

Abstract
It has been reported that cyclin-dependent kinase 5 (cdk5), a critical neuronal kinase, is hyperactivated in Alzheimer’s disease (AD) and may be, in part, responsible for the hallmark pathology of amyloid plaques and neurofibrillary tangles (NFTs). It has been proposed by several laboratories that hyperactive cdk5 results from the overexpression of p25 (a truncated fragment of p35, the normal cdk5 regulator), which, when complexed to cdk5, induces hyperactivity, hyperphosphorylated tau/NFTs, amyloid-β plaques, and neuronal death. It has previously been shown that intraperitoneal (i.p.) injections of a modified truncated 24-aa peptide (TFP5), derived from the cdk5 activator p35, penetrated the blood-brain barrier and significantly rescued AD-like pathology in 5XFAD model mice. The principal pathology in the 5XFAD mutant, however, is extensive amyloid plaques; hence, as a proof of concept, we believe it is essential to demonstrate the peptide’s efficacy in a mouse model expressing high levels of p25, such as the inducible CK-p25Tg model mouse that overexpresses p25 in CamKII positive neurons. Using a modified TFP5 treatment, here we show that peptide i.p. injections in these mice decrease cdk5 hyperactivity, tau, neurofilament-M/H hyperphosphorylation, and restore synaptic function and behavior (i.e., spatial working memory, motor deficit using Rota-rod). It is noteworthy that TFP5 does not inhibit endogenous cdk5/p35 activity, nor other cdks in vivo suggesting it might have no toxic side effects, and may serve as an excellent therapeutic candidate for neurodegenerative disorders expressing abnormally high brain levels of p25 and hyperactive cdk5.
Keywords: Alzheimer’s disease, cyclin-dependent kinase 5, hyperphosphorylation, synaptic function, TFP5
INTRODUCTION
Alzheimer’s disease (AD), a disease of aging, is manifest by a progressive loss of memory and cognitive function; its incidence increases with age as various aspects of brain physiology decline. A complex systemic network of functional changes, lesions, and somatic mutations that characterize brain aging results in mitochondrial dysfunction, accumulations of amyloid plaques, neurofibrillary tangles (NFTs), inflammation, and neuronal cell death. Among these changes is the hyperactivation of a neuronal, multifunctional serine/threonine kinase, cdk5 [1–5]. The activity of this kinase is critical for neuronal development and synaptic activity in the brain. Its normal, tightly regulated activity depends on the binding of neuron-specific, cyclin-related activators, p35 and p39 [6–9]. In normal development and synaptic function, the enzyme complex (cdk5/p35) targets many cytoskeletal and synaptic proteins and enzymes that are involved in neuronal migration, synaptogenesis, and synaptic function, among others [10]. It has been suggested that aberrant hyperactivation of cdk5 in AD brains is correlated with an increase in p25, derived as a proteolytic fragment of the normal p35 activator [5, 11–13]. Although this observation is controversial (see Discussion), several laboratories have continued to explore this hypothesis in various animal models of neurodegeneration. For example, oxidative stress, amyloid-β (Aβ) toxicity, inflammation, and excitotoxicity induce intracellular calcium flux, that activates proteases which cleave p35 into p25 and p10 fragments, producing a more stable hyperactive cdk5/p25 complex [3, 13–18]. A cdk5/p25 hyperactive complex, when induced in cortical neurons in culture, specifically hyper-phosphorylates tau and neurofilaments ultimately inducing cell death [19, 20]. Two peptides derived from p35, CIP, a large 126, amino acid (a.a.) sequence and a much smaller p5 (24a.a.), both of which specifically inhibit cdk5/p25 activity in situ and in brain lysates of AD model mice also prevent expression of AD-like phenotypes in vivo and in vitro in cultured cells [20–24]. In vivo studies rely on TFP5, a modified p5 peptide able to penetrate the blood-brain barrier (See Materials and Methods, Design and synthesis of TFP5). Accordingly, it has been hypothesized that hyperactivation of cdk5 via elevation of p25, is a potential therapeutic target in AD [25–28]. To more directly test this hypothesis, we have chosen a mouse mutant, CK-p25Tg, which can be induced by doxycycline withdrawal to overexpress p25 in forebrain CamKII positive neurons [16]. These mice exhibit robust p25 expression in various brain regions, elevated cdk5/p25 activity (but no cdk5/p35 activity) in brain lysates, extensive neuronal loss, and manifest AD-related phenotypes including neuronal tau and neurofilament hyperphosphorylation and microglia and astrocytes activation. Here we demonstrate that i.p. injections of TFP5 into overexpressing CK-p25Tg mutant mice (p25TgOE) restored synaptic activity (i.e., long term potentiation (LTP)), spatial working memory, and other behavioral deficits. These TFP5 treated mice show significant reduction in cdk5 hyperactivity, reduced inflammation, and tau/neurofilament hyperphosphorylation. The results are consistent with the hypothesis that hyperactive cdk5/p25 plays a role in the development of AD-like pathology and suggests that TFP5 may serve as an excellent therapeutic candidate worthy of further studies.
MATERIALS AND METHODS
Animals
In this study, we used bitransgenic (CK-p25Tg) mice (males and females) in which forebrain CK-p25 expression was tightly regulated by the tet on/off system facilitated by the CamKII (CK) promoter [16]. The tet-off system was induced by dietary alteration in doxycycline-supplemented food in which CK-p25 expression was suppressed in the presence of doxycycline (Fig. 1). Calmodulin-dependent protein kinase II (CaMKII or CK) is a promoter used to activate the expression of the CK-p25 gene in the transgenic mice. It was shown that CK-p25Tg mice develop significant neuronal loss in the cortex and hippocampus and display neurodegeneration and pathological tau hyperphosphorylation when p25 expression is induced. These mice are identified as p25Tg OE. High p25 expression and elevated hippocampal cdk5 activity has been observed within 2 weeks after induction.
Fig. 1.
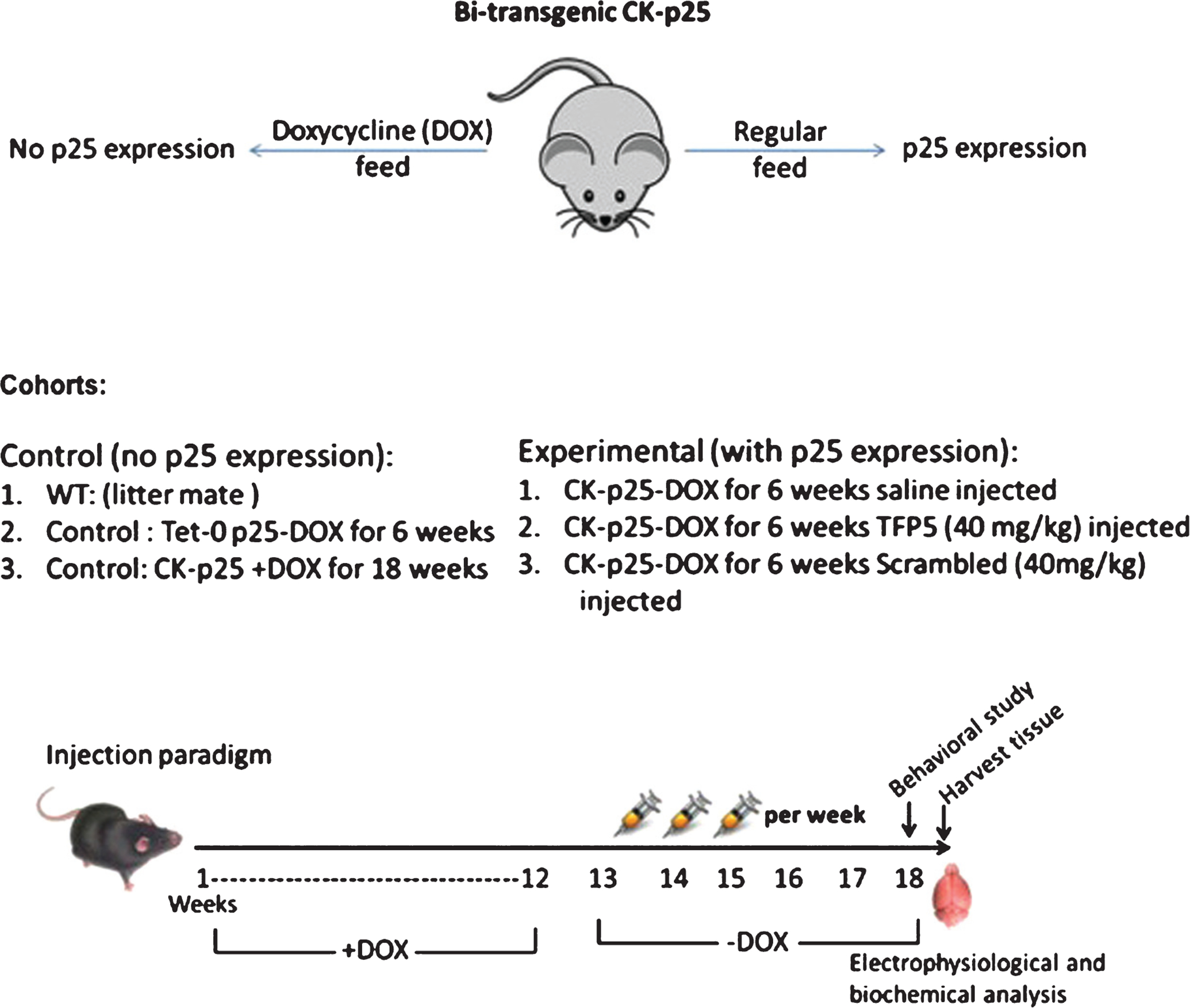
Controlled p25 expression in a p25tg AD mouse model. The transgenic line is initiated by crossing a p25-expressing transgenic mouse with a transgenic mouse expressing the CamKII promoter controlling a transactivator protein regulated by doxycycline intake. P25 mutant mice fed doxycycline do not express P25; in its absence high levels of P25 are expressed in the brain and the mice acquire an AD phenotype; i.e., hyperactive cdk5/p25 activity, Aβ accumulation, hyperphosphorylation of tau, inflammation, and behavioral defects. Cohorts and intraperitoneal injection paradigm: three control groups (left), expressing no p25 are compared to three experimental groups, off doxycycline, expressing p25. One injected with saline, a second injected with TFP5, and a third injected with scrambled peptide at the same dose.
