

ABSTRACT
Limited knowledge is available on the pharmacokinetics of rifampicin in children with tuberculous meningitis (TBM) and its penetration into brain tissue, which is the site of infection. In this analysis, we characterize the distribution of rifampicin in cerebrospinal fluid (CSF), lumbar (LCSF) and ventricular (VCSF), and brain extracellular fluid (ECF). Children with TBM were included in this pharmacokinetic analysis. Sparse plasma, LCSF, and VCSF samples were collected opportunistically, as clinically indicated. Brain ECF was sampled using microdialysis (MD). Rifampicin was quantified with liquid chromatography with tandem mass spectrometry in all samples, and 25-desacetyl rifampicin in the plasma samples. The data were interpreted with nonlinear mixed-effects modeling, with the CSF and brain ECF modeled as “effect compartments.” Data were available from 61 children, with median (min-max) age of 2 (0.3 to 10) years and weight of 11.0 (4.8 to 49.0) kg. A one-compartment model for parent and metabolite with first-order absorption and elimination via saturable hepatic clearance described the data well. Allometric scaling, maturation, and auto-induction of clearance were included. The pseudopartition coefficient between plasma and LCSF/VCSF was ~5%, while the value for ECF was only ~0.5%, possibly reflecting low recovery of rifampicin using MD. The equilibration half-life between plasma and LCSF/VCSF was ~4 h and between plasma and ECF ~2 h. Our study confirms previous reports showing that rifampicin concentrations in the LCSF are lower than in plasma and provides novel knowledge about rifampicin in the VCSF and the brain tissue. Despite MD being semiquantitative because the relative recovery cannot be quantified, our study presents a proof-of-concept that rifampicin reaches the brain tissue and that MD is an attractive technique to study site-of-disease pharmacokinetics in TBM.
KEYWORDS: WHO, brain extracellular fluid, cerebrospinal fluid, children, meningitis, microdialysis, population pharmacokinetics, rifampicin, tuberculosis
INTRODUCTION
Tuberculous meningitis (TBM) is the most severe manifestation of tuberculosis (TB) and results in high rates of death and disability. Children are among the most vulnerable populations and TBM remains the most common childhood meningitis in South Africa (1). The WHO-recommended regimen for TBM is the same as that for pulmonary TB but with a longer continuation phase (10 months for TBM versus 4 months for pulmonary TB) (2). Due to the poor blood-brain barrier (BBB) penetration of anti-TB drugs, the TBM regimen probably results in suboptimal concentrations in the central nervous system (CNS) (1). Drugs targeted at TBM should cross several barriers to reach their site of action in the CNS. The systemic circulation is separated from the CNS by the BBB and the blood-cerebrospinal fluid barrier (BCSFB). These barriers pose a therapeutic challenge by limiting entry of drugs into the CNS. Moreover, disease-specific changes in BBB permeability may have important implications for drug penetration into the brain (3). Recent reports, including a report from South Africa, investigated a shorter regimen with higher doses that proved successful (4) which led the WHO to include the short intensive regimen as an alternative in the latest update of their guidelines for TBM treatment in children (released March 21, 2022) (5).
Most TBM studies investigating CNS concentrations rely on lumbar CSF (LCSF) as a surrogate for anti-TB drug concentrations in the ventricular CSF (VCSF) and in the brain and for which repeated sampling cannot be performed. Microdialysis (MD) is a semi-invasive technique that uses a probe implanted in tissue, including the brain (6). It enables continuous measurement of the unbound concentration of molecules in the interstitial compartment of the brain over time. As such, MD enables sampling of the brain extracellular fluid (ECF) and provides complementary information on the drug distribution in the brain because repeated sampling can be performed (7). However, when used to obtain brain ECF samples, it is only semiquantitative because the extraction efficiency or relative recovery cannot be calculated (8).
Rifampicin is an important drug in the treatment of TBM owing to its potent antimicrobial effect (9). Following oral administration, rifampicin is readily absorbed, but administration with food makes absorption variable. After absorption, rifampicin enters the hepatoportal system, where it is converted primarily to 25-desacetyl rifampicin and mainly eliminated in the bile (10). Rifampicin induces its own clearance, with clearance approximately doubling after 2 weeks due to this autoinduction effect. Saturable elimination has been reported for rifampicin at higher doses due to saturation of the biliary transport mechanisms (11, 12). Rifampicin is widely distributed throughout the body and its plasma protein binding is about 80%, with most of the unbound fraction not ionized and diffusing freely into tissues (13). However, due to its lipophilicity and high molecular mass, rifampicin’s CSF penetration is suboptimal, particularly when meningeal inflammation is resolving (14, 15). Therefore, sampling brain compartments closest to the site of infection would provide useful information. In children, factors such as maturation of enzymes, body size, nutritional status, and disease severity affect children’s ability to absorb, metabolize, and eliminate the drug (16).
The objective of this study was to develop a population pharmacokinetic (PK) model for the characterization of rifampicin in plasma, LCSF, VCSF, and brain ECF for children diagnosed with definite or probable TBM. Additionally, simulations were performed to compare rifampicin exposures from the current and former WHO recommended dosing regimens.
RESULTS
Data.
A total of 61 children were enrolled in the study. The median age, weight, and weight-for-age z-score were 2 (0.3 to 10) years, 11 (4.8 to 49) kg, and −1.23 (−4.97 to 2.44), respectively. Height was missing in 25/61 (41%) of the children. The median (min - max) duration on rifampicin treatment before PK sampling was 4 (0 to 72) days. A summary of the baseline patient characteristics is shown in Table 1. A total of 307 samples were available, 91 plasma (8 below the limit of quantification [BLQ], 8.4%), 84 lumbar CSF (13 BLQ, 14.8%), 53 ventricular CSF (8 BLQ, 16.0%), and 79 brain ECF (17 BLQ, 21.5%).
TABLE 1.
