with the comparison method [9] as shown in Table 6.
Fig. 5.
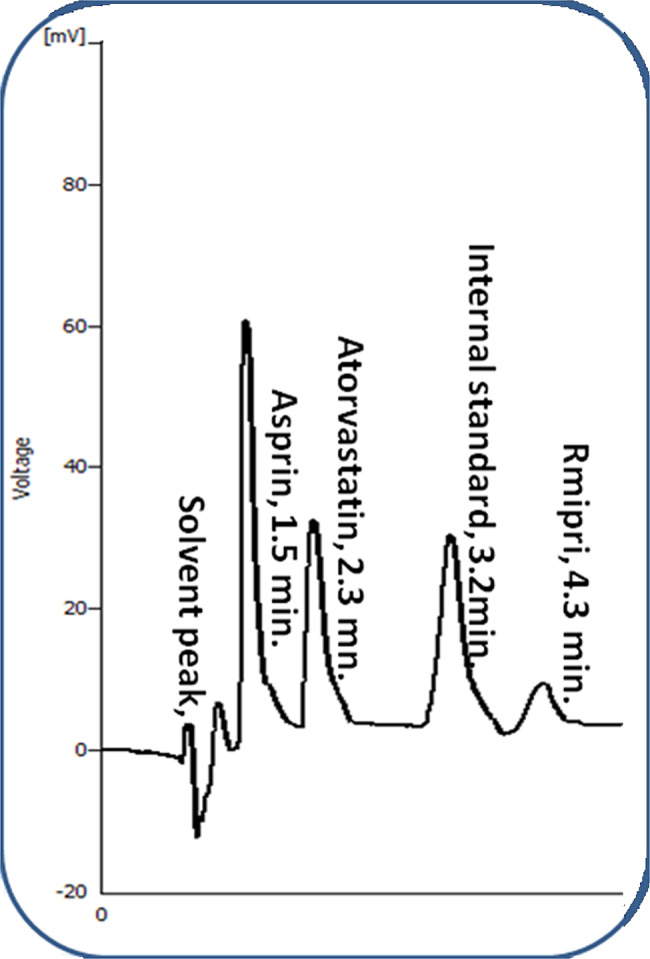
Typical chromatogram of 187.5 µg/mL ASP, 75 µg/mL ATR And 18.75 µg/mL RAM in the prepared tablet using 10.0 µg/mL VAL (I.S) using the optimum chromatographic conditions
Content uniformity
The suggested technique was ideally suitable for content uniformity testing of RAM. Table 10 showed that acceptance value (AV) was smaller than the maximum allowed acceptance value (L1). The results indicated excellent drug uniformity.
Table 10.
Results of content uniformity testing of RAM in Ttritace® tablets using the applied method
| Parameter | Tablet no. | Percentage of the label claim |
|---|---|---|
| Data | 1 | 102.49 g% |
| 2 | 101.21 g% | |
| 3 | 102.11 g% | |
| 4 | 98.52 g% | |
| 5 | 100.44 g% | |
| 6 | 98.59 g% | |
| 7 | 102.54 g% | |
| 8 | 98.18 g% | |
| 9 | 102.80 g% | |
| 10 | 99.85 g% | |
| Mean | 100.67 g% | |
| S.D. | 1.81 | |
| % RSD | 1.80 | |
| % Error | 0.569 | |
| Acceptance value (AV)[4] | 4.35 | |
| Max. allowed AV (L1)[4] | 15.0 | |
Tritace® tablets: Labeled to contain 1.25 mg ramipril per tablet; manufactured by Sanofi Aventis.
Conclusion
New, green, accurate and rapid micellar HPLC method was designed for the concurrent determination of ASP, ATR and RAM in their ternary mixtures. The correlation coefficients are 0.9998, 0.9999 and 0.9999 for ASP, ATR and RAM respectively. The recommended method had practical limits of detection of 0.19, 0.13 and 0.30 µg/mL and limits of quantitation of 0.63, 0.44 and 0.99 µg/mL, respectively. It could be applied to the assay of the three drugs in their combined tablets. It was also very suitable to be used in content uniformity testing for RAM. The suggested approach could be considered a suitable alternative for the previous harmful methods.
