

SUMMARY
Patients with Smoldering Multiple Myeloma (SMM) are observed until progression, but early treatment may improve outcomes. We conducted a Phase II trial of Elotuzumab, Lenalidomide, and Dexamethasone (EloLenDex) in patients with high-risk SMM and performed single-cell RNA and T cell Receptor (TCR) sequencing on 149 bone marrow (BM) and peripheral blood (PB) samples from patients and healthy donors (HD). We find that early treatment with EloLenDex is safe and effective and provide a comprehensive characterization of alterations in immune cell composition and TCR repertoire diversity in patients. Importantly, we show that the similarity of a patient’s immune cell composition to that of HD may have prognostic relevance at diagnosis and post-treatment, and the abundance of GZMK+ CD8+ effector memory T cells may be associated with treatment response. Lastly, we uncover similarities between immune alterations observed in the BM and PB, suggesting that PB-based immune profiling may have diagnostic and prognostic utility.
Keywords: Smoldering Multiple Myeloma, Bone marrow, Peripheral blood, Immune profiling, Single-cell RNA sequencing, Single-cell TCR sequencing, Immune biomarkers, Immunotherapy, Granzyme K
eTOC blurb
Sklavenitis-Pistofidis et al. report results of a Phase II trial of EloLenDex in patients with high-risk smoldering multiple myeloma and use single-cell RNA sequencing to identify biomarkers of outcome. They show that immune cell composition impacts progression-free survival and that blood-based immune profiling can detect immune alterations observed in the marrow.
Graphical Abstract
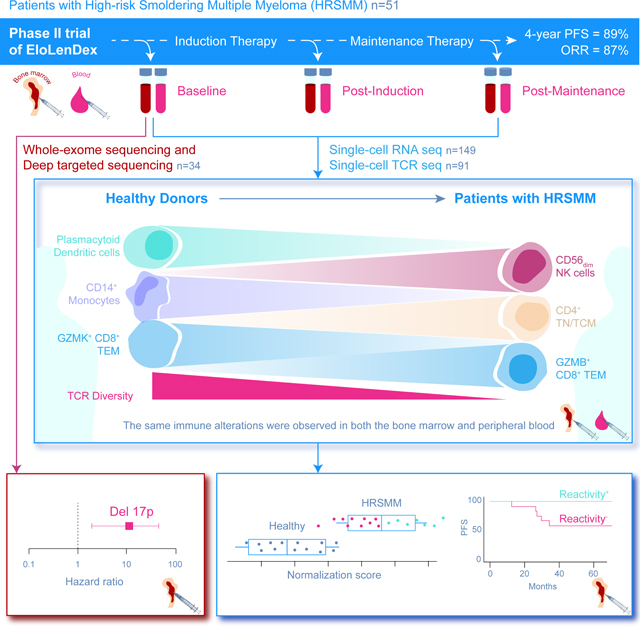
INTRODUCTION
Multiple Myeloma (MM) is an incurable plasma cell malignancy that resides in the bone marrow (BM) and is preceded by an asymptomatic condition called Smoldering MM (SMM) (Weiss et al., 2009). Approximately half of patients with SMM will progress within 5 years from diagnosis, but not all patients with SMM progress (Kyle et al., 2007; Mateos et al., 2020). Therefore, patients with SMM are observed until end-organ damage occurs, which signifies progression to overt MM, warranting treatment (Visram et al., 2021). Two Phase III clinical trials, testing either Lenalidomide/Dexamethasone (LenDex) or Lenalidomide alone (Len) versus observation, demonstrate that early treatment can significantly prolong progression-free survival (PFS) in patients with high-risk SMM (HRSMM) and, in one instance, sustainably prolong overall survival (OS) (Mateos et al., 2013; Mateos et al., 2022; Lonial et al., 2020). Nevertheless, identifying the patients who will benefit the most from early intervention remains an unmet clinical need (Kim et al., 2020; Mateos et al., 2015). Currently, patients at high risk of progression are identified using prediction models based on clinical variables (Kyle et al., 2007; Pérez-Persona et al., 2007), such as the “20–2-20” model developed by the International Myeloma Working Group (IMWG), which stratifies patients based on the BM plasma cell infiltration (>20%), the amount of M-protein in the serum (>2 g/dL), and the free light-chain ratio (>20) (Mateos et al., 2020). In recent years, it has become evident that genomic biomarkers detected through cytogenetics and sequencing, including translocations t(4;14) and t(14;16), gain or amplification of chr1q, del13q, del17p, translocations and copy number abnormalities (CNAs) involving MYC, and KRAS mutations, may be associated with faster progression in untreated patients with SMM (Mateos et al., 2020; Bustoros et al., 2020; Misund et al., 2019; Boyle et al., 2021; Bolli et al., 2018; Oben et al., 2021). Moreover, tumor-extrinsic microenvironmental factors are important for disease control at early stages, and the BM immune microenvironment is altered in both composition and functionality in patients with SMM (Zavidij et al., 2020; Bailur et al., 2019; Dhodapkar et al., 2003; Das et al., 2016). However, little is known about the role of BM-based immune profiling in patient prognostication and the utility of peripheral blood (PB) for the detection of immune alterations related to disease.
Here, we conducted a Phase II trial of the immunotherapeutic anti-SLAMF7 antibody, Elotuzumab, in combination with LenDex (EloLenDex, E-PRISM study; ClinicalTrials.gov Identifier: NCT02279394), to determine the utility and safety of early immunotherapy in patients with HRSMM. Moreover, we performed correlative DNA sequencing studies on 34 BM samples at baseline (BL), and single-cell RNA and T cell receptor (TCR) sequencing studies on 149 serial BM and PB samples from patients and healthy donors (HD) to identify genomic and immune biomarkers for optimal patient selection and monitoring of response to treatment.
RESULTS
Early treatment with Elotuzumab, Lenalidomide & Dexamethasone is safe and effective in patients with HRSMM
We conducted a Phase II trial of EloLenDex in patients with HRSMM (n=51). Enrollment in E-PRISM was based on the HRSMM criteria by Rajkumar et al. (Rajkumar et al., 2015). The primary objective was to determine the proportion of patients with HRSMM who were progression-free at 2 years post-treatment. Patients were initially randomized 1:1 to Arm A (EloLenDex, n=40) or Arm B (EloLen, n=11), but accrual to Arm B was halted early, as the difference in exposure to steroids was negligible between the two arms due to the premedication requirement for Elotuzumab infusion. Treatment was planned for 24 cycles followed by observation until progression to overt MM. Stem cell collection was allowed for eligible patients. The baseline characteristics of all patients are listed in Table 1, and the trial schema and CONSORT diagram are shown in Figures S1A and S1B.
Table 1:
Baseline characteristics of patients enrolled in the E-PRISM study.
| Treatment arm |
||||
|---|---|---|---|---|
| Total | A: Elo/Len/Dex | B: Elo/Len |
p-value
|
|
| n = 51 (%) | n = 40 (78) | n = 11 (22) | ||
|
| ||||
| Aae at registration | ||||
| Median (range) | 62 (29 – 79) | 62 (29 – 79) | 62 (44 – 75) | 0.65 |
|
| ||||
| Sex | ||||
| Female | 32 (63) | 26 (65) | 6 (55) | 0.73 |
| Male | 19 (37) | 14 (35) | 5 (45) | |
|
| ||||
| Race | ||||
| Black or African American | 7 (14) | 7 (18) | - | 0.59 |
| White | 42 (82) | 31 (78) | 11 (100) | |
|
| ||||
| Ethnicity | ||||
| Non-Hispanic | 48 (94) | 37 (92) | 11 (100) | |
|
| ||||
| ECOG PS | ||||
| 00 - Fully Active | 34 (67) | 27 (68) | 7 (64) | 0.94 |
| 01 - Restricted | 16 (31) | 12 (30) | 4 (36) | |
| 02 - Ambulatory | 1 (2) | 1 (2) | - | |
|
| ||||
| Heavy chain | ||||
| IgA | 14 (27) | 10 (25) | 4 (36) | > 0.99 |
| IgG | 29 (57) | 22 (55) | 7 (64) | |
|
| ||||
| Liaht chain | ||||
| Kappa Light Chain | 2 (4) | 2 (5) | - | > 0.99 |
| Lambda Light Chain | 3 (6) | 3 (8) | - | |
|
| ||||
| Mayo criteria 20-2-20 | ||||
| Low | 21 (41) | 17 (42) | 4 (36) | 0.47 |
| Intermediate | 15 (29) | 10 (25) | 5 (45) | |
| High | 15 (29) | 13 (32) | 2 (18) | |
|
| ||||
| Cytogenetics, FISH | ||||
| t(4;14) | ||||
| Yes | 6 (12) | 4 (10) | 2 (18) | |
| t(14;16) | ||||
| Yes | 2 (4) | 1 (2) | 1 (9) | |
| 17p deletion | ||||
| Yes | 4 (8) | 3 (8) | 1 (9) | |
| +1q amplification | ||||
| Yes | 11 (22) | 9 (22) | 2 (18) | |
|
| ||||
| High-risk cytogenetics | ||||
| No | 30 (59) | 25 (62) | 5 (45) | 0.70 |
| Yes | 17 (33) | 13 (32) | 4 (36) | |
Treatment-related Grade 3–5 adverse events with at least 10% frequency were hypophosphatemia (n=19/51; 37%), neutropenia (n=13/51; 26%), and lymphocytopenia (n=11/51; 22%). Thromboembolic events occurred in 6 patients (12%), and no secondary leukemias were observed. Treatment-related Grade 2 adverse events with at least 10% frequency and all Grade 3–5 events are summarized in Table 2 and Figure S1C. Overall, two deaths occurred: one due to uncontrolled diabetic ketoacidosis, bowel perforation, and septic shock at cycle 19, and another due to hypertensive crisis and myocardial infarction at cycle 24 (Figure S1B).
Table 2:
Highest grade toxicity experienced on treatment for n=51 patients. AEs grade 2+ in at least 10% of patients and all grade 4+.
| Worst grade |
|||||
|---|---|---|---|---|---|
| Total N=51 | 2 - Moderate | 3 - Severe | 4 - Life Threatening | 5 - Fatal | |
|
| |||||
| Upper respiratory infection | 22 (43) | 21 (41) | 1 (2) | - | - |
| Fatigue | 21 (41) | 18 (35) | 3 (6) | - | - |
| Hypophosphatemia | 21 (41) | 2 (4) | 16 (31) | 3 (6) | - |
| Neutropenia | 15 (29) | 2 (4) | 12 (24) | 1 (2) | - |
| Lymphocytopenia | 14 (27) | 3 (6) | 11 (22) | - | - |
| Hyperglycemia | 12 (24) | 9 (18) | 2 (4) | 1 (2) | - |
| Constipation | 10 (20) | 9 (18) | 1 (2) | - | - |
| Cytopenia | 10 (20) | 8 (16) | 2 (4) | - | - |
| Diarrhea | 10 (20) | 8 (16) | 2 (4) | - | - |
| Anemia | 8 (16) | 7 (14) | 1 (2) | - | - |
| Lung infection | 8 (16) | 3 (6) | 5 (10) | - | - |
| Hypertension | 7 (14) | 4 (8) | 3 (6) | - | - |
| Insomnia | 7 (14) | 6 (12) | 1 (2) | - | - |
| Thrombocytopenia | 7 (14) | 4 (8) | 2 (4) | 1 (2) | - |
| Rash maculopapular | 6 (12) | 4 (8) | 2 (4) | - | - |
| Cataract | 1 (2) | - | - | 1 (2) | - |
| Cholecystitis | 1 (2) | - | - | 1 (2) | - |
| Diabetic ketoacidosis | 1 (2) | - | - | 1 (2) | - |
| Lymphocytosis | 1 (2) | - | - | 1 (2) | - |
| Myocardial infarction | 1 (2) | - | - | - | 1 (2) |
| Sepsis | 1 (2) | - | - | - | 1 (2) |
Median follow-up for all 51 patients was 50 months (range 2–67). Six patients had PFS events defined as progression to myeloma-defining events (n=4) or death (n=2). Median PFS and OS were not reached for the 46 patients who were eligible for the study and received at least two full treatment cycles; PFS was 88.7% at 48 months (90% CI, 81.2–96.9%) and OS was 95.6% at 48 months (90% CI, 90.6–100%) (Figure S1D). The Kaplan-Meier curve of PFS for eligible patients who received at least two cycles is depicted in Figure 1A.
Figure 1.

Genomic dissection of response to early treatment with EloLenDex. A) Kaplan-Meier (KM) curve of Progression-Free Survival (PFS) in the E-PRISM cohort (n=46). B, C) Scatter plot of cancer cell fractions (CCF) at BL and EOT for progressor patients #1 and #2 (in red: mutational drivers and CNVs associated with risk of progression). All CNVs and mutational drivers are visualized. D) Comutation plot visualizing the genomic landscape of the E-PRISM cohort at BL (n=34). E) Univariate Cox regression forest plot of genomic variables present in at least 3 individuals. Hazard ratio, 95% confidence interval, and p-values were computed using Cox proportional hazards regression. F) KM curve of PFS in the E-PRISM cohort, stratified based on the presence of del17p (n=33). See also Figures S1, S2, and Table S1.
