

Abstract
We describe the Predicting Protein–Compound Interactions (PrePCI) database which comprises over 5 billion predicted interactions between 6.8 million chemical compounds and 19,797 human proteins. PrePCI relies on a proteome‐wide database of structural models based on both traditional modeling techniques and the AlphaFold Protein Structure Database. Sequence‐ and structural similarity‐based metrics are established between template proteins, T, in the Protein Data Bank that bind compounds, C, and query proteins in the model database, Q. When the metrics exceed threshold values, it is assumed that C also binds to Q with a likelihood ratio (LR) derived from machine learning. If the relationship is based on structural similarity, the LR is based on a scoring function that measures the extent to which C is compatible with the binding site of Q as described in the LT‐scanner algorithm. For every predicted complex derived in this way, chemical similarity based on the Tanimoto coefficient identifies other small molecules that may bind to Q. An overall LR for the binding of C to Q is obtained from Naive Bayesian statistics. The PrePCI database can be queried by entering a UniProt ID or gene name for a protein to obtain a list of compounds predicted to bind to it along with associated LRs. Alternatively, entering an identifier for the compound outputs a list of proteins it is predicted to bind. Specific applications of the database to lead discovery, elucidation of drug mechanism of action, and biological function annotation are described.
Keywords: chemical similarity, protein–compound interactions, protein–compound database, structural alignment
Abbreviations
- PCIs
protein–compound interactions
- PDB
Protein Data Bank
- TC
Tanimoto coefficient
- AF
AlphaFold
- CDD
Conserved Domain Database
1. INTRODUCTION
Protein‐small molecule interactions, termed here protein–compound interactions (PCIs), play essential roles at all biological levels (Cappelletti et al., 2021; Diether et al., 2019; Feng et al., 2014; Lempp et al., 2019; Milanesi et al., 2020). Delineation of PCIs for the human proteome is essential for developing a systems‐level understanding of biological networks and the molecular basis of therapeutic and off‐target effects of drugs. Recent advances in mass spectrometry have enabled high‐throughput identification of PCIs for focused sets of metabolites and drugs, uncovering many previously unreported PCIs (Diether et al., 2019; Lempp et al., 2019; Piazza et al., 2018, 2020), suggesting that much of protein–compound space remains to be discovered. We previously reported LT‐scanner (Hwang et al., 2017), a template‐based method that uses protein structural alignment between models of query proteins and experimentally determined protein–compound complexes to predict PCIs involving 26 K compounds in the Protein Data Bank (PDB; Berman et al., 2000). Here we describe Predicting Protein–Compound Interactions (PrePCI), which extends LT‐scanner by dramatically increasing the number of compounds and proteins considered. Calculating chemical similarity with the Tanimoto coefficient (TC; Bajusz et al., 2015) between molecular fingerprints of PDB ligands and compounds in the PubChem database (Kim et al., 2019, 2021) expands the chemical space explored to 6.8 million compounds. In addition, combining protein models from the PDB (Berman et al., 2000), the AlphaFold Protein Structure Database (Jumper et al., 2021; Varadi et al., 2022), and our in‐house homology database PrePMod (Garzón et al., 2016) provides almost complete structural coverage of the human proteome. PrePCI provides predictions for ~5 billion PCIs, and, as reported below, is extensively validated.
Many computational approaches have been developed to predict PCIs. Docking‐based methods generate poses of compounds in complex with a protein of interest which are subsequently scored to estimate binding affinities by functions typically based on physical forcefields (Bottegoni et al., 2009; Friesner et al., 2006; Miller et al., 2021; Murphy et al., 2016; Trott & Olson, 2009). Such strategies have enabled structure‐based virtual screening of hundreds of millions to billions of small molecules and have discovered novel protein chemotypes (Lyu et al., 2019; Sadybekov et al., 2022). However, the computational costs of pose generation and scoring currently prevent docking methods from being applied at a proteome scale. Ligand‐based approaches like Similarity Ensemble Approach (Keiser et al., 2007) and QSAR‐based methods (Nikolova & Jaworska, 2004) infer novel PCIs based on the similarity of query compounds to seed compounds that are already known to bind the query protein (Willett, 2010). Such methods are able to leverage increasingly large amounts of high‐throughput screening data and are generally rapid enough to use with large‐scale chemical libraries. However, since most proteins do not have a set of known binders against which to compare, ligand‐based methods have difficulty scaling to proteome‐wide applications. More recent approaches include (1) machine‐learned scoring functions (Ballester & Mitchell, 2010; Brown et al., 2021; Paggi et al., 2021; Wójcikowski et al., 2017; Zhu et al., 2020), (2) protein–ligand interaction fingerprints which compare predicted poses to experimentally determined complexes (Da & Kireev, 2014; Perez‐Nueno et al., 2009), and (3) convolutional neural networks which learn structural features directly from PCI structures (Ragoza et al., 2017; Stepniewska‐Dziubinska et al., 2018; Wallach et al., 2015).
In contrast, proteochemometric methods which infer PCIs using independent protein and compound features are potentially amenable to proteome‐scale PCI prediction as they do not require pose generation for each PCI. For example, algorithms such as REMAP (Lim, Poleksic, et al., 2016), COSINE (Lim, Gray, et al., 2016), NRLMF (Liu et al., 2016), and MDMF2A (Liu et al., 2022), formulate PCI prediction as a matrix factorization problem in which low rank matrices representing abstract protein and chemical features are derived from protein sequence similarity and chemical similarity, respectively (Lim, Poleksic, et al., 2016). 3D‐REMAP augments REMAP with ligand binding site similarity and binding affinities for compounds of interest (Lim et al., 2019).
LT‐scanner uses experimentally resolved protein–compound complexes from the PDB (Berman et al., 2000) to scan large databases of protein models to identify residues on their surfaces that are likely to bind similar ligands (Hwang et al., 2017). Use of a simple scoring function enables proteome‐wide application. Most closely related to PrePCI is the FINDSITE suite of programs which use protein threading to identify regions within a query protein which can be reasonably modeled using a PDB protein–compound complex as a template (Zhou et al., 2018, 2021). Ligands bound to identified binding sites are then used as seeds for ligand‐based virtual screening. Unique to PrePCI is the large proteome‐wide database of predicted PCIs described in this publication.
To illustrate potential applications of the PrePCI database we present a number of case studies where PrePCI is used to generate suggestions of novel lead compounds for cancer targets and possible targets underlying drug mechanisms of action and to annotate protein function based on interactions with cellular signaling molecules. Notably, in each of these cases, the predictions emphasize the contribution of structure to proteome‐scale PCI prediction. We anticipate that PrePCI will be a useful resource for generating hypotheses regarding protein–compound interactions in multiple applications. PrePCI predictions can be queried through the web‐hosted database application available at https://honiglab.c2b2.columbia.edu/prepci.html.
