

Abstract
The severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) nucleocapsid protein is the most abundantly expressed viral protein during infection where it targets both RNA and host proteins. However, identifying how a single viral protein interacts with so many different targets remains a challenge, providing the impetus here for identifying the interaction sites through multiple methods. Through a combination of nuclear magnetic resonance (NMR), electron microscopy, and biochemical methods, we have characterized nucleocapsid interactions with RNA and with three host proteins, which include human cyclophilin‐A, Pin1, and 14–3–3τ. Regarding RNA interactions, the nucleocapsid protein N‐terminal folded domain preferentially interacts with smaller RNA fragments relative to the C‐terminal region, suggesting an initial RNA engagement is largely dictated by this N‐terminal region followed by weaker interactions to the C‐terminal region. The nucleocapsid protein forms 10 nm ribonuclear complexes with larger RNA fragments that include 200 and 354 nucleic acids, revealing its potential diversity in sequestering different viral genomic regions during viral packaging. Regarding host protein interactions, while the nucleocapsid targets all three host proteins through its serine‐arginine‐rich region, unstructured termini of the nucleocapsid protein also engage host cyclophilin‐A and host 14–3–3τ. Considering these host proteins play roles in innate immunity, the SARS‐CoV‐2 nucleocapsid protein may block the host response by competing interactions. Finally, phosphorylation of the nucleocapsid protein quenches an inherent dynamic exchange process within its serine‐arginine‐rich region. Our studies identify many of the diverse interactions that may be important for SARS‐CoV‐2 pathology during infection.
Keywords: COVID‐19, host protein, nucleocapsid, RNA, SARS
1. INTRODUCTION
Coronaviruses (CoVs) have emerged as formidable human pathogens since their detection within human populations only 50 years ago (V'Kovski et al., 2020), leading to the recent epidemic caused by severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) and underlying the need to rapidly characterize their molecular mechanisms. CoVs comprise four structural proteins, which include the spike (S), the envelope (E), the membrane (M), and the nucleocapsid (N) protein. The N protein is the most abundantly expressed protein during infection where it serves multiple roles that include packaging RNA and modulating the host innate immune response through interactions with multiple host proteins (Bouhaddou et al., 2020; Lu et al., 2021; V'Kovski et al., 2020). Like most CoV N proteins, the SARS‐CoV‐2 N protein comprises two distinctly folded regions that include the N‐terminal domain (NTD) and the C‐terminal dimerization domain (CTD), which are flanked by inherently disordered regions (Figure 1). Disordered regions include an N‐terminal extension (NTE) of residues (1–49), centrally disordered regions that comprise the serine/arginine (SR) region, and a C‐terminal extension (CTE) of residues 365–419. However, the unique and diverse functions of CoV N proteins such as that of SARS‐CoV‐2 N have made standard structural methods that address their molecular interactions difficult. For example, such difficulties include their promiscuity in RNA binding, potentially comprising multiple host protein binding sites, inherently weak interactions, and the presence of relatively disordered regions thought to be critical for engaging RNA and host proteins (Cascarina & Ross, 2020; Chang et al., 2014). Solution‐based methods such as nuclear magnetic resonance (NMR) offer a solution to such challenges, as multiple binding modes that include weak interactions can be simultaneously characterized. Furthermore, disordered regions are amenable to NMR studies. Moreover, the modular nature of the N protein makes it possible to specifically interrogate interactions within each region of the protein.
FIGURE 1.
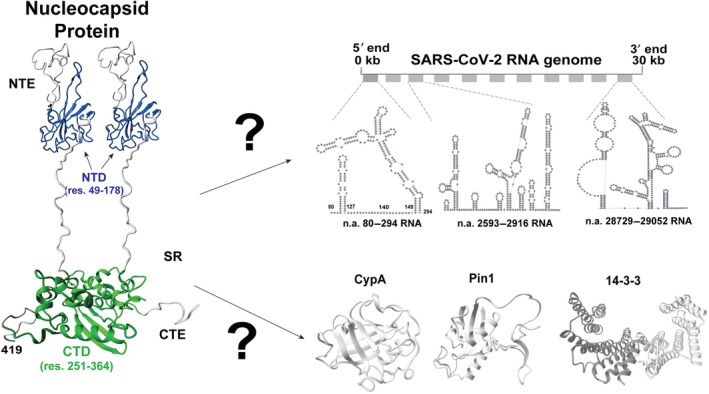
The multifunctional severe acute respiratory syndrome coronavirus‐2 (SARS‐CoV‐2) nucleocapsid protein. The N protein comprises two folded domains that include an N‐terminal domain (NTD; blue, PDB accession 6YI3) and C‐terminal domain (CTD) dimerization domain (green, PDB accession 6WJI), which are flanked by inherently disordered regions of the N‐terminal extension (NTE), serine/arginine (SR), and C‐terminal extension (CTE). Extended termini not found within PDB deposited structures were built in Chimera (Pettersen et al., 2004) and minimized in CHARMM (Brooks et al., 1983). The SR specifically includes residues 176–209 but residues 210–361 are also inherently disordered.
The primary role of the N protein is to package RNA within the mature virion (Figure 1, top), although there remain conflicting roles regarding the specific packaging signals involved (Masters, 2019). For SARS‐CoV‐2, cryo‐electron tomography (cryo‐ET) has revealed that viral ribonucleoprotein (vRNP) complexes form like “beads‐on‐a‐string” throughout the viral genome and often near membranes where the M protein likely contributes through interactions with the N protein (Klein et al., 2020; Yao et al., 2020). However, recent electron microscopy (EM) studies reveal N/RNA interactions alone are sufficient to form vRNPs and have concluded that there are likely multiple packaging signals that result in an array of different topologies (Carlson et al., 2022). Several reports have suggested that different regions may exhibit preferential interactions, such as experimental cross‐linking studies that have shown N protein enrichment within multiple RNA elements throughout the viral genome that include both its 5′‐untranslated region (UTR) and the encoding region for the N protein itself (Iserman et al., 2020). An elegant study employing all four structural proteins to produce viral‐like particles (VLPs) was shown to deliver many transcripts throughout the viral genome (Syed et al., 2021). These findings are consistent with recent theoretical studies that predict multiple packaging signals exist as ~400 nucleic acid repeats (Chechetkin & Lobzin, 2021; Chechetkin & Lobzin, 2022). Although NMR studies that include our own have confirmed that the SARS‐CoV‐2 N protein NTD and flanking regions bind to short RNAs (Dinesh et al., 2020; Redzic et al., 2021), whether the CTD and CTE have similar affinity for short RNAs remains unclear. Differential affinities could suggest a hierarchy in RNA binding whereby the N protein NTD binds first followed by other regions. For example, multiple regions that include the CTD have been shown to increase the affinity of SARS‐CoV‐1 N protein to a poly‐U model RNA (Chang et al., 2009), yet these may comprise secondary interactions. Thus, comparative studies on small RNA fragment binding to the NTD with its flanking regions and the CTD with the CTE could provide insight into the initial binding events, whereas studies with larger RNA fragments could address the variability in vRNP assembly.
Beyond packaging RNA, N protein interactions with host proteins are also potentially critical for infection (Figure 1, bottom). For example, the abundant host prolyl‐isomerase, cyclophilin‐A (CypA), has been known for many years to modulate infection and specifically viral replication in multiple CoVs (Carbajo‐Lozoya et al., 2014; Colpitts et al., 2020; Pfefferle et al., 2011). More recently, inhibitors that target CypA have been shown to reduce SARS‐CoV‐2 infection in multiple cellular models (Berthold et al., 2022; Softic et al., 2020) and another ubiquitous host prolyl‐isomerase, Pin1, has been shown to increase infection in a cellular model (Ino et al., 2022). While we have shown that human CypA binds to multiple proline residues within the disordered regions that flank the SARS‐CoV‐2 N protein NTD (Redzic et al., 2021), whether CypA also targets sites that flank the CTD remains unknown. Furthermore, in addition to Pin1 that binds phosphorylated serine or threonine residues proceeding a proline, the human chaperone family of 14–3–3 proteins also engage phosphorylated SARS‐CoV‐2 N protein at multiple sites, which could be evaluated by NMR (Ino et al., 2022). For example, expression of Pin1 enhances infection in cellular models by targeting a phosphorylated serine at S79–P80, yet other interaction sites exist considering that mutation of this serine still maintains an interaction with host Pin1 (Ino et al., 2022). Although multiple 14–3–3 protein family members were found to target the SARS‐CoV‐2 N protein at a phosphorylated S197 site, several other sites are likely also utilized (Tugaeva et al., 2021). Finally, N self‐association mediates a liquid–liquid phase separation (LLPS) responsible for the condensate formation, which is membrane‐less compartments (i.e., condensates) thought to be centers of viral replication (Iserman et al., 2020; Perdikari et al., 2021; Savastano et al., 2020). Thus, there appears to be a balance between host protein interactions along with direct N protein self‐association, and identifying such interaction sites could help inform mutations that can then be tested for infection.
Here, we combined biophysical and biochemical methods that include NMR, EM, fluorescence energy resonance transfer (FRET), and mass spectrometry (MS) to probe SARS‐CoV‐2 N protein interactions with RNA and three host proteins (Figure 1). For RNA interactions, we show that the NTD engages small RNA fragments much tighter than the CTD and CTE and that the full‐length N protein forms structurally diverse vRNPs with different regions of the viral genome. For protein interactions, we show that human CypA largely engages the N protein within its NTE and CTE while both human Pin1 and 14–3–3τ also engage multiple sites within the phosphorylated form of the N protein. The implications of such RNA interactions and host protein interactions are discussed.
2. RESULTS
2.1. Phosphorylation shifts a dynamic equilibrium within the N protein
Prior to probing N protein interactions, we sought a method that would facilitate N protein phosphorylation to enable us to probe for phosphorylation‐dependent interactions. A recently developed expression system that exploits co‐expression with the catalytic subunit of protein kinase‐A (PKA) has been used to phosphorylate the SARS‐CoV‐2 N protein (Tugaeva et al., 2021). This prior study has revealed micromolar affinities between the phosphorylated N protein and host cell 14–3–3 proteins (Tugaeva et al., 2021). We, therefore, utilized this co‐expression method with PKA together with NMR analysis to probe the phosphorylation‐dependent spectral changes induced, which could then serve as a foundation for further interrogating the solution interactions of the phosphorylated N protein presented in subsequent sections.
Initial co‐expression of two N protein constructs with PKA was used to identify phosphorylation‐dependent changes to 15 N‐HSQC spectra. These two constructs have previously been used to characterize the N protein (Redzic et al., 2021), which includes N 1–209 that comprises the NTD with flanking regions (NTE and SR) and N 251–419 that comprise the CTD and CTE. The presence of new phosphorylation‐dependent resonances was immediately obvious within both N 1–209 (Figure 2a) and N 251–419 (Figure S1a). Considering our previous discoveries that revealed most of the resonances within the entire SR region exhibit a temperature‐dependent exchange that leads to their complete disappearance at 35°C and that the SR region is implicated in multiple functions (Redzic et al., 2021), we focused our efforts on this region. Specifically, we assigned 13C,15 N‐labeled N 1–209 phosphorylated with PKA during expression that confirmed at least several new phosphorylation‐dependent resonances belong to the SR region that includes S193, S194, and S197 (Figure 2a, inset). Unfortunately, MS analysis of unlabeled N 1–209 and full‐length N protein is grown in the presence of PKA also confirmed heterogeneous mixtures of phosphorylated sites that complicated our efforts to fully assign the SR region (Figure S2). Specifically, there were limited correlations observed for many phosphorylation‐dependent resonances. Mutagenesis was also explored within the context of 15 N‐labeled N 49–209 constructs that included the WT, S187A, and S197A, which were co‐expressed with PKA in order to further identify resonances (Figure 2b). While these comparisons helped confirm that the new emerging resonances were from the phosphorylated SR region, phosphorylation patterns differed for many residues suggesting that the promiscuous PKA activity is dependent on phosphorylation of neighboring residues. Thus, the most important finding here is that the SR region is unobservable at 35°C until it is phosphorylated, which indicates that an inherent exchange (i.e., conformational sampling) on the intermediate timescale of μs‐ms is likely shifted in population upon phosphorylation or is suppressed.
