

Abstract
Alzheimer’s disease (AD) is the most common cause of dementia, characterized by a spectrum of symptoms associated with memory loss and cognitive decline with deleterious consequences in everyday life. The lack of specific drugs for the treatment and/or prevention of this pathology makes AD an ever-increasing economic and social emergency. Oligomeric species of amyloid-beta (Aβ) are recognized as the primary cause responsible for synaptic dysfunction and neuronal degeneration, playing a crucial role in the onset of the pathology. Several studies have been focusing on the use of small molecules and peptides targeting oligomeric species to prevent Aβ aggregation and toxicity. Among them, peptide fragments derived from the primary sequence of Aβ have also been used to exploit any eventual recognition abilities toward the full-length Aβ parent peptide. Here, we test the Aβ8-20 fragment which contains the self-recognizing Lys-Leu-Val-Phe-Phe sequence and lacks Arg 5 and Asp 7 and the main part of the C-terminus, key points involved in the aggregation pathway and stabilization of the fibrillary structure of Aβ. In particular, by combining chemical and biological techniques, we show that Aβ8-20 does not undergo random coil to β sheet conformational transition, does not form amyloid fibrils by itself, and is not toxic for neuronal cells. Moreover, we demonstrate that Aβ8-20 mainly interacts with the 4–11 region of Aβ1-42 and inhibits the formation of toxic oligomeric species and Aβ fibrils. Finally, our data show that Aβ8-20 protects neuron-like cells from Aβ1-42 oligomer toxicity. We propose Aβ8-20 as a promising drug candidate for the treatment of AD.
Keywords: Alzheimer, anti-aggregation, fibrils, oligomers
Introduction
Alzheimer’s disease (AD) is considered the most common form of dementia in the elderly population.1 Dementia is a significant contributor to loss of independence, disability, and care home placement and represents one of the costliest long-term pathologies to society, with 85% of costs related to family or social care.2 According to data from the World Alzheimer Report, over 46.8 million people were affected by dementia worldwide in 2015, with a prevision of a doubling of this number in the next 20 years.2−4 To date, despite the high interest of the scientific community, the etiopathogenesis of the disease is not fully understood, and no drugs are still available for the treatment despite the large number of clinical trials5 and the intense research activity on synthetic and/or natural compounds.6−11
AD is characterized by the appearance in the hippocampal region of the brain12 of two kinds of proteinaceous deposits: (i) one mainly constituted by fibrillar aggregates of phosphorylated tau protein inside neuronal cells, called neurofibrillary tangles,13 and (ii) one in the extracellular space, called amyloid plaques, mainly constituted by fibrillar aggregates of amyloid beta protein (Aβ).14 Amyloid plaque composition was demonstrated to be a complex mixture with the presence of 100s of proteins (∼500) and several non-proteinaceous components.15−17
Aβ is the final product of the cleavage of amyloid precursor protein (APP)18 operated by the sequential action of β and γ secretases. Although Aβs spanning 34 to 50 amino acid length are the most common, there are even shorter Aβ isoforms (Aβ1–17/18/19/20) that depend on γ-secretase, but the precise mechanism of their generation is unknown.19−21 It was proposed that an abnormally high concentration of Aβ1-40 and/or Aβ1-42, the two most common isoforms, could result in aggregation into a β-sheet-rich structure, the starting point of Aβ fibrillogenesis.22,23 Aβ aggregation is a complex mechanism which starts with the formation of oligomeric species, suggested to be the most toxic species for cells,24−31 which undergo conformational reorganization into protofibrils and fibrils. Monomeric and fibrillar forms have been demonstrated to be, respectively, protective32−34 and mostly inert35,36 for neuronal cells.
Many studies focused on the primary sequence of Aβ, trying to shed light on the role played by a single amino acid.37 Although a high amount of data is available, the literature is often contradictory. It has been shown that the mainly hydrophobic C-terminal region of Aβ plays a pivotal role in controlling Aβ structure stability and self-assembly.38−41 On the contrary, it is generally accepted that the N-terminal region of Aβ, Aβ1-16, is not able to aggregate and is not cytotoxic.42 Nevertheless, it was also shown that under particular conditions, Aβ1-16 can aggregate and form cytotoxic species containing β-turns.43 Moreover, the N-terminal region of Aβ was demonstrated to control the aggregation rate and fibrillar stability of amyloid fibers.44 In particular, residues Arg5, Asp7, and Ser8 were found to form important inter-molecular contacts stabilizing the overall fibril structure of three-fold symmetry.45
Aβ undergoes several post-translational modifications,19 including the formation of truncated species as the result of physiological enzymatic cleavage.46−49 Many truncated forms of Aβ have been identified in blood plasma samples and human cerebrospinal fluids of AD patients.50 Recently, Abedin et al. reported a structural and aggregation propensity study of seven Aβ fragments with the aim of identifying the region of Aβ which is able to inhibit fibrillogenesis.51 Generally, truncated forms of Aβ are of particular interest since they are known to affect the Aβ aggregation rate. Short Aβ fragments known as β-sheet breakers are able to recognize and tie to the same regions of the parent amyloid peptide, effectively inhibiting accumulation or promoting disaggregation of pre-existing fibrillar amyloids.52−58 In particular, β-sheet breakers Aβ17-21 and Aβ16-20 (KLVFF) have been shown to significantly inhibit amyloidogenic aggregation in vitro.59−63 Unfortunately, these peptide-based systems have a remarkable tendency to self-aggregate and short circulatory half-lives. For this reason, the inclusion of charged residues at the N-terminus could be thought of as a valuable strategy to enhance bioavailability. Here, we explore the ability of the Aβ fragment SGYEVHHQKLVFF (Aβ8-20) to prevent the aggregation and toxicity of Aβ1-40 and Aβ1-42. The choice of this peptide, which has not yet been found in vivo, arises from two main reasons:
-
(i)
the absence of Arg5 and Asp7 is important to prevent the stabilization of the fibril structure.45 Moreover, it is known that angiotensin-converting enzyme is a candidate enzyme for the formation of the 8-x Aβ species,64 although so far, there are no in vivo data supporting this pathway;
-
(ii)
the cleavage in position 20 removes a major part of the Aβ C-terminus region which is known to play an important role in the aggregation process. Moreover, it encompasses the Lys-Leu-Val-Phe-Phe (KLVFF) sequence which was demonstrated to recognize the analogous sequence in the full-length protein.65 Indeed, several studies were performed on KLVFF alone or embedded in the peptide sequence,6,52,66,67 indicating the ability of the KLVFF sequence to both recognize and prevent Aβ aggregation.
We used chemical and biochemical techniques to fully evaluate the behavior and the properties of Aβ8-20. We show that Aβ8-20 invariably maintains a random coil conformation and is not able to form amyloid aggregates by itself. Interestingly, Aβ8-20 suppresses the Aβ1-40 and Aβ1-42 random coil to β-sheet conformational transition and completely prevents their ability to form amyloid aggregates. We also demonstrate, through a combination of mass spectrometry and dot blot assay, that this peptide hampers the formation of Aβ1-42 oligomeric species, which are considered the most toxic species, probably by interacting in the 4–11 region of the protein. Finally, we show that Aβ8-20, which is not toxic per sé, protects neuronal-like cells from Aβ1-42 toxicity. Overall, our data indicate Aβ8-20 as a good candidate for the prevention of cell damage induced by Aβ in AD and help us improve our knowledge of the mechanism underlying the detrimental action of oligomeric and/or prefibrillar Aβ species.
Results and Discussion
Aβ8-20 Adopts a Stable Random Coil Conformation and Does Not Form Amyloid Aggregates
Several fragments of Aβ have been shown to undergo amyloidogenic aggregation.51,68 Although Aβ8-20 lacks the aggregation-prone C-terminus region and residues in the N-terminus responsible for amyloid fiber stabilization, we could not rule out the possibility that this fragment may form amyloid aggregates. To evaluate the aggregation properties of Aβ8-20, we initially performed a well-known thioflavin T (ThT) assay. Aβ8-20 in buffer solution (10 mM MOPS buffer, 100 mM NaCl, pH 7.4) does not aggregate over a time length of 48 h (Figure 1a, blue curve).
Figure 1.
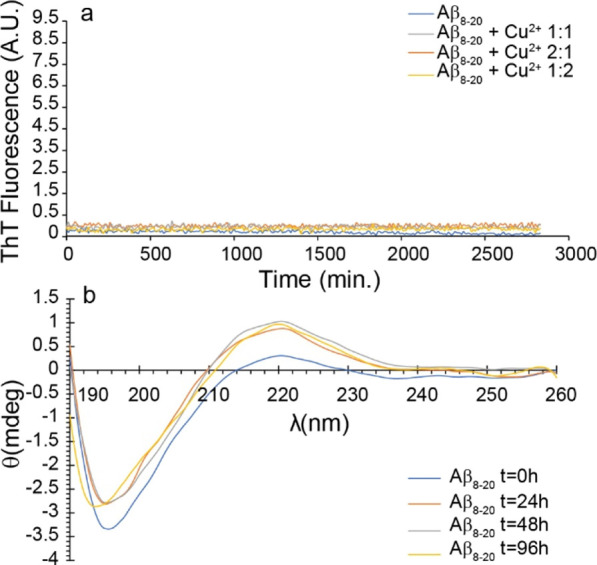
(a) Amyloid aggregation measured by the ThT assay of 10 μM Aβ8-20 (blue curve), Aβ8-20:Cu2+ 1:1 (gray curve), Aβ8-20:Cu2+ 2:1 (orange curve), and Aβ8-20:Cu2+ 1:2 (yellow curve). (b) Secondary structure measured by CD of 10 μM Aβ8-20 at t = 0 (blue curve), t = 24 h (orange curve), t = 48 h (gray curve), and t = 96 h (yellow curve). All the experiments were performed in 10 mM MOPS buffer and 100 mM NaCl, pH 7.4. ThT curves are the average of three independent experiments.
To further test the amyloidogenic properties of the peptide, we performed ThT experiments also under more complex fibrillogenic conditions. Interestingly, also the presence of the Cu2+ ion, which is well known to strongly modulate the aggregation of Aβ depending on the ion/protein ratio,69−73 in sub-stoichiometric (Figure 1a, orange curve), stoichiometric (Figure 1a, gray curve), and over-stoichiometric (Figure 1a, yellow curve) ratios does not induce any aggregation of Aβ8-20.