2. RESULTS
2.1. PrePCI overview
The PrePCI algorithm is depicted in Figure 1 and consists of three components. Figure 1a illustrates the sequence similarity component where a query protein sequence is matched to protein sequences from PDB template–compound complexes with BLAST. A query protein is predicted as a target of a compound which appears in a PDB complex based on the sequence alignment score of the query with the template protein (see Section 4). Figure 1b illustrates the LT‐scanner component in which a query protein is structurally aligned to a template complex in the PDB (Berman et al., 2000) with the Ska program (Yang & Honig, 2000), which calculates a protein structure distance (PSD) between the aligned structures. Ska emphasizes local versus global alignment with the effect of highlighting structural elements of likely functional relevance, enabling the comparison of the predicted query interface with the template binding site (Yang & Honig, 2000). The transformation that aligns the two proteins is used to place the PDB compound in the coordinate frame of the query. The LT‐scanner scoring function (Hwang et al., 2017) assesses the compatibility of the compound with the query protein by calculating a score based on the extent to which residues in the query binding site recapitulate the physicochemical interactions between the protein and compound in the template complex. The sequence similarity and LT‐scanner calculations are performed for all query protein sequences and models and against all PDB protein–compound templates. Figure 1c illustrates the chemical similarity component where PDB compounds are matched to topologically similar PubChem compounds. When the TC between a PDB compound and a Pubchem compound exceeds 0.5, the Pubchem compound is predicted to target the protein found by either sequence similarity (Figure 1a) or LT‐scanner (Figure 1b). A Bayesian procedure is used to integrate the sequence, structure, and chemical similarity scores for each query protein–compound prediction into a likelihood ratio derived from a true positive set of experimentally characterized PCIs. The scored PCI predictions comprise the PrePCI database (PrePCI/DB).
FIGURE 1.
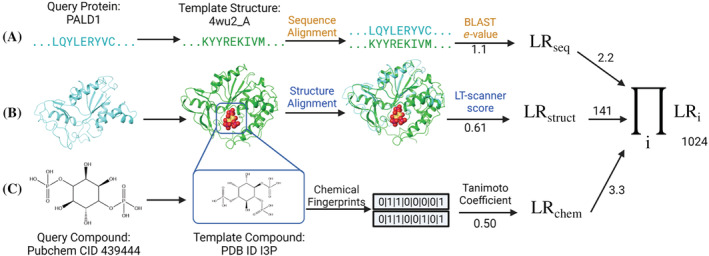
PrePCI algorithm for the prediction of protein‐compound interactions. PrePCI uses BLAST and LT‐scanner for the query protein (aqua sequence and model, left) to identify proteins within PDB protein‐compound complexes that have (a) sequence and/or (b) structural similarity to the query. (c) Compounds predicted to bind the query are identified using a Tanimoto coefficient (TC) chemical similarity search of fingerprints representing the PDB compound and compounds from PubChem. In this example, the sequence and model for Paladin (PALD1) are matched to the PDB complex (PDB ID 4wu2) of Selenomonas ruminatum myo‐inositol hexaphosphate phosphohydrolase bound to the PDB compound I3P with BLAST e‐value 1.1 and LT‐scanner score 0.61. A query compound from PubChem (CID 439444) has TC = 0.5 with the PDB compound. The LR for the interaction between PALD1 and the query compound is the product of the LRs from sequence, structure, and chemical similarity scores.
An essential component of LT‐scanner is a database of structural models for most query proteins and their constituent domains in the human proteome. To date, we have relied on our PrePMod database of homology models (Garzón et al., 2016). Currently, PrePMod contains models for 76,816 protein domains as defined by the Conserved Domain Database (CDD; Marchler‐Bauer et al., 2011) where 17,150 human proteins have at least one domain modeled. As described in Section 4, a database was constructed of models taken either directly from the AlphaFold Protein Structure Database (AF; Jumper et al., 2021; Varadi et al., 2022) or obtained after parsing AF models into CDD domains (AF/CDD; Marchler‐Bauer et al., 2011). AF/CDD contains 89,645 domain models covering 20,546 proteins while the union of AF/CDD and PrePMod contains models of 90,308 domains spanning 20,599 proteins. This constitutes a significant (~15%–20%) increase in structural coverage which now includes one representative for essentially every coding gene in the human proteome (Bateman et al., 2022).
2.2. PrePCI training and evaluation
To evaluate PrePCI's performance, a Naive Bayes Classifier was trained using 10‐fold cross‐validation with a true positive set of PCIs that have bioactivity data in PubChem (Kim et al., 2019, 2021). As described in Section 4, the true positive set consists of 285 K PCIs for 142 K compounds and 2,926 proteins. The negative set consists of 417 M hypothetical PCIs between the 142 K compounds and 2,926 proteins for which PubChem provides no bioactivity data. For each of the 10‐folds, PrePCI's performance was evaluated by ranking predictions by their likelihood ratio (LR) and computing the area under the receiver operator characteristic (ROC) curve (AUROC) and the average precision (or area under the precision–recall curve, AUPRC; Figure 2). The resulting ROC and precision–recall curves are highly concordant, with mean AUROC and average precision of 0.828 ± 0.001 and 0.168 ± 0.002, respectively. It is important to note that, due to the size of the negative set, the testing set is heavily imbalanced, and random precision would thus be 7 × 10−4. The average precision of 0.168, therefore, constitutes a substantial enrichment of true positive predictions and is likely an underestimate as many PCIs considered false positives presumably correspond to as yet undiscovered true interactions.
FIGURE 2.
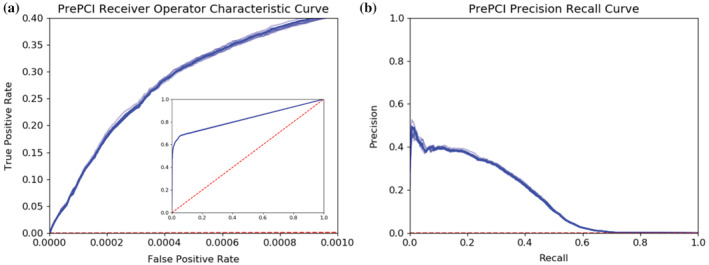
PrePCI performance on unbiased, all‐against‐all experimental protein–compound data from PubChem. (a) Receiver operating characteristic (ROC) curve and (b) Precision Recall curve for each of the 10‐folds of cross‐validation for training and testing PrePCI on experimentally observed PCIs from PubChem. Curves for the median area under the ROC curve (AUROC, a) and average precision (b) across all cross‐validation folds are darker, while curves for remaining folds (lighter blue) are included to display the range of results obtained from the individual folds. PrePCI's average AUROC and Average Precision on the PubChem dataset are 0.828 ± 0.001 and 0.168 ± 0.002, respectively.