Abbreviations
- ASP
Aspirin
- ATR
Atorvastatin
- RAM
Ramipril
- TEA
Triethylamine
- SDS
Sodium dodecyl sulfate
- VAL
Valsartan
- IS
Internal standard
- LOD
Limits of detection
- LOQ
Limits of quantitation
- ICH
International council for harmonisation
- ACE
Angiotensin-converting enzyme
- WHO
World health organization
- EML
Essential medicines list
- FDC
Fixed-dose combination.
Author contributions
N.A carried out the practical work. N.A. and M.E wrote the main manuscript. A.E and F.I. revised the manuscript and supervised the whole work. All authors reviewed the manuscript.
Funding
Not applicable.
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Data availability
Most of the data of this study are included within the article. Other supplementary data are available from the corresponding author upon request.
Competing interests
No confict of interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not Applicable.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Moffat AC, Osselton MD, Widdop B. Clarke’s analysis of drugs and poisons in Pharmaceuticals; body fluids and Postmortem Material. 4. London: Pharmaceutical press; 2011. [Google Scholar]
- 2.Brayfield AE, Martindale . The Complete Drug Reference, Online version. London: ed, Pharmaceutical Press; 2017. p. 39. [Google Scholar]
- 3.The British Pharmacopoeia . Electronic version [CD-ROM] London: The Stationery Office; 2012. [Google Scholar]
- 4.The United States Pharmacopeia (USP 40)., The National Formulary (NF 35), Online version. 2017.
- 5.World Health Organization (2007). Prevention of cardiovascular disease: Guidelines for assessment and management of total cardiovascular risk. (Last accessed 08.03.2023). https://apps.who.int/iris/handle/10665/43685
- 6.The Selection and Use of Essential Medicines (2017). TRS 1006, WHO Technical Report Series, No. 1006. (Last accessed 08.03.2023). https://www.who.int/publications/i/item/9789241210157
- 7.Panchal HJ, Suhagia BN, Patel NJ, Rathod IS, Patel BH. Simultaneous Estimation of Atorvastatin Calcium, Ramipril and Aspirin in Capsule Dosage Form by RP-LC. Chromatographia. 2009, 69, 91–95. (10.1365/s10337-008-0831-z)
- 8.Sharma R, Khanna S, Mishra GP. Development and Validation of RP-HPLC Method for Simultaneous Estimation of Ramipril, Aspirin and Atorvastatin in Pharmaceutical Preparations. E-Journal of Chemistry. 2012, 9. (10.1155/2012/891695)
- 9.Damle MS, Patole SM, Potale LV, Khodke AS. A validated HPLC method for analysis of Atorvastatin Calcium, Ramipril and Aspirin as the bulk drug and in combined capsule dosage forms. Int J Pharm Sci Rev Res. 2010;4:40–5. [Google Scholar]
- 10.Kasawar GB, Farooqui M. Development and validation of a stability indicating RP-HPLC method for the simultaneous determination of related substances of albuterol sulfate and ipratropium bromide in nasal solution. Journal of pharmaceutical and biomedical analysis. 2010, 1;52(1):19–29. [DOI] [PubMed]
- 11.Panchal H, Suhagia B, Patel N. Simultaneous HPTLC analysis of atorvastatin calcium, ramipril, and aspirin in a capsule dosage form. JPC-Journal of Planar Chromatography-Modern TLC. 2009, 1;22(4):265 – 71.
- 12.Sankar AK, Vetrichelvan T, Venkappaya D. Simultaneous estimation of ramipril, acetylsalicylic acid and atorvastatin calcium by chemometrics assisted UV-spectrophotometric determinations in capsules. Acta Pharmaceutica. 2011, 1;61(3):283 – 96. [DOI] [PubMed]
- 13.Keith LH, Gron LU, Young JL. Green analytical methodologies. Chem Rev. 2007;13(6):2695–708. doi: 10.1021/cr068359e. [DOI] [PubMed] [Google Scholar]
- 14.Welch CJ, Wu N, Biba M, Hartman R, Brkovic T, Gong X, Helmy R, Schafer W, Cuff J, Pirzada Z, Zhou L. Greening analytical chromatography. TrAC Trends in Analytical Chemistry. 2010, 1;29(7):667 – 80.