All mice were developed and raised in the presence of doxycycline (1 mg/g in food). To induce CK-p25 expression, the mice were fed a normal diet. To inhibit p25 production, the mice were again fed a doxycycline diet. All experimental cohorts were treated under the same conditions and fed doxycycline diet for the same period of time. Note that all animal procedures were performed in accordance with the NIH animal care committee’s regulations.
Design and synthesis of TFP5
TFP5 is a truncated portion of p35 (activator of Cdk5), which extends 24 aa residues in length (Lys254–Ala277) conjugated with an 11-aa modifying peptide derived from the trans-activator domain of TAT protein at the C terminus (to facilitate passage through the blood-brain barrier), while FITC, fluorescein isothiocyanate (a green fluorescent tag) with linker GGG, was attached at the N terminus (to serve as a marker). A scrambled peptide (Scb) was used as a control to TFP5 (sequence shown below). Peptide 2.0 (Chantilly, VA, USA) commercially synthesized both TFP5 and Scb peptides which were used after dissolving both in saline or double distilled water.
Sequences used were as follows:
TFP5, FITCGGGKEAFWDRCLSVINLMSSKM LQINAYARAARRAARR;
Scb peptide, FITCGGGGGGFWDRCLSGKGK MSSKGGGINAYARAARRAARR.
Intraperitoneal (i.p.) injection paradigm
Five cohorts of mice were used: vehicle-injected wild-type (WT), CK-p25Tg+DOX, CK-p25Tg-DOX, TFP5-injected CK-p25Tg-DOX, and Scb-injected CK-p25Tg-DOX (Fig. 1). After 12 weeks, the three mutant cohorts were taken off DOX to activate the expression of CK-p25. All five cohorts were age-matched and WT, CK-p25Tg+DOX, CK-p25Tg-DOX were treated with i.p. injection of vehicle, while another group of -DOX mutant mice was injected with 40 mg/kg/d TFP5. As an additional control, a cohort of -DOX mutants was injected with 40 mg/kg/d Scb peptide. All five cohorts were injected 3 days/week on weeks 13–17 for a total of 18 injections. All the mice were subjected to behavior analysis on week 18 and the mice were euthanized on week 19 when brain tissue was harvested for biochemical analysis. Based on the observation that injection of 200 mg/kg/day of TFP5 into WT mice in 5XFAD studies was not toxic, had no effect on body weight, behavior, appearance, and [23], we decided to limit the use of the expensive peptides and did not inject peptide into WT and CK-p25Tg+DOX cohorts as controls in the various experiments.
Sample preparation and Western blot analysis
Lysates were created from PBS-perfused hemibrains in 10% w/v homogenization buffer [20 mM Tris-HCl, pH 7.4; 150 mM NaCl; 1 mM EDTA; 1% Nonidet P-40; and complete mini protease inhibitor (Roche, Basel, Switzerland)]. Western blot analysis was performed according to standard protocols.
Immunoprecipitation and kinase assay
Cdk5 was immunoprecipitated from the respective brain lysates using the polyclonal C8 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and kinase assays were conducted using TFP5 and Scb as described. All kinase assays were carried out as double blind experiments. As reported for the p25TgOE mutant, cdk5 activity assayed in this manner reflected cdk5/p25 rather than cdk5/p35 or other kinases [16].
Synaptic activity: long term potentiation
Details of the procedures for recording LTP and its response to TFP5 in -DOX off animals are included in legend to Fig. 5.
Fig. 5.
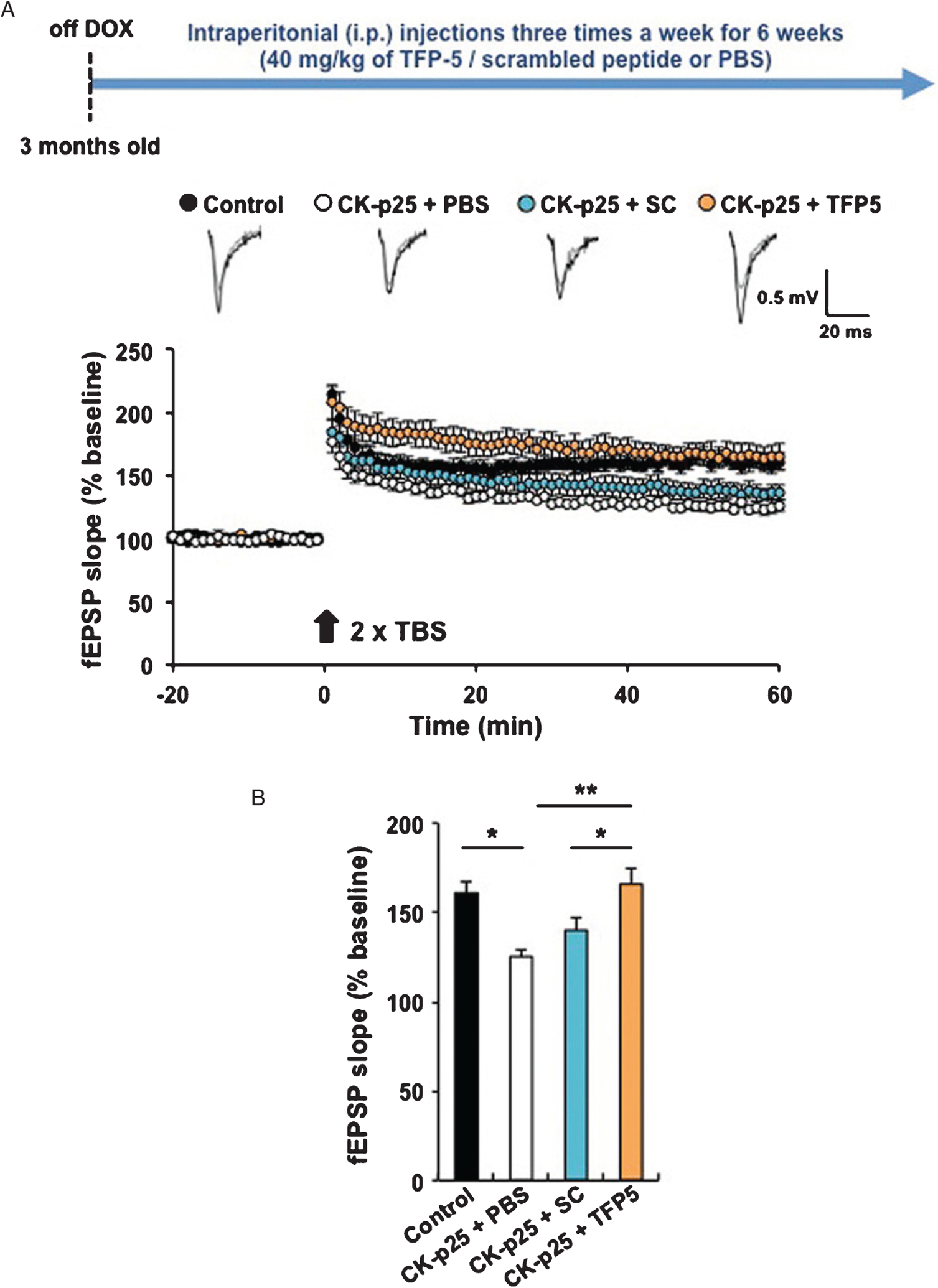
TFP5 restores normal synaptic activity and LTP. A) To record field excitatory postsynaptic potentials, transverse hippocampal slices were prepared from mice. In brief, the brain was rapidly removed and transferred to ice-cold, oxygenated (95% O2 and 5% CO2) cutting solution containing (mM) 211 sucrose, 3.3 KCl, 1.3 NaH2PO4, 0.5 CaCl2, 10 MgCl2, 26 NaHCO3, and 11 glucose. Hippocampal slices were cut with a Leica VT1000S vibratome (Leica) and transferred for recovery to a holding chamber containing oxygenated artificial cerebrospinal fluid (ACSF) consisting of (mM) 124 NaCl, 3.3 KCl, 1.3 NaH2PO4, 2.5 CaCl2, 1.5 MgCl2, 26 NaHCO3, and 11 glucose at 28–30°C for at least 1 h before recording. CA1 field potentials evoked by Schaffer collateral stimulation were measured. After recording of stable baseline (at least 20 min of stable responses), LTP was induced by two episodes of theta burst stimulation (TBS) with 10-s intervals. TBS consisted of ten bursts (each with four pulses at 100 Hz) of stimuli delivered every 200 ms. Recordings were performed using an AM-1800 Microelectrode amplifier (A-M systems) and a Digidata 1440A A-D converter (Axon Instruments). All data were digitized and analyzed by the use of pClamp 10 software (Axon Instruments). Sample traces represent fEPSPs at 1 min before (gray trace) and 1 h after (black trace) TBS. B) Bar graph: average slopes of fEPSP during the last 10 min of recording (percentage of baseline response) (*p < 0.05; **p < 0.01). Control (p25 only – Dox off): 160.6 ± 6.1 %, 6 slices from 4 mice; CK-p25 + PBS: 125.2 ± 3.7 %, 5 slices from 3 mice.
Behavioral analysis
As previously stated, the following behavioral studies were conducted with 18-week-old mice of five cohorts: vehicle-injected WT, CK-p25Tg+DOX, CK-p25Tg-DOX, TFP5-injected CK-p25Tg-DOX, and Scb-injected CK-p25Tg-DOX. The Scb-injected CK-p25Tg-DOX was used as a second control. For each group, ≥7–15 animals were tested. Note, all the behavioral studies were double blinded.
Spontaneous alternation Y-maze task