Moreover, experimentally known PCIs with low PrePCI scores, corresponding to the high false positive region of the ROC curve, are primarily cases where PrePCI could not identify a template compound that was both similar to the query compound and predicted to bind the query protein. Consequently, these PCIs could not be effectively scored, limiting the maximum AUROC obtained, as reflected by the sharp elbow in the ROC curve (Figure 2a). To evaluate PrePCI's performance on its meaningful predictions, we recomputed ROC and precision–recall curves for each of the 10‐folds, restricting the evaluation to PCIs where a template could be identified (see Section 4), which yielded improved performance in AUROC and AUPRC to 0.936 ± 0.001 and 0.235 ± 0.002 with a more constant precision over most of the recall range (see Figure S1 and related discussion).
2.3. PrePCI performance on an independent drug‐target interaction gold standard data set
To compare PrePCI to matrix factorization methods, we performed 10‐fold cross‐validation with four benchmark datasets originally created by Yamanishi et al. (2008) and recently updated by Liu et al. (2022), each corresponding to a separate class of protein targets: Enzymes, Nuclear Receptors, GPCRs, and Ion channels (Table S5 and Section 4). For each class, all possible protein–compound pairs were scored, and similar performance as with PubChem‐derived data was obtained (Table S6). Despite quite high AUROC scores, PrePCI performance is less impressive than the other methods which utilize many more tunable parameters and do not rely on the availability of PDB template complexes. Moreover, given its relative simplicity, its ability to provide structures for its predictions, and its proteome‐wide applicability, the good performance within specific protein classes underscores PrePCI's utility for more focused studies as illustrated below.
2.4. LT‐scanner and sequence similarity are synergistic
PrePCI performance was compared with the performance of classifiers using features based only on sequence similarity or LT‐scanner alone. Sequence similarity outperforms LT‐scanner and performs comparably to PrePCI (Figure S2) although PrePCI's use of both enables superior performance (Figure S2). In ROC curve analysis, PCIs present in the PDB were excluded from LT‐scanner testing. However, because a sequence identity cutoff was not implemented, many of the sequence similarity targets are likely to be obvious and, thus, underlie the performance of the sequence similarity classifier. The unique feature of LT‐scanner is its ability to identify non‐trivial relationships. Indeed, as can be seen in Table S1, LT‐scanner identifies many more relationships than available from sequence similarity alone. The combined use of sequence and structure yields the greatest coverage of true positive PCIs without impairing performance as each method identifies PCIs not detected by the other at comparable LRs (Figure S2).
2.5. The union of homology models and AlphaFold structures as targets increases PCI coverage
PrePCI performance was evaluated with predictions for query structures from PrepMod versus AF/CDD. As shown in Figure S3, performance is similar regardless of the query model database used. While the number of predictions is greater with AF/CDD versus PrePMod structures, the combination of the databases is synergistic and results in the highest number of PCI predictions (Table S2). For example, as depicted in the first row of Table S2, in cases where the query model aligns well with the template complex (LT‐scanner score ≥0.6), PrePCI‐PrePMod predicts 64 K PCIs and PrePCI‐AF/CDD predicts 77 K PCIs. The intersection of the two sets is 39 K PCIs and the union is 101 K PCIs.
2.6. The PrePCI database—PrePCI/DB
PrePCI predictions are available through a web‐hosted searchable database (PrePCI/DB) at https://honiglab.c2b2.columbia.edu/prepci.html. PrePCI/DB contains predictions for ~5 billion PCIs involving 6.8 M compounds and 75,643 CDD domains representing 19,797 human proteins. Users can query the database for proteins (with UniProt Accession ID or gene name) or for compounds (PDB compound ID, PubChem CID, or SMILES) to obtain PrePCI predictions for compounds and targets, respectively. Searching by protein will return a list of PDB compounds predicted to bind the protein by either LT‐scanner (Figure 1b), sequence similarity (Figure 1a), or both, along with the corresponding LT‐scanner scores, BLAST e‐values and PrePCI LRs (Figure 3). From our benchmarking results, we found that the LRs of 1,400,000, 15,000, and 190 correspond to FPR values of 10−4, 10−3, and 10−2, respectively. While predictions with higher LRs are more likely to be correct, predictions with lower LRs, particularly those with high LT‐scanner scores, are more likely to be evolutionarily conserved and thus constitute novel PCIs. The “Click to view PCI” icon will trigger the website to display interactive JSMol windows for exploration of the predicted binding interface as well the structural superposition of the query protein model and the PDB template complex. PDB‐formatted files for both the interaction model and the structural superposition can be downloaded for further analysis including more detailed docking studies, as described below. Additional similar compounds (Figure 1c) can be retrieved by clicking on the “Click to Find Other PCIs” icon which will open a new tab containing all PubChem compounds that are similar to the selected PDB compound (Figure 3). Together with the interaction visualization windows, this two‐step procedure allows the user to evaluate predicted PCI interfaces before considering additional compounds likely to bind in a similar mode. Alternatively, users can query the database for a compound in the form of a PDB ID, PubChem CID, or SMILES string. The following case studies illustrate how both strategies—querying PrePCI/DB for predicted targets and for compounds—can be used to discover novel therapeutically interesting lead compounds and generate novel biological hypotheses.
FIGURE 3.
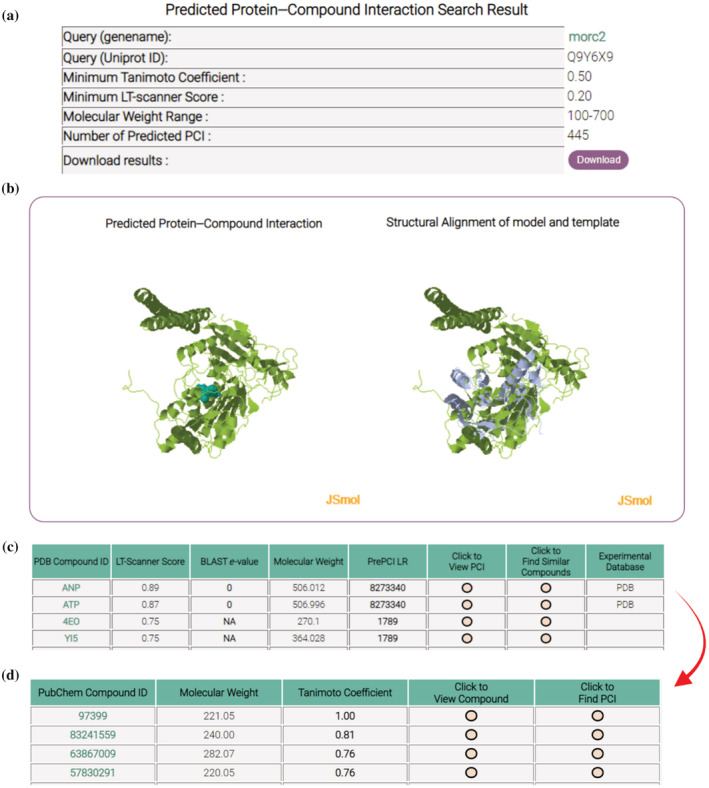
PrePCI webpage output. Protein query search: (a) The top of the webpage displays search criteria and the number of PCIs predicted. (b) Two JSmol windows display the query protein–compound interaction model (left) and query‐template superposition (right) which a user may manipulate. (c) A table displays the PDB compounds predicted to interact with the query protein and their corresponding LT‐scanner scores, BLAST e‐values, and PrePCI LRs. The “Experimental Database” column contains links to associated PDB and Pubchem pages for experimentally validated PCIs. PCIs for chemically similar compounds: The “Click to View PCI” triggers the webpage to display the interaction and superposition models (panel B) while “Click to Find Similar Compounds” opens (d) a new webpage listing PubChem compounds similar to the query PDB compound, which can in turn be used to search for target proteins via the “Click to Find PCI” button.