Baseline participant characteristics
| Participant characteristic | Median (min to max) or no. (%) |
|---|---|
| Total (N = 61) | |
| Sex: females/males | 32 (52.5%)/29 (47.5%) |
| Age (yrs) | 2 (0.3 to 10) |
| Wt (kg) | 11 (4.8 to 49) |
| Wt-for-age z-scorea | −1.23 (−4.97 to 2.44) |
| Htb (cm) | 81 (61 to 131) |
| Ht-for-age z-scorea,b | −0.996 (−7.12 to 1.74) |
| Days since start of treatment (days) | 4 (0 to 72) |
| Dose per body wt (mg/kg) | 18.0 (9.38 to 22.5) |
The WHO and CDC tables were used for the calculation of the z-scores.
Height and height-for-age z-scores were missing for 25/61 (41.0%) participants.
Population pharmacokinetic analysis.
The schematic representation of the final structural model is depicted in Fig. 1.
FIG 1.
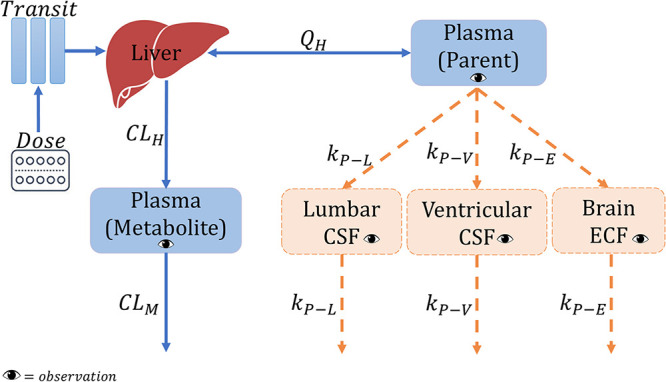
Schematic representation of the final model. CLH, hepatic clearance; CLM, metabolite clearance; kP−E, equilibration rate constant plasma-brain extracellular fluid; kP−L, equilibration rate constant plasma-lumbar cerebrospinal fluid (CSF); kP−V, equilibration rate constant plasma-ventricular CSF; QH, hepatic flow rate.
(i) Rifampicin in plasma joint parent-metabolite model. The final parent-metabolite model is a one-compartment disposition model with transit compartments absorption with the absorption rate constant, ka, fixed to be equal to the first-order transit rate constant (ktr) to simplify the model. Elimination was characterized with saturable hepatic clearance. The effect of body size on the disposition parameters of both the parent and the metabolite was included in the model using allometric scaling by weight as suggested by Anderson and Holford (17). This improved the model fit as shown by the drop in objective function value of 40 points. Allometric scaling using fat-free mass (FFM) was not tested because the height was missing for 41% of the participants. The effect of age on clearance was included by incorporating a maturation factor (MF). A MF as defined by Anderson and Holford (16) was added to the clearance as well because allometry alone cannot account for the differences in clearance between children and adults.
Estimates from Denti et al. (18) were used as priors to guide the estimation of intrinsic clearance (), central volume of distribution (), saturation and maturation of clearance and bioavailability parameters, while estimates from Chirehwa et al. (12) were used as priors for the autoinduction of clearance (ratio of at steady-state to at baseline [] and IND50). The uncertainty for all priors was 10%, except for and , which was 30%. The prior values used are shown in Table 2 footnotes. Uninformative priors, which carry the lowest weight or influence but still allow for good estimation, were used for between-subject variability in clearance. A metabolite compartment was added, where the fraction of parent converted to metabolite, was fixed to the reference value of 1 to make the model identifiable. The between-subject variability (BSV) in clearance of metabolite (CLM) (BSVCLM) was 100% correlated with the BSV in clearance. Thus, subjects were assumed to have the same random effect for both clearance and CLM, then BSVCLM was inflated by a factor that was estimated to be 2.92 as follows: BSVCLM = BSVCL x factor, therefore, allowing for more variability in BSVCLM. The effect of nasogastric tube administration was not statistically significant. The final parameter estimates are shown in Table 2.
TABLE 2.
Final population pharmacokinetic parameter estimates for rifampicin
| Parameter | Typical value (95% CI)a |
|---|---|
| Plasma (parent) parameters | |
| Vol of distribution of plasma, (L)b | 18.2 (15.7 to 23.0) |
| Mean transit time, (h)c | 0.251 Fixed |
| No. of absorption transit compartments, (_) | 0.269 (0.037 to 0.556) |
| Saturation of clearance | |
| Michaelis-Menten constant, (mg/L)d | 9.03 (7.45 to 11.2) |
| Hepatic vol of distribution, (L)e | 1 Fixed |
| Hepatic blood flow, (L/h)e | 90 Fixed |
| Unbound fraction, (_) | 0.2 Fixed |
| Clearance autoinduction | |
| Intrinsic clearance at steady-state, (L/h)b | 34.8 (27.3 to 43.1) |
| / Intrinsic clearance at baseline () (_)f | 1.60 (1.35 to 1.90) |
| Induction half-life, IND50 (days)f | 4.53 (3.69 to 5.52) |
| Maturation of clearance | |
| Postmenstrual age when maturation reaches 50%, TM50 (mo)d | 12.2 (10.1 to 14.4) |
| Shape factor for maturation, (_)d | 3.29 (2.73 to 3.97) |
| Bioavailability, (_) | 1 Fixed |
| Maturation of F | |
| at birth (_)d | 0.642 (0.544 to 0.776) |
| Age at which reaches full maturation, (yrs)d | 2.80 (2.26 to 3.43) |
| Between-subject variability in (%) | 46.3 (32.6 to 59.5) |
| Between-occasion variability in (%) | 34.9 (26.2 to 44.7) |
| Between-occasion variability in (%) | 179 (66 to 245) |
| Proportional error, parent (%) | 33.8 (27.1 to 42.6) |
| Additive error, parent (mg/L)g | 0.023 Fixed |
| Plasma (metabolite) parameters | |
| Vol of metabolite compartment, (L) | 7.76 (4.14 to 11.7) |
| Clearance of metabolite, (L/h) | 13.7 (8.56 to 20.5) |
| Fraction of parent drug metabolized, (_) | 1 Fixed |
| Factor for Between-subject variability in (-fold) | 2.92 (2.48 to 3.65) |
| Proportional error, metabolite (%) | 48.6 (40.1 – 60.3) |
| Additive error, metabolite (mg/L)g | 0.008 Fixed |
| Correlation between errors (parent and metabolite) (%) | 85.7 (75.8 to 91.8) |
Values in parentheses are the 95% confidence interval, computed with sampling importance resampling (SIR) on the final model. SIR was done sequentially; first for the plasma model then for the effect compartments model, while fixing the plasma model parameter estimates.