FIGURE 2.
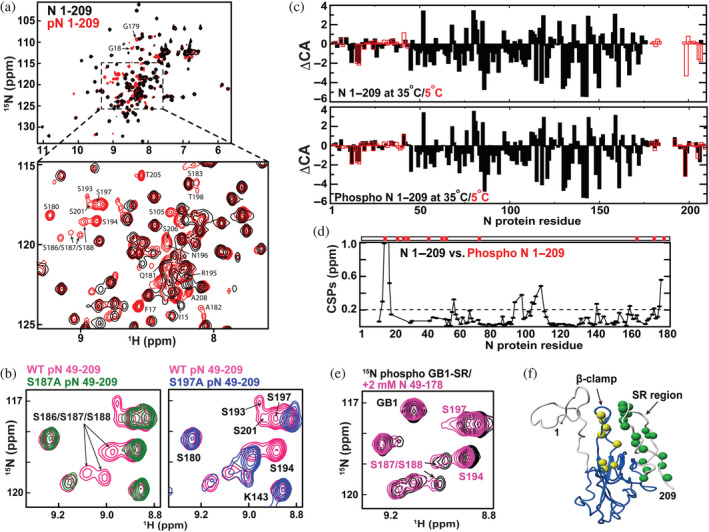
Phosphorylation‐dependent spectral changes to N protein constructs. (a) 15 N‐HSQC spectra of 15 N‐labeled N 1–209 (black) and the analogous phosphorylated form (red) at 35°C. Assignments of new resonances that appear upon phosphorylation are delineated. (b) 15 N‐HSQC spectra of wild‐type (WT) N 49–209 (pink), mutant S187A (left, green), and S197A phosphorylated in (right, blue) at 35°C. (c) CA deviations from their random coil resonances (ΔCA) at both 35°C (black, closed bar) and 5°C (red, open bar) for N 1–209 (top) and phosphorylated N 1–209 (bottom). (d) Amide chemical shift comparisons (CSPs) are shown between N 1–209 and phosphorylated N 1–209 with a dotted line indicating the average plus half the standard deviation (0.201 ppm) calculated by excluding T16 which shows the largest CSP (3.42 ppm) by direct phosphorylation. Phosphorylated residues confirmed by liquid chromatography/mass spectrometry are marked (top red bar). (e) 15 N‐labeled GB1‐serine/arginine alone at 500 μM (black) and in the presence of 2 mM unlabeled N 49–178 at 35°C. (f) Structural changes induced by phosphorylation are mapped onto N 1–209 that includes the folded NTD and flanking disordered regions modeled. Specifically, amide residues that exhibit high CSPs upon phosphorylation within the β‐hairpin (yellow) and resonances that emerge upon phosphorylation (green) are shown as spheres. All data were collected at 900 MHz with the indicated temperatures.
Secondary structure propensities monitored through CA chemical shifts indicate that there are no large structural changes upon phosphorylation (Figure 2c). Specifically, CA chemical shifts are routinely used to monitor local secondary structure by comparing CA chemical shift deviations to their random coil values (i.e., ΔCA). This is because the chemical environment is largely sensitive to the secondary structure where positive ΔCA values indicate the helical structure and negative values indicate beta strand structures. To determine whether there are phosphorylation‐dependent changes to the secondary structure, we calculated ΔCA values at 35°C where the SR region is not visible and 5°C where the SR region is visible and compared these to the previously assigned unphosphorylated form (Redzic et al., 2021). While phosphorylation leads to visible resonances of the SR region at the higher temperature described above, the secondary structure propensities reported by ΔCA values at both temperatures for the rest of the protein remain similar (Figure 2d, top compared with bottom). Thus, phosphorylation does not significantly alter the secondary structure of the N‐terminal region of the N protein.
Amide chemical shift comparisons (CSPs) between unphosphorylated and phosphorylated N 1–209 along with MS analysis of the phosphorylation sites reveal both direct and indirect changes due to phosphorylation. Specifically, there appears to exist an interaction between the phosphorylated SR region and the β‐hairpin of residues 91–105 within the folded NTD that are likely intermolecular and intramolecular (Figure 2d,e). This is because significant CSPs are observed within the β‐hairpin upon phosphorylation despite the absence of direct phosphorylation of the β‐hairpin itself (Figure 2d). We sought to further test this by phosphorylating the SR region to test whether it may intermolecularly interact with the folded NTD (i.e., N 49–178). While we were previously unable to recombinantly produce a peptide comprising the SR region alone (Redzic et al., 2021), here we recombinantly produced a construct that comprised GB1 attached to the SR region (i.e., GB1‐SR). The phosphorylated GB1‐SR recombinantly produced with PKA recapitulated a weak interaction with the NTD, as the addition of the unphosphorylated N 49–178 exhibited CSPs that confirmed a direct interaction (Figure 2e). Consistent with these findings was a comparative analysis of R1, R2, and R1rho relaxation rates between the unphosphorylated and phosphorylated N 1–209 (Figure S1b–d). Here, phosphorylation led to changes in both R1 and R2 relaxation rates within the β‐hairpin that suggests the dynamics of this region are altered upon phosphorylation of the distal SR region that induces their interaction. Overall, the ratio of R1rho to R1 relaxation rates indicates an increase in tumbling time from 9.0 to 9.7 ns upon phosphorylation, consistent with more self‐association. Thus, the phosphorylation of the SR region leads to changes in the inherent exchange of the SR region and induces a weak interaction between the phosphorylated SR region and the NTD core.
2.2. NMR and gel‐shift studies reveal some specificities in nucleotide binding
While we previously screened short nucleotide fragments to assess binding to N 1–209 (Redzic et al., 2021), here we extended such studies to also include N 251–419 to compare their affinities. Specifically, a small seven nucleotide RNA fragment derived from a linear portion of the 5′‐UTR of the SARS‐CoV‐2 genome, as well as a DNA fragment corresponding to this same sequence, were assessed to compare affinities through NMR titrations (Figure 3). Previous cross‐linking studies have shown that the N protein is enriched within this linear region (Iserman et al., 2020). When compared with CSPs for N 1–209 (Figure 3a,c), CSPs for N 251–419 were so small that they were comparable to a simple dilution of this same construct (Figure 3b,d in gray). Despite the small CSPs induced within N 251–419, residues that were impacted cluster into similar regions that suggest specificity with an estimated binding affinity of ~300–400 μM that is 2‐fold to 3‐fold lower in affinity than that previously reported for N 1–209 (Redzic et al., 2021). As predicted, phosphorylation of the tighter binding N 1–209 leads to reduced CSPs (Figure S3a). Thus, the N protein N‐terminal region engages short nucleotides much tighter than the C‐terminal region and such an interaction is reduced upon its phosphorylation.
FIGURE 3.
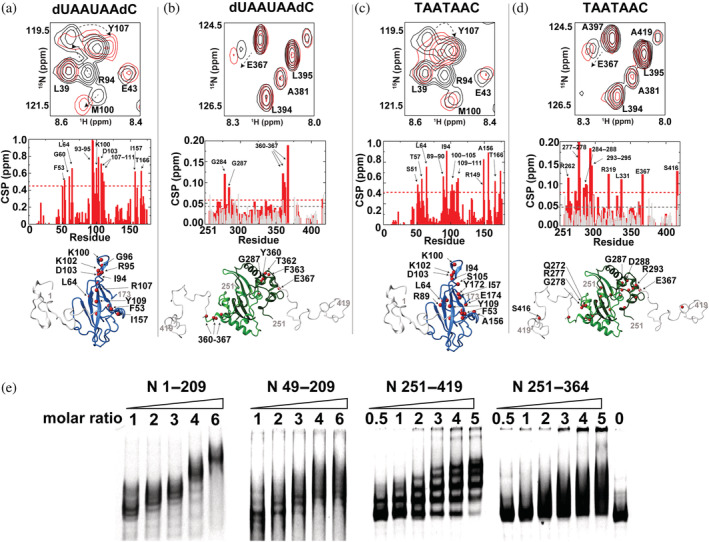
Nuclear magnetic resonance titration comparison of the N‐terminal and C‐terminal regions of the N protein with RNA and DNA fragments. Each sample comprised 100 μM 15 N‐labeled N 1–209, 200 μM N 251–419. (a) N 1–209 binding to dUAAUAAdC. Top: Section of 15 N‐HSQC of 100 μM N 1–209 alone (black) and with 400 μM dUAAUAAdC (red). Middle: Global chemical shift comparisons (CSPs) between these two spectra. Bottom: Amides that induce greater than the average plus 1 standard deviation (red spheres) are mapped onto the structure of the N‐terminal domain (NTD) with modeled flanking regions. (b) N 251–419 dimerization domain binding to dUAAUAAdC. Top: Section of 15 N‐HSQC of 200 μM N 251–419 alone (black) and with 400 μM dUAAUAAdC (red). Middle: Global CSPs between these two spectra (red) along with a dilution of free N 251–419 alone at 200 and 100 μM (gray). Bottom: Amides that induce greater than the average plus 1 standard deviation (red spheres) are mapped onto the structure of the C‐terminal domain (CTD) with modeled flanking regions. (c) N 1–209 binding to TAATAAC. Top: Section of 15 N‐HSQC of 100 μM N 1–209 alone (black) and with 400 μM TAATAAC (red). Middle: Global CSPs between these two spectra. Bottom: Amides that induce greater than the average plus 1 standard deviation (red spheres) are mapped onto the structure of the NTD with modeled flanking regions. (d) N 251–419 dimerization domain binding to TAATAAC. Top: Section of 15 N‐HSQC of 200 μM N 251–419 alone (black) and with 400 μM TAATAAC (red). Middle: Global CSPs between these two spectra (red) along with a dilution of free N 251–419 at 200 and 100 μM (gray). Bottom: Amides that induce greater than the average plus 1 standard deviation (red spheres) are mapped onto the structure of the CTD with modeled flanking regions. (e) Electrophoresis mobility shift assay performed with 5′ untranslated region n.a. 80–294 with increasing concentration of four different N protein constructs: N 1–209, N 49–209, N 251–419, and N 251–364. All gels comprised of 10% acrylamide with 2 mM MgCl2 and are stained with ethidium bromide.
As previous cross‐linking experiments indicated preferred N protein interactions with the 5′‐UTR (Iserman et al., 2020), we next probed N protein binding to a larger RNA fragment derived from this region of nucleotides 80–294 (n.a. 80–294) using electrophoresis mobility shift assays (EMSAs). Two N‐terminal constructs (N 1–209 and N 49–209) and two C‐terminal constructs (N 251–419 and N 251–364) co‐migrate with n.a. 80–294 indicating that multiple isolated regions from the N protein engage this large RNA fragment to form multimeric complexes that could lead to vRNPs (Figure 3e). Thus, nucleotide length enhances binding even for the C‐terminal region, as that the C‐terminal region barely engages small nucleotide fragments shown above via NMR. However, there are further distinctions in binding conferred by both the N‐terminus and C‐terminus as well as the role of the NTE and CTE that can be observed. First, N 1–209 forms a distinct complex as opposed to N 251–419 that displays a distribution in complexes as well as larger aggregates that do not migrate into the gel. Removal of the NTE‐disordered and CTE‐disordered regions leads to smeared distributions of complexes, suggesting that such regions enhance the specificity of vRNP formation and/or their stability. In conclusion from both NMR and EMSA studies, the N protein N‐terminal region confers higher specificity to small nucleotide fragments that lead to defined interactions with larger RNA, yet both the CTD and disordered regions contribute to binding. Finally, we note that the phosphorylated N 1–209 is still capable of engaging n.a. 80–294, albeit a slightly higher ratio of RNA:protein was necessary (Figure S3b), suggesting a weaker affinity as expected for the reduced electrostatic potential.