2.7. Applications of PrePCI/DB
2.7.1. Lead compound discovery
We used PrePCI to search for compounds predicted to bind to peroxisomal succinyl‐coenzyme A thioesterase (ACOT4), an enzyme involved in lipid metabolism. A recent study found that pancreatic ductal adenocarcinoma cells are dependent on free CoA generated by ACOT4 while knockdown and catalytic inactivation of ACOT4 impaired proliferation and tumor formation, suggesting ACOT4 as a possible therapeutic target (Ni et al., 2021). Notably, while no PCIs are predicted for ACOT4 on the basis of sequence similarity, structural alignment identified 41 PDB compounds with LT‐scanner scores ≥0.3. We chose to focus on C1E (Figure 4a) due to its relatively high LT‐scanner score (0.39) and the diversity of similar compounds in PubChem for screening (80 with TC <0.7). The C1E‐ACOT4 interaction was predicted based on the crystal structure of C1E complexed with the Burkholderia xenovorans C–C hydrolase (PDB ID 2RHT, Chain A). Glide (Friesner et al., 2004, 2006) was used to dock C1E into both ACOT4 and the template protein structure as control, which yielded favorable glide scores of −7.1 and −10.1 kcal/mol, respectively, indicating C1E is a reasonable lead for ACOT4. The 80 similar compounds were similarly analyzed and the best scoring ligand (−9.2 kcal/mol) was Pubchem CID 5367065 (Figure 4b,c). The predicted binding mode of CID 5367065 positions a benzaldehyde ring in a pose similar to the template while the remainder of the compound provides additional contacts and more fully occupies ACOT4's active site. We used a similar strategy to identify lead compounds for the ATPase MORC2 (Figure S4).
FIGURE 4.
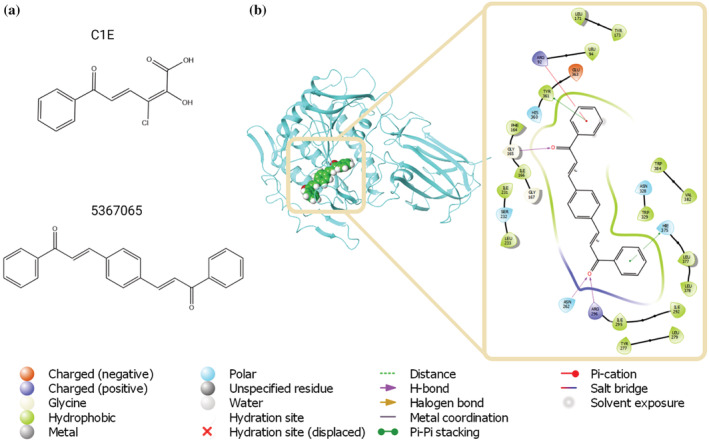
PrePCI guided structure‐based virtual screening. (a) PrePCI predicts 41 PDB compounds bind to ACOT4 with LT‐scanner score ≥0.3, including C1E (top). In silico screening of C1E and similar compounds identifies PubChem CID 5367065 as a possible binder (bottom). (b) The docking pose (left) is depicted as a blue backbone ribbon for the target (ACOT4) and space‐filling representation for the compound (5367065). The diagram (right) highlights atomic interactions predicted by docking.
2.7.2. Phosphoinositides
Many peripheral membrane proteins transiently associate with membrane surfaces by recognizing phosphatidylinositol phosphates, such as PI(4,5)P2 (PIP2) and PI(3,4,5)P3 (PIP3) (Mandal, 2020; Overduin & Kervin, 2021; Pemberton & Balla, 2019). Structural studies have elucidated the binding mode of a wide array of protein domains to the head groups of PIP2 and PIP3 (denoted here by their PDB IDs, I3P, and 4IP), which appear as non‐covalent ligands in 46 and 27 PDB complexes, respectively. PrePCI predicts over 400 targets for each (LT‐scanner score of at least 0.3), many of which are novel. Figure 1 provides an example of a novel I3P target. Paladin was previously annotated as an inactive protein phosphatase but is predicted by PrePCI to bind to I3P through its first protein‐tyrosine phosphatase‐like domain, suggesting Paladin may be a lipid phosphatase thus correcting the original annotation. Consistent with this prediction, Paladin was recently identified as a PIP2 phosphatase through a colorimetric screen for phosphate in the presence of PIP2 (Nitzsche et al., 2021). As depicted in Figure 1, PrePCI also predicts that Paladin binds Ins(1,4)P2 (PubChem CID 439444), a compound chemically similar to I3P which corresponds to the head group of PI4P. Since PI4P is the product of 5‐phosphatase activity against PI(4,5)P2, the prediction that Paladin binds the head groups of both reactant (PI(4,5)P2) and product (PI4P) suggests that Paladin may be a 5‐phosphatase. As a cautionary note, PrePCI predicts that Paladin also binds 4IP (Ins(1,3,4,5)P4) albeit with a lower LR (93 for 4IP vs. 315 for 3IP). Additional computational and experimental analysis is required to determine whether Paladin is a PIP2 phosphatase, a PIP3 phosphatase, or both.
The integration of PrePCI with high‐throughput lipidomic assays provides structural annotation of protein–lipid interactions, boosts confidence in the discovery of novel binders and, thus, expands phosphatidylinositol phosphate interactomes. Two studies used mass spectrometry‐based methods to identify PIP3‐binding proteins in HeLa cells (Jungmichel et al., 2014) and human platelets (Durrant et al., 2017). In both cases, PrePCI predicts 45% of the 30 highest scoring and 25% of all PIP3 binders as 4IP targets (Table S3). Of 21 proteins annotated as known binders (Jungmichel et al., 2014), PrePCI predicts all 21 with LRs ranging from 100 to 690 K. Mass spectrometry and PrePCI jointly identify 16 of the known PIP3 binders as well as an additional 70 potentially novel PIP3 interactors (Table S3).