Spontaneous alternation was measured using the Y-maze as previously described [29, 30]. The Y-maze test assesses hippocampal-dependent spatial working memory of the brain. Each mouse was placed at the center of the symmetrical Y-maze and was allowed to explore freely through the maze over an 8-min period. The sequence and total number of arms entered were recorded. Percentage alternation is measured as follows: number of triads containing entries into all three arms/maximum possible alternations. Each of the five experimental cohorts of animals were tested once after the i.p. injection regimen.
Open-field task
The open-field task was used to assess exploratory behavior and locomotor activity, as described previously [31]. The testing apparatus was made of PVC, with a square 42- × 42-cm surface area and 31-cm-high walls. Mice were placed individually in the square open field area for a 20-min session and were monitored by an automated tracking system (Digipro; Accuscan Instruments Inc., Columbus, OH, USA). The exploratory activity and anxiety of all five cohorts were examined. For each group, the animals were tested once after i.p. injections.
Rotarod task
The rotarod task assesses the ability of an animal to keep its balance on a rotating rod and measures the animal’s balance, coordination, and motor ability. Rotarod assay was performed on 18-week-old mutant and WT mice as previously described [32]. The animal was placed on a cylinder which begins to rotate at a slowly increasing speed. The duration of time before it fell was recorded. For each group, the animals were tested thrice after i.p. injections.
Statistics
All statistical analyses of behavioral experiments are reported as means ± SE, where n ≥ 7–15 for each treated condition, as previously stated. Student’s t test was used to make comparisons between control and TFP5-treated mice. Values of p ≤ 0.05 were considered statistically significant. Except where indicated all other statistical analyses were also as student T-tests.
RESULTS
Reduction in hyperactivation of CDk5 activity post TFP5 treatment
The proof of concept in this instance requires that the TFP5 peptide does, indeed, successfully and specifically target (i.e., inhibit) overexpression of p25-induced Cdk5 hyperactivity, reduce AD-like pathology, improve abnormal behavior and promote survival. Previous studies have shown that a regimen of i.p. injections of the TFP5 peptide successfully passed the blood-brain barrier and reduced cdk5/p25 hyperactivity and AD phenotypes in a 5XFAD mouse model. This transgenic mouse expresses five human mutant genes involved in AβPP processing which may confound the phenotype. The CK-p25Tg mouse, however, is a better model for p25 overexpression; within six weeks after p25 induction it develops significant neuronal loss in cortex and hippocampus and displays neurodegeneration and pathological tau hyperphosphorylation [16, 32]. The entire phenotype is a result of specific overexpression of p25. To assess the effect of the peptide, brain tissue lysates harvested from the five cohorts (induced and/or treated), were immunoprecipitated with a cdk5 antibody and assayed for cdk5 activity (Fig. 2A). The results show the cdk5 activity levels increase more than 2.5 fold after p25 induction (in Fig. 2B, compare lane 3 with lane 1, 2). This is consistent with the evidence of p25 overexpression as reported [16]. Treatment with TFP5, however, shows a significant reduction in activity (almost 40%; compare lane 4 with lane 3) which more closely resembles the CK-p25Tg+DOX cdk5 activity or that of the WT control; the scrambled peptide control, however, had no effect on the hyperactivity which was similar to the untreated p25TgOE mice. These data show that TFP5 treatment inhibits hyperactive cdk5 and suggest that TFP5 treatment of the p25TgOE mouse may also prevent the AD-like phenotypes induced by cdk5 hyperactivation.
Fig. 2.

Reduction in cdk5 activity post TFP5 treatment. A) Autoradiograph of kinase assays in brain lysates derived from WT control and p25 non-expressing transgenics compared to those from p25 expressing mutants with and without TFP5 treatment (i.e., scrambled peptide, S). B) Quantitation of pad assays of cdk5 kinase activities in brain lysates derived from identical cohorts as in A. TFP5 treatment produces a significant reduction in activity (compare lanes 2, 3, 4) while scrambled peptide treatment (lane 5) has no effect. (n = 4; *p < 0.05, **p < 0.01).
TFP5 inhibits tau and neurofilament hyperphosphorylation
Hyperphosphorylated tau and neurofilaments contribute to NFTs, a hallmark of AD pathology [1, 16, 33, 34]. Tau contains multiple target sites for phosphorylation by several kinases, the principal ones being cdk5 and GSK3-β [35–38]. Brains of CK-p25Tg mice, when induced, exhibit high levels of tau and neurofilament-H/M phosphorylated at PHF sites Ser202, Ser404, and Thr205 [16]. To determine whether the TFP5 peptide would affect tau phosphorylation, we prepared Western blots of brain lysates from treated and untreated cohort mice using tau site- specific phospho-antibodies (Fig. 3A, B). Antibody SMI31 detected phosphorylated neurofilaments (p-NF-H/M) and also detects phospho-tau at 64 kDa. Western blot results of the brain lysates from CK-p25Tg mice show that of the four cohorts only the untreated p25TgOE mutant (lane 3) displayed increased levels of p-NF-H/M and p-Tau (25–50%) as detected by SMI 31, AT8, and pTau 404 antibodies, respectively. Brain lysates from TFP5-treated p25TgOE mice, however, showed significant reduction in both p-NF-H/M and p-Tau levels (lane 4, Fig. 3A–B). Actin antibody was employed to verify equal loading of the samples.
