

Abstract
The Multiethnic Cohort Study has demonstrated that the risk for lung cancer in cigarette smokers among three ethnic groups is highest in Native Hawaiians, intermediate in Whites, and lowest in Japanese Americans. We hypothesized that differences in levels of DNA adducts in oral cells of cigarette smokers would be related to these differing risks of lung cancer. Therefore, we used liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry to quantify the acrolein-DNA adduct (8R/S)-3-(2’-deoxyribos-1’-yl)-5,6,7,8-tetrahydro-8-hydroxypyrimido[1,2-a]purine-10(3H)-one (ɣ-OH-Acr-dGuo, 1) and the lipid peroxidation-related DNA adduct 1,N6-etheno-dAdo (ɛdAdo, 2) in DNA obtained by oral rinse from 101 Native Hawaiians, 101 Whites, and 79 Japanese Americans. Levels of urinary biomarkers of nicotine, acrolein, acrylonitrile, and a mixture of crotonaldehyde, methyl vinyl ketone, and methacrolein were also quantified. Whites had significantly higher levels of ɣ-OH-Acr-dGuo than Japanese Americans and Native Hawaiians, after adjusting for age and sex. There was no significant difference in levels of this DNA adduct between Japanese Americans and Native Hawaiians, which is not consistent with the high lung cancer risk of Native Hawaiians. Levels of ɛdAdo were modestly higher in Whites and Native Hawaiians than Japanese Americans. The lower level of DNA adducts in the oral cells of Japanese American cigarette smokers than Whites is consistent with their lower risk for lung cancer. The higher levels of ɛdAdo, but not ɣ-OH-Acr-dGuo, in Native Hawaiian versus Japanese American cigarette smokers suggest that lipid peroxidation and related processes may be involved in their high risk for lung cancer, but further studies are required.
Graphical Abstract
INTRODUCTION
More than 900 million people in the world smoke cigarettes, the cause of about two thirds of all lung cancer, a disease which killed 1.8 million people worldwide in 2020 accounting for 18% of all cancer deaths.1, 2 The Multiethnic Cohort study (MEC), a prospective cohort study that recruited more than 215,000 men and women in 1993–1996, has investigated ethnic differences in lung cancer susceptibility among five ethnic groups: Native Hawaiians, Whites, Japanese Americans, African Americans, and Latinos. Results from the MEC have shown that Native Hawaiians and African Americans are at highest risk for lung cancer due to cigarette smoking, Whites are at intermediate risk, while Japanese Americans and Latinos are at lowest risk, when smoking dose is determined by cigarettes per day.3 In the study reported here, we investigated levels of DNA adducts in oral cells obtained from cigarette smokers recently recruited from 3 ethnic groups with differing risks for lung cancer: Native Hawaiians, Whites, and Japanese Americans.
There is no doubt that DNA adducts resulting from exposure to genotoxic compounds in cigarette smoke are central in the carcinogenic process. When certain DNA adducts persist and evade repair systems, they can cause miscoding in DNA leading to permanent mutations in oncogenes or tumor suppressor genes such as KRAS and TP53 involved in growth control.4–6 Thus, DNA adducts form the requisite link between carcinogen exposure and mutations in critical genes. In one study, somatic mutations were compared in over 5000 tumor samples from cigarette smokers and non-smokers, demonstrating clear increases in five mutational signatures in smokers compared to non-smokers.7
Analysis of oral cell DNA adducts is a practical approach to assessing DNA damage in cigarette smokers.8 Collection of oral mucosa cells is relatively simple, repeatable, and inexpensive. Some studies have demonstrated consistency between molecular changes in oral cells and bronchial cells.8 The oral mucosa is the first site of contact with cigarette smoke and it is reasonable to expect that DNA damage in oral cells would be similar to that observed in bronchial cells as the smoke and its constituents travel down the respiratory tract. Several studies have shown associations between molecular changes in the oral mucosa and bronchial cells from the same individuals, consistent with field carcinogenesis.8
We have recently described a quantitative liquid chromatography-nanoelectrospray ionization-high resolution tandem mass spectrometry (LC-NSI-HRMS/MS) method for the analysis of acrolein-DNA adducts and etheno-DNA adducts in human oral cell DNA.9 The results showed remarkably higher levels of these adducts in smokers than in non-smokers. Acrolein is one of the most toxic compounds in cigarette smoke, consistently generated at levels exceeding 100 μg per cigarette under Health Canada Intense conditions, and with a mean level of 177 μg per cigarette in brands from the U.S. market.10–12 The International Agency for Research on Cancer recently evaluated acrolein as “probably carcinogenic to humans” (Group 2A).12 This conclusion was based partially on sufficient evidence of cancer in experimental animals. Thus, exposure to acrolein by inhalation significantly increased the incidence of malignant lymphoma in female mice and caused rare rhabdomyoma and squamous cell carcinoma of the nasal cavity in female rats. These observations combined with strong mechanistic evidence, but inadequate evidence regarding cancer in humans, were the basis for the classification.12 With respect to lipid peroxidation, abundant evidence demonstrates the formation of ɛdAdo as well as other etheno-DNA adducts from peroxidation of lipids including 4-hydroxynonenal, in reactions that proceed via initial epoxidation followed by loss of the alkyl side chain.13–15 ɛdAdo can also be formed upon metabolism of vinyl chloride and ethyl carbamate,16 but levels of these compounds in cigarette smoke are quite low; vinyl choride (46 – 98 ng/cigarette, Canadian Intense conditions;17 ethyl carbamate, barely detectable).18
We have now applied our method to quantify the acrolein-DNA adduct (8R/S)-3-(2’-deoxyribos-1’-yl)-5,6,7,8-tetrahydro-8-hydroxypyrimido[1,2-a]purine-10(3H)-one (ɣ-OH-Acr-dGuo, 1) and the lipid peroxidation-related adduct 1,N6-etheno-dAdo (ɛdAdo, 2,Figure 1) in oral cell DNA obtained from Native Hawaiians, Whites, and Japanese American adult cigarette smokers living in Hawaii.
Figure 1.
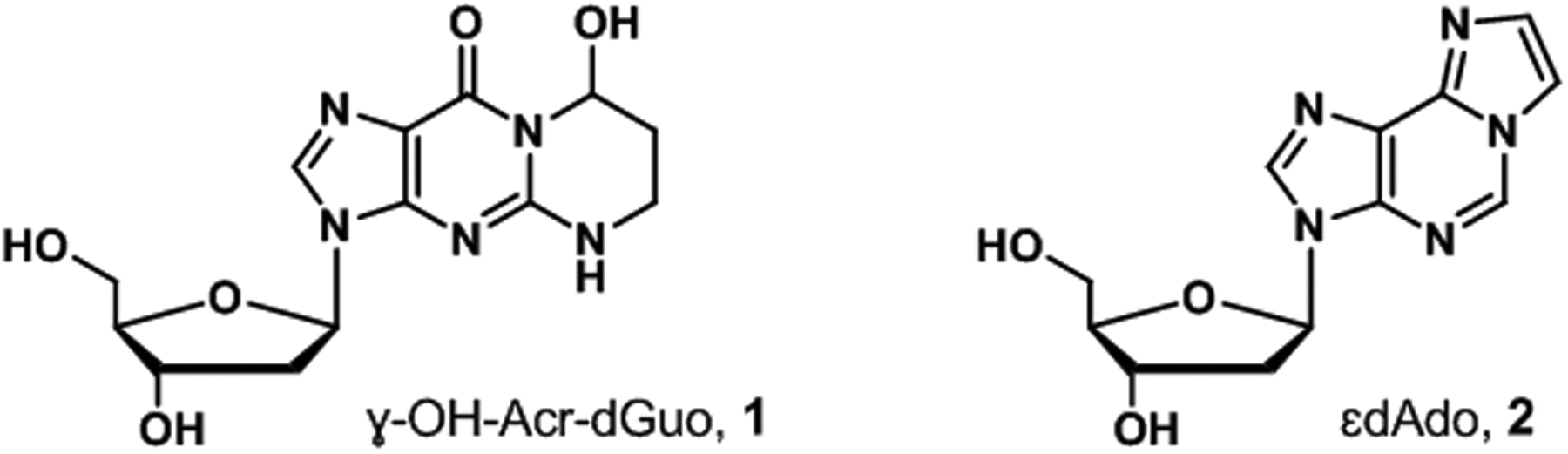
Structures of ɣ-OH-Acr-dGuo, 1 and ɛdAdo, 2.