The disposition parameters, CLint,max, V, CLM, and VM, were allometrically scaled by weight, and the values reported here refer to an 11-kg participant (the median weight in the cohort). Priors from Denti et al. (18) with typical values of 8.58 L/h for CLint,maxSS and 17.3 L for V were used with 30% uncertainty.
The value of MTT was estimated from the data, but was fixed in the final runs to improve the model stability.
Priors from Denti et al. (18) were used with 10% uncertainty. The values of the priors were 12.48 months for TM50, 3.22 for , 0.655 for F at birth, and 2.72 years for .
These values refer to a 70-kg adult and were scaled allometrically with weight.
Priors from Chirehwa et al. with typical values of 2 for CLint,maxSS/Clearance at baseline (CLint,max0) and 4.5 days for IND50 were used with 10% uncertainty.
The estimate of the additive component of the error was not significantly different from its lower boundary of 20% of LLOQ, so it was fixed to this value.
(ii) Rifampicin in CSF and brain model. Rifampicin concentrations in the CSF and brain ECF were linked to the plasma concentrations using pseudopartition coefficients (PPC) of 4.08% for the lumbar and ventricular CSF and 0.459% for brain ECF, and equilibration rate constants of 0.176 h−1 for CSF and 0.353 h−1 for ECF, which correspond to equilibration half-lives of 3.94 h for CSF and 1.96 h for ECF. It was not statistically significant to include in the model separate pseudopartition coefficients and equilibration rate constants for each of lumbar CSF and ventricular CSF separately. It is important to note that while the typical population PPC value was the same for both lumbar and ventricular CSF, on an individual level, each participant had a different PPC for lumbar and ventricular CSF. The final parameter estimates for the CSF and brain model are shown in Table 3.
TABLE 3.
Final population pharmacokinetic parameter estimates for rifampicin for the effect compartments
| Effect compartment model | Typical value (95% CI)a |
|---|---|
| Pseudopartition coefficient for lumbar CSF and ventricular CSF, (%) | 4.60 (3.77 to 5.24) |
| Pseudopartition coefficient for brain ECF, (%) | 0.435 (0.175 to 0.714) |
| Equilibration rate constant for lumbar CSF and ventricular CSF ()b | 0.163 (0.122 to 0.206) |
| Equilibration rate constant for brain ECF c | 0.330 (0.221 to 0.580) |
| Between-subject variability in (%) | 21.6 (4.00 to 27.9) |
| Between-subject variability in (%) | 114 (82.9 to 155) |
| Proportional error, lumbar CSF (%) | 78.6 (72.2 to 98.0) |
| Additive error, lumbar CSF (mg/L)d | 0.001 Fixed |
| Proportional error, ventricular CSF (%) | 55.4 (44.4 to 75.5) |
| Additive error, ventricular CSF (mg/L)d | 0.001 Fixed |
| Proportional error, brain ECF (%) | 51.1 (40.6 to 69.4) |
| Additive error, brain ECF (mg/L)d | 0.001 Fixed |
Values in parentheses are the 95% confidence interval, computed with sampling importance resampling (SIR) on the final model. SIR was done sequentially; first for the plasma model then for the effect compartments model, while fixing the plasma model parameter estimates.
The equilibration rate constant corresponds to an equilibration half-life of 4.23 h for lumbar CSF and ventricular CSF.
The equilibration rate constant corresponds to an equilibration half-life of 2.09 h for the brain ECF.
The estimate of the additive component of the error was not significantly different from its lower boundary of 20% of LLOQ, so it was fixed to this value.
The effect of CSF albumin, CSF total protein, and ratio of CSF:plasma albumin on the PPC were not statistically significant. The albumin and total protein values are shown in Table S1. Fig. 2 shows the visual predictive check (VPC) of the final model.
FIG 2.

Visual predictive check (log scale) for the final model stratified by observation. The lower, middle, and upper solid lines are the 2.5th, 50th, and 97.5th percentiles of the observed concentrations. The shaded areas are the 95% confidence intervals for the same percentiles. The circles represent the observed concentrations.
Simulations.
The typical child with TBM in this cohort was 2 years old and weighed 11 kg. According to the short intensive regimen in the 2022 WHO guidelines (5), this child would receive 4 HR 50/75 (isoniazid 50 mg / rifampicin 75 mg) dispersible tablets (i.e., total dose of rifampicin is 300 mg, 27.3 mg/kg) versus 3 tablets of RHZ 60/30/150 (rifampicin 60 mg / isoniazid 30 mg / pyrazinamide 150 mg) (i.e., total dose of rifampicin is 180 mg, 16.4 mg/kg) according to the 2014 WHO guidelines. The simulated typical concentration-time profiles in plasma, lumbar and ventricular CSF, and brain ECF are presented in Fig. 3, in which we can see that with the newer regimen, the exposures in both plasma and CSF are higher, which is particularly important in TBM because it is needed to attain high exposure early in treatment. Fig. 4 shows the simulated exposure in terms of area under the concentration-time curve from 0 to 24 hours (AUC0−24h) for the same compartments. Fig. S1 shows the simulated AUC0−24h for plasma, CSF, and ECF across all weight bands.
FIG 3.

Simulated typical concentration-time profiles for the typical participant (11 kg and 2 years of age) (a) plasma, (b) lumbar and ventricular cerebrospinal fluid, and (c) brain extracellular fluid shown for the 2014 WHO regimen and the 2022 WHO short intensive TB meningitis regimen, as well as for the first-dose versus the steady-state (please note that true rifampicin concentrations in the brain tissue are unknown as relative recovery cannot be calculated). The dashed lines represent the MIC levels of 0.2 and 0.4 mg/L (42).
