

SUMMARY
Advanced solid cancers are complex assemblies of tumor, immune, and stromal cells characterized by high intratumoral variation. We use highly multiplexed tissue imaging, 3D reconstruction, spatial statistics, and machine learning to identify cell types and states underlying morphological features of known diagnostic and prognostic significance in colorectal cancer. Quantitation of these features in high-plex marker space reveals recurrent transitions from one tumor morphology to the next, some of which are coincident with long-range gradients in the expression of oncogenes and epigenetic regulators. At the tumor invasive margin, where tumor, normal, and immune cells compete, T-cell suppression involves multiple cell types and 3D imaging shows that seemingly localized 2D features such as tertiary lymphoid structures are commonly interconnected and have graded molecular properties. Thus, while cancer genetics emphasizes the importance of discrete changes in tumor state, whole-specimen imaging reveals large-scale morphological and molecular gradients analogous to those in developing tissues.
Keywords: large-scale, intermixed molecular, cellular and morphological features
In-brief
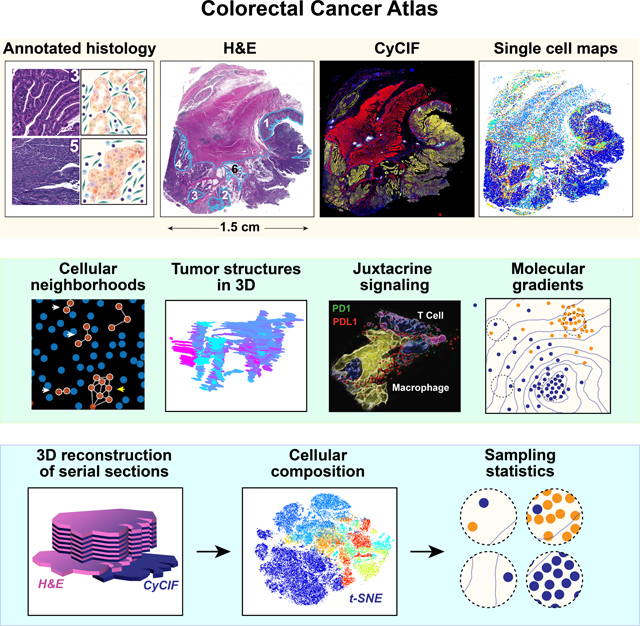
Multiplexed whole-slide imaging analysis characterizes intermixed and graded morphological and molecular features in human colorectal cancer samples, highlighting large-scale cancer characteristic structural features and variations in intra-tertiary lymphoid cellular compositions and structural patterning.
Graphical Abstract
INTRODUCTION
One hundred and fifty years of inspection of hematoxylin and eosin (H&E)-stained tissue sections by histopathologists, complemented for over eighty years by immunohistochemistry,1 has identified numerous recurrent tumor features with diagnostic or prognostic significance.2 However, these classical methods provide insufficient information for mechanistic studies and precision medicine. Spatial tumor atlases3 aim to build on this foundation and contemporary tumor genetics by collecting detailed molecular and morphological information on cells in a preserved 3D environment. Atlas construction is made possible by new highly-multiplexed tissue imaging methods4–11 that yield subcellular resolution images of 10–80 antigens. When segmented and quantified, these images generate single-cell data on cell types, states, and interactions that complement scRNA-seq.12–14 However, despite deep knowledge about the genomic drivers of cancer – from oncogenic mutations to chromosomal rearrangements – we do not yet know how the spatial arrangement of the tumor microenvironment (TME) impacts pathogenesis; for instance, which feature types and spatial scales are relevant, how disease-associated histological features relate to molecular states, and whether morphological differences are discrete (like mutations) or continuous (like morphogen gradients).
‘Bottom-up’ approaches to tissue analysis involve enumerating cell types, identifying cell-cell interactions, and generating local neighborhoods using spatial statistics. Such approaches leverage tools developed for dissociated single cell data (e.g., mass cytometry15 and scRNA-seq16). In contrast, “top-down” approaches involve annotating histopathologic features (histotypes) that are associated with a disease state or outcome2 followed by computation on the multiplexed data to identify underlying molecular patterns. Histopathology has long been challenged by striking spatial features that do not have prognostic or diagnostic value on follow-up, introducing a note of caution into ‘bottom-up’ analysis.17,18 At the same time, discoveries arising from ‘top-down’ analysis are strongly influenced by prior expectations. In this paper, we analyze colorectal cancer (CRC) using both approaches and compare the resulting insights.
Histological features of established significance in CRC include: (i) the degree of differentiation relative to normal epithelial and tumor cell morphology (e.g., cell shape, nuclear size, etc.) and the organization of cellular neighborhoods (e.g., glandular organization, hypercellularity, etc.)19; (ii) the position and morphology of the invasive margin10,20 including the presence of “tumor buds,” small clusters of tumor cells surrounded by stroma21 that are correlated with poor outcomes (i.e., increased risk of local recurrence, metastasis, and cancer-related death)22; (iii) the extent of T-cell infiltration23 and the presence of peritumoral tertiary lymphoid structures (TLS) (organized aggregates of B, T and other immune cell types24). In many cases, the origins and molecular basis of these histological features are not fully understood, although de-differentiation, “stemness”,25 epithelial-mesenchymal transition (EMT),26 changes in nuclear mechanics,27 and similar processes are involved.28
In this paper, we combined high-plex cyclic immunofluorescence (CyCIF)8 and H&E images of CRC with single-cell sequencing and micro-region transcriptomics. We show that accurate assessment of disease-relevant tumor structures requires the statistical power of whole-slide imaging, not the small specimens found in tissue microarrays (TMAs). Using 3D reconstruction of serial sections and supervised machine learning, we show that archetypical CRC histologic features are often graded and substantially larger than they appear in 2D. Thus, the TME is organized on spatial scales spanning 3–4 orders of magnitude, from subcellular organelles to cellular assemblies of hundreds of microns or more.
RESULTS
Overview of the specimens and data.
Multiplexed CyCIF and H&E imaging were performed on 93 FFPE CRC human specimens spanning histologic and molecular subtypes (Table S1) in three different formats (Figure 1A). CRC1 (Figures 1B–1E) was subjected to 3D analysis by imaging serial sections (see Methods), combined with scRNA-seq, and GeoMx transcriptomics29 (Figures 1A, S1A; Table S2). CRC1 is a poorly differentiated stage IIIB BRAFV600E adenocarcinoma (pT3N1bM0)30 with microsatellite instability (MSI-H) and a complex histomorphology. It has an extended front invading into underlying smooth muscle (muscularis propria) and connective tissue that includes a ‘budding invasive margin’ in submucosa adjacent to normal colonic mucosa (IM-A), a ‘mucinous invasive margin’ (IM-B), and a deep ‘pushing invasive margin’ (IM-C); the latter two regions invade the submucosa and muscularis (Figure 1B). 16 additional samples (CRC2–17) were acquired using 2D whole slide imaging (WSI). Finally, CRC2–17 plus 77 additional tumors (CRC18–93) were imaged as part of a TMA (Figure 1A). In each case, CyCIF was performed using various combinations of 102 lineage-specific antibodies against epithelial, immune, and stromal cell populations and markers of cell cycle state, signaling pathway activity, and immune checkpoint expression (antibodies for each panel in Table S3). MCMICRO software31 was used to segment images, quantify fluorescence intensities on a per-cell basis, and assign cell types based on lineage-specific marker expression (Figures 1C, S1B–S1C; Table S4). Overall, ~2 × 108 segmented cells were identified in 75 whole-slide images using different combinations of antibodies (~6TB of data).32 All data are available for download via the HTAN Portal and images of CRC1–17 are available for interactive online viewing through MINERVA.33,34
Figure 1. Data overview.
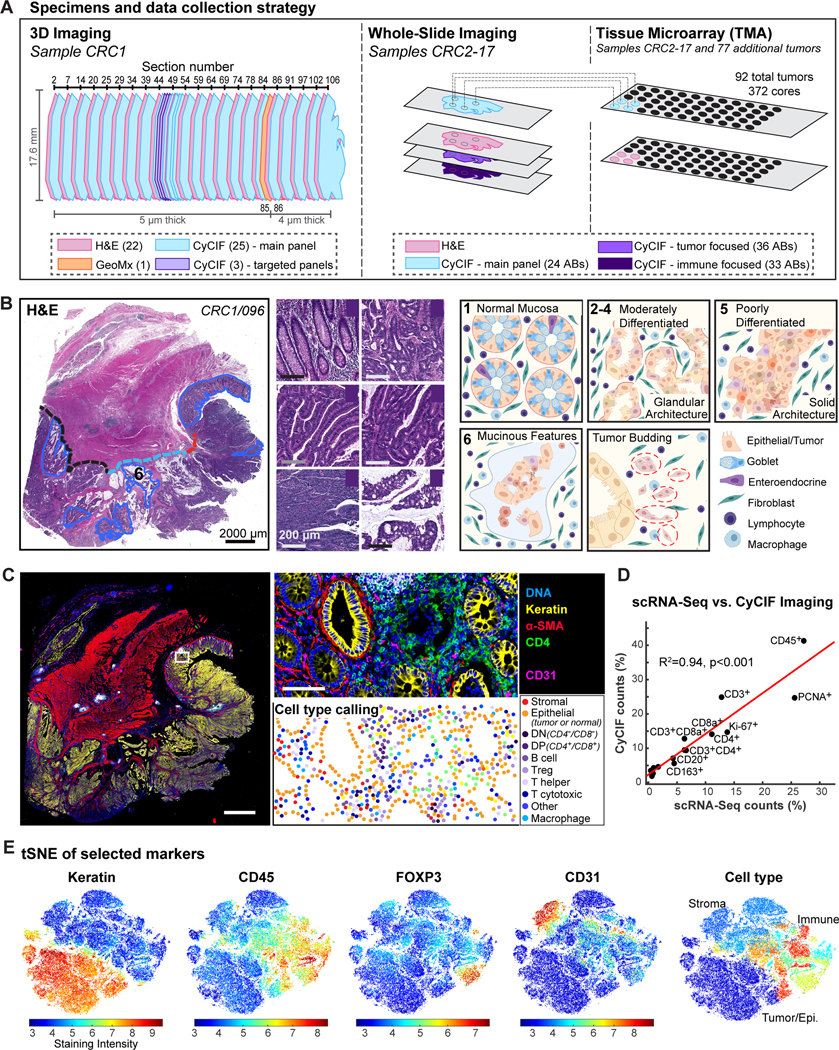
(A) Data collection strategy - 93 CRC specimens available as 3D stack, single whole-slides, and TMAs. (B) Histopathologic annotation of six ROIs and three invasive margins (A: budding, B: mucinous, C: pushing) on H&E (left). Representative images of ROIs (center). Schematic diagram of architectural features (right). (C) CyCIF whole-slide image and cell-type assignment. 21 cell types from 3 main categories (tumor, stroma, and immune; Table S4) were defined and locations mapped. (D) Comparison of cell-type percentages by scRNA-seq and CyCIF. (E) t-SNE of single-cell data (CRC1/097) generated using all markers; 50,000 randomly-sampled cells displayed. Cell-type plot (right) color code same as Figure S1C.
t-SNE on CyCIF data demonstrated a clear separation of cytokeratin-positive (CK+) epithelial cells (both normal and transformed) from CD31+ endothelial cells (primarily blood vessels), desmin+ stromal cells, and CD45+ immune cells (Figures S1B–S1D; Table S5). Immune cells were further divided into biologically important classes such as CD8+PD1+ cytotoxic T cells (Tc), CD4+ helper T cells, CD20+ B cells, CD68+ and/or CD163+ macrophages, as well as discrete sub-categories such as CD4+FOXP3+ T regulatory cells (Tregs) (Table S4). When scRNA-seq35 was performed on ~104 cells from an adjacent region of CRC1, estimated cell-type abundances exhibited a high degree of concordance with estimations from image data (R2 = 0.94; Figures 1D–1E, S1E–S1F).
Impact of spatial correlation on statistical power.
Most high-plex tissue imaging papers to date focus on TMAs or – in the case of mass spectrometry-based imaging methods (MIBI, IMC) – on fields of view (FOVs) of ~1 mm2 because less data is involved and it is easier to acquire tissue from cohorts. It is nonetheless well-established that the minimum dimension needed to accurately measure features within an image depends on the size of these features, which can be estimated from cell-to-cell correlation lengths.36 In CRC1–17, we observed correlation lengths ranging from ~80 μm for CD31 positivity to ~400 μm for keratin or CD20 positivity (Figures 2A–2D, S2A). These length scales were directly related to recurrent morphological features, including small capillaries for CD31+ cells, sheets of tumor for CK+ cells, and TLS for CD20+ cells (Figures 2C–2D), but were also similar in size to TMA cores. We therefore used empirical and first-principles approaches to study the impact of sample size on the accuracy and precision of statistical analysis of 3D, 2D WSI, and TMA data.
Figure 2. Spatial heterogeneity and estimation errors for regional sampling.

(A) Length scales for select markers across CRC1–17. (B) Spatial correlations of binarized staining intensities for CK+ (red), α-SMA+ (blue), and FOXP3+ (green) cells, and exponential fits. (C) CyCIF image showing CD20+ TLS (pink circle) and CD31+ blood vessel (yellow circle). (D) Spatial distribution of CD20+ cells (magenta dots, contours) and CD31+ cells (cyan dots); #1–6: annotated ROIs. (E) Virtual TMA cores from CRC1/097 and real TMA cores from CRC2–93. (F) Cell-type abundance estimates using vTMA cores or random sampling. (G) Estimation error of vTMAs summarized by fold-reduction in effective sample size, N/Neff, for marker log-intensities and cell-type compositions. (H) Correlation of select cell-type pairs amongst 10 nearest neighbors (I) Correlation functions of CK+ cells, estimated from vTMAs or random sampling. Estimates from four cores also shown. (J) Images of cores highlighted in (I). (K) Fraction of marker-positive cells across CRC2–17 whole-slide or TMA data, or TMAs from CRC18–93. Box plot displays data points and 1st-3rd quartiles, whiskers extend at most to 1.5x interquartile range, and proportions <0.0001 are denoted as a single data point along dotted line. Outliers labeled as crosses (F&H) or circles (A&K); medians are indicated.
First, we generated a “virtual TMA” (vTMA) comprising 1 mm diameter FOVs subsampled from an image of CRC1 section 097 (CRC1/097); each virtual core contained ~103 cells as compared to ~5 × 105 for WSI. Sampling was performed so that each vTMA core would primarily contain CK+ tumor or epithelial cells. CRC2–17 had been used, prior to the current work, to generate a real TMA (rTMA), allowing us to confirm that vTMA and rTMA cores were similar (Figure 2E). When we computed the abundance of CK+ cells (cell count divided by the total cell number) in each vTMA core we found that it varied 20-fold from 5–95%, whereas the true value determined by counting all cells in CRC1/097 was 45% (Figure 2F). Abundance estimates for α-SMA and FOXP3 positivity in vTMA cores were also imprecise, but to a lesser extent (Figure 2F). In contrast, when random samples of ~103 cells were drawn from the single cell data without regard to position in the specimen, the estimated abundance of CK+ cells was 45 ± ~1%, a good estimate of the actual value (Figure 2F). Thus, imprecision associated with computing cell abundance from a vTMA arises only when spatial arrangements are preserved.
These findings can be explained by the Central Limit Theorem for correlated data.37 The effective sample size (Neff) for correlated data is related to the sample size N for “dissociated cells” (cells chosen at random without regard to position in an image or drawn from a dissociated cell preparation as in scRNA-seq or flow cytometry) via a simple scaling law (see Methods for derivation):
| (EQ1) |
where is the spatial correlation strength, CAB(r) the length scale (e.g., ~400 μm for CK+) and average cell size . We observed a good match between CyCIF data and theory (R2 = 0.97; Figures 2G, S2B) corresponding to a reduction in effective sample size (N/Neff) of 10- to 1,000-fold depending on the marker identity (median value ~100). Thus, a 1 mm core containing ~103 spatially correlated cells constituted as few as 1 to 3 independent samples, which explains high variance in feature values. We conclude that the analysis of TMA cores and other similarly small FOVs is an inadequate means to accurately determine features as simple as cell abundance because the sample is too small relative to feature sizes.