Circular dichroism (CD) experiments, performed in a time range of 96 h (Figure 1b), reveal that Aβ8-20 adopts a random coil conformation over time. Interestingly, our results differ from those obtained by Abedin et al. for the Aβ11-20 fragment which shows, despite the high sequence homology, a high propensity to form a β-sheet-rich structure.51 The absence of any significant secondary structure transition, typical of amyloid fiber formation, supports well ThT results, although it is not possible to exclude the formation of amorphous aggregates. However, the stable intensity of ellipticity measured by CD experiments suggests that Aβ8-20 does not form any insoluble aggregates over time.
Aβ8-20 Prevents Aggregation and Random Coil to β-Sheet Transition of Both Aβ1-40 and Aβ1-42
Aggregation of both Aβ1-40 and Aβ1-42 peptides in buffer solution shows the typical sigmoidal ThT curve (Figure 2a black curve and Figure 2b black curve). CD spectra show, over time, a random coil to β-sheet secondary structure transition (Figure 2a inset and Figure 2b inset). Aβ1-42, which is considered the most toxic species,74,75 forms amyloid fibers faster than Aβ1-40, which, in turn, is the most abundant species in vivo76−79 but is considered less toxic. Noteworthy, the presence of Aβ8-20 in a 1:1 concentration ratio completely suppresses Aβ1-40 aggregation (Figure 2a red curve) and prevents the random coil to β-sheet transition (Figure 2a inset) after 24 h incubation. A similar effect was observed for samples containing Aβ1-42 (Figure 2b, black curve). Aβ1-42 in the presence of Aβ8-20 shows only a small residual increase in the ThT signal (Figure 2b, red curve). Interestingly, CD spectra (Figure 2b, inset) reveal a mixture of the random coil and β-sheet structure at t = 0 which evolves over 24 h into a random coil/α-helix. Thus, our data suggest that Aβ8-20 could interfere with the aggregation process of both Aβ1-40 and Aβ1-42 already at a 1:1 molar ratio, suggesting a higher efficiency than, for example, that of Aβ11-20.51
Figure 2.
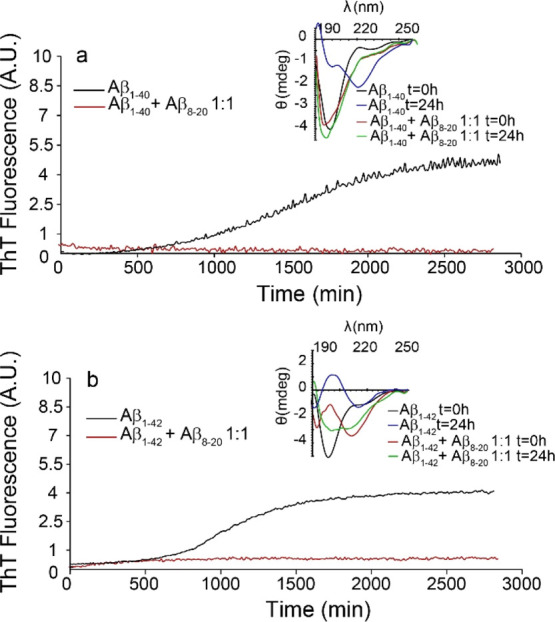
(a) Amyloid aggregation measured by ThT assay of 10 μM Aβ1-40 (black curve) and Aβ1-40:Aβ8-20 1:1 (red curve). Inset: CD spectra of 10 μM Aβ1-40 at t = 0 (black curve) and t = 24 h (blue curve) and Aβ1-40:Aβ8-20 1:1 at t = 0 (red curve) and t = 24 h (green curve). (b) Amyloid aggregation measured by ThT assay of 10 μM Aβ1-42 (black curve) and Aβ1-42:Aβ8-20 1:1 (red curve). Inset: CD spectra of 10 μM Aβ1-42 at t = 0 (black curve) and t = 24 h (blue curve) and Aβ1-42:Aβ8-20 1:1 at t = 0 (red curve) and t = 24 h (green curve). All the experiments were performed in 10 mM MOPS buffer and 100 mM NaCl, pH 7.4. ThT curves are the average of three independent experiments, and the single traces and error bar are reported in the Supporting Information (Figure S3).
To confirm ThT results, we acquired transmission electron microscopy (TEM) images for samples containing Aβ1-42 100 μM alone (Figure 3a) and Aβ1-42 100 μM: Aβ8-20 100 μM (Figure 3b) after 96 h incubation at 37 °C. β1-42 showed the classical fiber network, while the presence of the Aβ8-20 fragment almost completely inhibited the fiber formation of Aβ1-42 which were not detected over the entire surface of the grid. This result confirms what we observed by ThT experiments.
Figure 3.

TEM image of (a) Aβ1-42 100 μM after 96 h incubation and (b) Aβ1-42 100 μM: Aβ8-20 100 μM after 96 h incubation.
Aβ8-20 Reduces the Dimension of Aβ1-40 Soluble Aggregates
To evaluate the effect of the presence of Aβ8-20 on the size distribution of soluble species of Aβ, we resorted to dynamic light scattering (DLS) measurements. We chose the Aβ1-40 isoform since it is known to aggregate more slowly than Aβ1-42, giving us enough time to evaluate the dimension of soluble aggregated species. Thus, the growth of the Aβ1-40 aggregates was monitored in the absence and in the presence of Aβ8-20. Data collected at t = 0, 24 h, and 6 days as the analysis of the intensity (%) of scattering objects are reported in Figure 4. The analysis indicated that freshly prepared samples of Aβ1-40 and Aβ8-20 form structures with mean hydrodynamic diameter around 50 and 60 nm, respectively (Figure 4a, red and black curves, respectively). After 24 h, in the sample containing only Aβ1-40, aggregation phenomena generated a population of larger aggregates with size centered at 1505 nm (64%) in addition to a population centered at 190.9 nm (36%), whereas only a population centered at 116 nm was observed in the presence of Aβ8-20 (Figure 4b). After 6 days, two main populations with mean hydrodynamic diameter centered at 1663 (60%) and 102.7 nm (40%) and at 850.3 (33%) and 188.7 nm (67%) were observed for Aβ1-40 alone and in the presence of Aβ8-20, respectively (Figure 4c). The data collected clearly indicated the reduction of the dimension of Aβ1-40 soluble aggregates in the presence of the Aβ8-20 fragment.
Figure 4.
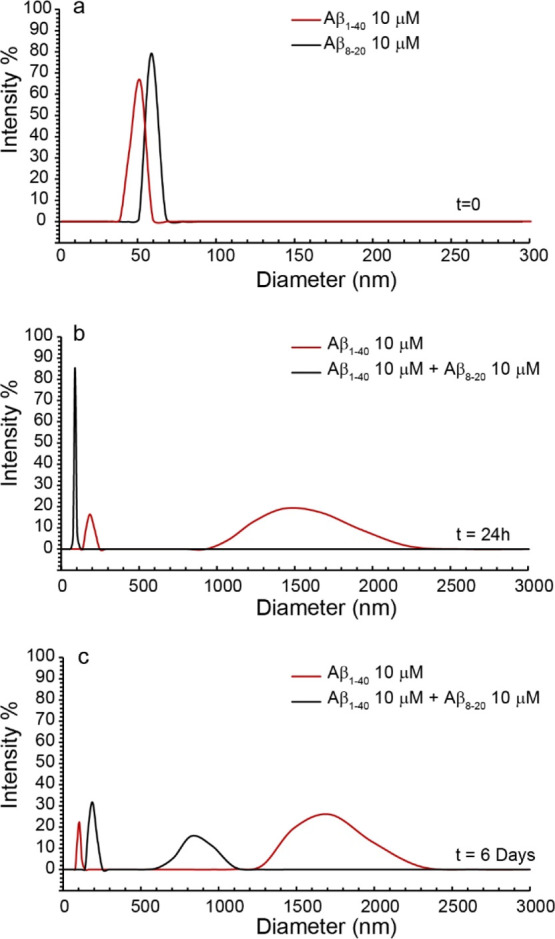
Intensity-weighted hydrodynamic diameter distribution for (a) 10 μM Aβ1-40 and Aβ8-20 at t = 0 and (b) 10 μM Aβ1-40 (red line) and Aβ1-40:Aβ8-20 1:1 concentration ratio (black line) at t = 24 h and (c) at t = 6 days. Curves are the average of three independent experiments. Measures were performed at 37 °C in 10 mM phosphate buffer.
Aβ8-20 Hampers Aβ1-42 Oligomer Formation
Since Aβ1-42 oligomers have been demonstrated to be the main species responsible for Aβ toxicity in vitro and in vivo,24−27 we resorted to matrix-assisted laser desorption ionization mass spectrometry (MALDI-MS) to investigate the effect of the Aβ8-20 peptide on the formation of Aβ1-42 oligomers. Indeed, MALDI-MS can acquire the m/z values of peptides and proteins predominately in the singly charge state, enabling a direct indication of the mass of Aβ1-42 oligomers. In particular, the m/z values reported in the mass spectra acquired by MALDI-MS give a direct indication of the mass of peptides and proteins, revealing the monomeric/multimeric composition of Aβ samples.80−82 Nevertheless, a drawback of the MALDI approach in the characterization of multimeric forms of Aβ1-42 was the use of organic solvent acetonitrile (ACN) during sample preparation. In particular, the ACN/H2O (1:1, v/v) solvent mixture, needed for both matrix dissolution and rapid evaporation of the solvent after the deposition of the sample on target plates (see the Materials and Methods section), prevents the hydrophobic interactions within oligomers affecting the oligomer composition.83 Therefore, we carried out MALDI experiments at Aβ1-42 concentrations higher (100 μM) than those generally used in MALDI investigations (5–10 μM) to aid the formation of high-molecular weight (MW) oligomers84 and prevent the complete dissolution of oligomers during deposition of the sample (Aβ/matrix mixture) on target plates.
An Aβ1-42 sample was analyzed by MALDI-time of flight (TOF) after 2, 4, 6, 8, and 24 h of incubation at 37 °C. The mass spectra acquired (Figure 5a) were compared with those recorded when an equimolar amount of the Aβ8-20 peptide was added to the Aβ1-42 sample solution (Figure 5b).
Figure 5.

MALDI-MS spectra acquired in the linear mode (m/z range = 7000–20,000) at different incubation times of (a) Aβ1-42 (c = 100 μM) in PBS buffer (5 mM) pH 7.8 and (b) Aβ1-42/Aβ8-20 in PBS buffer (5 mM) pH = 7.8 (cAβ = cAβ8-20 = 100 μM).