The overall response rate (Partial Response or better) for these patients was 87% (n=40). Three patients (7%) experienced a best overall response of Stringent Complete Response, 1 patient (2%) achieved Complete Response, 14 patients (30%) achieved Very Good Partial Response, 22 patients (48%) achieved Partial Response, and 6 patients (13%) achieved Minor Response (Table S1).
Overall, approximately 13% (n=6) of all patients and 20% (n=3) of patients defined as having HRSMM by the 20–2-20 criteria experienced progression or death within 4 years of follow-up (Figure S1E). Without treatment, approximately 80% of patients with HRSMM as defined by the 20–2-20 criteria are expected to progress within 5 years of follow-up (Mateos et al., 2020). Taken together, our data suggest that early treatment with EloLenDex was safe and effective in patients with HRSMM.
Genomic predictors of PFS in patients with HRSMM treated with EloLenDex
We observed three main categories of progression in our cohort: those who developed overt myeloma (n=4), those who were treated based on evolving biomarkers and the physician’s decision (n=4), and those who presented biochemical progression (n=19). In two of the four individuals who developed overt MM and had samples available for testing, we performed whole-exome sequencing (WES) on CD138+ BM cells taken at BL and end of treatment (EOT), to assess whether early treatment had selected for aggressive subclones with high-risk genetic abnormalities. Both individuals had high-risk genomic features at BL: progressor patient #1 had del17p and a TP53 single nucleotide variant (SNV), while progressor patient #2 had t(4;14), del1p, and TP53 and KRAS SNVs. We did not observe selection of subclones carrying abnormalities associated with increased risk of progression (Figure 1B, 1C). In progressor patient #1, several large-scale CNAs increased in frequency with treatment, although none are known to be associated with an increased risk of progression. In both patients, we observed a post-treatment contraction of potentially aggressive subclones that harbor amp1q (progressor patient #1), and del1p and a TP53 SNV (progressor patient #2), suggesting that in some instances, early treatment can alter the tumor’s clonal dynamics.
A total of 15 patients showed no evidence of clinical or biochemical progression, suggesting that our patient selection can improve further to identify patients with HRSMM who are more likely to benefit from early treatment. Although we were underpowered, we did not observe a significant association between 20–2-20 risk staging and PFS in our cohort or the treatment arm in Lonial et al. (Figure S2A and S2B) (Lonial et al., 2020). To search for genomic biomarkers of PFS, we performed WES (n=31) and deep targeted sequencing (DTS) (n=3) on BM samples collected at BL to detect coding genetic alterations and explore their association with patient outcomes (Figure 1D). Testing events present in at least 3 individuals, del17p, t(4;14), and del16q were significant predictors of PFS at the univariate level, while del17p was an independent predictor of progression at the multivariate level (Figure 1E, 1F). These results are consistent with our prior study showing that del17p is a risk factor for progression in untreated patients with SMM (Bustoros et al., 2020), and suggest that patients with SMM and del17p may need a different or longer regimen to prolong PFS, a hypothesis that requires validation in larger cohorts.
Comprehensive characterization of changes in the BM immune cell composition and TCR repertoire of patients with HRSMM
Next, we set out to explore whether immune profiling can help to identify patients with HRSMM who are more likely to benefit from EloLenDex. We performed single-cell RNA sequencing on 211,374 immune cells from 149 samples of 34 patients enrolled in E-PRISM [60 CD138- BM mononuclear cell samples at BL (n=28), cycle 9 day 1 (C9D1, n=16) and EOT (n=16), and 68 PB mononuclear cell (PBMC) samples at BL (n=33), cycle 4 day 1 (C4D1, n=1), C9D1 (n=13), and EOT (n=21)], and 21 age-matched HD [BM samples (n=11) and PBMCs (n=10)]. We also integrated E-PRISM data with our cohort of non-trial patients with MGUS (n=6), low-risk SMM (LRSMM, n=3), HRSMM (n=12), newly diagnosed MM (NDMM, n=9), and HD (n=11) (Zavidij et al., 2020), reaching a total of 190 samples and 231,608 cells (Figure 2A–L). For 9 samples, we thawed two cell vials and found that the inter-replicate divergence of immune cell composition was significantly lower compared to cross-replicate estimates, suggesting that the quantification of immune cell composition is reproducible (Figure S3A). To assess whether RNA-based quantification is consistent with protein-based estimates, we performed cytometry by time-of-flight (CyTOF) on 17 BM samples (10 drawn at BL, and 7 drawn at EOT) with matched single-cell RNA sequencing data and observed significant positive correlation between protein-based and RNA-based abundance estimates (Figure S3B). By comparing the BM immune cell composition of patients with HRSMM at BL (n=32) and HD (n=19) with at least 100 cells, we observed a significant increase in the abundance of naïve and memory CD4+ T cells (including Tregs, Th1, Th2, and Th17 cells), granzyme B (GZMB)-expressing CD8+ T cells, and CD56dim NK cells in patients, and a significant decrease in the abundance of CD14+ Monocytes, plasmacytoid DCs (pDCs), AXL+ SIGLEC6+ dendritic cells (AS-DCs), progenitor cells, and immature B cells (Figure 3A). Furthermore, we observed a relative decrease in the abundance of mucosa-associated invariant T cells (MAITs) in patients, a shift from CD14+ to CD16+ monocyte subpopulations, and a bias towards memory B cells (Figure S4A–C).
Figure 2.

BM and PB immune cell populations in the E-PRISM cohort. UMAP embeddings and heatmaps of gene expression markers (Mean Z-score of normalized expression) in T cells (A, B), NK cells (C, D), B cells (E, F), Monocytes (G, H), Dendritic cells (I, J), and Progenitor cells (K, L).
Figure 3.

Comprehensive profiling of changes in BM immune cell composition and TCR repertoire in patients with HRSMM. A) Volcano plot of proportion changes in the BM of patients with HRSMM (n=32) compared to HD (NBM, n=19) with at least 100 cells overall. P-values were computed with Wilcoxon’s rank-sum test and corrected using the Benjamini-Hochberg approach. Cell types with a q-value of <0.2 were marked with stars. B) Boxplots, violin plots, and scatter plots comparing BM TCR repertoire diversity, as assessed by the Chao index, between patients with HRSMM (n=14) and healthy individuals (NBM, n=11) given different numbers of downsampled cells. Each data point represents the average diversity estimate across 100 random samples of the given size for a single sample; the range of diversity estimates across all iterations for each sample is visualized in error bars (dotted line). Violin outline width represents density. P-values were computed with Wilcoxon’s rank-sum tests. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). C) Barplots showing the proportion of T cells in a given BL patient sample (P, n=14) or sample from a HD (n=11) that were determined to belong to one of four clone size categories (Rare: ≤1%; Small: >1% and <5%; Medium: ≥5% and <10%; Large: ≥10%) through iterative (n=100) downsampling of 100 cells. The average proportion per clone size category was visualized and the standard deviation across iterations was depicted in solid-line error bars. D) UMAP embedding of HD BM (NBM) and patient BM T cells at BL with matched TCR data. T cells belonging to rare clonotypes (with a frequency of ≤1%) were colored in blue, while T cells belonging to expanded clonotypes (with a frequency of >1%) were colored in red. E) Barplots showing the proportion of clonotypes in a given T cell subtype across all patients (n=14) or HD (n=11) that belonged to one of the four clone size categories. For each T cell subtype, 100 cells were randomly sampled 100 times from all patients or HD, and the proportion of expanded (1-Rare) clonotypes was compared between patients and HD using bootstrapping with 10,000 iterations. The average proportion per clone size category was visualized and the standard deviation across iterations was depicted in solid-line error bars. F) Volcano plot highlighting genes that are highly expressed in expanded GZMB+ CD8+ TEM cells from patients (n=743, in red) compared to expanded GZMB+ CD8+ TEM cells from HD (n=1,074, in blue). See also Figures S3 and S4.
Next, we set out to characterize changes in the TCR repertoire of patients compared to HD and performed single-cell TCR sequencing on 91 samples with available single-cell RNA sequencing data. Specifically, we profiled 70 BM and PB patient samples drawn before and after therapy [BM: 14 at BL, 3 at C9D1, 2 at EOT; PB: 26 at BL, 8 at C9D1, 17 at EOT], and 21 samples from HD [BM:11; PB:10]. To control for variability in T cell numbers which can impact diversity estimates, we randomly sampled a range of T cell numbers (n=75–300) from each sample over 100 iterations, and averaged repertoire diversity estimates across iterations. We observed that patient BM TCR repertoires were significantly less diverse compared to those of HD, as assessed by the Chao index, independent of the number of T cells sampled (Figure 3B). The observed decrease in repertoire diversity appeared to be driven by broad low-level clonal expansion, rather than a targeted expansion of a few large clones dominating the repertoire (Figure 3C). Regulatory T cells and GZMB+ CD8+ TEM cells from patient BM were more likely to be part of an expanded clone compared to those from HD BM, where clonal expansion was contained within cytotoxic populations (Figure 3D, 3E). This finding is consistent with the increase in Treg and GZMB+ CD8+ TEM cell abundance observed in patient BM and suggests that these cells may be expanding in an antigen-specific manner. To gain more insight into the phenotype of clonally expanded GZMB+ CD8+ TEM cells in patients, we compared their gene expression to that of clonally expanded GZMB+ CD8+ TEM cells from HD (Figure 3F). Compared to HD, clonally expanded cytotoxic T cells from patients expressed higher levels of ZNF683 (which encodes the tissue residence transcription factor Hobit), senescence markers KLRG1 and B3GAT1 (which encodes CD57), genes associated with effector function (ITGB1, ITGA4, ITGB2, ITGB7, S100A4, S100A10, S100A6, TNF, PRF1, CCL5, GZMH), and genes induced by interferon signaling (STAT1, XAF1, IFITM1, IFITM2), and lower levels of genes related to the induction of potent cytotoxic responses (GZMB, FGFBP2, TBX21, PRDM1, EOMES, IFNGR1) or granule formation and degranulation (VAMP2, OCD1, SRGN). These results suggest that, despite their increased abundance in patient BM, cytotoxic T cells may be dysfunctional. Further mechanistic studies are required to determine the impact of these changes on T cell functionality and tumor clearance.
Immune reactivity at BL and post-therapy immune normalization are associated with longer PFS in patients with HRSMM treated with EloLenDex
We hypothesized that the similarity of a patient’s BM immune cell composition to that of HD could be harnessed for prognostication. We trained a Naïve Bayes classifier with 10-fold cross-validation and 100 iterations on a training set of 41 BM samples from patients and HD, to predict the presence of malignancy based on the composition of the BM immune microenvironment; the classifier achieved 94% accuracy in the testing set (n=16, Figure S5A). We then computed a weighted sum score of the product of each cell type’s proportion and its corresponding signed importance to the classification (“normalization score”), and classified patients based on the median score at BL as reactive (least normal-like) or non-reactive (most normal-like) (Figure 4A). Patients who were classified as reactive at BL had significantly longer PFS (Figure 4B). Immune reactivity was independent of the patients’ risk stage but was significantly associated with the presence of del17p, although patients with del17p were only a fraction of the non-reactive group (Figure 4C, 4D). Plasmacytoid DCs and the pro-inflammatory Cytokine+ CD14+ Monocytes, which expressed genes related to neutrophil chemotaxis and acute inflammation, were among the top 3 cell types with the highest importance for a sample to be classified as normal-like. Aside from monocytes, this pro-inflammatory signaling was also active in DCs and pDCs (gene expression signature GEX-6), and together with pDC signaling (GEX-13), they were significantly associated with shorter PFS (Figure 4E, S5B–E). As expected, reactive patients had significantly lower abundance of pDCs and Cytokine+ CD14+ Monocytes (Figure S5F). By comparing gene expression levels between reactive and non-reactive patients, we observed a significant downregulation of exhaustion markers (TOX, TNFRSF9, TNFSF9, PDCD1, NR4A2, NR4A3) in GZMK+ CD8+ TEM cells of reactive patients, along with an upregulation of markers associated with long-lived memory effectors (IL7R and CD27), markers associated with terminal effectors (GZMB, GZMH, FCGR3A, FGFBP2, NKG7), and markers associated with functionality (IFNG, TNF), a profile consistent with long-lived memory effector cells of increased potency (Figure 4F). Moreover, in GZMB+ CD8+ TEM cells from reactive patients, we observed an upregulation of exhaustion markers (TIGIT, LAG3) and the senescence marker KLRG1, along with higher levels of IFNG and TNF, and downregulation of cytotoxicity markers (GZMB, FCGR3A, PRF1), a profile consistent with exhausted, senescent terminal effector cells (Figure 4G). Consistent with the hypothesis that these cells may be exhausted due to chronic antigenic stimulation, GZMB+ CD8+ TEM cells from reactive patients were more likely to be clonally expanded, compared to those from non-reactive patients (Figure 4H). Collectively, these results suggest that immune reactivity captures a patient subpopulation with decreased pro-inflammatory myeloid signaling, long-lived GZMK+ effector memory cells of increased potency, and clonally expanded, exhausted GZMB+ terminal effectors.