2.7.3. Drug mechanism of action
The DeMAND algorithm is a network‐based approach to elucidating drug mechanism of action as defined by a compound's direct and indirect targets (effectors and modulators) through the analysis of cellular perturbation gene expression profiles (Woo et al., 2015). The integration of DeMAND and PrePCI predictions identify direct targets and off‐targets of compounds on a genome‐wide scale in particular cellular contexts. For example, high‐scoring predictions in both DeMAND and PrePCI for methotrexate (a chemotherapy agent and immune‐system suppressant) and genistein (a flavonoid in clinical trials as a treatment for prostate cancer) recapitulate known targets and highlight potential off‐targets in diffuse large B cell lymphoma cells (Table S4). The WW domain‐containing oxidoreductase (WWOX) is predicted as a novel target of methotrexate. WWOX regulates susceptibility of squamous cell carcinoma to methotrexate, and small interfering RNA against WWOX blocked methotrexate‐mediated cell death (Tsai et al., 2013) supporting WWOX as a direct target. Polo‐like kinase 1 (PLK1) is predicted as an off‐target of genistein, which was shown to function as a mitotic blocker by directly inhibiting PLK1 activity in transformed cells (Shin et al., 2017) supporting PLK1 as a direct target.
3. DISCUSSION
We have presented the PrePCI algorithm, and a corresponding database PrePCI/DB, which integrates protein structure and chemical and sequence similarity to predict protein compound interactions (Figure 1). PrePCI is an extension of our template‐based PCI prediction algorithm, LT‐scanner (Hwang et al., 2017), which identifies protein targets of small molecules present in the PDB (Berman et al., 2000). The LT‐scanner query model database has been updated with structures from the AlphaFold Protein Structure Database providing essentially complete structural coverage for all human protein‐coding genes. In addition, PrePCI uses chemical compound similarity based on Tanimoto coefficients among chemical fingerprints to link PDB compounds to compounds in PubChem, which has the effect of increasing the number of compounds that can be explored by more than 200‐fold. The increased structural coverage of the human proteome and the expanded chemical space have enabled the prediction of over 5 billion PCIs, each with component scores and an overall likelihood ratio that allow users to prioritize predictions. While this constitutes a significant expansion over our previous work, it still excludes many compounds which are dissimilar from those in the PDB but for which non‐structural bioactivity data is available. Integration of PrePCI scores with machine learning methods (Table S6) could enable further expansion into chemical space while preserving structure‐based and proteome‐scale predictions. Finally, PrePCI achieves high precision for evaluation on large‐scale bioactivity data (PubChem, Figure 2) strongly indicating that many novel interactions may be present among the most highly ranked predictions in the negative PCI dataset.
There are, of course, significant limitations to PrePCI performance. First, because LT‐scanner compares the binding interfaces of individual protein chains, it can yield high‐scoring predictions for compounds which are observed to bind at protein–protein interfaces that are present in the template PDB complex. While LT‐scanner detects the high structural similarity with one chain in the template, it is currently unable to penalize the query PCI for lacking additional contacts provided by the second protein chain, and it can thus predict interactions that appear visually implausible in the absence of the partner protein. We speculate that this feature of LT‐scanner can be leveraged to identify compounds that can bind at protein–protein interfaces and act either as molecular glue or bias molecular complexes into specific conformations. Second, the protein models provided by PrePCI lack metal ions, cofactors, and compounds which can play central roles in ligand binding. Third, the Tanimoto coefficient measures overall topological similarity and may therefore identify compounds lacking critical interacting functional groups or with large changes in physical features such as net charge. The user is therefore encouraged to review the template PDB complex on which a prediction is based to verify whether the provided interaction is lacking features which could make the prediction more or less plausible.
We have demonstrated how PrePCI can be used for common medicinal chemistry tasks, such as the identification of small chemical fragments as lead compounds for structure‐based virtual screening (Figure 4) or elucidation of a drug's mechanism of action, as well as the generation of biological hypotheses (Figure 1) by detecting novel protein–ligand interactions. Importantly, for compounds present in the PDB (Berman et al., 2000), PrePCI generates 3D interaction models and predicts interfacial residues. Given how they are constructed, the models are expected to be crude but can be refined with various docking strategies as illustrated above. While an interaction model is not created for a PCI predicted by chemical similarity (Figure 1c), the predicted compound can be cross‐docked to the template PDB compound in the underlying LT‐scanner model (Figures 4 and S4). In this regard, it is important to emphasize that PrePCI is primarily intended for hypothesis generation. PrePCI/DB, which encompasses 5 billion predicted PCIs involving 6.8 million compounds and 19,797 protein targets, is a conveniently accessible structure‐informed resource to search for compounds that potentially bind a given protein or, alternatively, proteins that are potential targets of a given compound.
4. MATERIALS AND METHODS
4.1. Template and model selection
LT‐scanner requires databases of query protein structure models and experimentally resolved holo‐structures of protein–ligand co‐complexes. To select a representative set of template PDB holo‐structures, all PDB complexes identified in the PDB ligand expo (http://ligand-expo.rcsb.org/) were parsed to identify protein chains bound to ligands, and all chains were mapped to their respective UniProt IDs using the SIFTS database (Dana et al., 2019; Velankar et al., 2013). Chains with more than one corresponding UniProt ID, commonly chimeric fusion proteins, as well as chains that did not map to a UniProt ID were excluded. X‐ray crystal structures and cryo‐EM structures with resolution >4 and 4.5 Å, respectively, were removed, and, when a PCI was represented more than once, the highest resolution complex was retained. This procedure yielded 55,994 unique template PCIs between 17,705 proteins and 25,613 compounds after removing compounds with molecular weight <200 Da and fewer than six heavy atoms.
4.2. Model databases
For the human reference proteome (Jumper et al., 2021; Varadi et al., 2022; https://www.uniprot.org/proteomes/UP000005640), structural models for full‐length sequences and protein domains as defined by the conserved domain database (CDD; Marchler‐Bauer et al., 2011) were constructed as follows.
4.2.1. PrePMod
BLAST was used to identify proteins in the PDB with sequences similar to the query sequence. For BLAST e‐value ≤10−12, a homology model for the query sequence with the PDB structure as template was created with Nest (Petrey et al., 2003). If no template was identified, remote sequence homologs within the PDB were identified by HHblits (Remmert et al., 2012) with 5 iterations, and, for e‐value ≤10−12, a homology model was created with Nest (Petrey et al., 2003). Otherwise, a homology model for the query was not created. This process yielded PrePMod, a protein model database containing 76,816 domain models for 17,150 human proteins.
4.2.2. AlphaFold/CDD (AF/CDD)
Models for query proteins and their CDD domains were taken from the AlphaFold Protein Structure Database (AF). For proteins with more than 2700 residues, AF provides multiple sequence‐redundant models. In these cases, the pLDDT scores (the per‐residue confidence metric) were summed across the CDD (Marchler‐Bauer et al., 2011) domains identified, and the model with the largest total pLDDT was chosen. This procedure yielded 89,645 domain models for 20,526 proteins.
Altogether, the combination of PrePMod and AF/CDD provides 166,461 models for 90,308 domains for 20,599 proteins.