EXPERIMENTAL SECTION
Chemicals and Enzymes.
ɣ-OH-Acr-dGuo and [13C1015N5]ɣ-OH-Acr-dGuo were prepared by reaction of acrolein with dGuo or fully labeled dGuo (Toronto Research Chemicals). The resulting products were characterized and quantified by 1H NMR, using toluene as an internal standard, as previously described;19 εdAdo and [13C5]εdAdo (labeled with 13C in the five carbons of the deoxyribose ring) were purchased from Toronto Research Chemicals (North York, Ontario, Canada). Reagents for DNA isolation were obtained from Qiagen (Germantown, MD). Calf thymus DNA, micrococcal nuclease and phosphodiesterase II were purchased from Worthington Biochemical Co. (Lakewood, NJ). Alkaline phosphatase and other solvents were obtained from Sigma-Aldrich Chemical Co. (Milwaukee, WI).
Study Design and Oral Cell Collection.
Smokers were recruited through advertisements among residents of the island of Oahu, Hawaii. Inclusion criteria included: self-reported race/ethnicity (Native Hawaiians, Japanese Americans and Whites); smoking of more than five cigarettes per day over the past three months; fewer than 14 drinks of alcohol per week; and generally stable and good health. Exclusion criteria included: current use of other nicotine containing products > 1 time per month and no use of any nicotine-containing products except cigarettes for 1 week prior to the study; currently taking any medications affecting relevant metabolic enzymes or anti-inflammatory medications such as ibuprofen.
Native Hawaiians included individuals with at least one parent of Hawaiian descent; Japanese Americans had two parents of Japanese descent (or at least 3 grandparents of Japanese descent), and Whites had two parents of non-Hispanic white descent (or at least 3 grandparents of non-Hispanic White descent). Some of the subjects may have had admixture beyond these specific criteria reflecting current trends in Hawaii.
Participants provided a first morning void urine sample, and blood was collected at the visit. They were asked to brush their teeth and not to eat, drink, smoke or chew gum for 20 min prior to obtaining oral cells. They were then provided with a 15 mL mono-dose of saline and asked to swish the oral rinse throughout the mouth for 45 sec. Samples were processed and stored at −80 °C.
DNA Isolation from Oral Cells.
This was carried out essentially as described.9 DNA isolation used the DNA purification from oral rinse protocol from Qiagen (Qiagen, Valencia, CA) with modifications. Briefly, samples were thawed and centrifuged at 15,000g for 2 min to pellet the oral cells. The supernatant was discarded and cells were lysed using 1000 μL of cell lysis solution containing 1 mM glutathione (GSH) and incubated for 20 min at room temperature. This was followed by a 15 min treatment at room temperature with 10 μL of proteinase K, and a 30 min treatment at 37 °C with 6 μL of RNase . Then, 340 μL of protein precipitation solution was added and the samples were centrifuged at 16,000 g for 10 min. DNA was precipitated by pouring the oral rinse supernatant into a 5 mL tube containing 1460 μL of 100% ice-cold 2-propanol. The precipitated oral rinse DNA pellet was washed with 1000 μL of 70% ice-cold 2-propanol and then 1000 μL of 100% ice-cold 2-propanol. DNA was dried and stored at −20 °C until analysis.
DNA Hydrolysis and Sample Purification.
This was performed essentially as described previously.9 The isolated DNA (0.5 – 50 μg) was dissolved in 200 μL of 10 mM sodium succinate/5 mM CaCl2 buffer (pH 7.0) containing 5mM GSH to which 10 fmol of [13C1015N5]Acr-dGuo ([13C1015N5]1) and [13C5]εdAdo ([13C5]2) were added as internal standards. A buffer blank (200 μL) was prepared each time and used as a negative control to evaluate possible contamination; a sample of calf thymus DNA (20 μg in 200 μL of buffer) was also included in each set of samples as a positive control. The mixture was heated at 100 °C for 30 min and cooled to room temperature. Then, the DNA was enzymatically hydrolyzed by incubation with 7.5 units of micrococcal nuclease and 0.045 units of phosphodiesterase II at 37 °C overnight. Then, 15 units of alkaline phosphatase (from calf intestine) were added, and the mixture was incubated at 37 °C overnight. The resulting mixtures were partially purified by centrifugal filtration (Ultracel 10K, Millipore). A 10 μL aliquot was removed for dGuo quantitation, and the remaining hydrolysate was purified using a solid-phase extraction (SPE) cartridge [Strata-X, 33 μm, 30 mg/1 mL (Phenomenex, Torrance, CA)] activated with 3 mL of MeOH and 3 mL of H2O containing 0.1 mM GSH. After the samples were applied, the cartridges were washed with 6 mL of H2O containing 0.1 mM GSH and 1 mL 5% CH3OH in H2O containing 0.1mM GSH, and the analytes were eluted into high recovery glass vials (Thermo Scientific, San Jose, CA) with 1 mL 35% CH3OH in H2O containing 0.1 mM GSH. Prior to elution, 0.65 μL of 100 mM GSH was added in each vial. The eluants were evaporated to dryness and stored at 4 °C.
Analysis of ɣ-OH-Acr-dGuo (1) and ɛdAdo (2).
Samples were dissolved in 20 μL of H2O for LC-NSI-HRMS/MS analysis. The analysis was carried out on an Orbitrap Fusion Lumos instrument (Thermo Scientific, San Jose, CA) interfaced with a UPLC system (Ultimate 3000 RSLCnano UPLC, Thermo Scientific, Waltham, MA) using nanoelectrospray ionization. The UPLC was equipped with a 5 μL loop and the separation was performed using a capillary column (75 μm ID, 20 cm length, 15 μm orifice) prepared by hand packing a commercially available fused-silica emitter (New Objective, Woburn MA) with Luna C18 5μ bonded separation media (Phenomenex, Torrance, CA). The mobile phase consisted of 2 mM NH4OAc and CH3CN. The gradient started at 1% CH3CN for 6 min at a flow rate of 0.9 μL/min, increased to 13% CH3CN in 20 min at a flow rate of 0.3 μL/min and then to 30% CH3OH in 1 min, holding at this composition for 4 min. The gradient was then returned to 1% CH3CN in 1 min and the system was re-equilibrated at this mobile phase composition for 1 min at a flow rate of 0.9 μL/min before the next injection. The source temperature was set at 300 °C and the spray voltage was static at 2200 V. The maximum injection time was 400 ms and the normalized automatic gain control (AGC) was set at 50%. The precursor ions were isolated by the quadrupole with an isolation width of m/z 1.5 and fragmented by higher energy collisional dissociation (HCD) at 20% for ɣ-OH-Acr-dGuo (1), [13C1015N5]ɣ-OH-Acr-dGuo ([13C1015N5]1), εdAdo (2) and [13C5]εdAdo ([13C5]2), and the product ions were detected by the Orbitrap detector at a resolution of 60,000. For quantitation of adduct levels, the ion transitions monitored with accurate mass extracted were: m/z 324.1 → m/z 208.083 ([M + H]+ → [BH]+) for adduct 1, m/z 339.1 → m/z 218.085 for [13C1015N5]1, m/z 276.1 → m/z 160.062 for adduct 2, and m/z 281.1 → m/z 160.062 for [13C5]2. The ion transitions m/z 324.1 → m/z 164.057 for ɣ-OH-Acr-dGuo and m/z 339.1 → m/z 174.0586 for [13C1015N5]ɣ-OH-Acr-dGuo were also monitored as qualifying ions.