Figure 4.

Immune reactivity at BL and post-therapy immune normalization are associated with significantly longer PFS in patients with HRSMM under treatment. A) Barplot visualizing the signed importance of each cell type towards the classification. B) Kaplan-Meier (KM) curve of PFS in the E-PRISM cohort, stratified based on the median normalization score (Reactivity+: > median) (n=24). C, D) Barplots visualizing the frequency of 20–2-20 risk stages (C) and del17p (D) by reactivity status (LR: low-risk, IR: intermediate-risk, HR: high-risk). The p-value was computed with a Fisher’s exact test. E) UMAP embedding of lymphocytes and myeloid cells colored by the log-scaled activity of signature GEX-6. F, G) Volcano plots visualizing genes that are differentially expressed in GZMK+ CD8+ TEM (F) and GZMB+ CD8+ TEM (G) cells of reactive patients (n=12) compared to non-reactive (n=12). H) Barplot visualizing the proportion of clonotypes belonging to each of four clone size categories per cytotoxic T cell subtype in BM samples from reactive (n=7) or non-reactive patients (n=7). For each T cell subtype, 100 cells were randomly sampled 100 times from patients with or without reactivity, and the proportion of expanded (1-Rare) clonotypes was compared between the two using bootstrapping with 10,000 iterations. The average proportion per clone size category was visualized and the standard deviation across iterations was depicted in solid-line error bars. I) Boxplots, violin plots, and scatter plots visualizing normalization scores in patient BM samples drawn at BL (n=28) or EOT (n=16), and in HD BM samples (NBM, n=22). Violin outline width represents density. P-values were computed using a paired t-test for paired patient samples or Wilcoxon’s rank-sum test for comparisons between patients and HD. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). J) Boxplots, violin plots, and scatter plots visualizing paired normalization scores at BL and EOT from patients with HRSMM (n=12). Violin outline width represents density. Solid lines connect samples from patients classified as PIN+; dashed lined connect samples from patients classified as PIN. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). K) Histogram of the distribution of change in normalization scores between paired BL and EOT BM patient samples (n=12). The dashed line corresponds to the threshold used to determine the presence of PIN. L) KM curve of PFS in the E-PRISM cohort, stratified based on PIN status (n=12). See also Figure S5.
On average, patients showed significantly higher normalization scores at EOT, compared to BL (Figure 4I). Using a threshold of 0.1 based on the distribution of change in normalization scores at EOT, we defined a state of post-therapy immune normalization (PIN) and classified four patients who showed no or minimal increase in their normalization score at EOT as PIN- (Figure 4J, 4K). Patients who achieved PIN at EOT (PIN+) had significantly longer PFS (Figure 4L), suggesting that PIN may be important for prognostication. Of note, both patients who had del17p out of the 12 patients with matched BL and EOT BM samples were classified as PIN−, so the effect of PIN on PFS needs to be deconvolved from that of del17p in larger cohorts.
Higher abundance of Granzyme K-expressing CD8+ T cells is associated with longer PFS in patients with HRSMM treated with EloLenDex
We previously showed that patients with HRSMM have decreased abundance of memory-like GZMK+ CD8+ TEM cells in favor of more mature GZMB+ effectors and that lower levels of memory T cells in vivo are associated with shorter OS (Zavidij et al., 2020). Therefore, we hypothesized that higher levels of GZMK+ CD8+ TEM cells in the BM may be associated with prolonged PFS in our patients. Consistent with our prior findings, patients with HRSMM showed a significant shift from GZMK+ to GZMB+ CD8+ TEM cells in the BM, with a significant decrease in the abundance of GZMK+ CD8+ TEM cells relative to all cytotoxic T cells at BL compared to HD (Figure 5A). However, the average levels of GZMK expression in patient T cells increased significantly at EOT, reflecting a significant increase in GZMK-expressing cells, which was validated by CyTOF (Figure 5B, S6A). In the PB, where we were powered to investigate changes in T cell clonality at EOT, we found that GZMK+ CD8+ TEM cells from EOT samples were more likely to belong to expanded clones (Figure 5C). Together, these results suggest that this subpopulation may play a role in response to immunotherapy. Compared to GZMB+ CD8+ TEM cells in our cohort, GZMK+ CD8+ TEM cells showed higher expression of CXCR4 and lower expression of S1P receptors, suggesting preferential retainment of GZMK+ CD8+ TEM cells within the BM (Figure 5D). Indeed, by comparing the abundance of these cells between matched BM and PB patient samples, we observed enrichment of GZMK+ CD8+ TEM cells in the BM (Figure 5E). This enrichment was also observed in BM samples from HD (data not shown), suggesting that this may be a physiological bias. Furthermore, GZMK+ CD8+ TEM cells had higher expression of canonical exhaustion markers TOX, TIGIT, TNFRSF9 (encoding 4–1BB), and TNFSF9 (encoding 4–1BBL), as well as markers of long-lived memory effector cells, including TCF7 (encoding Tcf-1), CD27 and CD28, a phenotype consistent with progenitor exhausted T cells (TPEX) (Figure 5D). Indeed, at the protein level, GZMK+ CD8+ TEM cells were shown to be a major source of PD-1 expression within cytotoxic T cells in patients (Figure 5F, S6B), in accordance with the suspected TPEX phenotype. Progenitor exhausted T cells maintain their proliferative capacity, can differentiate into terminal effector cells, and are thought to mediate the clinical benefits observed in patients treated with immune checkpoint inhibitors (Galletti et al., 2020). In our cohort, patients with a higher abundance of GZMK+ CD8+ TEM cells had significantly longer PFS in response to EloLenDex (Figure 5G). Similarly, patients with higher average GZMK expression across their T cells had significantly longer PFS (Figure S6C). Of note, no association was observed between the abundance of GZMK+ CD8+ TEM cells and age (Wilcoxon, p=0.55), while age had no effect on PFS (Log-rank, p=0.7).
Figure 5.

A higher abundance of Granzyme K-expressing CD8+ T cells is associated with longer PFS in patients with HRSMM treated with EloLenDex. A) Boxplots, violin plots, and scatter plots visualizing the relative abundance of GZMK+ CD8+ TEM and GZMB+ CD8+ TEM out of all cytotoxic T cells in patient BM (n=33) at BL compared to samples from HD (NBM, n=22). Violin outline width represents density. P-values were computed with Wilcoxon’s rank-sum test. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). B) Boxplots, violin plots, and scatter plots visualizing the abundance of GZMK+ CD8+ TEM cells by CyTOF in patient BM samples drawn at BL (n=10) and EOT (n=7). Violin outline width represents density. The p-value was computed with Wilcoxon’s rank-sum test, as these samples were not paired. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). C) Barplot visualizing the proportion of clonotypes belonging to each of four clone size categories per cytotoxic T cell subtype in patient PB samples drawn at BL (n=22) or EOT (n=17). For each T cell subtype, 100 cells were randomly sampled 100 times from patients at BL and EOT, and the proportion of expanded (1-Rare) clonotypes was compared between the two using bootstrapping with 10,000 iterations. The average proportion per clone size category was visualized and the standard deviation across iterations was depicted in solid-line error bars. D) Volcano plot visualizing genes that are differentially expressed between GZMK+ CD8+ TEM and GZMB+ CD8+ TEM cells from patient BM samples drawn at BL (n=28). E) Boxplots, violin plots, and scatter plots comparing the abundance of GZMK+ CD8+ TEM cells between paired BM and PB samples drawn at BL from patients with HRSMM (n=22). Violin outline width represents density. Solid red lines connect samples that are enriched in the BM, while dashed orange lines connect samples that are enriched in the PB. The p-value was computed using a paired t-test. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). F) Scatter plot visualizing the correlation between the BL proportion of CD8+ T cells expressing PD-1 and those expressing GZMK by CyTOF (n=10). A regression line was fitted (in red) and the correlation coefficient and p-value were computed using Pearson’s approach. G) Kaplan-Meier curve of PFS in the E-PRISM cohort, stratified based on the median abundance of GZMK+ CD8+ TEM cells in patient BM samples at BL (n=26). See also Figure S6.
Collectively, these results suggest that GZMK+ CD8+ TEM cells may be important for disease control and response to therapy in patients with SMM.
PB-based immune profiling accurately detects alterations in immune cell composition and TCR repertoire diversity observed in the BM
Next, we set out to determine whether PB can be used reliably for immune profiling of patients with HRSMM, as BM biopsies are invasive and carry risk, which precludes regular patient testing. First, we compared the Jensen-Shannon divergence of immune cell composition between matched and unmatched PB and BM samples and observed significantly lower divergence in matched pairs, which was also low overall, suggesting that matched PB samples reflect BM composition well (Figure 6A). In PCA space, where PC1 captured the compositional changes observed in patient BM (Figure 6B), HD PB and BM samples clustered together on the right end of the axis, while patient PB and BM samples clustered together on the opposite side of PC1 (Figure 6C). This observation suggests that the compositional differences between patient and HD PB are similar to those observed in patient BM compared to HD BM. Indeed, when we compared PB samples between patients and HD, we recovered compositional changes originally discovered in our BM-based analysis (Figure 6D). However, patient PB showed a significantly lower abundance of CD16+ monocytes, which are enriched in patient BM.
Figure 6.

PB-based immune profiling accurately detects alterations in immune cell composition and TCR repertoire diversity observed in the BM. A) Boxplots, violin plots, and scatter plots showing the Jensen-Shannon divergence between matched BM and PB samples (n=22), compared to unmatched samples. Violin outline width represents density. The p-value was computed using Wilcoxon’s rank-sum test. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). B) Heatmap of Pearson’s correlation coefficient (r) between BM immune cell abundance and the first 10 principal components. The x-axis was sorted by decreasing PC1 value. The top panel shows the log2 fold-change in abundance between BM samples from patients with HRSMM (n=32) and those from HD (n=19). P-values were corrected using the Benjamini-Hochberg approach and stars correspond to pairs with significant (q<0.05) correlation. C) Two-dimensional density plot of BM and PB samples from patients (BM: n=26, PB: n=29) or HD (BM: n=22, PB: 10) according to PC1 and PC2. D) Volcano plot of proportion changes in the PB of patients with HRSMM (n=19) compared to HD (NPB, n=10) with at least 100 cells overall. P-values were computed with Wilcoxon’s rank-sum test and corrected using the Benjamini-Hochberg approach. Cell types with a q-value of <0.2 were marked with stars. E) Boxplots, violin plots, and scatter plots comparing PB TCR repertoire diversity, as assessed by the Chao index, between patients with HRSMM (n=22) and healthy individuals (NPB, n=10) given different numbers of downsampled cells. Each data point represents the average diversity estimate across 100 random samples of the given size for a single sample; the range of diversity estimates across all iterations for each sample is visualized in error bars (dotted line). Violin outline width represents density. P-values were computed with Wilcoxon’s rank-sum tests. (Box: 1st quartile, median, 3rd quartile; whiskers: +/− 1.5*IQR). F) Forest plot showing the effect of mean BL GEX-13 activity in the PB on PFS. Hazard ratio, 95% confidence interval, and p-value were computed using Cox proportional hazards regression. G) Confusion matrix visualizing the accuracy of a Naïve Bayes classifier trained on BM samples from patients and HD (training set, n=41) and tested in PB samples (n=29). See also Figure S7.
Contrary to HD where CD16+ monocytes are significantly enriched in the PB compared to the BM, in patients there was no significant difference between the PB and the BM, suggesting that these cells may be homing to the patient BM (Figure S7). Lastly, consistent with what we observed in patient BM, patient PB TCR repertoire showed significantly lower diversity compared to HD, suggesting that the observed effect on diversity may not be constrained to the tumor microenvironment (Figure 6E). These results suggest that certain immune biomarkers may be measurable in the PB. Consistent with what we saw in the BM, higher BL levels of signature GEX-13 in patient PB were associated with significantly shorter PFS (Figure 6F). Of note, neither signature GEX-6 nor immune reactivity were prognostic in PB, suggesting that PB-based immune profiling may not be able to replace BM-based profiling entirely.
Lastly, given the concordance of compositional changes between patient BM and PB, we tested whether immune profiling of PB samples alone could enable the diagnosis of an individual with HRSMM. Remarkably, a classifier that we trained on BM composition data (this time excluding progenitor cells, which are primarily encountered in the BM) to infer the presence of SMM was able to correctly classify nearly all our patient and HD PB samples with an accuracy of 97% (Figure 6G). Collectively, our findings demonstrate that PB-based immune profiling accurately detects compositional changes observed in the BM and may hold diagnostic and prognostic potential.
