

Abstract
Background:
Despite the importance of tumor-infiltrating T lymphocytes (TILs) in cancer biology, the relationship between TIL phenotypes and their prognostic relevance for localized non-small cell lung cancer (NSCLC) has not been well established.
Patients and Methods:
Fresh tumor and normal adjacent tissue was prospectively collected from 150 patients with localized NSCLC. Tissue was comprehensively characterized by high-dimensional flow cytometry of TILs integrated with immunogenomic data from multiplex immunofluorescence, TCR sequencing, exome sequencing, RNA sequencing, targeted proteomics, and clinicopathologic features.
Results:
While neither the magnitude of TIL infiltration nor specific TIL subsets were significantly prognostic alone, the integration of high-dimensional flow cytometry data identified two major immunotypes (IM1 and IM2) that were predictive of recurrence-free survival independent of clinical characteristics. IM2 was associated with poor prognosis and characterized by the presence of proliferating TILs expressing CD103, PD-1, TIM3, and ICOS. Conversely, IM1 was associated with good prognosis and differentiated by an abundance of CD8+ T cells expressing cytolytic enzymes, CD4+ T cells lacking the expression of inhibitory receptors, and increased levels of B cell infiltrates and tertiary lymphoid structures. While increased B cell infiltration was associated with good prognosis, the best prognosis was observed in patients with tumors exhibiting high levels of both B cells and T cells. These findings were validated in patient tumors from The Cancer Genome Atlas.
Conclusions:
Our study suggests that although the number of infiltrating T cells is not associated with patient survival, the nature of the infiltrating T cells, resolved in distinct TIL immunotypes, is prognostically relevant in NSCLC and may inform therapeutic approaches to clinical care.
Keywords: Lung Cancer, T cells, Biomarkers, Immune system, Microenvironment
One Sentence Summary:
Integrated immunogenomics analysis of localized non-small cell lung cancer reveals prognostic immunotypes.
INTRODUCTION
Lung cancer is the leading cause of cancer-related death worldwide 1–3. The most frequently diagnosed lung cancer type, non-small cell lung cancer (NSCLC), is a very heterogeneous disease comprised of multiple histologic subtypes including lung adenocarcinoma (L-ADCA), lung squamous cell carcinoma (L-SCCA), and lung large-cell carcinoma (L-LCC) 4,5. NSCLC is potentially immunogenic given its typically high tumor mutational burden (TMB) leading to an abundance of neoantigens for immune cell recognition 6,7. However, NSCLC is often highly immunosuppressive and capable of downregulating the antitumor immune responses through multiple mechanisms, including deficiencies in antigen processing and presentation, release of immunomodulatory cytokines, and recruitment of immunosuppressive cells such as a regulatory T cells (Tregs) 8,9 and myeloid-derived suppressor cells (MDSCs). Consistent with this, MDSCs have been associated with increased tumor burden in NSCLC patients 10. In addition to Tregs and MDSCs, neutrophils are the most prevalent immune cell type in adenocarcinoma 11 and their expansion has been associated with increased tumor burden and poor clinical prognosis in patients with resectable NSCLC 12. Previous comprehensive studies devoted to characterization of the immune landscape present in lung cancer demonstrated increased abundance of CD19+ B cells, FoxP3+ Tregs, CD8+ T cells, and particularly CD45RO+ memory CD8+ T cells, but with a significant absence of NK cells 11,13,14. Lizotte et al.13 identified an immunologically hot cluster of NSCLC patients whose tumors have abundant CD8+ T cells expressing high levels of PD-1 and TIM3 and an immunologically cold cluster with tumors characterized by lower relative abundance of CD8+ T cells and lower expression of inhibitory markers.
Despite the comprehensive profile of immune cell content and function in NSCLC, the relationship between TIL phenotypes and their prognostic relevance is not well established. Upregulation of PD-1 and TIM3 occurs after repeated antigen exposure, thus PD-1 expressing T cells in the tumor are believed to be enriched for anti-tumor activity, according to studies performed in melanoma 15. However, the lung is an organ under constant environmental (bacterial, viral, smoking, etc.) exposure. Examination of the T cell repertoire in NSCLC in comparison to normal adjacent uninvolved lung tissue shows a significant overlap in TCR sequences containing a high prevalence of TCR sequences predicted to recognize viral epitopes 16, suggesting that a considerable proportion of T cells in the lung may be irrelevant to tumor control. Thus, it is paramount to identify qualitative in lieu of quantitative differences in T cell infiltrate that may impact patient outcome. Furthermore, understanding of the TIL landscape in NSCLC has become increasingly critical with the advent of immune checkpoint inhibitors targeting PD-1/PD-L1 and CTLA-4 pathways, which have revolutionized the management of NSCLC. The Food and Drug administration (FDA) and/or European Medicines Agencies (EMA) approved the use of inhibitory antibodies to PD-1 (nivolumab and pembrolizumab), PD-L1 (atezolizumab and durvalumab), and CTLA-4 (ipilimumab) 17–22. However, a significant proportion of patients do not benefit from these therapies 23–25. A better understanding of the association between TIL abundance and phenotypes associated with patient outcome may provide novel insights to design novel immunotherapies.
In the current study, we performed immuno-genomic profiling by multiparameter flow cytometry, multiplex immunofluorescence (mIF), RNA sequencing (RNA-seq), whole-exome sequencing (WES), TCR sequencing (TCR-seq), and targeted proteomics on resected tumor tissue obtained from 150 patients with localized NSCLCs enrolled in the ImmunogenomiC prOfiling of early-stage NSCLC (ICON) study, a multigroup and cross-disciplinary prospective study carried out at the University of Texas MD Anderson Cancer Center. We correlated these data to clinical and pathological characteristics and discovered that while bulk TIL infiltration was not associated with patient outcomes, two distinct immunotype subgroups characterized by the relative enrichment of defined T cell markers were independent prognosticators of patient survival.
MATERIALS AND METHODS
Additional methods details can be found in the Supplementary Data.
Patients and Study Design.
This study was performed on NSCLC tissue and non-adjacent uninvolved lung tissue from the same patient. Patient samples were collected from NSCLC patients enrolled in the ImmunogenomiC prOfiling of early-stage NSCLC (ICON) project which began in April 2016 and enrolled the last patient in September 2018. Informed consent was obtained from all study participants approved by the University of Texas MD Anderson Cancer Center’s Institutional Review Board (IRB). Clinicopathologic features and subject characteristics of patients involved in this study are presented in Table 1. Tumor and matched uninvolved tissues were freshly collected from a cohort of 76 patients to perform in-depth analysis of immune cells using flow cytometry, multiplex immunofluorescence (mIF), TCR sequencing, RNA sequencing and whole exome sequencing (WES). An overview of all available samples and assays is shown in Table S1.
Table 1.