Fig. 3.

TFP5 inhibits tau and neurofilament hyperphosphorylation. A) Western blots of brain lysates derived from the standard four cohorts of TFP5-treated and untreated mice showing the expression of pNFH (Smi31 antibody) and hyperphosphorylated tau (pTau 404, AT8 antibodies). B) Quantitation of densities using NIH Image J software scanning protocol. Comparison of lanes 3 and 4 show significant reductions in pNFH and phospho tau expression as a consequence of TFP5 treatment. (n = 5; p < 0.05).
TFP5 reduces neuro-inflammation
The AD brain in humans and in AD mouse models is also characterized by neuro-inflammation involving activated astrocytes and microglia [39–41]. Activation of astrocytes parallels the accumulation of Aβ and microglia; the immune cells of the brain are activated when in proximity to senile plaques. Inflammation is a key early phenotype of the p25TgOE mutant after p25 induction [22, 42] and is ameliorated by the expression of transfected CIP, a larger truncated fragment containing the P5 sequence. Enhanced neuro-inflammation (almost five-fold) after p25 induction in brains of p25Tg OE mutants is significantly reduced (approximately 20–50%) upon TFP5 treatment as displayed in Western blots using GFAP antibody for activated astrocytes, Iba-1 for activated microglia and CD 86 for neuro-inflammation (Fig. 4 A, B). Equal loading of the samples was confirmed by immunoblotting with actin antibody.
Fig. 4.
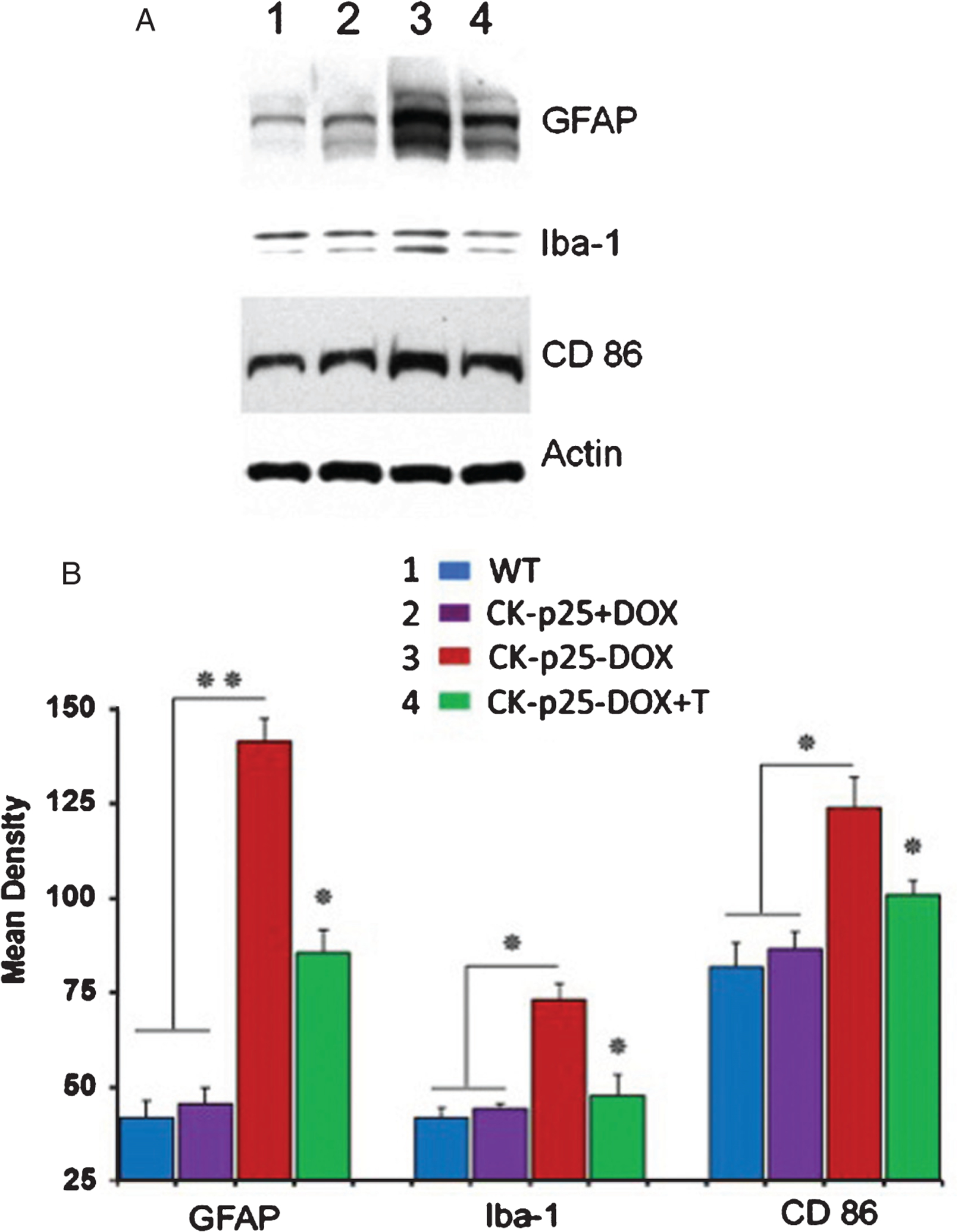
TFP5 administration reduces neuroinflammation. A) Western blots of brain lysates prepared from non-p25 expressing (lanes 1, 2) and p25-expressing animals (lanes 3, 4) treated with TFP5 according to the standard protocol. Antibodies to inflammatory cytokines in astrocytes and microglia show a significant reduction in inflammatory response as a result of the TFP5 injection protocol. B) Density scans of the blots with Image J software show a significant reduction of inflammatory response after TFP5 administration. (n = 5; p < 0.05).
TFP5 treatment restores normal synaptic activity and LTP
It has been shown that forebrain-specific overexpression of p25 results in the impairment of LTP in mouse hippocampus [32]. Moreover, a recent study indicates that chronic p25 expression is responsible for synaptic dysfunction in the 5XFAD mouse model of AD. Therefore, we sought to address whether TFP5 could affect p25-mediated synaptic dysfunction in the brain of the p25TgOE mouse. To do this, we examined LTP at Schaffer-collateral synapses in hippocampus of p25TgOE mice with TFP5, scrambled peptides or PBS administration. Three-month-old CKp25 male mice were fed with regular diet (off-DOX) for the onset of p25 expression. During the off-DOX period (6 weeks), we intraperitoneally injected the mice with TFP5, scrambled peptide (Scb), or PBS three times a week with 40 mg/kg of animal weight. Electrophysiological experiments were carried out at 18 weeks of age. After stable recording of baseline at least 20 min, we applied two times theta-burst stimulation (TBS) at 10-s intervals. Consistent with a previous study [43], we observed significantly impaired LTP in hippocampal slices from PBS-injected CK-p25Tg mice compared to littermate controls (Fig. 5A). We found that TFP5 administration completely restored hippocampal LTP whereas scrambled peptide failed to attenuate LTP deficits in these animals. The quantitation of these data shown in Fig. 5B indicates that TFP5 effectively inhibits p25-mediated synaptic dysfunction in the brain of the p25TgOE mouse.
TFP5 inhibits NMDA-induced cdk5/p25 activation in mouse hippocampal slices
It has been shown that glycine or NMDA stimulation of hippocampal slices induces elevated cdk5/p25 activity within a few minutes. The uptick in activity is presumed to result from increased availability of p25. If true, then our hypothesis predicts that treatment of hippocampal slices with TFP5 should successfully prevent hyperactive cdk5 under these conditions. The experiment, including scrambled peptide as control, is shown in Fig. 6A and indeed shows that TFP5 successfully inhibited cdk5 hyperactivity induced by stimulation.
Fig. 6.

TFP5 inhibits NMDA-induced cdk5/p25 activation in mouse hippocampal slices. A) Hippocampal slices from 3 to 5- month-old WT mice were pre-incubated with TFP5 or scrambled peptide (100 nM) for 30 min followed by cLTD induction (NMDA 50 μM for 5 min and recovery for 15 min), and then subjected to cdk5 kinase assay (n = 5 from 3 mice). Control: 100 ± 10.1, Scrambled + NMDA: 138.2 ± 7.3, TFP5 + NMDA 92.7 ± 10.0, ANOVA test; p = 0.0097 with post hoc Tukey HSD. B) Cdk5 activity in hippocampal slices from the Dp35KI calpain-resistant mutant, that fails to produce p25 endogenously or after stimulation, is significantly unaffected by TFP5 treatment. Slices were preincubated with TFP5 (100 nM for 30 min), then subjected to a kinase assay after cdk5 immunoprecipitation, Activity, though slightly less than the untreated control activity, was not significant (n = 3, p < 0.44 with post hoc Tukey). This is consistent with observations showing that endogenous brain cdk5/p35 is not inhibited by TFP5.