DISCUSSION
While early treatment has been shown to delay progression in patients with HRSMM, identifying those who will benefit the most remains an unmet clinical need (Mateos et al., 2015; Kim et al., 2020). Despite the availability of clinical and genomic biomarkers of progression (Kyle et al., 2007; Mateos et al., 2020; Pérez-Persona et al., 2007; Bustoros et al., 2020; Misund et al., 2019; Boyle et al., 2021; Bolli et al., 2018; Oben et al., 2021), little is known about the role of BM immune dysregulation in disease progression and response to treatment or the diagnostic and prognostic utility of PB-based immune profiling. Here, we conducted a Phase II trial to determine the utility and safety of early treatment with EloLenDex in patients with HRSMM and performed correlative DNA sequencing and single-cell RNA and TCR sequencing studies to develop genomic and immune biomarkers for optimal patient selection and monitoring of response to treatment. The overall response rate was 87%, PFS was 88.7% at 48 months (90% CI, 81.2–96.9%), and OS was 95.6% at 48 months (90% CI, 90.6–100%). Although this trial did not have an observation arm, this progression rate (~13% at 4 years) is substantially lower than that determined for patients with HRSMM in large observational studies which showed progression rates of 50% or more within 2 years from diagnosis and 70–80% within 5 years from diagnosis (Rajkumar et al., 2015; Kyle et al., 2007; Mateos et al., 2020). This was also true for patients in our trial who were high-risk by the 20–2-20 criteria and who showed a 20% progression rate within 4 years of follow-up. Treatment was well-tolerated with minimal toxicity of Grade 3 or higher, including hypophosphatemia and cytopenias, which were expected. As this was a single-arm study, we cannot comment on how the response rate and PFS of EloLenDex compares to those of LenDex alone, and future clinical trials randomizing these 2 regimens would be required to answer this. Overall, EloLenDex was deemed safe and effective, with no evidence of aggressive subclone selection post-treatment in two patients who progressed. In fact, in those patients, we observed a regression of subclones harboring either amp1q or del1p and a TP53 SNV. Although preliminary, these results suggest that in some instances early treatment could alter the clonal dynamics in patients who progress, which could potentially result in improved OS in the long-term. These findings would need to be validated in larger trials with more available samples pre- and post-therapy. Aggressive tumor biology, as determined by the presence of del17p, was a significant predictor of shorter PFS in patients treated with EloLenDex in our cohort. Therefore, patients with HRSMM and del17p may perhaps need different regimens or longer duration of therapy.
Patients with HRSMM showed increased abundance of naïve and memory CD4+ T cells, GZMB+ CD8+ TEM cells, and CD56dim NK cells, and decreased abundance of CD14+ monocytes, and pDCs compared to HD. Furthermore, their TCR repertoire diversity was significantly lower compared to HD, due to broad low-level clonal expansion, which was mostly observed in GZMB+ CD8+ TEM cells and Tregs. Further studies are required to determine the functionality and antigen specificity of these immune cell populations and their role in controlling or promoting disease progression.
Importantly, we found that the similarity of a patient’s BM immune microenvironment to that of HD may be useful for prognostication. At BL, patients with the least normal-like immune cell composition, presumed to be reactive to the tumor, had significantly longer PFS under treatment with EloLenDex. Reactive patients had lower proportions of pro-inflammatory myeloid cells and pDCs, and had long-lived GZMK+ CD8+ TEM cells of increased potency, and clonally expanded, exhausted GZMB+ CD8+ TEM cells. Granzyme K-expressing cells, which are normally enriched in the BM, had a phenotype compatible with progenitor exhausted cells, and clonally expanded post-treatment, suggesting that they may play a role in response to therapy. Patients with a higher abundance of GZMK+ CD8+ TEM cells in their BM showed significantly longer PFS. Furthermore, patients whose BM normalization scores increased post-therapy (Post-therapy Immune Normalization, PIN), had significantly longer PFS. This finding suggests that immune profiling may be useful to monitor response to treatment, although validation in larger cohorts and with different regimens is required.
Lastly, immune profiling currently requires a BM biopsy, which precludes its regular use for prognostication. Remarkably, by comparing PB from patients and HD, we identified nearly the same alterations in immune cell composition and TCR repertoire diversity as those seen in our BM-based comparisons. Moreover, a classifier trained to detect the presence of SMM based on BM immune profiling was able to correctly classify nearly all PB samples from patients with SMM and HD. Our results suggest that PB-based immune profiling may have diagnostic and prognostic utility, although it may not be able to entirely replace BM-based profiling.
Collectively, our results demonstrate the utility and safety of EloLenDex in patients with HRSMM, provide a comprehensive characterization of alterations in immune cell composition and TCR repertoire diversity in patient BM and PB, nominate immune biomarkers for optimal patient selection and assessment of response to treatment, and demonstrate that PB-based immune profiling may have diagnostic and prognostic utility in patients with SMM.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Irene Ghobrial (irene_ghobrial@dfci.harvard.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
Raw single-cell RNA and TCR sequencing data generated for this study have been deposited in dbGaP (phs002476.v1.p1). Raw single-cell RNA sequencing data generated in Zavidij et al. have been deposited in dbGaP (phs001323.v3.p1). Raw whole-exome sequencing data generated for this study have been deposited in dbGaP (phs001323.v3.p1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Clinical trial and patients
Study design
The primary objective of this trial was to determine the proportion of patients with high-risk SMM (HRSMM) who were progression-free at 2 years post-treatment. Secondary objectives included safety and toxicity, time to progression, overall response rate, duration of response, and overall survival.
From January 2015 to November 2016, patients were enrolled at Dana Faber Cancer Institute, Boston, Massachusetts; Beth Israel Deaconess Medical Center, Boston, Massachusetts; Carolina HC Levine Cancer Institute, Charlotte, North Carolina; Colorado Blood Cancer Institute, Denver, Colorado; Eastern Maine Medical Center, Bangor, Maine; Karmanos Cancer Institute, Detroit, Michigan; Marlene and Stewart Greenbaum Cancer Center, Baltimore, Maryland; Massachusetts General Hospital, Boston, Massachusetts; Newton-Wellesley Hospital, Newton, Massachusetts; St. Francis Hospital, Hartford, Connecticut; and University of Chicago, Chicago, Illinois.
Patients were eligible for this trial if they were 18 years of age or older, had an Eastern Cooperative Oncology Group performance status of 0 to 2, and were classified as HRSMM. HRSMM was defined according to Rajkumar et al. as the presence of BM clonal plasma cells ≥10% (but less than 60%) and at least one of the following: serum M protein ≥ 3.0 mg/dL; IgA SMM; immunoparesis with reduction of two uninvolved immunoglobulin isotypes; serum involved/uninvolved free light chain ration ≥ 8 (but less than 100); evolving type of SMM, i.e., progressive increase in M protein level; BM clonal plasma cells 50–60%; abnormal cell immunophenotype (≥ 95% of BM plasma cells are clonal) and reduction of one or more uninvolved immunoglobulin isotypes; t(4;14) or del17p or amp1q; increased circulating plasma cells; MRI with diffuse abnormalities or 1 focal lesion (≥ 5mm); PET-CT with one focal lesion (≥ 5mm) with increased uptake without underlying osteolytic bone destruction (Rajkumar et al., 2015). Furthermore, patients had to show no evidence of myeloma-defining events as described by the IMWG (Rajkumar et al., 2021), and the following laboratory values had to be shown within 28 days prior to registration: Absolute Neutrophil Count ≥ 1000/mm3, Platelets (PLT) ≥ 50,000/mm3, Total Bilirubin ≤ 2.0mg/dL, Aspartate and Alanine Aminotransferase (AST/ALT) ≤ 3 × ULN, and an estimated creatinine clearance > 50mL/min/1.73m2. Patients with other concurrent chemotherapy, immunotherapy, radiotherapy, or any ancillary therapy considered investigational, serious medical or psychiatric illness likely to interfere with participation, a diagnosis or treatment for another malignancy within 2 years of enrollment were excluded.
All patients provided informed consent. The review boards of all participating centers approved the study in accordance with the Declaration of Helsinki and the International Conference of Harmonization Guideline for Good Clinical Practice. This study was registered as a phase II study with ClinicalTrials.gov NCT02279394.
Study treatment
This phase II trial used a single-stage design to assess the efficacy of the combination of Elotuzumab and Lenalidomide with or without Dexamethasone in patients with HRSMM. Patients were randomized 1:1 to Arm A (EloLenDex) or Arm B (EloLen), based on the stratification factors described below.
In the treatment schema a cycle is defined as 28 consecutive days. In cycles 1 and 2, 10mg/kg of intravenous push Elotuzumab was administered on days 1, 8, 15, and 22. In cycles 3–4, it was administered on days 1 and 15. A 25mg dose of oral Lenalidomide was administered on days 1–21 of each cycle. In Arm A, 40mg of oral Dexamethasone was administered on days 1, 8, 15, and 22 in cycles 1 and 2 and on days 1, 8, and 15 in cycles 3–8. Patients on maintenance treatment (cycles 9–24) were administered 20mg/kg of intravenous push Elotuzumab on day 1 and 15mg oral Lenalidomide on days 1–21. All patients received thromboprophylaxis.
In consideration of the Mateos et al. study, investigators were given the option to allow high-dose Dexamethasone to be resumed during maintenance due to biochemical progression (progressive increase in serum M-spike by at least 25% with an absolute increase of at least 0.5 g/dL or urine M-spike by at least 25% with an absolute increase of at least 200 mg/24 hours, on two successive evaluations, as determined by the IMWG response criteria or documented progression by serum free light chain criteria in the absence of serum or urine involvement; Mateos et al., 2013). A total of 5 patients were given Dexamethasone during maintenance. The dosing schedule for this followed that of cycles 3–8, with 8mg intravenous push and 32mg oral Dexamethasone on days 1 and 15 and 40mg oral Dexamethasone on day 8.
Stratification Factors
Originally, the study aimed to secondarily determine whether Dexamethasone is beneficial or detrimental in this immunotherapy regimen for SMM. Therefore, the study was designed with two arms, meant to serve as two independent phase II studies to determine the activity of EloLen with or without Dexamethasone in the patient population. Patients were stratified by age (≥65 vs <65) and high-risk cytogenetics [t(4;14), t(14;16), del17p, TP53 mutation, amp1q] (high-risk, low-risk, FISH failure) before being randomly assigned to one of the two arms.
Arm B accrual
Arm B accrual was halted in April 2016 for two reasons: 1) treatment with Elotuzumab required premedication with high doses of steroids to avoid infusion reactions and the difference in Dexamethasone dosing between the two arms (approximately 4mg Dexamethasone per dosing day) for the first 2 cycles was not significant, and 2) a publication by Paiva et al. (Paiva et al., 2016). demonstrated that once weekly high-dose Dexamethasone does not have a detrimental effect on the immune system in patients with SMM in a clinical trial of Lenalidomide and Dexamethasone. The patients who were originally enrolled in Arm B remained in Arm B until the completion of therapy.
Cohort details
In total, 51 patients enrolled and received at least one dose of treatment. Three patients were deemed ineligible soon after they received treatment, due to misdiagnosis (overt myeloma with lesions on PET-CT, amyloidosis, and light chain deposition disease), and were excluded from further analysis. Two patients received less than 2 full cycles of treatment and were excluded from our overall response assessment and correlative studies but were included for toxicity assessment. Out of the 46 patients remaining in the cohort, 29 were female and 17 were male, while the median age at registration was 62 (range: 29–77).
Imaging
To assess the extent of disease involvement, imaging studies were performed at both BL and EOT. All patients had either an MRI with a skeletal survey or a PET/CT scan.
Efficacy and Safety Assessments
Toxicities were monitored throughout the study and for up to 30 days after the last dose of the study drug. Adverse events (AEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE; version 4.0; Bethesda, MD). In addition, neuropathy symptoms were assessed using the FACT/GOG-Neuropathy questionnaire (Version 4.0). Efficacy was measured using the IWMG criteria for response.
Treatment discontinuation and patient disposition
The median duration of treatment was 24 cycles (range= 1–24). In total, 40 of 51 patients received all 24 cycles. Of those, 30 (75%) were in arm A and 10 (25%) in arm B. Of the 10 patients in Arm A who did not complete all 24 cycles, 3 patients discontinued therapy during the induction cycles due to an incorrect initial diagnosis (one participant had myeloma lytic lesions on a PET scan that was originally misread at an outside institution and re-checked at DFCI afterward; another participant had amyloidosis that was missed at the time of enrollment but was discovered after 3 cycles of therapy—the patient achieved VGPR on therapy before being removed from the protocol and placed on active amyloidosis therapy with a proteasome inhibitor-based approach; one final patient had light chain deposition disease, which was identified on a kidney biopsy after one cycle of therapy—the patient was then removed from the protocol to start active therapy). Six other patients discontinued therapy due to toxicity and withdrawal of consent or physician decision. Patient disposition is visualized in Figure S1B.