Clinicopathological characteristics of the ICON cohort
| ICON cohort (N = 150) | |||
|---|---|---|---|
|
| |||
| Age | Years | Sex | n |
| Median | 68 | Male | 70 |
| Range | 38–85 | Female | 80 |
| Smoking history | n | Tumor size (cm) | |
| Ever | 125 | Median | 4 |
| Never | 25 | Range | 0–11.5 |
| Pathological stage (AJCC7) | n | Clinical stage (AJCC7) | n |
| Stage 0 | 2 | Stage 0 | 0 |
| Stage 1 | 58 | Stage 1 | 68 |
| Stage 2 | 52 | Stage 2 | 54 |
| Stage 3 | 36 | Stage 3 | 26 |
| Stage 4 | 2 | Stage 4 | 2 |
| Pretreatment | n | Histology | n |
| None | 112 | Adenocarcinoma | 97 |
| Neoadjuvant | 38 | Squamous cell carcinoma | 38 |
| Other | 15 | ||
Abbreviations: Ever, current or former smoker; Never, has never smoked.
RESULTS
TILs are activated and hyper-proliferative, but functionally impaired in early-stage resectable NSCLC
The ICON study prospectively enrolled 150 surgical patients to undergo integrated molecular characterization to identify key immunogenomic features of early-stage NSCLC, with clinicopathological features shown in Table 1. Multi-omic profiling was performed on both tumor and normal adjacent tissue, ranging from 76 profiled by high-dimensional flow cytometry to 139 profiled by multispectral immunofluorescence, subject to available tissue quantity (Table S1). Quantification of the T cells using mIF demonstrated that CD3+ T cell counts were significantly higher in tumors when compared to adjacent uninvolved lung tissues (Fig. 1A–B), which was orthogonally validated by sequencing of T cell receptor β (TCRβ) (Fig. 1C) and multiparameter flow cytometry analyses (Fig. 1D, S1). There was a good correlation between T-cell changes determined by flow cytometry and other metrics evaluated, supporting the robustness of the assays (Fig. S2). In addition, the percentages of CD8+ T cells, CD4+ T cells, and CD4+CD25hiFoxp3+ Tregs within the total CD45+ immune cells infiltrate were significantly higher in tumors compared to uninvolved lung tissues (Fig. 1E–G). Tumors showed a significantly lower CD8+ T/Tregs ratio compared to uninvolved lung tissues (Fig. 1H). CD8+ T cells in the tumor were found to have a higher proliferative capacity compared to the CD4+ T cells as measured by Ki67 expression, whereas no difference in proliferation was observed in uninvolved lung tissue (Fig. 1I).
Figure 1. T-cell frequency is increased in tumors.

(A, B) Representative image of immune cell infiltration in uninvolved lung tissue and tumor analyzed using multiplex immunofluorescence (mIF, A), and quantification of CD3+ cells (n = 139, B). (C) T-cell receptor sequencing examining the T-cell fraction in uninvolved lung tissue and tumor as defined by immunoSEQ (n = 55). (D-I) Percentages of T lymphocytes within CD45+ cells in uninvolved lung tissue and tumor, n = 58, as measured by flow cytometry. (D) Percentage of CD3+ T cells. (E) Percentage of CD8+ T cells. (F) Percentage of CD4+ T cells. (G) Percentage of CD4+CD25hiFoxP3+ Tregs. (H) Ratio of CD8/Tregs from uninvolved lung tissue and tumor. (I) Percentages of proliferating (Ki-67+) CD4+ and CD8+ T cells in uninvolved lung tissue and tumor. Differences between the two groups were calculated with a signed-rank (B-H) or one-way analysis of variance (I).
Phenotypically, tumor-infiltrating CD8+ T cells expressed significantly higher levels of T cell activation/co-inhibitory receptors including ICOS, PD-1, TIM3, and LAG3 compared to uninvolved lung tissues (Fig. 2A), enriched expression of the tissue resident memory (TRM) marker CD103 (Fig. 2B), and loss of cytolytic enzymes perforin and granzyme B (Fig. 2C). Expression of activation/co-inhibitory receptors was higher in the CD103+ CD8 T cell compartment compared to CD103−, although both CD103+ and CD103− CD8 T cells showed increased activation/co-inhibitory receptor expression in the tumor compared to uninvolved tissue (Fig. S3). Increased expression of T cell activation/co-inhibitory receptors and CD103 were observed in CD4+ T cells as well (Fig. 2D–E). Restricting our analysis to specific subsets of patients based on key clinicopathological features indicated that the differences in T cell infiltration levels and phenotypes between tumor and adjacent uninvolved tissue are conserved across patient subsets, with similar results observed regardless of smoking status, tumor histology, or if samples were collected after receiving neoadjuvant chemotherapy (Fig. S4). Collectively, our results corroborate observations that in NSCLC, TILs are abundant, activated, hyper-proliferative, but functionally impaired.
Figure 2. Tumor-infiltrating T lymphocytes are activated and express checkpoint molecules at higher levels.

(A) Percentage of ICOS+CD8+, PD1+CD8+, TIM3+CD8+, and LAG3+CD8+ T cells within CD3+ T cells in tumor and uninvolved matched tissues (n = 45). (B) Percentage of CD103+CD8+ T cells in tumor and uninvolved matched tissues (n = 45). (C) Percentage of Perforin+CD8+ and granzyme B+CD8+ T cells in tumor and uninvolved matched tissues (n = 66). (D) Percentage of ICOS+CD4+, PD1+CD4+, TIM3+CD4+, and LAG3+CD4+ T cells within CD3+ T cells in tumor and uninvolved matched tissues (n = 43). (E) Percentage of CD103+CD4+ T cells in tumor and uninvolved matched tissues (n = 43). All panels represent flow cytometry data on freshly disaggregated tissue. Paired Student’s t-test was used to determine significance for all comparisons.
TIL infiltration and individual subpopulations are not prognostic of survival
We next evaluated the relationship of T cell infiltrates with recurrence-free survival (RFS) in this cohort of early-stage surgical patients. Multiplex immunofluorescence data showed that high numbers of intratumoral CD3+ T cells were not associated with longer RFS (p = 0.32) (Fig. 3A). Similarly, TCR sequencing analysis demonstrated that high T cell fraction within the DNA isolated from the tumor tissue was not associated with longer RFS (p = 0.61) (Fig. 3B), consistent with our previous report 16. Furthermore, RNA sequencing data demonstrated the absence of association between CD3E expression and RFS (p = 0.90) (Fig. 3C). Multiplex immunofluorescence and RNA sequencing data also showed that the number of intratumoral CD8+ T cells and the expression of CD8A transcript were not associated with RFS (p = 0.77 and 0.89) (Fig. 3D–E). In addition, high dimensional flow cytometry on freshly disaggregated tumor tissue showed an absence of association between any of the 220 individual T cell subpopulations assessed and RFS (Fig. 3F). To validate these findings in external cohorts, we examined the survival in TCGA patients based on CD3E and CD8A transcripts. We confirmed the lack of association between CD3E and CD8A transcripts with progression-free survival (PFS) (Fig. 3G–H). Taken together, our results showed that neither total T cell infiltrates nor individual immune cell populations are prognostic of recurrence in NSCLC patients.