4.3. PrePCI
4.3.1. LT‐scanner
The LT‐scanner algorithm which uses structure alignment to relate query proteins to PDB template complexes has been described previously (Hwang et al., 2017). Briefly, the extent to which a query protein is able to recapitulate the intermolecular interactions formed between a template and its ligand is calculated by a similarity score, SIM. For each potential protein–compound interaction, the LT‐scanner score is defined as the maximal observed SIM score between the query protein and any structurally similar template in the holo‐structure database. LT‐scanner was applied to both the PrePMod and AF/CDD model databases. In cases where PrePMod and AF/CDD contain models for the same protein/domain, the query model that obtains the higher LT‐scanner score was included in the LT‐scanner evaluation analyses (Table S2).
4.3.2. Sequence similarity
For each of the structures in the holo‐structure template database, BLAST was run using the sequence of the PDB chain as a query against the human reference proteome (UP000005640). For a given PCI, the PDB template complex with the lowest e‐value was assigned an interaction sequence score of –ln(e‐value). The e‐values of 0 were re‐assigned to 1e−181, the smallest non‐zero e‐value obtained from the BLAST results. The sequence similarity component of PrePCI yields predictions for 17,864 proteins (Table S1).
4.3.3. Chemical similarity
Chemical structure data for ~110 M compounds and 26 K PDB compounds was obtained from PubChem (Kim et al., 2019, 2021) and the PDB in SMILES format (Weininger, 1988), Rdkit (RDKit) was used to compute 1024‐bit Morgan2 fingerprints (Rogers & Hahn, 2010) for each compound, and Tanimoto coefficients (Bajusz et al., 2015) were computed for each PDB‐PubChem compound pair. The reliability of inferring novel compounds from known compounds drops off at Tanimoto coefficients below 0.5 (Bajorath et al., 2013) so only those pairs of compounds with TC ≥0.5 were retained, yielding 6,835,528 compounds similar to at least one PDB compound. Overall, PrePCI provides predictions for 6.8 M compounds.
4.3.4. Naive Bayes integration
A Naive Bayes Classifier was trained to integrate scores into a single likelihood ratio (LR; Figure 1). For each query PCI, the reference PDB compound is the highest TC PDB compound predicted by either LT‐scanner or sequence similarity. The chemical, structural, and sequence scores are then defined as (1) the TC between the query compound and the reference compound, (2) the LT‐scanner score for the query protein‐reference compound pair, and (3) the sequence score between the query protein and the most similar template protein from among complexes with the reference compound, respectively. The number of bins was chosen as 10, 10, and 20 for chemical similarity, structural similarity, and sequence similarity, respectively, and the range of scores for each feature was divided into equal intervals. Likelihood ratios for each feature and bin were computed as
The final likelihood ratio for a PCI is defined as the product of the three component feature likelihood ratios:
4.4. PubChem benchmarking
All available protein bioactivity data for human proteins was downloaded as PubChem's “Tested Compounds” data from the “Chemicals and Bioactivities Data” section (Kim et al., 2019, 2021). The data was filtered to retain active, nonredundant experimental PCIs defined as “Active” for the “activity” feature or “<” or “=” for the “acqualifier” feature. This process yielded 1,122,699 PCIs involving 3,559 proteins and 642,498 compounds. Of the 642,498 Pubchem PubChem compounds, 142,490 (22%) have Tanimoto coefficient ≥0.5 with at least one PDB compound, and 2,926 of the 3,559 proteins have experimental evidence supporting an interaction with at least one of the 142,490 compounds. After filtering, the true positive set comprised 285,108 PCIs. The true negative set was defined as the remaining 416,640,632 protein‐compound pairwise combinations (2,926 * 142,490 – 285,108) not identified as true positives in Pubchem. We split both the positive and negative sets into 10 mutually disjoint subsets and, using each subset in turn as a test set, trained LRs using the PCIs from the remaining 9 sets as a training set. PCIs in the test set were scored and ranked by their composite LR, and the AUROC was computed.
To better evaluate the accuracy of PrePCI's predictions, we removed PCIs for which PrePCI does not make a prediction from the positive and negative sets which resulted in 204,919 true positive and 62,414,150 true negative PCIs which are 72% and 15% the size of the original PubChem benchmark set. We computed ROC and precision–recall curves for each of the 10‐folds using this restricted set of PCIs. Further, to evaluate PrePCI's performance using a more balanced dataset, we randomly sampled 2,049,190 of the 62,414,150 true negative interactions such that ratio of negatives to positives in each fold was 10:1 and generated ROC and precision–recall curves.
4.5. Benchmarking on an independent drug target interaction gold standard dataset
Updated versions of the protein class datasets compiled by Yamanishi et al. (2008) and updated in Liu et al. (2022) were obtained from https://github.com/intelligence-csd-auth-gr/DTI_MDMF2A/tree/main/datasets_mv. KEGG Compound IDs were mapped to SMILES strings using the Pubchem Chemical Identifier Exchange Service (https://pubchem.ncbi.nlm.nih.gov/idexchange/idexchange.cgi). KEGG protein IDs were mapped to Uniprot IDs using the Uniprot ID mapping tool (https://www.uniprot.org/id-mapping). PCIs present in each dataset were considered true positives while the remaining all‐on‐all protein–compound pairs were considered true negatives. We performed 10‐fold cross‐validation and calculated performance statistics.
4.6. Docking analysis and screening
An initial putative interaction model of the PCI was created by aligning the query protein to the template protein using ska (Yang & Honig, 2000). Protein structures were then prepared in the presence of the template ligand using the Protein Preparation Wizard in Maestro version 13.1 with default settings (Madhavi Sastry et al., 2013). Receptor grids were generated around the template ligand setting all neighboring groups as rotatable with other settings taken as the defaults. Ligand structures were prepared using Ligprep: C1E was prepared from its coordinates, and its chirality was inferred from 3D structure; screening compounds were prepared from SMILES strings and all combinations of chiral centers were generated to expand diversity of the screening pool. All docking was performed with flexible ligand sampling using the XP scoring function.
AUTHOR CONTRIBUTIONS
Stephen J. Trudeau: Conceptualization (equal); methodology (lead); software (lead); writing – review and editing (equal). Howook Hwang: Conceptualization (equal); methodology (equal). Deepika Mathur: Software (equal). Kamrun Begum: Software (equal). Donald Petrey: Software (supporting). Diana Murray: Methodology (equal); writing – review and editing (equal). Barry Honig: Conceptualization (equal); writing – review and editing (equal).
CONFLICT OF INTEREST STATEMENT
BH is a member of the SAB and consultant for Schrodinger Inc.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
This work was supported by the National Institute of Health (grant R35‐GM139585, U54‐CA209997, BH; grant T32‐GM008224, T32‐GM145440, SJT).
Trudeau SJ, Hwang H, Mathur D, Begum K, Petrey D, Murray D, et al. PrePCI: A structure‐ and chemical similarity‐informed database of predicted protein compound interactions. Protein Science. 2023;32(4):e4594. 10.1002/pro.4594
Review Editor: Nir Ben‐Tal
Funding information National Institute of Health, Grant/Award Numbers: R35 GM1395858, T32 GM008224, T32 GM145440, U54 CA209997
Contributor Information
Diana Murray, Email: dm527@cumc.columbia.edu.