The MALDI-TOF spectrum recorded after 8 h of incubation showed (Figure 5a) the formation of a series of signals corresponding to dimeric {[(Aβ1-42)2 + H]+m/z = 9029} and trimeric {[(Aβ1-42)3 + H]+m/z = 13540} oligomers (Figure 5a). These signals could be related to the formation of high-MW oligomers that were partially disrupted when the Aβ1-42 sample was mixed with matrix solution. Interestingly, the mass spectrum of the sample containing the Aβ1-42/Aβ8-20 mixture, recorded after 8 h of incubation (Figure 5b), showed a clear reduction of the signal’s intensity corresponding to the Aβ1-42 oligomers. The m/z signal corresponding to the Aβ1-42 dimer can be observed only in the mass spectrum acquired after 24 h of incubation.
These findings are in keeping with the results observed in ThT experiments and support the hypothesis that Aβ8-20 may interfere with Aβ aggregation by means of the formation of a noncovalent adduct with the amyloid peptide.
To further confirm the interaction between Aβ8-20 and Aβ1-42 and investigate oligomer formation, we performed gel electrophoresis after incubation of freshly prepared Aβ1-42 in the presence or in the absence of Aβ8-20 for 48 h at 4 °C. We tested three different molar ratios of Aβ1-42/Aβ8-20 (1:1; 1:5; and 1:10), and after incubation, each sample was characterized for its composition of Aβ aggregates. The samples were loaded onto a polyacrylamide gel and transferred onto a nitrocellulose membrane (Figure 6). As expected, we found that Aβ1-42 alone aggregates into small oligomers ranging from 8 to 16 kDa, representing dimers, trimers, and tetramers. In the presence of Aβ8-20, the signal observed was less intense when incubated at the molar ratio of 1:1 and became very weak at 1:5 and 1:10. These data suggest the ability of Aβ8-20 to bind the full-length peptide, inhibiting its aberrant aggregation by hampering oligomer formation.
Figure 6.
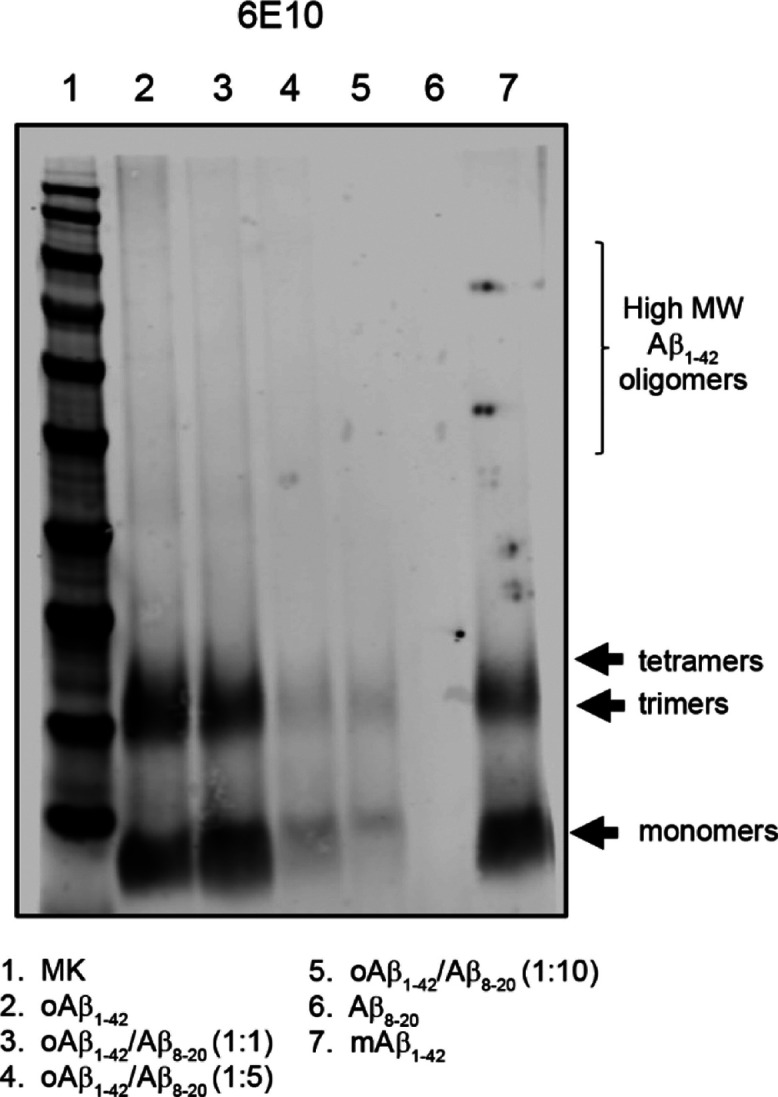
Representative western blot of Aβ oligomers prepared in the presence or absence of Aβ8-20. Samples were separated onto a 4–12% bis·tris SDS-PAGE gel and blotted with anti-Aβ N-terminal 1–16 mouse monoclonal antibody 6E10 (1:500).
Aβ8-20 Interacts with the N-Terminal Region of Aβ1-42
Noteworthily, it is clear from Figure 6 that samples containing Aβ1-42/Aβ8-20 in molar ratios 1:5 and 1:10 not only show a decrease in the intensity of trimer and tetramer signals, but also monomer bands seem to disappear. To better investigate this unexpected result, we spotted the co-incubated samples Aβ1-42/Aβ8-20 at the three different molar ratios (1:1; 1:5; 1:10) onto a nitrocellulose membrane that we probed with the 6E10 antibody (Figure 7a). We used Aβ1-42 oligomers and freshly prepared monomers as controls. Even in this case, we found that when Aβ8-20 was incubated with Aβ1-42 at more than the 1:1 molar ratio, a clear signal decrease was evident. Controls confirmed that the antibody was working properly, revealing the presence of Aβ in both incubated and freshly spotted samples.
Figure 7.

Dot blot analysis of Aβ oligomers prepared in the presence or absence of Aβ8-20. Samples were spotted after 48 h incubation at 4 °C under gentle rotation. Membranes were blotted with the following antibodies: anti-Aβ N-terminal 1–16 mouse monoclonal antibody 6E10 (1:100); anti-Aβ 17–24 mouse monoclonal antibody 4G8 (1:100); or anti-oligomer A11 rabbit polyclonal antibody (1:100).
As for electrophoresis data, these results suggest that the presence of the peptide strongly modulates Aβ1-42 self-assembly. To prove this, we used a different antibody, anti-Aβ 4G8 (Figure 7b), which is reported to react to amino acid residues 17–24 with the epitope lying within amino acids 18–22 of β-amyloid (VFFAE). Targeting a different epitope led to a different signal pattern in which we detected a clear staining even in the case of co-incubated samples, revealing that the lack of 6E10 signals previously observed could be due to the presence of the small peptide Aβ8-20 along the Aβ1-42 reactive sequence.
6E10 is, in fact, directed against amino acids 1–16 of the Aβ sequence. During the incubation time, the binding of Aβ8-20 to Aβ1-42 could hinder the interaction between the antibody and its target sequence (Scheme 1).
Scheme 1. Schematic Representation of Anti-Aβ Antibody Interaction with Aβ1-42 in the Presence of the Aβ8-20 Fragment.

We finally used the anti-oligomer antibody A11 to assess the conformation of each spotted sample and the ability of Aβ8-20 to effectively interfere with Aβ1-42 assembly. As expected, A11 strongly reacted with the Aβ incubated alone and very slightly with Aβ monomers freshly spotted. The increasing presence of the Aβ8-20 fragment during the incubation time leads to a decrease in the antibody signal underlying the reduction of the oligomeric species formed.
On the basis of these findings, we moved onto limited proteolysis experiments to better clarify the site of interaction between Aβ8-20 and Aβ1-42. Indeed, these interactions could occur at the peptide bonds involved in the proteolytic cleavage affecting, in turn, enzyme’s accessibility to the cleavage sites.
To this scope, we used α-chymotrypsin enzyme that selectively catalyzes the hydrolysis of peptide bonds at the C-terminal side of tyrosine, phenylalanine, tryptophan, and leucine residues. We analyzed by MALDI-TOF the Aβ1-42 peptide fragments generated at the initial stage, namely, after 10 min of α-chymotrypsin digestion, where the hydrolysis rate is higher, and small differences in the peptide interactions with Aβ1-42 would be more pronounced, as observed in our previous studies.6 The identified peptide fragments are indicated in Scheme 2. A quite similar proteolytic pattern was observed in the MALDI-TOF spectrum of Aβ1-42 digested in the presence of the Aβ8-20 peptide.
Scheme 2. Aβ1-42 Proteolytic Pattern after 10 min of α-Chymotrypsin Digestion.

Aβ1-42 (10 μM) in PBS buffer (5 mM) pH 7.8 and an enzyme/substrate ratio of 1:200 w/w.
Despite the low reproducibility of MALDI measurements, a comparative analysis of the signal intensity averaged over 15 replicate measurements (Figure 8) revealed some reasonable differences. In particular, the reduction of signal intensity of the peaks assigned to the peptide fragments Aβ5-42 and Aβ11-42, when the Aβ8-20 peptide was added to the Aβ1-42 sample solution (Figure 8, orange bar), indicates a lower hydrolysis rate at the cleavage sites of Phe4 and Tyr10. This may suggest a lower accessibility of these cleavage sites. Interestingly, the CD spectra of Aβ1-42 in the presence of Aβ8-20 show a mixture of the random coil and β-sheet structure at t = 0 h (Figure 2b inset, red curve) which evolves over 24 h into a random coil/α-helix conformation (Figure 2b inset, orange curve). The structuring effects within the polypeptide backbone can alter peptide chain flexibility of the Aβ1-42 N-terminal domain, affecting the cleavage of the peptide bonds by a protease.
Figure 8.
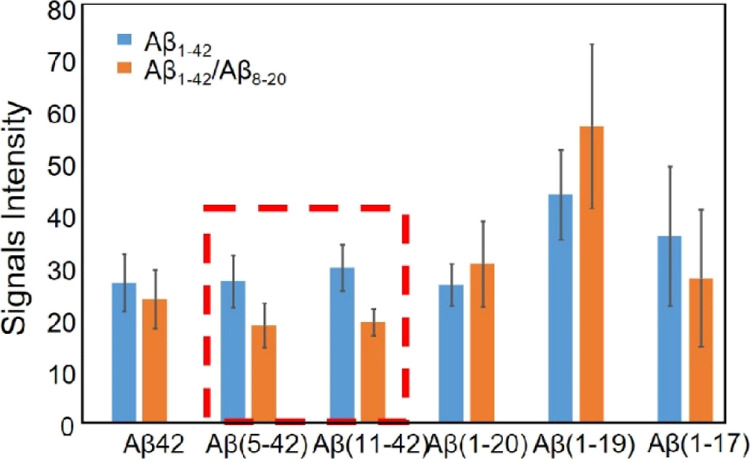
Signal intensities of digestion fragments of Aβ1-42 protein in Aβ1-42 (blue bar) and Aβ1-42/Aβ8-20 samples (orange bar), after 10 min of α-chymotrypsin digestion.