A more compelling negative control is illustrated in a knock-in mutant mouse model in which endogenous p35 is replaced by a calpain resistant mutant of p35 (Δp35KI) to prevent p25 generation. These mutants, however, exhibit normal endogenous cdk5/p35 activity. Unable to produce p25, these mice should show endogenous levels of cdk5/p35 activity unresponsive to TFP5 pretreatment (100 μM for 30 min) and in Fig. 6B we note that the modest reduction seen in activity after TFP5 treatment is not significant. We conclude that TFP5 selectively inhibits aberrant hyperactive cdk5/p25 activity but not cdk5/p35 physiological activity essential for neural development and function. It should be further noted that crossing the Δp35KI mutant with the 5XFAD AD mutant resulted in reduction of Aβ, synaptic depression, and cognitive impairment. Expression of the calpain resistant gene was sufficient to reduce the expression of p25 in the hybrid brain.
Spatial working memory deficit is rescued in p25TgOE mutants after TFP5 treatment
The mice overexpressing p25 (CK-p25Tg) display a spatial working memory deficit due to neuronal loss induced by AD-like pathology [16, 32]. Spatial working memory of CK-p25Tg and WT mice was tested measuring spontaneous alternation in the Y-maze (Fig. 7A). A spontaneous alternation value <45% indicates significant spatial working memory deficit.
Fig. 7.

Behavioral defects in CK-p25TgOE mice rescued after TFP5 treatment. Various CK-p25 cohorts were tested for behavior assays including Y maze, Rota-rod and Open field. A) Spatial memory deficit was measured by Y maze assay represented in the bar graph as average % alternation. The bi-transgenic CK-p25Tg performed poorly (less than 50%) and the poor performance was rescued in the CK-p25TgOE mice injected with TFP5. Scrambled peptide had no rescue affect. B) Motor deficits were accessed via rota-rod experiments. The average time (in seconds, as shown in the bar graph) for TFP5 injected CK-p25TgOE mice was comparable to the control cohorts, whereas PBS or scrambled injected mice could not balance themselves well on the rotating rod. C-D). Open-field test was carried out to observe the exploratory (total distance travelled over 20 min time) and anxiety behavior (time spent at the center of the box). Even though there was a significant decrease in the total distance travelled in the open field by TFP5 injected CK-p25TgOE mice compared with PBS or scrambled injected mice, there was no significant difference in the time spent at center of the open field box between the cohorts. Each column in the graph represents the mean ± SEM. (n = 10–16 and *p ≤ 0.05, **p ≤ 0.01).
The injection paradigm was amended for the behavioral experiments and the spontaneous alternation was studied for the five cohorts after 6 weeks off DOX (Fig. 7A, D). Using the second injection paradigm, WT mice displayed normal alternation performance in the Y maze, whereas p25TgOE mice performed poorly (<45%). The peptide- treated CK-p25Tg+DOX+T mice performed well with a spontaneous alternation value well over 50%, comparable in value to the WT and CK-p25Tg+DOX mice, while the untreated and Scb-treated groups showed similar poor spontaneous alternation performance (Fig. 7A).
TFP5 treatment improves other behavioral defects induced by p25 overexpression
Induction of p25 after DOX removal results in a 40% reduction in fall latency (roto-rod test) which is significantly restored after TFP5 treatment (Fig. 7B); again, we see that the scrambled peptide control has no effect on the abnormal behavior. The motor defects induced by p25 overexpression are prevented by TFP5 treatment. The results of the open field exploration test Fig. 7C) were unexpected; the p25 overexpressing animals (neurotoxic) exhibit hyperactive exploratory behavior compared to saline injected controls. This effect is not understood other than to suggest that some motor centers may be affected in response to high p25 levels. Nevertheless, TFP5 treatment succeeds in inhibiting that hyperactivity towards control values (compare red curve with green and blue). Here, too, scrambled peptide has no effect. It is interesting that the open field anxiety test (Fig. 7D) showed no significant difference among all the cohorts.
DISCUSSION
It has been reported that elevated levels of p25 in AD human brains and brains of AD rodent models specifically hyperactivate cdk5, a key neuronal kinase, which may contribute to the abnormal tau and Aβ pathology of AD [5, 16, 22, 23, 44–47]. Overexpression of p25 and hyperactivated cdk5 have also been noted in Parkinson’s disease [48] and Amyotrophic Lateral Sclerosis [4]. According to the model, p25 derived from cleavage of p35, the normal regulator of cdk5, accumulates in stressed or aging brains to form stable, activated cdk5/p25 complexes that contribute to AD etiology. The model claims that cdk5/p25 activity is significantly greater in vivo than the physiologically normal, cdk5/p35 activity. Consistent with this model is the demonstration that p25 induction leads to neurotoxicity [13]. Consequently, cdk5/p25 has been identified as a therapeutic target for AD [28].
It should be pointed out, however, that the role of p25 in the induction and evolution of the AD phenotype in humans (or in model systems) is quite controversial. The human brain data were questioned with evidence from two laboratories of a significant downregulation of p25 in frontal cortex of AD brains [49, 50]. In defense, the Patrick group repeated their study of human brains, showing a significant increase in the p25/p35 ratio [51]. Here, too, the results were doubtful, post-mortem changes complicate the interpretation of the data. The claim of cdk5/p25 hyperactivity exceeding cdk5/p35 activity has also been queried. In vitro, it has been shown that catalytic kinetics for phosphorylation of tau and histone is similar for cdk5/p25 and cdk5/p35 [52]. Finally, a more recent postmortem study only confirmed a reduction in both p25 and p35 in mild cases of AD, which continued unchanged during the more severe AD phenotype [53]. Although diverse postmortem changes may account for the differences [54], to date, there has been no resolution; in fact the situation has become even more confusing with the mixed results from other p25 mouse transgenics initiated at fertilization [1, 55]. Robust p25 expression and cdk5 activity at 4–5 months correlated with tau phosphorylation, axonopathy, neurodegeneration, and severe motor defects but no evidence of other AD phenotypes. Moreover, later studies of the doxycyclin-regulated p25Tg only added to the confusion; two roles for p25 were revealed a low-dose, positive physiological role in memory formation, while a higher sustained p25 elevation induced neurodegeneration and AD-like phenotypes [32].
In spite of the controversy, a number of laboratories have shown that specific inhibition of cdk5/p25 activity with compounds such as roscovitine do affect neuronal degeneration in vitro and in model mice. [15, 39, 56–62]. Roscovitine-like compounds are non-specific, however; they bind to the ATP binding site and inhibit cell cycle cdks and other kinases. Hence more specific inhibitors have been sought. P5, a 24aa peptide from p35, which, when modified as TFP5 (an N-terminal FITC fluorescent marker and a C-terminal TAT sequence to penetrate the blood-brain barrier) has been shown to specifically inhibit cdk5/p25 in a 5XFAD model mouse and alleviate AD-like phenotypes (including behavior) without affecting normal activity of cdk5/p35 [23]. Although a modest level of cdk5/p25 is produced in these mouse brains, it is insufficient as proof of the pathological role of p25, possibly because low, transient p25 levels are involved in normal synaptic activity [63]. Optimally, we require a model in which robust, sustained levels of p25 can be induced and regulated experimentally to test the efficacy of the TFP5 peptide. The experiments reported here, we believe, have successfully demonstrated in a p25 transgenic mutant CK-p25Tg [3, 16], that elevated p25 and hyperactive cdk5 initiate a cascade of AD-like biochemical, physiological and behavioral events that are ameliorated by specific TFP5 inhibition of cdk5 hyperactivity.
TFP5 is unique among a large number of cdk5 inhibitors. These are ATP analogues, hydrophobic, pass cell membranes, inhibit almost all the kinases, therefore, are toxic. TFP5 is not toxic and inhibits cdk5 hyperactivity (cdk5/p25) but not the physiological activity of cdk5 (cdk5/p35).