Figure 3. Tumor-infiltrating T-lymphocyte infiltration and individual subpopulations cannot predict survival.

(A-E) Kaplan–Meier curves and log-rank P values showing recurrence-free survival for non-small-cell lung cancer patients with high (upper tertile, red) and low levels (lower two tertiles, blue) of (A) CD3 T cells (cells/mm2) defined by multiplex immunofluorescence (mIF). (B) T-cell fraction defined by ImmunoSeq. (C) CD3E transcript defined by RNA Sequencing. (D) CD8 T cells (cells/mm2) defined by mIF. (E) CD8A transcript defined by RNA Sequencing. (F) Volcano plot showing hazard ratios for all immune populations determined by flow cytometry using Cox proportional hazards model considering stage as a covariate. Benjamini–Hochberg procedure used to correct for multiple comparisons. (G, H) Survival in The Cancer Genome Atlas (TGCA) patients based on CD3E (G) and CD8A (H) transcript levels split into high (upper tertile, red) and low (lower two tertiles, blue). Log-rank test was used to determine significance.
FDR, false discovery rate.
Integrated flow cytometry-based immunotype predicts patient outcomes
As no individual immune cell population was independently prognostic in our cohort of patients with NSCLC, we next asked if integration of all immune cell populations analyzed may provide more meaningful insight. We selected a subset of our cohort for high-dimensional analysis which had sufficient tissue for profiling by all 3 flow cytometry panels used in this study (Table S4), and sufficient events to allow for analysis of the majority of populations evaluated, resulting in a total of 47 samples. Using all flow cytometry populations with sufficient sample size, we performed t-distributed stochastic neighbor embedding (tSNE) (Fig. 4A), which divided patients into two immunotypes (IM1 and IM2) based on multiple clustering criteria (Fig. S5). In contrast to the analysis of single immune cell populations, RFS analysis indicated that immunotypes were strongly prognostic, with patients belonging to the IM1 immunotype having a significantly longer RFS (p=0.007) (Fig. 4B). Multivariate analysis controlling for tumor stage indicated immunotype was independently associated with RFS, with a numerically larger hazard ratio than tumor stage itself (HRstage = 1.26, HRIM2 = 2.12) (Fig. 4C). Repeating this procedure to define immunotypes based on analysis of flow cytometry data of the uninvolved tissue revealed no prognostically relevant cluster (Fig. S6), suggesting that the association between T cell phenotype and outcome is specific to the tumor immune infiltration.
Figure 4. Immunotype determined by tumor-infiltrating T lymphocyte composition is prognostic in non-small-cell lung cancer (NSCLC).
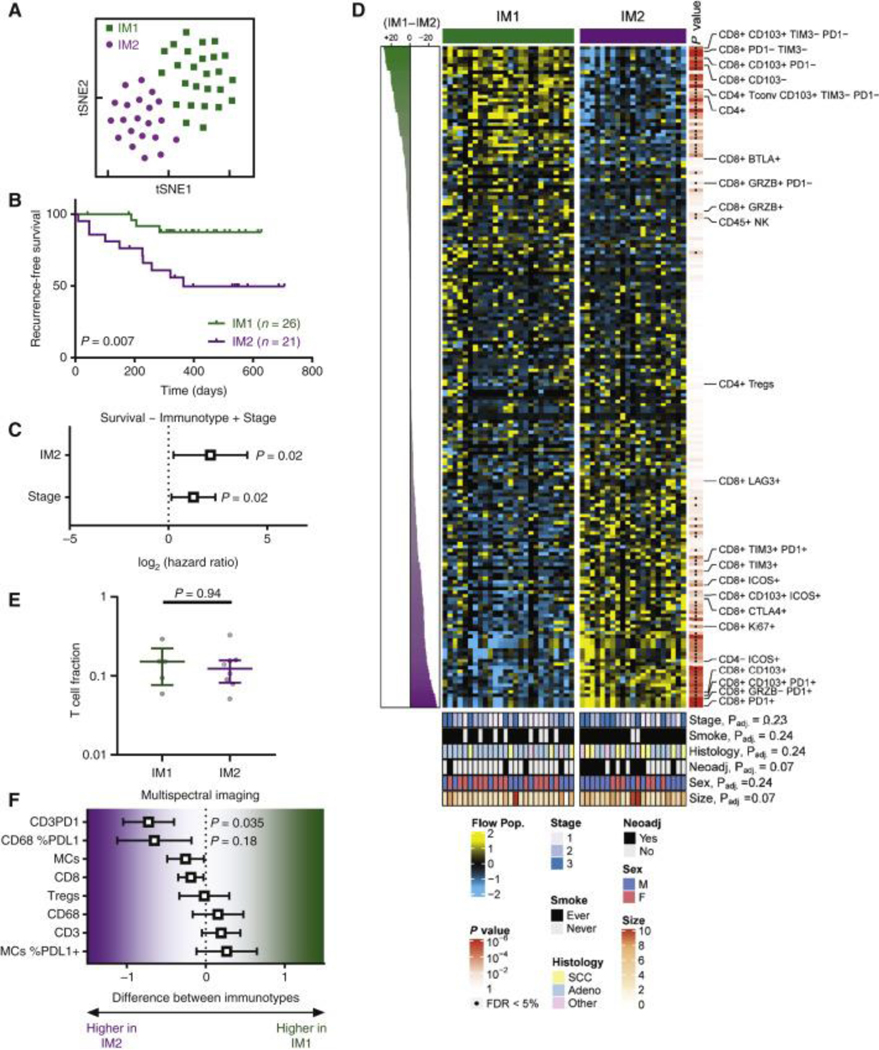
(A) tSNE showed that tumors from NSCLC patients (n = 47) could be assigned to two major immunotypes (IM1 and IM2). (B) Kaplan–Meier curves showing recurrence-free survival (RFS) in IM1 (green, n = 26) and IM2 (purple, n = 21) tumors. (C) Multivariate Cox proportional hazards model for RFS based on immunotype and tumor stage. (D) Heat map showing all immune populations measured by flow cytometry, with significance indicated to the right of the heat map and clinical parameters indicated at the bottom of the heat map. (E) T-cell fraction determined by immunoSEQ in IM1 and IM2 patients. Median with interquartile range. Rank-sum P value is shown. (F) Malignant cells (MCs), CD8, regulatory T cells (CD3+CD8−Foxp3+), CD68 (macrophages), CD3, CD3PD1, CD68 %PDL1, and MCs %PDL1 expression were analyzed using multiplex immunofluorescence in IM1 and IM2 patients. Values are shown as mean difference with 95% confidence interval.
PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; Tregs, regulatory T cells.
While we found no significant differences in clinical variables between IM1 and IM2, we identified 77 immune populations differentially enriched at under 5% false discovery rate (FDR) (Fig. 4D, Table S2). Notable immune cell populations with greater prevalence in poor prognosis IM2 immunotype included CD8+ T cells co-expressing both the TRM marker CD103 and inhibitory receptor PD-1. In addition to CD103 and PD-1, IM2 CD8+ T cells expressed high levels of the activation marker ICOS, inhibitory receptors TIM3, LAG3, and the proliferation marker Ki67. Similar to the CD8+ T cell compartment, the CD4+ T cell infiltrate was significantly enriched in PD-1+ and ICOS+ cells in IM2. Conversely, IM1, which was associated with better outcome, was differentiated by an abundance of NK cells, CD8+ T cells expressing BTLA, Granzyme B, and CD103 in the absence of PD-1. IM1 immunotype was also enriched in total CD4+ T cells, specifically those expressing CD103 and lacking the expression of inhibitory receptors PD-1 and TIM3. Consistent with our observations that T cell infiltration was not independently prognostic (Fig. 3), we observed no difference in T cell fraction derived from TCR sequencing data between IM1 and IM2 (Fig. 4E). Orthogonal analysis of immune infiltrates using multispectral immunofluorescence (Fig. S7) confirmed there was no alterations in total T cell numbers, Tregs (CD3+CD8−Foxp3+), or CD8+ T cells between tumors assigned to IM1 and IM2, and also confirmed the increase in PD-1+ T cells observed in IM2 (Fig. 4F). Multispectral immunofluorescence detected no difference in total or PD-L1+ malignant cells (MCs) or macrophages (CD68+ cells) between IM1 and IM2 tumors (Fig. 4F).
To further confirm that clinicopathological differences between patients in IM1 and IM2 were not driving the observed immunological differences between two immunotypes, we next sought to systematically compare the differences between IM1 and IM2 across different clinically relevant sub-populations. We hypothesized that if a given clinicopathological feature was unduly influencing the identified immunotypes, the observed differences in immune cell populations between IM1 and IM2 would be mitigated by only analyzing that subset of samples. To assess this, we first compared the frequency of PD1+ CD8+ T cells between IM1 and IM2 in various patient subsets, finding that the increased prevalence of PD1+ CD8+ T cells was observed in IM2 for all clinicopathological subsets (Fig. 5A). To generalize this analysis, we determined the difference in frequency between IM1 and IM2 for all immune cell populations either in all samples (plotted along the x-axis) or various indicated sub-populations (plotted along the y-axis) (Fig. 5B). This analysis found that the differences in frequency of each immune cell populations between IM1 and IM2 are highly concordant regardless of the subset analyzed (mean correlation coefficient = 0.93, 95% CI = 0.89–0.97) and did not deviate from the expected correlation coefficient obtained by random sub-sampling (r = 0.92), suggesting no single patient subset is disproportionately influencing the observed immunotype clusters. Altogether these results suggest that patients can be stratified according to the degree of activation of their TIL independently of tumor features and patient clinical characteristics.
Figure 5. Immunotype changes are conserved across different patient subsets.

(A) Comparison of percentage of CD8 T cells positive for PD1 between IM1 and IM2 in indicated subsets of patients. White dot indicates median value, Δ indicates difference in %PD1+ CD8+ T cells between groups. Rank-sum test. (B) Plots comparing the difference (Δ) in all immune cell populations between IM1 and IM2 observed in the entire set of patients analyzed (x-axis) and to the difference observed in indicated subsets of patients (y-axis). Pearson correlation coefficient (r) and corresponding P value (inset).
N0, no evidence of regional node involvement; NX, any evidence of lymph node involvement; SCC, squamous cell carcinoma.
Molecular differences between IM1 and IM2
To identify potential molecular alterations that may be causing the differences observed between IM1 and IM2, we analyzed differences at the DNA, RNA, and protein levels. At the DNA level, we did not observe significant differences in TMB (Fig. 6A). To ensure TMB analysis was not confounded by differences in tumor purity, we analyzed the correlation between tumor purity determined from WES and TMB and found no relationship between the two (Fig. S8A), nor a difference in tumor purity between IM1 and IM2 (Fig. S8B). In addition to equivalent TMB between IM1 and IM2, we also found no enrichment for any specific mutations (Fig. 6B). We next analyzed mutational signatures 6,26,27, which evaluate mutational patterns associated with different mutagenic processes, detecting a total of 11 out of 30 mutational signatures in our samples. Comparison of these between IM1 and IM2 revealed an enrichment in mutational signature 6 in IM1, which is typically associated with mismatch repair deficient tumors, as well as mutational signature 1 in IM2, which is typically correlated with age and is thought to be due to spontaneous cytosine deamination (Fig. 6C) 6. As mismatch repair defects can promote formation of both single nucleotide variants and small indels28, the latter of which can be particularly antigenic29, we assessed if IM1 harbored more indels, but found no significant difference (Fig. S9A). Increased levels of indels were detected in tumors positive for mutational signature 6 or 15 (both of which are associated with mismatch repair defects) (Fig. S9B), but not mutational signature 6 alone (Fig. S9C). This difference in mutational signatures could imply that tumors with different immunotypes are associated with different evolutionary trajectories.
Figure 6. Molecular characterization of IM1 versus IM2 in the ICON cohort.

(A) Tumor mutation burden in IM1 (n = 21) and IM2 (n = 20) tumors. Median with interquartile range and P value by rank-sum test. (B) Comparison of mutation frequency for specific genes between IM1 (n = 21) and IM2 (n = 20) by Fisher’s exact test. Odds ratio shown with 95% confidence interval. (C) Volcano plot showing difference in various mutational signatures between IM1 (n = 21) and IM2 (n = 20) tumors. Rank-sum test with Benjamini–Hochberg false discovery rate (FDR). Dotted line indicates 10% FDR. (D) Gene set enrichment analysis of differential gene expression between IM1 (n = 15) and IM2 (n = 13) tumors. Pathways enriched in IM1 have positive scores and pathways enriched in IM2 have negative scores. (E) Volcano plot showing proteins differentially expressed between IM1 (n = 24) and IM2 (n = 21) tumors. Rank-sum test with Benjamini–Hochberg FDR. Dotted line indicates 10% FDR.
MMR, mismatch repair.