Barry Honig, Email: bh6@columbia.edu.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are openly available in the following locations: Pubchem at https://pubchem.ncbi.nlm.nih.gov/, references 11,12; The Human Proteome Sequence database at https://www.uniprot.org/proteomes/UP000005640, references 13,14; Directory of Useful Decoys‐Enhanced (DUD‐E) at http://dude.docking.org/db/subsets/all/all.tar.gz, reference 42. The subset corresponding to human proteins used in this study is available in table S3; Demanding Evaluation Kits for Objective In Silico Screening 2.0 (DEKOIS 2.0) at http://www.pharmchem.uni-tuebingen.de/dekois/, reference 43. The subset corresponding to human proteins used in this study is available in Table S4; SIFTS database at https://www.ebi.ac.uk/pdbe/docs/sifts/quick.html, references 50,51; Template/Model proteins and PDB/Pubchem Compounds used in PrePCI are accessible via PrePCI/DB website interface described above.
REFERENCES
- Bajorath J, Hu Y, Stumpfe D. Advancing the activity cliff concept. F1000Res. 2013;2:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajusz D, Rácz A, Héberger K. Why is Tanimoto index an appropriate choice for fingerprint‐based similarity calculations? J Cheminform. 2015;7:1–13. 10.1186/s13321-015-0069-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester PJ, Mitchell JBO. A machine learning approach to predicting protein‐ligand binding affinity with applications to molecular docking. Bioinformatics. 2010;26:1169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin M‐J, Orchard S, Magrane M, Ahmad S, Alpi E, et al. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res. 2022;51:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The protein data bank. Nucleic Acids Res. 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottegoni G, Kufareva I, Totrov M, Abagyan R. Four‐dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J Med Chem. 2009;52:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BP, Mendenhall J, Geanes AR, Meiler J. General purpose structure‐based drug discovery neural network score functions with human‐interpretable pharmacophore maps. J Chem Inf Model. 2021;61:603–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappelletti V, Hauser T, Piazza I, Pepelnjak M, Malinovska L, Fuhrer T, et al. Dynamic 3D proteomes reveal protein functional alterations at high resolution in situ. Cell. 2021;184:545–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da C, Kireev D. Structural protein‐ligand interaction fingerprints (SPLIF) for structure‐based virtual screening: method and benchmark study. J Chem Inf Model. 2014;54:2555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana JM, Gutmanas A, Tyagi N, Qi G, O'Donovan C, Martin M, et al. SIFTS: updated structure integration with function, taxonomy and sequences resource allows 40‐fold increase in coverage of structure‐based annotations for proteins. Nucleic Acids Res. 2019;47:D482–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diether M, Nikolaev Y, Allain FH, Sauer U. Systematic mapping of protein‐metabolite interactions in central metabolism of Escherichia coli . Mol Syst Biol. 2019;15:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant TN, Hutchinson JL, Heesom KJ, Anderson KE, Stephens LR, Hawkins PT, et al. In‐depth PtdIns(3,4,5)P3 signalosome analysis identifies DAPP1 as a negative regulator of GPVI‐driven platelet function. Blood Adv. 2017;1:918–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, de Franceschi G, Kahraman A, Soste M, Melnik A, Boersema PJ, et al. Global analysis of protein structural changes in complex proteomes. Nat Biotechnol. 2014;32:1036–44. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein‐ligand complexes. J Med Chem. 2006;49:6177–96. [DOI] [PubMed] [Google Scholar]
- Garzón JI, Deng L, Murray D, Shapira S, Petrey D, Honig B. A computational interactome and functional annotation for the human proteome. Elife. 2016;5:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Dey F, Petrey D, Honig B. Structure‐based prediction of ligand‐protein interactions on a genome‐wide scale. Proc Natl Acad Sci U S A. 2017;114:13685–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungmichel S, Sylvestersen KB, Choudhary C, Nguyen S, Mann M, Nielsen ML. Specificity and commonality of the phosphoinositide‐binding proteome analyzed by quantitative mass spectrometry. Cell Rep. 2014;6:578–91. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Roth BL, Armbruster BN, Ernsberger P, Irwin JJ, Shoichet BK. Relating protein pharmacology by ligand chemistry. Nat Biotechnol. 2007;25:197–206. [DOI] [PubMed] [Google Scholar]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 2019;47:D1102–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. 2021;49:D1388–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempp M, Farke N, Kuntz M, Freibert SA, Lill R, Link H. Systematic identification of metabolites controlling gene expression in E. coli . Nat Commun. 2019;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Gray P, Xie L, Poleksic A. Improved genome‐scale multi‐target virtual screening via a novel collaborative filtering approach to cold‐start problem. Sci Rep. 2016;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, He D, Qiu Y, Krawczuk P, Sun X, Xie L. Rational discovery of dual‐indication multi‐target pde/kinase inhibitor for precision anti‐cancer therapy using structural systems pharmacology. PLoS Comput Biol. 2019;15:e1006619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim H, Poleksic A, Yao Y, Tong H, He D, Zhuang L, et al. Large‐scale off‐target identification using fast and accurate dual regularized one‐class collaborative filtering and its application to drug repurposing. PLoS Comput Biol. 2016;12(10):1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Papadopoulos D, Malliaros FD, Tsoumakas G, Papadopoulos AN. Multiple similarity drug–target interaction prediction with random walks and matrix factorization. Brief Bioinform. 2022;23(5):1–9. [DOI] [PubMed] [Google Scholar]
- Liu Y, Wu M, Miao C, Zhao P, Li XL. Neighborhood regularized logistic matrix factorization for drug‐target interaction prediction. PLoS Comput Biol. 2016;12:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu J, Wang S, Balius TE, Singh I, Levit A, Moroz YS, et al. Ultra‐large library docking for discovering new chemotypes. Nature. 2019;566:224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–34. [DOI] [PubMed] [Google Scholar]
- Mandal K. Review of PIP2 in cellular signaling, functions and diseases. Int J Mol Sci. 2020;21:1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchler‐Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese‐Scott C, et al. CDD: a conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011;39:D225–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milanesi R, Coccetti P, Tripodi F. The regulatory role of key metabolites in the control of cell signaling. Biomolecules. 2020;10:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EB, Murphy RB, Sindhikara D, Borrelli KW, Grisewood MJ, Ranalli F, et al. Reliable and accurate solution to the induced fit docking problem for protein‐ligand binding. J Chem Theory Comput. 2021;17:2630–9. [DOI] [PubMed] [Google Scholar]