Regulated by doxycycline, levels of p25 in CK-p25 transgenics can be selectively expressed to induce the pathology and abnormal behavior of AD. Animals, maintained on doxycycline during development and grown to 12 weeks of age, produce no p25; removal of doxycycline, however, induces a robust expression of brain p25. Description of the ensuing phenotype depends on the number of time points taken for biochemical or behavioral assay, most of which were between 4- and 12-weeks post induction [3, 16]. For example, robust expression of p25 with upregulated cdk5 activity was detected at 5-weeks post induction [16], whereas cdk5 hyperactivity (2X) was noted 1-week post induction in the same mutant [42]. Although an amyloid response was noted at 2 weeks [3], in the latter study the earliest event correlating with cdk5 activity was inflammation at one week (astrogliosis, i.e., GFAP expression, PLA2 upregulation, LPC production) followed by microgliosis (cytokine expression) at 4 weeks. Here, 4–5 weeks also marked the expression of tau hyperphosphorylation which increased three fold at 12 weeks, clearly later than the inflammation response. During this period neuronal loss was 25–40% by 12 weeks.
Based on a more detailed time-line provided by Sundaram et al. [42], which includes underlying mechanisms, we note that 6-week TFP5 peptide treatment post induction, covers the key events in development of the AD-like phenotype. Hence, our behavioral, physiological and biochemical analyses, though representing a single time point (6-weeks post induction), evaluate the impact of the peptide treatment during the peak of the early response to p25 activation. We suggest that this is, indeed, a “proof of concept” test of the hypothesis outlined above.
The peptide, TFP5, was injected at high dose (120 mg/kg per week for 6 weeks) into activated transgenics (−DOX). Among the control cohorts were activated transgenics receiving an equivalent dose of scrambled peptide. It is clear that at 6-weeks post induction, cdk5 activity is still 3 X WT controls in transgenics, whereas activity in TFP5-treated mice is restored almost to control levels. No reduction is seen in scrambled peptide controls, however. Moreover, in response to reduced cdk5 activity, a similar pattern of recovery is evident in expression of hyperphosphorylated neurofilament and tau. It is also noteworthy that the early inflammatory response induced simultaneously with the uptick of hyperactive cdk5/p25 at one week, is sustained at high levels (3X controls) at 6 weeks in untreated transgenics. Nevertheless, TFP5 treatment succeeds in significantly reducing cytokine overexpression 20–50%, marking an improvement in inflammation.
Within six weeks after p25 expression the p25TgOE mouse displays a significant loss of neurons coupled to behavioral defects. A memory loss detected in Y maze tests is most dramatic, joined by defects in open field exploration and rotorod motility. No significant change occurred in the open field anxiety test, however. Here, too, sustained TFP5 treatment in the weeks prior to testing succeeded in restoring normal behavior in most animals. A similar injection regimen with the scrambled peptide failed to improve any behavioral defect. Consistent with these results is the successful restoration of normal LTP activity in the activated p25 hippocampus after 6 weeks of peptide treatment. One would expect synaptic defects in hippocampal neurons to contribute to the abnormal behavior at 6-weeks post induction. Phosphorylated tau and Aβ upregulation, which are detected at 4–5 weeks, may be responsible for behavioral defects but it is more likely that the longer period of persistent astrogliosis and microgliosis is a prime factor in neurodegeneration [64–66]. Inflammation does correlate with neurodegeneration in a related p25 transgenic also controlled by doxycycline [41]. In this mutant, removal of doxycycline at 6 weeks of age induces high levels of neuronal p25 within a week, without affecting cdk5 catalytic subunit expression. Surprisingly, after 5 months, in spite of a 44% reduction in brain weight, the mice appeared in good health and behaved normally and different simple tests including clasping, walking, and rotorod, revealed no major clinical problem. After analysis, however, it was concluded that progressive neuronal loss in response to p25 expression was not due to tau phosphorylation, amyloid deposition, nor apoptosis but was evoked by intensive astrogliosis and microgliosis in cortex and hippocampus as documented by a rapid rise of GFAP and CD45 markers, barely 2 weeks after induction [41]. Preliminary data suggested that inflammation was induced by activation of astrocytes and microglia by a soluble factor released from p25-overexpressing and dying neurons.
Subsequent studies confirmed this hypothesis and provided additional molecular details as to the mechanism initiating inflammation [42]. Briefly, they have shown that hyperactive cdk5/p25 in neurons activates cytosolic phospholipase A2 (cPLA2) which hydrolyzes phosphatidylcholine to produce lysophosphatidylcholine (LPC). Soluble LPC is released from neurons and activates glial synthesis of various chemokines and cytokines that trigger inflammation. They, too, noted the early rise of GFAP one-week post p25 induction and the sustained inflammation peaking at 6 weeks. One should note, therefore, that in our experiments, TFP5 treatment was intense throughout this period, inhibited cdk5 activity and at 6 weeks we could show a significant down-regulation of inflammation.
Variability in the timing of events in development of the phenotype in the CK-p25 Tg model mouse, as reported in three different laboratories, is not surprising. The time points at which sampling occurs either for biochemical or histochemical assay are different in each laboratory as is the specific bias adopted as relevant to the onset of the pathology. Future studies of the kinetics of inhibition by these peptides will require a thorough pharmacokinetic and pharmacodynamic analysis of the fate of the peptide after injection. How much penetrates the brain, in what form, and for how long need to be determined. Its precise mechanism of action in vivo may then be accessible for analysis.
It is becoming evident that p25 is essential in increasing synaptic density during spatial memory formation under normal physiological conditions [32, 63, 67–71]. This is probably true for transient, low levels of p25 expression occurring during NMDA-induced long term depression [72], or glycine evoked long-term potentiation [71]. Presumably, these Ca-mediated synaptic events activate calpain cleavage of p35 to p25, which, in turn, initiate synthesis of key synaptic proteins [53].
Cdk5 has been identified as a key player in synaptic activity underlying plasticity and memory formation [73, 74]. Moreover, cdk5 phosphorylation of the NR2A subunit of the NMDA receptor is involved in regulation of NMDAR receptors [75]. More recently, inhibition of cdk5 phosphorylation of the NR2B receptor improves synaptic plasticity and increases expression of spines at the dendritic surface [76]. Activation and/or inhibition of cdk5 phosphorylation of the NMDA receptor at serine 1116, regulates synaptic plasticity and memory formation; inhibition of activity with a small interfering peptide targeting the NR2B sequence affected cdk5 activity, promoted synaptic transmission and enhanced memory [76]. We see that synaptic stimulation that elevates p25 at the synapse should engage a network underlying LTP and memory regulation. Our study with TFP5 peptide carried out in a model mouse exhibiting an unusually robust overexpression of p25 affected the later stages of neurodegeneration when synaptic function is seriously compromised.
ACKNOWLEDGMENTS
We would like to thank Dr. Avi Nath for his critically reviewing the manuscript and suggestions.
This work was supported by the intramural research program of the DHHS, U.S. National Institutes of Health (NIH), NINDS and by the NIH RO1 grant (5R01NS051874) to L.-H.T.
Authors’ disclosures available online (http://j-alz.com/manuscript-disclosures/16-0916r1).