Calibration curves (reported in the Supporting Information, Figure S1) were prepared using standard solutions of adducts 1, [13C1015N5]1, and 2 and [13C5]2. A constant amount of [13C1015N5]1 and [13C5]2 (200 amol/μL) was mixed with different amounts of adducts 1 and 2 (5–500 amol/μL). Internal standard [13C1015N5]1 was prepared by reaction of [13C1015N5]dGuo with acrolein, and therefore also contained the [13C1015N5]α-Acr-dGuo adducts (see Figure 2F).
Figure 2.
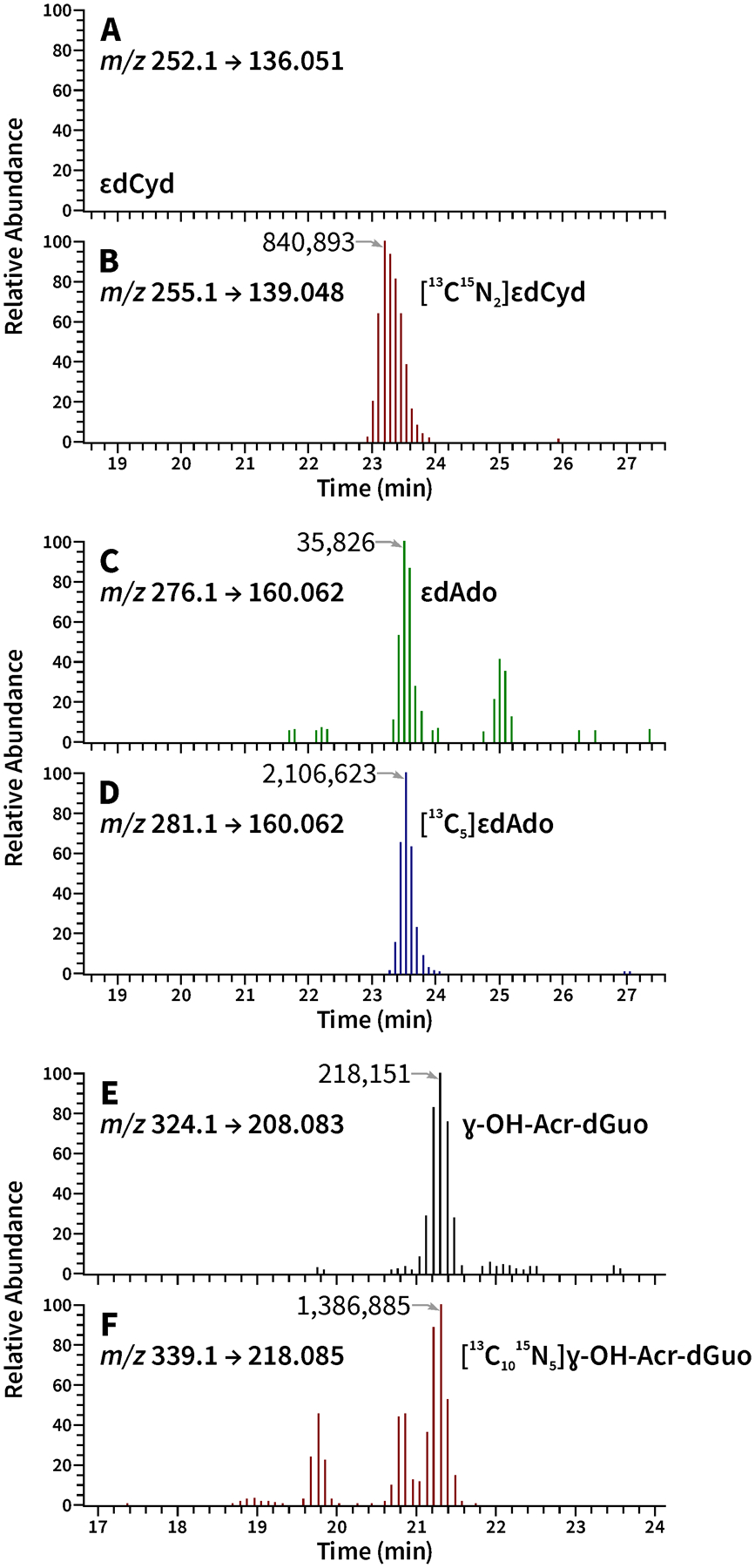
Representative LC-NSI-HRMS/MS chromatograms obtained upon analysis of human oral cell DNA from an oral rinse sample. SRM was carried out at (A) m/z 252.1→ 136.051 for ɛdCyd, (B) m/z 255.1 → 139.048 for [13C15N2]ɛdCyd , (C) m/z 276.1 → 160.062 for ɛdAdo, (D) m/z 281.1 → 160.062 for [13C5]ɛdAdo, (E) m/z 324.1 → 208.083 for ɣ-OH-Acr-dGuo, (F) m/z 339.1 → 218.085 for [13C1015N5]ɣ-OH-Acr-dGuo. This internal standard also includes peaks for [13C1015N5]α-OH-Acr-dGuo, retention times 19.8 and 20.8 min.
Concentrations of adducts in DNA were expressed as adducts per 109 nucleotides. Analysis of the adducts in calf thymus DNA, 2 samples per assay run, was used as the positive control. Limits of detection and quantitation for adducts 1 and 2 were previously established and reported,9 An example of traces corresponding to the limit of quantitation is reported in the Supporting Information (Figure. S2)
Analysis of dGuo.
Quantitation of dGuo was similar as that reported previously,20 and was performed using a TSQ Vantage triple-quadrupole mass spectrometer (Thermo Scientific, Waltham, MA) interfaced with a Dionex Ultimate 3000 UHPLC system (NCS-3500RS pump and WPS-3000PL autosampler). Analysis was performed on a Luna C18 column (5 μm, 150 × 0.5 mm, Phenomenex) at a flow rate of 10 μl/min at room temperature. Sample injection volume was 4 μl. The mobile phase consisted of 5 mM NH4OAc and CH3CN with a linear gradient from 3 to 40% CH3CN over 20 min, followed by ramping to 90% CH3CN within 1 min and holding at this composition for 4 min. The gradient was then returned to 3% CH3CN in 1 min followed by 10 min re-equilibration. The ESI source was operated in positive ion mode, monitoring m/z 268.1 [M+H]+ → m/z 152.1 [C5H6N5O]+ for dGuo and the corresponding transition m/z 283.1 → 162.1 for [13C1015N5]dGuo. The collision gas was Ar at 1 mTorr with a collision energy of 15 eV. The S-lense was set at 85. The quadrupoles Q1 and Q2 were both operated at a resolution of 0.7 Da.
Urinary biomarker analysis.