FIG 4.

Expected exposures in terms of area under the curve () for the typical participant (11 kg and 2 years of age) in (a) plasma, (b) lumbar and ventricular CSF, and (c) brain ECF for the 2014 WHO regimen and the 2022 WHO short intensive TB meningitis dosing recommendations, as well as, for the first-dose versus the steady-state (please note that true rifampicin concentrations in the brain tissue are unknown as relative recovery cannot be calculated). The box represents the median and interquartile range and the whiskers correspond to the 5th and 95th percentiles.
DISCUSSION
In this study, we characterized the pharmacokinetics of rifampicin in in plasma, lumbar and ventricular CSF, and brain ECF children with TBM. The population equilibration delay was 4 h between plasma and lumbar as well as ventricular CSF and was 2 h between plasma and brain ECF. As described by the PPC, the extent of rifampicin’s penetration was 5% for the lumbar and ventricular CSF and 0.4% for the brain ECF. Importantly, our study is the first to report ventricular and ECF, which significantly adds to the limited knowledge available on rifampicin’s distribution into the central nervous system. These sites are more likely to reflect drug concentrations at the site of TBM infection than the plasma and lumbar (spinal) CSF, of previously published pharmacokinetic models, and therefore extend our understanding of rifampicin exposure in the brain. Increasing evidence from protein, inflammatory mediator, and transcriptomic data suggest that spinal CSF and ventricular (brain) CSF are different (19, 20), in part perhaps because of the frequency of spinal disease in TBM patients that may affect CSF dynamics (21) but also potentially because of barrier permeability differences between brain and spinal cord capillaries.
Recognizing the limits of spinal CSF to study brain penetration of drugs, the use of MD to interpret drug concentrations in the brain is reported as a gold standard technique for quantitative and time-resolved analysis of brain PK studies (22). However, the interpretation of drug concentrations using MD has some limitations. First, it is difficult to determine the relative recovery rate in the microdialysate from tissue ECF, i.e., what percentage of the actual concentration in the tissue is retrieved in the MD samples. Second, TBM is characterized by perfusion differences due to variable small and large vessel disease; therefore, distribution of drug in the brain may be highly variable (21, 23). Nevertheless, our recovery of rifampicin in the brain ECF provides proof-of-concept that rifampicin does penetrate the brain tissue—its targeted site of action in pediatric TBM patients. Furthermore, under static conditions, the method provides the first demonstration of repeated measures of rifampicin in the brain, i.e., the relative values provide multiple measures in a single patient over several time points postdose. The simulations show that the short intensive regimen recently included in the 2022 WHO operational handbook provides drug exposures that are higher and more equal across the different weight bands.
The difference in the plasma-brain ECF and plasma-CSF equilibration rate could be attributed to changes in CSF volume which may affect pressure equilibrium and the concentration gradient between brain and CSF (24). Furthermore, previous simulations showed that a change in CSF dynamics changed the CSF pharmacokinetic profiles but not necessarily the brain ECF profiles regardless of the drug’s physiochemical properties (25). Pathologically, CSF dynamics will be affected by hydrocephalus (HCP), where CSF volume in the ventricles is larger (which will affect dilution of drug entering through the choroid plexus or brain interstitium) and spinal subarachnoid disease (which will affect circulation between brain and spinal CSF) (25). Finally, differences in the blood-spinal cord barrier (compared to the BBB) (26) and the influence of reduced CSF flow on permeability must also be taken into account (27). Collectively, these findings reiterate that caution must be applied when interpreting lumbar CSF as a proxy for brain ECF concentrations in CNS diseases (25).
Due to the sparseness of the plasma data collected, priors from previous models (12, 18) were used to stabilize the estimation of the plasma model’s parameter estimates. is somewhat lower than the values from Denti et al. (18), Garcia-Prats et al. (Opti-Rif trial) (28), and Zvada et al. (29) in South African children with pulmonary tuberculosis. This could be attributed to the fact that children with TBM are typically sicker than those with pulmonary TB, and particularly the children in this study cohort who were mostly in the intensive care unit (ICU) and had HCP requiring intervention. In addition to surgical interventions, critically ill patients have pathophysiological changes that may affect drug pharmacokinetics (30). The plasma and CSF concentrations obtained in our cohort are similar to those from studies in children with TBM receiving doses between 15 and 20 mg/kg (31, 32). In the study by Panjasawatwong et al. (32), in children with TBM, the pseudopartition coefficient estimated was 17% versus 4% in our model. In their model, the lumbar CSF observations were modeled as a CSF compartment linked to the plasma compartment (33). Their model relies on an empirical formula to calculate the CSF compartment volume. We get around the need to estimate a CSF volume by modeling the CSF as an effect compartment. Svensson et al. (30) and Savic et al. (31) have published models in adults with TBM, where the pseudopartition coefficients and equilibration half-lives were 5% and 2 h and 8% and 6 h, respectively. In addition, in previously published animal models, the brain tissue:plasma rifampicin concentrations ratios were reported to be between 0.02 and 0.05 (23, 34). Both studies found the rifampicin CSF concentrations to be lower than those in the brain tissue/lesion. However, comparing ECF concentrations to either the plasma or the CSF in our study should be cautiously interpreted since the absolute concentrations in the brain ECF samples obtained with MD cannot be calculated. Levels of CSF protein or albumin are expected to correlate with the pseudopartition coefficient because these are often considered markers of the degree of inflammation of the blood-brain barrier, and hence, its permeability to drugs. Svensson et al. (33) found a linear relationship between the CSF protein level and rifampicin penetration into the lumbar CSF; with each 10-fold change in protein levels, the penetration coefficient increases by 63%. In Panjasawatwong et al. (32), there is an exponential relationship between CSF protein concentration and the PC where an increase of 1 g/L in CSF protein concentration resulted in a 1.28-fold increase in PC. In our analysis, we did not find a significant effect for either the levels of albumin or total protein, which could be due to fewer samples and narrower range of CSF protein values in our cohort. However, we previously demonstrated differences in biomarkers, inflammatory mediators, and differential gene expression in time-linked samples in patients with TBM. Combined with the recognition that reduced spinal CSF may increase protein concentrations in spinal CSF compared to ventricular CSF (27), these factors may contribute to the explanation of lack of effect.