Overall, two deaths occurred. One patient had Type I Diabetes and was hospitalized for Diabetic ketoacidosis (cycle 19). This was at a time when the patient was not receiving Dexamethasone. The patient developed diverticulitis, bowel perforation, and sepsis and died due to sepsis. The second patient was on the last cycle of therapy when they experienced uncontrolled hypertension due to non-compliance with their hypertension medications. They then developed heart failure and died of hypertensive crisis.
METHOD DETAILS
Sample processing
Bone marrow (BM) aspirate and peripheral blood (PB) samples were collected in EDTA tubes, and mononuclear cells (BMMCs & PBMCs) were isolated using Red Blood Cell Lysis Buffer (ThermoFisher). Tumor and immune cells were then isolated by magnetic enrichment for CD138 (Miltenyi Biotec), according to the manufacturer’s instructions, and cryopreserved in FBS with 10% DMSO.
Whole-Exome and Deep Targeted Sequencing
We performed WES on baseline BM aspirate specimens from 37 study participants. Tumor DNA was extracted from CD138+ cells from the patients’ BM. For germline control, DNA was obtained from PBMCs. Genomic DNA was extracted using the QIAamp DNA mini kit (QIAGEN) according to the manufacturer’s protocols, and the DNA concentration was quantified using PicoGreen dsDNA Assay kit (Life Technologies). WES libraries were prepared by Agilent SureSelect XT2 Target Enrichment kit (V5+UTR probes). For three individuals, libraries for deep targeted sequencing (DTS) were prepared using SureSelect XT Reagent Kits (Agilent) and an in-house bait set targeting 117 genes, comprising pan-cancer driver genes and frequently mutated genes in MM (Bustoros et al., 2020). Lastly, for two out of four patients who progressed to overt myeloma, tumor samples were available at the time of progression and were also studied by WES (Roche MedExome+) to characterize the landscape of clonal evolution under treatment. All sequencing was performed on the Illumina HiSeq 4000 platform at the Genomics Platform of the Broad Institute of MIT & Harvard. Six WES samples were excluded from analysis due to low tumor purity (<15%). Our final baseline cohort comprised 31 WES samples and 3 DTS samples.
Single-cell RNA sequencing
We performed single-cell RNA sequencing on 149 CD138− BMMC and PBMC samples from 34 patients enrolled in the E-PRISM trial. Specifically, we sequenced 60 CD138− BMMC samples at BL (n=28), C9D1 (n=16), and EOT (n=16), 68 PBMC samples at BL (n=33), C9D1 (n=13), C4D1 (n=1), and EOT (n=21)], 11 CD138− BMMC samples from age-matched HD, and 10 PBMC samples from age-matched HD. For 23 patients, BL BM and PB samples were matched; for 8 patients, C9D1 BM and PB samples were matched; for 13 patients EOT BM and PB samples were matched (Figure S8A). Of note, in contrast to our patient samples, HD BM and PB samples were from different individuals.
For library preparation, we used Chromium Single Cell 3’ Gene Expression (n=40) and Chromium Single Cell 5’ Gene Expression kits (n=109) by 10X Genomics. Specifically, we performed 3’ single-cell RNA sequencing on 40 CD138− BMMC samples drawn at BL (n=11), C9D1 (n=13) or EOT (n=9), and PBMC samples drawn at BL(n=4) and C9D1 (n=3). We performed 5’ single-cell RNA sequencing on 98 CD138- BMMC samples drawn at BL (n=17), C9D1 (n=3) or EOT (n=7), and PBMC samples drawn at BL (n=29), C4D1 (n=1), C9D1 (n=10) or EOT (n=21). We also performed 5’ single-cell RNA sequencing on 11 CD138- BMMC samples and 10 PBMC samples from HD. All 3’ libraries were sequenced on a HiSeq2500 instrument and all 5’ libraries were sequenced on a NovaSeq 6000 instrument at the Genomics Platform of the Broad Institute of MIT & Harvard.
Reproducibility of cell type abundance quantification via single-cell RNA sequencing
To assess the reproducibility of our cell type quantification, we thawed two cell vials for each of 9 samples; for five of them, both replicate libraries were prepared using the same technology (3’- or 5’-end sequencing) (technical replicates); for four of them, each replicate library was prepared with a different technology (technology replicates).
Single-cell TCR sequencing
We performed single-cell TCR sequencing on 91 samples with available single-cell RNA sequencing data (Figure S8A). Specifically, we profiled 70 BM and PB patient samples drawn before and after therapy [BM: 14 at BL, 3 at C9D1, 2 at EOT; PB: 26 at BL, 8 at C9D1, 17 at EOT], and 21 samples from HD [BM:11; PB:10]. Complementary DNA generated from barcoded CD138− immune cells using Chromium Single Cell 5’ Gene Expression and V(D)J enrichment kits by 10X Genomics was subjected to V(D)J enrichment and library preparation and sequenced on a NovaSeq 6000 instrument at the Genomics Platform of the Broad Institute of MIT & Harvard.
Cytometry by Time-of-Flight (CyTOF)
Metal-tagged antibodies used for the mass cytometry panel are listed in the Key Resources table. In-house conjugation of Granzyme K antibody was performed with Maxpar labeling kit per manufacturer instructions (Fluidigm). All antibodies were used per the manufacturer’s recommendation (Fluidigm).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Human CD45 (HI30)-89Y—100 Tests | Fluidigm | Cat#3089003B |
| Anti-Human CD196/CCR6 (G034E3)-141Pr—50 Tests | Fluidigm | Cat#3141003A |
| Anti-Human CD19 (HIB19)-142Nd—100 Tests | Fluidigm | Cat#3142001B |
| Anti-Human CD45RA (HI100)-143Nd—100 Tests | Fluidigm | Cat#3143006B |
| Anti-Human CD69 (FN50)-144Nd—100 Tests | Fluidigm | Cat#3144018B |
| Anti-Human CD4 (RPA-T4)-145Nd—100 Tests | Fluidigm | Cat#3145001B |
| Anti-Human IgD (IA6–2)-146Nd—100 Tests | Fluidigm | Cat#3146005B |
| Anti-Human CD20 (2H7)-147Sm—100 Tests | Fluidigm | Cat#3147001B |
| Anti-Human CD14 (RMO52)-148Nd—100 Tests | Fluidigm | Cat#3148010B |
| Anti-Human CD56/NCAM (NCAM16.2)-149Sm—100 Tests | Fluidigm | Cat#3149021B |
| Anti-Human CD138/Syndecan-1 (DL-101)-150Nd—25 tests | Fluidigm | Cat#3150012C |
| Anti-Human TNFa (mab11)-152Sm—100 Tests | Fluidigm | Cat#3152002B |
| Anti-Human TIGIT (MBSA43)-153Eu—100 Tests | Fluidigm | Cat#3153019B |
| Anti-Human CD3 (UCHT1)-154Sm—100 Tests | Fluidigm | Cat#3154003B |
| Anti-Human CD27 (L128)-155Gd—100 Tests | Fluidigm | Cat#3155001B |
| Anti-Human IFNg (B27)-158Gd—100 Tests | Fluidigm | Cat#3158017B |
| Anti-Human CD161 (HP-3G10)-159Tb—100 Tests | Fluidigm | Cat#3159004B |
| Anti-Human/Mouse MIP1beta (D21–1351)-160Gd—100 Tests | Fluidigm | Cat#3160013B |
| Anti-Human/Mouse Tbet (4B10)-161Dy—100 Tests | Fluidigm | Cat#3161014B |
| Anti-Human FoxP3 (259D/C7)-162Dy—50 Tests | Fluidigm | Cat#3162024A |
| Anti-Human CD183/CXCR3 (G025H7)-163Dy—100 Tests | Fluidigm | Cat#3163004B |
| Anti-Human IL-17A (N49–653)-164Dy—100 Tests | Fluidigm | Cat#3164002B |
| Anti-Human CD279/PD-1 (EH12.2H7)-165Ho—100 Tests | Fluidigm | Cat#3165042B |
| Anti-Human CD197/CCR7 (G043H7)-167Er—50 Tests | Fluidigm | Cat#3167009A |
| Anti-Human CD8 (SK1)-168Er—100 Tests | Fluidigm | Cat#3168002B |
| Anti-Human CD25 (2A3)-169Tm—100 Tests | Fluidigm | Cat#3169003B |
| Anti-Human Granzyme B (GB11)-171Yb—100 Tests | Fluidigm | Cat#3171002B |
| Anti-Human CD57 (HCD57)-172Yb—100 Tests | Fluidigm | Cat#3172009B |
| Anti-Human HLA-DR (L243)-173Yb—100 Tests | Fluidigm | Cat#3173005B |
| Anti-Human CD94 (HP-3D9)-174Yb—100 Tests | Fluidigm | Cat#3174015B |
| Anti-Human Perforin (B-D48)-175Lu—100 Tests | Fluidigm | Cat#3175004B |
| Anti-Human CD127/IL-7Ra (A019D5)-176Yb—100 Tests | Fluidigm | Cat#3176004B |
| Anti-Human CD16 (3G8)-209Bi—100 Tests | Fluidigm | Cat#3209002B |
| Anti-Granzyme K antibody [GM-24C3] (ab3771) | Abcam | Cat#ab3771 |
| Chemicals, peptides, and recombinant proteins | ||
| 1X Phosphate-Buffered Saline without Calcium & Magnesium | Mediatech | Cat #21040CV |
| 10X RBC Lysis Buffer | BioLegend | Cat #420301 |
| 0.4% Trypan Blue Solution | Life Technologies | Cat#15250061 |
| autoMACS Running Buffer - MACS Separation Buffer | Miltenyi Biotec | Cat#130091221 |
| CD138 MicroBeads, Human | Miltenyi Biotec | Cat#130051301 |
| DMSO | Sigma Aldrich | Cat #D2650 |
| PBS, pH 7.4 (1X) | Gibco | Cat#10010023 |
| Bovine Serum Albumin (BSA), 30% ± 2% in 0.85% NaCl | Sigma Aldrich | Cat#A7284 |
| Sodium Azide, 10% (w/v) solution in Ultra-Pure H2O | Teknova | Cat#S0209 |
| RPMI 1640 (1X) Medium | Gibco | Cat#11875093 |
| Fetal Bovine Serum, heat inactivated, qualified (FBS) | Gibco | Cat#10438026 |
| Antibiotic-Antimycotic (100X) | Gibco | Cat#15240096 |
| UltraPure™ DNAse/RNAse-Free Distilled Water | Invitrogen | Cat#10977023 |
| ViaStain™ AOPI Staining Solution | Nexcelom Biosciences |
Cat#CS2–0106-5mL |
| Cell-IDTM Intercalator-Rh—500 μM | Fluidigm | Cat#201103A |
| Cell-IDTM Intercalator-Ir—125 μM | Fluidigm | Cat#201192A |
| Maxpar® Cell Acquisition Solution | Fluidigm | Cat#201237 |
| Maxpar® Cell Staining Buffer j? | Fluidigm | Cat#201068 |
| EQTM Four Element Calibration Beads | Fluidigm | Cat#201078 |
| eBioscienceTM Permeabilization Buffer (10X) | Invitrogen | Cat#00833356 |
| eBioscience™ Fixation/Permeabilization Concentrate | Invitrogen | Cat#00512343 |
| eBioscienceTM Fixation/Permeabilization Diluent | Invitrogen | Cat#00522356 |
| Human TruStain FcXTM (FcR Blocking Solution) | BioLegend | Cat#422302 |
| PierceTM 16% Formaldehyde Solution (w/v), Methanol-free | Thermo Scientific | Cat#28906 |
| Critical commercial assays | ||
| Maxpar® X8 Antibody Labeling Kit, 166Er—4 Rxn | Fluidigm | Cat#201166A |
| Software and algorithms | ||
| FlowJo | Becton Dickinson & Company | https://www.flowjo.com/solutions/flowjo/downloads/ |
| CyTOF Software v7.0.8493 | Fluidigm | https://www.fluidigm.com/software |
| Biological Samples | ||
| Peripheral blood and bone marrow samples collected | This paper; E-PRISM trial | ClinicalTrials.gov: NCT02279394 |
| Deposited Data | ||
| Single-cell RNA sequencing (n=149) and TCR sequencing (n=91) | This paper; E-PRISM trial | phs002476.v1.p1 |
| Single-cell RNA sequencing | Zavidij et al., 2020 | phs001323.v3.p1 |
| Whole-exome sequencing | Bustoros et al., 2020 | phs001323.v3.p1 |
Cryopreserved patient CD138− BM Mononuclear Cell (BMMC) samples were thawed, counted using AO/PI, and pelleted by centrifugation at 400g for 10 minutes. Cells were then incubated in 103Rh viability stain for 15 minutes, washed in CyFACS, and incubated with undiluted Human TruStain FcX for 10 min for Fc receptor blocking. Antibody master mix was applied to the cell suspension for 30 minutes, washed, and fixed/permeabilized with FoxP3 Fixation/Permeabilization Concentrate and Diluent following manufacturer’s guidelines (eBioscience). A mix of intracellular antibodies prepared with 1X Perm Wash was added to each sample and incubated for 30 minutes. Next, cells were washed with 1X Perm Wash, and incubated overnight at 4°C in FoxP3 Fixation/Permeabilization Concentrate and Diluent, containing 191/193Ir DNA Intercalator. Prior to acquisition, samples were transferred to 5 mL round-bottom polystyrene tubes with cell strainer caps, washed and filtered with Cell Staining Buffer (CSB), Cell Acquisition Solution (CAS), and resuspended in CAS supplemented with EQ Four Element Calibration Beads (1:10).