2.3. Synergy in N protein/RNA interactions with a disparity in vRNPs topologies
Tryptophan fluorescence (TF) was used to quantify full‐length N protein binding to n.a. 80–294 with findings that indicate synergy in binding specific elements of this region of the 5′‐UTR. Again, we probed this particular region of the SARS‐CoV‐2 genome based on previous cross‐linking experiments (Iserman et al., 2020), which could indicate the presence of a packaging signal. Six RNA constructs were produced from n.a. 80–294 in order to address the contributions to N protein binding for both its stem‐loops and the linear region that connects these stem‐loops (see Figure 1 and Table 1). Affinity was first quantified for the full n.a. 80–294 along with two constructs that comprise the linear region and only one stem‐loop, n.a. 80–148 and n.a. 127–294 (Figure 4a). Dissociation constants could be fit using the Hill equation for a two‐site model of binding with one high‐affinity and one low‐affinity dissociation constant. This is an obvious estimation, as higher stoichiometries can be loaded onto each RNA with observable gel shifts. All three of these initial constructs exhibited similar affinities, suggesting that a single stem‐loop and the linear region are sufficient to dictate binding. To further dissect the role of the linear portion and the stem‐loops, each stem‐loop alone and the entire linear portion alone were also used to assess N protein binding. Regardless of the stem‐loop used, when the linear portion was included the affinity was dramatically increased (Figure 4b,c). As these results indicate either stem‐loop along with the linear portion recapitulates binding of the entire n.a. 80–294, we questioned whether the multimeric complexes, referred to herein as vRNPs, form similarly using native gel analysis. Interestingly, n.a. 80–148 forms higher‐order structures with less N protein relative to n.a. 127–294 (Figure 4d,e). In contrast, n.a. 127–294 forms similar stoichiometries with the N protein to that of full n.a. 80–294 (Figure 4e,f). Thus, the linear portion connecting the stem loops within n.a. 80–294 synergistically contributes to N protein binding and the second stem‐loop dictates specificity for this particular interaction.
TABLE 1.
N protein binding affinities to both full‐length and subfragments of SARS‐CoV‐2 n.a. 80–294.
| 5′ UTR fragments | K d (nM) |
|---|---|
| 80–294 | 40.01 ± 3.7 |
| 80–148 | 35.4 ± 8.4 |
| 127–294 | 16.9 ± 4.1 |
| 123–152 | 199.4 ± 62.1 |
| 80–127 | 325.7 ± 61.2 |
| 148–294 | 220 ± 52.1 |
Note: Although each binding isotherm was fit to a multi‐site binding isotherm, only the well‐determined high‐affinity dissociation constant is reported here.
Abbreviations: SARS‐CoV‐2, severe acute respiratory syndrome coronavirus‐2; UTR, untranslated region.
FIGURE 4.
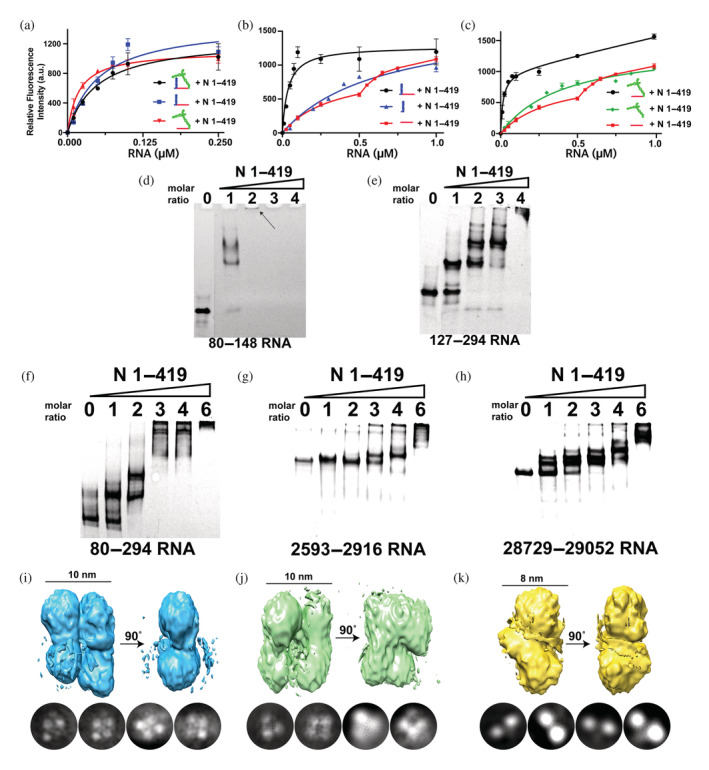
Quantifying and comparing binding and overall topologies of viral ribonucleoprotein (vRNP) complexes. Intrinsic tryptophan fluorescence binding assays for N protein binding to n.a. 80–294 and its subfragments, which include the following: (a) 5′‐untranslated region (UTR) 80–294 (black), 80–148 (blue), and 127–294 (red), (b) 80–148 (black), 80–127 (blue), and 123–153 (red), (c) 127–294 (black), 148–294 (green) and 123–153 (red). Electrophoresis mobility shift assays were performed with n.a. 80–293 and its subfragments along with two potential RNA packaging signals with increasing stoichiometries of the N protein for the following: (d) N protein with 5′‐UTR n.a. 80–148, (e) N protein with 5′‐UTR n.a. 127–294, (f) N protein with 5′‐UTR n.a. 80–294, (g) N protein with n.a. 2593–2916, (h) N protein with n.a. 28,729–29,052. 3D electron microscopy reconstructions and representative 2D class averages of N protein complexes for the following vRNPs: (i) N protein with 5′‐UTR n.a. 80–294 estimated resolution of 5.9 Å, (j) N protein with n.a. 2593–2916 estimated resolution of 7.1 Å, (k) N protein with n.a. 28,729–29,052 estimated resolution of 5.2 Å.
To begin addressing whether different regions of the viral genome may form similar or distinct vRNPs, we produced two additional large RNA fragments in addition to n.a. 80–294 for native gel analysis and EM studies. We selected these two additional large RNA fragments based on recent reports that indicate 54 n.a. repeats are present throughout the SARS‐CoV‐2 genome that are proposed to form multiple packaging signals (Chechetkin & Lobzin, 2021; Chechetkin & Lobzin, 2022). Each of these new RNA fragments was designed to have 6 of these repeats and thus, comprised 324 nucleic acids that correspond to the viral genome of n.a. 2593–2916 and n.a. 28,729–29,052. The formation of multi‐protein complexes was confirmed via native gels for all three of these large RNA fragments, as shown here for n.a. 80–294 (Figure 4f), n.a. 2593–2916 (Figure 4g), and n.a. 28,729–29,052 (Figure 4h). However, the stoichiometry of the N protein necessary to initiate vRNP formation differed between these RNA fragments, as n.a. 80–294 began forming larger complexes with a stoichiometry of 1:1 of protein:RNA. Furthermore, n.a. 80–294 was saturated by a 6:1 stoichiometry of protein:RNA, whereas the other 324 nucleotide repeats required more RNA to form larger complexes. Finally, we directly compared the vRNPs formed by each of these large RNA fragments through EM using saturating concentrations of the N protein. Interestingly, while similar topologies of vRNPs for both n.a. 80–294 (Figure 4i) and n.a. 2593–2916 (Figure 4j) were observed, a distinctly different topology was observed for n.a. 28,729–29,052 (Figure 4k). Although it is currently difficult to determine the exact stoichiometry within these vRNPs due to their low resolutions, such studies may suggest the formation of different topologies that are specific for different RNA segments.
2.4. Host CypA specifically engages multiple N protein binding sites
NMR titration experiments here reveal that the host protein CypA binds to multiple proline residues within the disordered regions of the N protein with preferences for those within the NTE and CTE. Two binding sites at the NTE were previously identified (Redzic et al., 2021) and shown here to exhibit CSPs upon the addition of human CypA (Figure 5a,b). Residues 210–250 do not comprise a proline residue that is the target site of proline isomerases like CypA and thus, this region was not considered. In order to assess whether host CypA may bind to the C‐terminal region of the N protein, assignments of the CTD alone (N 251–364) and the CTD with the CTE (N 251–419) were made under our buffer conditions (Figure S4a). 15 N‐labeled N 251–419 titrated with CypA reveal CSPs adjacent to both P381 and P396 within the CTE (Figure 5c,d, left). These binding isotherms determined from the CSPs indicate that host CypA engages the NTE and CTE relatively weakly within the high micromolar to millimolar range that is consistent with most CypA binding affinities. N protein CSPs for both N 1–209 (Figure 5e) and N 251–419 (Figure 5f) clearly show that there are at least 5 primary proline sites targeted by human CypA, which include P6, P13, P42, P383, and P396. Thus, the N protein comprises multiple CypA binding sites, as CypA targets at least 5 residues from 56 total within the N protein. However, these interactions are specific, as reverse titrations monitoring 15 N‐labeled CypA with each of these constructs, N 1–209 and N 251–419, indicate that only amides within the CypA active site exhibit CSPs (Figure S5).
FIGURE 5.
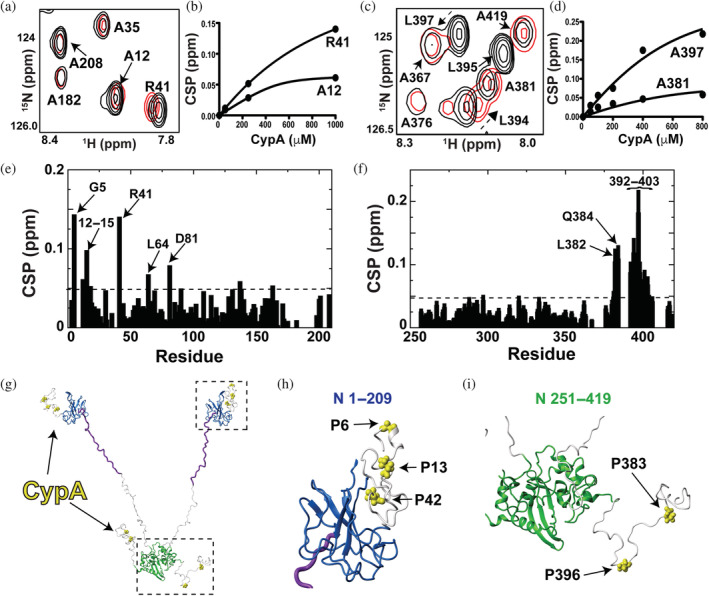
Identifying cyclophilin‐A (CypA) interaction sites within the N protein. Titrations were all conducted on a Varian 900 spectrometer at 35 °C. (a) Section of 15 N‐HSQC spectra of 200 μM 15 N‐labeled N 1–209 alone (black) and the addition of 1.0 mM CypA (red). (b) Binding isotherm for 15 N‐labeled N 1–209 with the addition of CypA. (c) 15 N‐HSQC of 200 μM 15 N‐labeled N 251–419 alone (black) and the addition of 0.8 mM CypA (red). (d) Binding isotherm for 15 N‐labeled N 251–419 with the addition of CypA. (e) Chemical shift comparisons (CSPs) between 15 N‐labeled 1–209 alone and in the presence of CypA with the dashed line (0.048 ppm) delineating the sum of the average CSP (0.025 ppm) plus 1 standard deviation (0.023 ppm). (f) CSPs between 15 N‐labeled 251–419 alone and in the presence of CypA with the dashed line (0.064 ppm) delineating the sum of the average CSP (0.031 ppm) plus 1 standard deviation (0.033 ppm). (g) Prolines within the N protein targeted by CypA are mapped onto a full‐length model of the N protein (yellow bonds). This N protein model comprises the folded N‐terminal domain and C‐terminal domain with the remaining regions built in Chimera (purple is the serine/arginine region and the remaining disordered regions in white). (h) Blow up of the N‐terminal region with CypA‐target sites (yellow bonds). (i) Blow up of the C‐terminal region with CypA‐target sites (yellow bonds).