- Murphy RB, Repasky MP, Greenwood JR, Tubert‐Brohman I, Jerome S, Annabhimoju R, et al. WScore: a flexible and accurate treatment of explicit water molecules in ligand‐receptor docking. J Med Chem. 2016;59:4364–84. [DOI] [PubMed] [Google Scholar]
- Ni C, Zheng K, Gao Y, Chen Y, Shi K, Ni C, et al. ACOT4 accumulation via AKT‐mediated phosphorylation promotes pancreatic tumourigenesis. Cancer Lett. 2021;498:19–30. [DOI] [PubMed] [Google Scholar]
- Nikolova N, Jaworska J. Approaches to measure chemical similarity ‐ a review. QSAR Comb Sci. 2004;22:1006–26. [Google Scholar]
- Nitzsche A, Pietilä R, Love DT, Testini C, Ninchoji T, Smith RO, et al. Paladin is a phosphoinositide phosphatase regulating endosomal VEGFR2 signalling and angiogenesis. EMBO Rep. 2021;22:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overduin M, Kervin TA. The phosphoinositide code is read by a plethora of protein domains. Expert Rev Proteomics. 2021;18:483–502. [DOI] [PubMed] [Google Scholar]
- Paggi JM, Belk JA, Hollingsworth SA, Villanueva N, Powers AS, Clark MJ, et al. Leveraging nonstructural data to predict structures and affinities of protein‐ligand complexes. Proc Natl Acad Sci U S A. 2021;118:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pemberton JG, Balla T. Polyphosphoinositide‐binding domains: insights from peripheral membrane and lipid‐transfer proteins. In: Crusio WE, Dong H, Radeke HH, Rezaei N, Steinlein O, Xiao J, editors. Advances in experimental medicine and biology. Volume 1111. Springer, Switzerland; 2019. p. 77–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Nueno VI, Rabal O, Borrell JI, Teixido J. APIF: a new interaction fingerprint based on atom pairs and its application to virtual screening. J Chem Inf Model. 2009;49:1245–60. [DOI] [PubMed] [Google Scholar]
- Petrey D, Xiang Z, Tang CL, Xie L, Gimpelev M, Mitros T, et al. Using multiple structure alignments, fast model building, and energetic analysis in fold recognition and homology modeling. Proteins. 2003;53:430–5. [DOI] [PubMed] [Google Scholar]
- Piazza I, Beaton N, Bruderer R, Knobloch T, Barbisan C, Chandat L, et al. A machine learning‐based chemoproteomic approach to identify drug targets and binding sites in complex proteomes. Nat Commun. 2020;11:1–13. 10.1038/s41467-020-18071-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza I, Kochanowski K, Cappelletti V, Fuhrer T, Noor E, Sauer U, et al. A map of protein‐metabolite interactions reveals principles of chemical communication. Cell. 2018;172:358–72. 10.1016/j.cell.2017.12.006 [DOI] [PubMed] [Google Scholar]
- Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein−ligand scoring with convolutional neural networks. J Chem Inf Model. 2017;57:942–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RDKit: Open‐source chemoinformatics . http://www.rdkit.org
- Remmert M, Biegert A, Hauser A, Söding J. HHblits: lightning‐fast iterative protein sequence searching by HMM‐HMM alignment. Nat Methods. 2012;9:173–5. [DOI] [PubMed] [Google Scholar]
- Rogers D, Hahn M. Extended‐connectivity fingerprints. J Chem Inf Model. 2010;50:742–54. [DOI] [PubMed] [Google Scholar]
- Sadybekov AA, Sadybekov A v, Liu Y, Iliopoulos‐Tsoutsouvas C, Huang XP, Pickett J, et al. Synthon‐based ligand discovery in virtual libraries of over 11 billion compounds. Nature. 2022;601:452–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin S‐B, Woo S‐U, Chin Y‐W, Jang Y‐J, Yim H. Sensitivity of TP53‐mutated cancer cells to the phytoestrogen Genistein is associated with direct inhibition of Plk1 activity. J Cell Physiol. 2017;232:2818–28. [DOI] [PubMed] [Google Scholar]
- Stepniewska‐Dziubinska MM, Zielenkiewicz P, Siedlecki P. Development and evaluation of a deep learning model for protein–ligand binding affinity prediction. Bioinformatics. 2018;34:3666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009;31:455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai CW, Lai FJ, Sheu HM, Lin YS, Chang TH, Jan MS, et al. WWOX suppresses autophagy for inducing apoptosis in methotrexate‐treated human squamous cell carcinoma. Cell Death Dis. 2013;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. AlphaFold protein structure database: massively expanding the structural coverage of protein‐sequence space with high‐accuracy models. Nucleic Acids Res. 2022;50:D439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velankar S, Dana JM, Jacobsen J, van Ginkel G, Gane PJ, Luo J, et al. SIFTS: structure integration with function, taxonomy and sequences resource. Nucleic Acids Res. 2013;41:D483–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach I, Dzamba M, Heifets A. AtomNet: a deep convolutional neural network for bioactivity prediction in structure‐based drug discovery. arXiv. 2015;1510.02855:1–11. [Google Scholar]
- Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J Chem Inf Comput Sci. 1988;28:31–6. [Google Scholar]
- Willett P. Similarity searching using 2D structural fingerprints. Methods Mol Biol. 2010;672:133–58. [DOI] [PubMed] [Google Scholar]
- Wójcikowski M, Ballester PJ, Siedlecki P. Performance of machine‐learning scoring functions in structure‐based virtual screening. Sci Rep. 2017;7:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell. 2015;162:441–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug‐target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24:i232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AS, Honig B. An integrated approach to the analysis and modeling of protein sequences and structures. I. Protein structural alignment and a quantitative measure for protein structural distance. J Mol Biol. 2000;301:665–78. [DOI] [PubMed] [Google Scholar]
- Zhou H, Cao H, Skolnick J. FINDSITEcomb2.0: a new approach for virtual ligand screening of proteins and virtual target screening of biomolecules. J Chem Inf Model. 2018;58:2343–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Cao H, Skolnick J. FRAGSITE: a fragment‐based approach for virtual ligand screening. J Chem Inf Model. 2021;61:2074–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Zhang X, Allen JE, Jones D, Lightstone FC. Binding affinity prediction by pairwise function based on neural network. J Chem Inf Model. 2020;60:2766–72. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are openly available in the following locations: Pubchem at https://pubchem.ncbi.nlm.nih.gov/, references 11,12; The Human Proteome Sequence database at https://www.uniprot.org/proteomes/UP000005640, references 13,14; Directory of Useful Decoys‐Enhanced (DUD‐E) at http://dude.docking.org/db/subsets/all/all.tar.gz, reference 42. The subset corresponding to human proteins used in this study is available in table S3; Demanding Evaluation Kits for Objective In Silico Screening 2.0 (DEKOIS 2.0) at http://www.pharmchem.uni-tuebingen.de/dekois/, reference 43. The subset corresponding to human proteins used in this study is available in Table S4; SIFTS database at https://www.ebi.ac.uk/pdbe/docs/sifts/quick.html, references 50,51; Template/Model proteins and PDB/Pubchem Compounds used in PrePCI are accessible via PrePCI/DB website interface described above.