To identify pathways that may exhibit differential activation, we next used RNAseq gene expression data to perform gene set enrichment analysis (GSEA). As shown in Fig. 6D, we found the top 4 pathways enriched in IM1 were largely focused on B cell activation, whereas the top 4 pathways enriched in IM2 were focused on cell proliferation and hypoxia. Increased proliferation markers in IM2 were also confirmed at the protein level using reverse phase protein array (RPPA) data, including Cyclin B1, 4EBP1, and phospho-Rb (Fig. 6E). Consistent with our flow cytometry results, reverse phase protein array (RPPA) analysis also showed that protein levels of CD4 were enriched in IM1, along with increased levels of phosphorylated STING, Granzyme B, and phosphorylated STAT3.
Generalization of immunotypes to TCGA data
To assess whether the immunotypes observed within our ICON cohort were generalizable, we used our previously published IRAPS algorithm to develop an immunotype gene expression signature30 (Fig. S10A). Briefly, we randomly divided the 28 tumors from the ICON cohort with both immunotype and tumor RNAseq data into a training cohort of 19 tumors (N = 9 IM1, N = 10 IM2) to develop the gene expression signature and a testing cohort of 9 tumors (N = 4 IM1, N = 5 IM2) to validate the gene expression signature’s accuracy. From the training cohort, we defined a 70 gene IM1 gene expression signature (Table S3) that robustly predicted immunotype both in the training cohort (AUC = 0.91) as well as the testing cohort (AUC = 0.95) (Fig. S10B–D). We further validated the IM1 gene expression signature by applying it to the remaining ICON samples for which no flow cytometry data was available (N = 24), finding the signature robustly recapitulated the prognostic difference observed between immunotypes (Fig. S10E–G).
To validate our findings in external larger cohorts, we next determined immunotype scores in NSCLC tumors from TCGA (LUAD, N=515) using the 70 gene IM1 signature and available TCGA RNAseq data. Upon dividing patients from TCGA into IM1 and IM2, we observed that immunotype was prognostic both with univariate analysis (Fig. 7A), and multivariate survival analysis accounting for tumor stage (Fig. 7B). At the gene expression level, we found that TCGA patients closely mirrored ICON patients with strong enrichment in B cell pathways in IM1, and cell cycle/hypoxia pathways in IM2 (Fig. 7C). Similar to what was observed in the ICON cohort for the mutational signatures, we found that in TCGA patients assigned to IM1 there was a large increase in mutational signature 6, although whereas IM2 patients did not show an increase in mutational signature 1 (Fig. 7D).
Figure 7. Validation of IM1 and IM2 phenotypes in The Cancer Genome Atlas (TCGA) patients.

(A) Overall survival in TCGA patients based on predicted immunotype. Log-rank P value. (B) Multivariate survival analysis controlling for tumor stage using Cox proportional hazards model in TCGA patients. (C) Gene set enrichment analysis plots for pathways previously identified in the ICON cohort (Figure 6D). (D) Differences in mutational signatures identified as differentially enriched in the ICON cohort (Figure 6C). IM1, n = 228; IM2, n = 242. Rank-sum test. Line indicates median value.
FDR, false discovery rate; NES, net enrichment score.
Improved patient prognosis is associated with dual enrichment for B and T cells
Based on the enrichment in B cell gene expression pathways in IM1 patients with good prognosis in both the ICON (Fig. 6D) and TCGA (Fig. 7C) cohorts, we hypothesized that B cell infiltrates may be associated with improved patient outcomes. Quantification of B cell marker CD20 staining on FFPE tissue in the ICON cohort indicated that high levels of B cells was associated with a favorable prognosis (Fig. 8A). In patients from the TCGA cohort, we utilized protein levels determined from RPPA to interrogate the presence of B cells and confirmed that high CD20 expression was associated with improved prognosis (Fig. 8B). We hypothesized that while abundance of T cells alone is not prognostic, dual enrichment for tumor infiltrating B cells and T cells may contribute to better prognosis for patients with NSCLCs. We further divided the ICON cohort into 4 groups including patients with high B and T cell infiltration (both high), high B cells infiltration only, high T cells infiltration only, and low B and T cell infiltration and found that improved prognosis associated with high B cells infiltration required concordant high levels of T cells, as neither high level of B or T cells alone were significantly associated with improved RFS (Fig. 8C). To confirm this result using the TCGA data, we merged CD20 protein expression determined by RPPA with T cell levels determined by RNA-seq and again found that patients with high levels of both B cells and T cells exhibited the best prognosis (HR = 0.56, P = 0.002) (Fig. 8D). In contrast to the ICON cohort, TCGA patients low for both markers also displayed statistically worse survival than those low for only one marker (HR = 2.2, P = 0.0006) (Fig. 8D), which may be attributed to either differences in the precision of the analysis between ICON (mIF) and TCGA (RPPA/RNAseq) or differences in patient characteristics. Recent studies demonstrated that T-B cell aggregates and tertiary lymphoid structures (TLS), ectopic lymphoid tissue composed of T-B cell aggregates that develop secondary to chronic inflammation, were associated with improved survival in multiple types of cancers 31–33. Consistent with the observation that high levels of both B and T cells are required for improved patient prognosis, quantification of tertiary lymphoid structures by histology in the ICON cohort revealed significantly more immune aggregates in IM1 than IM2 (Fig. 8E). Taken together, these results suggest complex immune cell interactions are required for optimal clinical outcomes.
Figure 8. Best prognosis is observed with high levels of both B cells and T cells.

(A) Recurrence-free survival (RFS) of patients from the ICON cohort with high versus low abundance of CD20+ B cells determined by multiplex immunofluorescence (mIF). (B) Progression-free survival (PFS) of patients from The Cancer Genome Atlas (TCGA) stratified by CD20 protein levels determined using reverse-phase protein array (RPPA). (C) RFS of patients from the ICON cohort with high levels of both CD20+ B cells and CD3+ T cells, high levels of only one immune cell population, or low levels of both populations as determined by mIF. (D) PFS of patients from TCGA stratified by high levels of both CD20 protein determined by RPPA and CD3 expression determined by RNAseq, high levels of only one immune cell population, or low levels of both immune cell populations. (E) Number of tertiary lymphoid structures per mm2 in IM1 and IM2 tumors. Rank-sum test. For all survival analyses, patients were divided at median level of immune cell infiltrates, and survival assessed using the log-rank test. Hazard ratio is given with 95% confidence interval.
TIL, tumor-infiltrating T lymphocyte.