We have previously shown that the N protein NTE mediates self‐association through binding to the NTD (Redzic et al., 2021), which suggests that self‐association and CypA interactions occur simultaneously. However, titrations monitoring 15 N‐labeled CTD with recombinantly produced CTE suggest that a similar interaction within this region is barely detectable (Figure S4b,c). Thus, in contrast to NTE‐mediated self‐association that CypA could compete with or potentially modulate, CypA interactions with the N protein CTE would dominate.
2.5. N protein interactions with 14–3–3
As we are able to monitor resonances from phosphorylated N protein constructs, we probed N protein interactions with the 14–3–3 molecular chaperone system recently shown to be dependent on N protein phosphorylation (Tugaeva et al., 2021). Specifically, human 14–3–3 proteins engage phosphorylated serine and/or threonine residues. Such residues are especially rich within the N‐terminal region of the N protein that includes the SR region (Tugaeva et al., 2021). To this end, recombinant 14–3–3τ was purified to perform NMR titration experiments using N 1–209. We specifically chose the τ isoform of 14–3–3 based on its projected weaker affinity relative to other isoforms (Tugaeva et al., 2021; Tugaeva et al., 2022), as its relatively weak interaction could result in fast exchange amenable to follow resonances during titrations. However, as described below, 14–3–3τ binding still led to the disappearance of resonances within 15 N‐labeled N 1–209, suggesting that affinity was still on the slow exchange timescale and relatively tight once the N protein was phosphorylated. Our observed disappearance of phosphorylated N 1–209 resonances is likely due to the formation of a larger complex that culminates in line‐broadening, as the stoichiometry of 14–3–3 isoforms to the N protein has been shown to be 2:2 (i.e., each N protein dimer engages one 14–3–3 dimer), which was shown through size‐exclusion‐chromatography (Tugaeva et al., 2021). However, the presence of multiple binding sites on the N protein for 14–3–3 proteins likely also contributes to the loss of intensities upon binding. Thus, for 14–3–3τ titrations, in addition to CSPs that can report on changing chemical environments under fast exchange, we also monitored resonance intensity changes that diminish upon complex formation under slow exchange. Finally, CSPs and intensity changes are reported here for subsaturating conditions of 14–3–3τ, as stoichiometric concentrations of both unphosphorylated and phosphorylated N 1–209 titrations resulted in the nearly complete disappearance of all resonances.
First, we probed for potential interactions between unphosphorylated 15 N‐labeled N 1–209 and 14–3–3τ. CSPs were largely centralized around the N protein β‐hairpin residues 91–105 (Figure 6a, bottom), indicating that this region in the unphosphorylated N protein interacts with 14–3–3τ. Resonance intensities also diminished for the NTD (Figure 6a, top), which is consistent with an interaction with 14–3–3τ. Intensities were diminished to a lesser degree for the β‐hairpin itself likely due to increased flexibility for this region in its free state that would dominate relaxation under fast exchange with 14–3–3τ (Redzic et al., 2021). An example of such CSPs is shown here that could also be used under such fast exchange to monitor the affinity of unphosphorylated N 1–209 to 14–3–3τ (Figure 6b), which was relatively weak with a dissociation constant of 400 ± 130 μM. However, caution should be taken for overinterpreting the observation of interaction of unphosphorylated N protein with 14–3–3τ considering that the N protein is highly basic (pI~10) and 14–3–3 proteins are highly acidic (pI~4). Therefore, we next probed 14–3–3τ binding to phosphorylated N 1–209.
FIGURE 6.
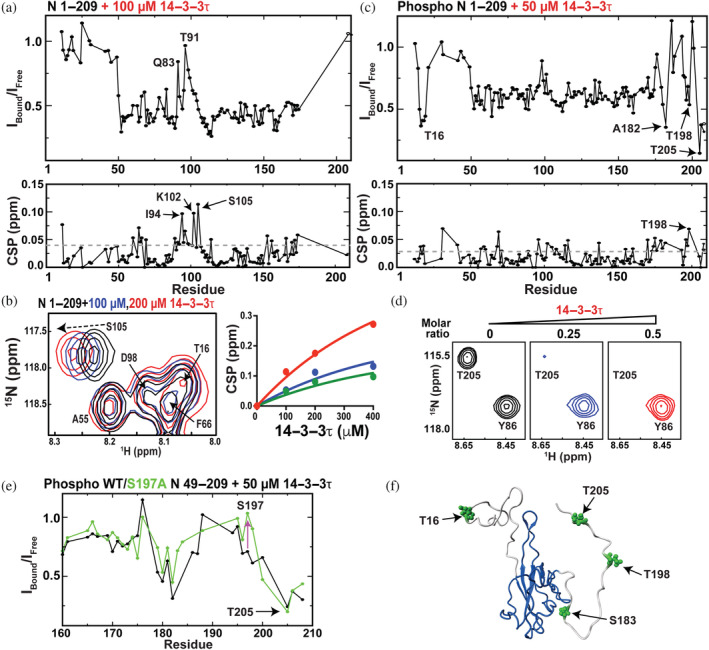
Identifying 14–3–3τ interaction sites within the N protein N 1–209. (a) Top: Per residue intensity of 200 μM unphosphorylated N 1–209 in the presence of 100 μM 14–3–3τ over the intensity of N 1–209 alone. Bottom: chemical shift comparisons (CSPs) between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.040 ppm). (b) Left: Section of 15 N‐HSQC titration of 200 μM unphosphorylated N 1–209 with 100 μM 14–3–3τ (blue) and 200 μM 14–3–3τ (red). Right: Binding isotherm using amides of G60 (blue), S105 (red), and Y172 (green). (c) Top: Per residue intensity of 200 μM phosphorylated N 1–209 in the presence of 50 μM 14–3–3τ over the intensity of N 1–209 alone. Bottom: CSPs between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.030 ppm). (d) Section of 15 N‐HSQC spectra of 200 μM phosphorylated N 1–209 (left, black) in the presence of 50 μM 14–3–3τ (middle, blue) and 100 μM 14–3–3τ (right, red). (e) Overlay of the ratio of the intensities for 200 μM phosphorylated N 49–209 in the presence of 50 μM 14–3–3τ normalized by phosphorylated N 49–209 alone (black) or the same using the mutant phosphorylated N 49–209 S197A (green bonds). (f) Phosphorylated sites within N 1–209 that interact with 14–3–3τ based on CSPs are highlighted (green bonds) onto the structure of the N‐terminal domain with flanking regions.
Phosphorylated 15 N‐labeled N 1–209 titrated with 14–3–3τ led to the disappearance of resonances much faster than that with unphosphorylated N 1–209, which is consistent with the increased binding affinity recently reported for all 14–3–3 proteins (Tugaeva et al., 2021). For example, most of the resonance intensities diminish to nearly 50% of their initial intensity even with the first titration point of 50 μM 14–3–3τ (Figure 6c, top), while few CSPs are observed under such slow exchange considering that resonance positions are not averaged between free and bound (Figure 6c, bottom). Importantly, several resonances within phosphorylated N 1–209 diminish in intensity faster than neighboring residues, which include resonances that surround T16 and several resonances within the SR region that includes T205 (Figure 6d). Such differential intensity changes are due to line‐broadening from the increased size of the bound region to 14–3–3τ. While these results are consistent with the phosphorylated SR region being a target of 14–3–3 proteins (Tugaeva et al., 2021), these titrations also suggest that T205 is the primary site within the SR region and highlight a novel interaction with T16 within the disordered NTE. Consistent with T205 being the dominant‐target site within the SR region for 14–3–3τ, intensity changes at this site were similar for a phosphorylated 15 N‐labeled S197A mutant of N 49–209 (Figure 6e). We repeated titration experiments with phosphorylated N 49–209 with only the SR region and phosphorylated N 1–178 with only the NTE to further validate such interactions (Figure S6a,b). Indeed, these differential intensity changes were once again observed within these trimmed constructs, highlighting interactions of 14–3–3τ with both disordered phosphorylated regions at T16 and T205 within the NTE and SR regions, respectively. Thus, there is a “switch” from a weak and potentially nonspecific fast exchange with unphosphorylated N 1–209 to slow exchange with phosphorylated N 1–209 that culminate in specific interactions with the NTE and the SR regions (Figure 6f).
2.6. PKA‐phosphorylated N 1–209 weakly interacts with Pin1
Recent studies have revealed that human Pin1, a proline isomerase that targets phosphorylated Ser‐Pro or Thr‐Pro sequences (Lin et al., 2015), also mediates infection through unknown mechanisms that are dependent on SARS‐CoV‐2 N interactions (Ino et al., 2022; Yamamotoya et al., 2021). The N protein comprises five potential interaction sites for Pin1, which include S79–P80, S106–P107, T141–P142, T198–P199, and S206–P207 that are all within the N protein construct N 49–209. Thus, titrations of phosphorylated 15 N‐labeled N 49–209 with Pin1 were performed. CSPs indicated that the SR region once again provides a binding site to host Pin1 (Figure 7a), as illustrated by CSPs to T198 (Figure 7b). Although several other amides exhibit CSPs during such titrations that include S180, most of these other sites do not comprise a canonical Pin1 binding site suggesting that they may be non‐specific. Some of these sites, such as the N protein D81, could mimic the negative charge and therefore bind Pin1, although S180 is followed by glutamine that is less likely to do so. Unfortunately, the current co‐expression system used with PKA did not lead to phosphorylation of a key serine within the N protein sequence S79–P80 that has been recently shown to provide for a Pin1 binding site (Ino et al., 2022). This is not unexpected, as S79 does not comprise a canonical PKA phosphorylation motif (NTNSS*P with S79 in bold) and PKA has recently been shown to be ineffective in phosphorylating this site (Tugaeva et al., 2021). In vivo, it has been suggested that other kinases phosphorylate Pin1 S79, which includes CDK, GSK3, MAPK, and CLK (Ino et al., 2022). Nonetheless, data presented here do indicate that the N protein binds to Pin1 through its SR region weakly, which may be enhanced by tighter binding to other sites that include S79.
FIGURE 7.
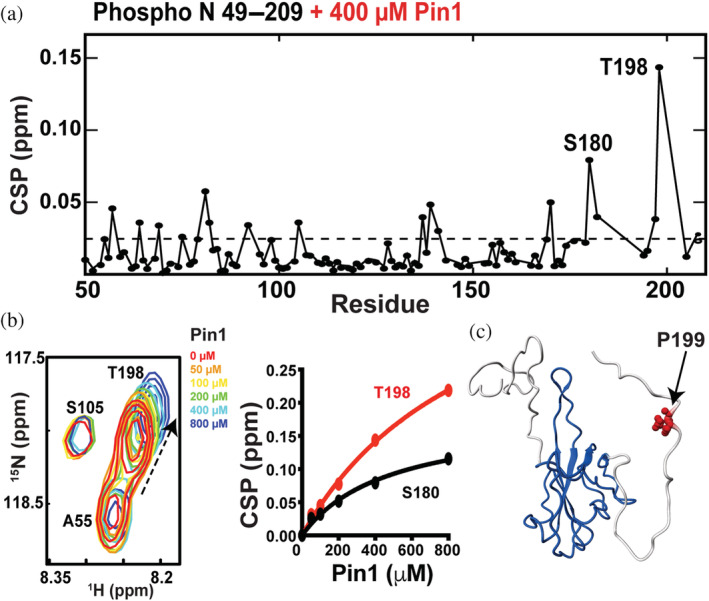
Identifying Pin1 interaction sites within the N protein. (a) Chemical shift comparisons (CSPs) observed between 100 μM phosphorylated N 49–209 alone and in the presence of 400 μM Pin1. (b) Section of 15 N‐HSQC spectra of 100 μM phosphorylated N 49–209 alone (red) and in the presence of 50 μM (orange), 100 μM (yellow), 200 μM (green), 400 μM (cyan), and 800 μM (blue) Pin1 with binding isotherms of both S180 and T198. (c) The likely interaction site of P199 (red bonds) plotted onto the NTD with flanking regions.