Both strengths and limitations in our study are related to the use of MD sampling technique. MD provides the unique opportunity to obtain rifampicin samples from the brain tissue, which is the site of infection/drug action, in addition to, the opportunity for serial/continuous sampling. On the other hand, the MD conditions, e.g., flow rate, semipermeable membrane pore size, may affect the drug’s recovery in addition to the limitations mentioned above. The exact location of MD catheter placement in the brain tissue may also affect the concentrations measured; however, there is currently no method that accomplishes both spatial and temporal resolution in studying the brain. It has also been suggested that the CSF collection method may influence drug concentrations; however, the CSF drains were in the lateral ventricle which is likely the best location for CSF sampling as it is the most proximal location based on current knowledge on where the bulk of CSF is produced and on CSF flow dynamics (35, 36). Comparison of ventricular CSF and lumbar CSF is limited by the opportunistic nature of sampling and not all patients had time-linked samples for comparison; there may be bias in why some participants had ventricular or lumbar sampling and not both. Lastly, these data—like the existing literature on rifampicin pharmacokinetics in TBM—report only total rifampicin concentrations, while only the unbound fraction that is active. Our use of the 100 kDa membrane reflects total drug concentration rather than unbound drug. Unfortunately, there are no data available for rifampicin protein binding in the CSF. CSF protein binding dynamics is expected to be different than that of the plasma because the CSF protein levels are different and are more variable between patients.
To overcome some of the methodological limitations of MD, suggested future research include in vitro experiments to optimize the MD method. In terms of study design, because oral absorption may be variable, particularly in critically ill patients, the use of intravenous administration of rifampicin would be an attractive alternative to eliminate the variability in rifampicin absorption following oral administration and improve our description of plasma PK, as well as to mitigate the challenges of administering drugs to children, e.g., crushing tablets and administration by an oral syringe. If more CSF and brain ECF samples could be obtained, a more complex mechanistic model that accounts for drug transfer between the LCSF, VCSF, and brain ECF could be tested. Still, there is a substantial amount of the underlying physiology in humans that require better definition and some uncertainties that still exist, largely because of lack of human data and methodological limitations in the existing animal data. For example, the flux between the ECF and ventricular CSF has been predicted but not yet fully understood or quantified (22, 25). The flux with the spinal CSF also needs better definition because of the influence of relative blocks of CSF flow in pathology (27) and the increased permeability of the tight junctions of capillaries supplying the spinal cord (37).
In conclusion, we show that rifampicin’s extent of penetration into both the lumbar and ventricular CSF is ~5% and provide proof-of-concept that rifampicin reaches the brain tissue. Still, further studies need to be conducted to better define antitubercular drug exposure in CSF and brain in children with TBM, including unbound drug, and treatment strategies to increase this exposure.
MATERIALS AND METHODS
Study participants.
Children presenting to the Red Cross War Memorial Children’s Hospital with definite or probable TBM were enrolled between January 2017 and September 2019. They were enrolled regardless of how long they had been on rifampicin-based treatment. All patients were managed according to standard institutional protocol (38) and no procedures were carried out for research. The patients were categorized into two groups: (i) patients who underwent neurosurgical procedures to manage TBM HCP, and (ii) patients who presented with lowered levels of consciousness, had invasive monitoring with MD, and had placement of external ventricular drain (EVD) that was part of the institutional protocol for urgent decrease of intracranial pressure. Brain MD was used in critically ill patients to guide therapy based on changes in brain chemistry due cerebral ischemia (38). Further details about the clinical use of MD are included in the supplemental material. Remnant ECF fluid (after bedside clinical analysis was performed) was frozen at −80°C and used for rifampicin assays. Ethical approval was obtained from the University of Cape Town human research ethics committee (HREC 564/2012 and 070/2018). Hospital clearance was obtained from the Western Cape Department of Health. In addition, the parents or legal guardians of all participants provided informed consent on their behalf.
Drug administration.
All TBM patients were treated according to the Western Cape regimen that is typically used in South Africa which consists of rifampicin (20 mg/kg), isoniazid (15 to 20 mg/kg), pyrazinamide (40 mg/kg), and ethionamide (20 mg/kg) for 2 months followed by a 4-month continuation phase of rifampicin and isoniazid, according to weight-band based dosing. A hospital nurse administered the anti-TB drugs orally or via nasogastric tube.
Sample collection. (i) Plasma.
Three serial blood samples (0.6 mL) were drawn on a single day in the first and second week, and where possible at 2-, 4-, and 6-h postdose.
(ii) Lumbar and Ventricular CSF. Clinically indicated CSF samples were collected at random time points, from procedures in published protocols (38), up to 24-h postdose into sterile 15-mL tubes and kept on ice. Ventricular CSF samples were taken either at placement of a ventriculoperitoneal shunt or an EVD, or when clinically indicated from an indwelling EVD. Lumbar CSF samples were taken either as a diagnostic procedure or for medical therapy of raised intracranial pressure related to HCP.
(iii) Brain ECF using MD technique. An MD catheter with a 100-kDa membrane was placed concurrently with the primary ventricular drain and monitor into the same region (usually right frontal white matter) or sometimes on the side where hypodensity was more prominent on the initial head computed tomography scan. MD vials were changed at hourly intervals and analyzed at the bedside for clinical purposes. As MD volumes are typically small, remnant hourly samples were pooled over 2- to 3-h epochs to ensure sufficient volumes for analysis. Concentrations of substances in the microdialysate are a percentage of the true concentrations in the ECF and not absolute concentrations. This percentage is termed relative recovery. More details on sample collection and bedside protocols can be found in the supplemental material.
Drug quantification.