DISCUSSION
NSCLC is a heterogeneous disease on both molecular and immunological levels 13,34–38. In this study, we integrated high dimensional tumor-infiltrating T cell characterization with genomic and clinicopathological data to gain insights into functional T cell markers correlating with outcome. We have identified two prognostic immunotypes, IM1 and IM2, that characterized tumor microenvironment and defined patient outcome independently of clinicopathological features of the patient cohort, with IM1 being the immunotype associated with significantly improved RFS. While the abundance of T cell infiltration was not predictive of outcome, increased expression of key markers on CD8+ T cells including BTLA, and Granzyme B, high NK infiltration, and the co-existence of specific T cell subsets with lower checkpoint molecule expression were key features associated to IM1. To exclude the possibility that the differences observed between IM1 and IM2 immunotypes were driven by TMB, a predictor of immunotherapy outcome in NSCLC 49–51, we analyzed mutational changes at the DNA level and found no significant differences in tumor mutation burden or enrichment for any specific mutation between immunotypes, further supporting the notion that immunotype-based stratification of outcome was independent of tumor features.
Consistent with previous findings11,13,14, we have also confirmed that TILs are abundant, activated, and hyper-proliferative, but functionally impaired in NSCLC. Higher density of total CD103+ cells in the tumor has been found to correlate with survival in lung cancer 34,39. In vitro experimentation has revealed that CD8+ CD103+ TIL expanded from NSCLC exhibit anti-tumor activity, and that the CD103 molecule itself, an integrin binding E-cadherin on the surface of tumor cells, plays an important role in the cytotoxic activity of the CD8+ TRM subset40,41. Our analysis indicated that the CD8+CD103+ TIL population was lower in IM1 (mean expression of 19% versus 45% in IM2). Conversely, the CD103+ T cell populations present in IM2, the immunotype with the poorer outcome, shared some phenotypic characteristics with exhausted or/and dysfunctional CD8+ T cells, including the expression of inhibitory receptors PD-1, TIM3 and LAG335,42, altogether suggesting a context-dependent prognostic relevance of CD103 expression.
PD-1 expression suggests antigen experience, and repeated antigen exposure. However, CD8+PD-1+ tumor-infiltrating lymphocytes have been regarded as functionally exhausted rather than activated because of persistent antigenic exposure from either viral infection or the tumor microenvironment42. PD-1 tends to be expressed on clonally expanded T cells in solid tumors, and the clonal expansion of T cells in the tumor has been associated with anti-tumor activity44,45. TCR repertoire analysis of early-stage NSCLC demonstrated a great degree of overlap between TCR present in the tumor and adjacent uninvolved tissue, and greater clonal expansion of T cells in the normal adjacent uninvolved lung, many of which recognize viral epitopes16. Such a situation may arise in response to the constant viral challenge occurring in the lung tissue and suggests that PD-1 expression in lung tumor tissue is not intimately associated with T cells recognizing tumor antigens but in large part dependent on viral antigen recognition. Our study identifies subpopulations of PD-1 negative effector T cells associated with better prognosis in early-stage NSCLC, which likely represent the true anti-tumor fraction as opposed to passive infiltrates. Orthogonal studies in NSCLC resected patients support a more favorable clinical outcome for patients with CD8 lymphocytes lacking PD-1 compared to those harboring CD8 lymphocytes expressing PD-143,47. Conversely, in the context of immune checkpoint inhibitors targeting PD-1/PD-L1 pathway, studies suggest that increased CD8+PD-1+ tumor-infiltrating lymphocytes predict the clinical efficacy of PD-1 blockade therapies and correlates with favorable clinical outcome42,46. Taking together the combined evidence that high levels of CD8+PD-1+ immune infiltrates are associated with worse general prognosis but increased response to anti-PD-1/PD-L1, these studies could support an improved, tailored treatment model for early-stage NSCLC. Quantification of CD8+PD-1+ immune infiltrates upon resection may first be used to identify high-risk patients, and then this increased risk may be mitigated by adjuvant anti-PD-1/PD-L1 therapy.
Another important finding in our study is that good prognosis is dependent neither on total T cell infiltration nor on individual T cell phenotype. It is conceivable this could be due to intratumor heterogeneity, as prior studies have shown that immune cell infiltration can be heterogeneous even within the same tumor48. However, we found that T cell infiltration was generally correlated across different assays (and consequentially, different tumor regions), suggesting that the bulk T cell level, the magnitude of the infiltrate was typically similar throughout the tumor in our cohort. While some outliers were observed, the use of different pieces of the same tissue in multiple assays, each with their own degree of variability, prevents deconvolution of technical assay-dependent variation from actual heterogeneity to explain any discordance. However, both the robust concordance between assays (CD3+ from mIF, flow cytometry, RNAseq and T cell fraction from TCRseq data) and lack of any prognostic signature across assays, strongly suggests bulk T cell infiltration has minimal prognostic relevance in NSCLC. Altogether, these observations support the idea that the careful analysis of the functional status of TIL populations is key to help predict the course of the disease.
Crucially, this study identified increased infiltration of B cells along with T cells not expressing checkpoint molecules in IM1, suggesting that a productive immune response where B cells could act as antigen presenting cells to properly activate T cells is likely taking place. Moreover, the increased presence of immune cell aggregates or tertiary lymphoid structures in IM1 suggests greater interaction and co-localization of immune cells which favors anti-tumor immune response. These observations are consistent with previous studies showing the important role of B cells in the immuno-biology of NSCLC 52–54, and supporting the notion that enrichment of B cells in TLS is predictive of response to checkpoint blockade 31–33.
In summary, our study has identified two major prognostically relevant immunotypes in NSCLC which are independent of both tumor features and clinical characteristics of the patients and has demonstrated that the engagement of complex immune cell interactions within the tumor microenvironment is required for tumor control and improved clinical outcome. This work also provides important insights to the development of cell-based biomarker signatures prognostic of outcome in NSCLCs, suggesting a model whereby increased risk for recurrence in patients with high levels of CD8 T cells expressing PD-1 is addressed by adjuvant immune checkpoint blockade. This therapeutic approach, as well as how exposure to immune checkpoint blockade modulates our observed immunotypes, warrants exploration in future studies.
Supplementary Material
Table S1. Assays and samples included in this study.
Fig. S1. Flow cytometry gating strategy for the lymphocyte characterization in uninvolved lung tissue and tumors.
Fig. S2. Correlation between assays.
Fig. S3. Expression of co-stimulatory/checkpoint proteins by CD103+ status.
Fig. S4. Comparison of tumor and adjacent uninvolved tissue stratified by clinicopathological features.
Fig. S5. Metrics for determining optimal number of clusters for immunotypes.
Fig. S6. tSNE analysis of uninvolved lung tissue and prognostic relevance.
Fig. S7. Exemplary multispectral immunofluorescence images.
Fig. S8. Tumor purity does not influence TMB.
Fig. S9. Relationship between indels and dMMR mutational signatures.
Fig. S10. Overview of IRAPS and immunotype signature validation.
Table S2: Immunotypes and clinical annotations.
Table S3. Immunotype gene expression signature.
Table S4. Antibodies for flow cytometry.