All mass cytometry data was collected on a HeliosTM Mass Cytometer (Fluidigm). The instrument was tuned using CyTOF Tuning Solution according to the Helios User Guide (Fluidigm, p. 60–68). A brief overview of tuning steps includes Pre-XY Optimization, Mass Calibration, XY Optimization, DV Calibration, Dual Calibration, Gases/Current Calibration, and QC report. EQ Four Element Calibration Beads (1:10 in CAS) were used according to the manufacturer protocol before and during acquisition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Clinical trial statistical analysis & biomarker discovery
Patient baseline characteristics were summarized as the number of patients or median values. Responses to study treatment were reported as proportions with 90% exact binomial confidence intervals.
We assessed the proportion of patients alive and progression-free at 2 years, compared to the previously published rate for HRSMM—a median time to progression of only 1.9 years according to the Kyle et al. model (Kyle et al., 2007). Therefore, we decided that for this study, a 2-year progression-free survival rate of 50% would not be considered promising; instead, a progression-free survival rate of 70% or higher would be a promising result.
This phase II study used a single-stage binomial design. With a sample size of 39 patients, the probability of concluding that the treatment is effective if the true rate is 50% was 0.10 and was >0.9 if the true rate is 70%. Assuming an ineligibility rate of 5%, we aimed to accrue 41 patients per Arm to the trial. Time to response (TTR) was measured from treatment initiation to the date the response was first observed. Duration of response was measured from response to progressive disease (PD) or death, censored at the date of last disease assessment for those who did not progress. Time-to-progression (TTP), and progression-free survival (PFS) were measured from the time of treatment initiation to event (PD for TTP; PD or death for PFS). Patients without event were censored at the date of last disease assessment for both TTP and PFS. If non-protocol therapy, excluding erythropoietin, was added prior to an event, patients were censored in the time-to-event analyses at the initiation of non-protocol therapy. Six total patients had progression-free survival events defined as progressive disease to myeloma-defining events (n=4) or death (n=2). Another four patients (n=4) were treated based on worsening biomarkers and the physician’s decision and were considered progressors for our correlative studies only. The Kaplan-Meier method was used to estimate time to event and the log-rank test was used to compare time to event. Cox proportional hazards regression was employed to assess the significance of variable associations with outcome. Continuous variables, such as cell type proportions, were tested as such and were dichotomized based on the median for Kaplan-Meier curves. Statistical analyses were performed using R version 4.1.3. All results with p-values < 0.05 were considered statistically significant. The Benjamini-Hochberg method was used to correct for multiple hypothesis testing, when appropriate. Our data cut-off was November 7, 2020.
ECOG Randomized Trial of Lenalidomide versus Observation in patients with HRSMM
Due to the unavailability of patient-level information, survival data were recapitulated from published Kaplan-Meier (KM) plots in Lonial et al (Lonial et al., 2020). The curves and at-risk tables were extracted as raster images, and their x and y coordinates were digitized using the commercial software DigitizeIt (https://www.digitizeit.xyz/). Using the algorithm proposed by Guyot et al., the coordinates and number of patients at-risk during each time interval were used to estimate censored, time-to-event data (Guyot et al., 2012). With the patient-level data, we were able to re-generate the survival curves, at-risk tables, log-rank tests, etc. In particular, we used the KM method to explore the effect of the 20–2-20 risk stratification system on PFS in the Lenalidomide arm of the ECOG trial and the log-rank test to compute the p-value.
WES analysis
The output from Illumina software was processed by the Picard data processing pipeline to yield BAM files containing well-calibrated, aligned reads. We utilized the Getz Lab CGA WES Characterization pipeline developed at the Broad Institute to call, filter, and annotate somatic mutations and copy number variation. The pipeline employs the following tools: MuTect, ContEst, Strelka, Orientation Bias Filter, DeTiN, AllelicCapSeg, MAFPoNFilter, RealignmentFilter, ABSOLUTE, GATK, PicardTools, Variant Effect Predictor, Oncotator (Cibulskis et al., 2013; Cibulskis et al., 2011; Saunders et al., 2012; Costello et al., 2013; Taylor-Weiner et al., 2018; Landau et al., 2013; Lawrence et al., 2014; Carter et al., 2012; McKenna et al., 2010; McLaren et al., 2016; Ramos et al., 2015). SNVs and CNVs were further cleaned with a custom PoN made of matched normal samples (Lawrence et al., 2014), and a bait bias filter was developed to correct for observed artifacts in the SNV data, as described before (Bustoros et al., 2020). ABSOLUTE was applied to estimate sample purity, ploidy, and absolute somatic copy numbers, which were used to infer the CCFs of point mutations and CNVs from the WES data, following the framework previously described (Carter et al., 2012).
Single-cell RNA sequencing analysis
Analysis workflow
CellRanger v.5.0.1. was used for FASTQ file extraction and generation of count matrices (Zheng et al., 2017). We integrated E-PRISM data with our cohort of non-trial patients with MGUS (n=6), low-risk SMM (LRSMM, n=3), HRSMM (HRSMM, n=12), newly diagnosed MM (NDMM, n=9) and HD (n=11) (Zavidij et al., 2020). Non-trial patients with SMM were risk-stratified based on the original Mayo Clinic criteria (Kyle et al., 2007), and NDMM was diagnosed based on the CRAB criteria.
We performed ambient RNA correction with SoupX; doublet detection with Scrublet, scDblFinder, and SCDS; normalization with Scran; and integration with Harmony (correcting for sample ID and technology) (Young et al., 2020; Wolock et al., 2019; Germain et al., 2021; Bais et al., 2019; Lun et al., 2016; Korsunsky et al., 2019). Samples prepared with 3’-end or 5’-end technology mixed well in PCA, demonstrating successful integration across technologies (Figure S8B). Droplets were deemed to be doublets when at least 2 out of four methods classified them as such and they were only removed from consideration when they clustered together and co-expressed markers of multiple cell types. Droplets containing dying cells with more than 15% mitochondrial gene expression were removed before clustering. Clusters with higher mitochondrial and ribosomal gene expression that clearly separated from well-annotated clusters and lacked interpretable expression markers were removed downstream. In total, we profiled 211,374 immune cells (~1,419 cells/sample), comprising T cells, NK cells, B cells, Monocytes, Dendritic cells, and progenitor populations. For each cell population (T cells, NK cells, B cells, Monocytes, Dendritic cells, progenitor cells), feature selection, dimensionality reduction, and clustering were also performed separately, to identify cell subpopulations (see below for annotation strategy).
Cell type/subtype proportions were calculated as the fraction of cells belonging to said type/subtype out of all immune cells unless otherwise specified. For comparisons of cell type proportions, samples with fewer than 100 cells were removed from consideration. Proportion changes were assessed with Wilcoxon’s rank-sum tests or paired t-tests when comparing serial timepoints within the same patients or matched BM and PB samples. When multiple hypotheses were tested, p-values were corrected using the Benjamini-Hochberg approach. For survival analysis, cell type abundance was discretized based on the median.
Differential expression was performed using Wilcoxon’s rank-sum tests at the single-cell level and DESeq2 at the pseudobulk level (Love et al., 2014). Genes with more than 3 counts in at least 10 cells were considered for differential expression. For pseudobulk analyses, genes with more than 10 counts in at least 25% of samples were considered for differential expression. P-values were corrected for multiple hypotheses testing using the Benjamini-Hochberg approach.
Cell type and subtype annotation
Gene expression markers used for cell type and subtype annotation can be found in Figure 2. Scran-normalized expression values were Z-scored and the mean Z-score per cluster was used to plot the heatmaps.
Within the T cell compartment, we identified naïve CD8+ (CD8+ TN) and CD4+ T cells (CD4+ TN) (LEF1, TCF7, SELL, CCR7), central memory CD8+ (CD8+ TCM) and CD4+ T cells (CD4+ TCM) (FAS, IL7R, HNRNPLL), helper type 1 T cells (Th1) (CD4, CXCR3, GZMA, GZMK, LYAR, CCL5), helper type 2 T cells (Th2) (CD4, GATA3, KRT1, CCR4), helper type 17 T cells (Th17) (CD4, RORA, KLRB1, CCR6, TNFRSF4), regulatory T cells (Treg) (CD4, IL2RA, FOXP3, CTLA4, RTKN2, IKZF2), mucosa-associated invariant T cells (MAIT) expressing a mixture of Th17 and Th1 markers, as well as the specific marker SLC4A10 (MAIT annotation was confirmed by invariant TCR chain usage in overlapping TCR sequencing data), two effector memory CD8+ T cell subpopulations, GZMK+ CD8+ TEM cells (GZMK, CMC1, XCL1, XCL2) and terminal effector cells (GZMB+ CD8+ TEM) (GZMB, GZMH, FGFBP2, PRF1, FCGR3A, GNLY). Furthermore, we identified a subpopulation of CD4+ TN cells expressing genes related to response to interferon-gamma (IFN+ CD4+ TN) (MX1, ISG15, IFI6, IFI44L), two separate subpopulations expressing genes related to response to interferon type-I (IFN-I+ CD4+ TEM & IFN-I+ CD8+ TEM) (IFIT1, IFIT2, IFIT3, TNF), and two activated CD4+ subpopulations expressing genes of the AP-1 module and general activation markers (aCD4+ TN & aCD4+ TCM) (JUN, FOS, FOSB, CD69, DUSP1, TSC22D3). Lastly, we identified a second CD4+ regulatory subpopulation, clustering adjacent to Tregs and expressing high levels of CTLA4 and MHC-II-encoding genes, a profile most consistent with effector Tregs, which we annotated as eTregs.
Within the NK cell compartment, we identified CD56bright NK cells (CD56br NK) (NCAM1), CD2, XCL2, KLRC1, IL7R, SELL, GZMK), CD160+ NK cells expressing markers of CD56bright NK cells as well as CD160, SPRY2, and TOX2, CD56dim NK cells (CD56dim NK) (GZMB, GZMH, FGFBP2, PRF1, NKG7, CX3CR1, GNLY, B3GAT1) and gamma-delta T cells (Tgd) (CD3D, TRGV9, TRDV2). Furthermore, we identified a subpopulation of NK cells expressing genes related to response to interferon-gamma (IFN+ NK) (MX1, ISG15, IFI6, IFI44L), a separate subpopulation expressing genes related to response to interferon type-I (IFN-I+ NK) (IFIT1, IFIT2, IFIT3, TNF), and an activated subpopulation with characteristics of both CD56bright and CD56dim NK cells (aNK), expressing genes of the AP-1 module and general activation markers (FOS, JUN, JUNB, CD69, CXCR4, DUSP1, NFKB1, NFKB2).
Within the B cell compartment, we identified immature B cells (IBC) (MME, CD19, CD38, IGHM, SOX4, TCL1A, RAG1, RAG2, VPREB1, IGLL1), transitional B cells (TBC) (CD19, MS4A1, CD38, IGHM, IGHD, TCL1A, IL4R, SELL), naïve B cells (NBC) (CD19, MS4A1, IGHM, IGHD, IL4R, SELL, CCR7, FCER2, ABCB1), a population expressing higher levels of CXCR5, CD83, and TNFSF9 (encoding 4–1BBL), called germinal center B cells (GBC), marginal zone B cells (MZB) (CD1C, IGHM, PLD4, LINC01857), atypical B cells (ABC) (TBX21, ITGAX, FCRL5, ENC1, TNFRSF1B, SOX5, MPP6), and two populations of class-switched memory B cells: one expressed higher levels of TCF7 and was called resting memory B cells (BRM), while the other expressed higher levels of FAS, CD86, ITGB1, S100A10, and TOX, and was called effector memory B cells (BEM). Lastly, we identified a subpopulation of B cells expressing genes related to response to interferon-gamma (IFN+ BC) (MX1, ISG15, IFI6, IFI44L).