3. DISCUSSION
3.1. Implications for N protein‐mediated RNA packaging
NMR titration experiments using small nucleotide fragments indicate that the N‐terminal region that includes the NTD binds small nucleotides tighter than the C‐terminal region that includes the CTD (Figure 8a). This indicates some degree of specificity, even though both regions are extremely basic (pI~10) and may have implications for the hierarchy of events during the vRNP formation. Specifically, the N protein N‐terminal region could dictate the initial binding events for viral genomic RNA followed by the N protein CTD engaging RNA. This is consistent with previous models for SARS‐CoV‐1 where RNA has been proposed to wrap around the CTD and bridge multimers of the N protein (Chen et al., 2007). A preference for the NTD for RNA binding is also consistent with the high degree of aromatics within the NTD region that could stabilize RNA via intercalation, as observed within the bacteriophage MS2 virion solved by cryo‐EM (Dai et al., 2017). For example, CSPs observed by others and our group within the NTD upon titrations of small nucleotide fragments are consistent with the involvement of aromatic residues (Dinesh et al., 2020; Redzic et al., 2021).
FIGURE 8.
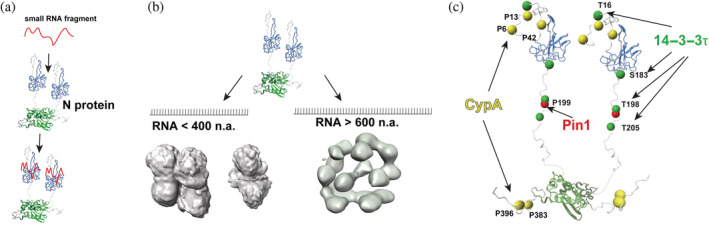
RNA and host protein interactions. (a) The N protein N‐terminal domain (blue) selectively binds small nucleotide fragments (red cartoon) based on nuclear magnetic resonance studies. (b) RNA length dictates viral ribonucleoprotein (vRNP) formation, as suggested from electron microscopy studies here of fragments that are shorter than 400 nucleic acids (left) compared with longer fragments likely more than 600 nucleic acids identified through in situ studies (right). Previously determined vRNP are from cryo‐electron tomography density (EMD‐30429, right) by Yao et al. (2020) (c) Host protein interaction sites are mapped onto a model of the N protein structure for cyclophilin‐A (yellow), 14–3–3τ (green) and Pin1 (red).
The use of both fluorescence and EM studies here to probe N protein interactions with longer RNA fragments reveals a synergistic role in binding for a linear region within the 5′‐UTR along with heterogeneity in the architecture of all three vRNPs studied here. While this is consistent with increased N protein cross‐linking observed within linear regions throughout the viral genome (Iserman et al., 2020), the structural organization observed within these vRNPs appears different than those observed in situ (Klein et al., 2020; Yao et al., 2020). Specifically, seminal cryo‐ET studies have characterized an average of 35–40 vRNPs per virion that each span 14–15 nm (Klein et al., 2020; Yao et al., 2020), which have a different morphology than the ~10 nm particles observed within this study. It is possible that the vRNPs observed here represent sampling of immature particles due to the smaller sizes of the RNA fragments used, which were 214 nucleotides for n.a. 80–294 and 354 nucleotides for both n.a. 2593–2916 and n.a. 28,729–29,052. For example, as 35–40 vRNPs are observed within each virion within the SARS‐CoV‐2 genome of 30,000 nucleotides (Klein et al., 2020; Yao et al., 2020), this means that on average there are roughly 800 nucleotides per vRNP that are bigger than the longer RNA fragments used here for EM. It is likely that mature vRNPs comprise <800 nucleotides, as the initial cryo‐ET data support a “beads‐on‐a‐string” model where the vRNPs are the beads and the viral genome is the string. This would require some spacer RNA to connect each vRNP. In fact, while our studies were in progress, an in vitro study reported that vRNPs similar to that found in situ can form with RNA fragments of 600 nucleotides or longer, but that 400 nucleotide fragments produced heterogeneous mixtures such as those observed here (Carlson et al., 2022). Thus, while smaller RNA fragments <600 nucleotides may form vRNPs, there may be a threshold for producing the mature 14–15 nm vRNPs observed in situ (Figure 8b).
Finally, it should be noted that vRNP formation in the context of the full virus is a complicated process likely orchestrated by multiple components. For example, a role of the M protein along with the lipid membrane has been shown to influence vRNP formation (Masters, 2019; Zhang et al., 2022) and a recent study using VLPs has revealed differences in delivery for transcripts within the RNA genome that illustrate RNA sequence‐dependent vRNP formation (Syed et al., 2021). It is therefore interesting to speculate that vRNPs such as those formed here with fragments shorter than 600 nucleotides could undergo multiple interactions to then form mature vRNPs (Carlson et al., 2022).
3.2. Implications for N protein interactions with the host
Using NMR, we have shown that the SARS‐CoV‐2 N protein directly binds host CypA, 14–3–3τ, and Pin1 through relatively weak but specific interactions (Figure 8c). Such viral/host interactions may not necessarily need to be high affinity considering the high expression of these host proteins and the fact that the N protein itself is the most highly expressed viral protein during infection (Bouhaddou et al., 2020). For example, CypA has been reported to comprise nearly 0.5% of the total cellular protein (Ryffel et al., 1991) and 14–3–3 proteins are expressed at even higher levels according to the Protein Abundance Database (Wang et al., 2015). Thus, such low affinities may still be enough for these host interactions to be exploited by the virus. CypA enhances viral replication for CoVs through unknown mechanisms (Carbajo‐Lozoya et al., 2014) and CypA inhibitors block SARS‐CoV‐2 infection (Softic et al., 2020). One potential mode that host CypA could play in viral replication that would be consistent with the weak interactions identified here may lie in recent studies that have defined the role of the N protein in LLPS (Iserman et al., 2020; Perdikari et al., 2021; Savastano et al., 2020). Specifically, membrane‐less compartments that are formed by N protein condensates are also centers for viral replication (Savastano et al., 2020). Thus, CypA could play an indirect role in viral replication through its interactions with the N protein. Interestingly, the role of CypA in SARS‐CoV‐2 infection is likely as complex as in other viruses, considering that extracellular CypA has recently been shown to directly bind the S protein and block its interaction with the host ACE2 receptor (Sekhon et al., 2022). Meaning, in some contexts host CypA is protective but SARS‐CoV‐2 has adapted to utilize intracellular CypA to increase infection. Lessons should also be learned from the roles of host CypA in HIV‐1 infection. For example, an initial high‐affinity interaction with the HIV‐1 capsid is followed by secondary interactions observed only in multimeric capsid hexamers that remained elusive for decades after the initial discovery that host CypA was hijacked for infection (Liu et al., 2016; Ni et al., 2020). Thus, further roles of host CypA must still be explored beyond its interactions with the N protein. In general, it is difficult to predict which of the five proline sites on the N protein that we have identified here for CypA are preferred (P6, P13, P42, P383, and P396), considering that CypA binding shows only a slight preference for the preceding residue (Harrison & Stein, 1990). However, CypA binding to relatively flexible regions of viral proteins appears to be a wide‐spread theme, such as the HIV‐1 capsid protein (Braaten et al., 1995) and the hepatitis C Virus nonstructure‐5A (NS5A) protein (Ngure et al., 2016). Our findings here that CypA engages both disordered termini of the SARS‐CoV‐2 N protein is therefore consistent with observations in multiple viruses that have evolved to exploit their disordered regions to engage host CypA.
NMR studies here using phosphorylated N protein constructs also reveal a change in the inherent exchange of the N protein SR region and identified novel binding sites for the phosphorylation‐dependent host proteins, 14–3–3τ and Pin1. The N protein SR region is a hot spot for mutations that result in higher infectivity (Rahman et al., 2020) Although the exact mechanism remains unknown, mutations within the SR region result in more effective transcript delivery in a model system (Syed et al., 2021), which suggests a direct involvement in coordinating RNA packaging. Considering that our findings here have shown that phosphorylation of the SR region leads to the appearance of its resonances within NMR spectra, this means that phosphorylation quenches an inherent dynamic exchange process within the unphosphorylated protein that we have previously reported (Redzic et al., 2021). Interestingly, recent studies indicate SR phosphorylation reduces condensates that are foci for transcription and replication (Savastano et al., 2020). Another study showed that phosphorylation results in a more dynamic condensate (i.e., more liquid‐like; Carlson et al., 2020), which also induces template switching (Wu et al., 2014). Thus, our findings here suggest that phosphorylation of the SR region reduces a dynamic exchange process within these membrane‐less compartments that lead to observed changes in LLPS. Finally, both 14–3–3τ and Pin1 associate with the phosphorylated N protein at discrete sites. Although N protein constructs were not phosphorylated by the recombinant system used here at S79 shown to induce relatively tight binding to Pin1 (Ino et al., 2022), we both confirmed and extended the identification of phosphorylation‐dependent interactions with 14–3–3τ. Specifically, the primary interaction sites of 14–3–3 proteins were identified as S197 and T205 that are both within the SR region (Tugaeva et al., 2021; Tugaeva et al., 2022). NMR titrations here indicate that T205 is preferred over S197 and that T16 also serves as an interaction site, which means that 14–3–3τ and likely other 14–3–3 family members engage multiple sites within the intrinsically disordered regions of the N protein.
In summary, the SARS‐CoV‐2 N protein uses multiple interaction sites within its disordered regions to engage ubiquitously expressed host proteins that include both CypA and 14–3–3 proteins through multiple interactions. Although the relevance to SARS‐CoV‐2 infection of these viral/host interactions remains unknown and may play roles in LLPS for condensate formation, recent discoveries in innate immune signaling may also provide clues. Specifically, the retinoic acid‐inducible gene I (RIG‐1) pathway serves as a crucial innate immune response pathway to RNA viruses that culminate in an interferon response where both cellular CypA and 14–3–3 proteins are involved (Fan & Jin, 2019; Liu et al., 2017). Here, RNA binding to TRIM25 promotes its E3 ligase activity to poly‐ubiquitinated RIG‐1 at K172 through ubiquitin K63‐linkages. Poly‐ubiquitinated RIG‐1 is then shuttled to mitochondrial membranes by 14–3–3 proteins for its next signaling step and host CypA stabilizes the activated RIG‐1 complex. Thus, in addition to roles in LLPS, it stands to reason that the highly expressed SARS‐CoV‐2 N protein may interfere in RIG‐1 signaling by targeting both CypA and 14–3–3 proteins through specific interactions identified here in order to downregulate innate immunity.