Analysis of higher-order spatial features, such as cell proximity (Figures 2H, S2C), was also strongly impacted by sampling under spatial correlation. For example, vTMA data were less precise than random sampling when computing the correlation of CK+ (tumor) cell frequency with neighboring α-SMA+ (stromal) cell frequency as a function of distance (compare blue and green in Figure 2H; note that distance is plotted as the number of neighboring cells, which is proportional to distance squared). The same was true when we searched for neighborhoods containing CD45+ immune cells and CD31+ endothelial cells, which represent areas of perivascular inflammation. Inspection of underlying images showed that these differences related to common forms of variation in tissue morphologies and spatial arrangements (Figures 2I, 2J, S2D).
To compare the magnitude of biological (patient-to-patient) variability with sampling error, we computed cell abundances for single markers and biologically-relevant marker combinations (e.g., CD68+PDL1+ macrophages) and observed a 3- to 10-fold variation across CRC2–17 (Figure 2K, red). However, inter-core variance from any single specimen obtained from rTMAs was substantially greater (Figure 2K, blue & teal). Only one TMA-derived measurement, Ki-67 positivity in CK+ cells, exhibited inter-patient variability (18–61%) greater than sampling error between cores (~30%) (Figures 2K, S2E–S2F). Moreover, sampling error is sufficient in magnitude that it can lead to false associations with patient outcome in Kaplan-Meier analysis (Figures S2G–S2H).
To determine whether 2D WSI adequately samples a 3D specimen we computed cell abundances and spatial correlations for 24 Z-sections from CRC1 and compared this to patient-to-patient variability estimated from whole-slide images of specimens CRC2–17 (compare red and blue in Figures S2I–S2J). For all but a few markers, we found that variance between Z-sections was substantially smaller than patient-to-patient variability. We conclude that 2D whole-slide imaging of a 3D specimen does not, in general, suffer from the same subsampling problem as TMAs or small FOV. As we show below, however, many mesoscale tumor features can only be detected in 3D data.
Morphological and molecular gradients involving tumor phenotypes.
To link high-plex image features to histological features with established prognostic value in CRC, such as the degree of tumor differentiation (well, moderate, poor), grade (low, high), subtype (mucinous, signet ring cell, etc.),30 two board-certified pathologists annotated regions of interest (ROI) from all 22 H&E sections of CRC1 and then transferred the annotations to adjacent CyCIF images for single-cell analysis. Annotations included normal colonic mucosa (ROI1); moderately differentiated invasive adenocarcinoma with glandular morphology involving the luminal surface (ROI2), submucosa (ROI3) or the muscularis propria at the deep invasive margin (ROI4); regions of poorly differentiated (high-grade) adenocarcinoma with solid and/or signet ring cell architecture (ROI5); and regions of invasive adenocarcinoma with prominent extracellular mucin pools (ROI6) (Figure 1B). A region with prominent tumor budding (TB) near margin IM-A was also annotated. Excluding muscle, CyCIF data showed that solid adenocarcinoma (RO15) had the highest proportion of CK+ tumor cells (~70%), whereas adjacent normal epithelium (ROI1) had the fewest CK+ (~25%) and the most stromal and immune cells.
To identify molecular features corresponding to each histology, k-nearest neighbor (kNN) classifiers were trained using molecular features (CyCIF intensities) on pathology labels; the CyCIF data comprised only cell positions (centroids) and integrated marker intensities, not morphological or neighborhood information. For simplicity, we consolidated the ROIs into four classes with half of the cells in each class used for training and half for validation. A different classifier was generated for each pair of CyCIF and H&E images for CRC1–17. We observed high confidence predictions from the trained kNN classifier (Shannon entropy near zero) on the validation set (Figures 3A, S3A) showing that the classifier had encoded disease-relevant morphology using marker intensity alone. However, no single molecular marker was unique to a specific ROI or tissue morphology implying that morphology is encoded in hyperdimensional intensity features.
Figure 3. Correlation and prediction of morphologic and molecular tumor phenotypes.

(A) Example ROIs corresponding to four tumor morphologies used for training and non-adjacent regions predicted with high confidence. kNN classifiers were trained and validated separately for each section to evaluate model reproducibility. (B) Prediction confidence for assignment of kNN classes as measured by Shannon entropy (0 corresponds to perfect certainty; 2 indicates random assignment (equal mixing). (C) Posterior probability that each CK+ cell belongs to the given tumor class. Annotation reflects classifier gradients corresponding to morphologic phenotype. (D) Left: Sample tumor region that transitions from normal to abnormal glandular features coinciding with transition from E-cadherin expression to PCNA (CyCIF, bottom). Contours describe averaged local epithelial cell expression of PCNA. Center and right: Additional examples of transition regions. (E) PCA of 31 spatially-resolved GeoMx transcriptomics regions (areas in Figure S1A). (F) Cumulative distribution of single-cell classification entropy of CRC1–17. Patients with only two classes had only normal epithelial and a tumor morphology class. Different CRC1 sections used different markers for classification. (G) Examples of marker gradients; whole tumor sections. White circles denote TMA cored regions.
Unexpectedly, kNN classifiers scored most regions of CRC1 outside of the training and validation data as comprising a mixture of morphological classes (as quantified by the posterior probability) with spatial transitions from one class to another. In many regions, Shannon entropy values approached two, demonstrating an equal mixture of all four classes (red in Figures 3B, S3B). This was not a limitation of the markers used for classification, because similar results were obtained with combinations of ~100 antibodies used to stain CRC1 sections 044–047 (Figures S3C–S3D; Table S3). When tumor regions with high Shannon entropy values were examined in H&E, we found that they corresponded to transitions between classical morphologies (Figure 3D), including ones from mucinous to glandular, mucinous to solid, and glandular to solid. Transitions recurred multiple times in spatially separated tumor areas on dimensions ranging from a few cell diameters (~50 μm) to the whole image (~1 cm) (Figure 3C).
When we performed principal component analysis (PCA) on 31 spatially resolved GeoMx transcriptomic microregions (with each microregion sorted into CK+ or CK− cells) we also observed gradations in molecular state for both the tumor/epithelial (CK+; Figure 3E, circles) and immune/stromal (CK−; squares) compartments. PC1, the dominant source of variance, correlated with histologic subtype and grade while PC2 correlated with epithelial vs. stromal compartment. In support of kNN models of CyCIF data, we observed a graded transition along PC1 from glandular/mucinous (low-grade) to fragmented/budding (high-grade) histologies in both the epithelial/tumor and stromal/immune compartments.
Across all 17 tumors, analysis of CyCIF data revealed intermixing of histologies to a greater or lesser extent with some tumors exhibiting contiguous blocks of a single morphology (e.g., CRC5) as compared to CRC1-like intermixing in others (e.g., CRC14; Figures 3F, S3B). There was no obvious correlation between the degree of intermixing and MSI-H status (which promotes genome instability). Thus, the highly characteristic histological phenotypes routinely used for pathology grading are present in both discrete and intermixed forms in CRCs, most likely due to epigenetic rather than genetic heterogeneity.
We also found that CyCIF markers exhibited intensity gradients that in some cases encompassed an entire tumor and in others coincided with local morphological gradients. Four examples are shown: a normal-glandular transition corresponding to E-cadherin and PCNA gradients that are inversely correlated (Figure 3D; left); a mucinous-solid transition coinciding with inversely correlated cytokeratin 20 and cytokeratin 18 gradients (Figure 3D; center); alternating glandular-solid transitions (Figure 3D; right, yellow curved arrow); and a glandular-solid transition coinciding with a graded transition in the levels of histone acetylation (H3K27ac) vs. trimethylation (H3K27me3) (Figure 3D; right, white arrow; also visible in CRC4, CRC5 in Figure 3G). H3K27ac and H3K27me3 epigenetic marks are known to play complementary roles in transcriptional regulation,38 providing further evidence of organized epigenetic states in the TME. Graded expression of the tumor suppressor p53 and oncogene EGFR – two genes important for CRC biology – was also observed (Figure 3G). Of note, the white circles in Figure 3G are regions of tissue removed for rTMA construction (4 or 5 cores per specimen) that we find to lie along a staining gradient. Such variation between TMAs from a single specimen is often attributed to random heterogeneity rather than molecular and physical gradients, even though these are known to play essential roles in normal tissue development.39
Tumor budding and molecular transitions at the deep invasive front.
For diagnostic purposes, tumor buds are defined by the International Tumor Budding Consensus Conference (ITBCC) as clusters of ≤4 tumor cells surrounded by stroma and lying along the invasive front,21 or, less commonly, the non-marginal ‘internal’ tumor mass.40 Using ITBCC criteria, a pathologist identified a total ~7 × 103 budding cells in 10 of 17 CRC specimens examined (representing ~0.01% of all tumor cells; Figure 4A, arrows and boxes highlight examples on H&E, yellow outlines on CyCIF images indicate segmented budding cells, Figure S4A). In CRC1, buds were largely confined to one ~2.0 × 0.7 × 0.4 mm region of the invasive front (region IM-A, Figure 1B) near normal colonic epithelium and interspersed with T cells (Figure 4B). In 3D we found that these “ITBCC buds” were frequently connected to each other and to the main tumor mass (Figures 4C–4D, S4B, Video S1). Thus buds as classically defined appeared to be predominantly cross-sectional views of these fibrillar structures, as previously suggested from H&E imaging.41
Figure 4. Tumor budding is a distributed phenomenon associated with graded molecular and morphologic transitions.
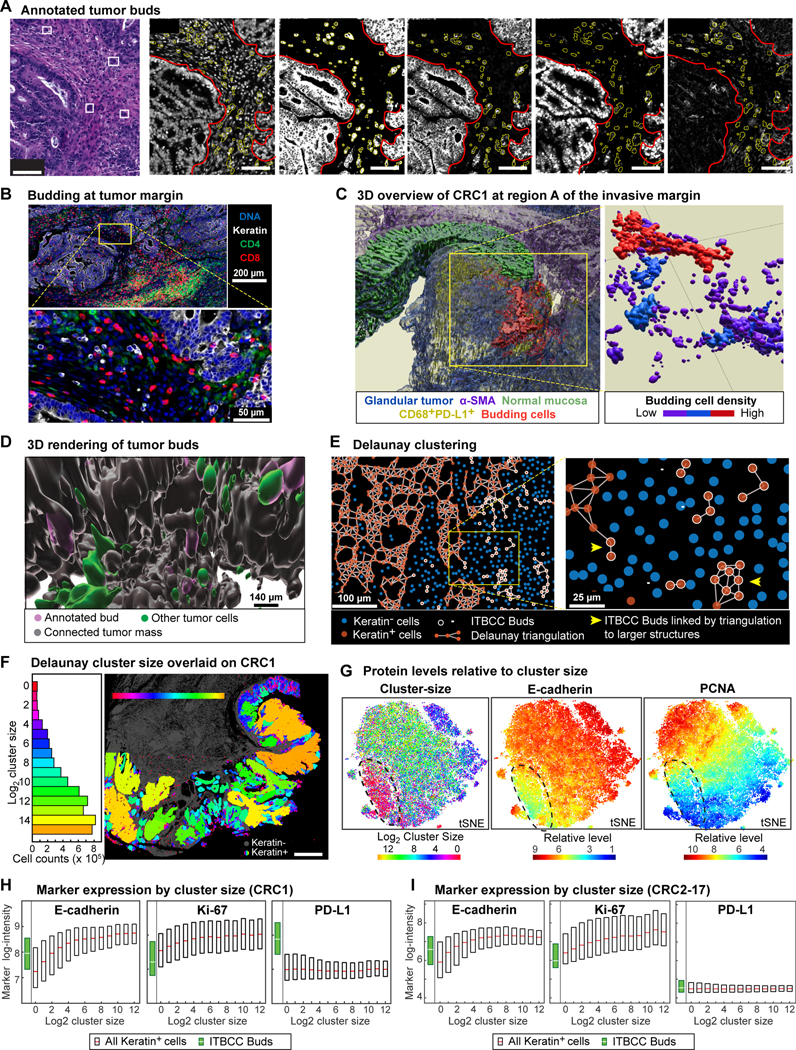
(A) Left: H&E FOV from CRC1/096 IM-A (Figure 1B); budding cells indicated by boxes/arrowheads. Right: Corresponding CyCIF (CRC1/097). Outlines indicate main tumor mass (red) and canonical tumor buds (yellow). (B) Different magnifications of annotated budding region (CRC1/097). (C) CRC1 IM-A 3D overview. Left: Surface renderings of glandular tumor (blue), α-SMA+ stroma (purple), normal mucosa (green), CD68+PDL1+ cells (yellow), budding cells (red). Right: All annotated buds colored by budding cell density showing interconnected fibril-like networks of budding cells. (D) 3D visualization of annotated buds (purple) relative to connected tumor mass (gray) and cells with uncertain connectivity (green). Corresponding regions in 2D images shown in Figure S4B. (E) Delaunay clusters of CK+ cells in a local FOV (CRC1/097). CK+ cell neighborhoods are denoted by edges, along with CK− cells (blue) and pathology annotated buds (white). (F) Cluster sizes (log2) in CRC1. Left: Histogram across all 25 sections. Right: Mapped onto section 097. (G) Left: t-SNE of cluster size. Color represents log2 cluster size; black outline denotes small clusters (including annotated buds). Center and right: t-SNE of CK+ cell expression of indicated marker intensity. (H-I) Marker intensity and cluster-size. Annotated buds in green. Box plots show 1st-3rd quartiles; points beyond not shown. Each box represents ~105-106 tumor cells.
To analyze these structures objectively, we used Delaunay triangulation42 to identify CK+ cells (i.e., tumor and normal epithelium) that were immediately adjacent to each other (Figure 4E). The smallest Delaunay clusters corresponded to ITBCC buds with 1–4 contiguous tumor cells surrounded by stroma (Figure 4F; red), whereas the largest clusters contained >104 cells and mapped to regions of poorly differentiated adenocarcinoma with solid architecture (primarily tumor cells; yellow and orange). The widest range of cluster sizes was observed in differentiated regions with glandular architecture (Figure 4F; blue green). A key feature of tumor budding cells is that they express low levels of cell-tocell adhesion proteins (e.g., E-cadherin, CD44, Ep-CAM)43 and have a low proliferative index.44,45 We confirmed that buds matching ITBCC criteria had reduced expression of adhesion and proliferation markers (Figure S4C). Moreover, a t-SNE representation of all single cell data labeled by Delaunay cluster size showed that CK+ cells in the smallest clusters expressed the lowest E-cadherin levels and that proliferation markers (e.g., PCNA) were also expressed at low levels (Figure 4G, circled region). However, tumors in our cohort did not contain a discrete population of E-cadherin/proliferation-low budding cells, instead, the expression of E-cadherin, Na-K ATPase, PCNA, and Ki-67 varied continuously with cluster size in CRC1 (Figures 4H, S4D) and other CRC tumors (Figures 4I, S4E).
Inspection of the underlying images (Figures 5A–5B) showed that regions of cohesive glandular tumor (which were associated with large Delaunay clusters and a PCNAhigh state) were often fragmented into fibrillar structures comprised of smaller clusters with a PCNAlow state. At the terminal tips of these fibrillar structures we found ‘bud-like’ structures exhibiting the lowest PCNA expression and surrounded by stroma (Figure 5A) or mucin (Figure 5B; mucins are large glycoproteins that protect the gastrointestinal epithelium). Analogous transitions between tumor masses and small Delaunay clusters were observed throughout the tumor both at the invasive front (IM-A in CRC1), in mucinous spaces (IM-B), and along the luminal surface of the tumor in regions corresponding to discohesive growth with focal signet ring cell morphology (ROI5, Figure 1B).46 The small Delaunay clusters found in mucin pools were not distinguishable in size or marker expression from classically-defined buds (Figures 4I, S4E), even though the ITBCC definition encompasses only clusters in fibrous stroma. Moreover, GeoMx RNA expression data (Figure 3E) confirmed that regions with ITBCC buds (brown dots), fragmented tumor and budding (orange), and budding into mucinous spaces (yellow) were similar to each other and distinct from other tumor morphologies (Figure 3E). All three bud-like morphologies expressed elevated levels of genes in the EMT Hallmark gene set (GSEA M5930; Figure 5C, orange, yellow, brown) consistent with the idea that loss of cell cohesion occurs frequently across tumors, is associated with an EMT-like process, and may be driven by a similar epigenetic program.28 In 2D views, mucin surrounding bud-like structures is found in pools that appear isolated from each other (Figure 5D arrowheads).47 In 3D however, these mucin pools were frequently continuous with each other and the colonic lumen up to 1 cm away; in CRC1 this is most prominent in the central region involving invasive margin IM-B (Figure 5E). Thus, both the buds and mucin pools visible as isolated structures are in fact commonly inter-connected in 3D; moreover, large mucin-containing structures can connect to the lumen and its microbiome.