HIGHLIGHTS:
Overall T cell infiltration did not predict prognosis in localized NSCLC
Analysis of high-dimensional TIL flow cytometry data identified two immunotypes predictive of patient outcomes
Good prognosis subgroup was associated with T cells lacking inhibitory receptors & presence of tertiary lymphoid structures
Acknowledgments:
The authors are grateful to Beatriz Sanchez-Espiridion, Sandesh Subramanya and Karen Millerchip for effort coordination; Renganayaki Krishna Pandurengan and Shanyu Zhang for data consolidation; Mei Jiang and Auriole Tamegnon for mIF staining; and Wei Lu and Jianling Zhou for IHC staining, as well as Kyle Mitchell, Erin Bayley, Emily Roarty, Lara Landry, and May Celestino for additional contributions making these studies possible.
Funding:
This work was supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program, the Gil and Dody Weaver Foundation and Bill and Katie Weaver Charitable Trust, Rexanna’s Foundation for Fighting Lung Cancer and the MD Anderson Cancer Center Support Grant P30 CA01667. Additional support came from NIH CCSG Award (CA016672 (Institutional Tissue Bank (ITB) and Research Histology Core Laboratory (RHCL)), and the Translational Molecular Pathology-Immunoprofiling Lab (TMP-IL) at the Department Translational Molecular Pathology, The University of Texas MD Anderson Cancer. D.J.M. was supported by NCI grant K99CA240689. P.O.G. was supported by the Fonds de Recherche Québec–Santé’s (FRQS) Resident Physician Health Research Career Training Program (32667).
CB has received research funding from Iovance Biotherapeutics and has participated in advisory committees for Myst Therapeutics and Turnstone Biologics. DLG has received research funding from AstraZeneca, Astellas, Janssen, Ribon Therapeutics and Takeda and has participated in advisory boards for AstraZeneca Journal Pre-proof 24 and Sanofi. CH served on the advisory board for Briacell. SS has participated in advisory committees for Ethicon and for the Peter MacCallum Cancer Center. JVH has received research support from AstraZeneca, Bayer, GlaxoSmithKline, and Spectrum; participated in advisory committees for AstraZeneca, Boehringer Ingelheim, Exelixis, Genentech, GlaxoSmithKline, Guardant Health, Hengrui, Lilly, Novartis, Specrtum, EMD Serono, and Synta; and received royalties and/or licensing fees from Spectrum. TC has received speaker’s fees from the Society for Immunotherapy of Cancer and Bristol-Myers Squibb and research funding to MD Anderson Cancer Center from Boehringer Ingelheim and Bristol-Myers Squibb. JZ served on advisory board for AstraZeneca and Geneplus and received speaker’s fees from BMS, Geneplus, OrigMed, Innovent, grants from Merck, Johnson and Johnson from outside the submitted work.
Footnotes
Competing interests: No potential conflicts of interest are disclosed by the other authors.
References and Notes
- 1.Boloker G, Wang C. & Zhang J. Updated statistics of lung and bronchus cancer in United States (2018). Journal of thoracic disease 10, 1158–1161, doi: 10.21037/jtd.2018.03.15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD & Jemal A. Cancer statistics, 2019. CA: a cancer journal for clinicians 69, 7–34, doi: 10.3322/caac.21551 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Torre LA et al. Global cancer statistics, 2012. CA: a cancer journal for clinicians 65, 87–108, doi: 10.3322/caac.21262 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Davidson MR, Gazdar AF & Clarke BE The pivotal role of pathology in the management of lung cancer. Journal of thoracic disease 5 Suppl 5, S463–478, doi: 10.3978/j.issn.2072-1439.2013.08.43 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langer CJ, Besse B, Gualberto A, Brambilla E. & Soria JC The evolving role of histology in the management of advanced non-small-cell lung cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 28, 5311–5320, doi: 10.1200/jco.2010.28.8126 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Alexandrov LB et al. Signatures of mutational processes in human cancer. Nature 500, 415–421, doi: 10.1038/nature12477 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chalmers ZR et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome medicine 9, 34, doi: 10.1186/s13073-017-0424-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mellman I, Coukos G. & Dranoff G. Cancer immunotherapy comes of age. Nature 480, 480–489, doi: 10.1038/nature10673 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo EY et al. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. Journal of immunology (Baltimore, Md. : 1950) 168, 4272–4276, doi: 10.4049/jimmunol.168.9.4272 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Srivastava MK et al. Myeloid suppressor cells and immune modulation in lung cancer. Immunotherapy 4, 291–304, doi: 10.2217/imt.11.178 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kargl J. et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nature communications 8, 14381, doi: 10.1038/ncomms14381 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitchell KG et al. Neutrophil expansion defines an immunoinhibitory peripheral and intratumoral inflammatory milieu in resected non-small cell lung cancer: a descriptive analysis of a prospectively immunoprofiled cohort. J Immunother Cancer 8, doi: 10.1136/jitc-2019-000405 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lizotte PH et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI insight 1, e89014, doi: 10.1172/jci.insight.89014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lavin Y. et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell 169, 750–765.e717, doi: 10.1016/j.cell.2017.04.014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gros A. et al. PD-1 identifies the patient-specific CD8⁺ tumor-reactive repertoire infiltrating human tumors. The Journal of clinical investigation 124, 2246–2259, doi: 10.1172/jci73639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reuben A. et al. Comprehensive T cell repertoire characterization of non-small cell lung cancer. Nature communications 11, 603, doi: 10.1038/s41467-019-14273-0 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dang TO, Ogunniyi A, Barbee MS & Drilon A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert review of anticancer therapy 16, 13–20, doi: 10.1586/14737140.2016.1123626 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garon EB et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England journal of medicine 372, 2018–2028, doi: 10.1056/NEJMoa1501824 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Lim SH et al. Pembrolizumab for the treatment of non-small cell lung cancer. Expert opinion on biological therapy 16, 397–406, doi: 10.1517/14712598.2016.1145652 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Sul J. et al. FDA Approval Summary: Pembrolizumab for the Treatment of Patients With Metastatic Non-Small Cell Lung Cancer Whose Tumors Express Programmed Death-Ligand 1. The oncologist 21, 643–650, doi: 10.1634/theoncologist.2015-0498 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brahmer J. et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. The New England journal of medicine 373, 123–135, doi: 10.1056/NEJMoa1504627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hellmann MD et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 381, 2020–2031, doi: 10.1056/NEJMoa1910231 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Kaufman HL et al. The promise of Immuno-oncology: implications for defining the value of cancer treatment. Journal for immunotherapy of cancer 7, 129, doi: 10.1186/s40425-019-0594-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Topalian SL et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine 366, 2443–2454, doi: 10.1056/NEJMoa1200690 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brahmer JR et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. The New England journal of medicine 366, 2455–2465, doi: 10.1056/NEJMoa1200694 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexandrov LB et al. Clock-like mutational processes in human somatic cells. Nature genetics 47, 1402–1407, doi: 10.1038/ng.3441 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Helleday T, Eshtad S. & Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nature reviews. Genetics 15, 585–598, doi: 10.1038/nrg3729 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belfield EJ et al. DNA mismatch repair preferentially protects genes from mutation. Genome research 28, 66–74, doi: 10.1101/gr.219303.116 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turajlic S. et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. The Lancet. Oncology 18, 1009–1021, doi: 10.1016/s1470-2045(17)30516-8 (2017). [DOI] [PubMed] [Google Scholar]