Aβ8-20 Prevents Aβ1-42 Toxicity in Differentiated SH-SY5Y Cells
To have a functional readout of the data, we investigated the protective activity of Aβ8-20 both per sé and toward the toxicity of Aβ1-42 oligomers by using the well-known viability test, 3-(4 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. We used a neuronal-like model obtained by the neuroblastoma cell line, SH-SY5Y, fully differentiated with all-trans-retinoic acid (RA). Aβ8-20 did not show any toxic activity. Even at higher concentrations and after 48 h exposure, Aβ8-20 was not significantly toxic for the cell, whose viability was comparable to that of controls (Figure 9).
Figure 9.
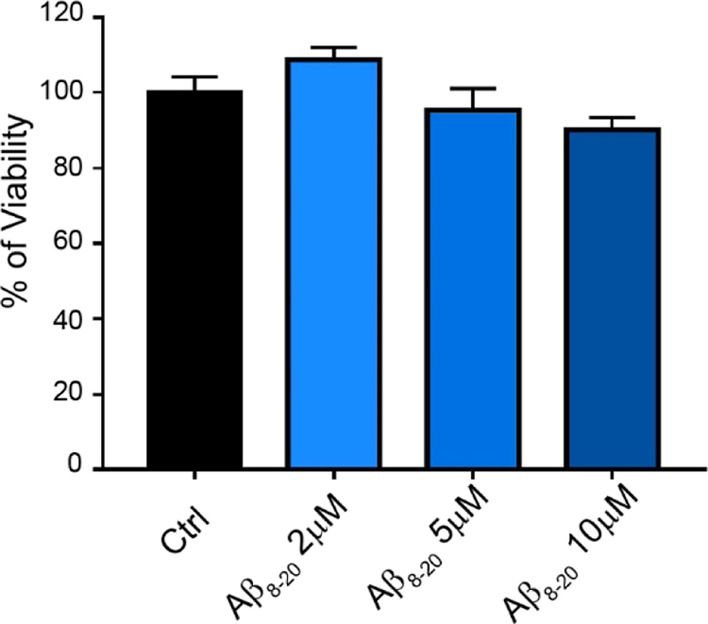
MTT assay of fully differentiated SH-SY5Y cells treated for 48 h with increasing concentrations of Aβ8-20 (2, 5, and 10 μM). Bars represent means ± SEM of three independent experiments with n = 3 each.
The ability of Aβ8-20 to prevent oligomer toxicity was tested by incubating Aβ1-42 for 48 h at 4 °C alone or in combination with Aβ8-20, added at the molar ratios of 1:1; 1:5; and 1:10, previously used for dot blot analysis. After incubation, cells were exposed to oligomers at the final concentration of 2 μM, and the resulting toxicity was compared to the effects of the other co-incubated solutions (Figure 10). As expected, after 48 h of treatments, oligomers were toxic, affecting the cell viability by approximately a 30% reduction. Unlike the lower (2 μM) concentration, both 1:5 and 1:10, corresponding, respectively, to 10 and 20 μM Aβ8-20, were able to counteract oligomer toxicity.
Figure 10.
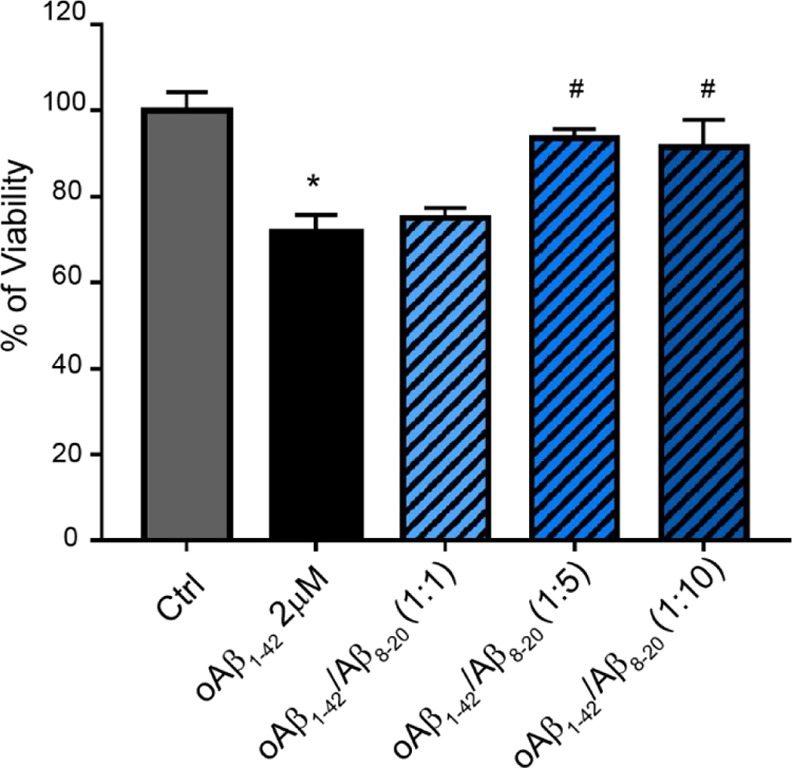
MTT assay of fully differentiated SH-SY5Y cells treated for 48 h with Aβ oligomers prepared in the presence or absence of different molar ratios of Aβ8-20 (1:1; 1:5; and 1:10). Samples were incubated at 4 °C under gentle rotation for 48 h. Bars represent means ± SEM of three independent experiments with n = 3 each. ***P < 0.001 vs Ctrl by one-way ANOVA + Tukey test and # <0.001 vs Aβ1-42 by one-way ANOVA + Tukey test.
Conclusions
Over the past years, the anti-aggregating properties of several natural compounds, synthetic derivatives, and peptides have attracted the attention of the scientific community as potential drugs for the treatment of AD. Furthermore, the growing evidence supporting a role for Aβ in neuronal physiology32,34,85 has highlighted the need to find novel potential drugs capable of blocking the progression of the disease and preserving the natural functions of Aβ, which are normally lost following its self-assembly. Although difficult due to their transient nature, oligomers represent the main targets to be addressed, while the activity of Aβ is still a matter of investigation and only partially clarified. To date, none of the proposed molecules have been successfully proven to halt and/or prevent the pathology.
In this paper, we present data about a newly synthesized fragment of Aβ, encompassing residues 8–20 (Aβ8-20), which has shown promising features to be considered in AD therapy.
Aβ8-20 freshly solubilized has a hydrodynamic diameter of ∼60 nm (Figure 4a) and assumes a random coil conformation which does not evolve into a β-sheet-rich structure over time (Figure 1b) according to the absence of any amyloid structure as evidenced by ThT assay under several conditions (Figure 1a). Thus, as expected, the lack of the hydrophobic C-terminus and residues Arg5 and Asp7 involved in fiber stabilization in the N-terminal region of the protein hampers the formation of the amyloid structure. Notably, this peptide is not toxic toward cells (Figure 9) up to a concentration of 10 μM. Aβ8-20 almost completely inhibits fiber formation of both Aβ1-40 and Aβ1-42 (Figure 2a,b, respectively, and Figure 3) which mostly remains in a random coil conformation over time (Figure 2a inset and Figure 2b inset, respectively). Moreover, the presence of Aβ8-20 significantly reduces the dimension of soluble aggregated species of Aβ1-40 over time (Figure 4b,c), suggesting a strong modulation of oligomer formation. This hypothesis was corroborated by MALDI experiments combined with dot blot analysis which clearly show that Aβ8-20 efficiently reduced the formation of the Aβ1-42 dimer and trimer species (Figure 5) in a concentration-dependent way (Figure 6). The presence of the self-recognizing KLVFF sequence suggests that Aβ8-20/Aβ1-42 interaction should occur in the N-terminal region as confirmed by the dot blot analysis (Figure 7) which shows that the interaction of Aβ8-20 with Aβ1-42 hinders the link between the 6E10 antibody and its target sequence (1–16) on Aβ. Limited proteolysis experiments confirm these data, indicating the 4–11 region as the most involved in the interaction (Figure 8). Finally, Aβ8-20 proved to completely protect, in a dose-dependent way, SH-SY5Y cells from the toxicity of Aβ1-42 oligomers (Figure 10).
The whole of our results clearly indicates Aβ8-20 as an interesting Aβ fragment that combines the required β-sheet breaker activity with promising features such as the lack of toxicity on neuronal cultures and the effective protective properties against oligomer-mediated cellular death.
These properties make the fragment a good candidate for a potential disease-modifying drug in AD therapy, encouraging a more in-depth study on the biological and molecular features of the peptide.
Materials and Methods
Reagents
Aβ 1–40 (Aβ1-40) and Aβ 1–42 (Aβ1-42) were purchased from Bachem (Bubendorf, Switzerland) with a purity of ≥95%. Amyloid fragments 8–20 (Aβ8-20) were synthesized and purified in our laboratory with a purity of ≥98%. ThT, ascorbic acid, NaCl, and all other salts were purchased from Sigma-Aldrich (St. Louis, MO, USA). The MALDI matrices 3,5-dimethoxy-4- hydroxycinnamic acid (sinapinic acid, SIN) and α-cyano-4-hydroxycinnamic acid (α-CHCA) were purchased from Sciex and used without further purification. Bovine serum albumin, immunoglobulin G, and peptide mass standard calibration kits were purchased from Sciex. ACN and trifluoroacetic acid (TFA) (mass spectrometry-grade) were purchased from Fisher Scientific. Alpha-chymotrypsin was purchased from Sigma-Aldrich. All aqueous solutions were prepared using a Barnstead NanoPure system with a 0.2 mm membrane filter (Thermo Scientific).