Figure 5. Small, isolated tumor and mucin structures in 2D are large, connected networks in 3D.
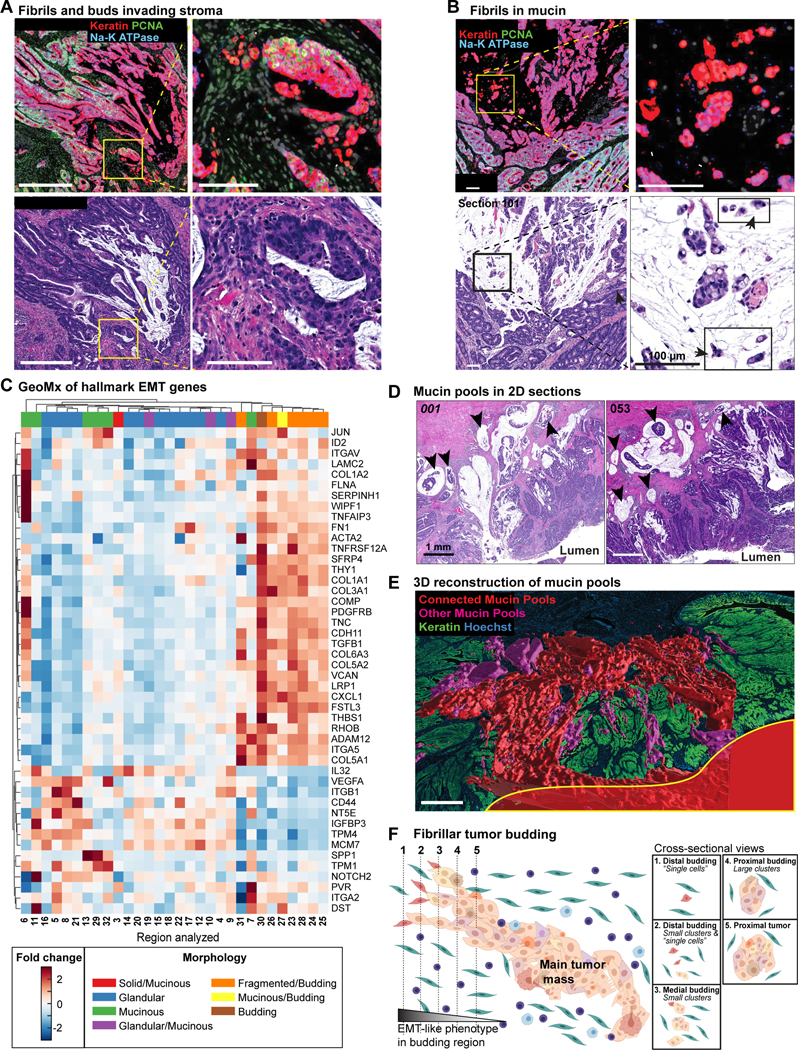
(A) Example transition from main tumor mass into fibrils and ‘bud-like’ cells in stroma; CyCIF (top), H&E (bottom). Na-K ATPase and PCNA decrease with cluster size from main tumor mass to fibril tips (arrows, budding cells). Image oversaturated for visualization. (B) Analogous budding structures in mucinous tumor regions, with fibrils and budding cells (arrowheads) extending into mucin pools. (C) GeoMx data heatmap for selected EMT hallmark genes. Columns correspond to analyzed region from one tissue section (Figure S1A); morphology indicated. (D) Two H&E FOVs from different regions of reconstructed mucin structure with apparently isolated pools in 2D sections (arrowheads). (E) Connectivity of mucin pools across serial sections. Largest contiguous mucin network (red) extends to lumen surface (yellow outline). Image mirrored along Z relative to Figure 1B. (F) Schematic depicting serial sectioning through fibrils at invasive margin, illustrating contiguous 3D structures appearing as isolated cells/small clusters in 2D.
We conclude that EMT-like transitions and tumor budding in CRC1 is characterized not by the formation of isolated spheres of cells, as first described by Weinberg and colleagues in tissue culture,48 but instead by the formation of large fibrillar structures that appear to be small buds when viewed in cross-section at their distal tips. Fibrils can invade into several different environments, including stroma and mucin and we speculate that their formation is driven by a gradual (not abrupt) breakdown in cell adhesion associated with a graded EMT-like transition (Figure 5F).
Networks of tertiary lymphoid structures and their composition.
Anti-tumor immunity involves innate and adaptive mechanisms that mediate the expansion and activation of cytotoxic T cells and the production of antibodies by B cells (plasma cells). Adaptive immunity occurs within secondary lymph organs (SLO; e.g., Peyer’s patches in colonic mucosa)49 and TLS, which develop in non-lymphoid tissues such as tumors and other sites of chronic inflammation. The presence of TLS is associated with good prognosis and immune checkpoint inhibitor (ICI) responsiveness.50,51 Pathology inspection of 47 individual sections of CRC1 (22 H&E and 25 CyCIF) identified over 900 distinct SLO and TLS domains in 2D (Figures 6A, S5A). However, we found that many of these domains were interconnected, forming larger 3D structures; for example, seven large networks (Figure 6B, Video S2) each spanning >12 sections and several millimeters laterally, could be assembled from 20–200 individual 2D domains (the final assembly included 133 additional smaller SLO/TLS networks; Figures 6C, S5B). These large tertiary lymphoid structure networks (TLSNs) were found along the invasive fronts (networks A, B, D), inside tumor (F, G), or in layers of the muscularis (E) or subserosa (C; the subserosa is peri-colonic fibroadipose tissue external to the muscularis).
Figure 6. 3D TLS structure and cell compositions.
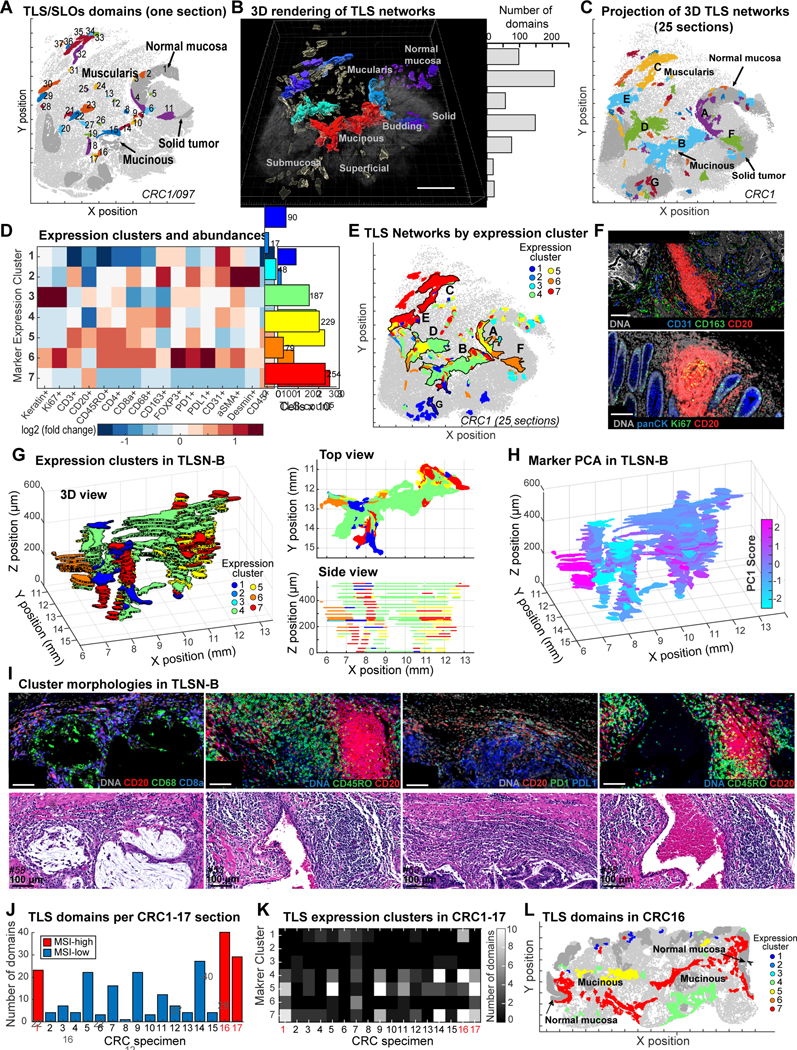
(A) 2D TLS domains (CRC1/097); numbers indicate individual TLS/SLO domains in this section. (B) 3D rendering of TLS networks (TLSNs); CRC1. 7 largest TLSNs (A-G) - histogram shows number of individual TLS identified in 2D sections from each. (C) 3D TLSNs projected onto XY-surface. (D) TLS domain clustering by kNN (left) and number of domains in each cluster (right). (E) TLS cluster distribution in CRC1; 7 largest TLSNs are outlined/labeled. (F) Example CyCIF images of TLS clusters 1 and 3. (G) Left: 3D view of TLSN-B from CRC1 with each TLS domain colored by cluster. Right: Cross-sectional views of XY (top) and XZ (bottom) show TLS domains in TLSN-B. (H) 3D view of TLSN-B, colored by principal component 1. (I) Example CyCIF and H&E images of TLS clusters 4, 5, 6, 7. (J) TLS domain counts in CRC1–17 (section 097 for CRC1). (K) TLS cluster heatmap from CRC1–17. (L) 2D TLS domains of CRC16, colored by clusters.
To study the cellular composition of TLSNs, we performed K-means clustering on CyCIF intensity data (with k = 7 to match the number of large networks, Figure 6D) and recovered clusters with the properties of SLOs (cluster 3) near normal mucosa (as expected for Peyer’s patches) and typical TLS-like lymphoid-aggregates within the tumor itself (cluster 1, Figures 6E–6F, S5C–S5D). TLS undergo maturation and are expected to differ from one another, but when we mapped marker expression clusters onto the physical organization of TLSNs, we found that some were relatively homogenous, containing cells from one expression cluster, whereas others were heterogenous. For example, TLSN-C, which was predominantly located in the subserosa, was >96% composed of expression cluster 7, showed a marked predominance of CD45+CD20+ B cells with little enrichment of other populations; TLSN-F, which was found immediately adjacent to the region of tumor budding, was 95% comprised of cluster 6, a cluster involving B cells, numerous PD1+ cytotoxic T cells, FOXP3+ Tregs, and PDL1+ myeloid cells. In contrast, TLSN-A, -B, and -D contained mixtures of expression clusters (Figures 6E, S5C).
To study an intermixed TLSN in greater detail, we projected marker clusters onto a 3D reconstruction of TLSN-B (Figure 6G), which involved the greatest number of individual 2D domains (206) (Figures 6B, S5B). We observed enrichment of myeloid cells (CD68+CD163+; cluster 4, green) on the mucinous side of TLSN-B, with enrichment of T cells (CD3+, CD45RO+, CD4+; cluster 5, yellow) and B cells (CD20+CD45+; cluster 7, red) along the stromal side (Figure 6G). Inspection of corresponding H&E images revealed numerous discrete B-cell aggregates with associated T cells Figure 6I). The impression of graded composition was confirmed when we performed PCA on marker intensities and mapped principal component scores onto the TLSN-B structure (Figures 6H, S5E).
To extend this analysis, we superimposed marker-based clustering from CRC1 onto CRC2–17 (Figure S5F) and found that the prevalence of individual marker clusters varied from tumor to tumor but was similar for CRC1 and CRC2–17 in aggregate (Figures 6J, 6K). Like CRC1, CRC16 and CRC17 are MSI-H tumors with rich TLS networks. In CRC16 the area surrounding mucin pools and TLS were enriched in cells from marker clusters 4, 5 and 7 – as in CRC1 (Figure 6L) From these data, we conclude that our single 3D reconstruction of a TLS in CRC1 is a reasonable exemplar of our overall cohort in showing that: (i) TLS form interconnected 3D networks rather than the isolated structures observed in 2D sections, (ii) TLS networks within a single tumor can have different cellular compositions, and (iii) variation in cell types and functional markers within a single large TLS network is graded, implying intra-TLS patterning and communication.
Immune profiling of the invasive margin.
The immune response at the tumor margin strongly influences disease progression and ICI responsiveness.52 Among the three morphologies found at the CRC1 invasive margin, IM-A, the region with tumor budding and poorly differentiated morphology, had the greatest immune cell density (Figure 7A) but was also strongly immunosuppressive, with abundant CD4+FOXP3+ Tregs partially-localized with CD8+ cytotoxic T cells (Figure 7B). While PDL1+ cells were found both inside the tumor and stroma (Figure 7C), interactions between PDL1+ and PD1+cells were enriched near buds in the stroma (Figure 7D). IM-B exhibited the least immune cell infiltration, consistent with a role for mucins in immune evasion or sequestration.53 IM-C was rich in Tregs but had very few PDL1+ cells as compared to IM-A (Figures 7C, 7D).
Figure 7. Immune landscape of CRC and its invasive margins.

Abundance and distribution of (A) CD45+, (B) CD4+FOXP3+(Treg), CD8+(Tc), and (C) PDL1+ cells; TB (tumor budding); labels correspond to Figure 1B. (D) Co-occurrence of PDL1+ and PD1+ using a 20 μm distance cutoff. Panels A-C and K depict CRC1/097. (E) LDA topics and relative abundancies along the tumor margin. (F) PDL1 expression in indicated cell types. Top panel represents relative fractions of PDL1+ cells over indicated populations, while bottom panel shows absolute fractions of PDL1+ or double-marker positive cells. (G) Representative images of PDL1+CK+ cells in CRC1 (top) and CRC17 (lower). (H) Plot of PDL1+CK+ (top) or PDL1+CD68+ cell fractions in MSI-H or MSI-L samples from TMA data (CRC2–93). (I-J) Fraction of PDL1:PD1 interaction (20 μm) within CK+ (top) and CD45+ (bottom) cells; P-values from pairwise t-test shown (n=25). (I) In CRC1 (all 25 sections) or (J) CRC1–17 (n=17). (K) Co-occurrence maps using 20 μm distance cutoff. (L) High-resolution 3D imaging of PDL1:PD1 interaction among tumor and myeloid cells. Top: maximum intensity projections. Bottom: 3D rendering, Imaris software. (M) Schematic illustrating tumor-immune interactions at different types of invasive margins. Boxplots 25%−75% with whiskers at 5% and 95%; medians indicated. Outliers labeled crosses (F&H), circles (E).
To quantify relationships between tumor margin morphologies and molecular properties we used Latent Dirichlet Allocation (LDA), a probabilistic modeling method that reduces complex structures into distinct component communities (“topics”) while accounting for uncertainty and missing data.54–56 We annotated invasive margins in CRC1–17 for i) infiltration with tumor budding, ii) deepest invasion, and iii) all other morphologies (mucinous fronts were too infrequent to represent their own category) then performed LDA on CyCIF data (33-plex immune panel; Figure S6A).14 We found that LDA topic frequencies varied significantly in different regions of the invasive margin (Figures 7E, S6B–S6C). Margins with tumor budding were significantly associated with CD4+ and CD8+ T cells (Figure 7E, topic 1), the deep invasive front with tumor cell proliferation (Ki-67+ CK+ cells; topic 9), and the remainder of the front with podoplanin positivity (PDPN+; topic 7). PDPN is a short transmembrane protein implicated in cell migration, invasion, and metastasis.57 Fibroblasts secrete abundant cytokines and growth factors, potentially explaining the activation of signal transduction (i.e., phosphotyrosine (pTyr) and phospho-SRC positivity; topic 10) along this portion of the tumor margin. In contrast, myeloid cells were ubiquitous, and their frequency (topics 5 and 12) did not significantly associate with any specific margin morphology. Thus, morphologically distinguishable domains of the CRC invasive margin have differing levels of tumor cell proliferation (low in buds and high in deep invasive margins), activation of signaling pathways (pTyr levels), and immune suppression.
Cell types involved in presenting PDL1 to PD1+ T cells.