Total rifampicin was assayed in all samples: plasma, lumbar CSF, ventricular CSF, and brain ECF. Additionally, rifampicin’s primary metabolite, 25-desacetyl rifampicin, was assayed in the plasma samples. High-performance liquid chromatography with tandem mass spectrometry detection methods were developed and validated at the Division of Clinical Pharmacology, University of Cape Town for the assays. The method was validated over the ranges 0.117 to 30.0 μg/mL for rifampicin and 0.0391 to 10.0 μg/mL for 25-desacetyl-rifampicin in plasma, and 5 to 2,500 ng/mL for the rifampicin in CSF and ECF assay. A more detailed description of the analytical methods can be found in the supplemental material.
Population PK analysis.
A population pharmacokinetic model was developed to describe rifampicin pharmacokinetics in plasma, CSF, and brain ECF using nonlinear mixed-effects modeling in NONMEM 7.5.0 and the algorithm first-order conditional estimation with eta-epsilon interaction (FOCE-I). Pirana was used for model management, and Perl-speaks-NONMEM (PsN) 4.9.0 and R 4.0.3 were used for postprocessing NONMEM results and generating figures (39). Modeling was done sequentially, first developing the joint parent-metabolite model in plasma, including variability and residual error, then developing a full model incorporating the CSF and brain ECF observations, while fixing the population parameter estimates for the plasma joint model.
(i) Rifampicin in plasma joint parent-metabolite model. Previously published rifampicin models in children with pulmonary TB by Denti et al. (18) and Zvada et al. (29) were used as a starting point for modeling the rifampicin in plasma data. Thereafter, the parent PK model was extended to include the metabolite. From these data, it is not mathematically possible to estimate both the FM and VM simultaneously (i.e., the model is structurally unidentifiable). In order to solve this identifiability issue, two methods were tested: the first was to assume that the parent is fully converted to the metabolite, the second was to assume that the volume of distribution of the metabolite is equal to that of the parent.
Therefore, FM was fixed to 1 and the and CLM were estimated. A correction factor was included in the model to adjust for the difference in molecular weight at the conversion of rifampicin (822.94 g/mol) to 25-desacetyl rifampicin (780.90 g/mol). Because rifampicin and its metabolite were quantified simultaneously, a correlation of measurement error factor was included.
(ii) Rifampicin in CSF and brain model. The concentrations obtained from the LCSF, VCSF, and brain ECF were modeled using three separate “effect compartments” in a manner similar to this described by Sheiner et al. (40) with the following equation:
where is the first-order equilibration rate constant between rifampicin in the central compartment (i.e., plasma) and the effect compartment, and it can also be expressed as equilibration half-life. is the pseudopartition coefficient which describes the extent of partitioning or penetration of rifampicin between the plasma and each of the effect compartments. is the predicted rifampicin concentration in plasma at time . is the concentration in the effect compartment. Effect compartments are assumed to have a negligible volume compared to the central compartment, with negligible drug transfer between the two compartments.
The effects of CSF and plasma albumin, CSF total protein, CSF glucose, and CSF:plasma albumin ratio were tested as covariates on the , as well as the effects of body weight, age, weight-for-age z-score, duration of rifampicin treatment, and route of administration (oral versus nasogastric tube) on the plasma model parameters. More details regarding the population pharmacokinetic modeling are in the supplemental material.
Simulations.
Simulations were done in Berkeley Madonna version 10.1.2 using the final model to simulate rifampicin exposures in the different matrices for the typical TBM pediatric patient in the cohort. For all matrices, concentration-time profiles were simulated and AUC0−24h were calculated. Additionally, Monte-Carlo Simulations were carried out in NONMEM to compare rifampicin exposures following administration according to the newly released updated WHO dosing guidelines on March 21, 2022 (5) versus the previous WHO guidelines, in addition to comparing exposures on the first day of rifampicin treatment versus at steady-state. The was compared for all weight bands for the updated dosing guidelines using the virtual pediatric population in Wasmann et al. (41). Simulations were repeated 10,000 times.
Data availability.
The data that support this research can be made available upon reasonable request by contacting Paolo Denti (paolo.denti@uct.ac.za).
ACKNOWLEDGMENTS
We thank all families/guardians for participation in this study, and the whole study team. Computations were performed using facilities provided by the University of Cape Town’s ICTS High Performance Computing team: https://ucthpc.uct.ac.za.
A.F. is supported by the NRF SARChI Chair of Clinical Neurosciences, which also provided for support for the clinical components of the work. M.(P).T. was supported by a NRF postgraduate student grant and the Oppenheimer Memorial Trust for postgraduate study. H.M. is supported by the Wellcome Trust (206379/Z/17/Z). The University of Cape Town Clinical PK Laboratory is supported in part via the Adult Clinical Trial Group (ACTG), by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health under award numbers UM1 AI068634, UM1 AI068636, and UM1 AI106701; as well as the Infant Maternal Pediatric Adolescent AIDS Clinical Trials Group (IMPAACT), funding provided by National Institute of Allergy and Infectious Diseases (U01 AI068632), the Eunice Kennedy Shriver National Institute of Child Health and Human Development, and National Institute of Mental Health grant AI068632. M.T.A. received training in research that was supported by the Fogarty International Center of the National Institutes of Health under Award Number D43 TW010559.