REFERENCES
- [1].Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD (2000) Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci U S A 97, 2910–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Cheung ZH, Ip NY (2004) Cdk5: Mediator of neuronal death and survival. Neurosci Lett 361, 47–51. [DOI] [PubMed] [Google Scholar]
- [3].Cruz JC, Kim D, Moy LY, Dobbin MM, Sun X, Bronson RT, Tsai LH (2006) p25/cyclin-dependent kinase 5 induces production and intraneuronal accumulation of amyloid beta in vivo. J Neurosci 26, 10536–10541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nguyen MD, Lariviere RC, Julien JP (2001) Deregulation of Cdk5 in a mouse model of ALS: Toxicity alleviated by perikaryal neurofilament inclusions. Neuron 30, 135–147. [DOI] [PubMed] [Google Scholar]
- [5].Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH (1999) Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. [DOI] [PubMed] [Google Scholar]
- [6].Dhavan R, Greer PL, Morabito MA, Orlando LR, Tsai LH (2002) The cyclin-dependent kinase 5 activators p35 and p39 interact with the alpha-subunit of Ca2+/calmodulin-dependent protein kinase II and alpha-actinin-1 in a calcium-dependent manner. J Neurosci 22, 7879–7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Humbert S, Dhavan R, Tsai L (2000) p39 activates cdk5 in neurons, and is associated with the actin cytoskeleton. J Cell Sci 113(Pt 6), 975–983. [DOI] [PubMed] [Google Scholar]
- [8].Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, Tsai LH (2001) p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J Neurosci 21, 6758–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB (1996) Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci U S A 93, 11173–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lim AC, Qu D, Qi RZ (2003) Protein-protein interactions in Cdk5 regulation and function. Neurosignals 12, 230–238. [DOI] [PubMed] [Google Scholar]
- [11].Patrick GN, Zukerberg L, Nikolic M, de La Monte S, Dikkes P, Tsai LH (2001) reply: Neurobiologyp25 protein in neurodegeneration. Nature 411, 764–765. [DOI] [PubMed] [Google Scholar]
- [12].Patzke H, Tsai LH (2002) Cdk5 sinks into ALS. Trends Neurosci 25, 8–10. [DOI] [PubMed] [Google Scholar]
- [13].Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH (2000) Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature 405, 360–364. [DOI] [PubMed] [Google Scholar]
- [14].Bu B, Li J, Davies P, Vincent I (2002) Deregulation of cdk5, hyperphosphorylation, and cytoskeletal pathology in the Niemann-Pick type C murine model. J Neurosci 22, 6515–6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Camins A, Verdaguer E, Folch J, Canudas AM, Pallas M (2006) The role of CDK5/P25 formation/inhibition in neurodegeneration. Drug News Perspect 19, 453–460. [DOI] [PubMed] [Google Scholar]
- [16].Cruz JC, Tseng HC, Goldman JA, Shih H, Tsai LH (2003) Aberrant Cdk5 activation by p25 triggers pathological events leading to neurodegeneration and neurofibrillary tangles. Neuron 40, 471–483. [DOI] [PubMed] [Google Scholar]
- [17].Hashiguchi M, Saito T, Hisanaga S, Hashiguchi T (2002) Truncation of CDK5 activator p35 induces intensive phosphorylation of Ser202/Thr205 of human tau. J Biol Chem 277, 44525–44530. [DOI] [PubMed] [Google Scholar]
- [18].Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T (2002) Cleavage of the cyclin-dependent kinase 5 activator p35 to p25 does not induce tau hyperphosphorylation. Biochem Biophys Res Commun 298, 693–698. [DOI] [PubMed] [Google Scholar]
- [19].Zheng YL, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 24, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zheng YL, Amin ND, Hu YF, Rudrabhatla P, Shukla V, Kanungo J, Kesavapany S, Grant P, Albers W, Pant HC (2010) A 24-residue peptide (p5), derived from p35, the Cdk5 neuronal activator, specifically inhibits Cdk5-p25 hyperactivity and tau hyperphosphorylation. J Biol Chem 285, 34202–34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Zheng Y, Kesavapany S, Gravell M, Hamilton RS, Schubert M, Amin N, Albers W, Grant P, Pant HC (2005) A Cdk5 inhibitory peptide reduces tau hyperphosphorylation and apoptosis in neurons. EMBO J 24, 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sundaram JR, Poore CP, Sulaimee NH, Pareek T, Asad AB, Rajkumar R, Cheong WF, Wenk MR, Dawe GS, Chuang KH, Pant HC, Kesavapany S (2013) Specific inhibition of p25/Cdk5 activity by the Cdk5 inhibitory peptide reduces neurodegeneration in vivo. J Neurosci 33, 334–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Shukla V, Zheng YL, Mishra SK, Amin ND, Steiner J, Grant P, Kesavapany S, Pant HC (2013) A truncated peptide from p35, a Cdk5 activator, prevents Alzheimer’s disease phenotypes in model mice. FASEB J 27, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kesavapany S, Zheng YL, Amin N, Pant HC (2007) Peptides derived from Cdk5 activator p35, specifically inhibit deregulated activity of Cdk5. Biotechnol J 2, 978–987. [DOI] [PubMed] [Google Scholar]
- [25].Cuny GD (2009) Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Des 15, 3919–3939. [DOI] [PubMed] [Google Scholar]
- [26].Glicksman MA, Cuny GD, Liu M, Dobson B, Auerbach K, Stein RL, Kosik KS (2007) New approaches to the discovery of cdk5 inhibitors. Curr Alzheimer Res 4, 547–549. [DOI] [PubMed] [Google Scholar]
- [27].Lau LF, Seymour PA, Sanner MA, Schachter JB (2002) Cdk5 as a drug target for the treatment of Alzheimer’s disease. J Mol Neurosci 19, 267–273. [DOI] [PubMed] [Google Scholar]
- [28].Tsai LH, Lee MS, Cruz J (2004) Cdk5, a therapeutic target for Alzheimer’s disease? Biochim Biophys Acta 1697, 137–142. [DOI] [PubMed] [Google Scholar]
- [29].Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4, 97–100. [DOI] [PubMed] [Google Scholar]
- [30].Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- [31].Bolognin S, Blanchard J, Wang X, Basurto-Islas G, Tung YC, Kohlbrenner E, Grundke-Iqbal I, Iqbal K (2012) An experimental rat model of sporadic Alzheimer’s disease and rescue of cognitive impairment with a neurotrophic peptide. Acta Neuropathol 123, 133–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Fischer A, Sananbenesi F, Pang PT, Lu B, Tsai LH (2005) Opposing roles of transient and prolonged expression of p25 in synaptic plasticity and hippocampus-dependent memory. Neuron 48, 825–838. [DOI] [PubMed] [Google Scholar]
- [33].Chen YG (2005) Specific tau phosphorylation sites in hippocampus correlate with impairment of step-down inhibitory avoidance task in rats. Behav Brain Res 158, 277–284. [DOI] [PubMed] [Google Scholar]
- [34].Rudrabhatla P, Grant P, Jaffe H, Strong MJ, Pant HC (2010) Quantitative phosphoproteomic analysis of neuronal inter-mediate filament proteins (NF-M/H) in Alzheimer’s disease by iTRAQ. FASEB J 24, 4396–4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Imahori K, Hoshi M, Ishiguro K, Sato K, Takahashi M, Shiurba R, Yamaguchi H, Takashima A, Uchida T (1998) Possible role of tau protein kinases in pathogenesis of Alzheimer’s disease. Neurobiol Aging 19, S93–S98. [DOI] [PubMed] [Google Scholar]
- [36].Muyllaert D, Kremer A, Jaworski T, Borghgraef P, Devijver H, Croes S, Dewachter I, Van Leuven F (2008) Glycogen synthase kinase-3beta, or a link between amyloid and tau pathology? Genes Brain Behav 7(Suppl 1), 57–66. [DOI] [PubMed] [Google Scholar]
- [37].Sato S, Tatebayashi Y, Akagi T, Chui DH, Murayama M, Miyasaka T, Planel E, Tanemura K, Sun X, Hashikawa T, Yoshioka K, Ishiguro K, Takashima A (2002) Aberrant tau phosphorylation by glycogen synthase kinase-3beta and JNK3 induces oligomeric tau fibrils in COS-7 cells. J Biol Chem 277, 42060–42065. [DOI] [PubMed] [Google Scholar]
- [38].Takashima A, Murayama M, Yasutake K, Takahashi H, Yokoyama M, Ishiguro K (2001) Involvement of cyclin dependent kinase5 activator p25 on tau phosphorylation in mouse brain. Neurosci Lett 306, 37–40. [DOI] [PubMed] [Google Scholar]