The following biomarkers of exposure were quantified as previously described: total nicotine equivalents (TNE);21 3-hydroxypropyl mercapturic acid (3-HPMA, biomarker of acrolein);22 cyanoethyl mercapturic acid (CEMA, biomarker of acrylonitrile),23 hydroxymethylpropyl mercapturic acids22 (HMPMA, combined biomarker of crotonaldehyde, methyl vinyl ketone, and methacrolein).24
Genotyping.
DNA was extracted from blood leukocytes by lysing any red blood cells present in the sample with Qiagen RBC lysis solution prior to using a QIAmp Blood Mini Extraction kit (Qiagen, Germantown, MD). GSTT1 and GSTM1 gene deletions were genotyped for each sample using a predesigned TaqMan GSTT1 copy number assay (Hs00010004_cn) and run on the 7900HT Fast Real-Time System (Life Technologies, Foster City, CA). Copy number counts were calculated using Life Technologies CopyCaller v2.0 software.
Statistical Analysis.
Outliers, defined as those in the bottom (ɛdAdo <4 and ɣ-OH-Acr-dGuo <13) and top 1% (ɛdAdo > 823 and ɣ-OH-Acr-dGuo > 4880) of the distribution, were removed from the analysis. Pearson’s partial correlation for TNE, CEMA, HMPMA and 3-HPMA for each of the DNA adducts (ɛdAdo and ɣ-OH-Acr-dGuo) were calculated adjusting for age at biospecimen collection and sex. All urinary biomarkers and DNA adducts were transformed by taking the natural log to better meet model assumptions. Multivariable linear models regressed the urinary smoking biomarkers with the DNA adducts on the following predictors: age at time of biospecimen collection (continuous), sex (when results were not stratified by sex), and race/ethnicity (when results were not stratified by race/ethnicity) (Model A). To examine racial/ethnic differences, covariate-adjusted geometric means were computed for each racial/ethnic group at the mean covariate vector. We further adjusted for TNE and 3-HPMA to evaluate whether the associations remained independent of smoking dose and the metabolite of acrolein, respectively. We also examined whether the geometric means of these DNA adducts differed by the GSTT1 and GSTM1 polymorphism. The GST copy number polymorphisms were modeled by category of number of gene copies (i.e. 2 or 1 copies or 0 copy) and adjusted for the previously mentioned predictors.
RESULTS
Table 1 presents demographics and levels of selected urinary biomarkers for the 281 participants (101 Whites, 79 Japanese Americans and 101 Native Hawaiians) for whom ɣ-OH-Acr-dGuo and ɛdAdo were measured. Whites had the highest unadjusted mean TNE levels (73 ± 52 nmol/ml), followed by Native Hawaiians (60 ± 43 nmol/ml) and Japanese Americans (50 ± 37 nmol/ml). This pattern remained after adjusting for age and sex, and was not globally statistically significant (p=0.25) (Table S1, Supporting Information). This same racial/ethnic pattern was observed for CEMA, a urinary biomarker of combusted tobacco product use, independent of TNE (Table S1, Supporting Information).
Table 1.
Descriptive statistics for sex, age, urinary biomarkers, DNA adducts, and GST status.
| Overall | Whites | Japanese Americans | Native Hawaiians | |||||
|---|---|---|---|---|---|---|---|---|
| N | 281 | 101 | 79 | 101 | ||||
| Sex (M/F): N | 153/128 | 52/49 | 53/26 | 48/53 | ||||
| Age (years): Mean [sd] | 50.1 | 17.1 | 47.5 | 15.7 | 59.2 | 14.6 | 45.5 | 17.7 |
| TNE (nmol/mL): Mean [sd] | 61.97 | 45.67 | 72.63 | 52.31 | 50.35 | 37.01 | 60.41 | 42.58 |
| CEMA (pmol/mL): Mean [sd] | 757.1 | 701.9 | 864.1 | 790.6 | 599.7 | 581.3 | 773.2 | 677.8 |
| HMPMA (pmol/mL): Mean [sd] | 4527 | 4952 | 5213 | 7014 | 4024 | 3157 | 4235 | 3309 |
| 3-HPMA (pmol/mL): Mean [sd] | 7077 | 6764 | 7324 | 7809 | 7027 | 6966 | 6868 | 5400 |
| ɣ-OH-Acr-dGuo (per 109 nucleotides): Mean [sd] | 84.0 | 92.9 | 111 | 131 | 70.8 | 62.2 | 66.9 | 52.6 |
| ɛdAdo (per 109 nucleotides): Mean [sd] | 13.8 | 11.8 | 14.0 | 10.5 | 13.9 | 14.8 | 13.5 | 10.4 |
| N | % | N | % | N | % | N | % | |
| GSTM1 – frequency | ||||||||
| 0 | 159 | 57.0 | 57 | 57.0 | 39 | 49.4 | 63 | 63.0 |
| 1 | 108 | 38.7 | 37 | 37.0 | 38 | 48.1 | 33 | 33.0 |
| 2 | 12 | 4.30 | 6 | 6.00 | 2 | 2.53 | 4 | 4.00 |
| Missing | 2 | 1 | 0 | 1 | ||||
| GSTT1 – frequency | ||||||||
| 0 | 78 | 28.1 | 17 | 17.0 | 37 | 46.8 | 24 | 24.2 |
| 1 | 139 | 50.0 | 58 | 58.0 | 34 | 43.0 | 47 | 47.5 |
| 2 | 61 | 21.9 | 25 | 25.0 | 8 | 10.1 | 28 | 28.3 |
| Missing | 3 | 1 | 0 | 2 | ||||
Most oral rinse collections provided 0.5–50 μg DNA. Typical LC-NSI-HRMS/MS chromatograms obtained upon analysis of oral rinse DNA from a subject in this study are illustrated in Figure 2. Clear and reproducible chromatograms were obtained for ɛdAdo (Figure 2C,D) and ɣ-OH-Acr-dGuo (Figure 2E,F) and their internal standards, and these were used for quantitation of adduct levels. In Figure 2F, the internal standard [13C1015N5]ɣ-OH-Acr-dGuo (retention time 21.3 min) also includes both of the α-OH-Acr-dGuo isomers (retention times 19.8 and 20.8 min). In addition to the traces shown in Figure 2E,F, we also monitored the characteristic product ion transitions m/z 324.1 → 164.0567 for ɣ-OH-Acr-dGuo and m/z 339.1 → 174.05863 for [13C1015N5]ɣ-OH-Acr-dGuo (retention times 21.3 min), as qualifying ions, which were clearly observed in each subject sample. The LC-NSI-HRMS/MS response for the α-OH-Acr-dGuo isomers (Figure 2E, F) and ɛdCyd (Figure 2A,B) in the subject samples were either below the detection limit or too low to quantify in most chromatograms. The relatively low abundance of α-OH-Acr-dGuo compared to ɣ-OH-Acr-dGuo is consistent with the results of our previous study of these DNA adducts in human oral cells from cigarette smokers.9 Two samples of calf thymus DNA were included as positive controls in each batch; the overall coefficients of variation for ɣ-OH-Acr-dGuo and ɛdAdo in these samples were 18.8% (N=26) and 10.0% (N=26), respectively.
Correlations of urinary biomarkers with DNA adduct levels are presented in Table S2, Supporting Information. When considering all subjects, each urinary biomarker was significantly correlated with levels of ɣ-OH-Acr-dGuo (P<0.0001) but not with ɛdAdo, indicating that the latter is not a biomarker of exposure. Stratifying by ethnicity, the correlations with ɣ-OH-Acr-dGuo remained highly significant for all biomarkers in Whites, and for all except 3-HPMA in Japanese Americans, but only for CEMA in Native Hawaiians.