The immunosuppressive interaction between PD1+ and PDL1+ can be targeted therapeutically in CRC58 and is therefore clinically significant. Across CRC1–17, the fraction of PD1+cells varied 4-fold (from 3–12% of all cells), and these cells were >80% CD4+ or CD8+ T cells (Figures S6D, S7). The fraction of PDL1+ cells in the same specimens varied 12-fold (3–40%) (Figure S6E) and correlated with the number of PD1+ cells (r=0.52, p=0.034; Z test). While a small minority (1–5%) of tumor cells expressed PDL1, the cells most likely to be PDL1+ were CD68+ (14–51% positive) and CD11c+ myeloid cells (10–88% positive); PDL1+ myeloid cells were also ~6.5-fold more abundant on average than PDL1+ tumor cells (Figures 7F, S6E). The sole exception to this rule was CRC17, with >40% of tumor cells strongly PDL1 positive; this tumor was also high-grade with extensive necrosis and poorly differentiated solid architecture. t-SNE showed it to be a clear outlier in our cohort with respect to composition (Figures 7G; S7A–S7C). Immunotherapy is indicated for MSI-H CRCs because they are highly immunogenic59 and we found that MSI-H tumors in our cohort (n=16 of 93; see methods) had 5fold more PDL1+ tumor cells and 6-fold more PDL1+ myeloid cells on average than MSI-L tumors (p=0.044 and 0.002 two-sided t-test, Figure 7H), but the latter still outnumbered the former ~4-fold. Moreover, ~80% of MSI-H tumors had more PDL1+ myeloid cells than the average MSI-L tumor (Figure 7H). Across the CRC cohort, we found that single positive CD68+CD11c− or CD68−CD11c+ and double positive CD68+CD11c+ cells were commonly PDL1+, although the relative abundance of each myeloid subset varied several fold (Figures S6F–S6G). We do not have the markers in our panels to more precisely subtype PDL1+ myeloid populations, but our interpretation is that they include variable proportions of macrophages, dendritic cells, and other mononuclear phagocytes.
Functionally, it is not the prevalence of PDL1+ cells that is relevant for T-cell suppression but rather which cells are close enough for PDL1:PD1 binding. To study this, we performed proximity analysis using a 20 μm cutoff and found that, across 24 CRC1 sections, cells interacting with PD1+ cells were strongly enriched for CD45+ and depleted for CK+ (p<0.001 pairwise t-test, two-sided), showing that PD1+ T cells interact with PDL1+ immune cells more commonly than PDL1+ tumor cells. This was also true of CRC2–16, with CRC17 representing the sole exception (Figure 7J, red lines). Cells interacting with PD1+ cells were also significantly more likely to be CD44+ (an adhesion receptor60) and HLA-A+ than non-interacting cells. Co-localization of CD68+PDL1+ myeloid cells with PD1+CD8+ T cells was also confirmed by co-occurrence mapping in CRC1 (Figure 7K, upper panel). Finally, high resolution optical sectioning of 12-plex CyCIF provided direct evidence of PDL1+ on myeloid cells co-localizing with PD1+ T cells at the tumor margin, consistent with formation of functional cell-cell interactions (Figure 7L). We conclude that immunosuppression of PD1+ T cells in our CRC cohort most commonly involves PDL1+ myeloid, not tumor cells. Nevertheless, PDL1-expressing tumor cells may be involved in immune suppression in some tumors: the 3% of tumor cells that express PDL1 in CRC1 are concentrated at the budding margin near T cells (Figure 7K, lower panel; Figure 7M).
DISCUSSION
Understanding intra-tumor heterogeneity (ITH) is essential for improving our knowledge of tumor biology and for optimizing diagnosis and therapy.61 The image-based single cell analysis described in this paper supports two broad conclusions about the nature and organization of ITH in CRC. First, molecular states (protein markers) and tissue morphologies (histotypes) are often graded, with phenotypic transitions spanning spatial scales from a few cell diameters to many millimeters. For example, gradients in the epigenetic markers H3K27me3 and H3K27ac can span several centimeters along an entire tissue specimen. These proteins play complementary roles in regulating transcription,38 and we find that their levels are commonly anti-correlated. In other cases, changes in cellular phenotypes are graded or recur in a semi-periodic manner, reminiscent of the “reaction-diffusion” morphogen gradients observed in embryonic development,62 by imaging,63 and by mass spectrometry of human tissue.64 Second, cellular communities most commonly studied in 2D at a local level are often organized into large interconnected 3D structures. These structures include: (i) 1–4 cell tumor buds, which are cross-sectional views of fibrillar structures41 that express progressively lower levels of cell adhesion and proliferation markers as the fibrils narrow along the proximal-to-distal axis; (ii) intertumoral mucin pools, which are surrounded by tumor in 2D but comprise 3D networks that can connect to the intestinal lumen and its microbiome; (iii) TLS, which are strongly implicated in anti-tumor immunity65 and form 3D interconnected networks with graded molecular and cellular composition. The presence of large and small-scale gradients is consistent with control of tissue development66 but contrasts in cancer biology with an emphasis on enumerating discrete cell states and mutations using single-cell sequencing.
When a machine learning (kNN) model involving high-plex intensity data was trained by a pathologist to distinguish morphologies such as glandular vs. solid and high vs. low-grade tumor, we found archetypal morphologies used in diagnosis were graded and intermixed across different specimens. The degree of intermixing did not appear to correspond to MSI-H (hypermutant) vs. MSI-L status, suggesting that epigenetics play a greater role than genetics in this form of ITH. We also found that differences in morphology did not map to differences in single markers, but instead to hyperdimensional features involving combinations of multiple proteins. We therefore speculate that the morphologic gradients observed in tissue specimens result from the aggregate action of several underlying molecular gradients, which may include epigenetic regulators, oncogenes, cytokines and nutrients.
Graded changes in protein expression along tumor cell fibrils are one setting in which molecular and morphological gradients are likely related. The diagnostic criterion for a tumor bud is the presence of 1–4 cell clusters at the tumor invasive margin, surrounded by stroma,21 and expressing EMT-like signatures consistent with a role in infiltration and metastasis.48 However, like an earlier H&E study,41 we find that buds in CRC1 are most likely cross-sectional views of the narrow distal tips of fibrillar structures projecting from a tumor mass. By quantifying these structures with Delaunay triangulation, we observe progressively lower E-cadherin and Ki-67 levels from the widest (proximal) to the narrowest (distal) fibril segments, as well as morphologically similar fibrils in other regions of the tumor, including as projections into the mucin network. This recurrence of morphological transitions is consistent with an epigenetic origin for bud-like states.67,68
Ensuring adequate spatial power for tissue imaging.
To date, most analysis of high-plex tissue images has focused on reconstructing small neighborhoods of cells, particularly from tissue microarrays and small FOVs. However, we find that even local proximity analysis is confounded by poor statistical power due to spatial correlation, which arises from the spatial organization of the structures we seek to characterize with high-plex imaging. Whereas the number of independent samples in a set of dissociated cells (e.g., in scRNA-seq) is equal to the number of cells (N), the Central Limit Theorem tells us that the effective sample size (Neff) for spatially correlated data will always be smaller.37 In CRCs we observe correlation length scales up to ~500 μm, making Neff 100 to 1000-fold smaller than N. Thus TMAs and mm-scale FOVs often contain only a one or a few instances of a feature of interest, resulting in measurement error that is substantially greater than the patient-to-patient variability. This “spatial power” penalty is even more severe for complex properties such as neighborhood inclusion and exclusion and is sufficient to generate spurious correlations with Kaplan-Meier survival estimators.
In contrast, 2D WSI (~105 cells per specimen) largely overcomes this problem (Neff >100) for characterization of local neighborhoods. WSI is also the standard in conventional pathology69 and is regarded by the FDA as a diagnostic necessity.70,71 The argument for WSI has not conventionally had a statistical foundation and is instead justified by the need to view cell morphologies in the overall context of the tumor and adjacent normal tissue as part of TNM classification,2 the performance of which is only rarely exceeded by the addition of molecular data. However, the two arguments are fundamentally similar. Our data show that 3D reconstruction provides additional insight into the large-scale connectivity of biological structures, but for relatively straightforward tasks such as cell-type enumeration, 2D WSI is often adequate. A requirement for WSI in a research and diagnostic setting comes with substantial cost: per-patient data sets are >102-fold larger than with TMAs, cohorts are more difficult to acquire (whole blocks must be accessed and recut), and data is substantially more challenging.
Immunology of the CRC invasive margin.
The morphology and depth of invasion of a tumor margin has high prognostic value30 and differences between infiltrative and well-delineated pushing margins are commonly used for patient management.72 We find that the immune environment can vary substantially within a single tumor and recurrently with margin morphology across specimens. Budding regions are the most T-cell rich, but also the most immunosuppressive (with abundant Tregs and PDL1-expressing cells). Whereas tumor buds have few proliferating cells, tumor cells in deep invasive margins are highly proliferative and have fewer immediately adjacent immune cells. Because MSI-H CRC is often treated with ICIs, the mechanism of PDL1-mediated suppression of T cells at the tumor margin is particularly relevant.58 In all but one of the 17 CRCs we examined, PDL1-expressing myeloid cells outnumbered PDL1-expressing tumor cells 4-fold or more; high resolution imaging also showed that myeloid cells frequently form PDL1:PD1 mediated contacts with PD1+ T cells. These findings are consistent with recent data from mouse models of colon cancer showing that dendritic cells are a primary source of immunosuppressive PDL173 and with a general role for dendritic cells in tolerization. However, the relative abundance of PDL1+ cells proximate to T cells varies from tumor to tumor, suggesting that dendritic cells are not the only relevant PDL1+ myeloid population. Moreover, although PDL1+ tumor cells were rare in all but CRC17, these cells may also play an immunosuppressive role because they are often concentrated in regions of tumor budding. An obvious question requiring follow-up studies is whether the type of cell presenting PDL1 to T cells plays a role in responsiveness to ICIs.
Limitations of this study.
Only one CRC has as-yet been reconstructed in 3D, largely because the process remains manual and slow and many of the features we describe in 3D – tumor budding fibrils, TLS networks, and invasive margins – would benefit from deeper molecular profiling to better identify cell types and states. There are many spatial relationships among the 2 × 108 cells in our dataset that we have not yet explored. Moreover, the state of the art in image segmentation and cell-type calling continues to improve, arguing for future reprocessing of primary images using the best available methods. To mitigate these and other limitations, all images described in this study have been released in multiple formats.
STAR METHODS
RESOURCE AVAILABILITY
MATERIALS AVAILABILITY
This manuscript contains no unique reagents or resources; all antibodies are available commercially (see Table S3 and Key Resources file).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 |
Thermo Fisher | RRID: AB_2535794 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 |
Thermo Fisher | RRID: AB_162543 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 |
Thermo Fisher | RRID: AB_162542 |
| Anti-CD3 antibody [CD3-12] | Abcam | RRID: AB_2889189 |
| Na,K-ATPase α1 (D4Y7E) Rabbit mAb | Cell Signaling Technology | Cat#: 23565 RRID: Pending |
| Monoclonal Mouse Anti-Human CD45R0 | Dako | RRID: AB_2237910 |
| Ki-67 (D3B5) Rabbit mAb (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | RRID: AB_2687824 |
| Pan Cytokeratin Monoclonal Antibody (AE1/AE3), eFluor 570, eBioscience™ | Thermo Fisher/ eBioscience | RRID: AB_11218704 |
| Alpha-Smooth Muscle Actin Monoclonal Antibody (1A4), eFluor 660, eBioscience™ | eBioscience | RRID: AB_2574362 |
| Recombinant Anti-CD4 antibody [EPR6855] (Alexa Fluor® 488) | Abcam | RRID: AB_2889191 |
| PE anti-human CD45 Antibody | Biolegend | RRID: AB_2562057 |
| Recombinant Anti-PD1 antibody [EPR4877(2)] (Alexa Fluor® 647) | Abcam | RRID: AB_2728811 |
| CD20 Monoclonal Antibody (L26), Alexa Fluor 488, eBioscience™ | eBioscience | RRID: AB_10734357 |
| CD68 (D4B9C) XP® Rabbit mAb (PE Conjugate) | Cell Signaling Technology | RRID: AB_2799935 |
| CD8a Monoclonal Antibody (AMC908), eFluor 660, eBioscience™ | eBioscience | RRID: AB_2574149 |
| Recombinant Anti-CD163 antibody [EPR14643-36] – C-terminal (Alexa Fluor® 488) | Abcam | RRID: AB_2889155 |
| FOXP3 Monoclonal Antibody (236A/E7), eFluor 570, eBioscience™ | eBioscience | RRID: AB_2573609 |
| PD-L1 (E1L3N®) XP® Rabbit mAb (Alexa Fluor® 647 Conjugate) | Cell Signaling Technology | RRID: AB_2728832 |
| E-Cadherin (24E10) Rabbit mAb (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | RRID: AB_10691457 |
| Vimentin (D21H3) XP® Rabbit mAb (Alexa Fluor® 555 Conjugate) | Cell Signaling Technology | RRID: AB_10859896 |
| Recombinant Alexa Fluor® 647 Anti-CDX2 antibody [EPR2764Y] | Abcam | RRID: AB_2728786 |
| Lamin A/C (4C11) Mouse mAb (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | RRID: AB_10997529 |
| Recombinant Alexa Fluor® 488 Anti-Lamin B1 antibody [EPR8985(B)] - Nuclear Envelope Marker |
Abcam | RRID: AB_2728786 |
| Recombinant Alexa Fluor® 555 Anti-Desmin antibody [Y66] - Cytoskeleton Marker |
Abcam | RRID: AB_2890164 |
| Recombinant Anti-CD31 antibody [EPR3094] (Alexa Fluor® 647) | Abcam | RRID: AB_2857973 |
| PCNA (PC10) Mouse mAb (Alexa Fluor® 488 Conjugate) | Cell Signaling Technology | RRID: AB_11178664 |
| Ki-67 Monoclonal Antibody (20Raj1), eFluor 570, eBioscience™ | eBioscience | RRID: AB_11220088 |
| Collagen IV Monoclonal Antibody (1042), Alexa Fluor 647, eBioscience™ |
Thermo Fisher/ eBioscience | RRID: AB_10854267 |
| CD11c (D3V1E) XP® Rabbit mAb #45581 | Cell Signaling Technology | RRID:AB_2799286 |
| Granzyme B (Concentrate) clone GrB-7 | Agilent | RRID:AB_2114697 |
| Recombinant Alexa Fluor® 647 Anti-HLA A antibody [EP1395Y] (ab199837) |
Abcam | RRID:AB_2728798 |
| Phospho-Rb (Ser807/811) (D20B12) XP® Rabbit mAb (Alexa Fluor® 555 Conjugate) #8957 | Cell Signaling Technology | RRID:AB_2728827 |
| Phospho-Tyrosine Mouse mAb (P-Tyr-100) (Alexa Fluor® 647 Conjugate) #9415 | Cell Signaling Technology | RRID:AB_10693160 |
| Alexa Fluor® 647 antiPodoplanin (Lymphatic Endothelial Marker) Antibody | Biolegend | RRID:AB_2810816 |
| CD44 (156-3C11) Mouse mAb (PE Conjugate) #8724 | Cell Signaling Technology | RRID:AB_10829611 |
| p53 Protein (Concentrate) Clone DO-7 | Agilent | RRID:AB_2206626 |
| EGF Receptor (D38B1) XP® Rabbit mAb (Alexa Fluor® 488 Conjugate) #5616 |
Cell Signaling Technology | RRID:AB_10691853 |
| CDX2 (D11D10) Rabbit mAb (Alexa Fluor® 555 Conjugate) #84638 | Cell Signaling Technology | RRID:AB_10691853 |
| Tri-Methyl-Histone H3 (Lys27) (C36B11) Rabbit mAb (PE Conjugate) #40724 | Cell Signaling Technology | RRID:AB_2799182 |
| Recombinant Alexa Fluor® 647 Anti-Histone H3 (acetyl K27) antibody [EP16602] (ab245912) | Abcam | Cat# ab245912, RRID: pending |
| Purified anti-TIF1β (KAP-1, TRIM28) Phospho (Ser473) Antibody | Biolegend | RRID:AB_2563298 |
| CD11b Monoclonal Antibody (C67F154), Alexa Fluor™ 488, eBioscience™ | Thermo Fisher | RRID:AB_2637200 |
| Alexa Fluor® 488 anti-human CD15 (SSEA-1) Antibody | Biolegend | RRID:AB_493257 |
| Anti-CD14 antibody [EPR3653] (Alexa Fluor® 647) | Abcam | RRID:AB_2890135 |
| Collagen IV Monoclonal Antibody (1042), Alexa Fluor 647, eBioscience™ |
Thermo Fisher/eBioscience | RRID: AB_10854267 |
| Biological samples | ||
| FFPE tissue block and frozen tissue (CRC1) | Cooperative Human Tissue Network, Western Division | N/A |
| FFPE tissue blocks (CRC2-93) | Department of Pathology, Brigham and Women’s Hospital | N/A |
| Software and algorithms | ||
| MCMICRO pipeline (de0d76d7cf0870f1ed979722a465de0fc246b90b) | https://doi.org/10.1101/2021.03.15.435473 | https://github.com/labsyspharm/mcmicro |
| ImageJ (1.53c) | doi:10.1038/nmeth.2019 | https://imagej.nih.gov/ij/ |
| MATLAB 2019b | Mathworks Inc. | https://www.mathworks.com/products/matlab.html |
| Minerva Story | doi: 10.1038/s41551-02100789-8 and doi: 10.21105/joss.02579 | https://github.com/labsyspharm/minerva-story |
| Deposited Data | ||
| Human Tumor Atlas Network | https://humantumoratlas.org/ | https://humantumoratlas.org/explore |
LEAD CONTACT
Requests for further information should be directed and will be fulfilled by Lead Contact, Peter Sorger (peter_sorger@hms.harvard.edu).