4. MATERIALS AND METHODS
4.1. Protein expression and purification
Six N protein constructs were engineered that include the full‐length N protein of residues 1–419, N 48–178, N 1–178, N 1–209, N 49–209, N 251–364, and N 251–419, which were designed with a C‐terminal 6xhis‐tag and cloned into the pET29 expression vector purchased from Twist Bioscience (San Francisco, California). The plasmids for all mutant N 49–209 were engineered by polymerase chain reaction (PCR) amplification using terminal primers containing a single mutation. N protein constructs were expressed and purified as previously reported (Redzic et al., 2021). Briefly, both unlabeled and labeled N proteins were expressed in Escherichia coli BL21(DE3) cells, and expression was induced with 0.1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) at an OD600 of 0.8 for 3 h at 37°C with typical growth volumes of 2–4 L. Pellets were lysed via sonication in Ni buffer (50 mM Na2HPO4, pH 7.0, 500 mM NaCl, 10 mM imidazole), centrifuged, and supernatants applied to a 20 mL Ni affinity column (Sigma). Eluted proteins were dialyzed against ion exchange buffer (50 mM Tris, pH 7.0, 150 mM NaCl, 1 mM EDTA) and applied to a 20 mL Q fast‐flow column to collect the flow‐through that was followed by a 40 mL SP fast‐flow column to collect the elution (Cytiva). Eluted protein from the SP column was concentrated to 3–4 mL and applied to a Superdex‐75 column (Cytiva, 120 mL total bed volume), which was equilibrated in NMR buffer (50 mM HEPES, pH 7.0, 150 mM NaCl). Proteins were concentrated and frozen until further use.
Phosphorylated N protein constructs were co‐expressed with a catalytic subunit of mouse PKA similar to that previously described in a low expression vector, pACYC‐PKA, which is chloramphenicol resistant (Tugaeva et al., 2021). N protein constructs and pACYC‐PKA were co‐transformed simultaneously with both kanamycin and chloramphenicol selection. Cells were induced with 1 mM of IPTG at an OD600 of 0.7 and grown overnight at 25°C. Purification was analogous to that described above.
Plasmids and purifications for both 14–3–3τ and Pin1 were previously described and both proteins were purified identically here (Born et al., 2019; Tugaeva et al., 2021). Briefly, for 14–3–3τ the plasmid pProExHtb was used with an N‐terminal 6xhis‐tag and for Pin1 the pET28 plasmid was used also with an N‐terminal 6xhis‐tag. Cells were harvested and lysed in Ni A buffer and applied to a Ni affinity column. Eluted proteins were concentrated to 3 mL and applied to a Superdex‐200 column in NMR buffer.
CypA was expressed and purified as we have previously described (Schlegel et al., 2009). However, the final purification step of size‐exclusion‐chromatography using a Superdex‐75 column was performed within the same NMR buffer as all proteins described above.
All final samples for NMR studies below comprised 330 μL total volume and were supplemented with 10% D2O.
4.2. Nucleotide reagents
RNA fragments (dUAAUAAdC) were purchased from Horizon Discovery (Boulder, Colorado), and DNA (TAATAAC) from IDT (Coralville, Iowa).
Viral genomic RNAs including 5′‐UTR n.a. 80–294, n.a. 2594–2916, and n.a. 28,729–29,052 were in vitro transcribed. Specifically, purchased gblocks for all DNA templates (Integrated DNA Technologies, San Diego, California) were inserted into the pUC19 vector using In‐Fusion HD cloning (Takara Bio). DNA templates were linearized with EcoR1 (NEB) and purified with phenol/chloroform extraction. In vitro transcription was performed by standard T7 RNA polymerase in 2 mL which contains 50 μg of DNA template, 4 mM NTPs, 25 mM MgCl2, 26 mg/mL T7 RNA polymerase, and 20 μL of RNase inhibitor in 1× reaction buffer (30 mM tris, pH 8, 0.01% vol/vol Triton X‐100, 0.01% wt/vol Spermidine‐HCl). After 1 h of incubation at 37°C, RNAs were purified by 12% urea‐polyacrylamide gel electrophoresis (PAGE) gel and extracted from gels in RNA elution buffer (0.5 M NaOAc, pH 5.2, 5 mM EDTA, and 2% phenol) for subsequent phenol/chloroform extraction. RNAs were dissolved in diethyl polycarbonate (DEPC) water and quantified at 260 nm for storage at −80°C until further use.
4.3. NMR spectroscopy
Phosphorylated N 1–209, unphosphorylated N 251–364, and N 251–419 were all assigned here while all other constructs were previously assigned (Redzic et al., 2021). For backbone assignment of phosphorylated N 1–209, standard HNCA, HNCACB, and CBCA(co)NH spectra were all collected on 500 μM 13C,15 N‐labeled samples at both 35°C and 5°C on a Bruker Avance Neo 600 MHz equipped with cryo‐probe. These experiments were used for the backbone assignments of unphosphorylated N 251–364 and N 251–419 all collected at 35°C at 800 MHz equipped with a cryo‐probe at the National High Magnetic Field Laboratory, Tallahassee, Florida. Assignments for the phosphorylated N 1–209 at these conditions, unphosphorylated N 251–364, and unphosphorylated N 251–419 have been deposited into the Biological Magnetic Resonance Data Bank (BMRB ID 51710, 51,709, respectively). For both titration and relaxation data, all data were collected on the Varian 900 MHz equipped with a cryo‐probe at the Rocky Mountain 900 MHz at the University of Colorado Anschutz Medical School, Aurora, CO. For both R2 and R1ρ relaxation rates on the phosphorylated N 1–209 proteins, mixing times of 0.01, 0.03, 0.05, 0.09, 0.11, 0.13, and 0.15 s were used and for R1 relaxation rates, mixing times of 0.01, 0.1, 0.3, 0.5, 0.9, 1.1, 1.3, and 1.5 s were used. All 15 N‐HSQC spectra were conducted at 35 °C except for 15 N‐labeled CypA. All ligand titration experiments to N protein were collected at 35°C and titration of N protein to CypA was collected at 25°C. All spectra were processed using NMRPipe (Delaglio et al., 1995) and all data were analyzed using CCPNmr software (Vranken et al., 2005). Finally, all binding affinities were derived from the indicated titration data and using GraphPad Prism version 4.0 (GraphPad Software Inc., San Diego, California).
4.4. Electrophoretic mobility shift assay
RNA stocks were diluted to 20 μM with reaction buffer (50 mM HEPES, 150 mM NaCl, 2 mM MgCl2, pH 7) and denatured at 95°C for 2 min to reanneal at 37°C for 15 min. Reannealed RNA was mixed with full‐length N protein in the reaction buffer in a final volume of 10 μL. After 5 min incubations at room temperature, the reactions were analyzed in 6%–10% non‐denaturing PAGE gel containing 2 mM MgCl2, prepared in 1× TB buffer (89 mM Tris, 89 mM boric acid, pH 8.2). The prerun was performed at 70 V for 30 min, and the sample was run at 70 V for 90 min. The gel was stained in ethidium bromide (Sigma) for 10 min and imaged on UV light.
4.5. TF assay
TF measurements were performed using a Fluoromax‐3 spectrofluorometer at 35°C. Spectra were recorded with 0.5 μM full‐length N protein with increasing concentrations of 5′‐UTR RNAs in buffer containing 50 mM HEPES, 150 mM NaCl, pH 8. Samples were excited at 295 nm and emission spectra were obtained from 320 to 360 nm with 0.5 nm step size and a 1 s integration time. K d values were determined using the Hill equation with multisite binding in GraphPad Prism. The K d values were averaged over three separate experiments and the error was calculated from the variation between experiments.
4.6. Negative stain EM
Samples for EM comprised 0.3 μM of RNA mixed with 3 μM of full‐length N protein and incubated for 2 hours at room temperature. After incubation, 3 μL of RNA protein complex was applied to a glow‐discharged 300‐mesh carbon grid using PELCO easiGlow Discharge system (Ted Pella Inc, USA). After 60 seconds to allow for absorption, the sample was stained with a 2% uranyl acetate solution. Data were acquired on Talos L120C transmission electron microscope (Thermoscientific) operating at 120 keV that resulted in a pixel size of 1.964 Å per pixel at the specimen level. Collected EM micrographs were processed in Cryosparc, generating the final 3D models for each complex. A total of 50,682, 28,969, and 195,896 particles were used for the final reconstruction for 5′‐UTR n.a. 80–294, n.a. 2593–2916, and n.a. 28,729–29,052, respectively.
4.7. Mapping sites of phosphorylation on SARS‐CoV‐2 nucleocapsid protein using MS
PKA phosphorylated and nonphosphorylated SARS‐CoV‐2 Nucleocapsid protein (~50 μg) was denatured, reduced, and alkylated in 5% (wt/vol) sodium dodecyl sulfate, 10 mM tris (2‐carboxyethyl) phosphine hydrochloride (TCEP‐HCl), 40 mM 2‐chloroacetamide, 50 mM Tris pH 8.5 and boiled at 95°C for 10 min. Samples were prepared for MS analyses using the SP3 method (Hughes et al., 2014). Carboxylate‐functionalized speedbeads (GE Life Sciences) were added to protein samples. Acetonitrile was added to 80% (vol/vol) to precipitate protein and bind it to the beads. The protein‐bound beads were washed twice with 80% (vol/vol) ethanol and twice with 100% acetonitrile. LysC/Trypsin mix (Promega) was added for 1:50 protease to protein ratio in 50 mM Tris pH 8.5 and incubated rotating at 37°C overnight. Tryptic digests were cleaned‐up using a Water Oasis HLB 1 cc (10 mg) cartridge according to the manufacturer and dried using a speed‐vac rotatory evaporator and resuspended in 0.1% trifluoroacetic acid (TFA), 3% acetonitrile in water to peptides on the column for liquid chromatography/MS (LC/MS) analyses. Peptides were directly injected onto a Waters M‐class column (1.7 μm, 120A, rpC18, 75 μm × 250 mm) and gradient eluted from 2% to 20% acetonitrile over 40 min at 0.3 μL/min using a Thermo Ultimate 3000 ultra performance liquid chromatography (UPLC) (Thermo Scientific). Peptides were detected with a Thermo Q‐Exactive HF‐X mass spectrometer (Thermo Scientific) scanning MS1 at 120,000 resolution from 350 to 1550 m/z with a 50 ms fill time and 3E6 AGC target. The top 12 most intense peaks were isolated with 1.4 m/z window with a 100 ms fill time and 1E5 AGC target and 27% HCD collision energy for MS2 spectra scanned at 15,000 resolution. Dynamic exclusion was enabled for 5 s. Fragmentation spectra were interpreted using Peaks X Pro (v. 10.6) against the UniProt sequence for the SARS‐CoV‐2 nucleocapsid protein. Precursor mass tolerance was set to 10 ppm and fragment ion mass tolerance was set to 0.02 Da. Cysteine carbamidomethylation was set as a fixed modification, whereas methionine oxidation, protein N‐terminal, and phosphorylation at serine, threonine, and tyrosine were set as variable modifications.
Supporting information
FIGURE S1. Phosphorylation‐dependent changes to the N protein. (a) 15 N‐HSQC spectra of 15 N‐labeled N 251–419 (black) and phosphorylated N 251–419 (red). Inset highlights resonances that emerge only after phosphorylation. (b) R1 relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). (c) R2 relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). (d) R1ρ relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). All data were collected at 900 MHz at 35°C.
FIGURE S2. N protein phosphorylation sites identified via mass spectrometric analysis with recombinant co‐expression of protein kinase‐A. (a) The position of phosphorylated amino acids in N 1–419 protein is highlighted in red and bold letters. Sequence coverage includes 93.3% of the 419 amino acids and 14 out of 15 phosphorylated Ser/Thr within the SR region. (b) Liquid Chromatography with tandem mass spectrometry (LC)–MS/MS analysis of the β‐hairpin region of the N‐terminal domain (NTD). (c) LC–MS/MS analysis of the SR region that is positioned between the NTD and C‐terminal domain. The assigned b and y ions which are from fragmentation of each peptide are labeled in blue and red, respectively. Phosphorylated residues are highlighted in red within the peptide sequence.