The results of the LC-NSI-HRMS/MS analyses of 281 oral rinse DNA samples for ɣ-OH-Acr-dGuo and ɛdAdo are presented in Tables 2A, B and 3. The overall geometric mean levels of ɣ-OH-Acr-dGuo were 53.7 adducts/109 nucleotides, while for ɛdAdo the corresponding figure was 10.5 adducts/109 nucleotides (Table 2A). Men had significantly higher levels of ɛdAdo than women when adjusted for age, sex, TNE and 3-HPMA (p=0.007, Table 3). Although TNE was accounted for, to confirm that this difference was not a result of residual confounding by smoking intensity, as men smoke more than women, the geometric means for ɛdAdo per TNE were also examined (Table S3, Supporting Information). The difference across sex remained (p’s=0.02–0.0005).
Table 2.
Geometric means for ɣ-OH-Acr-dGuo and ɛdAdo , overall and by race/ethnicity
| Part A. Adduct amounts | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DNA Adduct | Overall | Whites | Japanese Americans | Native Hawaiians | |||||||||
| N | Geometric means (95% CI) |
N | Geometric means (95% CI) |
N | Geometric means (95% CI) |
N | Geometric means (95% CI) |
||||||
| ɣ-OH-Acr-dGuo, per 109 nucleotidesa | 281 | 53.7 | (48.0–60.1) | 101 | 67.1 | (55.7–80.7) | 79 | 46.5 | (37.2–58.0) | 101 | 49.7 | (41.2–59.9) | 0.0204 |
| ɣ-OH-Acr-dGuo, per 109 nucleotidesb | 281 | 53.8 | (48.7–59.4) | 101 | 64.4 | (54.5–76.2) | 79 | 47.9 | (39.3–58.4) | 101 | 50.5 | (42.7–59.6) | 0.0433 |
| εdAdo, per 109 nucleotidesa | 281 | 10.5 | (9.75–11.4) | 101 | 11.3 | (9.94–12.9) | 79 | 9.16 | (7.83–10.7) | 101 | 11.3 | (9.90–12.9) | 0.0854 |
| εdAdo, per 109 nucleotidesb | 281 | 10.6 | (9.76–11.4) | 101 | 11.3 | (9.87–12.8) | 79 | 9.22 | (7.89–10.8) | 101 | 11.3 | (9.94–12.9) | 0.101 |
| Part B. p-Values comparing groups | |||
|---|---|---|---|
| DNA Adduct | Whites>Japanese Americans | Whites>Native Hawaiians | Native Hawaiians>Japanese Americans |
| ɣ-OH-Acr-dGuoa | 0.0139 | 0.0246 | 0.656 |
| ɣ-OH-Acr-dGuob | 0.0272 | 0.0417 | 0.697 |
| εdAdoa | 0.0434 | 0.973 | 0.0505 |
| εdAdob | 0.0591 | 0.938 | 0.0520 |
Model adjusted for age, sex, (and race/ethnicity)
Model adjusted for age, sex, log-TNE, log-3HPMA, (and race/ethnicity)
presents the global p-value across race/ethnicity
Table 3.
Geometric means for ɣ-OH-Acr-dGuo and ɛdAdo , overall and by sex
| DNA Adduct | Men | Women | |||||
|---|---|---|---|---|---|---|---|
| N | Geometric means (95% CI) |
N | Geometric means (95% CI) |
||||
| ɣ-OH-Acr-dGuo (per 109 nucleotides)a | 153 | 55.5 | (47.8 – 64.5) | 128 | 52.0 | (44.0 – 61.4) | 0.566 |
| ɣ-OH-Acr-dGuo (per 109 nucleotides)b | 153 | 56.0 | (49.0 – 64.1) | 128 | 51.7 | (44.5 – 60.0) | 0.434 |
| ɛdAdo (per 109 nucleotides)a | 153 | 11.8 | (10.6 – 13.1) | 128 | 9.46 | (8.40 – 10.6) | 0.00748 |
| ɛdAdo (per 109 nucleotides)b | 153 | 11.8 | (10.6 – 13.1) | 128 | 9.46 | (8.41 – 10.6) | 0.00753 |
Model adjusted for age, sex, (and race/ethnicity)
Model adjusted for age, sex, log-TNE, log-3HPMA, (and race/ethnicity)
By race/ethnicity, Whites (67.1 adducts/109 nucleotides) had significantly higher levels of ɣ-OH-Acr-dGuo than Japanese Americans (46.5 adducts/109 nucleotides; p=0.01) and Native Hawaiians (49.7 adducts/109 nucleotides; p= 0.02), after adjusting for age and sex. There was no significant difference between Japanese Americans and Native Hawaiians (Table 2B). This pattern remained even after accounting for TNE and 3-HPMA (Model b in Table 2A,B). The geometric mean levels of ɛdAdo were modestly higher in Whites (11.3 adducts/109 nucleotides for both Models a and b) and Native Hawaiians (11.3 adducts/109 nucleotides), than Japanese Americans (9.16–9.22 adducts/109 nucleotides) for both Models a and b; p’s=0.04–0.06; (Table 2A,B). No difference in levels of ɛdAdo between Whites and Native Hawaiians was detected.
There were no significant effects of GSTM1 or GSTT1 copy numbers on ɣ-OH-Acr-dGuo levels (Table S4, Supporting Information). However ɛdAdo adducts were almost 20% lower in smokers who were GSTM1 null (p = 0.0082). This effect appears to be driven predominantly by the data for Japanese Americans.
DISCUSSION
This is the largest study published to date quantifying Acr-dGuo adducts in oral cells using MS-based methods. There has been only one previously reported study of this size, conducted by Tsou et al using a slot blot assay.25 Similarly, no previous study of this size has quantified ɛdAdo in human DNA.