- [39].Kitazawa M, Oddo S, Yamasaki TR, Green KN, LaFerla FM (2005) Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J Neurosci 25, 8843–8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mastrangelo MA, Bowers WJ (2008) Detailed immune-histochemical characterization of temporal and spatial progression of Alzheimer’s disease-related pathologies in male triple-transgenic mice. BMC Neurosci 9, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Muyllaert D, Terwel D, Kremer A, Sennvik K, Borghgraef P, Devijver H, Dewachter I, Van Leuven F (2008) Neurodegeneration and neuroinflammation in cdk5/p25-inducible mice: A model for hippocampal sclerosis and neocortical degeneration. Am J Pathol 172, 470–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sundaram JR, Chan ES, Poore CP, Pareek TK, Cheong WF, Shui G, Tang N, Low CM, Wenk MR, Kesavapany S (2012) Cdk5/p25-induced cytosolic PLA2-mediated lysophosphatidylcholine production regulates neuroinflammation and triggers neurodegeneration. J Neurosci 32, 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Graff J, Rei D, Guan JS, Wang WY, Seo J, Hennig KM, Nieland TJ, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai LH (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kanungo J, Zheng YL, Amin ND, Pant HC (2009) Targeting Cdk5 activity in neuronal degeneration and regeneration. Cell Mol Neurobiol 29, 1073–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kesavapany S, Li BS, Amin N, Zheng YL, Grant P, Pant HC (2004) Neuronal cyclin-dependent kinase 5: Role in nervous system function and its specific inhibition by the Cdk5 inhibitory peptide. Biochim Biophys Acta 1697, 143–153. [DOI] [PubMed] [Google Scholar]
- [46].Nikolic M, Tsai LH (2000) Activity and regulation of p35/Cdk5 kinase complex. Methods Enzymol 325, 200–213. [DOI] [PubMed] [Google Scholar]
- [47].Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M (2001) Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett 489, 46–50. [DOI] [PubMed] [Google Scholar]
- [48].Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O’Hare MJ, Callaghan S, Slack RS, Przedborski S, Anisman H, Park DS (2003) Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci U S A 100, 13650–13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Tandon A, Yu H, Wang L, Rogaeva E, Sato C, Chishti MA, Kawarai T, Hasegawa H, Chen F, Davies P, Fraser PE, Westaway D, St George-Hyslop PH (2003) Brain levels of CDK5 activator p25 are not increased in Alzheimer’s or other neurodegenerative diseases with neurofibrillary tangles. J Neurochem 86, 572–581. [DOI] [PubMed] [Google Scholar]
- [50].Yoo BC, Lubec G (2001) p25 protein in neurodegeneration. Nature 411, 763–764; discussion 764–765. [DOI] [PubMed] [Google Scholar]
- [51].Tseng HC, Zhou Y, Shen Y, Tsai LH (2002) A survey of Cdk5 activator p35 and p25 levels in Alzheimer’s disease brains. FEBS Lett 523, 58–62. [DOI] [PubMed] [Google Scholar]
- [52].Peterson DW, Ando DM, Taketa DA, Zhou H, Dahlquist FW, Lew J (2010) No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proc Natl Acad Sci U S A 107, 2884–2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Engmann O, Hortobagyi T, Thompson AJ, Guadagno J, Troakes C, Soriano S, Al-Sarraj S, Kim Y, Giese KP (2011) Cyclin-dependent kinase 5 activator p25 is generated during memory formation and is reduced at an early stage in Alzheimer’s disease. Biol Psychiatry 70, 159–168. [DOI] [PubMed] [Google Scholar]
- [54].Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S (2000) Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem 275, 17166–17172. [DOI] [PubMed] [Google Scholar]
- [55].Bian F, Nath R, Sobocinski G, Booher RN, Lipinski WJ, Callahan MJ, Pack A, Wang KK, Walker LC (2002) Axonopathy, tau abnormalities, and dyskinesia, but no neurofibrillary tangles in p25-transgenic mice. J Comp Neurol 446, 257–266. [DOI] [PubMed] [Google Scholar]
- [56].Chen PC, Chen JC (2005) Enhanced Cdk5 activity and p35 translocation in the ventral striatum of acute and chronic methamphetamine-treated rats. Neuropsychopharmacology 30, 538–549. [DOI] [PubMed] [Google Scholar]
- [57].Crews L, Patrick C, Adame A, Rockenstein E, Masliah E (2011) Modulation of aberrant CDK5 signaling rescues impaired neurogenesis in models of Alzheimer’s disease. Cell Death Dis 2, e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hamdane M, Bretteville A, Sambo AV, Schindowski K, Begard S, Delacourte A, Bertrand P, Buee L (2005) p25/Cdk5-mediated retinoblastoma phosphorylation is an early event in neuronal cell death. J Cell Sci 118, 1291–1298. [DOI] [PubMed] [Google Scholar]
- [59].Jamsa A, Backstrom A, Gustafsson E, Dehvari N, Hiller G, Cowburn RF, Vasange M (2006) Glutamate treatment and p25 transfection increase Cdk5 mediated tau phosphorylation in SH-SY5Y cells. Biochem Biophys Res Commun 345, 324–331. [DOI] [PubMed] [Google Scholar]
- [60].Kerokoski P, Suuronen T, Salminen A, Soininen H, Pirttila T (2002) Influence of phosphorylation of p35, an activator of cyclin-dependent kinase 5 (cdk5), on the proteolysis of p35. Brain Res Mol Brain Res 106, 50–56. [DOI] [PubMed] [Google Scholar]
- [61].Lefevre K, Clarke PG, Danthe EE, Castagne V (2002) Involvement of cyclin-dependent kinases in axotomy-induced retinal ganglion cell death. J Comp Neurol 447, 72–81. [DOI] [PubMed] [Google Scholar]
- [62].Lopes JP, Oliveira CR, Agostinho P (2007) Role of cyclin-dependent kinase 5 in the neurodegenerative process triggered by amyloid-Beta and prion peptides: Implications for Alzheimer’s disease and prion-related encephalopathies. Cell Mol Neurobiol 27, 943–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Seo J, Giusti-Rodriguez P, Zhou Y, Rudenko A, Cho S, Ota KT, Park C, Patzke H, Madabhushi R, Pan L, Mungenast AE, Guan JS, Delalle I, Tsai LH (2014) Activity-dependent p25 generation regulates synaptic plasticity and Abeta-induced cognitive impairment. Cell 157, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Marchetti B, Abbracchio MP (2005) To be or not to be (inflamed)–is that the question in anti-inflammatory drug therapy of neurodegenerative disorders? Trends Pharmacol Sci 26, 517–525. [DOI] [PubMed] [Google Scholar]
- [65].Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog Neurobiol 76, 77–98. [DOI] [PubMed] [Google Scholar]
- [66].Wyss-Coray T (2006) Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat Med 12, 1005–1015. [DOI] [PubMed] [Google Scholar]
- [67].Angelo M, Plattner F, Giese KP (2006) Cyclin-dependent kinase 5 in synaptic plasticity, learning and memory. J Neurochem 99, 353–370. [DOI] [PubMed] [Google Scholar]
- [68].Giese KP, Ris L, Plattner F (2005) Is there a role of the cyclin-dependent kinase 5 activator p25 in Alzheimer’s disease? Neuroreport 16, 1725–1730. [DOI] [PubMed] [Google Scholar]
- [69].Ris L, Angelo M, Plattner F, Capron B, Errington ML, Bliss TV, Godaux E, Giese KP (2005) Sexual dimorphisms in the effect of low-level p25 expression on synaptic plasticity and memory. Eur J Neurosci 21, 3023–3033. [DOI] [PubMed] [Google Scholar]
- [70].Angelo M, Plattner F, Irvine EE, Giese KP (2003) Improved reversal learning and altered fear conditioning in transgenic mice with regionally restricted p25 expression. Eur J Neurosci 18, 423–431. [DOI] [PubMed] [Google Scholar]
- [71].Giese KP (2014) Generation of the Cdk5 activator p25 is a memory mechanism that is affected in early Alzheimer’s disease. Front Mol Neurosci 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Abadi AH, Abou-Seri SM, Abdel-Rahman DE, Klein C, Lozach O, Meijer L (2006) Synthesis of 3-substituted-2-oxoindole analogues and their evaluation as kinase inhibitors, anticancer and antiangiogenic agents. Eur J Med Chem 41, 296–305. [DOI] [PubMed] [Google Scholar]
- [73].Cheng K, Ip NY (2003) Cdk5: A new player at synapses. Neurosignals 12, 180–190. [DOI] [PubMed] [Google Scholar]
- [74].Cheung ZH, Fu AK, Ip NY (2006) Synaptic roles of Cdk5: Implications in higher cognitive functions and neurodegenerative diseases. Neuron 50, 13–18. [DOI] [PubMed] [Google Scholar]
- [75].Li BS, Sun MK, Zhang L, Takahashi S, Ma W, Vinade L, Kulkarni AB, Brady RO, Pant HC (2001) Regulation of NMDA receptors by cyclin-dependent kinase-5. Proc Natl Acad Sci U S A 98, 12742–12747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Plattner F, Hernandez A, Kistler TM, Pozo K, Zhong P, Yuen EY, Tan C, Hawasli AH, Cooke SF, Nishi A, Guo A, Wiederhold T, Yan Z, Bibb JA (2014) Memory enhancement by targeting Cdk5 regulation of NR2B. Neuron 81, 1070–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]