- 30.McGrail DJ et al. Improved prediction of PARP inhibitor response and identification of synergizing agents through use of a novel gene expression signature generation algorithm. NPJ systems biology and applications 3, 8, doi: 10.1038/s41540-017-0011-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Helmink BA et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 577, 549–555, doi: 10.1038/s41586-019-1922-8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petitprez F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560, doi: 10.1038/s41586-019-1906-8 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Cabrita R. et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 577, 561–565, doi: 10.1038/s41586-019-1914-8 (2020). [DOI] [PubMed] [Google Scholar]
- 34.Ganesan AP et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nature immunology 18, 940–950, doi: 10.1038/ni.3775 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghorani E. et al. The T cell differentiation landscape is shaped by tumour mutations in lung cancer. Nature Cancer 1, 546–561, doi: 10.1038/s43018-020-0066-y (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imielinski M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120, doi: 10.1016/j.cell.2012.08.029 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bindea G. et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795, doi: 10.1016/j.immuni.2013.10.003 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Gentles AJ et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature medicine 21, 938–945, doi: 10.1038/nm.3909 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Djenidi F. et al. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J Immunol 194, 3475–3486, doi: 10.4049/jimmunol.1402711 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Le Floc’h A. et al. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cgamma-dependent pathway. Cancer Res 71, 328–338, doi: 10.1158/0008-5472.CAN-10-2457 (2011). [DOI] [PubMed] [Google Scholar]
- 41.Gauthier L. et al. Paxillin binding to the cytoplasmic domain of CD103 promotes cell adhesion and effector functions for CD8+ resident memory T cells in tumors. Cancer Res, doi: 10.1158/0008-5472.CAN-17-1487 (2017). [DOI] [PubMed]
- 42.Thommen DS et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nature medicine 24, 994–1004, doi: 10.1038/s41591-018-0057-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazzaschi G. et al. Low PD-1 Expression in Cytotoxic CD8(+) Tumor-Infiltrating Lymphocytes Confers an Immune-Privileged Tissue Microenvironment in NSCLC with a Prognostic and Predictive Value. Clin Cancer Res 24, 407–419, doi: 10.1158/1078-0432.CCR-17-2156 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Gros A. et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 124, 2246–2259, doi: 10.1172/JCI73639 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gros A. et al. Recognition of human gastrointestinal cancer neoantigens by circulating PD-1+ lymphocytes. J Clin Invest 129, 4992–5004, doi: 10.1172/JCI127967 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumagai S. et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nature immunology 21, 1346–1358, doi: 10.1038/s41590-020-0769-3 (2020). [DOI] [PubMed] [Google Scholar]
- 47.Koh J. et al. Clinicopathologic analysis of programmed cell death-1 and programmed cell death-ligand 1 and 2 expressions in pulmonary adenocarcinoma: comparison with histology and driver oncogenic alteration status. Modern Pathology 28, 1154–1166, doi: 10.1038/modpathol.2015.63 (2015). [DOI] [PubMed] [Google Scholar]
- 48.Rosenthal R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485, doi: 10.1038/s41586-019-1032-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rizvi NA et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, N.Y.) 348, 124–128, doi: 10.1126/science.aaa1348 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Snyder A. et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. The New England journal of medicine 371, 2189–2199, doi: 10.1056/NEJMoa1406498 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van Allen EM et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science (New York, N.Y.) 350, 207–211, doi: 10.1126/science.aad0095 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kinoshita T. et al. Prognostic value of tumor-infiltrating lymphocytes differs depending on histological type and smoking habit in completely resected non-small-cell lung cancer. Annals of oncology : official journal of the European Society for Medical Oncology 27, 2117–2123, doi: 10.1093/annonc/mdw319 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Germain C. et al. Presence of B cells in tertiary lymphoid structures is associated with a protective immunity in patients with lung cancer. American journal of respiratory and critical care medicine 189, 832–844, doi: 10.1164/rccm.201309-1611OC (2014). [DOI] [PubMed] [Google Scholar]
- 54.Faruki H. et al. Lung Adenocarcinoma and Squamous Cell Carcinoma Gene Expression Subtypes Demonstrate Significant Differences in Tumor Immune Landscape. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 12, 943–953, doi: 10.1016/j.jtho.2017.03.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parra ER et al. Validation of multiplex immunofluorescence panels using multispectral microscopy for immune-profiling of formalin-fixed and paraffin-embedded human tumor tissues. Scientific reports 7, 13380, doi: 10.1038/s41598-017-13942-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Posch F. et al. Maturation of tertiary lymphoid structures and recurrence of stage II and III colorectal cancer. Oncoimmunology 7, e1378844, doi: 10.1080/2162402x.2017.1378844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Favero F. et al. Sequenza: allele-specific copy number and mutation profiles from tumor sequencing data. Annals of oncology : official journal of the European Society for Medical Oncology 26, 64–70, doi: 10.1093/annonc/mdu479 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Subramanian A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550, doi: 10.1073/pnas.0506580102 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knijnenburg TA et al. Genomic and Molecular Landscape of DNA Damage Repair Deficiency across The Cancer Genome Atlas. Cell reports 23, 239–254.e236, doi: 10.1016/j.celrep.2018.03.076 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Assays and samples included in this study.
Fig. S1. Flow cytometry gating strategy for the lymphocyte characterization in uninvolved lung tissue and tumors.
Fig. S2. Correlation between assays.
Fig. S3. Expression of co-stimulatory/checkpoint proteins by CD103+ status.
Fig. S4. Comparison of tumor and adjacent uninvolved tissue stratified by clinicopathological features.
Fig. S5. Metrics for determining optimal number of clusters for immunotypes.
Fig. S6. tSNE analysis of uninvolved lung tissue and prognostic relevance.
Fig. S7. Exemplary multispectral immunofluorescence images.
Fig. S8. Tumor purity does not influence TMB.
Fig. S9. Relationship between indels and dMMR mutational signatures.
Fig. S10. Overview of IRAPS and immunotype signature validation.
Table S2: Immunotypes and clinical annotations.
Table S3. Immunotype gene expression signature.
Table S4. Antibodies for flow cytometry.