DATA AND CODE AVAILABILITY
All full resolution images, derived image data (e.g., segmentation masks) and all cell count tables are available via the NCI-sponsored repository for Human Tumor Atlas Network (HTAN; https://humantumoratlas.org/) at Sage Synapse. A version of this data is available at https://www.synapse.org/#!Synapse:syn18434611/wiki/597418.
Several of the figure panels in this paper are available with text and audio narration for anonymous on-line browsing using MINERVA software 34, as are images of CRC2–17; see https://www.tissue-atlas.org/atlas-datasets/lin-wang-coy-2021/.
scRNA-seq data is available in the Gene Expression Omnibus (GEO accession: GSE166319).
All software used in this manuscript is freely available via GitHub as described in 31 and references therein and in https://github.com/labsyspharm/CRC_atlas_2022.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Participants
The tumor and adjacent normal tissue in CRC1 was collected from a resection of the cecum of a 69-year old male; the medical reports indicated that the tumor was a poorly differentiated stage IIIB adenocarcinoma (pT3N1bM0) 34 with microsatellite instability (MSI-H) and a BRAFV600E (c.1799T>A) mutation. Additional colon adenocarcinoma specimens were retrieved from the archives of the Department of Pathology at Brigham and Women’s Hospital (BWH) with Institutional Review Board (IRB) approval (IRB21–0656) as part of a discarded/excess tissue protocol (Table S1). 92 different tumor samples (CRC2–93) were used to construct a tissue microarray (HTMA 402; four 0.6 mm diameter cores were extracted from the FFPE donor blocks per patient and assembled into a recipient TMA block). The average patient age was 58.7 years (range 25–98), including 46 males (49.5%) and 47 females (50.5%), with no known relevant underlying pathologic conditions (e.g., inflammatory bowel disease, Lynch syndrome, polyposis syndromes). The cohort included 88 primarily diagnosed tumors (94.6%), and 5 recurrent tumors (5.4%). Whole-slide sections of 16 of these colon adenocarcinoma specimens (CRC2–17) were also analyzed, after the four cores were removed. Clinical metadata was abstracted from the BWH medical record and clinical and biospecimen metadata for CRC1 was provided by the CHTN.
METHOD DETAILS
Tissue samples
Unfixed (fresh) tissue from a resection of a colon adenocarcinoma (CRC1) was isolated by the Cooperative Human Tissue Network (CHTN) for single cell RNA-sequencing. A portion of the sample was formalin-fixed and paraffin-embedded (FFPE) and tissue sections were generated by the CHTN as outlined in Table S2. Data related to CRC1 indicates section number as CRC1/section#. For CRC1, 106 serial sections were cut from an ~1.7 × 1.7 cm piece of FFPE tissue and 22 H&E and 25 CyCIF images were collected, skipping some sections to increase the total dimension along the Z-axis. Histopathology review showed that the tumor had a broad front invading into underlying muscle (muscularis propria) and connective tissue giving rise to a ‘budding margin’ (IM-A) adjacent to an area of normal colon mucosa (ROI1), a ‘mucinous margin’ in the middle of the specimen (IM-B), and a deep ‘pushing margin’ (IM-C) (these three margins are denoted “A”, “B” and “C” in Figure 1B).
CyCIF protocol
Tissue-based cyclic immunofluorescence (CyCIF) was performed as previously described.8 The detailed protocol is available in protocols.io (dx.doi.org/10.17504/protocols.io.bjiukkew). In brief, the BOND RX Automated IHC/ISH Stainer was used to bake FFPE slides at 60°C for 30 minutes, to dewax the sections using the Bond Dewax solution at 72°C, and for antigen retrieval using Epitope Retrieval 1 (Leica™) solution at 100°C for 20 minutes. Slides underwent multiple cycles of antibody incubation, imaging, and fluorophore inactivation. All antibodies were incubated overnight at 4°C in the dark. Slides were stained with Hoechst 33342 for 10 minutes at room temperature in the dark following antibody incubation in every cycle. Coverslips were wet-mounted using 200 μL of 10% Glycerol in PBS prior to imaging. Images were acquired using a 20x objective (0.75 NA) on a CyteFinder slide scanning fluorescence microscope (RareCyte Inc. Seattle WA). Fluorophores were inactivated using a 4.5% H2O2, 24 mM NaOH/PBS solution and an LED light source for 1 hour.
Single-cell RNA-sequencing
Samples for scRNA-seq were processed according to the HTAN publication.35 Surgical tissues were removed and placed into RPMI solution and transported directly to the processing laboratory within 10 minutes. Tissue samples were immediately minced to approximately 4 mm2 and washed with DPBS. The samples were then incubated in chelation buffer (4 mM EDTA, 0.5 mM DTT) at 4°C for 1 hour and 15 minutes. Then, the resulting suspensions were dissociated with cold protease and DNAse I for 25 minutes. The suspensions were triturated throughout the process, every 10 minutes, then washed three times with DPBS before encapsulation. Single cells were encapsulated and barcoded using the inDrop scRNA-seq platform as previously described,74 targeting about 2,500 cells. Sequencing libraries were prepared using TruDrop library structure.75 Sequencing was performed on the NovaSeq 6000 (150 bp paired end) at a depth of approximately 150 million reads per sample.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image processing and data quantification
Image analysis was performed with the Docker-based NextFlow pipeline MCMICRO)31 and with customized scripts in Python, ImageJ and MATLAB. All code is available in GitHub (https://github.com/labsyspharm/CRC_atlas_2022). Briefly, after raw images were acquired, stitching and registration of the different tiles and cycles was performed with MCMICRO using the ASHLAR module.32 The assembled OME.TIFF files from each slide were then passed through quantification modules. For background subtraction, a rolling ball algorithm with 50-pixel radius was applied using ImageJ/Fiji. For segmentation and quantification, UNMICST2 was used31,76 supplemented by customized ImageJ scripts8 to generate single-cell data. More details and source code can be found at www.cycif.org and as listed in the software availability section.
Single-cell data quality control for CyCIF
Single-cell data for multiplexed images was passed through several quality control (QC) steps during generation of the cell feature table. Initial QC was done simultaneously with segmentation and quantification, so that cells lost from the specimen in the later cycles would not be included in the output. Next, single-cell data was filtered based on the mean Hoechst staining intensity across cycles; cells with coefficient of variation (CV) greater than three standard deviations from the mean were discarded as were any objects identified by segmentation as “cells” but having no DNA intensity. These steps are designed to eliminate cells in which the nuclei are not included as a result of sectioning. Highly autofluorescent (AF) cells (measured in cycle 1 or 2) were also removed from the analysis, using a customized MATLAB script that applied a Gaussian Mixture Model (GMM) to identify high-AF populations. More details and scripts are available at https://github.com/labsyspharm/CRC_atlas_2022.
Cell-type identification using CyCIF data
Multiparameter single-cell intensity data was used for generating binary gates. For the main CyCIF panels, 16 measurements (cytokeratin, Ki-67, CD3, CD20, CD45RO, CD4 CD8a, CD68 CD163, FOXP3, PD1, PDL1, CD31, α-SMA, desmin, and CD45) were subjected to binary gating. All samples and markers were gated independently. A customized MATLAB script was used to apply 2-component Gaussian Mixture Modeling and generate the initial gate, followed by human-inspection and adjustment. Double or triple gates were also generated via Boolean operation in single-cell data. For hierarchal cell-type identification, a modified SYLARAS algorithm77 was applied with these datasets, and a total of 21 different cell types were assigned using the 16 markers described above. Additional markers (e.g., E-cadherin) were considered to be continuous variables and used for analysis but not cell-type assignment. The completed cell dictionary for cell-type identification can be found in Table S4.
Pathology annotation of histologic features
Hematoxylin and eosin (H&E) stained tissue sections from all specimens (CRC1–17) were evaluated by two board-certified pathologists (S.C., S.S.). For each case, 6 principle regions of interest (ROI) corresponding to histopathologic regions or morphologic variations defined in the pathologic evaluation of CRC were defined when present for all 22 H&E Z-levels, including: (1) normal mucosa; (2) moderately differentiated invasive adenocarcinoma (glandular, typical morphology) involving the luminal surface, (3) submucosa (corresponding to ‘pT2’ depth by TNM staging), and (4) muscularis propria (corresponding to ‘pT3’ by TNM staging); (5) poorly differentiated invasive adenocarcinoma (solid, signet ring cells, corresponding to ‘high-grade’ histology); and (6) moderately-poorly differentiated invasive adenocarcinoma with mucinous features and extracellular mucin pooling (6). Regions of ITBCC-defined tumor budding (i.e., clusters of ≤4 cells apparently detached from the main tumor mass surrounded by stroma at the tumor invasive front) were also annotated in CRC2–17 and on all 22 H&E Z-levels of CRC1. For CRC2–17, additional histologic features that were not present in CRC1 were also annotated when present, including: adenoma (tubular), tumor necrosis, comedo necrosis, squamoid, pleomorphic, and extensive signet ring cell tumor morphology, and perineural or lymphovascular invasion by tumor. In cases with clear anatomic orientation, the deep invasive tumor front was initially delineated as a band with an approximate width of 5–10 cell diameters (50–100 μm) at the deep edge of the tumor. In cases with multiple histologic subtypes present at the invasion margin, each type was annotated separately; in CRC1, this included IM-A (budding/infiltrative), IM-B (mucinous), and IM-C (pushing) margins, with similar notation used in other cases. Tertiary lymphoid structures were defined in each case by identifying aggregates of lymphoid cells on H&E and correlating with CD20, CD4, and CD8 immunofluorescence (CyCIF) to identify discrete aggregates of B cells with adjacent or intermixed T-cell populations, including both immature/early TLS without histologic evidence of well-formed germinal centers, and more mature TLS with germinal center formation 78.
Pathologist-annotated budding cells and Delaunay cluster-sizes of cytokeratin+ cells
Using ITBCC criteria, a trained pathologist annotated budding regions in CRC1 (n = 25) and CRC2–17 (n = 16) from both CyCIF and H&E images. These selected ROIs were used in the data analysis, and CK+ cells in these areas were labelled as “budding tumor cells.” In cluster size analyses, a neighborhood graph was constructed for all segmented cell centroids using Delaunay triangulation, removing edges whose lengths were greater than 20 μm. Then, the CK+ neighborhood graph was defined as the subgraph restricted to the CK+ cells (i.e., removing all nodes and edges connected to CK− cells). The cluster size of each CK+ cell was defined as the number of nodes in its connected component of the subgraph. For quantification of marker expression dependence on cluster-size, cells annotated as normal colon mucosa (ROI1) were removed from the CK+ subgraph. In the 25 CRC1 Z-sections, cells in the upper-left corner of the image (1 cm x 1 cm) were also removed; this region contained CK+ cells of reactive, benign, and mesothelial origin, as opposed to tumor cells of interest.
Biased downsampling based on cluster-size for t-SNE visualization
By definition, most tumor cells have a large cluster-size. Therefore, to visualize the cluster-size dependence of marker expression with t-SNE, we downsampled cells in Figure 4G by stochastically rejecting cells at frequency , for cluster-size . The power of 4 was chosen empirically to balance the representation of various cluster sizes. Final t-SNE plots were made by further subsampling 1,000 cells from each section uniformly. The t-SNE plots in Figure 4G were computed using the following markers: Na-K ATPase, Ki-67, cytokeratin, PDL1, E-cadherin, vimentin, CDX2, lamin ABC, desmin, and PCNA.
kNN-classification of epithelial cell morphologies trained on pathologist annotations
To develop a kNN classifier for pathologist-annotated regions of interest (ROIs), epithelial cells were defined by gating using a univariate, 2-component Gaussian Mixture Model on the relevant marker (cytokeratin, cytokeratin 19, cytokeratin 18, or E-cadherin) in each section. A kNN-classifier was trained on the annotated, epithelial cells using CyCIF marker expression as predictors, and annotated ROI labels as responses. Markers that exhibited unexpected optical artefacts or significant tissue loss were not used (see below for specific markers that were excluded). Learning and prediction were performed using MATLAB’s fitchnn() and functions, with k = 40 neighbors. The prior probability of each label was set as uniform. In each section, there were at least 2,000 annotated cells for each label. Annotated cells were split 50/50 into training and validation sets. Posterior probability colors in Figure S3C (panels in right column) were visualized based on its vector of classification posterior probabilities (, , ,), for 1: normal, 2: glandular classes, 3: solid, and 4: mucinous. The RGB-values of each cell were then defined as:
to capture the relative weight of each class.
For the sections in the primary CRC1 dataset (e.g., section 044), the following markers were used as predictors: Na-K ATPase, Ki-67, keratin, PDL1, E-cadherin, vimentin, CDX2, lamin, desmin, PCNA, autofluorescence; see paragraph below for further details on included and excluded markers. For CRC1 section 046, which was stained with an extended antibody panel, the following markers were used as predictors: cyclin B1, cytokeratin 20, cytokeratin 18, NUP98, cytokeratin 8, PDL1, acetyl-tubulin, p62, pan-cytokeratin, lamin A/C, tubulin. For sections CRC1 sections 045 and 047, which were also stained with different extended antibody panels, we used all artefact-free markers (totaling 29 and 36 respectively). For CRC2–17, the entire antibody panel was used.
In the primary dataset, for kNN classification we excluded Hoechst, CD3, CD4, CD20, CD163, CD45, CD68, FOXP3, CD45RO, α-SMA, PD1, CD8a, CD31, collagen, and autofluorescence as being irrelevant to tumor-intrinsic feature expression. The Ki-67 (D3B5) Rabbit mAb was included because it showed superior staining to another Ki67 antibody (Ki67_570) which was excluded. For CRC1 section 045, we excluded Hoechst and autofluorescence. CK17 was excluded due to staining artefacts. CK14, alternate pERK, Cyclin B1, Perforin, MAP2, GFAP, Cyclin A2, p-mTOR, Cyclin E were excluded due to tissue loss in the final cycles. For CRC1 section 046, we excluded Hoechst, autofluorescence, CD3, CD4, CD57, CD163, IBA1, CD16, CD11c, CD45, CD68, CD11b, CD11a, CD1a, Granzyme B, CD14, PD1, HLA-A, CD8a, and CD31 as irrelevant to tumor extrinsic programs. PAX5, POLR2A, NFATc1, PAX8, and phospho-BTK were excluded due to tissue loss in late cycles. VEGFR2 was excluded due to the presence of staining artefacts. For CRC1 section 047, we excluded Hoechst, autofluorescence, and CD20 as irrelevant to tumor expression. EZH2, phospho-CDK, E2F1, FOXA2 were excluded due to staining artefacts.
Contour plots of epithelial cell marker expression gradients
Contours represent level sets for the average marker expression of the 400 nearest tumor cells, and were computed using the MATLAB contour() function.
3D registration of CRC1 serial sections
All CyCIF sections were registered using a custom script written in MATLAB 2018 (MathWorks). Briefly, each section was first registered using a rigid transformation followed by elastic deformations starting at section 012 and cascading towards the top and bottom sections. For the rigid transformation, an early cycle Hoechst signal with minimal artefacts from each section was selected. All channels were padded by an equivalent of 1,600 pixels along all borders when registering at full resolution. Rigid transformation required consistent landmarks across all sections. Therefore, we identified two such features: the edge of the mucosa section and a point where it transitions into the stromal region. This region was annotated on several downsampled sections, providing training data for a UNet model to estimate fuzzy locations of the transition point and the mucosal edge. Starting from section 012 and taking the centroid of each fuzzy estimate as that section’s transition point, all 25 sections were aligned by translation. Each section was then rotated around the transition point until the fuzzy estimates for the edge of the mucosa region overlapped maximally between sections. For subsequent elastic deformation, we manually selected between 25–35 control points across each section. Most control points were located near the site of budding cells. Then, using local weighted means with these control points via the fitgeotrans() MATLAB function, we applied a deformation starting from section 012 towards section 001 and 025. Finally, we applied Demon’s algorithm to refine registration further. Images were downsampled by a factor of 0.25 and histogram matched, before applying the imregdemons() MATLAB function with an accumulated field smoothing of 1.5 and downsampling with 7 pyramid levels. Demon’s algorithm was applied starting from section 12.