The funders had no role in the study design, data collection, data analysis, data interpretation, or writing of this report.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Wilkinson RJ, Rohlwink U, Misra UK, Van Crevel R, Mai NTH, Dooley KE, Caws M, Figaji A, Savic R, Solomons R, Thwaites GE, Aarnoutse R, Van Laarhoven A, Dian S, Bahr NC, Boulware DR, Cronan MR, Tobin D, Dunstan S, Feng GD, Shi X, Wang T, Marais S, McIlleron H, Meintjes G, Rizal A, Ruslami R, Garg RK, Gupta M, Gupta RK, Gupta S, Heemskerk AD, Nguyên TTT, Nguyên MTH, Srinivasan V, Trân TTB, Vân Trân TT, Trân ATN, Vo THY, Wolbers M, Kalita J, Misra UK, Lai R, Marais BJ, Trinh MQ, Nguyên BD, Nguyên YB, Patel V, Pouplin T, Ramakrishnan L. Tuberculous Meningitis International Research Consortium, et al. . 2017. Tuberculous meningitis. Nat Rev Neurol 13:581–598. 10.1038/nrneurol.2017.120. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. 2014. Guidance for national tuberculosis programmes on the management of tuberculosis in children, second edition. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 3.Nau R, Sörgel F, Eiffert H. 2010. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin Microbiol Rev 23:858–883. 10.1128/CMR.00007-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Toorn R, Schaaf HS, Laubscher JA, van Elsland SL, Donald PR, Schoeman JF. 2014. Short intensified treatment in children with drug-susceptible tuberculous meningitis. Pediatr Infect Dis J 33:248–252. 10.1097/INF.0000000000000065. [DOI] [PubMed] [Google Scholar]
- 5.WHO operational handbook on tuberculosis. 2022. Module 5: management of tuberculosis in children and adolescents. World Health Organization, Geneva, Switzerland. [PubMed] [Google Scholar]
- 6.Yamamoto Y, Danhof M, de Lange ECM. 2017. Microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J 19:891–909. 10.1208/s12248-017-0050-3. [DOI] [PubMed] [Google Scholar]
- 7.Mindermann T, Zimmerli W, Gratzl O. 1998. Rifampin concentrations in various compartments of the human brain: a novel method for determining drug levels in the cerebral extracellular space. Antimicrob Agents Chemother 42:2626–2629. 10.1128/AAC.42.10.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ungerstedt U, Rostami E. 2004. Microdialysis in neurointensive care. Curr Pharm Des 10:2145–2152. 10.2174/1381612043384105. [DOI] [PubMed] [Google Scholar]
- 9.Gumbo T, Louie A, Deziel MR, Liu W, Parsons LM, Salfinger M, Drusano GL. 2007. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob Agents Chemother 51:3781–3788. 10.1128/AAC.01533-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Acocella G. 1983. Pharmacokinetics and metabolism of rifampin in humans. Clin Infect Dis 5:S428–S432. 10.1093/clinids/5.Supplement_3.S428. [DOI] [PubMed] [Google Scholar]
- 11.Svensson RJ, Aarnoutse RE, Diacon AH, Dawson R, Gillespie SH, Boeree MJ, Simonsson USH. 2018. A population pharmacokinetic model incorporating saturable pharmacokinetics and autoinduction for high rifampicin doses. Clin Pharmacol Ther 103:674–683. 10.1002/cpt.778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chirehwa MT, Rustomjee R, Mthiyane T, Onyebujoh P, Smith P, McIlleron H, Denti P. 2016. Model-based evaluation of higher doses of rifampin using a semimechanistic model incorporating autoinduction and saturation of hepatic extraction. Antimicrob Agents Chemother 60:487–494. 10.1128/AAC.01830-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Acocella G. 1978. Clinical pharmacokinetics of rifampicin. Clin Pharmacokinet 3:108–127. 10.2165/00003088-197803020-00002. [DOI] [PubMed] [Google Scholar]
- 14.Kaojarern S, Supmonchai K, Phuapradit P, Mokkhavesa C, Krittiyanunt S. 1991. Effect of steroids on cerebrospinal fluid penetration of antituberculous drugs in tuberculous meningitis. Clin Pharmacol Ther 49:6–12. 10.1038/clpt.1991.2. [DOI] [PubMed] [Google Scholar]
- 15.Nau R, Sörgel F, Prange HW. 1994. Lipophilicity at pH 7.4 and molecular size govern the entry of the free serum fraction of drugs into the cerebrospinal fluid in humans with uninflamed meninges. J Neurol Sci 122:61–65. 10.1016/0022-510x(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 16.Anderson BJ, Holford NHG. 2009. Mechanistic basis of using body size and maturation to predict clearance in humans. Drug Metab Pharmacokinet 24:25–36. 10.2133/dmpk.24.25. [DOI] [PubMed] [Google Scholar]
- 17.Anderson BJ, Holford NHG. 2008. Mechanism-based concepts of size and maturity in pharmacokinetics. Annu Rev Pharmacol Toxicol 48:303–332. 10.1146/annurev.pharmtox.48.113006.094708. [DOI] [PubMed] [Google Scholar]
- 18.Denti P, Wasmann RE, van Rie A, Winckler J, Bekker A, Rabie H, Hesseling AC, van der Laan LE, Gonzalez-Martinez C, Zar HJ, Davies G, Wiesner L, Svensson EM, McIlleron HM. 2022. Optimizing dosing and fixed-dose combinations of rifampicin, isoniazid, and pyrazinamide in pediatric patients with tuberculosis: a prospective population pharmacokinetic study. Clin Infect Dis 75:141–151. 10.1093/cid/ciab908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rohlwink UK, Figaji A, Wilkinson KA, Horswell S, Sesay AK, Deffur A, Enslin N, Solomons R, Van Toorn R, Eley B, Levin M, Wilkinson RJ, Lai RPJ. 2019. Tuberculous meningitis in children is characterized by compartmentalized immune responses and neural excitotoxicity. Nat Commun 10:3767. 10.1038/s41467-019-11783-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rohlwink UK, Mauff K, Wilkinson KA, Enslin N, Wegoye E, Wilkinson RJ, Figaji AA. 2017. Biomarkers of cerebral injury and inflammation in pediatric tuberculous meningitis. Clin Infect Dis 65:1298–1307. 10.1093/cid/cix540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rohlwink UK, Kilborn T, Wieselthaler N, Banderker E, Zwane E, Figaji AA. 2016. Imaging features of the brain, cerebral vessels and spine in pediatric tuberculous meningitis with associated hydrocephalus. Pediatr Infect Dis J 35:e301–e310. 10.1097/INF.0000000000001236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Lange ECM. 2013. Utility of CSF in translational neuroscience. J Pharmacokinet Pharmacodyn 40:315–326. 10.1007/s10928-013-9301-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruiz-Bedoya CA, Mota F, Tucker EW, Mahmud FJ, Reyes-Mantilla MI, Erice C, Bahr M, Flavahan K, de Jesus P, Kim J, Foss CA, Peloquin CA, Hammoud DA, Ordonez AA, Pardo CA, Jain SK. 2022. High-dose rifampin improves bactericidal activity without increased intracerebral inflammation in animal models of tuberculous meningitis. J Clin Invest 132. 10.1172/JCI155851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Lange EC, Danhof M, de Boer AG, Breimer DD. 1997. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood–brain barrier. Brain Res Brain Res Rev 25:27–49. 10.1016/S0165-0173(97)00014-3. [DOI] [PubMed] [Google Scholar]