FIGURE S3. DNA binding and viral ribonucleoprotein formation with phosphorylated N protein constructs. (a) Overlay of 15 N‐HSQCs for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red) and corresponding chemical shift comparisons (CSPs) for the entire construct are shown collected at 900 MHz at 35°C. (b) Electrophoresis mobility shift assay of phosphorylated N 1–209 and the 5′‐untranslated region (UTR) 80–294 RNA with increasing concentrations of the latter.
FIGURE S4. Assignments of the C‐terminal region of the N protein and probing potential interactions between the disordered C‐terminal extension (N 365–419) and folded C‐terminal domain (CTD; N 251–364). (a) CA deviations from their random coil resonances (ΔCA) deviations are shown for both N 251–419 (black, closed) and N 251–364 (red, open bar). Secondary structure elements from the x‐ray crystal structure of N 251–364 are also shown for comparison. (b) Left: A representative portion of the 15 N‐HSQC comparison of 500 μM N 251–364 alone (red), in the presence of 2 mM N 361–419 (blue), and in the presence of 6 mM N 361–419 (red). chemical shift comparisons (CSPs) of N 251–364 upon the addition of 6 mM of N 361–419 (right) with the dashed line (0.10 ppm) delineates the sum of the average CSP (0.06 ppm) plus 1 standard deviation (0.04 ppm). Amides that have larger CSPs than the average plus 1 standard deviation are mapped onto the x‐ray crystal structure of the CTD with flanking regions (red spheres). (c) Left: 15 N‐HSQC comparison of N 251–364 (gray) and N 251–419 (red). Right: CSPs between N 251–364 and 251–419 with the dashed line (0.11 ppm) delineates the sum of the average CSP (0.04 ppm) plus 1 standard deviation (0.07 ppm). Residues that have larger CSPs than the average plus 1 standard deviation are mapped onto the x‐ray crystal structure of the C‐terminal folded domain (red spheres). All data were collected at 900 MHz at 35°C.
FIGURE S5. Identifying weak N protein interaction sites within cyclophilin‐A (CypA). (a) A representative section of the 15 N‐HSQC spectrum is shown for 500 μM 15 N‐CypA alone (black) and in the presence of 1.2 mM N 1–209 (red). (b) A representative section of the 15 N‐HSQC spectrum is shown for 500 μM 15 N‐CypA alone (black) and in the presence of 1.2 mM N 251–419 (red). (c) Binding isotherm derived from 15 N‐CypA with the addition of N 1–209 results in a dissociation constant of 0.9 ± 0.1 mM. (d) Binding isotherm derived from 15 N‐CypA with the addition of N 251–419 results in a dissociation constant of 1.1 ± 0.1 mM. (e) Chemical shift comparisons (CSPs) between 15 N‐CypA alone and in the presence of 1.2 mM N 1–209 with the dashed line (0.15 ppm) delineating the sum of the average CSP (0.07 ppm) plus 1 standard deviation (0.08 ppm). (f) CSPs between 15 N‐CypA alone and in the presence of 1.2 mM N 251–419 with the dashed line (0.16 ppm) delineating the sum of the average CSP (0.07 ppm) plus 1 standard deviation (0.09 ppm). (g) CSPs greater than the average plus 1 standard deviation are shown for the addition of N 1–209 (red spheres). (h) CSPs greater than the average plus 1 standard deviation are shown for the addition of N 251–419 (red spheres). All experiments probing 15 N‐labeled CypA were conducted at 900 MHz at 25°C.
FIGURE S6. Identifying 14–3–3τ interaction sites within the N protein N 49–209 and N 1‐178. (a) Top: Per residue intensity of 200 μM phosphorylated N 49–209 in the presence of 50 μM 14–3–3τ over the intensity of N 1–209 alone. Bottom: Chemical shift comparisons (CSPs) between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.024 ppm). (b) Top: Per residue intensity of 200 μM phosphorylated N 1–178 in the presence of 50 μM 14–3–3τ over the intensity of N 1–178 alone. Bottom: CSPs between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.028 ppm). All data were collected at 900 MHz at 35°C
ACKNOWLEDGMENTS
Eunjeong Lee, Jasmina S. Redzic, and Elan Eisenmesser were supported by NIH R21 AI146295 and R01 GM139892. Nikolai N. Sluchanko is grateful to Kristina V. Tugaeva for providing the recombinant co‐expression system with PKA and for the Russian Science Federation for funding. Rui Zhao and Xueni Li were supported by R35GM145289. Natalie Ahn was supported by NIH R35GM136392.
Lee E, Redzic JS, Saviola AJ, Li X, Ebmeier CC, Kutateladze T, et al. Molecular insight into the specific interactions of the SARS‐Coronavirus‐2 nucleocapsid with RNA and host protein. Protein Science. 2023;32(4):e4603. 10.1002/pro.4603
Review Editor: Hideo Akutsu
Funding information Division of Intramural Research, National Institute of Allergy and Infectious Diseases, Grant/Award Number: 5R21AI166292; NIH, Grant/Award Numbers: R35GM136392, R35GM145289, R01 GM139892, R21 AI146295; PKA‐Russian Science Federation, Grant/Award Number: 19‐74‐10031
DATA AVAILABILITY STATEMENT
Data is available upon request.
REFERENCES
- Berthold EJ, Ma‐Lauer Y, Chakraborty A, von Brunn B, Hilgendorff A, Hatz R, et al. Effects of immunophilin inhibitors and non‐immunosuppressive analogs on coronavirus replication in human infection models. Front Cell Infect Microbiol. 2022;12:958634. 10.3389/fcimb.2022.958634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born A, Nichols PJ, Henen MA, Chi CN, Strotz D, Bayer P, et al. Backbone and side‐chain chemical shift assignments of full‐length, apo, human Pin1, a phosphoprotein regulator with interdomain allostery. Biomol NMR Assign. 2019;13:85–9. 10.1007/s12104-018-9857-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M, Memon D, Meyer B, White KM, Rezelj VV, Marrero MC, et al. The global phosphorylation landscape of SARS‐CoV‐2 infection. Cell. 2020;182:685–712.e19. 10.1016/j.cell.2020.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braaten D, Franke EK, Luban J. Gag and cyclophilin‐A in Hiv‐1 replication. AIDS Res Hum Retroviruses. 1995;11:S88–8. [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. Charmm ‐ a program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- Carbajo‐Lozoya J, Ma‐Lauer Y, Malesevic M, Theuerkorn M, Kahlert V, Prell E, et al. Human coronavirus NL63 replication is cyclophilin A‐dependent and inhibited by non‐immunosuppressive cyclosporine A‐derivatives including Alisporivir. Virus Res. 2014;184:44–53. 10.1016/j.virusres.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CR, Adly AN, Bi M, Howard CJ, Frost A, Cheng Y, et al. Reconstitution of the SARS‐CoV‐2 ribonucleosome provides insights into genomic RNA packaging and regulation by phosphorylation. J Biol Chem. 2022;298:102560. 10.1016/j.jbc.2022.102560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CR, Asfaha JB, Ghent CM, Howard CJ, Hartooni N, Safari M, et al. Phosphoregulation of phase separation by the SARS‐CoV‐2 N protein suggests a biophysical basis for its dual functions. Mol Cell. 2020;80:1092–1103.e4. 10.1016/j.molcel.2020.11.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascarina SM, Ross ED. A proposed role for the SARS‐CoV‐2 nucleocapsid protein in the formation and regulation of biomolecular condensates. FASEB J. 2020;34:9832–42. 10.1096/fj.202001351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Ck, Hou M‐H, Chang C‐F, Hsiao C‐D, Huang T‐H. The SARS coronavirus nucleocapsid protein ‐ forms and functions. Antiviral Res. 2014;103:39–50. 10.1016/j.antiviral.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CK, Hsu YL, Chang YH, Chao FA, Wu MC, Huang YS, et al. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for Ribonucleocapsid protein packaging. J Virol. 2009;83:2255–64. 10.1128/jvi.02001-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chechetkin VR, Lobzin VV. Evolving ribonucleocapsid assembly/packaging signals in the genomes of the human and animal coronaviruses: targeting, transmission and evolution. J Biomol Struct Dyn. 2021;40:11239–63. 10.1080/07391102.2021.1958061 [DOI] [PubMed] [Google Scholar]
- Chechetkin VR, Lobzin VV. Ribonucleocapsid assembly/packaging signals in the genomes of the coronaviruses SARS‐CoV and SARS‐CoV‐2: detection, comparison and implications for therapeutic targeting. J Biomol Struct Dyn. 2022;40:508–22. 10.1080/07391102.2020.1815581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Chang CK, Chang YW, Sue SC, Bai HI, Riang L, et al. Structure of the SARS coronavirus nucleocapsid protein RNA‐binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J Mol Biol. 2007;368:1075–86. 10.1016/j.jmb.2007.02.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colpitts CC, Ridewood S, Schneiderman B, Warne J, Tabata K, Ng CF, et al. Hepatitis C virus exploits cyclophilin a to evade PKR. Elife. 2020;9:e52237. 10.7554/eLife.52237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai XH, Li ZH, Lai M, Shu S, Du YS, Zhou ZH, et al. In situ structures of the genome and genome‐delivery apparatus in a single‐stranded RNA virus. Nature. 2017;541:112–6. 10.1038/nature20589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. Nmrpipe ‐ a multidimensional spectral processing system based on Unix pipes. J Biomol NMR. 1995;6:277–93. [DOI] [PubMed] [Google Scholar]
- Dinesh DC, Chalupska D, Silhan J, Koutna E, Nencka R, Veverka V, et al. Structural basis of RNA recognition by the SARS‐CoV‐2 nucleocapsid phosphoprotein. PLoS Pathog. 2020;16(12):e1009100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan XJ, Jin TC. In: Jin T, Yin Q, editors. Structural immunology vol. 1172 advances in experimental medicine and biology. Singapore: Springer, 2019. p. 157–88. [DOI] [PubMed] [Google Scholar]
- Harrison RK, Stein RL. Substrate specificities of the peptidyl prolyl cis‐trans isomerase activities of cyclophilin and FK‐506 binding‐protein ‐ evidence for the existence of a family of distinct enzymes. Biochemistry. 1990;29:3813–6. 10.1021/bi00468a001 [DOI] [PubMed] [Google Scholar]
- Hughes CS, Foehr S, Garfield DA, Furlong EE, Steinmetz LM, Krijgsveld J. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014;10:757. 10.15252/msb.20145625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ino Y, Nishi M, Yamaoka Y, Miyakawa K, Jeremiah SS, Osada M, et al. Phosphopeptide enrichment using Phos‐tag technology reveals functional phosphorylation of the nucleocapsid protein of SARS‐CoV‐2. J Proteomics. 2022;255:104501. 10.1016/j.jprot.2022.104501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iserman C, Roden CA, Boerneke MA, Sealfon RSG, McLaughlin GA, Jungreis I, et al. Genomic RNA elements drive phase separation of the SARS‐CoV‐2 nucleocapsid. Mol Cell. 2020;80(6):1078–1091.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein S, Cortese M, Winter SL, Wachsmuth‐Melm M, Neufeldt CJ, Cerikan B, et al. SARS‐CoV‐2 structure and replication characterized by in situ cryo‐electron tomography. Nat Commun. 2020;11:5885. 10.1038/s41467-020-19619-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Li HY, Lee YC, Calkins MJ, Lee KH, Yang CN, et al. Landscape of Pin1 in the cell cycle. Exp Biol Med. 2015;240:403–8. 10.1177/1535370215570829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Perilla JR, Ning JY, Lu MM, Hou GJ, Ramalho R, et al. Cyclophilin a stabilizes the HIV‐1 capsid through a novel non‐canonical binding site. Nat Commun. 2016;7:10714. 10.1038/ncomms10714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Li J, Zheng WN, Shang YL, Zhao ZD, Wang SS, et al. Cyclophilin A‐regulated ubiquitination is critical for RIG‐I‐mediated antiviral immune responses. Elife. 2017;6:e24425. 10.7554/eLife.24425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S, Ye QZ, Singh D, Cao Y, Diedrich JK, Yates JR, et al. The SARS‐CoV‐2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane‐associated M protein. Nat Commun. 2021;12:502. 10.1038/s41467-020-20768-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters PS. Coronavirus genomic RNA packaging. Virology. 2019;537:198–207. 10.1016/j.virol.2019.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngure M, Issur M, Shkriabai N, Liu HW, Cosa G, Kvaratskhelia M, et al. Interactions of the disordered domain II of hepatitis C virus NS5A with cyclophilin a, NS5B, and viral RNA show extensive overlap. ACS Infect Dis. 2016;2:839–51. 10.1021/acsinfecdis.6b00143 [DOI] [PubMed] [Google Scholar]