Amyloid Fragment 8–20 Synthesis
The Aβ 8–20 (Aβ8-20) were synthesized by a fully automated microwave-assisted solid-phase peptide synthesis following the Fmoc/tBu strategy on a liberty peptide synthesizer (CEM) starting from Rink amide AM resin (substitution 0.59 mmol/g). After the resin swelling in dimethylformamide (DMF), all orthogonally protected Fmoc amino acids were introduced according to the following N,N′-Diisopropylcarbodiimide (DIC)/Oxyma activation method consisting of (1) Fmoc deprotections (20% piperidine in DMF); (2) washes (3×) with DMF; (3) couplings with protected amino acids (5 equiv, 0.2 M in DMF), Oxyma pure (5 equiv, 1 M in DMF), and DIC (5 equiv, 0.5 M in DMF) prepared in separate bottles; and (4) washes (3×) with DMF. The following instrumental conditions were used for each coupling cycle: (a) 220 W, 65 °C, 30 s and (b) 25 W, 90 °C, 90 s. The instrumental conditions used for the deprotection cycle were (a) 220 W, 70 °C, 30 s and (b) 25 W, 75 °C, 30 s. After the last Fmoc deprotection, N-terminal acetylation was carried out with Ac2O (100 μL/200 mg of resin) in DMF (2 × 10 min). The cleavage of the peptide from the resin, with concomitant deprotection of acid-labile amino acid side-chains, was achieved by treatment of peptide–resin with TFA/Triisopropylhydrosilane/H2O (95:2.5:2.5 v/v/v, 10 mL) for 2.5 h at room temperature and with magnetic stirring. The resin was filtered, and the crude peptide was recovered by precipitation with freshly distilled diisopropyl ether. The purification of crude Aβ8-20 was carried out by preparative reversed-phase high-performance liquid chromatography (RP-HPLC) using a SHIMADZU LC-20A chromatography system equipped with an SPD-M20A photodiode array detector with detection at 222 and 254 nm. A Jupiter 10u Proteo C12 250 × 21.2 mm (90 Å pore size, AXIA Packed) column was used. The peptides were eluted at a flow rate of 10 mL/min according to the following protocol: from 0 to 3 min, isocratic conditions in 90% solvent A (H2O containing 0.1% TFA) followed by a 3 min linear gradient from 10 to 45% B (CH3CN containing 0.1% TFA) and then a 4 min linear gradient from 45 to 50% B and finally 5 min isocratic conditions in 50% B. Fractions containing the desired product were collected and lyophilized. The purity of the peptide was checked by analytical RP-HPLC using a Phenomenex Kinetex XB- C18 analytical column (pore size: 100 A, particle size: 5 μm, column length: 250 mm, and internal diameter: 4,60 mm). A linear gradient of ACN (containing 0.1% TFA) and water (containing 0.1% TFA) (90:10 water–acetonitrile to 0:100 water–acetonitrile over 15 min and at a flow rate of 1 mL min–1) was used. Sample identity was confirmed by MALDI-MS. Calculated mass 1632.81; observed: [M + H]+ = 1632.96; [M + Na]+ = 1653.87; [M + K]+ = 1670.85.
Peptide Preparation
To prevent the presence of any preformed aggregates, Aβ1-40, Aβ1-42, and Aβ8-20 were initially dissolved in hexafluoroisopropanol (HFIP) at a concentration of 1 mg/mL and then lyophilized overnight. To be used for the experiments, the lyophilized powder was initially dissolved in 1 mM NaOH to obtain a stock solution with a final concentration of 100 μM. Each stock solution was used immediately after preparation by diluting it in the opportune buffer solution to reach the concentration needed for experiments.
Thioflavin T Assay
Kinetics of Aβ1-40, Aβ1-42, and Aβ8-20 fiber formation was measured using ThT assay. Samples were prepared by diluting, in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer, stock solution of Aβ8-20, Aβ1-40, Aβ1-42, or a combination to reach the final concentration. Copper, where present, was added from a stock solution at the indicated concentration. ThT was then added to a final concentration of 20 μM. Experiments were carried out in Corning 96-well non-binding surface plates. Time traces were recorded using a Varioskan (Thermo Fisher, Walham, MA) plate reader using a λex of 440 nm and a λem of 485 nm at 37 °C, shaking the samples for 10 s before each read. All ThT curves represent the average of three independent experiments.
Circular Dichroism
CD spectra were acquired using a J-810 spectrometer (Jasco, Japan) under a constant flow of N2 at room temperature. The CD spectra were recorded for Aβ1-40 and Aβ1-42 (10 μM) monomers in the absence and presence of Aβ8-20 (10 μM) in the 1:1 molar ratio. The lyophilized samples were dissolved in 1 mM NaOH and then diluted to obtain a concentration of 10 μM for Aβ alone and the mixture. The CD measurements were carried out in aqueous solution (1 × 10–3 M MOPS buffer and 0.05 M NaF). A 0.5 cm path length quartz cuvette was used to acquire the far-UV CD spectra (190–260 nm), at a scan speed of 50 nm/min. 10 scans were collected. The measurements were performed in triplicate. The CD intensities were expressed as θ(mdeg).
Transmission Electron Microscopy
Samples were prepared by incubating at 37 °C for 96 h 100 μL of phosphate buffer solutions 10 mM and 100 mM NaCL, pH 7.4 containing Aβ1-42 100 μM or Aβ1-42 100 μM: Aβ8-20 100 μM. After incubation, 3 μL of each sample was deposited onto a copper grid and allowed to adsorb for 5 min before the grid was rinsed with H2O twice. Samples were dried overnight and then stained with uranyl acetate. TEM micrographs were acquired using JEOL JEM 2010F using a 2 K × 2 K Gatan ORIUS camera. Samples have been observed in low magnification using the image formed inside the transmitted beam on the focal plane. This method is useful in low-magnification detection of weak objects, without any change in the focus configuration of the microscope. By switching between the out-of-focus image in the focal plane and the in-focus image in the image plane, it is possible to scan a large area of the grid with enhanced detection capability and easily come back to the normal image configuration.
Dynamic Light Scattering
DLS measurements were carried out on a Zetasizer NanoZS90 Malvern Instrument (UK) equipped with a 633 nm laser at a scattering angle of 90° and at 37 °C. The samples of Aβ1-40 and Aβ8-20 (5 μM) were prepared under the same experimental conditions as those described above. Each measurement was performed three times.
Mass Spectrometry
MALDI mass spectra were obtained using a 5800 MALDI-TOF/TOF mass spectrometer (Sciex) equipped with an automated single-plate sample-loading system, 1 kHz OptiBeam On-Axis Laser Nd/YAG 349 nm wavelength, delayed extraction (DE), two acceleration regions, a two-stage reflector mirror, and a 1000 MHz digitizer. The instrument was operated in the reflectron mode (m/range: 800–5000) and linear mid-molecular weight mode (m/range: 7000–20,000). When operating in the linear mode, the instrument’s acquisition parameters were set to optimize detection sensitivity of Aβ oligomers. In particular, the lowest possible laser intensity was used to minimize dissociation and enable the detection of Aβ1-42 oligomers. Moreover, the detection of high-MW oligomers was hindered by the presence of the monomers. Therefore, mass spectra were acquired starting at higher m/z values to enhance the sensitivity for the large-MW species. DE was applied, and the delay time was set according to the MW of the analytes to optimize resolution of their molecular ion. Mass spectra were acquired by averaging 300 to 600 shots. Sinapinic acid and α-CHCA were prepared by dissolving 10 mg of matrices in 1 mL of 50% ACNe in 0.05% TFA and 1 mL of 30% ACN in 0.1% TFA, respectively. Standard kits were used to calibrate the mass scale of the MALDI mass spectrometer. The peptide mass standard kit includes des-Arg1-Bradikynin, angiotensin I, Glu1-Fibrinopeptide B, adrenocorticotropic hormone (ACTH) (clip 1–17), ACTH (clip 18–39), and ACTH (clip 7–38), and it was used to cover a mass range of 800 to 4000 Da. Bovine insulin, E. coli thioredoxin, and horse apomyoglobin were used to cover a mass range from 4000 to 20,000 Da. Aβ1-42 samples were monomerized to remove any preformed aggregates using the procedure described above. Stock 1 solutions of Aβ1-42 and Aβ8-20 were prepared by dissolving 0.1 mg of each lyophilized peptide in HFIP (stock 1 = 1.5 mM). An opportune amount of each stock solution was diluted in phosphate buffer solution (5 mM, pH 7.8) to a concentration of 50 μM (dtock 2) and mixed to obtain stock solutions to be used for the experiments. The Aβ1-42 sample and the equimolar mixtures of Aβ1-42/Aβ8-20 were prepared from stock 2 solutions and stock 1 solution for a final concentration of 5 μM for limited proteolysis experiments and 100 μM for oligomer experiments, respectively. For limited proteolysis experiments, a fresh stock of α-chymotrypsin (1.0 mg/mL) was made with HCl (1 × 10–3 mol dm–3), and then, an appropriate volume of the enzyme stock solution was added to Aβ1-42 and Aβ1-42/Aβ8-20 samples for a final enzyme/substrate ratio of 1:200 w/w. Solutions were incubated at 25 °C for 10 min. For MALDI-TOF measurements, samples were analyzed using the dried-droplet preparation methods. In particular, 1 to 2 μL of the sample and 1 to 2 μL of matrix solution were mixed into a 0.5 mL tube, and 1 μL of this mixture was deposited on a stainless steel 384-well plate. The mixture samples on the plate were dried by evaporation of the solvent at room temperature till a thin microcrystalline layer of the sample/matrix occurred. All the samples were spotted in three different wells of the plate (triplicate), and five mass spectra were recorded for every spot. MS data were imported into freely available open-source software mMass (http://www.mmass.org). Mass spectra acquired for each sample (15 spectra) were averaged, and monoisotopic peaks were automatically picked. Theoretical m/z values of Aβ1-42, Aβ8-20, and peptides resulting from in silico digestion of amyloid protein were compared with the m/z values assigned to experimental mass spectra. Peptides matching successfully, within a tolerance of 0.05 Da, were annotated. Moreover, mass spectra were exported as a peak list and processed using Excel (Microsoft) software to evaluate the 95% confidence interval of each signal intensity assigned.
Dot Blot Analysis
The Aβ1-42 sample (100 μM) was incubated for 48 h at 4 °C under gentle rotation in the presence or absence of different molar ratios of Aβ8-20 (1:1; 1:5; and 1:10). Then, samples were spotted onto a nitrocellulose membrane. The membrane was blocked with Odyssey blocking buffer (LiCor, Biosciences) at room temperature for 1 h. After blocking, the membrane was probed overnight at 4 °C and with gentle shaking with the following antibodies: anti-Aβ N-terminal 1–16 mouse monoclonal antibody 6E10 (1:100) (BioLegend), anti-Aβ 17–24 mouse monoclonal antibody 4G8 (1:100) (BioLegend), or anti-oligomer A11 rabbit polyclonal antibody (1:100) (Invitrogen, Thermo Fisher). Finally, the membrane was repeatedly washed and exposed to the anti-mouse antibody labeled with the IRDye secondary antibody (1:20.000 Li-Cor Biosciences) for 45 min at room temperature. Hybridization signals were detected with the Odyssey CLx Infrared Imaging System (LI-COR Biosciences).