3D visualization of registered CRC1 serial sections
Using Imaris, images were Gaussian-blurred, and an intensity threshold was applied to define regions (e.g., CK+). Connectivity of buds or mucin pools were defined on blurred, thresholded voxels.
Virtual TMA cores and fold-change in effective sample size N/Neff
Virtual TMAs (vTMA) were constructed from whole-slide sections by randomly selecting a central cell and including all cells within 500 μm of the central cell’s centroid as one core. For each vTMA core, a matching, uniform random sample was generated from the whole-slide section with an equal number of cells. The standard-errors of the mean from vTMA (i.e., regional) sampling () or random sampling () were estimated from the means of 1,000 cores and their matched, random samples. The effective sample size N/Neff was defined as the square of the standard-errors’ ratios:
Spatial correlation functions and predicting standard-error of regional sampling
For each sample (whole-slide, virtual TMA core, or real TMA core), spatial correlation functions () were calculated for a pair of variables , and a nearest-neighbor index . Specifically, was given by the Pearson correlation between cells’ -values and their – nearest neighbors’ -values. Each index was associated to the average, inter-cell-centroid distance of all – nearest neighbors in a sample. Correlations were computed up to . Each was fit to an exponential exp for parameters , , over the range of to avoid spurious correlations between adjacent cells that may arise from image segmentation errors. Correlation strength was defined as 𝑐0, and length scale . Fits were performed with the fit() MATLAB function with default options. We subsequently estimated the standard-error of the mean of a variable for a regional sample of correlated cells as follows. First, we computed the matrix of inter-cellular distances , and then computed the correlation matrix between cells using the fit of the spatial correlation function CAA(). By the Central Limit Theorem for weakly-dependent variables 79, we expect the standard- error of the mean for N samples to be , for the sum of all entries in .
Scaling analysis of fold-change in effective sample size N/Neff
For a variable with variance , the fold-change N/Neff is defined as:
The final term can be interpreted as the sum of correlations between an average cell and all other cells in the sample region . Choosing a coordinate system with an average cell at the origin, we approximate the sum as an integral:
Where is the density of cells, and is the spatial dimension of the regional sample. If we assume a uniform density for a cell length scale , and change variables in the integral to eliminate the length scale , we have:
which gives us a scaling relation with which we can roughly estimate N/Neff from parameters:
Variance between patient TMAs due to sampling error and an optimal score
For any given cell-type’s %-composition, we computed the variance of estimates from the whole-slide tumor regions of each patient, , and the variance of estimates from TMA cores,. We considered to be the biological variance of , and remaining variance to be residual error from sampling,. Percent of variance explained by sampling was given by . For the hypothetical scenario of averaging 4 cores, would be 4-fold lower, and percent variance explained was given by . Outliers in each distribution, as indicated in each boxplot, were excluded from the variance calculations.
Immune profiling, LDA analysis, and PDL1:PD1 interaction
For CRC1–17 whole-slide sections stained with the immune panel, multiparameter single-cell intensity data was used to generate binary gates (for 30 of 33 markers). LDA analysis for spatial topic analysis was performed using MATLAB “fitlda” function. In brief, the single-cell data of each sample was split into 200 microns x 200 microns grids, and the positive frequency for each marker was calculated for each grid. The pooled frequencies of all samples were used to train the final LDA model, and 16 topics were isolated. To determine PDL1:PD1 interactions in single-cell data, the cell neighbors within 20 microns were identified with a k-nearest searching algorithm. The PDL1+ cells with PD1+ cells in proximity were labeled as “PD1+ interactors.” The marker expression of PD1+ interactors and other PDL1+ cells were compared as described. In Figure 7F (top panel), number PDL1+ cells with indicated subsets (any, CK+, CD68+, and CD11c+) were divided by the total cell number in the given subset. In Figures 7I and 7J, the positive ratios were calculated by the positive cell number of indicated markers (CK+, CD45+, HLA-A+, and CD44+) normalized with the PDL1+ cells in either interacting or non-interacting groups.
scRNA-seq data analysis
Following sample demultiplexing from the sequencer, reads were filtered, sorted by their barcode of origin, and aligned to the reference transcriptome to generate a counts matrix using the DropEst pipeline.80 Barcodes containing cells were identified using dropkick.81 Batches were combined and consensus nonnegative matrix factorization (cNMF82) was performed to identify metagenes in the resulting cell matrix, assigning “usage” scores for each factor to all cells. The factors or metagenes contain gene loadings that rank detected genes by their contribution to each factor, which are shown on UMAP embeddings in descending order. CytoTRACE 83 was also run using the web portal at https://cytotrace.stanford.edu/ to calculate “stemness” or cellular plasticity scores based on genetic diversity. Leiden clustering84 and PAGA85 graph construction was performed on principal component analysis of the normalized and arcsinh-transformed raw counts matrix (PMID: 32375029, PMID: 33982010). A two-dimensional UMAP86 embedding was then generated using SCANPY87 based on principal component analysis and initial cluster positions determined by PAGA.
GeoMx RNA spatial transcriptomics
We used the GeoMx® Cancer Transcriptome Atlas (CTA) to profile RNA expression levels of ~1,800 genes from 32 selected regions (Figure S1A) from an FFPE tissue section of CRC1 using methods described by the manufacturer (NanoString Technologies, Seattle, WA). Probes were collected separately from CK+ and CK− cells and processed using cDNA library preparation methods. The library was then sent for sequencing with Illumina NovaSeq 6000. QC was performed using vendor-provided software. 31 of the 32 samples passed QC, and these datasets were used for downstream analysis. Probe counts were normalized with the total counts in each condition and used for principal component analysis and hierarchical clustering.
Schematic diagrams
Schematics in Figure 1B, Figure 5F, and Figure 7M were made with BioRender.
Supplementary Material
Figure S1. Overview of dataset and connection between cell-type calling and underlying morphologies. Related to Figure 1. Design of GeoMx experiment. (A) 32 regions were selected from one tissue section of CRC1 (31 passed quality control and analyzed). Representative images of the regions are shown (right panels). Scale bars: 2 mm in left panel, 200 μm in the zoomed in views and 200 μm in the right panels showing representative morphologies. (B) Representative images of main antibody panel from CRC1. (Blue: DNA stain with Hoechst 33342). Scale bars, 100 μm. (C) Cell-types mapped across CRC1, section 097. Cell-type definitions and main classification markers are as indicated. A detailed marker/reference dictionary is presented in Table S4. (D) Variation in composition of each annotated ROI across all sections of CRC1 for the same three main classes of cell-types as listed in Figure S1C (tumor epithelium, stroma, and immune). Boxplot represents 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers and medians are shown as dots. (E) UMAP plot of scRNA-seq data generated from CRC1, and cell-types identified by Leiden clustering (see Methods). (F) Marker-guided sub-clustering was performed as described in Methods. Positive cells are highlighted in yellow.
Figure S2. Impact of TMA sampling error. Related to Figure 2. (A) FOV portraying four different correlation length scales and strengths for CK+, FOXP3+, α-SMA+, and CD163+ cells. Four circles with radii denoting the length scale parameters. (B) Scaling law estimates of N/Neff for CK+, FOXP3+, and α-SMA+ based on the scaling law in Equation 1 (shaded colored boxes represent lcell = 7-13 μm, l from fits) compared to 0.6 mm vTMA cores (colored bars). (C) Correlation of select cell-type pairs amongst 10 nearest neighbors. (D) Correlation functions between select cell-type pairs as estimated from virtual tissue microarrays (vTMAs) or random sampling, overlaid with the correlation functions from four cores (from Figure 2I). (E) Percent variance in real TMA (rTMA) estimates of cell-type abundance that can be attributed to sampling error, after removing outliers. Expected improvement from sampling four cores per tumor is shown in yellow. (F) FOVs of patient sections with low and high Ki-67+ cell abundance. Circles show the length scale of Ki-67+ cells. (G-H) Kaplan-Meier (KM) curves for progression-free survival (patients CRC2-17), calculated from TMAs (left) and whole-slide images (WSI, right). (G) KM curves generated from data stratified with α-SMA+ percentage (cutoff 40%) in each patient sample. (H) KM curves generated from data stratified with mean CD4 expression level (cutoff: 3,500 AFU) in each patient sample. (I) Variation of cell-type composition between sections of a single tumor (CRC1) and sections from different patient tumors (CRC2-17). Section sampling error is typically a minority of the variance between patient sections. (J) Variation of cell-type spatial correlation strengths and length scales across CRC1 Z-sections (blue) and across patients CRC2-17 (red). In most cases, variation within a patient is smaller than that between patients and shows no signs of bias. Boxplots in (C), (I) and (J) represent 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers are labeled as dots or crosses and medians shown as dots or lines.
Figure S3. kNN-classification of epithelial histology. Related to Figure 3. (A) Precision and recall of morphology classifiers trained on CRC1 sections. (B) Normalized Shannon entropy of cells in the CRC sections indicated in Figure 3F. (C) (Left) Shannon entropy of kNN-classification for cells in CRC1. Normal cells from normal colon epithelium (ROI1) have low-entropy, indicating high-confidence classification. Regions used for training were also high confidence, as expected by definition. Most tumor regions were classified as being between classes, i.e., having high entropy. (Right) The relative weight of each class is visualized by hue. (D) Dimensional reduction of subsampled single-cell expression from CK+ cells by t-SNE, with pathologist annotations indicated by color. Each of the four marker panels provide enough information to cluster normal epithelial cells (black) separately from tumor cells, despite limited overlap in markers between panels (indicated by Venn diagram). Different annotations roughly occupy different regions of expression space, indicating that expression and morphology are correlated, but tumor cells largely form a continuous distribution, supporting the existence of mixed morphologies.
Figure S4. Tumor bud characterization. Related to Figure 4. (A) Proportion of pathology annotated budding cells amongst CK+ cells across each of the sections. (B) CyCIF image with location of 3D viewpoints corresponding to Figure 4C and 4D. Arrow represents approximate viewing angle in those figures. (C) Differential expression of markers in cells annotated as tumor buds. The relative expression of indicated markers is represented in the heatmap as the log2 ratio of budding tumor cells to all tumor cells. (D) Log-intensity of markers and their dependency on cluster-size in CRC1 tumor cells across all 25 sections (as in Figure 4H). Expression of annotated buds shown in green for reference. Box plots show 1st-3rd quartiles; points beyond are not shown. Each box represents ~105-106 cells. (E) Log-intensity of markers and their dependency on cluster-size for tumor cells in CRC2-17 (as in Figure 4I).
Figure S5. 3D TLS structures and clusters. Related to Figure 6. (A) 2D projection of TLS networks (TLSN) across all sections of CRC1 (left panel), and the section-by-section view of TLS networks in nine selected sections (right panels). (B) The number of TLS per 3D TLS network (top) and the number of the total of 25 slides in which a particular TLS network was identified (bottom). (C) TLS domain cluster composition in each section of CRC1. (D) Representative H&E images of TLS domain clusters 1 and 3, the same regions as shown in Figure 6F (serial section shown). (E) 3D view of TLSN-B, colored by principal component 2 (PC2) (PC1 shown in Figure 6H). (F) t-SNE plots of all TLS domain clusters from CRC1 (25 sections) and CRC2-17 (16 sections), colored by samples (left panel) or TLS domain clusters (right panel).
Figure S6. LDA analysis of immune composition. Related to Figure 7. (A) 16 LDA topics from CRC1-17, immune panel (33 antibodies). Representative markers are shown in red and black text (the size of label for each marker is proportional to its probability within each of the topics). (B) Two-way hierarchal clustering between LDA topics and pathology annotated regions. The cell/topic counts from all pathologist-annotated ROIs as well as marker-defined ROIs (*) were clustered with full lineage and Euclidean distancing. (C) The fractions of topics in three selected regions of the invasive margin across all samples (a subset of the topics is shown in Figure 7E). Boxplots represent 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers and medians are labeled as dots. (D) Fractions of PD1+ cells in selected populations. The percentage of PD1+ cells in total/any cells or cell groups selected with indicated markers were plotted sample by sample (CRC1-17). (E) Fractions of PDL1+ cells in selected populations. The percentage of PDL1+ cells in total/any cells or cell groups selected with indicated markers were plotted per CRC sample. (F) The fractions of PDL1+ cells in myeloid subsets. Box plot showing the percentage of PDL1+ cells in CD68+CD11c-, CD68-CD11c+, and CD68+CD11c+ per sample (boxes indicate 1st-3rd quartiles and whiskers extend at most to 1.5x interquartile range; red lines indicate median). (G) Relative frequency of PDL1+ myeloid subsets in each sample. The numbers of CD68+CD11c-, CD68-CD11c+ and CD68+CD11c+ cells in PDL1+ population were calculated, and the relative abundancy (divided by sum) of each subset is shown.
Figure S7. Cell composition in CRC2-17. Related to Figure 7. (A) t-SNE plots based on CyCIF data for specimens CRC1-17 (excluding data from the DNA staining). Cell types are shown at the bottom of the figure. Tumor/epithelium (T/E), stroma (S) and immune (I) populations are outlined in black. The t-SNE plot for CRC1 is reproduced from Figure 1E for reference. $$PARABREAKHERE$$(B) t-SNE of CRC1-17 labelled by specimen identity with labelling by general cell type in upper right. $$PARABREAKHERE$$(C) Cell-type composition for CRC1-17 shown as stacked bar graphs with the same color code as in panel A.
Video S1: CRC1 3D view of budding structures, related to Figure 4.
Video S2: CRC1 3D view of TLS/SLO structures, related to Figure 6.
Table S1. Clinical information for colorectal cancer cohort, related to Figure 1 and STAR Methods. Demographic and diagnostic information for all patient-derived specimens in this study. CRC1 was analyzed in 3D and CRC2-17 in whole-slide 2D and TMA 2D. Other specimens (CRC18-93) were imaged as TMA cores, as described in the text and Figure 1.
Table S3. Antibody panels, related to Figure 1, 3, 4, 7 and STAR Methods. Table includes six (overlapping) antibody panels used throughout the study: tab 1) The main CyCIF panel includes antibodies used to stain all CRC1 sections (except 044-046), CRC2-17 sections, and TMAs (see Figure 1 for graphical overview); tab 2) three additional panels for CRC1 sections 45, 46, and 47, respectively; tab 3) an immune-focused panel used for whole-slide images (WSI) of CRC2-17; tab 4) a tumor-focused antibody panel used for WSI of CRC2-17. CST refers to Cell Signaling Technologies (Beverley MA USA); RRID refers to the Research Resource Identifier available at https://scicrunch.org/resources.
Table S4. Cell-type dictionary, related to Figure 1 & S1 and STAR Methods.
Cell-type assignments based on marker intensities. The first tab shows the primary discriminating markers and tabs 2 and 3 show assignments based on all markers in the panel.
Table S5. Cell-type composition for regions of interest in CRC1, related to Figure 6 and STAR Methods.
Cell-type composition for pathologist-defined regions of interest (see Figure 1B) across all sections processed for CyCIF. Cell-type definitions as in Table S4.
Highlights.
Multiplexed analysis shows intermixed tumor morphologies and molecular gradients
Various cancer characteristic cellular features are large, interconnected structures
3D tertiary lymphoid structure (TLS) networks show intra-TLS patterning variation
PD1-PDL1 interactions are primarily between T and myeloid cells in this CRC cohort
ACKNOWLEDGEMENTS
This publication is part of the HTAN (Human Tumor Atlas Network) Consortium paper package. A list of HTAN members is available at humantumoratlas.org/htan-authors. We thank Juliann Tefft, Alyce Chen, Raquel Arias-Camison, Zoltan Maliga, and Jeremy Muhlich. Alan Simmons, Austin Southard-Smith, Qi Liu. This work was supported by NIH grants U54-CA225088 (PKS, SS), U2C-CA233280 (PKS, SS), U2C-CA233262 (PKS, SS), U2C-CA233291 (CNH, KSL), R01-DK103831 (CNH, KSL), NIH training grant T32-GM007748 (SC), P30-CA06516 (for histology), Ludwig Cancer Research, the Gray Foundation, and the David Liposarcoma Research Initiative.