- Ni T, Gerard S, Zhao GP, Dent K, Ning JY, Zhou J, et al. Intrinsic curvature of the HIV‐1 CA hexamer underlies capsid topology and interaction with cyclophilin a. Nat Struct Mol Biol. 2020;27:855–62. 10.1038/s41594-020-0467-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perdikari TM, Murthy AC, Ryan VH, Watters S, Naik MT, Fawzi NL. SARS‐CoV‐2 nucleocapsid protein phase‐separates with RNA and with human hnRNPs. EMBO J. 2021;39:e106478. 10.15252/embj.2020106478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF chimera ‐ a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- Pfefferle S, Schopf J, Kogl M, Friedel CC, Muller MA, Carbajo‐Lozoya J, et al. The SARS‐coronavirus‐host interactome: identification of cyclophilins as target for pan‐coronavirus inhibitors. PLoS Pathog. 2011;7:e1002331. 10.1371/journal.ppat.1002331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MS, Islam MR, Ul Alam A, Islam I, Hoque MN, Akter S, et al. Evolutionary dynamics of SARS‐CoV‐2 nucleocapsid protein and its consequences. J Med Virol. 2020;93:2177–95. 10.1002/jmv.26626 [DOI] [PubMed] [Google Scholar]
- Redzic JS, Lee E, Born A, Issaian A, Henen MA, Nichols PJ, et al. The inherent dynamics and interaction sites of the SARS‐CoV‐2 nucleocapsid N‐terminal region. J Mol Biol. 2021;433:167108. 10.1016/j.jmb.2021.167108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryffel B, Woerly G, Greiner B, Haendler B, Mihatsch MJ, Foxwell BMJ. Distribution of the cyclosporine binding‐protein cyclophilin in human tissues. Immunology. 1991;72:399–404. [PMC free article] [PubMed] [Google Scholar]
- Savastano A, de Opakua AI, Rankovic M, Zweckstetter M. Nucleocapsid protein of SARS‐CoV‐2 phase separates into RNA‐rich polymerase‐containing condensates. Nat Commun. 2020;11:6041. 10.1038/s41467-020-19843-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlegel J, Armstrong GS, Redzic JS, Zhang FL, Eisenmesser EZ. Characterizing and controlling the inherent dynamics of cyclophilin‐a. Protein Sci. 2009;18:811–24. 10.1002/pro.89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekhon SS, Shin WR, Kim SY, Jeong DS, Choi W, Choi BK, et al. Cyclophilin A‐mediated mitigation of coronavirus SARS‐CoV‐2. Bioeng Transl Med. 2022;e10436. 10.1002/btm2.10436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Softic L, Brillet R, Berry F, Ahnou N, Nevers Q, Morin‐Dewaele M, et al. Inhibition of SARS‐CoV‐2 infection by the cyclophilin inhibitor Alisporivir (Debio 025). Antimicrob Agents Chemother. 2020;64:e00876‐20. 10.1128/aac.00876-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AM, Taha TY, Tabata T, Chen IP, Ciling A, Khalid MM, et al. Rapid assessment of SARS‐CoV‐2‐evolved variants using virus‐like particles. Science. 2021;374:1626–32. 10.1126/science.abl6184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugaeva KV, Hawkins D, Smith JLR, Bayfield OW, Ker DS, Sysoev AA, et al. The mechanism of SARS‐CoV‐2 nucleocapsid protein recognition by the human 14‐3‐3 proteins. J Mol Biol. 2021;433:166875. 10.1016/j.jmb.2021.166875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tugaeva KV, Sysoev AA, Kapitonova AA, Smith JLR, Zhu P, Cooley RB, et al. Human 14‐3‐3 proteins site‐selectively bind the mutational hotspot region of SARS‐CoV‐2 nucleoprotein modulating its Phosphoregulation. J Mol Biol. 2022;435:167891. 10.1016/j.jmb.2022.167891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- V'Kovski P, Kratzel A, Steiner S, Stalder H, Thiel V. Coronavirus biology and replication: implications for SARS‐CoV‐2. Nature Reviews Microbiology. 2020;19:155–70. 10.1038/s41579-020-00468-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranken WF, Boucher W, Stevens TJ, Fogh RH, Pajon A, Llinas P, et al. The CCPN data model for NMR spectroscopy: development of a software pipeline. Proteins. 2005;59:687–96. [DOI] [PubMed] [Google Scholar]
- Wang MC, Herrmann CJ, Simonovic M, Szklarczyk D, von Mering C. Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell‐lines. Proteomics. 2015;15:3163–8. 10.1002/pmic.201400441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CH, Chen PJ, Yeh SH. Nucleocapsid phosphorylation and RNA helicase DDX1 recruitment enables coronavirus transition from discontinuous to continuous transcription. Cell Host Microbe. 2014;16:462–72. 10.1016/j.chom.2014.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamotoya T, Nakatsu Y, Kanna M, Hasei S, Ohata Y, Encinas J, et al. Prolyl isomerase Pin1 plays an essential role in SARS‐CoV‐2 proliferation, indicating its possibility as a novel therapeutic target. Sci Rep. 2021;11:18581. 10.1038/s41598-021-97972-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao HP, Song YT, Chen Y, Wu NP, Xu JL, Sun CJ, et al. Molecular architecture of the SARS‐CoV‐2 virus. Cell. 2020;183:730–738.e13. 10.1016/j.cell.2020.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZK, Nomura N, Muramoto Y, Ekimoto T, Uemura T, Liu KH, et al. Structure of SARS‐CoV‐2 membrane protein essential for virus assembly. Nat Commun. 2022;13:4399. 10.1038/s41467-022-32019-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Phosphorylation‐dependent changes to the N protein. (a) 15 N‐HSQC spectra of 15 N‐labeled N 251–419 (black) and phosphorylated N 251–419 (red). Inset highlights resonances that emerge only after phosphorylation. (b) R1 relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). (c) R2 relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). (d) R1ρ relaxation rates are shown for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red). All data were collected at 900 MHz at 35°C.
FIGURE S2. N protein phosphorylation sites identified via mass spectrometric analysis with recombinant co‐expression of protein kinase‐A. (a) The position of phosphorylated amino acids in N 1–419 protein is highlighted in red and bold letters. Sequence coverage includes 93.3% of the 419 amino acids and 14 out of 15 phosphorylated Ser/Thr within the SR region. (b) Liquid Chromatography with tandem mass spectrometry (LC)–MS/MS analysis of the β‐hairpin region of the N‐terminal domain (NTD). (c) LC–MS/MS analysis of the SR region that is positioned between the NTD and C‐terminal domain. The assigned b and y ions which are from fragmentation of each peptide are labeled in blue and red, respectively. Phosphorylated residues are highlighted in red within the peptide sequence.
FIGURE S3. DNA binding and viral ribonucleoprotein formation with phosphorylated N protein constructs. (a) Overlay of 15 N‐HSQCs for unphosphorylated N 1–209 (black) and phosphorylated N 1–209 (red) and corresponding chemical shift comparisons (CSPs) for the entire construct are shown collected at 900 MHz at 35°C. (b) Electrophoresis mobility shift assay of phosphorylated N 1–209 and the 5′‐untranslated region (UTR) 80–294 RNA with increasing concentrations of the latter.
FIGURE S4. Assignments of the C‐terminal region of the N protein and probing potential interactions between the disordered C‐terminal extension (N 365–419) and folded C‐terminal domain (CTD; N 251–364). (a) CA deviations from their random coil resonances (ΔCA) deviations are shown for both N 251–419 (black, closed) and N 251–364 (red, open bar). Secondary structure elements from the x‐ray crystal structure of N 251–364 are also shown for comparison. (b) Left: A representative portion of the 15 N‐HSQC comparison of 500 μM N 251–364 alone (red), in the presence of 2 mM N 361–419 (blue), and in the presence of 6 mM N 361–419 (red). chemical shift comparisons (CSPs) of N 251–364 upon the addition of 6 mM of N 361–419 (right) with the dashed line (0.10 ppm) delineates the sum of the average CSP (0.06 ppm) plus 1 standard deviation (0.04 ppm). Amides that have larger CSPs than the average plus 1 standard deviation are mapped onto the x‐ray crystal structure of the CTD with flanking regions (red spheres). (c) Left: 15 N‐HSQC comparison of N 251–364 (gray) and N 251–419 (red). Right: CSPs between N 251–364 and 251–419 with the dashed line (0.11 ppm) delineates the sum of the average CSP (0.04 ppm) plus 1 standard deviation (0.07 ppm). Residues that have larger CSPs than the average plus 1 standard deviation are mapped onto the x‐ray crystal structure of the C‐terminal folded domain (red spheres). All data were collected at 900 MHz at 35°C.
FIGURE S5. Identifying weak N protein interaction sites within cyclophilin‐A (CypA). (a) A representative section of the 15 N‐HSQC spectrum is shown for 500 μM 15 N‐CypA alone (black) and in the presence of 1.2 mM N 1–209 (red). (b) A representative section of the 15 N‐HSQC spectrum is shown for 500 μM 15 N‐CypA alone (black) and in the presence of 1.2 mM N 251–419 (red). (c) Binding isotherm derived from 15 N‐CypA with the addition of N 1–209 results in a dissociation constant of 0.9 ± 0.1 mM. (d) Binding isotherm derived from 15 N‐CypA with the addition of N 251–419 results in a dissociation constant of 1.1 ± 0.1 mM. (e) Chemical shift comparisons (CSPs) between 15 N‐CypA alone and in the presence of 1.2 mM N 1–209 with the dashed line (0.15 ppm) delineating the sum of the average CSP (0.07 ppm) plus 1 standard deviation (0.08 ppm). (f) CSPs between 15 N‐CypA alone and in the presence of 1.2 mM N 251–419 with the dashed line (0.16 ppm) delineating the sum of the average CSP (0.07 ppm) plus 1 standard deviation (0.09 ppm). (g) CSPs greater than the average plus 1 standard deviation are shown for the addition of N 1–209 (red spheres). (h) CSPs greater than the average plus 1 standard deviation are shown for the addition of N 251–419 (red spheres). All experiments probing 15 N‐labeled CypA were conducted at 900 MHz at 25°C.
FIGURE S6. Identifying 14–3–3τ interaction sites within the N protein N 49–209 and N 1‐178. (a) Top: Per residue intensity of 200 μM phosphorylated N 49–209 in the presence of 50 μM 14–3–3τ over the intensity of N 1–209 alone. Bottom: Chemical shift comparisons (CSPs) between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.024 ppm). (b) Top: Per residue intensity of 200 μM phosphorylated N 1–178 in the presence of 50 μM 14–3–3τ over the intensity of N 1–178 alone. Bottom: CSPs between the same samples with gray‐dashed line delineating the average plus 0.5 standard deviation (0.028 ppm). All data were collected at 900 MHz at 35°C
Data Availability Statement
Data is available upon request.