Western Blot Analysis
Aβ1-42 (100 μM) alone and in combination with different molar ratios of Aβ8-20 (1:1; 1:5; and 1:10) was incubated at 4 °C for 48 h to form Aβ oligomers. After incubation, the amount and size of Aβ aggregates were determined by Western blot analysis. A volume of 25 μL of each unheated sample was loaded onto a precast Bis-Tris gel (Bolt 4–12%, Life Technologies) with 2-morpholin-4-yl ethanesulfonic acid. Samples were transferred onto a nitrocellulose membrane (0.2 mm, Hybond ECL, Amersham Italia) by using a wet transfer unit Mini Blot Module (Life Technologies). Membranes were blocked in Odyssey blocking buffer (Li-COR Biosciences) and incubated at 4 °C overnight with anti-Aβ N-terminal 1–16 mouse monoclonal antibody 6E10 (1:500) (BioLegend). A secondary goat anti-mouse antibody labeled with infrared dye (1:20.000) was used at room temperature for 45 min. Hybridization signals were detected with the Odyssey CLx infrared imaging system (LI-COR Biosciences, Lincoln, NE).
Cell Culture and MTT Assay
The neuroblastoma cell line, SH-SY5Y, was maintained in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (Gibco, Thermo Fisher) supplemented with 10% heat-inactivated (HI) fetal calf serum (Gibco, Thermo Fisher), 100 mg/mL penicillin and streptomycin (Gibco, Thermo Fisher), and 2 mM l-glutamine at 37 °C and 5% CO2. Two weeks before experiments, 5 × 103 cells were plated on 96-well plates in DMEM-F12 with 5% HI fetal calf serum. The percentage of serum was gradually decreased until it was 1% of the total. All-trans-RA (Sigma), 5 μM, was used to promote neuronal differentiation, and medium-containing RA was changed every 3 days. Fully differentiated SH-SY5Y cells were treated with increasing concentrations of Aβ8-20 (2, 5, and 10 μM). After 48 h treatment, cultures were incubated with MTT (5 mg/mL) for 2 h at 37 °C and then lysed with dimethyl sulfoxide (DMSO), and the formazan production was evaluated in a plate reader through the absorbance at 570 nm.
Anti-Oligomerization Activity
To prepare Aβ1-42 oligomers, 1 mg of Aβ1-42 (HFIP-treated) was first dissolved in 5 mM DMSO. A solution of 100 μM Aβ1-42 in ice-cold DMEM F-12 without phenol red was prepared and allowed to oligomerize for 48 h at 4 °C according to the Lambert protocol86 with some modifications as previously described.32 To evaluate the ability of Aβ8-20 to inhibit toxic Aβ oligomerization, Aβ1-42 was incubated in the presence or absence of different molar ratios of Aβ8-20 (1:1; 1:5; and 1:10). After 48 h incubation at 4 °C under gentle rotation, Aβ1-42/Aβ8-20 samples were applied to the differentiated SH-SY5Y cells at the final concentration of 2 μM Aβ1-42. The ability of Aβ8-20 to prevent Aβ oligomer formation and toxicity was evaluated by measuring cell viability after 48 h treatment by MTT assay.
Acknowledgments
The authors thank Dr. Corrado Bongiorno from CNR- Istituto per la Microelettronica e i Microsistemi, Catania, Italy, for acquiring the TEM images and for his precious help in analyzing data. The authors thank CNR for partial financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.2c00720.
Chromatographic profile of Aβ8-20; FT-MS spectra of Aβ8-20; and single ThT traces of Aβ1-40 and Aβ1-42. (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Goedert M.; Spillantini M. G. A Century of Alzheimer’s Disease. Science 2006, 314, 777–781. 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- World Alzheimer Report 2015, The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends - Executive Summary. 24.
- World Alzheimer Report 2013, Journey of Caring: An Analysis of Long-Term Care for Dementia. 92.
- Cheignon C.; Tomas M.; Bonnefont-Rousselot D.; Faller P.; Hureau C.; Collin F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L.-K.; Chao S.-P.; Hu C.-J. Clinical Trials of New Drugs for Alzheimer Disease. J. Biomed. Sci. 2020, 27, 18. 10.1186/s12929-019-0609-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca M. F. M.; Di Natale G.; Milardi D.; Pappalardo G. Tau/Aβ chimera peptides: A Thioflavin-T and MALDI-TOF study of Aβ amyloidosis in the presence of Cu(II) or Zn(II) ions and total lipid brain extract (TLBE) vesicles. of Aβ Amyloidosis in the Presence of Cu(II) or Zn(II) Ions and Total Lipid Brain Extract (TLBE) Vesicles. Chem. Phys. Lipids 2021, 237, 105085. 10.1016/j.chemphyslip.2021.105085. [DOI] [PubMed] [Google Scholar]
- Sciacca M. F. M.; Naletova I.; Giuffrida M. L.; Attanasio F. Semax, a Synthetic Regulatory Peptide, Affects Copper-Induced Abeta Aggregation and Amyloid Formation in Artificial Membrane Models. ACS Chem. Neurosci. 2022, 13, 486–496. 10.1021/acschemneuro.1c00707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciacca M. F.; Romanucci V.; Zarrelli A.; Monaco I.; Lolicato F.; Spinella N.; Galati C.; Grasso G.; D’Urso L.; Romeo M.; Diomede L.; Salmona M.; Bongiorno C.; Di Fabio G.; La Rosa C.; Milardi D. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. 10.1021/acschemneuro.7b00110. [DOI] [PubMed] [Google Scholar]
- Velander P.; Wu L.; Henderson F.; Zhang S.; Bevan D. R.; Xu B. Natural Product-Based Amyloid Inhibitors. Biochem. Pharmacol. 2017, 139, 40–55. 10.1016/j.bcp.2017.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Viñuales S.; Ahmed R.; Sciacca M. F.; Lanza V.; Giuffrida M. L.; Zimbone S.; Romanucci V.; Zarrelli A.; Bongiorno C.; Spinella N.; Galati C.; Di Fabio G.; Melacini G.; Milardi D. Trehalose Conjugates of Silybin as Prodrugs for Targeting Toxic Aβ Aggregates. ACS Chem. Neurosci. 2020, 11, 2566–2576. 10.1021/acschemneuro.0c00232. [DOI] [PubMed] [Google Scholar]
- Walsh D. M.; Townsend M.; Podlisny M. B.; Shankar G. M.; Fadeeva J. V.; Agnaf O. E.; Hartley D. M.; Selkoe D. J. Certain Inhibitors of Synthetic Amyloid -Peptide (A ) Fibrillogenesis Block Oligomerization of Natural A and Thereby Rescue Long-Term Potentiation. J. Neurosci. 2005, 25, 2455–2462. 10.1523/JNEUROSCI.4391-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minati L.; Edginton T.; Grazia Bruzzone M.; Giaccone G. Reviews: Current Concepts in Alzheimer’s Disease: A Multidisciplinary Review. J. Alzheimers Dis. Dementiasr 2009, 24, 95–121. 10.1177/1533317508328602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundke-Iqbal I.; Iqbal K.; Tung Y. C.; Quinlan M.; Wisniewski H. M.; Binder L. I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau (Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. 1986, 83, 4913–4917. 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner G. G.; Wong C. W. Alzheimer’s Disease: Initial Report of the Purification and Characterization of a Novel Cerebrovascular Amyloid Protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Lashuel H. A. Rethinking protein aggregation and drug discovery in neurodegenerative diseases: Why we need to embrace complexity?. Curr. Opin. Chem. Biol. 2021, 64, 67–75. 10.1016/j.cbpa.2021.05.006. [DOI] [PubMed] [Google Scholar]
- Lutz B. M.; Peng J. Deep Profiling of the Aggregated Proteome in Alzheimer’s Disease: From Pathology to Disease Mechanisms. Proteomes 2018, 6, 46. 10.3390/proteomes6040046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao L.; Cheng D.; Wang J.; Duong D. M.; Losik T. G.; Gearing M.; Rees H. D.; Lah J. J.; Levey A. I.; Peng J. Proteomic Characterization of Postmortem Amyloid Plaques Isolated by Laser Capture Microdissection. J. Biol. Chem. 2004, 279, 37061–37068. 10.1074/jbc.M403672200. [DOI] [PubMed] [Google Scholar]
- Jakob-Roetne R.; Jacobsen H. Alzheimer’s Disease: From Pathology to Therapeutic Approaches. Angew. Chem., Int. Ed. 2009, 48, 3030–3059. 10.1002/anie.200802808. [DOI] [PubMed] [Google Scholar]
- Kummer M. P.; Heneka M. T. Truncated and modified amyloid-beta species. Ther. 2014, 6, 28. 10.1186/alzrt258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beher D.; Wrigley J. D. J.; Owens A. P.; Shearman M. S. Generation of C-Terminally Truncated Amyloid-β Peptides Is Dependent on γ-Secretase Activity. J. Neurochem. 2002, 82, 563–575. 10.1046/j.1471-4159.2002.00985.x. [DOI] [PubMed] [Google Scholar]
- Portelius E.; Price E.; Brinkmalm G.; Stiteler M.; Olsson M.; Persson R.; Westman-Brinkmalm A.; Zetterberg H.; Simon A. J.; Blennow K. A Novel Pathway for Amyloid Precursor Protein Processing. Neurobiol. Aging 2011, 32, 1090–1098. 10.1016/j.neurobiolaging.2009.06.002. [DOI] [PubMed] [Google Scholar]
- Ding F.; Borreguero J. M.; Buldyrey S. V.; Stanley H. E.; Dokholyan N. V. Mechanism for the α-Helix to β-Hairpin Transition. Proteins Struct. Funct. Genet. 2003, 53, 220–228. 10.1002/prot.10468. [DOI] [PubMed] [Google Scholar]