Footnotes
DECLARATION OF INTERESTS
PKS is a member of the BOD of Glencoe Software and Applied Biomath, a member of the SAB for RareCyte, NanoString, and Montai Health and a consultant for Merck. YC consults for RareCyte. Other authors declare no outside interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Coons AH, Creech Hugh J, Jones Norman, and Berliner Ernst (1942). The demonstration of pneumococcal antigen in tissues by the use of fluorescent antibody. J Immunol. 45, 159–170. [Google Scholar]
- 2.Amin MB, Greene FL, Edge SB, Compton CC, Gershenwald JE, Brookland RK, Meyer L, Gress DM, Byrd DR, and Winchester DP (2017). The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA. Cancer J. Clin. 67, 93–99. 10.3322/caac.21388. [DOI] [PubMed] [Google Scholar]
- 3.Rozenblatt-Rosen O, Regev A, Oberdoerffer P, Nawy T, Hupalowska A, Rood JE, Ashenberg O, Cerami E, Coffey RJ, Demir E, et al. (2020). The Human Tumor Atlas Network: Charting Tumor Transitions across Space and Time at Single-Cell Resolution. Cell 181, 236–249. 10.1016/j.cell.2020.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angelo M, Bendall SC, Finck R, Hale MB, Hitzman C, Borowsky AD, Levenson RM, Lowe JB, Liu SD, Zhao S, et al. (2014). Multiplexed ion beam imaging of human breast tumors. Nat. Med. 20, 436–442. 10.1038/nm.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, et al. (2013). Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc. Natl. Acad. Sci. U. S. A. 110, 11982–11987. 10.1073/pnas.1300136110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giesen C, Wang HAO, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, Schüffler PJ, Grolimund D, Buhmann JM, Brandt S, et al. (2014). Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 11, 417–422. 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- 7.Goltsev Y, Samusik N, Kennedy-Darling J, Bhate S, Hale M, Vazquez G, Black S, and Nolan GP (2018). Deep Profiling of Mouse Splenic Architecture with CODEX Multiplexed Imaging. Cell 174, 968-981.e15. 10.1016/j.cell.2018.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin J-R, Izar B, Wang S, Yapp C, Mei S, Shah PM, Santagata S, and Sorger PK (2018). Highly multiplexed immunofluorescence imaging of human tissues and tumors using t-CyCIF and conventional optical microscopes. eLife 7. 10.7554/eLife.31657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saka SK, Wang Y, Kishi JY, Zhu A, Zeng Y, Xie W, Kirli K, Yapp C, Cicconet M, Beliveau BJ, et al. (2019). Immuno-SABER enables highly multiplexed and amplified protein imaging in tissues. Nat. Biotechnol. 37, 1080–1090. 10.1038/s41587-019-0207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schürch CM, Bhate SS, Barlow GL, Phillips DJ, Noti L, Zlobec I, Chu P, Black S, Demeter J, McIlwain DR, et al. (2020). Coordinated Cellular Neighborhoods Orchestrate Antitumoral Immunity at the Colorectal Cancer Invasive Front. Cell 182, 1341–1359.e19. 10.1016/j.cell.2020.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner J, Rapsomaniki MA, Chevrier S, Anzeneder T, Langwieder C, Dykgers A, Rees M, Ramaswamy A, Muenst S, Soysal SD, et al. (2019). A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 177, 1330–1345.e18. 10.1016/j.cell.2019.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burger ML, Cruz AM, Crossland GE, Gaglia G, Ritch CC, Blatt SE, Bhutkar A, Canner D, Kienka T, Tavana SZ, et al. (2021). Antigen dominance hierarchies shape TCF1+ progenitor CD8 T cell phenotypes in tumors. Cell 184, 4996–5014.e26. 10.1016/j.cell.2021.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaglia G, Kabraji S, Rammos D, Dai Y, Verma A, Wang S, Mills CE, Chung M, Bergholz JS, Coy S, et al. (2022). Temporal and spatial topography of cell proliferation in cancer. Nat. Cell Biol. 24, 316–326. 10.1038/s41556-022-00860-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nirmal AJ, Maliga Z, Vallius T, Quattrochi B, Chen AA, Jacobson CA, Pelletier RJ, Yapp C, Arias-Camison R, Chen Y-A, et al. (2022). The spatial landscape of progression and immunoediting in primary melanoma at single cell resolution. Cancer Discov., candisc.1357.2021. 10.1158/2159-8290.CD-211357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendall SC, Simonds EF, Qiu P, Amir ED, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, et al. (2011). Single-Cell Mass Cytometry of Differential Immune and Drug Responses Across a Human Hematopoietic Continuum. Science 332, 687–696. 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luecken MD, and Theis FJ (2019). Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746. 10.15252/msb.20188746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazer BL, Homer RJ, and Rimm DL (2019). False-positive pathology: improving reproducibility with the next generation of pathologists. Lab. Invest. 99, 1260–1265. 10.1038/s41374-019-0257-2. [DOI] [PubMed] [Google Scholar]
- 18.Voskuil J (2015). How difficult is the validation of clinical biomarkers? F1000Research 4, 101. 10.12688/f1000research.6395.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fleming M, Ravula S, Tatishchev SF, and Wang HL (2012). Colorectal carcinoma: Pathologic aspects. J. Gastrointest. Oncol. 3, 153–173. 10.3978/j.issn.2078-6891.2012.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cianchi F, Messerini L, Palomba A, Boddi V, Perigli G, Pucciani F, Bechi P, and Cortesini C (1997). Character of the invasive margin in colorectal cancer: does it improve prognostic information of Dukes staging? Dis. Colon Rectum 40, 1170–1175; discussion 1175–1176. 10.1007/BF02055162. [DOI] [PubMed] [Google Scholar]
- 21.Lugli A, Kirsch R, Ajioka Y, Bosman F, Cathomas G, Dawson H, El Zimaity H, Fléjou J-F, Hansen TP, Hartmann A, et al. (2017). Recommendations for reporting tumor budding in colorectal cancer based on the International Tumor Budding Consensus Conference (ITBCC) 2016. Mod. Pathol. 30, 1299–1311. 10.1038/modpathol.2017.46. [DOI] [PubMed] [Google Scholar]
- 22.Rogers AC, Winter DC, Heeney A, Gibbons D, Lugli A, Puppa G, and Sheahan K (2016). Systematic review and meta-analysis of the impact of tumour budding in colorectal cancer. Br. J. Cancer 115, 831–840. 10.1038/bjc.2016.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bruni D, Angell HK, and Galon J (2020). The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat. Rev. Cancer 20, 662–680. 10.1038/s41568-020-0285-7. [DOI] [PubMed] [Google Scholar]
- 24.Di Caro G, Bergomas F, Grizzi F, Doni A, Bianchi P, Malesci A, Laghi L, Allavena P, Mantovani A, and Marchesi F (2014). Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 20, 2147–2158. 10.1158/1078-0432.CCR-13-2590. [DOI] [PubMed] [Google Scholar]
- 25.Aponte PM, and Caicedo A (2017). Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017. 10.1155/2017/5619472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kalluri R, and Weinberg RA (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest. 119, 1420–1428. 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Uhler C, and Shivashankar GV (2018). Nuclear Mechanopathology and Cancer Diagnosis. Trends Cancer 4, 320–331. 10.1016/j.trecan.2018.02.009. [DOI] [PubMed] [Google Scholar]
- 28.Centeno I, Paasinen Sohns A, Flury M, Galván JA, Zahnd S, Koelzer VH, Sokol L, Dawson HE, Lugli A, Cathomas G, et al. (2017). DNA profiling of tumor buds in colorectal cancer indicates that they have the same mutation profile as the tumor from which they derive. Virchows Arch. Int. J. Pathol. 470, 341–346. 10.1007/s00428-017-2071-9. [DOI] [PubMed] [Google Scholar]
- 29.Zollinger DR, Lingle SE, Sorg K, Beechem JM, and Merritt CR (2020). GeoMx™ RNA Assay: High Multiplex, Digital, Spatial Analysis of RNA in FFPE Tissue. Methods Mol. Biol. Clifton NJ 2148, 331–345. 10.1007/978-1-0716-0623-0_21. [DOI] [PubMed] [Google Scholar]
- 30.Weiser MR (2018). AJCC 8th Edition: Colorectal Cancer. Ann. Surg. Oncol. 25, 1454–1455. 10.1245/s10434-018-6462-1. [DOI] [PubMed] [Google Scholar]
- 31.Schapiro D, Sokolov A, Yapp C, Chen Y-A, Muhlich JL, Hess J, Creason AL, Nirmal AJ, Baker GJ, Nariya MK, et al. (2022). MCMICRO: a scalable, modular image-processing pipeline for multiplexed tissue imaging. Nat. Methods 19, 311–315. 10.1038/s41592-021-01308-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muhlich JL, Chen Y-A, Yapp C, Russell D, Santagata S, and Sorger PK (2022). Stitching and registering highly multiplexed whole slide images of tissues and tumors using ASHLAR. Bioinforma. Oxf. Engl, btac544. 10.1093/bioinformatics/btac544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoffer J, Rashid R, Muhlich JL, Chen Y-A, Russell DPW, Ruokonen J, Krueger R, Pfister H, Santagata S, and Sorger PK (2020). Minerva: a light-weight, narrative image browser for multiplexed tissue images. J. Open Source Softw. 5, 2579. 10.21105/joss.02579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rashid R, Chen Y-A, Hoffer J, Muhlich JL, Lin J-R, Krueger R, Pfister H, Mitchell R, Santagata S, and Sorger PK (2022). Narrative online guides for the interpretation of digital-pathology images and tissue-atlas data. Nat. Biomed. Eng. 6, 515–526. 10.1038/s41551-021-00789-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen B, Scurrah CR, McKinley ET, Simmons AJ, Ramirez-Solano MA, Zhu X, Markham NO, Heiser CN, Vega PN, Rolong A, et al. (2021). Differential pre-malignant programs and microenvironment chart distinct paths to malignancy in human colorectal polyps. Cell 184, 6262–6280.e26. 10.1016/j.cell.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rajaram S, Heinrich LE, Gordan JD, Avva J, Bonness KM, Witkiewicz AK, Malter JS, Atreya CE, Warren RS, Wu LF, et al. (2017). Sampling strategies to capture single-cell heterogeneity. Nat. Methods 14, 967–970. 10.1038/nmeth.4427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lavrakas PJ (2008). Encyclopedia of Survey Research Methods (SAGE Publications; ). [Google Scholar]
- 38.Zhao W, Xu Y, Wang Y, Gao D, King J, Xu Y, and Liang F-S (2021). Investigating crosstalk between H3K27 acetylation and H3K4 trimethylation in CRISPR/dCas-based epigenome editing and gene activation. Sci. Rep. 11, 15912. 10.1038/s41598-021-95398-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oudin MJ, and Weaver VM (2016). Physical and Chemical Gradients in the Tumor Microenvironment Regulate Tumor Cell Invasion, Migration, and Metastasis. Cold Spring Harb. Symp. Quant. Biol. 81, 189–205. 10.1101/sqb.2016.81.030817. [DOI] [PubMed] [Google Scholar]
- 40.Lugli A, Vlajnic T, Giger O, Karamitopoulou E, Patsouris ES, Peros G, Terracciano LM, and Zlobec I (2011). Intratumoral budding as a potential parameter of tumor progression in mismatch repair-proficient and mismatch repair-deficient colorectal cancer patients. Hum. Pathol. 42, 1833–1840. 10.1016/j.humpath.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 41.Bronsert P, Enderle-Ammour K, Bader M, Timme S, Kuehs M, Csanadi A, Kayser G, Kohler I, Bausch D, Hoeppner J, et al. (2014). Cancer cell invasion and EMT marker expression: a three-dimensional study of the human cancer-host interface: 3D cancer-host interface. J. Pathol. 234, 410–422. 10.1002/path.4416. [DOI] [PubMed] [Google Scholar]
- 42.Delaunay BN (1934). Sur la sphère vide. Bull. Académie Sci. URSS VII Sér. 1934, 793–800. [Google Scholar]
- 43.Gosens MJEM, van Kempen LCL, van de Velde CJH, van Krieken JHJM, and Nagtegaal ID (2007). Loss of membranous Ep-CAM in budding colorectal carcinoma cells. Mod. Pathol. Off. J. U. S. Can. Acad. Pathol. Inc 20, 221–232. 10.1038/modpathol.3800733. [DOI] [PubMed] [Google Scholar]
- 44.Rubio CA (2007). Further studies on the arrest of cell proliferation in tumor cells at the invading front of colonic adenocarcinoma. J. Gastroenterol. Hepatol. 22, 1877–1881. 10.1111/j.1440-1746.2007.04839.x. [DOI] [PubMed] [Google Scholar]
- 45.Rubio CA (2008). Arrest of cell proliferation in budding tumor cells ahead of the invading edge of colonic carcinomas. A preliminary report. Anticancer Res. 28, 2417–2420. [PubMed] [Google Scholar]
- 46.Sung CO, Seo JW, Kim K-M, Do I-G, Kim SW, and Park C-K (2008). Clinical significance of signet-ring cells in colorectal mucinous adenocarcinoma. Mod. Pathol. 21, 1533–1541. 10.1038/modpathol.2008.170. [DOI] [PubMed] [Google Scholar]
- 47.Bresalier RS (2002). Intestinal mucin and colorectal cancer: It’s not just goo. Gastroenterology 123, 648–649. 10.1053/gast.2002.1230648. [DOI] [PubMed] [Google Scholar]
- 48.Mani SA, Guo W, Liao M-J, Eaton E.Ng., Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schumacher TN, and Thommen DS (2022). Tertiary lymphoid structures in cancer. Science 375, eabf9419. 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- 50.Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, Johansson I, Phung B, Harbst K, Vallon-Christersson J, et al. (2020). Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565. 10.1038/s41586-019-1914-8. [DOI] [PubMed] [Google Scholar]
- 51.Helmink BA, Reddy SM, Gao J, Zhang S, Basar R, Thakur R, Yizhak K, Sade-Feldman M, Blando J, Han G, et al. (2020). B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555. 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Paijens ST, Vledder A, de Bruyn M, and Nijman HW (2021). Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 18, 842–859. 10.1038/s41423-020-00565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhatia R, Gautam SK, Cannon A, Thompson C, Hall BR, Aithal A, Banerjee K, Jain M, Solheim JC, Kumar S, et al. (2019). Cancer-associated mucins: role in immune modulation and metastasis. Cancer Metastasis Rev. 38, 223–236. 10.1007/s10555-018-09775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blei DM, Ng AY, and Jordan MI (2003). Latent dirichlet allocation. J. Mach. Learn. Res. 3, 993–1022. [Google Scholar]
- 55.Jackson HW, Fischer JR, Zanotelli VRT, Ali HR, Mechera R, Soysal SD, Moch H, Muenst S, Varga Z, Weber WP, et al. (2020). The single-cell pathology landscape of breast cancer. Nature 578, 615–620. 10.1038/s41586-019-1876-x. [DOI] [PubMed] [Google Scholar]
- 56.Valle D, Baiser B, Woodall CW, and Chazdon R (2014). Decomposing biodiversity data using the Latent Dirichlet Allocation model, a probabilistic multivariate statistical method. Ecol. Lett. 17, 1591–1601. 10.1111/ele.12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krishnan H, Rayes J, Miyashita T, Ishii G, Retzbach EP, Sheehan SA, Takemoto A, Chang Y, Yoneda K, Asai J, et al. (2018). Podoplanin: An emerging cancer biomarker and therapeutic target. Cancer Sci. 109, 1292–1299. 10.1111/cas.13580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.André T, Cohen R, and Salem ME (2022). Immune Checkpoint Blockade Therapy in Patients With Colorectal Cancer Harboring Microsatellite Instability/Mismatch Repair Deficiency in 2022. Am. Soc. Clin. Oncol. Educ. Book, 233–241. 10.1200/EDBK_349557. [DOI] [PubMed] [Google Scholar]
- 59.Boland CR, and Goel A (2010). Microsatellite Instability in Colorectal Cancer. Gastroenterology 138, 2073–2087.e3. 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Senbanjo LT, and Chellaiah MA (2017). CD44: A Multifunctional Cell Surface Adhesion Receptor Is a Regulator of Progression and Metastasis of Cancer Cells. Front. Cell Dev. Biol. 5, 18. 10.3389/fcell.2017.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marusyk A, Almendro V, and Polyak K (2012). Intra-tumour heterogeneity: a looking glass for cancer? Nat. Rev. Cancer 12, 323–334. 10.1038/nrc3261. [DOI] [PubMed] [Google Scholar]
- 62.Turing AM (1952). The Chemical Basis of Morphogenesis. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 237, 37–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kondo H, Ratcliffe CDH, Hooper S, Ellis J, MacRae JI, Hennequart M, Dunsby CW, Anderson KI, and Sahai E (2021). Single-cell resolved imaging reveals intra-tumor heterogeneity in glycolysis, transitions between metabolic states, and their regulatory mechanisms. Cell Rep. 34, 108750. 10.1016/j.celrep.2021.108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Randall EC, Lopez BGC, Peng S, Regan MS, Abdelmoula WM, Basu SS, Santagata S, Yoon H, Haigis MC, Agar JN, et al. (2020). Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 80, 1258–1267. 10.1158/0008-5472.CAN-19-0638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Edin S, Kaprio T, Hagström J, Larsson P, Mustonen H, Böckelman C, Strigård K, Gunnarsson U, Haglund C, and Palmqvist R (2019). The Prognostic Importance of CD20 + B lymphocytes in Colorectal Cancer and the Relation to Other Immune Cell subsets. Sci. Rep. 9, 19997. 10.1038/s41598-019-56441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rogers KW, and Schier AF (2011). Morphogen gradients: from generation to interpretation. Annu. Rev. Cell Dev. Biol. 27, 377–407. 10.1146/annurev-cellbio-092910-154148. [DOI] [PubMed] [Google Scholar]