Using the LC-NSI-HRMS/MS method employed here, we previously demonstrated that levels of ɣ-OH-Acr-dGuo were 27 times higher in oral cells obtained by oral rinse from cigarette smokers compared to non-smokers, consistent with the strong relationship of this DNA adduct to acrolein exposure from cigarette smoking, which delivers more than 100 μg of acrolein per cigarette.9, 10 The results of the present study, the first to investigate ethnic differences in oral cell DNA adduct levels among cigarette smokers, clearly demonstrate that levels of ɣ-OH-Acr-dGuo were significantly lower in oral cells of Japanese American than White smokers. This is consistent with the lower risk of Japanese American than White smokers for lung cancer when smoking dose is determined by cigarettes per day.26 The lower risk of Japanese American smokers can be explained by the preponderance of low activity forms of the highly polymorphic and primary nicotine metabolizing enzyme CYP2A6 in Japanese Americans, leading to less efficient nicotine metabolism and consequent lower need for intense smoking, as presented in two recent reviews.27, 28 Consistent with this proposal, when TNE, a robust measure of nicotine dose, is included in a lung cancer risk model for cigarette smokers, the risk of Japanese Americans is no longer lower than that of Whites.3 We note however that in the present study the levels of ɣ-OH-Acr-dGuo remained significantly lower in Japanese Americans even after correction for TNE and 3-HPMA (Table 2B). One possible explanation for this is that ɣ-OH-Acr-dGuo is a superior biomarker of smoking dose than urinary TNE or acrolein dose as measured by 3-HPMA, the latter also being affected by endogenous factors. Reasons for the potential superiority of ɣ-OH-Acr-dGuo as a biomarker of smoking dose include the following: the oral mucosa is the first site of exposure to cigarette smoke; there is no metabolism involved in formation of ɣ-OH-Acr-dGuo; and a steady state and persistent level of DNA adducts will result from continued smoking. In contrast, urinary TNE or 3-HPMA may be affected by tissue distribution, rates of excretion, daily fluctuation in extents of smoking, and other factors. Thus, oral cell DNA, which includes bacterial DNA, is acting as an effective in vivo endogenous trap for highly reactive acrolein. Additional unidentified factors such as differences in DNA repair or oral mucosa protein or amino acid concentrations may also contribute to the significantly lower levels of ɣ-OH-Acr-dGuo in oral cells of Japanese American smokers than in Whites.29 We also note that our earlier studies did not find differences in acrolein-DNA adduct levels in DNA from lung tissue or leukocytes of smokers vs. non-smokers, so further studies are required to fully understand the details of mechanisms pertaining to the possible role of acrolein in lung cancer etiology in cigarette smokers.19, 30
While levels of acrolein-DNA adducts in oral cells were significantly lower in Japanese Americans than in Whites, consistent with their differing risks for lung cancer, levels in Native Hawaiians, who have a higher risk for lung cancer than either Whites or Japanese Americans, were lower than in Whites and not significantly different from those in Japanese Americans. Similar results have been obtained in comparisons of TNE in Native Hawaiians with the other groups.21 As in this study, their levels of TNE were intermediate between those of Whites and Japanese Americans, indicating that factors other than extents of carcinogen and toxicant exposure (as represented by TNE) may be involved in the relatively high risk of Native Hawaiian cigarette smokers for lung cancer. The present results demonstrating similar and high levels of ɛdAdo in both Whites and Native Hawaiians compared to Japanese Americans suggest that oxidative stress, inflammation, lipid peroxidation and perhaps other endogenous processes leading to ɛdAdo DNA adducts may be important in the relatively high risk of Native Hawaiian cigarette smokers for lung cancer.31 Previous studies demonstrated that ɛdAdo is a product of lipid peroxidation, being formed when 2,3-epoxy-4-hydroxynonanal reacts with dAdo in DNA.13–15 We also note that the correlations of ɣ-OH-Acr-dGuo with urinary mercapturic acid exposure biomarkers were weaker in Native Hawaiians than in Whites or Japanese Americans (Table S2, Supporting Information), suggesting that inflammation and other endogenous processes may be relatively important in determining levels of ɣ-OH-Acr-dGuo in Native Hawaiians. Overall, the combination of genotoxic DNA damage as represented by ɣ-OH-Acr-dGuo and downstream effects as represented by ɛdAdo recapitulates the initiation/promotion model of tobacco carcinogenesis that has been well established in studies with laboratory animals.9, 32
Neither GSTT1 nor GSTM1 copy number affected the level of the acrolein-DNA adducts in oral cells. These data are consistent with the detoxification of acrolein by glutathione occurring predominately by a non-enzymatic mechanism.33 Consistent with this assumption was the lack of a relationship of the GSTT1 locus with the mercapturic acid of acrolein in our GWAS study in the MEC.22 This was in contrast to the level of the mercapturic acid metabolite of benzene, S-phenyl mercapturic acid, in the same study.34 It is unclear what drives the lower level of ɛdAdo adducts in individuals who are GSTM1 null. GSTM1 is not the only or most efficient GST involved in the detoxification of lipid peroxidation products.35–37 GSH is a critical molecule in the detoxification of many toxicants, including lipid peroxidation products, and its levels and the activity of GSTs and glutathione peroxidases influence these pathways. The interplay of the regulation of these pathways is complicated and their influence on the level of ɛdAdo adducts is unclear.
Previous studies of acrolein-DNA adducts in oral cells have recently been reviewed.12 Weng et al used an immunochemical method to measure ɣ-OH-Acr-dGuo and reported levels ranging from 0.2–2.5 adducts/105 dGuo, with significantly higher levels in smokers than non-smokers.38 These levels are considerably higher than those reported by MS-based methods. Nath and Chung used a 32P-postlabelling method to quantify ɣ-OH-Acr-dGuo in gingival tissue and reported levels of 1.36 ± 0.9 μmol/mol G in smokers (N=11) and 0.46 ± 0.26 μmol/mol G in non-smokers (N=12), a significant difference.39 Bessette et al used a data-dependent constant neutral loss MS3 method and estimated levels of ɣ-OH-Acr-dGuo and α-OH-Acr-dGuo to be above 5 adducts per 107 DNA bases in 6 smokers.40 Chen and Lin used LC-NSI-MS/MS and reported mean adduct levels of 104 ± 50 per 108 nucleotides in human salivary DNA.41 Wang et al estimated Acr-dGuo adducts using an immunoassay which was applied to oral cell DNA from subjects after they consumed commercial fried food. They did not report DNA adduct levels, only relative differences.42 Similarly, Tsou et al used a slot blot assay to investigate levels of Acr-dGuo adducts in buccal cells of patients with oral squamous cell carcinoma and in healthy subjects with a history of cigarette smoking and/or betel quid chewing, but reported only relative levels which were 1.4 fold higher in patients with oral cancer than in healthy subjects.25 None of these studies reported using glutathione to prevent artifact formation, shown by Chung and co-workers to be a potential problem in analysis of Acr-dGuo adducts.43 Our recent study using artifact protection with glutathione showed significantly higher levels of ɣ-OH-Acr-dGuo and α-OH-Acr-dGuo (27 times and 6.5 times higher, respectively) in oral cells of smokers than in non-smokers, consistent with previous studies, although the reported levels in smokers (163 ± 227 and 12.1 ± 17.9 adducts per 109 nucleotides , respectively) are lower than in previous studies.9
The U.S. Food and Drug Administration has classified biomarkers into two main groups: biomarkers of exposure (BOE) and biomarkers of potential harm (BOPH). The available data clearly indicate that acrolein DNA adducts in oral cell DNA are BOE, since they are consistently higher in smokers than non-smokers. But are they also BOPH? This is plausible because DNA adducts are on the pathway to induction of mutations in critical genes, leading to cancer.44 The IARC review concluded that “there is consistent and coherent evidence that acrolein exhibits key characteristics of carcinogens.”12 . In support of this, they cite, in addition to multiple studies demonstrating increased exposure to acrolein in smokers, the preferential formation of acrolein-DNA adducts at lung cancer TP53 mutational hot spots, its genotoxicity, its ability to induce oxidative stress and alter DNA repair, as well as other factors. Therefore, ɣ-OH-Acr-dGuo has the potential to be a BOPH, but further studies in which these adducts are incorporated into molecular epidemiology investigations are required.
Taken together with previous studies from our program on CYP2A6 and nicotine metabolism,27, 28 our data provide further mechanistic support for the lower lung cancer risk of Japanese Americans than Whites or Native Hawaiians by demonstrating significant differences in DNA damage in oral cells of these ethnic groups. But there are limitations to our study. The data do not provide a clear rationale for the higher lung cancer risk of Native Hawaiians than Whites, as observed in the MEC.3, 26 Furthermore, the relationship of oral cell DNA damage to corresponding effects in the lung are unclear, and further studies are required to determine whether lower levels of ɣ-OH-Acr-dGuo and ɛdAdo would also be seen in lung DNA of Japanese American versus White and Native Hawaiian smokers. These studies are challenging because, in contrast to oral cell DNA, bronchial brushings and sputum can be difficult to obtain.
In summary, in one of the largest DNA adduct studies yet performed by mass spectrometric methods, we report the first data on ethnic differences in levels of oral cell DNA adducts resulting from exposure to acrolein and lipid peroxidation due to cigarette smoking. The data demonstrate significantly higher levels of ɣ-OH-Acr-dGuo in oral cell DNA of Whites compared to Japanese American and Native Hawaiian smokers while levels of ɛdAdo were lowest in Japanese Americans among the three groups. These results are consistent with the relatively low risk for lung cancer of Japanese American cigarette smokers. The results also add to growing evidence that factors other than direct carcinogen exposure are important in the relatively high risk of Native Hawaiian smokers for lung cancer.