- Mett J.; Lauer A. A.; Janitschke D.; Griebsch L. V.; Theiss E. L.; Grimm H. S.; Koivisto H.; Tanila H.; Hartmann T.; Grimm M. O. W. Medium-Chain Length Fatty Acids Enhance Aβ Degradation by Affecting Insulin-Degrading Enzyme. Cells 2021, 10, 2941. 10.3390/cells10112941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forloni G.; Artuso V.; La Vitola P. L.; Balducci C. Oligomeropathies and Pathogenesis of Alzheimer and Parkinson’s Diseases. Mov. Disord. 2016, 31, 771–781. 10.1002/mds.26624. [DOI] [PubMed] [Google Scholar]
- Deshpande A.; Mina E.; Glabe C.; Busciglio J. Different Conformations of Amyloid β Induce Neurotoxicity by Distinct Mechanisms in Human Cortical Neurons. J. Neurosci. 2006, 26, 6011–6018. 10.1523/JNEUROSCI.1189-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R.; Head E.; Thompson J. L.; McIntire T. M.; Milton S. C.; Cotman C. W.; Glabe C. G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Glabe C. Common Mechanisms of Amyloid Oligomer Pathogenesis in Degenerative Disease. Neurobiol. Aging 2006, 27, 570–575. 10.1016/j.neurobiolaging.2005.04.017. [DOI] [PubMed] [Google Scholar]
- Nguyen P. H.; Ramamoorthy A.; Sahoo B. R.; Zheng J.; Faller P.; Straub J. E.; Dominguez L.; Shea J.-E.; Dokholyan N. V.; De Simone A.; Ma B.; Nussinov R.; Najafi S.; Ngo S. T.; Loquet A.; Chiricotto M.; Ganguly P.; McCarty J.; Li M. S.; Hall C.; Wang Y.; Miller Y.; Melchionna S.; Habenstein B.; Timr S.; Chen J.; Hnath B.; Strodel B.; Kayed R.; Lesné S.; Wei G.; Sterpone F.; Doig A. J.; Derreumaux P. Amyloid Oligomers: A Joint Experimental/Computational Perspective on Alzheimer’s Disease, Parkinson’s Disease, Type II Diabetes, and Amyotrophic Lateral Sclerosis. Chem. Rev. 2021, 121, 2545–2647. 10.1021/acs.chemrev.0c01122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan G. J. Transient Disorder along Pathways to Amyloid. Biophys. Chem. 2022, 281, 106711. 10.1016/j.bpc.2021.106711. [DOI] [PubMed] [Google Scholar]
- Fatafta H.; Kav B.; Bundschuh B. F.; Loschwitz J.; Strodel B. Disorder-to-Order Transition of the Amyloid-β Peptide upon Lipid Binding. Biophys. Chem. 2022, 280, 106700. 10.1016/j.bpc.2021.106700. [DOI] [PubMed] [Google Scholar]
- Kotler S. A.; Walsh P.; Brender J. R.; Ramamoorthy A. Differences between Amyloid-β Aggregation in Solution and on the Membrane: Insights into Elucidation of the Mechanistic Details of Alzheimer’s Disease. Chem. Soc. Rev. 2014, 43, 6692–6700. 10.1039/C3CS60431D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida M. L.; Caraci F.; Pignataro B.; Cataldo S.; De Bona P. D.; Bruno V.; Molinaro G.; Pappalardo G.; Messina A.; Palmigiano A.; Garozzo D.; Nicoletti F.; Rizzarelli E.; Copani A. β-Amyloid Monomers Are Neuroprotective. J. Neurosci. 2009, 29, 10582–10587. 10.1523/JNEUROSCI.1736-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H.; Shin H.; Hong S. H.; Kim Y. Physiological Roles of Monomeric Amyloid-β and Implications for Alzheimer’s Disease Therapeutics. Exp. Neurobiol. 2022, 31, 65–88. 10.5607/en22004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Lu J. G.; Siddu A.; Wernig M.; Südhof T. C. Synaptogenic Effect of APP-Swedish Mutation in Familial Alzheimer’s Disease. Sci. Transl. Med. 2022, 14, eabn9380 10.1126/scitranslmed.abn9380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Liu Q.; Sun X.; Yu J.; Zhao D.; Yu Y.; Luo J.; Hu J.; Yu Z.; Zhao Y.; Li Y. Fibrillar Seeds Alleviate Amyloid-β Cytotoxicity by Omitting Formation of Higher-Molecular-Weight Oligomersr. Biochem. Biophys. Res. Commun. 2013, 439, 321–326. 10.1016/j.bbrc.2013.08.088. [DOI] [PubMed] [Google Scholar]
- Lee S. J. C.; Nam E.; Lee H. J.; Savelieff M. G.; Lim M. H. Towards an Understanding of Amyloid-β Oligomers: Characterization, Toxicity Mechanisms, and Inhibitors. Chem. Soc. Rev. 2017, 46, 310–323. 10.1039/C6CS00731G. [DOI] [PubMed] [Google Scholar]
- Williams A. D.; Shivaprasad S.; Wetzel R. Alanine Scarnning Mutagenesis of Abeta(1-40) Amyloid Fibril Stability. J. Mol. Biol. 2006, 357, 1283–1294. 10.1016/j.jmb.2006.01.041. [DOI] [PubMed] [Google Scholar]
- Zheng X.; Wu C.; Liu D.; Li H.; Bitan G.; Shea J.-E.; Bowers M. T. Mechanism of C-Terminal Fragments of Amyloid β-Protein as Aβ Inhibitors: Do C-Terminal Interactions Play a Key Role in Their Inhibitory Activity?. J. Phys. Chem. B 2016, 120, 1615–1623. 10.1021/acs.jpcb.5b08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgourakis N. G.; Yan Y.; McCallum S.; Wang C.; Garcia A. E. The Alzheimer’s Peptides Aβ40 and 42 Adopt Distinct Conformations in Water: A Combined MD / NMR Study. J. Mol. Biol. 2007, 368, 1448–1457. 10.1016/j.jmb.2007.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.; Xu T.; Yan Y.; Zhou Y.; Jiang Y.; Melcher K.; Xu H. E. Amyloid beta: structure, biology and structure-based therapeutic development. Sin. 2017, 38, 1205–1235. 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos N.; Suelves N.; Perrin F.; Vadukul D. M.; Vrancx C.; Constantinescu S. N.; Kienlen-Campard P. Structural Determinant of β-Amyloid Formation: From Transmembrane Protein Dimerization to β-Amyloid Aggregates. Biomedicines 2022, 10, 2753. 10.3390/biomedicines10112753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R.; McAllister C.; Lyubchenko Y.; Sierks M. R. Proteolytic Antibody Light Chains Alter β-Amyloid Aggregation and Prevent Cytotoxicity. Biochemistry 2004, 43, 9999–10007. 10.1021/bi0492354. [DOI] [PubMed] [Google Scholar]
- Du X.; Wang L.; Wang Y.; Andreasen M.; Zhan D.; Feng Y.; Li M.; Zhao M.; Otzen D.; Xue D.; Yang Y.; Liu R. Aβ 1-16 Can Aggregate and Induce the Production of Reactive Oxygen Species, Nitric Oxide, and Inflammatory Cytokines. J. Alzheimers Dis. 2011, 27, 401–413. 10.3233/JAD-2011-110476. [DOI] [PubMed] [Google Scholar]
- Brännström K.; Öhman A.; Nilsson L.; Pihl M.; Sandblad L.; Olofsson A. The N-Terminal Region of Amyloid β Controls the Aggregation Rate and Fibril Stability at Low PH Through a Gain of Function Mechanism. J. Am. Chem. Soc. 2014, 136, 10956–10964. 10.1021/ja503535m. [DOI] [PubMed] [Google Scholar]
- Söldner C. A.; Sticht H.; Horn A. H. C. Role of the N-Terminus for the Stability of an Amyloid-β Fibril with Three-Fold Symmetry. PLOS ONE 2017, 12, e0186347 10.1371/journal.pone.0189238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancolio K.; Dumanchin C.; Barelli H.; Warter J. M.; Brice A.; Campion D.; Frébourg T.; Checler F. Unusual Phenotypic Alteration of β Amyloid Precursor Protein (ΒAPP) Maturation by a New Val-715 → Met ΒAPP-770 Mutation Responsible for Probable Early-Onset Alzheimer’s Disease. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 4119–4124. 10.1073/pnas.96.7.4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergeant N.; Bombois S.; Ghestem A.; Drobecq H.; Kostanjevecki V.; Missiaen C.; Wattez A.; David J.-P.; Vanmechelen E.; Sergheraert C.; Delacourte A. Truncated Beta-Amyloid Peptide Species in Pre-Clinical Alzheimer’s Disease as New Targets for the Vaccination Approach. J. Neurochem. 2003, 85, 1581–1591. 10.1046/j.1471-4159.2003.01818.x. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Overman M. J.; Cotman C. W. Amino-Terminal Deletions Enhance Aggregation of Beta-Amyloid Peptides in Vitro. J. Biol. Chem. 1995, 270, 23895–23898. 10.1074/jbc.270.41.23895. [DOI] [PubMed] [Google Scholar]
- Sahoo B. R.; Panda P. K.; Liang W.; Tang W.-J.; Ahuja R.; Ramamoorthy A. Degradation of Alzheimer’s Amyloid-β by a Catalytically Inactive Insulin-Degrading Enzyme. J. Mol. Biol. 2021, 433, 166993. 10.1016/j.jmb.2021.166993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunys J.; Valverde A.; Checler F. Are N- and C-Terminally Truncated Aβ Species Key Pathological Triggers in Alzheimer’s Disease?. J. Biol. Chem. 2018, 293, 15419–15428. 10.1074/jbc.R118.003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedin F.; Kandel N.; Tatulian S. A. Effects of Aβ-Derived Peptide Fragments on Fibrillogenesis of Aβ. Sci. Rep. 2021, 11, 19262. 10.1038/s41598-021-98644-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viet M. H.; Ngo S. T.; Lam N. S.; Li M. S. Inhibition of Aggregation of Amyloid Peptides by Beta-Sheet Breaker Peptides and Their Binding Affinity. J. Phys. Chem. B 2011, 115, 7433–7446. 10.1021/jp1116728. [DOI] [PubMed] [Google Scholar]
- Bartolini M.; Andrisano V. Strategies for the Inhibition of Protein Aggregation in Human Diseases. ChemBioChem 2010, 11, 1018–1035. 10.1002/cbic.200900666. [DOI] [PubMed] [Google Scholar]
- Brender J. R.; Ghosh A.; Kotler S. A.; Krishnamoorthy J.; Bera S.; Morris V.; Sil T. B.; Garai K.; Reif B.; Bhunia A.; Ramamoorthy A. Probing Transient Non-Native States in Amyloid Beta Fiber Elongation by NMR. Chem. Commun. 2019, 55, 4483–4486. 10.1039/C9CC01067J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapadia A.; Sharma K. K.; Maurya I. K.; Singh V.; Khullar M.; JainStructuralMechanistic R. Insights into the Inhibition of Amyloid-β Aggregation by Aβ39-42 Fragment Derived Synthetic Peptides. Eur. J. Med. Chem. 2021, 212, 113126. 10.1016/j.ejmech.2020.113126. [DOI] [PubMed] [Google Scholar]