- 67.Black JRM, and McGranahan N (2021). Genetic and non-genetic clonal diversity in cancer evolution. Nat. Rev. Cancer. 10.1038/s41568-021-00336-2. [DOI] [PubMed] [Google Scholar]
- 68.Sharma A, Merritt E, Hu X, Cruz A, Jiang C, Sarkodie H, Zhou Z, Malhotra J, Riedlinger GM, and De S (2019). Non-Genetic Intra-Tumor Heterogeneity Is a Major Predictor of Phenotypic Heterogeneity and Ongoing Evolutionary Dynamics in Lung Tumors. Cell Rep. 29, 2164–2174.e5. 10.1016/j.celrep.2019.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghaznavi F, Evans A, Madabhushi A, and Feldman M (2013). Digital imaging in pathology: whole-slide imaging and beyond. Annu. Rev. Pathol. 8, 331–359. 10.1146/annurev-pathol-011811-120902. [DOI] [PubMed] [Google Scholar]
- 70.Aeffner F, Zarella MD, Buchbinder N, Bui MM, Goodman MR, Hartman DJ, Lujan GM, Molani MA, Parwani AV, Lillard K, et al. (2019). Introduction to Digital Image Analysis in Whole-slide Imaging: A White Paper from the Digital Pathology Association. J. Pathol. Inform. 10, 9. 10.4103/jpi.jpi_82_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Health, C. for D. and R. (2019). Technical Performance Assessment of Digital Pathology Whole Slide Imaging Devices. US Food Drug Adm. http://www.fda.gov/regulatory-information/search-fda-guidance-documents/technical-performance-assessment-digital-pathology-whole-slide-imaging-devices. [Google Scholar]
- 72.Koelzer VHM, and Lugli AM (2014). The Tumor Border Configuration of Colorectal Cancer as a Histomorphological Prognostic Indicator. Front. Oncol. 4. 10.3389/fonc.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Oh SA, Wu D-C, Cheung J, Navarro A, Xiong H, Cubas R, Totpal K, Chiu H, Wu Y, CompsAgrar L, et al. (2020). PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 1, 681–691. 10.1038/s43018-020-0075-x. [DOI] [PubMed] [Google Scholar]
- 74.Banerjee A, Herring CA, Chen B, Kim H, Simmons AJ, Southard-Smith AN, Allaman MM, White JR, Macedonia MC, Mckinley ET, et al. (2020). Succinate Produced by Intestinal Microbes Promotes Specification of Tuft Cells to Suppress Ileal Inflammation. Gastroenterology 159, 2101–2115.e5. 10.1053/j.gastro.2020.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Southard-Smith AN, Simmons AJ, Chen B, Jones AL, Ramirez Solano MA, Vega PN, Scurrah CR, Zhao Y, Brenan MJ, Xuan J, et al. (2020). Dual indexed library design enables compatibility of inDrop single-cell RNA-sequencing with exAMP chemistry sequencing platforms. BMC Genomics 21, 456. 10.1186/s12864-020-06843-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yapp C, Novikov E, Jang W-D, Vallius T, Chen Y-A, Cicconet M, Maliga Z, Jacobson CA, Wei D, Santagata S, et al. (2022). UnMICST: Deep learning with real augmentation for robust segmentation of highly multiplexed images of human tissues. 2021.04.02.438285. 10.1101/2021.04.02.438285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baker GJ, Muhlich JL, Palaniappan SK, Moore JK, Davis SH, Santagata S, and Sorger PK (2020). SYLARAS: A Platform for the Statistical Analysis and Visual Display of Systemic Immunoprofiling Data and Its Application to Glioblastoma. Cell Syst. 11, 272–285.e9. 10.1016/j.cels.2020.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fridman WH, Meylan M, Petitprez F, Sun C-M, Italiano A, and Sautès-Fridman C (2022). B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 19, 441–457. 10.1038/s41571-022-00619-z. [DOI] [PubMed] [Google Scholar]
- 79.Ibragimov IA (1962). Some Limit Theorems for Stationary Processes. Theory Probab. Its Appl. 7, 349–382. 10.1137/1107036. [DOI] [Google Scholar]
- 80.Petukhov V, Guo J, Baryawno N, Severe N, Scadden DT, Samsonova MG, and Kharchenko PV (2018). dropEst: pipeline for accurate estimation of molecular counts in droplet-based single-cell RNA-seq experiments. Genome Biol. 19, 78. 10.1186/s13059-018-1449-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Heiser CN, Wang VM, Chen B, Hughey JJ, and Lau KS (2020). Automated quality control and cell identification of droplet-based single-cell data using dropkick. bioRxiv, 2020.10.08.332288. 10.1101/2020.10.08.332288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kotliar D, Veres A, Nagy MA, Tabrizi S, Hodis E, Melton DA, and Sabeti PC (2019). Identifying gene expression programs of cell-type identity and cellular activity with single-cell RNA-Seq. eLife 8, e43803. 10.7554/eLife.43803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, Ilagan F, Kuo AH, Hsieh RW, Cai S, et al. (2020). Single-cell transcriptional diversity is a hallmark of developmental potential. Science 367, 405–411. 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, and Theis FJ (2019). PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59. 10.1186/s13059-019-1663-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McInnes L, Healy J, and Melville J (2020). UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. ArXiv180203426 Cs Stat. [Google Scholar]
- 87.Wolf FA, Angerer P, and Theis FJ (2018). SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15. 10.1186/s13059-017-1382-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Overview of dataset and connection between cell-type calling and underlying morphologies. Related to Figure 1. Design of GeoMx experiment. (A) 32 regions were selected from one tissue section of CRC1 (31 passed quality control and analyzed). Representative images of the regions are shown (right panels). Scale bars: 2 mm in left panel, 200 μm in the zoomed in views and 200 μm in the right panels showing representative morphologies. (B) Representative images of main antibody panel from CRC1. (Blue: DNA stain with Hoechst 33342). Scale bars, 100 μm. (C) Cell-types mapped across CRC1, section 097. Cell-type definitions and main classification markers are as indicated. A detailed marker/reference dictionary is presented in Table S4. (D) Variation in composition of each annotated ROI across all sections of CRC1 for the same three main classes of cell-types as listed in Figure S1C (tumor epithelium, stroma, and immune). Boxplot represents 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers and medians are shown as dots. (E) UMAP plot of scRNA-seq data generated from CRC1, and cell-types identified by Leiden clustering (see Methods). (F) Marker-guided sub-clustering was performed as described in Methods. Positive cells are highlighted in yellow.
Figure S2. Impact of TMA sampling error. Related to Figure 2. (A) FOV portraying four different correlation length scales and strengths for CK+, FOXP3+, α-SMA+, and CD163+ cells. Four circles with radii denoting the length scale parameters. (B) Scaling law estimates of N/Neff for CK+, FOXP3+, and α-SMA+ based on the scaling law in Equation 1 (shaded colored boxes represent lcell = 7-13 μm, l from fits) compared to 0.6 mm vTMA cores (colored bars). (C) Correlation of select cell-type pairs amongst 10 nearest neighbors. (D) Correlation functions between select cell-type pairs as estimated from virtual tissue microarrays (vTMAs) or random sampling, overlaid with the correlation functions from four cores (from Figure 2I). (E) Percent variance in real TMA (rTMA) estimates of cell-type abundance that can be attributed to sampling error, after removing outliers. Expected improvement from sampling four cores per tumor is shown in yellow. (F) FOVs of patient sections with low and high Ki-67+ cell abundance. Circles show the length scale of Ki-67+ cells. (G-H) Kaplan-Meier (KM) curves for progression-free survival (patients CRC2-17), calculated from TMAs (left) and whole-slide images (WSI, right). (G) KM curves generated from data stratified with α-SMA+ percentage (cutoff 40%) in each patient sample. (H) KM curves generated from data stratified with mean CD4 expression level (cutoff: 3,500 AFU) in each patient sample. (I) Variation of cell-type composition between sections of a single tumor (CRC1) and sections from different patient tumors (CRC2-17). Section sampling error is typically a minority of the variance between patient sections. (J) Variation of cell-type spatial correlation strengths and length scales across CRC1 Z-sections (blue) and across patients CRC2-17 (red). In most cases, variation within a patient is smaller than that between patients and shows no signs of bias. Boxplots in (C), (I) and (J) represent 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers are labeled as dots or crosses and medians shown as dots or lines.
Figure S3. kNN-classification of epithelial histology. Related to Figure 3. (A) Precision and recall of morphology classifiers trained on CRC1 sections. (B) Normalized Shannon entropy of cells in the CRC sections indicated in Figure 3F. (C) (Left) Shannon entropy of kNN-classification for cells in CRC1. Normal cells from normal colon epithelium (ROI1) have low-entropy, indicating high-confidence classification. Regions used for training were also high confidence, as expected by definition. Most tumor regions were classified as being between classes, i.e., having high entropy. (Right) The relative weight of each class is visualized by hue. (D) Dimensional reduction of subsampled single-cell expression from CK+ cells by t-SNE, with pathologist annotations indicated by color. Each of the four marker panels provide enough information to cluster normal epithelial cells (black) separately from tumor cells, despite limited overlap in markers between panels (indicated by Venn diagram). Different annotations roughly occupy different regions of expression space, indicating that expression and morphology are correlated, but tumor cells largely form a continuous distribution, supporting the existence of mixed morphologies.
Figure S4. Tumor bud characterization. Related to Figure 4. (A) Proportion of pathology annotated budding cells amongst CK+ cells across each of the sections. (B) CyCIF image with location of 3D viewpoints corresponding to Figure 4C and 4D. Arrow represents approximate viewing angle in those figures. (C) Differential expression of markers in cells annotated as tumor buds. The relative expression of indicated markers is represented in the heatmap as the log2 ratio of budding tumor cells to all tumor cells. (D) Log-intensity of markers and their dependency on cluster-size in CRC1 tumor cells across all 25 sections (as in Figure 4H). Expression of annotated buds shown in green for reference. Box plots show 1st-3rd quartiles; points beyond are not shown. Each box represents ~105-106 cells. (E) Log-intensity of markers and their dependency on cluster-size for tumor cells in CRC2-17 (as in Figure 4I).
Figure S5. 3D TLS structures and clusters. Related to Figure 6. (A) 2D projection of TLS networks (TLSN) across all sections of CRC1 (left panel), and the section-by-section view of TLS networks in nine selected sections (right panels). (B) The number of TLS per 3D TLS network (top) and the number of the total of 25 slides in which a particular TLS network was identified (bottom). (C) TLS domain cluster composition in each section of CRC1. (D) Representative H&E images of TLS domain clusters 1 and 3, the same regions as shown in Figure 6F (serial section shown). (E) 3D view of TLSN-B, colored by principal component 2 (PC2) (PC1 shown in Figure 6H). (F) t-SNE plots of all TLS domain clusters from CRC1 (25 sections) and CRC2-17 (16 sections), colored by samples (left panel) or TLS domain clusters (right panel).
Figure S6. LDA analysis of immune composition. Related to Figure 7. (A) 16 LDA topics from CRC1-17, immune panel (33 antibodies). Representative markers are shown in red and black text (the size of label for each marker is proportional to its probability within each of the topics). (B) Two-way hierarchal clustering between LDA topics and pathology annotated regions. The cell/topic counts from all pathologist-annotated ROIs as well as marker-defined ROIs (*) were clustered with full lineage and Euclidean distancing. (C) The fractions of topics in three selected regions of the invasive margin across all samples (a subset of the topics is shown in Figure 7E). Boxplots represent 25%−75% and whiskers extend at most to 1.5x interquartile range. Outliers and medians are labeled as dots. (D) Fractions of PD1+ cells in selected populations. The percentage of PD1+ cells in total/any cells or cell groups selected with indicated markers were plotted sample by sample (CRC1-17). (E) Fractions of PDL1+ cells in selected populations. The percentage of PDL1+ cells in total/any cells or cell groups selected with indicated markers were plotted per CRC sample. (F) The fractions of PDL1+ cells in myeloid subsets. Box plot showing the percentage of PDL1+ cells in CD68+CD11c-, CD68-CD11c+, and CD68+CD11c+ per sample (boxes indicate 1st-3rd quartiles and whiskers extend at most to 1.5x interquartile range; red lines indicate median). (G) Relative frequency of PDL1+ myeloid subsets in each sample. The numbers of CD68+CD11c-, CD68-CD11c+ and CD68+CD11c+ cells in PDL1+ population were calculated, and the relative abundancy (divided by sum) of each subset is shown.
Figure S7. Cell composition in CRC2-17. Related to Figure 7. (A) t-SNE plots based on CyCIF data for specimens CRC1-17 (excluding data from the DNA staining). Cell types are shown at the bottom of the figure. Tumor/epithelium (T/E), stroma (S) and immune (I) populations are outlined in black. The t-SNE plot for CRC1 is reproduced from Figure 1E for reference. $$PARABREAKHERE$$(B) t-SNE of CRC1-17 labelled by specimen identity with labelling by general cell type in upper right. $$PARABREAKHERE$$(C) Cell-type composition for CRC1-17 shown as stacked bar graphs with the same color code as in panel A.
Video S1: CRC1 3D view of budding structures, related to Figure 4.
Video S2: CRC1 3D view of TLS/SLO structures, related to Figure 6.
Table S1. Clinical information for colorectal cancer cohort, related to Figure 1 and STAR Methods. Demographic and diagnostic information for all patient-derived specimens in this study. CRC1 was analyzed in 3D and CRC2-17 in whole-slide 2D and TMA 2D. Other specimens (CRC18-93) were imaged as TMA cores, as described in the text and Figure 1.
Table S3. Antibody panels, related to Figure 1, 3, 4, 7 and STAR Methods. Table includes six (overlapping) antibody panels used throughout the study: tab 1) The main CyCIF panel includes antibodies used to stain all CRC1 sections (except 044-046), CRC2-17 sections, and TMAs (see Figure 1 for graphical overview); tab 2) three additional panels for CRC1 sections 45, 46, and 47, respectively; tab 3) an immune-focused panel used for whole-slide images (WSI) of CRC2-17; tab 4) a tumor-focused antibody panel used for WSI of CRC2-17. CST refers to Cell Signaling Technologies (Beverley MA USA); RRID refers to the Research Resource Identifier available at https://scicrunch.org/resources.
Table S4. Cell-type dictionary, related to Figure 1 & S1 and STAR Methods.
Cell-type assignments based on marker intensities. The first tab shows the primary discriminating markers and tabs 2 and 3 show assignments based on all markers in the panel.
Table S5. Cell-type composition for regions of interest in CRC1, related to Figure 6 and STAR Methods.
Cell-type composition for pathologist-defined regions of interest (see Figure 1B) across all sections processed for CyCIF. Cell-type definitions as in Table S4.
Data Availability Statement
All full resolution images, derived image data (e.g., segmentation masks) and all cell count tables are available via the NCI-sponsored repository for Human Tumor Atlas Network (HTAN; https://humantumoratlas.org/) at Sage Synapse. A version of this data is available at https://www.synapse.org/#!Synapse:syn18434611/wiki/597418.
Several of the figure panels in this paper are available with text and audio narration for anonymous on-line browsing using MINERVA software 34, as are images of CRC2–17; see https://www.tissue-atlas.org/atlas-datasets/lin-wang-coy-2021/.
scRNA-seq data is available in the Gene Expression Omnibus (GEO accession: GSE166319).
All software used in this manuscript is freely available via GitHub as described in 31 and references therein and in https://github.com/labsyspharm/CRC_atlas_2022.