Supplementary Material
Figure S1. Calibration curves used for the quantitation of (A) ɣ-OH-Acr-dGuo and (B) ɛdAdo.
Figure S2. Example of a chromatogram showing traces corresponding to amounts of ɛdAdo and ɣ-OH-Acr-dGuo (3.1 and 5.7 per 109 nucleotides, respectively) close to the assay limit of quantitation.9
Table S1. Geometric means for urinary biomarkers of total nicotine equivalents (TNE) and mercapturic acids
Table S2. Pearson’s correlation coefficient – partial correlations adjusted for age and sex
Table S3. Geometric means for ɣ-OH-Acr-dGuo and ɛdAdo, per TNE, by sex
Table S4. Relationship of GSTM1 and GSTT1 status to biomarkers
ACKNOWLEDGMENTS.
We thank Viviana Paiano for her contributions to the isolation of DNA and to the method development for analysis of DNA adducts in this study, and Naomi Hee, Wileen Mau and Shelle Santoki for assistance with data and biospecimen collection.
Funding
This study was funded by Grant P01-CA-138338 from the National Cancer Institute. Mass spectrometry was carried out in the Analytical Biochemistry Shared Resource of the Masonic Cancer Center, supported in part by Cancer Center Support Grant CA-077598. Salary support for the Head of the Shared Resource, Dr. Peter Villalta, was provided by National Cancer Institute Grant R50-CA-211256.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/journal/crtoec
The authors declare no competing financial interest.
REFERENCES
- (1).GBD 2015 Tobacco Collaborators. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: A systematic analysis from the Global Burden of Disease Study 2015. Lancet 2017, 389 (10082), 1885–1906. DOI: 10.1016/S0140-6736(17)30819-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sung H; Ferlay J; Siegel RL; Laversanne M; Soerjomataram I; Jemal A; Bray F Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin 2021, 71 (3), 209–249. DOI: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- (3).Stram DO; Park SL; Haiman CA; Murphy SE; Patel Y; Hecht SS; Le Marchand L Racial/ethnic differences in lung cancer incidence in the multiethnic cohort study: An update. J. Natl. Cancer Inst 2019, 111, 811–819. DOI: 10.1093/jnci/djy206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hecht SS Lung carcinogenesis by tobacco smoke. Int J. Cancer 2012, 131 (12), 2724–2732. DOI: 10.1002/ijc.27816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Phillips DH; Venitt S DNA and protein adducts in human tissues resulting from exposure to tobacco smoke. Int J. Cancer 2012, 131 (12), 2733–2753. [DOI] [PubMed] [Google Scholar]
- (6).Stewart BW Mechanisms of Carcinogenesis: From Initiation and Promotion to the Hallmarks. In IARC Scientific Publication No. 165, Tumour Site Concordance and Mechanisms of Carcinogenesis, Baan RA, Stewart BW, Straif K Eds.; IARC, 2019; pp 93–106. [PubMed] [Google Scholar]
- (7).Alexandrov LB; Ju YS; Haase K; Van Loo P; Martincorena I; Nik-Zainal S; Totoki Y; Fujimoto A; Nakagawa H; Shibata T; Campbell PJ; Vineis P; Phillips DH; Stratton MR Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354 (6312), 618–622. DOI: 10.1126/science.aag0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hecht SS Oral cell DNA adducts as potential biomarkers for lung cancer susceptibility in cigarette smokers. Chem. Res. Toxicol 2017, 30 (1), 367–375. DOI: 10.1021/acs.chemrestox.6b00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Paiano V; Maertens L; Guidolin V; Yang J; Balbo S; Hecht SS Quantitative liquid chromatography-nanoelectrospray ionization-high-resolution tandem mass spectrometry analysis of acrolein-DNA adducts and etheno-DNA adducts in oral cells from cigarette smokers and nonsmokers. Chem. Res. Toxicol 2020, 33 (8), 2197–2207. DOI: 10.1021/acs.chemrestox.0c00223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bodnar JA; Morgan WT; Murphy PA; Ogden MW Mainstream smoke chemistry analysis of samples from the 2009 US cigarette market. Regul. Toxicol. Pharmacol 2012, 64 (1), 35–42. DOI: 10.1016/j.yrtph.2012.05.011. [DOI] [PubMed] [Google Scholar]
- (11).Haussmann HJ Use of hazard indices for a theoretical evaluation of cigarette smoke composition. Chem. Res. Toxicol 2012, 25 (4), 794–810. DOI: 10.1021/tx200536w. [DOI] [PubMed] [Google Scholar]
- (12).International Agency for Research on Cancer. Acrolein, Crotonaldehyde, and Arecoline; IARC, 2021. [PubMed] [Google Scholar]
- (13).Yu Y; Cui Y; Niedernhofer LJ; Wang Y Occurrence, biological consequences, and human health relevance of oxidative stress-induced DNA damage. Chem. Res. Toxicol 2016, 29 (12), 2008–2039. DOI: 10.1021/acs.chemrestox.6b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chung FL; Chen HJC; Nath RG Lipid peroxidation as a potential source for the formation of exocyclic DNA adducts. Carcinogenesis 1996, 17, 2105–2111. [DOI] [PubMed] [Google Scholar]
- (15).Bartsch H; Nair J; Owen RW Exocyclic DNA adducts as oxidative stress markers in colon carcinogenesis: potential role of lipid peroxidation, dietary fat and antioxidants. Biol. Chem 2002, 383 (6), 915–921. DOI: 10.1515/BC.2002.098. [DOI] [PubMed] [Google Scholar]
- (16).Barbin A Etheno-adduct-forming chemicals: From mutagenicity testing to tumor mutation spectra. Mutat. Res 2000, 462 (2–3), 55–69. DOI: 10.1016/s1383-5742(00)00014-4. [DOI] [PubMed] [Google Scholar]
- (17).Pazo DY; Moliere F; Sampson MM; Reese CM; Agnew-Heard KA; Walters MJ; Holman MR; Blount BC; Watson CH; Chambers DM Mainstream smoke levels of volatile organic compounds in 50 U.S. Domestic cigarette brands smoked with the ISO and Canadian Intense Protocols. Nicotine Tob. Res 2016, 18 (9), 1886–1894. DOI: 10.1093/ntr/ntw118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Oldham MJ; DeSoi DJ; Rimmer LT; Wagner KA; Morton MJ Insights from analysis for harmful and potentially harmful constituents (HPHCs) in tobacco products. Regul. Toxicol. Pharmacol 2014, 70 (1), 138–148. DOI: 10.1016/j.yrtph.2014.06.017. [DOI] [PubMed] [Google Scholar]