- 15.Smith RM. Superheated water chromatography–A green technology for the future. J Chromatogr A. 2008;141184(1–2):441–55. doi: 10.1016/j.chroma.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 16.Rainville PD, Simeone JL, McCarthy SM, Smith NW, Cowan D, Plumb RS. Investigation of microbore UPLC and nontraditional mobile phase compositions for bioanalytical LC–MS/MS. Bioanalysis. 2012;4(11):1287–97. doi: 10.4155/bio.12.78. [DOI] [PubMed] [Google Scholar]
- 17.(Last accessed 08.03.2023). https://osha.europa.eu/en/legislation/directives/exposure-to-chemical-agents-and-chemical-safety/osh-related-aspects/58
- 18.Anastas PT, Warner JC. Principles of green chemistry.Green chemistry: Theory and practice. 1998;29.
- 19.Yoshimura T, Nishinomiya T, Honma Y, Murabayashi M. Green Chemistry—Aim for the zero emission-chemicals. Tokyo: Sankyo; 2001. [Google Scholar]
- 20.Thomas DP, Foley JP. Improved efficiency in micellar liquid chromatography using triethylamine and 1-butanol as mobile phase additives to reduce surfactant adsorption. J Chromatogr A. 2008;1205:36–45. doi: 10.1016/j.chroma.2008.07.082). [DOI] [PubMed] [Google Scholar]
- 21.ICH Harmonized Tripartite Guideline, Validation of Analytical Procedures: Text and Methodology, Q2(R1). (Last accessed 08.03.2023). https://www.ich.org/page/quality-guidelines
- 22.Miller JC, Miller JN. Statistics and Chemometrics for Analytical Chemistry. 5. Harlow: Pearson Education Limited; 2005. [Google Scholar]
- 23.Tobiszewski M, Marć M, Gałuszka A, Namieśnik J. Green chemistry metrics with special reference to green analytical chemistry. Molecules. 2015, 12;20(6):10928-46. [DOI] [PMC free article] [PubMed]
- 24.Gałuszka A, Migaszewski ZM, Konieczka P, Namieśnik J. Analytical Eco-Scale for assessing the greenness of analytical procedures. TRAC Trends Anal Chem. 2012;1:61–72. doi: 10.1016/j.trac.2012.03.013. [DOI] [Google Scholar]
- 25.Ibrahim FA, El-Brashy AM, El-Awady MI, Abdallah NA. Assessment of the greenness of spectrophotometric and micellar liquid chromatographic methods using two approaches: application to pharmaceutical analysis of hydrochlorothiazide and telmisartan. Microchem J. 2019 Jul;148(1):197–205.
- 26.Ibrahim FA, El-Brashy AM, El-Awady MI, Abdallah NA. Fast simultaneous quantitation of valsartan and amlodipine besylate using an eco-friendly micellar HPLC-UV method: application to spiked human plasma and content uniformity testing for amlodipine. Anal Methods. 2018;10(43):5227–35. doi: 10.1039/C8AY01263F. [DOI] [Google Scholar]
- 27.Abdalah NA, Fathy ME, Tolba MM, El-Brashy AM, Ibrahim FA. Green spectrofluorimetric assay of dantrolene sodium via reduction method: application to content uniformity testing. Royal Soc Open Sci. 2021 Jul;14(7):210562. [DOI] [PMC free article] [PubMed]
- 28.Płotka-Wasylka J, Wojnowski W. Complementary green analytical procedure index (ComplexGAPI) and software. Green Chem. 2021;23(21):8657–65. doi: 10.1039/D1GC02318G. [DOI] [Google Scholar]
- 29.Pena-Pereira F, Wojnowski W, Tobiszewski M. AGREE—Analytical GREEnness metric approach and software. Anal Chem. 2020;15(14):10076–82. doi: 10.1021/acs.analchem.0c01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Most of the data of this study are included within the article. Other supplementary data are available from the corresponding author upon request.