Within the monocyte compartment, we identified three main subpopulations of classical (CD14+) monocytes: a subpopulation expressing higher levels of SELL, VCAN, S100A8, S100A9, and S100A12 (SELL+ CD14+ Mono), a subpopulation expressing higher levels of genes encoding MHC-II peptides (HLA-DR++ CD14+ Mono), and a subpopulation expressing pro-inflammatory factors, such as IL1B, CXCL8, CXCL2, CCL3, CCL4, and CEBPD (Cytokine+ CD14+ Mono). A fourth subpopulation expressed CD14, SELL, VCAN, S100A8, S100A9, S100A12, as well as MPO, AZU1, ELANE, PRTN3, and RNASE2, and was called “Neutrophil-like Mono”. Furthermore, we identified non-classical CD16+ Monocytes (FCGR3A, MS4A7, CSF1R, CDKN1C, RHOC), intermediated CD14+ CD16+ Monocytes expressing CD14, FCGR3A (which encodes CD16), and high levels of MHC-II-encoding genes, a CD16+ subpopulation of macrophages expressing high levels of complement factor C1q (C1QA, C1QB, C1QC, SELENOP, SDC3), and a population of monocytes expressing genes related to response to interferon-gamma (IFN+ Mono) (MX1, ISG15, IFI6, IFI44L).
Within the dendritic cell compartment, we identified canonical dendritic cells type 2 (cDC2) (FCER1A, CLEC10A, CD1C and MHC-II encoding genes), canonical dendritic cells type 1 (cDC1) (CLEC9A, C1orf54, IDO1, CADM1, CLMK, BATF3), monocyte-derived dendritic cells (MoDC) (FCER1A, CLEC10A, CD1C, CD14), plasmacytoid dendritic cells (pDCs) (LILRA4, IL3RA, GZMB, IRF4, SERPINF1), and AXL+ SIGLEC6+ dendritic cells (AS-DCs) (AXL, SIGLEC6, ADAM33, LTK). Furthermore, we identified cycling subpopulations of pDCs and cDC2s (cpDC, ccDC2), activated subpopulations expressing the pro-inflammatory markers IL1B, CXCL8, and CCL3 (acDC2, apDC), and a MoDC subpopulation expressing genes related to response to interferon-gamma (IFN+ MoDC) (MX1, ISG15, IFI6, IFI44L).
Lastly, within the progenitor cell compartment, we identified hematopoietic stem cells (HSC) (CD34, CD164, BEX1, BEX2, AVP, CRHBP, HLF), multi-potent progenitor cells (MPP) (CD34, CD33, MPO, FLT3, MZB1), granulocyte-monocyte progenitor cells (GMP) (MPO, AZU1, PRTN3, ELANE, LYZ), monocyte-dendritic cell progenitors (MDP) (MPO, LYZ, IRF8, LY86, RUNX2, LILRB4), megakaryocyte-erythroid progenitors (MEP) (GATA2, FCER1A, ITAG2B, CSF2RB), erythroid progenitors (EP) (GATA1, EPOR, CA1, CA2, EPCAM, KLF1, BLVRB, APOC1, APOE), megakaryocyte progenitors (MKP) (GATA2, FCER1A, ITGA2B, PLEK, GP1BB, PPBP, PF4, GP9), basophil-mast cell progenitors (BMP) (MS4A2, MS4A3, TPSAB1, TPSB2, HDC, CLC, PRG2), common lymphoid progenitors (CLP) (FLT3, TRBC2, MZB1, LTB, JCHAIN, ADA, BCL2), pro-B cells (Pro-B) (DNTT, MME, PAX5, RAG1, RAG2), and pre-B cells (Pre-B) (VPREB1, IGLL1, JCHAIN, TCL1A). Within this compartment, we also identified a small population of stroma cells (CXCL12, LEPR, KITLG, VCAM1, COL1A1, COL1A2, CCL2).
Gene expression signature discovery & annotation
A Bayesian version of Non-negative Matrix Factorization (NMF) was performed using SignatureAnalyzer on the count matrix of the top 5,000 highly variable genes for gene expression signature discovery (Kasar et al., 2015; Kim et al., 2016). Half-normal priors were imposed for both the W (gene by signature) and the H matrix (cell by signature), as we reasoned that a single gene could contribute towards multiple expression programs and a single cell could be described by multiple expression programs. The tool was run 30 times and the run that resulted in a K (i.e., number of signatures) equal to the mode of the K distribution and had the lowest objective was selected for downstream analysis (Figure S5B). Gene expression signature markers were nominated by (i) multiplying the W matrix by the sum of signature activity across all cells in the H matrix, (ii) calculating the fraction of each signature’s activity for each gene (matrix F) and (iii) ranking genes based on the product of W (i.e., how strongly each gene contributes to the signature) and F (i.e., how strongly each signature contributes to the gene). A total of 26 gene expression (GEX) signatures were extracted (Figure S5C): GEX-1 corresponded to cytokine signaling associated with GZMK-expressing subpopulations (XCL1, XCL2, CCL3, CCL4); GEX-2 captured naïve T cells (TCF7, LEF1, TXNIP, IL7R, LTB, NOSIP); GEX-3 captured dendritic cells (CLEC10A, FCER1A, CD74, HLA-DRA, HLA-DRB1); GEX-4 captured B cells (CD19, MS4A1, IGHM, IGHD, CD79A, CD79B); GEX-5 was associated with ferritin heavy and light chain expression (FTH1, FTL) and was primarily active in myeloid cells; GEX-6 corresponded to pro-inflammatory signaling (IL1B, CXCL8, CXCL2, CEBPD) and was primarily active in myeloid cells; GEX-7 corresponded to genes induced by interferon-gamma (MX1, STAT1, XAF1, IFI6, IFI44L, ISG15) and was active in T cells, B cells, NK cells, and monocytes; GEX-8 was associated with thymosin expression and the alpha and beta chains of the TCR (TMSB4X, TMSB10, TRAC, TRBC1), and was primarily active in T cells; GEX-9 was associated with histone 1 gene expression (HIST1H1C, HIST1H1D, HIST1H1E) and was primarily active in dendritic cells; GEX-10 was associated with metabolic programming (GLUL, SLC43A2, SLC25A37) and hypoxia-related signaling (HIF1A, TIMP1, NEAT1), and was primarily active in myeloid cells; GEX-11 corresponded to CD14+ monocytes (CD14, MS4A6A, FCN1, LYZ); GEX-12 corresponded to immature B cells (ACSM3, IGLL1, VPREB1, TCL1A, MME, SOX4); GEX-13 corresponded to pDCs (LILRA4, JCHAIN, SERPINF1, PTGDS, ITM2C, IRF4, IRF8); GEX-14 corresponded to a different program within classical monocytes (S100A8, S100A9, S100A12, VCAN); GEX-15 corresponded to Th17 cells and MAITs (RORA, KLRB1, CCR6); GEX-16 was associated with interferon type-I signaling (IFIT1, IFIT2, IFIT3, PMAIP1, OASL, TNF, HERC5, ZC3HAV1); GEX-17 was associated with T cell surface antigens (CD5, CD6, CD7); GEX-18 and GEX-19 were associated with genes of the AP-1 module (JUN, FOS, FOSB) and general activation markers (DUSP1, TSC22D3, NFKBIA); GEX-20 corresponded to CD16+ monocytes (FCGR3A, CDKN1C, SERPINA1, MS4A7); GEX-21 corresponded to cytotoxic T cells (GZMK, GZMH, NKG7, CD8A, CD8B); GEX-22 corresponded to GZMB+ cytotoxic T cells and NK cells (GZMB, GNLY, SPON2, CLIC3, FGFBP2); GEX-23 was associated with genes frequently expressed by myeloid cells (AHNAK, TLR2, CYBB) and was primarily active in monocytes; GEX-24 was associated with memory CD4+ T cell expression markers (GATA3, CD28, PRDM1, ITGB1, ANXA1) and was primarily active in those populations; GEX-25 and GEX-26 were associated with cytoskeleton-related genes (ACTB, ACTG1, PFN1, CFL) and were primarily active in myeloid cells.
For survival analysis, each sample was assigned its mean signature activity across cells, and mean activity was discretized based on the median.
Naïve Bayes Classifier and Normalization Scores
We trained a Naïve Bayes classifier using the R package caret on a training set (n=41) comprising BM samples from patients with HRSMM and HD, using 10-fold cross-validation and 100 iterations, following a parameter sweep (Kuhn et al., 2008). The input to the classifier was the composition matrix of cell type proportions and included data on 63 subpopulations. The accuracy in the testing set (n=16) was 94% (95% CI: 69.8%−99.8%, p=0.0057) (Figure S5A). For each cell type, we compute its importance to the classification using the varImp() function, which employs an ROC approach, and signed it based on whether the cell type had a higher mean abundance in disease (minus) or HD (plus). We then computed the weighted sum of the product of each cell type’s proportion and its signed importance (normalization score), and classified patients as reactive (least normal-like) or non-reactive (most normal-like), based on the median normalization score of patients at BL.
Single-cell TCR sequencing analysis
CellRanger v5.0.1 was used to extract FASTQ files and produce clonotype matrices (Zheng et al., 2017). When multiple alignments were called for a single chain, the alignment with the most UMIs was selected, and when multiple chains were called for a single cell barcode, the top two chains in terms of UMI counts were selected.
To control for variability in T cell numbers across samples, we downsampled a range of T cell numbers (n=75–500) from each sample with 100 iterations and averaged repertoire diversity estimates across iterations. Repertoire diversity was assessed using the Chao index, as implemented in the vegan R package. To estimate the proportion of clonotypes belonging to a given clone size (Rare: ≤1%; Small: >1% and <5%; Medium: ≥5% and <10%; Large: ≥10%) in a given sample, we downsampled 100 T cells from each sample with 100 iterations, converted clonotype counts into frequencies, computed the frequency of each clone size, and subsequently averaged the frequency of each clone size across iterations and renormalized those frequencies for plotting. To estimate the probability of a given T cell subtype to be clonally expanded in patients or HD, we first computed stable clone sizes for each clonotype in each sample, by downsampling 100 T cells with 100 iterations, converting clonotype counts into frequencies, computing each clonotype’s clone size, and subsequently assigning each clonotype the mode of its clone size distribution across all iterations. Next, for each T cell subtype, we randomly sampled 100 cells across all patients (or HD), and for each iteration, we computed the frequency of rare (proportion of singletons) and expanded (1-rare) clonotypes. For visualization purposes, the average frequencies were renormalized and plotted. P-values were computed by bootstrapping with 10,000 iterations.
CyTOF analysis
The data was normalized using the FCS Processing tab of the Fluidigm CyTOF Software 7.0.8493. Data analysis was manually performed using FlowJo 10.7.1. For initial data clean-up, cell events were gated to remove dead cells and debris through biaxial plots of Time vs. Event Length, Beads (for removal of the EQ Calibration Beads), and Gaussian-derived parameters (Residual, Width, Offset). The viability stain 103Rh was used to gate out dead cells on PBMC populations. All viable cells were backgated on both DNA parameters (191Ir and 193Ir) to ensure no doublets were included. Please refer to Hallisey et al. for the detailed protocol (Hallisey et al., 2022).
ADDITIONAL RESOURCES
The clinical trial study design and record can be found at: https://clinicaltrials.gov/ct2/show/NCT02279394
Supplementary Material
HIGHLIGHTS.
EloLenDex is associated with 4-year PFS of 89% in patients with high-risk SMM.
Immune reactivity and post-therapy immune normalization (PIN) are associated with longer PFS.
Higher abundance of GZMK+ cytotoxic T cells is associated with longer PFS.
Blood-based immune profiling detects immune dysregulation associated with disease.
ACKNOWLEDGEMENTS
The authors would like to thank the patients who participated in the E-PRISM trial. Additionally, the authors would like to thank Anna V. Justis, PhD, for medical editing support, and Sarah Nersersian, MSc, and Tiana Issa for illustration support. This research was supported by a Stand Up To Cancer-Bristol-Meyers Squibb Catalyst Research Grant (Grant Number: SU2C-AACR-CT05-17). Stand Up To Cancer is a division of the Entertainment Industry Foundation. Research Grants are administered by the American Association for Cancer Research, the Scientific Partner of SU2C. Furthermore, this research was supported in part by the National Institutes of Health (NIH R01 CA205954), the Leukemia and Lymphoma Society (LLS, Career Development Program 2316-17), the Multiple Myeloma Research Foundation (MMRF), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation. R.S.P. is supported by the MMRF Research Fellowship Award, the International Waldenstrom’s Macroglobulinemia Foundation’s Robert A. Kyle Award, the Claudia Adams-Barr Award for Innovative Basic Cancer Research, and the International Myeloma Society. G.G. is partially supported by the Paul C. Zamecnik Chair in Oncology at Massachusetts General Hospital Cancer Center. S.Z.U. was supported by a career development award from the LLS.