- (19).Zhang S; Villalta PW; Wang M; Hecht SS Detection and quantitation of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human lung by liquid chromatography-electrospray ionization-tandem mass spectrometry. Chem. Res. Toxicol 2007, 20, 565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Cheng G; Guo J; Carmella SG; Lindgren B; Ikuemonisan J; Jensen J; Hatsukami DK; Balbo S; Hecht SS Increased acrolein-DNA adducts in buccal brushings of e-cigarette users. Carcinogenesis 2022. DOI: 10.1093/carcin/bgac026 From NLM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Murphy SE; Park S-SL; Thompson EF; Wilkens LR; Patel Y; Stram DO; Le Marchand L Nicotine N-glucuronidation relative to N-oxidation and C-oxidation and UGT2B10 genotype in five ethnic/racial groups. Carcinogenesis 2014, 35, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Park SL; Carmella SG; Chen M; Patel Y; Stram DO; Haiman CA; LeMarchand L; Hecht SS Mercpaturic acids derived from the toxicants acrolein and crotonaldehyde in the urine of cigarette smokers from five ethnic groups with differing risks for lung cancer. PLoS One 2015, 10, e0124841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chen M; Carmella SG; Sipe C; Jensen J; Luo X; Le CT; Murphy SE; Benowitz NL; McClernon FJ; Vandrey R; Allen SS; Denlinger-Apte R; Cinciripini PM; Strasser AA; al’Absi M; Robinson JD; Donny EC; Hatsukami D; Hecht SS Longitudinal stability in cigarette smokers of urinary biomarkers of exposure to the toxicants acrylonitrile and acrolein. PLoS One 2019, 14 (1), e0210104. DOI: 10.1371/journal.pone.0210104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chen M; Carmella SG; Li Y; Zhao Y; Hecht SS Resolution and quantitation of mercapturic acids derived from crotonaldehyde, methacrolein, and methyl vinyl ketone in the urine of smokers and nonsmokers. Chem. Res. Toxicol 2020, 33 (2), 669–677. DOI: 10.1021/acs.chemrestox.9b00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Tsou HH; Hu CH; Liu JH; Liu CJ; Lee CH; Liu TY; Wang HT Acrolein is involved in the synergistic potential of cigarette smoking- and betel quid chewing-related human oral cancer. Cancer Epidemiol. Biomarkers Prev 2019, 28 (5), 954–962. DOI: 10.1158/1055-9965.EPI-18-1033. [DOI] [PubMed] [Google Scholar]
- (26).Haiman CA; Stram DO; Wilkens LR; Pike MC; Kolonel LN; Henderson BE; Le Marchand L Ethnic and racial differences in the smoking-related risk of lung cancer. N. Engl. J. Med 2006, 354 (4), 333–342. [DOI] [PubMed] [Google Scholar]
- (27).Murphy SE Biochemistry of nicotine metabolism and its relevance to lung cancer. J. Biol. Chem 2021, 296, 100722. DOI: 10.1016/j.jbc.2021.100722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Murphy SE; Park SL; Balbo S; Haiman CA; Hatsukami DK; Patel Y; Peterson LA; Stepanov I; Stram DO; Tretyakova N; Hecht SS; Le Marchand L Tobacco biomarkers and genetic/epigenetic analysis to investigate ethnic/racial differences in lung cancer risk among smokers. NPJ Precis. Oncol 2018, 2, 17. DOI: 10.1038/s41698-018-0057-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chung FL; Wu MY; Basudan A; Dyba M; Nath RG Regioselective formation of acrolein-derived cyclic 1,N2-propanodeoxyguanosine adducts mediated by amino acids, proteins, and cell lysates. Chem. Res. Toxicol 2012, 25 (9), 1921–1928. DOI: 10.1021/tx3002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Zhang S; Balbo S; Wang M; Hecht SS Analysis of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human leukocyte DNA from smokers and nonsmokers. Chem. Res. Toxicol 2011, 24 (1), 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Gentile F; Arcaro A; Pizzimenti S; Daga M; Cetrangolo GP; Dianzani C; Lepore A; Graf M; Ames PRJ; Barrera G DNA damage by lipid peroxidation products: implications in cancer, inflammation and autoimmunity. AIMS Genet. 2017, 4 (2), 103–137. DOI: 10.3934/genet.2017.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hecht SS Tobacco Smoke Carcinogens and Lung Cancer. In Chemical Carcinogenesis, Penning TM Ed.; Springer, 2011; pp 53–74. [Google Scholar]
- (33).Stevens JF; Maier CS Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res 2008, 52 (1), 7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Haiman CA; Patel YM; Stram DO; Carmella SG; Chen M; Wilkens L; Le Marchand L; Hecht SS Benzene uptake and glutathione S-transferase T1 status as determinants of S-phenylmercapturic acid in cigarette smokers in the Multiethnic Cohort. PLoS One 2016, 11 (3), e0150641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Berhane K; Widersten M; Engstrom A; Kozarich JW; Mannervik B Detoxication of base propenals and other alpha, beta-unsaturated aldehyde products of radical reactions and lipid peroxidation by human glutathione transferases. Proc. Natl. Acad. Sci. U.S.A 1994, 91 (4), 1480–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Balogh LM; Atkins WM Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab. Rev 2011, 43 (2), 165–178. DOI: 10.3109/03602532.2011.558092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Pacholak LM; Kern R; de Oliveira ST; Lucio LC; Amarante MK; Guembarovski RL; Watanabe MAE; Panis C Effects of GSTT1 and GSTM1 polymorphisms in glutathione levels and breast cancer development in Brazilian patients. Mol. Biol. Rep 2021, 48 (1), 33–40. DOI: 10.1007/s11033-020-06107-w. [DOI] [PubMed] [Google Scholar]
- (38).Weng MW; Lee HW; Park SH; Hu Y; Wang HT; Chen LC; Rom WN; Huang WC; Lepor H; Wu XR; Yang CS; Tang MS Aldehydes are the predominant forces inducing DNA damage and inhibiting DNA repair in tobacco smoke carcinogenesis. Proc. Natl. Acad. Sci. U.S.A 2018, 115 (27), E6152–E6161. DOI: 10.1073/pnas.1804869115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Nath RG; Ocando JE; Guttenplan JB; Chung FL 1,N2-Propanodeoxyguanosine adducts: Potential new biomarkers of smoking-induced DNA damage in human oral tissue. Cancer Res. 1998, 58, 581–584. [PubMed] [Google Scholar]
- (40).Bessette EE; Goodenough AK; Langouet S; Yasa I; Kozekov ID; Spivack SD; Turesky RJ Screening for DNA adducts by data-dependent constant neutral loss-triple stage mass spectrometry with a linear quadrupole ion trap mass spectrometer. Anal. Chem 2009, 81 (2), 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Chen HJ; Lin WP Quantitative analysis of multiple exocyclic DNA adducts in human salivary DNA by stable isotope dilution nanoflow liquid chromatography-nanospray ionization tandem mass spectrometry. Anal. Chem 2011, 83 (22), 8543–8551. [DOI] [PubMed] [Google Scholar]
- (42).Wang TW; Liu JH; Tsou HH; Liu TY; Wang HT Identification of acrolein metabolites in human buccal cells, blood, and urine after consumption of commercial fried food. Food Sci. Nutr 2019, 7 (5), 1668–1676. DOI: 10.1002/fsn3.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Emami A; Dyba M; Cheema AK; Pan J; Nath RG; Chung FL Detection of the acrolein-derived cyclic DNA adduct by a quantitative 32P-postlabeling/solid-phase extraction/HPLC method: Blocking its artifact formation with glutathione. Anal. Biochem 2008, 374 (1), 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Hecht SS Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst 1999, 91, 1194–1210. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Calibration curves used for the quantitation of (A) ɣ-OH-Acr-dGuo and (B) ɛdAdo.
Figure S2. Example of a chromatogram showing traces corresponding to amounts of ɛdAdo and ɣ-OH-Acr-dGuo (3.1 and 5.7 per 109 nucleotides, respectively) close to the assay limit of quantitation.9
Table S1. Geometric means for urinary biomarkers of total nicotine equivalents (TNE) and mercapturic acids
Table S2. Pearson’s correlation coefficient – partial correlations adjusted for age and sex
Table S3. Geometric means for ɣ-OH-Acr-dGuo and ɛdAdo, per TNE, by sex
Table S4. Relationship of GSTM1 and GSTT1 status to biomarkers