- Bansal S.; Maurya I. K.; Yadav N.; Thota C. K.; Kumar V.; Tikoo K.; Chauhan V. S.; Jain R. C-Terminal Fragment, Aβ32–37, Analogues Protect Against Aβ Aggregation-Induced Toxicity. ACS Chem. Neurosci. 2016, 7, 615–623. 10.1021/acschemneuro.6b00006. [DOI] [PubMed] [Google Scholar]
- Armiento V.; Spanopoulou A.; Kapurniotu A. Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration. Angew. Chem., Int. Ed. 2020, 59, 3372–3384. 10.1002/anie.201906908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal D.; Shuaib S.; Mann S.; Goyal B. Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci. 2017, 19, 55–80. 10.1021/acscombsci.6b00116. [DOI] [PubMed] [Google Scholar]
- Minicozzi V.; Chiaraluce R.; Consalvi V.; Giordano C.; Narcisi C.; Punzi P.; Rossi G. C.; Morante S. Computational and Experimental Studies on β-Sheet Breakers Targeting Aβ1-40 Fibrils. J. Biol. Chem. 2014, 289, 11242–11252. 10.1074/jbc.M113.537472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjernberg L. O.; Callaway D. J. E.; Tjernberg A.; Hahne S.; Lilliehöök C.; Terenius L.; Thyberg J.; Nordstedt C. A Molecular Model of Alzheimer Amyloid β-Peptide Fibril Formation*. J. Biol. Chem. 1999, 274, 12619–12625. 10.1074/jbc.274.18.12619. [DOI] [PubMed] [Google Scholar]
- Härd T.; Lendel C. Inhibition of Amyloid Formation. J. Mol. Biol. 2012, 421, 441–465. 10.1016/j.jmb.2011.12.062. [DOI] [PubMed] [Google Scholar]
- Gazit E. Mechanisms of Amyloid Fibril Self-Assembly and Inhibition. FEBS J. 2005, 272, 5971–5978. 10.1111/j.1742-4658.2005.05022.x. [DOI] [PubMed] [Google Scholar]
- Bunce S. J.; Wang Y.; Stewart K. L.; Ashcroft A. E.; Radford S. E.; Hall C. K.; Wilson A. J. Molecular Insights into the Surface-Catalyzed Secondary Nucleation of Amyloid-Β40 (Aβ40) by the Peptide Fragment Aβ16–22. Sci. Adv. 2019, 5, eaav8216 10.1126/sciadv.aav8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J.; Igarashi A.; Kamata M.; Nakagawa H. Angiotensin-converting Enzyme Degrades Alzheimer Amyloid β-Peptide (Aβ); Retards Aβ Aggregation, Deposition, Fibril Formation; and Inhibits Cytotoxicity. J. Biol. Chem. 2001, 276, 47863–47868. 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- Lowe T. L.; Strzelec A.; Kiessling L. L.; Murphy R. M. Structure-Function Relationships for Inhibitors of Beta-Amyloid Toxicity Containing the Recognition Sequence KLVFF. Biochemistry 2001, 40, 7882–7889. 10.1021/bi002734u. [DOI] [PubMed] [Google Scholar]
- Sciacca M. F.; Di Natale G.; Tosto R.; Milardi D.; Pappalardo G. Tau/Aβ Chimera Peptides: Evaluating the Dual Function of Metal Coordination and Membrane Interaction in One Sequence. J. Inorg. Biochem. 2020, 205, 110996. 10.1016/j.jinorgbio.2020.110996. [DOI] [PubMed] [Google Scholar]
- Watanabe K.; Nakamura K.; Akikusa S.; Okada T.; Kodaka M.; Konakahara T.; Okuno H. Inhibitors of Fibril Formation and Cytotoxicity of Beta-Amyloid Peptide Composed of KLVFF Recognition Element and Flexible Hydrophilic Disrupting Element. Biochem. Biophys. Res. Commun. 2002, 290, 121–124. 10.1006/bbrc.2001.6191. [DOI] [PubMed] [Google Scholar]
- Liao M. Q.; Tzeng Y. J.; Chang L. Y. X.; Huang H. B.; Lin T. H.; Chyan C. L.; Chen Y. C. The Correlation between Neurotoxicity, Aggregative Ability and Secondary Structure Studied by Sequence Truncated Abeta Peptides. FEBS Lett. 2007, 581, 1161–1165. 10.1016/j.febslet.2007.02.026. [DOI] [PubMed] [Google Scholar]
- Huang X.; Atwood C. S.; Moir R. D.; Hartshorn M. A.; Tanzi R. E.; Bush A. I. Trace Metal Contamination Initiates the Apparent Auto-Aggregation, Amyloidosis, and Oligomerization of Alzheimer’s Aβ Peptides. JBIC J. Biol. Inorg. Chem. 2004, 9, 954–960. 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- Atwood C. S.; Moir R. D.; Huang X.; Scarpa R. C.; Bacarra N. M. E.; Romano D. M.; Hartshorn M. A.; Tanzi R. E.; Bush A. I. Dramatic Aggregation of Alzheimer Aβ by Cu(II) Is Induced by Conditions Representing Physiological Acidosis*. J. Biol. Chem. 1998, 273, 12817–12826. 10.1074/jbc.273.21.12817. [DOI] [PubMed] [Google Scholar]
- Yoshiike Y.; Tanemura K.; Murayama O.; Akagi T.; Murayama M.; Sato S.; Sun X.; Tanaka N.; Takashima A. New Insights on How Metals Disrupt Amyloid β-Aggregation and Their Effects on Amyloid-β Cytotoxicity*. J. Biol. Chem. 2001, 276, 32293–32299. 10.1074/jbc.M010706200. [DOI] [PubMed] [Google Scholar]
- Sarell C. J.; Wilkinson S. R.; Viles J. H. Substoichiometric Levels of Cu2+ Ions Accelerate the Kinetics of Fiber Formation and Promote Cell Toxicity of Amyloid-β from Alzheimer Disease*. J. Biol. Chem. 2010, 285, 41533–41540. 10.1074/jbc.M110.171355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen J. T.; Østergaard J.; Rozlosnik N.; Gammelgaard B.; Heegaard N. H. H. Cu(II) Mediates Kinetically Distinct, Non-Amyloidogenic Aggregation of Amyloid-β Peptides*. J. Biol. Chem. 2011, 286, 26952–26963. 10.1074/jbc.M111.220863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser P. E.; Nguyen J. T.; Surewicz W. K.; Kirschner D. A. PH-Dependent Structural Transitions of Alzheimer Amyloid Peptides. Biophys. J. 1991, 60, 1190–1201. 10.1016/s0006-3495(91)82154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike C. J.; Burdick D.; Walencewicz A. J.; Glabe C. G.; Cotman C. W. Neurodegeneration Induced by Beta-Amyloid Peptides in Vitro: The Role of Peptide Assembly State. J. Neurosci. Off. J. Soc. Neurosci. 1993, 13, 1676–1687. 10.1523/jneurosci.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N.; Cheung T. T.; Cai X. D.; Odaka A.; Otvos L.; Eckman C.; Golde T. E.; Younkin S. G. An Increased Percentage of Long Amyloid Beta Protein Secreted by Familial Amyloid Beta Protein Precursor (Beta APP717). Mutants. Science 1994, 264, 1336–1340. 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- Vigo-Pelfrey C.; Lee D.; Keim P.; Lieberburg I.; Schenk D. B. Characterization of Beta-Amyloid Peptide from Human Cerebrospinal Fluid. J. Neurochem. 1993, 61, 1965–1968. 10.1111/j.1471-4159.1993.tb09841.x. [DOI] [PubMed] [Google Scholar]
- Dovey H. F.; Suomensaari-Chrysler S.; Lieberburg I.; Sinha S.; Keim P. S. Cells with a Familial Alzheimer’s Disease Mutation Produce Authentic Beta-Peptide. Neuroreport 1993, 4, 1039–1042. 10.1097/00001756-199308000-00011. [DOI] [PubMed] [Google Scholar]
- Seubert P.; Vigo-Pelfrey C.; Esch F.; Lee M.; Dovey H.; Davis D.; Sinha S.; Schiossmacher M.; Whaley J.; Swindlehurst C.; McCormack R.; Wolfert R.; Selkoe D.; Lieberburg I.; Schenk D. Isolation and Quantification of Soluble Alzheimer’s Beta-Peptide from Biological Fluids. Nature 1992, 359, 325–327. 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- Ştefănescu R.; Stanciu G. D.; Luca A.; Caba I. C.; Tamba B. I.; Mihai C. T. Contributions of Mass Spectrometry to the Identification of Low Molecular Weight Molecules Able to Reduce the Toxicity of Amyloid-β Peptide to Cell Cultures and Transgenic Mouse Models of Alzheimer’s Disease. Molecules 2019, 24, 1167. 10.3390/molecules24061167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor N. E.; Moss M. A.; Hestekin C. N. Unraveling the Early Events of Amyloid-β Protein (Aβ) Aggregation: Techniques for the Determination of Aβ Aggregate Size. Int. J. Mol. Sci. 2012, 13, 3038–3072. 10.3390/ijms13033038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. S.-H.; Whitehead S. N.; Yeung K. K.-C. Detection of Amyloid Beta (Aβ) Oligomeric Composition Using Matrix-Assisted Laser Desorption Ionization Mass Spectrometry (MALDI MS). J. Am. Soc. Mass Spectrom. 2018, 29, 786–795. 10.1007/s13361-018-1896-z. [DOI] [PubMed] [Google Scholar]
- Kheterpal I.; Zhou S.; Cook K. D.; Wetzel R. Abeta Amyloid Fibrils Possess a Core Structure Highly Resistant to Hydrogen Exchange. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13597–13601. 10.1073/pnas.250288897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung S. Y.; Keyes C.; Duhamel J.; Chen P. Concentration Effect on the Aggregation of a Self-Assembling Oligopeptide. Biophys. J. 2003, 85, 537–548. 10.1016/s0006-3495(03)74498-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuffrida M. L.; Tomasello M.; Pandini G.; Caraci F.; Battaglia G.; Busceti C.; Di Pietro P.; Pappalardo G.; Attanasio F.; Chiechio S.; Bagnoli S.; Nacmias B.; Sorbi S.; Vigneri R.; Rizzarelli E.; Nicoletti F.; Copani A. Monomeric SS-Amyloid Interacts with Type-1 Insulin-like Growth Factor Receptors to Provide Energy Supply to Neurons. Front. Cell. Neurosci. 2015, 9, 00297. 10.3389/fncel.2015.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M. P.; Barlow A. K.; Chromy B. A.; Edwards C.; Freed R.; Liosatos M.; Morgan T. E.; Rozovsky I.; Trommer B.; Viola K. L.; Wals P.; Zhang C.; Finch C. E.; Krafft G. A.; Klein W. L. Diffusible, Nonfibrillar Ligands Derived from Aβ1–42 Are Potent Central Nervous System Neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 1998, 95, 6448–6453. 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.