N.J.H. is a consultant for Constellation Pharmaceuticals. F.A. is an employee of Illumina Inc. O.Z. is an employee of Ikena Oncology and a stockholder in Ikena Oncology and Morphosys AG. G.G. receives research funds from IBM and Pharmacyclics and is an inventor on patent applications filed by the Broad Institute related to MSMuTect, MSMutSig, POLYSOLVER, SignatureAnalyzer-GPU, and MSIDetect. He is also a founder, consultant, and holds privately held equity in Scorpion Therapeutics. I.M.G. has a consulting or advisory role with AbbVie, Adaptive, Amgen, Aptitude Health, Bristol Myers Squibb, GlaxoSmithKline, Huron Consulting, Janssen, Menarini Silicon Biosystems, Oncopeptides, Pfizer, Sanofi, Sognef, Takeda, The Binding Site, and Window Therapeutics; has received speaker fees from Vor Biopharma and Veeva Systems, Inc.; and her spouse is the CMO and equity holder of Disc Medicine. S.M. has a consulting role with Abbvie, Adaptive Biotechnology, Amgen, Celgene/BMS, GlaxoSmithKline, Janssen, Novartis, Oncopeptides, Regeneron, Roche, Takeda, and has received research funding from Abbvie, Adaptive Biotechnology, Amgen, Celgene/BMS, GlaxoSmithKline, Janssen, Novartis, Oncopeptides, Regeneron, Roche, Takeda. A.J.Y. has a consulting role with Adaptive Biotechnologies, Amgen, BMS, Celgene, GSK, Janssen, Karyopharm, Oncopeptides, Sanofi, Takeda, and has received research funding from Amgen, Janssen, and Takeda. M.B. is a consultant for Sanofi, Genzyme, and Janssen, and has received research funding from MedImmune, Janssen, Legend Biotech, Amgen, Celularity, Bristol Myers Squibb, Celgene, Bluebird bio, Millennium, Takeda, Cerecor (currently Avalo therapeutics), and C4 Therapeutics. M.B has an advisory role and received honoraria from Bristol Myers Squibb, Takeda, Janssen, and Menarini. T.H.M. received advisory board fees from Legend Biotech. R.S.P., G.G., and I.M.G. are co-inventors on a patent application related to this work (PCT/US22/74839).
Footnotes
DECLARATION OF INTERESTS
M.P.A., R.A.R., J.B., M.H., A.S., S.V., D.H.M., S.U., N.K.S., E.D.L., S.T., T.W., L.F., S.A., J.M.R., J.Z., J.J.V., A.B., S.U., J.M., A.J., J.L., O.N., P.R., A.Z.B., M.V.M., and L.T. declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic and/or gender minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. We support inclusive, diverse, and equitable conduct of research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Weiss BM, Abadie J, Verma P, Howard RS, and Kuehl WM (2009). A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 113, 5418–5422. 10.1182/blood-2008-12-195008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, Larson DR, Plevak MF, Jelinek DF, Fonseca R, et al. (2007). Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med 356, 2582–2590. 10.1056/NEJMoa070389. [DOI] [PubMed] [Google Scholar]
- Mateos MV, Kumar S, Dimopoulos MA, Gonzalez-Calle V, Kastritis E, Hajek R, De Larrea CF, Morgan GJ, Merlini G, Goldschmidt H, et al. (2020). International Myeloma Working Group risk stratification model for smoldering multiple myeloma (SMM). Blood Cancer J 10, 102. 10.1038/s41408-020-00366-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visram A., Cook J., and Warsame R. (2021). Smoldering multiple myeloma: evolving diagnostic criteria and treatment strategies. Hematology Am Soc Hematol Educ Program 2021, 673–681. 10.1182/hematology.2021000304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos MV, Hernandez MT, Giraldo P, de la Rubia J, de Arriba F, Lopez Corral L, Rosinol L, Paiva B, Palomera L, Bargay J, et al. (2013). Lenalidomide plus dexamethasone for high-risk smoldering multiple myeloma. N Engl J Med 369, 438–447. 10.1056/NEJMoa1300439. [DOI] [PubMed] [Google Scholar]
- Mateos MV, Hernandez MT, Salvador C, Rubia J, de Arriba F, Lopez-Corral L, Rosinol L, Paiva B, Palomera L, Bargay J, et al. (2022). Lenalidomide-dexamethasone versus observation in high-risk smoldering myeloma after 12 years of median follow-up time: A randomized, open-label study. Eur J Cancer 174, 243–250. 10.1016/j.ejca.2022.07.030. [DOI] [PubMed] [Google Scholar]
- Lonial S, Jacobus S, Fonseca R, Weiss M, Kumar S, Orlowski RZ, Kaufman JL, Yacoub AM, Buadi FK, O’Brien T, et al. (2020). Randomized Trial of Lenalidomide Versus Observation in Smoldering Multiple Myeloma. J Clin Oncol 38, 1126–1137. 10.1200/JCO.19.01740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EB, Yee AJ, and Raje N.(2020). Treatment of Smoldering Multiple Myeloma: Ready for Prime Time? Cancers (Basel) 12. 10.3390/cancers12051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateos MV, and San Miguel JF (2015). Smoldering multiple myeloma: when to observe and when to treat? Am Soc Clin Oncol Educ Book, e484–492. 10.14694/EdBook_AM.2015.35.e484. [DOI] [PubMed] [Google Scholar]
- Perez-Persona E., Vidriales MB., Mateo G., Garcia-Sanz R., Mateos MV., de Coca AG., Galende J., Martin-Nunez G., Alonso JM., de Las Heras N., et al. (2007). New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of BM plasma cells. Blood 110, 2586–2592. 10.1182/blood-2007-05-088443. [DOI] [PubMed] [Google Scholar]
- Bustoros M, Sklavenitis-Pistofidis R, Park J, Redd R, Zhitomirsky B, Dunford AJ, Salem K, Tai YT, Anand S, Mouhieddine TH, et al. (2020). Genomic Profiling of Smoldering Multiple Myeloma Identifies Patients at a High Risk of Disease Progression. J Clin Oncol 38, 2380–2389. 10.1200/JCO.20.00437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misund K, Keane N, Stein CK, Asmann YW, Day G, Welsh S, Van Wier SA, Riggs DL, Ahmann G, Chesi M, et al. (2020). MYC dysregulation in the progression of multiple myeloma. Leukemia 34, 322–326. 10.1038/s41375-019-0543-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle EM, Deshpande S, Tytarenko R, Ashby C, Wang Y, Bauer MA, Johnson SK, Wardell CP, Thanendrarajan S, Zangari M, et al. (2021). The molecular make up of smoldering myeloma highlights the evolutionary pathways leading to multiple myeloma. Nat Commun 12, 293. 10.1038/s41467-020-20524-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, Martincorena I, Dawson KJ, Samur MK, Zamora J, et al. (2018). Genomic patterns of progression in smoldering multiple myeloma. Nat Commun 9, 3363. 10.1038/s41467-018-05058-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oben B, Froyen G, Maclachlan KH, Leongamornlert D, Abascal F, Zheng-Lin B, Yellapantula V, Derkach A, Geerdens E, Diamond BT, et al. (2021). Whole-genome sequencing reveals progressive versus stable myeloma precursor conditions as two distinct entities. Nat Commun 12, 1861. 10.1038/s41467-021-22140-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavidij O., Haradhvala NJ., Mouhieddine TH., Sklavenitis-Pistofidis R., Cai S., Reidy M., Rahmat M., Flaifel A., Ferland B., Su NK., et al. (2020). Single-cell RNA sequencing reveals compromised immune microenvironment in precursor stages of multiple myeloma. Nat Cancer 1, 493–506. 10.1038/s43018-020-0053-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailur JK, McCachren SS, Doxie DB, Shrestha M, Pendleton K, Nooka AK, Neparidze N, Parker TL, Bar N, Kaufman JL, et al. (2019). Early alterations in stem-like/resident T cells, innate and myeloid cells in the BM in preneoplastic gammopathy. JCI Insight 5. 10.1172/jci.insight.127807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhodapkar MV, Krasovsky J, Osman K, and Geller MD (2003). Vigorous premalignancy-specific effector T cell response in the BM of patients with monoclonal gammopathy. J Exp Med 198, 1753–1757. 10.1084/jem.20031030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Strowig T, Verma R, Koduru S, Hafemann A, Hopf S, Kocoglu MH, Borsotti C, Zhang L, Branagan A, et al. (2016). Microenvironment-dependent growth of preneoplastic and malignant plasma cells in humanized mice. Nat Med 22, 1351–1357. 10.1038/nm.4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, Landgren O, and Mateos MV (2015). Smoldering multiple myeloma. Blood 125, 3069–3075. 10.1182/blood-2014-09-568899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galletti G, De Simone G, Mazza EMC, Puccio S, Mezzanotte C, Bi TM, Davydov AN, Metsger M, Scamardella E, Alvisi G, et al. (2020). Two subsets of stem-like CD8(+) memory T cell progenitors with distinct fate commitments in humans. Nat Immunol 21, 1552–1562. 10.1038/s41590-020-0791-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajkumar SV, Dimopoulos MA, Palumbo A, Blade J, Merlini G, Mateos MV, Kumar S, Hillengass J, Kastritis E, Richardson P, et al. (2014). International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 15, e538–548. 10.1016/S1470-2045(14)70442-5. [DOI] [PubMed] [Google Scholar]
- Paiva B., Mateos MV., Sanchez-Abarca LI., Puig N., Vidriales MB., Lopez-Corral L., Corchete LA., Hernandez MT., Bargay J., de Arriba F., et al. (2016). Immune status of high-risk smoldering multiple myeloma patients and its therapeutic modulation under LenDex: a longitudinal analysis. Blood 127, 1151–1162. 10.1182/blood-2015-10-662320. [DOI] [PubMed] [Google Scholar]
- Guyot P, Ades AE, Ouwens MJ, and Welton NJ (2012). Enhanced secondary analysis of survival data: reconstructing the data from published Kaplan-Meier survival curves. BMC Med Res Methodol 12, 9. 10.1186/1471-2288-12-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G.(2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, and Getz G.(2011). ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602. 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CT, Wong WS, Swamy S, Becq J, Murray LJ, and Cheetham RK (2012). Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817. 10.1093/bioinformatics/bts271. [DOI] [PubMed] [Google Scholar]
- Costello M., Pugh TJ., Fennell TJ., Stewart C., Lichtenstein L., Meldrim JC., Fostel JL., Friedrich DC., Perrin D., Dionne D., et al. (2013). Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67. 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Weiner A, Stewart C, Giordano T, Miller M, Rosenberg M, Macbeth A, Lennon N, Rheinbay E, Landau DA, Wu CJ, and Getz G.(2018). DeTiN: overcoming tumor-in-normal contamination. Nat Methods 15, 531–534. 10.1038/s41592-018-0036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Carter SL, Stojanov P, McKenna A, Stevenson K, Lawrence MS, Sougnez C, Stewart C, Sivachenko A, Wang L, et al. (2013). Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726. 10.1016/j.cell.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G.(2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat Biotechnol 30, 413–421. 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, and DePristo MA (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, and Cunningham F.(2016). The Ensembl Variant Effect Predictor. Genome Biol 17, 122. 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, and Getz G.(2015). Oncotator: cancer variant annotation tool. Hum Mutat 36, E2423–2429. 10.1002/humu.22771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GX., Terry JM., Belgrader P., Ryvkin P., Bent ZW., Wilso R., Ziraldo SB., Wheeler TD., McDermott GP., Zhu J., et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat Commun 8, 14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, and Behjati S.(2020). SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience 9. 10.1093/gigascience/giaa151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock SL, Lopez R, and Klein AM (2019). Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291 e289. 10.1016/j.cels.2018.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain PL, Lun A, Garcia Meixide C, Macnair W, and Robinson MD (2021). Doublet identification in single-cell sequencing data using scDblFinder. F1000Res 10, 979. 10.12688/f1000research.73600.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bais AS, and Kostka D.(2020). scds: computational annotation of doublets in single-cell RNA sequencing data. Bioinformatics 36, 1150–1158. 10.1093/bioinformatics/btz698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun AT, McCarthy DJ, and Marioni JC (2016). A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res 5, 2122. 10.12688/f1000research.9501.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I., Millard N., Fan J., Slowikowski K., Zhang F., Wei K., Baglaenko Y., Brenner M., Loh PR., and Raychaudhuri S. (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296. 10.1038/s41592-019-0619-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S.(2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, Lawrence MS, Kiezun A, Fernandes SM, Bahl S, et al. (2015). Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun 6, 8866. 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D’Andrea A, and Getz G.(2016). Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 48, 600–606. 10.1038/ng.3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M.(2008). Building Predictive Models in R Using the caret Package. Journal of Statistical Software, 28(5), 1–26. 10.18637/jss.v028.i0527774042 [DOI] [Google Scholar]
- Hallisey M, Dennis J, Abrecht C, Pistofidis RS, Schork AN, Lightbody ED, Heilpern-Mallory D, Severgnini M, Ghobrial IM, Hodi FS, and Baginska J.(2022). Mass cytometry staining for human BM clinical samples. STAR Protoc 3, 101163. 10.1016/j.xpro.2022.101163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw single-cell RNA and TCR sequencing data generated for this study have been deposited in dbGaP (phs002476.v1.p1). Raw single-cell RNA sequencing data generated in Zavidij et al. have been deposited in dbGaP (phs001323.v3.p1). Raw whole-exome sequencing data generated for this study have been deposited in dbGaP (phs001323.v3.p1).
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.