- 25.Saleh MAA, Loo CF, Elassaiss-Schaap J, De Lange ECM. 2021. Lumbar cerebrospinal fluid-to-brain extracellular fluid surrogacy is context-specific: insights from LeiCNS-PK3.0 simulations. J Pharmacokinet Pharmacodyn 48:725–741. 10.1007/s10928-021-09768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chopra N, Menounos S, Choi JP, Hansbro PM, Diwan AD, Das A. 2021. Blood-spinal cord barrier: its role in spinal disorders and emerging therapeutic strategies. NeuroSci 3:1–27. 10.3390/neurosci3010001. [DOI] [Google Scholar]
- 27.Reiber H. 2003. Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci 21:79–96. [PubMed] [Google Scholar]
- 28.Garcia-Prats AJ, Svensson EM, Winckler J, Draper HR, Fairlie L, van der Laan LE, Masenya M, Schaaf HS, Wiesner L, Norman J, Aarnoutse RE, Karlsson MO, Denti P, Hesseling AC. 2021. Pharmacokinetics and safety of high-dose rifampicin in children with TB: the Opti-Rif trial. J Antimicrob Chemother 76:3237–3246. 10.1093/jac/dkab336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zvada SP, Denti P, Donald PR, Schaaf HS, Thee S, Seddon JA, Seifart HI, Smith PJ, McIlleron HM, Simonsson USH. 2014. Population pharmacokinetics of rifampicin, pyrazinamide and isoniazid in children with tuberculosis: in silico evaluation of currently recommended doses. J Antimicrob Chemother 69:1339–1349. 10.1093/jac/dkt524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blot SI, Pea F, Lipman J. 2014. The effect of pathophysiology on pharmacokinetics in the critically ill patient–concepts appraised by the example of antimicrobial agents. Adv Drug Deliv Rev 77:3–11. 10.1016/j.addr.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 31.Donald PR. 2010. Cerebrospinal fluid concentrations of antituberculosis agents in adults and children. Tuberculosis (Edinb) 90:279–292. 10.1016/j.tube.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 32.Panjasawatwong N, Wattanakul T, Hoglund RM, Bang ND, Pouplin T, Nosoongnoen W, Ngo VN, Day JN, Tarning J. 2020. Population pharmacokinetic properties of antituberculosis drugs in Vietnamese children with tuberculous meningitis. Antimicrob Agents Chemother 65. 10.1128/AAC.00487-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Svensson EM, DIan S, Te Brake L, Ganiem AR, Yunivita V, Van Laarhoven A, Van Crevel R, Ruslami R, Aarnoutse RE. 2020. Model-based meta-analysis of rifampicin exposure and mortality in Indonesian tuberculous meningitis trials. Clin Infect Dis 71:1817–1823. 10.1093/cid/ciz1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tucker EW, Guglieri-Lopez B, Ordonez AA, Ritchie B, Klunk MH, Sharma R, Chang YS, Sanchez-Bautista J, Frey S, Lodge MA, Rowe SP, Holt DP, Gobburu JVS, Peloquin CA, Mathews WB, Dannals RF, Pardo CA, Kannan S, Ivaturi VD, Jain SK. 2018. Noninvasive 11C-rifampin positron emission tomography reveals drug biodistribution in tuberculous meningitis. Sci Transl Med 10:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Battal B, Kocaoglu M, Bulakbasi N, Husmen G, Tuba Sanal H, Tayfun C. 2011. Cerebrospinal fluid flow imaging by using phase-contrast MR technique. Br J Radiol 84:758–765. 10.1259/bjr/66206791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Di Paolo A, Gori G, Tascini C, Danesi R, Del Tacca M. 2013. Clinical pharmacokinetics of antibacterials in cerebrospinal fluid. Clin Pharmacokinet 52:511–542. 10.1007/s40262-013-0062-9. [DOI] [PubMed] [Google Scholar]
- 37.Bartanusz V, Jezova D, Alajajian B, Digicaylioglu M. 2011. The blood-spinal cord barrier: morphology and clinical implications. Ann Neurol 70:194–206. 10.1002/ana.22421. [DOI] [PubMed] [Google Scholar]
- 38.Figaji AA, Fieggen AG. 2010. The neurosurgical and acute care management of tuberculous meningitis: evidence and current practice. Tuberculosis (Edinb) 90:393–400. 10.1016/j.tube.2010.09.005. [DOI] [PubMed] [Google Scholar]
- 39.Keizer RJ, Karlsson MO, Hooker A. 2013. Modeling and simulation workbench for NONMEM: tutorial on Pirana, PsN, and Xpose. CPT Pharmacometrics Syst Pharmacol 2:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham JS, Francisco C. 1979. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d-tubocurarine. [DOI] [PubMed]
- 41.Wasmann RE, Svensson EM, Walker AS, Clements MN, Denti P. 2021. Constructing a representative in-silico population for paediatric simulations: application to HIV-positive African children. Br J Clin Pharmacol 87:2847–2854. 10.1111/bcp.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rastogi N, Labrousse V, Goh KS. 1996. In vitro activities of fourteen antimicrobial agents against drug susceptible and resistant clinical isolates of Mycobacterium tuberculosis and comparative intracellular activities against the virulent H37Rv strain in human macrophages. Curr Microbiol 33:167–175. 10.1007/s002849900095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material. Download aac.01474-22-s0001.pdf, PDF file, 0.3 MB (280.4KB, pdf)
Data Availability Statement
The data that support this research can be made available upon reasonable request by contacting Paolo Denti (paolo.denti@uct.ac.za).